

Abstract
Background
Currently, meat cut traits are integrated in pig breeding objectives to gain extra profit. However, little is known about the heritability of meat cut proportions (MCP) and their correlations with other traits. The aims of this study were to assess the heritability and genetic correlation of MCP with carcass and meat quality traits using single nucleotide polymorphism chips and conduct a genome-wide association study (GWAS) to identify candidate genes for MCP.
Results
Seventeen MCP, 12 carcass, and seven meat quality traits were measured in 2012 pigs from four populations (Landrace; Yorkshire; Landrace and Yorkshire hybrid pigs; Duroc, and Landrace and Yorkshire hybrid pigs). Estimates of the heritability for MCP ranged from 0.10 to 0.55, with most estimates being moderate to high and highly consistent across populations. In the combined population, the heritability estimates for the proportions of scapula bone, loin, back fat, leg bones, and boneless picnic shoulder were 0.44 ± 0.04, 0.36 ± 0.04, 0.44 ± 0.04, 0.38 ± 0.04, and 0.39 ± 0.04, respectively. Proportion of middle cuts was genetically significantly positively correlated with intramuscular fat content and backfat depth. Proportion of ribs was genetically positively correlated with carcass oblique length and straight length (0.35 ± 0.08 to 0.45 ± 0.07) and negatively correlated with backfat depth (− 0.26 ± 0.10 to − 0.45 ± 0.10). However, weak or nonsignificant genetic correlations were observed between most MCP, indicating their independence. Twenty-eight quantitative trait loci (QTL) for MCP were detected by GWAS, and 24 new candidate genes related to MCP were identified, which are involved with growth, height, and skeletal development. Most importantly, we found that the development of the bones in different parts of the body may be regulated by different genes, among which HMGA1 may be the strongest candidate gene affecting forelimb bone development. Moreover, as previously shown, VRTN is a causal gene affecting vertebra number, and BMP2 may be the strongest candidate gene affecting hindlimb bone development.
Conclusions
Our results indicate that breeding programs for MCP have the potential to enhance carcass composition by increasing the proportion of expensive cuts and decreasing the proportion of inexpensive cuts. Since MCP are post-slaughter traits, the QTL and candidate genes related to these traits can be used for marker-assisted and genomic selection.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12711-023-00817-y.
Background
For a long time, the traditional pork sales model in China consisted in cutting the carcass into pieces following customers’ preferences [1, 2]. However, in recent years, many breeding firms have implemented centralized slaughtering, standardized quarantine, carcass cutting, and cold chain transportation to control the spread of African swine fever and other epidemic diseases. In addition, fast-paced and high-quality lifestyles and high-standard consumption concepts have established new market requirements [3]. As a result, precut and prepackaged cold meat is becoming increasingly popular. The prices of different cuts vary greatly, with up to two to three times differences between the highest and lowest price products. Notably, Qin [4] calculated the sum of sales of carcass cuts based on the average selling price of meat cuts and compared it with the total sale price of carcasses. He found that the sale of meat cuts represents at least 19% more economic revenue than the sale of carcasses, without considering costs such as labor and the depreciation of machines. As a result, breeders and scientists have become interested in investigating breeding programs to improve meat cut proportions (MCP) and in the genetic mechanisms underlying these traits.
As early as the 1970s, Whiteker et al. [5, 6] had already evaluated the weight and yield of different meat cuts. More recently, Overholt et al. [7] compared the differences in pork carcass cuts between barrows and gilts. Álvarez-Rodríguez and Teixeira [8] investigated the effect of slaughter weight and sex on carcass cuts based on three sequential target slaughter body weights of 17, 32, and 79 kg in Bisaro pigs and found that slaughter weight had a stronger impact on carcass cuts than sex. In our previous study, we observed that carcass weight, sex, and breed composition have significant effects on the weight and proportion of most meat cuts [9]. In addition, the coefficient of variation for MCP ranged from 4.2 to 25.3%, which indicates substantial heterogeneity between individuals [9]. Selecting elite pigs, including those with a higher proportion of high-price meat cuts, would benefit the pig industry. To implement a breeding program for improving MCP, it is necessary to first evaluate the genetic parameters and investigate the genetic architecture of these traits. Several studies have reported genetic parameters for meat cuts and found heritability estimates for carcass traits that were moderate to high [10–12]. However, these studies mainly focused on meat cuts under the standards of the United Nations Economic Commission for Europe (UNECE) and The Meat Buyer’s Guide from the North American Meat Processors Association (NAMP). In this study, the cut products followed the standards of Fresh and Frozen Pork and Pig By-Products (GB/T 9959) from the Standardization Administration of China, and included those reported in previous studies [10–12], as well as some novel meat cuts such as arm bones, scapula bone, chine bones, leg bones, and tail and pelvis bone. Between 2017 and 2018, the wholesale price for these bone meat cuts ranged from 11.5 to 17.7 yuan/kg, which is slightly lower than the wholesale price for the whole carcass (ranging from 14.5 to 19.4 yuan/kg), which is popular with consumers in China. Furthermore, the heritability estimates previously reported for meat cuts were mainly for commercial pigs, but estimates are also needed for purebred pigs.
In this study, we compared the heritability estimates for meat cuts between different breeds and crossbred populations and investigated their genetic correlations with carcass and meat quality traits, which will benefit decision-making in breeding programs for meat cut traits. Finally, we also performed a genome-wide association study (GWAS) to identify quantitative trait loci (QTL) for meat cut traits and, for the first time, the associated genes.
Methods
Ethics statement
All procedures involving animals followed the guidelines for the care and use of experimental animals approved by the State Council of the People’s Republic of China. The ethics committee of Jiangxi Agricultural University specifically approved this study.
Animals and phenotype data
In total, 2012 pigs from Muyuan Food Co., Ltd. (Henan, China) were randomly sampled for the meat cut evaluation experiment, as described in Lei et al. [9]. In brief, four populations were used: 265 Landrace (LD, 95 sows and 170 barrows), 698 Yorkshire (YK, 435 sows and 263 barrows), 689 Landrace and Yorkshire hybrid (LY, 402 sows and 287 barrows), and 258 Duroc × Landrace × Yorkshire hybrid (DLY, 115 sows and 143 barrows). All pigs were raised under consistent feeding and nutritional conditions and were given the same commercial diets and access to water ad libitum, which were formulated to meet the recommendations of the Chinese National Feeding Standard (GB, 2004) at different growth phases. After 180 days of age, they were centrally slaughtered following standard pig slaughtering processes.
