

Abstract
Herein, we describe studies showing that Lewis acid co-catalysts can significantly broaden the scope of alkenes that can be incorporated into the photosensitized visible-light De Mayo reaction. Mechanistic studies suggest the primary benefit of the Lewis acid is not on substrate sensitization but rather on bond-forming steps downstream of energy transfer, highlighting the diverse effects that Lewis acids can have on sensitized photoreactions.
Graphical Abstract
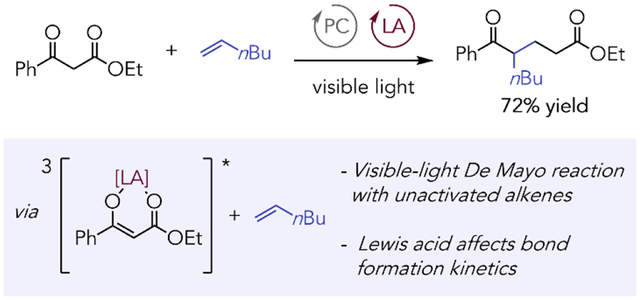
The De Mayo reaction generates 1,5-dicarbonyl compounds by formal insertion of an alkene into a 1,3-dicarbonyl compound. The classical mechanism of this photochemical transformation involves an initial [2+2] photocycloaddition of the enol tautomer of the 1,3-dicarbonyl with an alkene;1,2 retro-aldol ring-opening of the resulting cyclobutanol affords the 1,5-dicarbonyl product (Figure 1A). The photonic efficiency of the De Mayo reaction using standard UV irradiation, however, is relatively low, often resulting in lengthy reaction times of up to seven days.3,4,5 Triplet photosensitizers have been utilized to overcome this problem. For example, in developing a ring-expanding De Mayo reaction under UV-A light, Guldi and Glorius found that addition of an Ir photocatalyst resulted in significant product formation within just 24 h, whereas no product was detected in its absence.6 Triplet photosensitizers have been further leveraged to furnish De Mayo products under mild visible-light irradiation, allowing for increased functional group compatibility. Marzo and König first pioneered this approach, enabling the reaction of 1,3-dicarbonyls with styrenes and silyl enol ethers (Figure 1B).7 However, unactivated alkenes proved to be unreactive under these conditions, representing a limitation in the scope of De Mayo products available using visible light photosensitization.
Figure 1.

(A) Original De Mayo reaction and mechanism. (B) Seminal visible-light De Mayo reaction. (C) Strategy for reaction with unactivated alkenes under UV irradiation. (D) This work.
Our laboratory has a long-standing interest in understanding the influence of Lewis acids on photosensitized reactions, and we wondered if the incorporation of a Lewis acidic co-catalyst might enhance the reactivity of simple aliphatic alkenes in the sensitized visible-light De Mayo reaction. Lewis acids can have multiple effects in sensitized photoreactions. They can increase the efficiency of both electron- and energy-transfer activation processes.8,9,10 They can also influence the dynamics of bond-forming processes that occur after the initial photocatalytic activation step.11 The effect of Lewis acids on De Mayo reactions was previously investigated by Chow, who noted that although dibenzoylmethane (9) does not react productively with aliphatic olefins upon direct irradiation, the corresponding preformed BF2 adduct 10 reacts smoothly to afford De Mayo product 11 (Figure 1C).12 A very recent study that appeared during the preparation of this manuscript similarly demonstrated that the direct irradiation of catalytically generated Zr(IV) dibenzoylmethane complexes with visible light enables De Mayo reactions with a wide range of both activated and aliphatic alkene reaction partners.13 Given the beneficial effect of Lewis acids on the scope of De Mayo reactions initiated by direct photoexcitation, we hypothesized the addition of a Lewis acid co-catalyst might likewise expand the scope of sensitized De Mayo reactions to less-reactive aliphatic alkene partners.
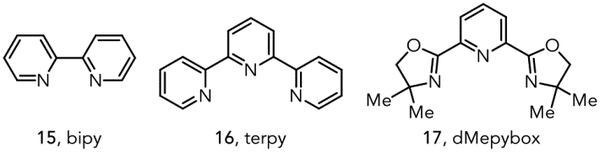
Our investigations commenced with a study of the reaction of ethyl benzoylacetate (12a) with 1-hexene (2a) in the presence of imidazole and Ir(dFCF3ppy)2(dtbbpy)PF6 (14) as the photocatalyst. A screen of Lewis acid additives revealed that De Mayo product 13a was observed only when a Lewis acid was included, with Sc(OTf)3 standing out as a promising co-catalyst (Table 1, entries 1–8). Notably, 13a was formed as a single regioisomer. Although addition of bidentate ligands such as 15 did not improve the yield, tridentate ligands 16 and 17 improved both the yield and overall mass balance (entries 9–11). On preparative scale, 13a was obtained in 72% yield at increased reaction concentration but with reduced loadings of Lewis acid, ligand, and base (entry 12). Control experiments indicated that the photocatalyst (entry 13) and light (entry 14) are critical to reaction success. The product is generated in the absence of imidazole, albeit in diminished yield (entry 15). Further details of the optimization studies can be found in the Supporting Information.