In the slaughterhouse, 12 carcass traits were measured, including carcass straight length (CSL), carcass oblique length (COL), thoracic length (THL), lumbar length (LUL), thoracic number (THN), lumbar number (LUN), single lumbar length (SLUL), shoulder backfat depth (SBD), 6th to 7th rib backfat depth (RBD), waist backfat depth (WBD), hip backfat depth (HBD), and mean of backfat depth (MBD). These traits were measured on the complete carcass, which was hung vertically by fixing the hind legs. Backfat depth refers to the thickness of the subcutaneous fat on the back of the carcass. After 24 h of acid excretion, the carcass was divided into three primal cuts (SC: shoulder cut, MC: middle cut, LC: leg cut), and 14 commercial meat cuts (BBS: boneless Boston shoulder, BPS: boneless picnic shoulder, FR: front ribs, AB: arm bones, SB: scapula bones, LO: loin, BE: belly, ribs, CB: chine bones, BF: backfat, BL: boneless leg, TL: tenderloin, LB: leg bones, TPB: tail and pelvis bone). The three primal and 14 commercial meat cuts obtained by dissecting the carcass in accordance with the processing standard (GB/T 9959) are called carcass cuts or meat cuts. Details of the carcass cutting process and the appearance of the meat cuts are described in our previous study [9]. Each cut was carefully weighed and measured by our investigators and the MCP were calculated by dividing the weight of the meat cut by the weight of the carcass. Evaluation of the pork loin meat quality followed the same method as outlined in [13, 14]. At 1-d postmortem, seven meat quality traits, namely, pH, L* for lightness (LIL), a* for redness (REA), b* for yellowness (YEB), the proportion of fat areas in the image (PFAI), visual marbling score (VMS) and loin muscle area (LMA), were measured on the longissimus dorsi muscle between the 11th rib and the first lumbar vertebra.
Genotyping
As pedigree information was not available for most individuals, all individuals were genotyped. Genomic DNA was isolated from the muscle tissue of each animal using the routine phenol/chloroform extraction method. The CC1 PorcineSNP50 BeadChip (51,368 SNPs) was used to genotype individuals in accordance with the manufacturer’s protocol. Our previous study [13] provided information on marker density and accuracy of the CC1 PorcineSNP50 BeadChip. All samples had a call rate higher than 90%. SNPs with a call rate lower than 95%, a minor allele frequency (MAF) lower than 5%, or that were in Hardy–Weinberg disequilibrium (P < 10E−5) were filtered out using the PLINK (v1.90b6.24) software [15]. In total, 2012 animals and 40,016 SNPs passed quality control and were included for further statistical analysis.
Model and estimation of variance components
The following two-trait animal model was used in the analyses:
where and are the vectors of the phenotypes for the two traits to be analyzed; and are the vectors of the fixed effects of traits 1 and 2, respectively, including sex, population (LD, YK, LY, and DLY), and slaughter batch (two slaughter batches, containing 1054 and 958 pigs respectively); and are the vectors of the random additive genetic effects, which were assumed to follow the multivariate normal distribution where is the kinship matrix calculated from the genome-wide SNP genotypes, and are the additive genetic variance for traits 1 and 2, respectively, and is the additive genetic covariance; and 1 and 2 are the vectors of residuals, which were assumed to follow the multivariate normal distribution with being an identity matrix; and are the variance of the residuals for traits 1 and 2, respectively, and is the number of individuals phenotyped; and are the corresponding incidence matrices for the effects in and respectively.
The variance–covariance matrix of (the vector of phenotypes for a single-trait) can be expressed as:
The genomic relationship matrix () was constructed as in VanRaden [16]:
where is the matrix of centered SNP genotype dosages, with dimensions equal to the number of individuals and the number of SNPs, and is the frequency of the reference allele at the -th SNP. These allele frequencies were computed based on the method of Gengler et al. [17].
The heritability was then estimated as where is the phenotypic variance and the sum of and .
Genetic correlations were calculated as follows:
Heritabilities and genetic correlations for all traits were estimated by restricted maximum likelihood (REML) methodology using the GCTA software (version 1.93.1beta) [18]. Estimates of heritability for all traits in the combined population were confirmed using the REML method in the DMUAI module of the DMU software (version 6) [19]. As the sample sizes for the LD and DLY populations were small and the heritability estimates for these populations were very different from those for the YK and LY populations, we only estimated heritabilities for the YK, LY, and combined populations.
Genome-wide association analyses
Genome-wide association studies (GWAS) were performed using the GEMMA software [20] and the following linear mixed model:
where is the vector of phenotypes; is the indicator matrix of the fixed effects, including sex, population, and slaughter batch; is the vector of the corresponding fixed effects; is the genotype indicator matrix; is the vector of SNP substitution effects; is the vector of the random polygenic effects, which was assumed to follow a multivariate normal distribution , where is the kinship matrix calculated from the genome-wide SNP genotypes; is the additive genetic variance; is the corresponding incidence matrix for the effects in ; is the vector of residual effects, which was assumed to follows a multivariate normal distribution , where is an identity matrix; is the variance of the residual; and is the number of individuals phenotyped.
For GWAS, statistical significance was set using the Bonferroni correction method. The genome-wide significance threshold was set at 0.05/N, with 1/N indicating a suggestive significance threshold [21, 22], where N is the number of SNPs (40,016) that passed quality control for GWAS, which resulted in p value thresholds of 1.25 × 10–6 and 2.50 × 10–5, respectively.
Results
Phenotypes and their heritabilities
Based on our previous results, carcass weight, sex, and breed composition had significant effects on the majority of the meat cut, meat quality, and carcass traits [9]. The coefficient of variation of these traits varied largely from 4.2% for the proportion of LC to 25.3% for the proportion of BF. Here, the heritability was estimated using the GCTA software after adjusting for the effects of carcass weight, sex, and breed composition for the YK, LY, and combined populations (Fig. 1 and Table 1). In addition, we used the DMU software to evaluate the heritability for each trait in the combined population and found that the results were similar to those obtained with the GCTA software (Fig. 1).
Fig. 1.

Estimates of heritabilities for all evaluated traits in the four populations. YK, LY and CP represent heritabilities for all traits estimated using the GCTA software in Yorkshire, Landrace Yorkshire and combined populations, respectively. CP_DMU represents heritabilities for all traits estimated using the DMU software in the combined population
Table 1.