Table 1.
Reaction optimization.

| |||
|---|---|---|---|
| entry | Lewis acid | ligand | % yielda |
| 1 | none | none | 0 |
| 2 | 20 mol% Sc(OTf)3 | none | 34 |
| 3 | 20 mol% Mg(OTf)2 | none | 14 |
| 4 | 20 mol% Cu(OTf)2 | none | 8 |
| 5 | 20 mol% Zn(OTf)2 | none | 19 |
| 6 | 20 mol% Fe(OTf)3 | none | 7 |
| 7 | 20 mol% La(OTf)3 | none | 4 |
| 8 | 20 mol% Yb(OTf)3 | none | 7 |
| 9 | 20 mol% Sc(OTf)3 | 30 mol% bipy | 14 |
| 10 | 20 mol% Sc(OTf)3 | 30 mol% terpy | 50 |
| 11 | 20 mol% Sc(OTf)3 | 30 mol% dMepybox | 47 |
| 12 b | 10 mol% Sc(OTf) 3 | 15 mol% terpy | 72 |
| 13c | 20 mol% Sc(OTf)3 | 30 mol% terpy | 0 |
| 14d | 20 mol% Sc(OTf)3 | 30 mol% terpy | 0 |
| 15e | 20 mol% Sc(OTf)3 | 30 mol% terpy | 24 |
| |||
Unless otherwise noted, reactions were conducted using 0.1 mmol 12a, 0.5 mmol 2a, 2 mol% 14, and 2 mL solvent.
Yields were determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as an internal standard.
0.3 mmol scale, 3 mL CH2Cl2, 25 mol% imidazole, isolated yield.
No photocatalyst.
Run in the dark.
No imidazole.
Experiments probing the scope of the reaction under these optimized conditions are summarized in Figure 2. Alkene substrates bearing a variety of functional groups reacted in moderate to good yield (13a–13g), though steric bulk proximal to the alkene led to diminished yield (13h). Alkenes activated by both electron-donating and electron-withdrawing functional groups furnished De Mayo products in good yields (13i–13k). Gratifyingly, although 12a can undergo Michael addition with N,N-dimethylacrylamide (2j) under the reaction conditions, De Mayo product 13j was formed in good yield. Unprotected alcohols are also compatible with this method but can lead to transesterified products: in CH2Cl2, the reaction with allyl alcohol (2l) primarily yielded lactone 13l, though switching the solvent to MeCN enabled isolation of the expected alcohol 13m. In addition, diverse alkene substitution patterns were permitted in the reaction (13n–13t). Product 13p was isolated predominantly as the unexpected anti diastereomer, suggesting that the initially generated syn product epimerizes under the reaction conditions. The reaction is amenable to scale-up, allowing 13q to be isolated in 60% yield at 1.0 mmol scale. Although reaction with 2-methyl-2-butene afforded the expected linear product 13r, reaction with other bulky tri- and tetrasubstituted olefins yielded cyclobutanol products 13s and 13t rather than the typical ring-opened products, consistent with previous reports.6 Notably, complex polycyclic products 13o and 13s were isolated as single diastereomers. With regard to the 1,3-dicarbonyl coupling partner, at least one aryl ketone was required for successful De Mayo reaction, as malonate esters proved unreactive. However, a variety of aromatic β-ketoesters reacted smoothly (13u–13ab). Both electron-donating and electron-withdrawing groups were tolerated. In addition to β-ketoesters, 1,3-diketones (18–19) and a β-keto amide substrate (20) successfully underwent the desired De Mayo reaction under the standard reaction conditions.
Figure 2.

Reaction scope. a40 h reaction time. b2 equiv allyl alcohol. cMeCN used as reaction solvent. dFrom cis-4-octene. e1.0 mmol scale.
In addition to improving the scope of the sensitized De Mayo reaction, we were intrigued by the mechanistic role of the Lewis acid co-catalyst. We first wondered whether addition of a Lewis acid could activate a photoredox pathway orthogonal to established direct excitation and energy transfer mechanisms by enabling redox events within the potential range of the photocatalyst.8 In such a photoredox mechanism, 22 could be oxidized by the excited photocatalyst to generate radical 23 that would then react with alkene 2 (Figure 3). Indeed, photoredox oxidation of enolates has been previously reported, including for substrate 12a.14 A previous study used cyclic voltammetry to estimate a peak potential at +0.75 V vs Fc/Fc+ for 12a.14 This oxidation should be well within the excited-state redox potential of Ir photocatalyst 14 (Ered PC*/PC− = +1.42 V vs Fc/Fc+)15 without any Lewis acid additive. However, replacing photocatalyst 14 with the highly oxidizing pyrylium photocatalysts Br-TPT (26, Ered PC*/PC− = +1.85 V vs Fc/Fc+)16 or Me-TPT (27, Ered PC*/PC− = +1.63 V vs Fc/Fc+)17 resulted in no observable reactivity, yielding only returned starting material in >75% yield (Figure 3A, entries 1 and 2). In contrast, using thioxanthone (28) as a photocatalyst resulted in a 16% yield of 13a (entry 3). Given thioxanthone’s modest excited-state redox potential (Ered PC*/PC− = +0.78 V vs Fc/Fc+)18 but high triplet energy (ET = 65.5 kcal/mol)18 comparable to that of 14 (ET = 60.1 kcal/mol),15 these data support the hypothesis that the Lewis acid-assisted De Mayo reaction is a triplet-state reaction rather than a photoredox radical process.
Figure 3.

Experiments probing the mechanistic role of Sc(OTf)3. (A) Screening organic photocatalysts to probe electron- and energy-transfer mechanisms. (B) Stern–Volmer luminescence quenching. (C) Keto-enol equilibrium experiments. (D) Alkene isomerization experiments.
We next examined the Lewis acid’s effect on the rate of triplet energy transfer. We previously reported that Lewis acid coordination can dramatically increase the rate of Dexter energy transfer to Lewis basic substrates.9 In the present system, however, a Stern–Volmer analysis revealed the rate of photocatalyst quenching by 12a increased only 1.5-fold upon addition of one equivalent of Sc(OTf)3 (Figure 3B). Moreover, a 1H NMR study examining the keto–enol tautomer ratio of 12a showed that the enol(ate) content increased five-fold upon addition of Sc(OTf)3 (Figure 3C). Thus, the modest increase in luminescence quenching in the presence of Lewis acid seems reflective of the Lewis acid’s ability to increase enol(ate) content. Given that no De Mayo product is observed without Lewis acid, it seems unlikely that the increase in Stern–Volmer quenching could rationalize the drastically different reaction outcome.
We conclude, therefore, that the Lewis acid principally influences the rate of bond-forming steps that occur after Dexter energy transfer. This conclusion is supported by experiments probing the configuration of the alkene component after the De Mayo reaction has occurred (Figure 3D). If the first bond-forming step of the stepwise diradical [2+2] cycloaddition is reversible, we imagined that alkene stereochemistry could be eroded during bond rotation of biradical intermediate 24. Indeed, when stereochemically pure cis-4-octene (2q-cis) was reacted with ketoester 12a under standard reaction conditions, alkene stereochemical purity was diminished (entry 1). Separate control reactions omitting the Lewis acid and 12a resulted in substantially lower degrees of isomerization (entries 2 and 3), suggesting that both of these components are involved in the major isomerization-promoting pathway. These data are consistent with the hypothesis that Lewis acid coordination directly facilitates initial bond formation by increasing the reactivity of the triplet diradical towards unactivated alkenes. Whether the increased reactivity can be attributed to increased lifetimes of triplet intermediates or to inhibition of deactivation pathways remains unclear at present.
In summary, the addition of a Lewis acid co-catalyst expands the scope of the sensitized visible-light De Mayo reaction to include unactivated alkenes. Mechanistic experiments indicate that the principal effect of the Lewis acid is to promote bond-forming step(s) downstream of sensitization. This study adds to the growing body of literature demonstrating that Lewis acids can have diverse beneficial effects on the outcomes of photocatalytic reactions. We hope these findings will be applied to expand the scope of other excited-state reactions in the future.