Estimates of heritabilities with their standard error (SE) for carcass cut proportions, carcass traits and meat quality of the longissimus dorsi muscle in the LY, YK, and combined (CP) populations
| Trait name | Acronym | n | Estimates of heritability ± SE | ||
|---|---|---|---|---|---|
| LY | YK | CP | |||
| Number of animals evaluated | 689 | 698 | 2012 | ||
| Carcass cut proportions | |||||
| Primal cuts | |||||
| Shoulder cut | SC | 1959 | 0.12 ± 0.06 | 0.13 ± 0.06 | 0.14 ± 0.03 |
| Middle cut | MC | 1978 | 0.22 ± 0.07 | 0.29 ± 0.07 | 0.27 ± 0.04 |
| Leg cut | LC | 1964 | 0.19 ± 0.07 | 0.39 ± 0.07 | 0.31 ± 0.04 |
| Shoulder cuts | |||||
| Boneless Boston shoulder | BBS | 1964 | 0.63 ± 0.07 | 0.23 ± 0.07 | 0.33 ± 0.04 |
| Boneless picnic shoulder | BPS | 1966 | 0.37 ± 0.07 | 0.46 ± 0.07 | 0.39 ± 0.04 |
| Front ribs | FR | 1966 | 0.16 ± 0.07 | 0.19 ± 0.06 | 0.15 ± 0.04 |
| Arm bones | AB | 1967 | 0.33 ± 0.08 | 0.46 ± 0.07 | 0.38 ± 0.04 |
| Scapula bones | SB | 1965 | 0.59 ± 0.07 | 0.63 ± 0.07 | 0.44 ± 0.04 |
| Middle cuts | |||||
| Loin | LO | 1985 | 0.52 ± 0.07 | 0.42 ± 0.07 | 0.36 ± 0.04 |
| Belly | BE | 1993 | 0.30 ± 0.07 | 0.36 ± 0.07 | 0.32 ± 0.04 |
| Ribs | RI | 1974 | 0.31 ± 0.07 | 0.35 ± 0.07 | 0.37 ± 0.04 |
| Chine bones | CB | 1976 | 0.33 ± 0.08 | 0.17 ± 0.06 | 0.23 ± 0.04 |
| Back fat | BF | 1984 | 0.38 ± 0.08 | 0.45 ± 0.07 | 0.44 ± 0.04 |
| Leg cuts | |||||
| Boneless leg | BL | 1988 | 0.28 ± 0.08 | 0.40 ± 0.07 | 0.34 ± 0.04 |
| Tenderloin | TL | 1876 | 0.33 ± 0.08 | 0.40 ± 0.07 | 0.27 ± 0.04 |
| Leg bones | LB | 1988 | 0.40 ± 0.08 | 0.47 ± 0.07 | 0.38 ± 0.04 |
| Tail and pelvis bone | TPB | 1724 | 0.03 ± 0.06 | 0.13 ± 0.06 | 0.13 ± 0.04 |
| Carcass traits | |||||
| Carcass weight | CW | 1997 | 0.49 ± 0.08 | 0.45 ± 0.07 | 0.39 ± 0.05 |
| Half carcass weight | HCW | 1997 | 0.46 ± 0.07 | 0.47 ± 0.07 | 0.40 ± 0.04 |
| Carcass straight length | CSL | 2008 | 0.45 ± 0.07 | 0.47 ± 0.07 | 0.45 ± 0.04 |
| Carcass oblique length | COL | 2008 | 0.50 ± 0.07 | 0.48 ± 0.07 | 0.45 ± 0.04 |
| Thoracic length | THL | 2011 | 0.47 ± 0.07 | 0.44 ± 0.07 | 0.43 ± 0.04 |
| Lumbar length | LUL | 2010 | 0.07 ± 0.05 | 0.24 ± 0.07 | 0.18 ± 0.04 |
| Thoracic number | THN | 2012 | 0.48 ± 0.07 | 0.45 ± 0.07 | 0.48 ± 0.04 |
| Lumbar number | LUN | 2010 | 0.09 ± 0.06 | 0.12 ± 0.06 | 0.12 ± 0.03 |
| Single lumbar length | SLUL | 2010 | 0.12 ± 0.06 | 0.23 ± 0.06 | 0.24 ± 0.04 |
| Backfat depth | |||||
| Shoulder backfat depth | SBD | 2012 | 0.31 ± 0.08 | 0.23 ± 0.06 | 0.26 ± 0.04 |
| 6th_7th rib backfat depth | RBD | 2012 | 0.33 ± 0.08 | 0.33 ± 0.07 | 0.32 ± 0.04 |
| Waist backfat depth | WBD | 2012 | 0.32 ± 0.08 | 0.20 ± 0.06 | 0.23 ± 0.04 |
| Hip backfat depth | HBD | 2012 | 0.23 ± 0.07 | 0.25 ± 0.07 | 0.32 ± 0.04 |
| Mean of backfat depth | MBD | 2012 | 0.39 ± 0.08 | 0.41 ± 0.07 | 0.43 ± 0.04 |
| Meat quality | |||||
| pH | 2010 | 0.40 ± 0.08 | 0.29 ± 0.06 | 0.28 ± 0.04 | |
| Lightness, L* | LIL | 2000 | 0.40 ± 0.08 | 0.40 ± 0.07 | 0.34 ± 0.04 |
| Redness, a* | REA | 2000 | 0.50 ± 0.07 | 0.39 ± 0.07 | 0.34 ± 0.04 |
| Yellowness, b* | YEB | 2000 | 0.35 ± 0.07 | 0.31 ± 0.07 | 0.30 ± 0.04 |
| PFAI | 1705 | 0.17 ± 0.08 | 0.23 ± 0.07 | 0.20 ± 0.04 | |
| Visual marbling score | VMS | 1705 | 0.19 ± 0.08 | 0.27 ± 0.07 | 0.20 ± 0.04 |
| Loin muscle area | LMA | 1949 | 0.45 ± 0.07 | 0.35 ± 0.07 | 0.29 ± 0.04 |
LY Landrace Yorkshire, YK Yorkshire, CP combined population, PFAI proportion of fat areas in the image
The estimates of heritability (± SE) for MCP ranged from 0.13 ± 0.04 for TPB to 0.44 ± 0.04 for SB in the combined population (Fig. 1 and Table 1). The estimates of heritability for SC, FR, and TPB were low across all populations, and in the combined population they were equal to 0.14 ± 0.03, 0.15 ± 0.04, and 0.13 ± 0.04, respectively. The estimates of heritability for SB, LO, BF, LB, BPS, and BBS in the combined population were 0.44 ± 0.04, 0.36 ± 0.04, 0.44 ± 0.04, 0.38 ± 0.04, 0.39 ± 0.04, and 0.33 ± 0.04, respectively, which are among the highest estimates of heritability found in this study. Overall, the estimates of the heritability for most MCP were moderate to high, which indicates that these traits can be improved in swine breeding programs.
Genetic parameters for carcass traits and meat quality have been investigated for a long time [10–12]. Most studies have reported moderate to high heritability estimates for most carcass traits and low to moderate heritability estimates for meat quality traits. As expected, the heritability estimates for carcass traits ranged from 0.39 ± 0.05 for carcass weight in the combined population to 0.50 ± 0.07 for carcass oblique length in the LY population, and those for meat quality ranged from 0.20 ± 0.04 for VMS to 0.34 ± 0.04 for LIL in the combined population (Fig. 1 and Table 1). Our results confirm the findings of most of the genetic parameter estimates previously reported for carcass and meat quality traits.
Genetic correlations between MCP, meat quality, and carcass traits
In the combined population, we estimated the genetic correlations between MCP, meat quality, and carcass traits (Fig. 2 and [see Additional file 1: Table S1]). Among these, BF was positively correlated with BE, with a genetic correlation estimate of 0.40 ± 0.08, whereas BF and BE had negative correlation estimates with other meat cuts (except BBS, ranging from -0.26 ± 0.10 between BE and TL to − 0.67 ± 0.06 between BF and LB). In addition, BF and BE were genetically positively correlated with backfat depth (ranging from 0.49 ± 0.09 to 0.84 ± 0.04), PFAI (ranging from 0.36 ± 0.11 to 0.38 ± 0.13), and VMS (ranging from 0.32 ± 0.11 to 0.36 ± 0.12), suggesting that selection to increase the proportion of BE may increase backfat depth and the proportion of BF, which would be detrimental to carcass sales. In addition, BPS was positively genetically correlated with the proportion of RI (0.36 ± 0.08), but negatively correlated with TPB (− 0.34 ± 0.13), CB (− 0.44 ± 0.09), SB (− 0.56 ± 0.07), AB (− 0.24 ± 0.09), LO (− 0.44 ± 0.08), BBS (− 0.71 ± 0.07), and BF (− 0.20 ± 0.08) [Fig. 2 and (see Additional file 1: Table S1)]. Rib proportion was moderately to highly positively genetically correlated with COL (0.45 ± 0.07), CSL (0.35 ± 0.08), THL (0.50 ± 0.07), and THN (0.42 ± 0.08), and negatively correlated with backfat depth (ranging from − 0.26 ± 0.10 to − 0.45 ± 0.10). The proportion of LO was highly positively genetically correlated with LMA (0.51 ± 0.08) but negatively correlated with pH (− 0.34 ± 0.13). Moreover, TL was positively correlated with BL, with a genetic correlation estimate of 0.36 ± 0.10, but was negatively correlated with backfat depth (ranging from − 0.24 ± 0.11 between TL and WBD to − 0.53 ± 0.1 between TL and SBD). However, BL was genetically slightly unfavorably correlated with carcass traits (ranging from − 0.21 ± 0.11 to − 0.35 ± 0.08).
Fig. 2.

Estimates of genetic correlations of meat cut proportions with carcass and meat quality traits in the combined population
It is worth noting that positive genetic correlations were estimated among the bone carcass cuts of LB, FR, TPB, CB, SB, and AB (ranging from 0.20 ± 0.13 between TPB and LB to 0.69 ± 0.14 between TPB and CB) but negative genetic correlations were estimated for these traits with BF, BE, BPS, and BL [Fig. 2 and (see Additional file 1: Table S1)]. These bone carcass cuts were also estimated to be negatively genetically correlated with backfat depth (ranging from − 0.21 ± 0.09 between SB and WBD to − 0.66 ± 0.06 between LB and MBD), except for the positive genetic correlation estimate between TPB and SBD (0.35 ± 0.16). The results indicate that, in the pig, the growth and development of bones in different parts of the body are synchronized.
As shown in Fig. 2 and Additional file 1: Table S1, we found that the genetic correlations between most meat cut traits were low (correlations lower than 0.3) or not significant (p value > 0.05). In addition, the majority of the meat cut traits had low or nonsignificant genetic correlations with carcass and meat quality traits. This is consistent with previously reported results [9] of low or nonsignificant phenotypic correlations between meat cut, carcass, and meat quality traits (Fig. 2).
Genome-wide association analysis of carcass cut traits
Based on 40,016 SNPs and 2012 individuals, we identified 28 QTL that were associated with 15 carcass cut traits and reached the suggestive significance threshold (Table 2). There was no population stratification, as shown in the Q-Q plots in Additional file 2: Fig. S1. Among these QTL, six had a top SNP that exceeded the genome-wide significance threshold (P = 1.25 × 10–6).
Table 2.
Single nucleotide polymorphisms that are significantly associated with carcass cut proportions detected in the single-trait genome-wide association studies
| Traits | TOP | Chr | Pos (bp) | p value | Nearest gene | Dis (bp) | Candidate gene |
|---|---|---|---|---|---|---|---|
| BBS | rs1501030 | 15 | 44,344,760 | 1.72E−05 | TENM3 | Within | |
| BPS | rs0702610 | 7 | 97,615,897 | 9.33E−06 | VRTN | Within | |
| AB | rs0700767 | 7 | 30,100,555 | 1.37E−08 | GRM4 | 78,750 | HMGA1, NUDT3, GRM4 |
| AB | rs0501169 | 5 | 66,142,277 | 2.19E−06 | CCND2 | 27,706 | |
| AB | rs1705053 | 17 | 15,896,846 | 2.25E−05 | BMP2 | 135,651 | HAO1, BMP2 |
| SB | rs1501163 | 15 | 50,915,343 | 5.05E−06 | UNC5D | 112,675 | |
| SC | rs0702610 | 7 | 97,615,897 | 2.76E−06 | VRTN | Within | |
| SC | rs0702516 | 7 | 121,223,217 | 2.01E−05 | WARS1 | Within | DLK1 |
| SC | rs0201138 | 2 | 60,064,913 | 1.19E−05 | MAP1S | 35,048 | |
| LO | rs0501038 | 5 | 57,616,938 | 9.18E−06 | ERP27 | Within | |
| BE | rs1806217 | 18 | 10,455,423 | 1.20E−05 | UBN2 | 4168 | |
| BE | rs0200123 | 2 | 6,851,449 | 2.45E−05 | SLC25A45 | 168 | |
| RI | rs0702610 | 7 | 97,615,897 | 4.18E−16 | VRTN | Within | VRTN |
| CB | rs0702038 | 7 | 97,618,073 | 6.94E−08 | VRTN | Within | VRTN |
| CB | rs1402627 | 14 | 125,929,866 | 2.77E−06 | ATRNL1 | Within | |
| CB | rs1500252 | 15 | 9,945,936 | 1.24E−05 | LRP1B | Within | |
| CB | rs1705053 | 17 | 15,896,846 | 1.53E−05 | BMP2 | 135,651 | BMP2 |
| BF | rs1100173 | 11 | 9,429,262 | 8.14E−06 | KL | Within | |
| MC | rs0702610 | 7 | 97,615,897 | 4.47E−13 | VRTN | Within | |
| BL | rs1704784 | 17 | 2,382,922 | 7.70E−06 | SGCZ | Within | |
| TL | rs1303072 | 13 | 148,117,239 | 1.08E−05 | CD96 | Within | |
| TL | rs0602330 | 6 | 123,281,566 | 2.04E−05 | - | - | |
| LB | rs1705079 | 17 | 16,788,523 | 2.67E−14 | HAO1 | Within | BMP2, BMP2 |
| LB | rs0501167 | 5 | 66,103,958 | 5.84E−08 | CCND2 | Within | |
| LB | rs0700777 | 7 | 30,235,367 | 3.80E−06 | GRM4 | Within | HMGA1, NUDT3, GRM4 |
| LB | rs1300094 | 13 | 3,477,035 | 1.02E−05 | RFTN1 | Within | |
| LC | rs1603764 | 16 | 32,867,067 | 1.58E−05 | NDUFS4 | 7383 | |
| LC | rs1806864 | 18 | 42,102,045 | 2.00E−05 | MINDY4 | Within |
For shoulder cut traits, two genome-wide significant SNPs and 20 suggestive significant (2.50 × 10–5) SNPs were identified to be associated with the proportion of BBS, BPS, AB and SC. Among the SNPs in the region between 29.99 and 30.57 Mb on the Sus scrofa chromosome (SSC) 7, two genome-wide significant SNPs were identified to be associated with the proportion of AB (Fig. 3a). The most significant SNP associated with the proportion of AB was located at position 30,100,555 bp on SSC7 (rs0700767) with a p value of 1.37 × 10–8, and was in strong linkage disequilibrium (D' = 0.94) with SNP rs0700777 (Fig. 3b). Phenotypes were corrected using the linear mixed model for GWAS, and the phenotypes of individuals were grouped into three classes according to their genotypes (AA, AG, and GG) at this locus (30,100,555 bp). Student’s t-test was performed between each pair of the three classes (Fig. 3d). The pairwise t-test p value were 0.268, 1.19 × 10–3, and 1.63 × 10–11 for AA vs. AG, AA vs. GG, and AG vs. GG, respectively (Fig. 3d). Although the difference between individuals with genotypes AA and AG was nonsignificant, this might be due to a smaller number of individuals with the AA genotype. Notably, the 29.99–30.57 Mb region on SSC7 includes four genes that have been previously reported as potential candidates for limb bone length [23] and body height in pigs, namely GRM4, HMGA1, RPS10 and NUDT3 [24, 25], and also with body mass index and height in humans [26, 27].
Fig. 3.

Genome-wide association study for the proportion of arm bones. a The Manhattan plot shows the associations of 40,016 SNPs with proportion of arm bones in the combined population. The red dots represent SNPs that reach the genome-wide significance threshold (P = 1.25 × 10–6). b A haplotype view of all polymorphic sites within the 500-kb interval before and after the top significant SNP. c Box plots showing the differences in proportion of arm bones according to genotype
For middle cut traits, multiple QTL in a 6.11-Mb region (from 97.61 to 103.72 Mb) on SSC7 that included 12, 4, and 10 genome-wide significant SNPs were identified to be associated with RI, CB, and MC proportions, respectively (Table 2). A previous study identified a causal mutation (g.19034 A > C) in the VRTN gene located within this region that affects thoracic vertebrae number and performed a series of functional validation experiments [28]. The number of ribs and the THL change with THN and we also identified QTL that were significantly associated with THN and THL in the same genomic region (see Additional file 3: Fig. S2a, b). The most significant SNP for THN was located at position 97,614,602 bp (rs0702609), with a p value of 2.02 × 10–107, and this locus corresponded exactly to the causal mutation g.19034 A > C. The most significant SNP for THL (P = 1.77 × 10–46), located within the VRTN gene at position 97,618,073 bp (rs0702038), was also the most significant SNP for CB (P = 6.94 × 10–8, Fig. 4a), and was in complete linkage disequilibrium with rs0702689 (see Additional file 3: Fig. S2c). The most significant SNP for both RI and MC proportions was rs0702610 (PRI = 4.18 × 10–16, PMC = 4.47 × 10–13, Fig. 4b, c, located within the VRTN gene at position 97,615,897 bp, which is in complete linkage disequilibrium with rs0702689 and rs0702609 (see Additional file 3: Fig. S2c). This suggests that alterations in THN and rib number due to causal variants within the VRTN gene can influence RI, CB and MC proportions.
Fig. 4.
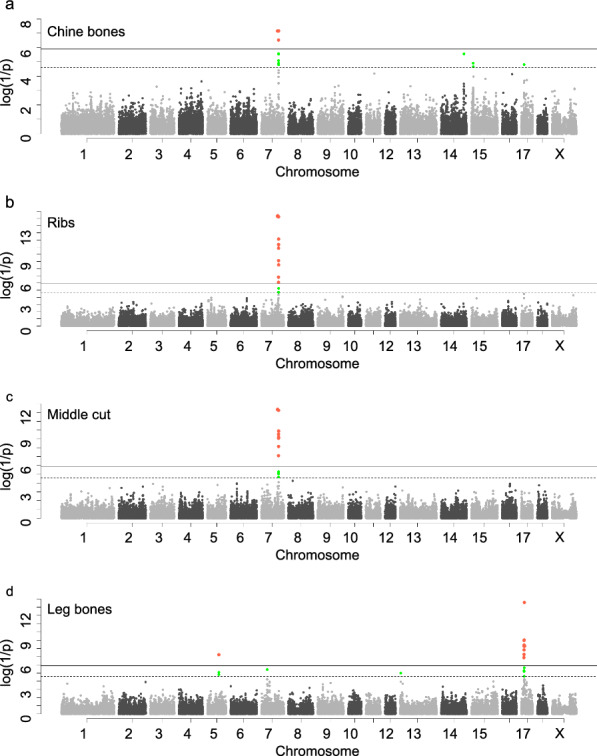
Genome-wide association study for proportion of chine bones, ribs, middle cut, and leg bones. a–d The Manhattan plot shows the associations of 40,016 SNPs with proportion of chine bones, ribs, middle cut and leg bones, respectively, in the combined population
For leg cut traits, we identified nine genome-wide significant SNPs and 14 suggestive significant SNPs that are associated with the proportion of BL, TL, LB, and LC (Table 2 and Fig. 4d). Specifically, one SNP on SSC5 and eight SNPs on SSC17 were significantly (P < 1.25 × 10–6) associated with LB proportion (Fig. 4d). The region containing the eight significant SNPs on SSC17 covers the BMP2 gene, a member of the bone morphogenetic protein (BMP) family, which is a candidate gene for LB proportion. Abnormal expression of BMP2 results in failure of some chondrogenic condensates to form in the limbs, which affects vertebrate limb development and skeletogenesis [29, 30]. The most significant SNP for LBP was located at position 16,788,523 bp on SSC17 (rs0700767), with a p value of 2.67 × 10–14, explaining 2.7% of the phenotypic variation. The other top SNP associated with LBP on SSC5 was located at position 66,103,958 bp (rs0501167) with a p value of 5.84 × 10–8, and was within the CCND2 gene. In previous studies, the CCND2 gene has been shown to regulate the cell cycle progression of hematopoietic stem cells [31], and SNPs in CCND2 have been found to be significantly associated with human height [32]. Moreover, An et al. [33] identified CCND2 as a candidate gene for hip height, body height, and body length in Chinese Wagyu beef cattle, while Le et al. [34] reported that CCND2 participates in growth-related processes and regulates conformational traits in pigs. These results suggest that BMP2 and CCND2 are candidate genes for LB.
Discussion
Cutting carcasses into different types of cut products can efficiently increase their economic value. As a result, an increasing number of academics and breeding companies are interested in selecting for carcass traits. Scholars have investigated the process of carcass cuts and yield segmentation production since the 1970s [5, 6]. Recent studies [7, 8], including our previous study [9], have investigated the effects of sex, breed, carcass weight, and other factors on meat cut traits. Using pedigree information, Newcom et al. [35], van Wijk et al. [12], and Miar et al. [11] estimated the heritability of such traits as well as their genetic correlations with other carcass features and with meat quality traits. They found that the heritability for the majority of meat cut traits is moderate and that the genetic correlations between these traits are low. Here, we constructed a kinship matrix using the genotypes for 40,016 SNPs and evaluated the genetic parameters of meat cut traits in various breeds. Our results are comparable to those of previous studies. In addition, based on the GB/T 9959 carcass cutting standards, we evaluated the genetic parameters of other meat cut traits than those that were evaluated in most previous reports, including AB, SB, CB, LB, and TPB cut proportions, which are favored by Chinese consumers.
The study of the heritability of meat cut traits from different breeds revealed that most of them had moderate to high heritabilities, which suggests that there is great potential to improve these traits in breeding programs. However, when planning breeding for meat cut traits, the genetic correlations of such traits of interest with other economically important traits must also be considered. Although the middle cut has a higher retail value than other primal cuts and the proportion of MC has a favorable positive genetic correlation with both BE proportion and intramuscular fat content, it also has a positive genetic correlation with BF proportion and backfat depth. Therefore, breeding to improve the proportion of MC may lead to an undesirable increase in backfat. In addition, rib proportion, which has the greatest retail value, was favorably positively correlated with carcass oblique length, straight length, thoracic number, and thoracic length and was also negatively correlated with backfat. Loin, which is a popular meat cut among consumers, shows a favorable genetic positive correlation with loin muscle area and a negative correlation with backfat depth. Selection for these two cut traits would also benefit fatness traits, which is consistent with results of previous studies [10–12]. In general, the majority of meat cuts had low or nonsignificant genetic correlations with carcass and meat quality traits, which indicates that breeding selection for carcass cuts has a limited impact on carcass and meat quality traits.
Meat cuts are post-slaughter carcass composition traits that are challenging to evaluate in vivo. Therefore, marker-assisted selection and genomic selection would be optimal methods to select for carcass cuts, as these strategies can shorten the generation interval through early selection and increase the accuracy of the predicted breeding values, especially for complex traits with a low heritability and traits that are difficult to measure [36–39]. However, few studies have dissected the genetic mechanisms that underlie meat cut traits. In this study, we report the detection of 28 QTL that are significantly associated with meat cuts through GWAS. For example, the causal SNP (g.19034 A > C) in the VRTN gene that is associated with thoracic vertebrae number [28] is also significantly associated with the proportions of middle cut, chine bones, ribs, shoulder cut and picnic shoulder. In addition, in previous reports, several candidate genes located near these QTL that are significantly associated with the proportions of arm bone and leg bone such as BMP2, GRM4, HMGA1, CCND2, HAO1, and NUDT3, have all been found to be associated with skeletal development [26, 27, 29, 30, 32] and height in humans [26, 27, 32], and with hip height, body height, and body length of pigs [23, 25, 34]. Overall, the QTL and candidate genes identified here for meat cut traits are relevant for the selective breeding for meat cut traits and provide an essential basis for further research of the genetic mechanism underlying meat cut traits.
The body shape of pigs includes two dimensions: body length (spine) and body height (forelimbs and hindlimbs). The carcass cuts in this study were divided based on the pig’s bone structure and commercial value, including the three types of bones: arm bones (forelimb), chine bones (spine), and leg bones (hindlimbs). We identified three candidate genes that are significantly associated with these three types of skeletal development, namely HMGA1, VRTN, and BMP2 [26–30, 32]. Among these, HMGA1 may be the strongest candidate gene for forelimb bone development, VRTN is a causal gene for vertebra number, as we have previously shown, and BMP2 may be the strongest candidate gene for hindlimb bone development. Therefore, it seems that bone development in different parts of the body is regulated by different genes. Analysis of the genetic architecture of meat cut traits will help analyze the molecular mechanism that underlies bone development of the forelimbs, spine and hindlimbs and lays a research foundation for the selection of the best shapes in different parts of the body.
Conclusions
This study evaluated the genetic parameters of 17 carcass cut traits, 12 carcass traits, and six meat quality traits in four populations of pigs. Genetic parameters for some of these novel meat cut traits have not been reported before. We found that some high economic value meat cuts, such as ribs, belly, loin, and tenderloin, have moderate to high heritabilities, which indicates that improvement of these traits through breeding programs is possible. This could increase the market price of the carcass by selecting on key meat cut traits. Estimates of genetic correlations revealed weak or nonsignificant correlations between many meat cut, carcass, and meat quality traits. Rib proportion was favorably correlated with carcass oblique length, straight length, thoracic number, and thoracic length, and negatively correlated with backfat depth. In addition, the GWAS identified 28 QTL and several candidate genes for meat cut traits for the first time. These findings provide a key reference for marker-assisted selection and genomic selection for meat cut traits.
Supplementary Information
Additional file 1: Table S1. Estimates of genetic correlations between carcass cut proportions with carcass traits and meat quality traits.
Additional file 2: Figure S1. Q-Q plot and lambda values of genome-wide association study (GWAS) of meat cut traits and carcass traits. Lambda values are normally used to characterize population stratification in multibreed GWAS.
Additional file 3: Figure S2. Genome-wide association study (GWAS) for thoracic number and thoracic length. a, b The Manhattan plots show the associations of 40,016 SNPs with thoracic number and thoracic length, respectively, in the combined population. c A haplotype view of all polymorphic sites within the 500-kb interval before and after the top significant SNP (rs0702038).
Acknowledgements
The authors would like to acknowledge Muyuan Food Co., Ltd. (Henan, China) and Longda Muyuan Meat Food Co., Ltd. (Henan, China) for providing the pig and meat samples.
Author contributions
LH, ZZ and SX conceived the study, designed the experiment, and supervised the study. LX, JQ, XT and DC carried out the phenotyping and sample collection. LC, LR, LX and JQ carried out the genotyping. TY provided software and analytical methods. LX performed the formal analysis and developed the methodology and was a major contributor in writing the manuscript. LH, ZZ, SX and LX contributed to interpretating the results and manuscript review and editing. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China [grant number 32160782].
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Lei Xie, Email: XieLstone@hotmail.com.
Jiangtao Qin, Email: 609826646@qq.com.
Tianxiong Yao, Email: 1159266279@qq.com.
Xi Tang, Email: 875069912@qq.com.
Dengshuai Cui, Email: 1305568175@qq.com.
Liqing Chen, Email: 2542321250@qq.com.
Lin Rao, Email: 1814348289@qq.com.
Shijun Xiao, Email: shjx_jxau@hotmail.com.
Zhiyan Zhang, Email: bioducklily@hotmail.com.
Lusheng Huang, Email: LushengHuang@hotmail.com.
References
- 1.Gong SL, Yang YS, Shen H, Wang XY, Guo HP, Bai L. Meat handling practices in households of mainland china. Food Control. 2011;22:749–755. doi: 10.1016/j.foodcont.2010.11.009. [DOI] [Google Scholar]
- 2.Liu H, Sun D. China pork consumption situation and outlook. Agric Outlook. 2010;52:35–38. [Google Scholar]
- 3.Grunert KG. Future trends and consumer lifestyles with regard to meat consumption. Meat Sci. 2006;74:149–160. doi: 10.1016/j.meatsci.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 4.Qin J. Evaluation of the value gain on pork economical carcass cutting and its related breeding technology development. Nanchang: Jiangxi Agricultural University; 2019. [Google Scholar]
- 5.Mandigo RW, Thompson TL, Weiss GM. Commercial accelerated pork processing: yields of fresh carcass cuts. J Food Sci. 1979;44:138–139. doi: 10.1111/j.1365-2621.1979.tb10026.x. [DOI] [Google Scholar]
- 6.Mandigo RW, Thompson TL, Weiss GM. Commercial accelerated pork processing: yields of cured ham, bacon and loins. J Food Sci. 1977;42:898–899. doi: 10.1111/j.1365-2621.1977.tb12632.x. [DOI] [Google Scholar]
- 7.Overholt MF, Arkfeld EK, Mohrhauser DA, King DA, Wheeler TL, Dilger AC, et al. Comparison of variability in pork carcass composition and quality between barrows and gilts. J Anim Sci. 2016;94:4415–4426. doi: 10.2527/jas.2016-0702. [DOI] [PubMed] [Google Scholar]
- 8.Alvarez-Rodríguez J, Teixeira A. Slaughter weight rather than sex affects carcass cuts and tissue composition of bisaro pigs. Meat Sci. 2019;154:54–60. doi: 10.1016/j.meatsci.2019.04.012. [DOI] [PubMed] [Google Scholar]
- 9.Xie L, Qin J, Rao L, Cui D, Tang X, Xiao S, et al. Effects of carcass weight, sex and breed composition on meat cuts and carcass trait in finishing pigs. J Integr Agr. 2023;22(5):1489–1501. doi: 10.1016/j.jia.2022.08.122. [DOI] [Google Scholar]
- 10.Khanal P, Maltecca C, Schwab C, Gray K, Tiezzi F. Genetic parameters of meat quality, carcass composition, and growth traits in commercial swine. J Anim Sci. 2019;97:3669–3683. doi: 10.1093/jas/skz247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miar Y, Plastow GS, Moore SS, Manafiazar G, Charagu P, Kemp RA, et al. Genetic and phenotypic parameters for carcass and meat quality traits in commercial crossbred pigs. J Anim Sci. 2014;92:2869–2884. doi: 10.2527/jas.2014-7685. [DOI] [PubMed] [Google Scholar]
- 12.van Wijk HJ, Arts DJG, Matthews JO, Webster M, Ducro BJ, Knol EF. Genetic parameters for carcass composition and pork quality estimated in a commercial production chain. J Anim Sci. 2005;83:324–333. doi: 10.2527/2005.832324x. [DOI] [PubMed] [Google Scholar]
- 13.Xie L, Qin J, Rao L, Tang X, Cui D, Chen L, et al. Accurate prediction and genome-wide association analysis of digital intramuscular fat content in longissimus muscle of pigs. Anim Genet. 2021;52:633–644. doi: 10.1111/age.13121. [DOI] [PubMed] [Google Scholar]
- 14.Zheng M, Huang Y, Ji J, Xiao S, Ma J, Huang L. Effects of breeds, tissues and genders on purine contents in pork and the relationships between purine content and other meat quality traits. Meat Sci. 2018;143:81–86. doi: 10.1016/j.meatsci.2018.04.022. [DOI] [PubMed] [Google Scholar]
- 15.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. Plink: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanraden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–4423. doi: 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- 17.Gengler N, Mayeres P, Szydlowski M. A simple method to approximate gene content in large pedigree populations: application to the myostatin gene in dual-purpose belgian blue cattle. Animal. 2007;1:21–28. doi: 10.1017/S1751731107392628. [DOI] [PubMed] [Google Scholar]
- 18.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common snps explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Madsen P, Jensen J. A user's guide to DMU. A package for analysing multivariate mixed models. Version 6, release 5.2. Tjele: University of Aarhus; 2013. [Google Scholar]
- 20.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–824. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo Y, Huang Y, Hou L, Ma J, Chen C, Ai H, et al. Genome-wide detection of genetic markers associated with growth and fatness in four pig populations using four approaches. Genet Sel Evol. 2017;49:21. doi: 10.1186/s12711-017-0295-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X, Xiong X, Yang J, Zhou L, Yang B, Ai H, et al. Genome-wide association analyses for meat quality traits in chinese erhualian pigs and a western Duroc × (Landrace × Yorkshire) commercial population. Genet Sel Evol. 2015;47:44. doi: 10.1186/s12711-015-0120-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Li N, Liu X, Liang J, Yan H, Zhao K, et al. A genome-wide association study of limb bone length using a large white × minzhu intercross population. Genet Sel Evol. 2014;46:56. doi: 10.1186/s12711-014-0056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J, Fu Y, Hu Y, Yin L, Tang Z, Yin D, et al. Whole genome variants across 57 pig breeds enable comprehensive identification of genetic signatures that underlie breed features. J Anim Sci Biotechnol. 2020;11:115. doi: 10.1186/s40104-020-00520-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Zhang L, Yan H, Liu X, Li N, Liang J, et al. Genome-wide association studies identify the loci for 5 exterior traits in a Large White × Minzhu pig population. PLoS One. 2014;9:e103766. doi: 10.1371/journal.pone.0103766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soranzo N, Rivadeneira F, Chinappen-Horsley U, Malkina I, Richards JB, Hammond N, et al. Meta-analysis of genome-wide scans for human adult stature identifies novel loci and associations with measures of skeletal frame size. PLoS Genet. 2009;5:e1000445. doi: 10.1371/journal.pgen.1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho YS, Go MJ, Kim YJ, Heo JY, Oh JH, Ban H, et al. A large-scale genome-wide association study of asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet. 2009;41:527–534. doi: 10.1038/ng.357. [DOI] [PubMed] [Google Scholar]
- 28.Duan Y, Zhang H, Zhang Z, Gao J, Yang J, Wu Z, et al. VRTN is required for the development of thoracic vertebrae in mammals. Int J Biol Sci. 2018;14:667–681. doi: 10.7150/ijbs.23815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosen V. BMP2 signaling in bone development and repair. Cytokine Growth Factor Rev. 2009;20:475–480. doi: 10.1016/j.cytogfr.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 30.Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2:e216. doi: 10.1371/journal.pgen.0020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamatani Y, Matsuda K, Okada Y, Kubo M, Hosono N, Daigo Y, et al. Genome-wide association study of hematological and biochemical traits in a japanese population. Nat Genet. 2010;42:210–215. doi: 10.1038/ng.531. [DOI] [PubMed] [Google Scholar]
- 32.Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet. 2021;53:1415–1424. doi: 10.1038/s41588-021-00931-x. [DOI] [PubMed] [Google Scholar]
- 33.An B, Xia J, Chang T, Wang X, Xu L, Zhang L, et al. Genome-wide association study reveals candidate genes associated with body measurement traits in chinese wagyu beef cattle. Anim Genet. 2019;50:386–390. doi: 10.1111/age.12805. [DOI] [PubMed] [Google Scholar]
- 34.Le TH, Christensen OF, Nielsen B, Sahana G. Genome-wide association study for conformation traits in three danish pig breeds. Genet Sel Evol. 2017;49:12. doi: 10.1186/s12711-017-0289-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newcom DW, Baas TJ, Mabry JW, Goodwin RN. Genetic parameters for pork carcass components. J Anim Sci. 2002;80:3099–3106. doi: 10.2527/2002.80123099x. [DOI] [PubMed] [Google Scholar]
- 36.Meuwissen TH, Hayes BJ, Goddard ME. Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001;157:1819–1829. doi: 10.1093/genetics/157.4.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.García-Ruiz A, Cole JB, Vanraden PM, Wiggans GR, Ruiz-López FJ, Van Tassell CP. Changes in genetic selection differentials and generation intervals in us holstein dairy cattle as a result of genomic selection. Proc Natl Acad Sci USA. 2016;113:E3995–4004. doi: 10.1073/pnas.1519061113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nejati-Javaremi A, Smith C, Gibson JP. Effect of total allelic relationship on accuracy of evaluation and response to selection. J Anim Sci. 1997;75:1738–1745. doi: 10.2527/1997.7571738x. [DOI] [PubMed] [Google Scholar]
- 39.Meuwissen T, Goddard M. Accurate prediction of genetic values for complex traits by whole-genome resequencing. Genetics. 2010;185:623–631. doi: 10.1534/genetics.110.116590. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Estimates of genetic correlations between carcass cut proportions with carcass traits and meat quality traits.
Additional file 2: Figure S1. Q-Q plot and lambda values of genome-wide association study (GWAS) of meat cut traits and carcass traits. Lambda values are normally used to characterize population stratification in multibreed GWAS.
Additional file 3: Figure S2. Genome-wide association study (GWAS) for thoracic number and thoracic length. a, b The Manhattan plots show the associations of 40,016 SNPs with thoracic number and thoracic length, respectively, in the combined population. c A haplotype view of all polymorphic sites within the 500-kb interval before and after the top significant SNP (rs0702038).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.