Supplementary Material
ACKNOWLEDGMENTS
Funding for this project was provided by the NIH (GM144129) and the Institute for Basic Science (IBS-R010-A1) in Korea. NMR facilities at UW−Madison used in this work were supported by NSF grant CHE-1048642 and by a generous gift from Paul J. and Margaret M. Bender. The mass spectrometry facility at UW−Madison was supported by NIH award 1S10 OD020022-1.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
General experimental procedures; characterization data; spectral data for all new compounds (PDF)
The authors declare no competing financial interest.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
REFERENCES
- 1.Wu Y-J de Mayo Reaction. In Name Reactions for Carbocyclic Ring Formations, 1st ed.; Li JJ, Ed.; John Wiley & Sons, Inc.: Hoboken, New Jersey, 2010; pp. 451–488. [Google Scholar]
- 2.De Mayo P; Takeshita H; Sattar ABMA The Photochemical Synthesis of 1,5-Diketones and their Cyclisation: A New Annulation Process. Proc. Chem. Soc 1962, 119. [Google Scholar]
- 3.Disanayaka BW; Weedon AC Application of the de Mayo Reaction to the Preparation of Tricyclo[6.3.0.02,6]undecanes: A Photochemical Synthesis of (±)-Hirsutene. J. Org. Chem 1987, 52, 2905–2910. [Google Scholar]
- 4.Minter DE; Winslow CD A Photochemical Approach to the Galanthan Ring System. J. Org. Chem 2004, 69, 1603–1606. [DOI] [PubMed] [Google Scholar]
- 5.Nozaki H; Kurita M; Mori T; Noyori R Photochemical Behaviour of Enolic β-Diketones towards Cycloolefins. Tetrahedron 1968, 24, 1821–1828. [Google Scholar]
- 6.Paulisch TO; Mai LA; Strieth-Kalthoff F; James MJ; Henkel C; Guldi DM; Glorius F Dynamic Kinetic Sensitization of β-Dicarbonyl Compounds—Access to Medium-Sized Rings by De Mayo-Type Ring Expansion. Angew. Chem., Int. Ed 2022, 61, e202112695. [DOI] [PubMed] [Google Scholar]
- 7.Martinez-Haya R; Marzo L; König B Reinventing the De Mayo reaction: synthesis of 1,5-diketones or 1,5-ketoesters via visible light [2+2] cycloaddition of β-diketones or β-ketoesters with styrenes. Chem. Commun 2018, 54, 11602–11605. [DOI] [PubMed] [Google Scholar]
- 8.Du J; Skubi KL; Schultz DM; Yoon TP A Dual-Catalysis Approach to Enantioselective [2 + 2] Photocycloadditions Using Visible Light. Science 2014, 344, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blum TR; Miller ZD; Bates DM; Guzei IA; Yoon TP Enantioselective photochemistry through Lewis acid–catalyzed triplet energy transfer. Science 2016, 354, 1391–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones BA; Solon P; Popescu MV; Du J-Y; Paton R; Smith MD Catalytic Enantioselective 6π Photocyclization of Acrylanilides. J. Am. Chem. Soc 2023, 145, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo R; Adak S; Bellotti P; Gao X; Smith WW; Le SN; Ma J; Houk KN; Glorius F; Chen S; Brown MK Photochemical Dearomative Cycloadditions of Quinolines and Alkenes: Scope and Mechanism Studies. J. Am. Chem. Soc 2022, 144, 17680–17691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chow YL; Cheng X Photocycloadditions and Photosensitizations Promoted by Electron Transfer: β-Diketonatoboron Difluorides as Electron Acceptors. J. Chem. Soc., Chem. Commun 1990, 1043–1045. [Google Scholar]
- 13.Zhang W; Luo S Visible-light promoted de Mayo Reaction by zirconium catalysis. Chem. Commun 2022, 58, 12979–12982. [DOI] [PubMed] [Google Scholar]
- 14.Li T; Liang K; Zhang Y; Hu D; Ma Z; Xia C Three-Component Minisci Reaction with 1,3-Dicarbonyl Compounds Induced by Visible Light. Org. Lett 2020, 22, 2386–2390. [DOI] [PubMed] [Google Scholar]
- 15.Singh A; Teegardin K; Kelly M; Prasad KS; Krishnan S; Weaver JD Facile Synthesis and Complete Characterization of Homoleptic and Heteroleptic Cyclometalated Iridium(III) Complexes for Photocatalysis. J. Organomet. Chem 2015, 776, 51–59. [Google Scholar]
- 16.Martiny M; Steckhan E; Esch T Cycloaddition Reactions Initiated by Photochemically Excited Pyrylium Salts. Chem. Ber 1993, 126, 1671–1682. [Google Scholar]
- 17.Gesmundo NJ; Nicewicz DA Cyclization–Endoperoxidation Cascade Reactions of Dienes Mediated by a Pyrylium Photoredox Catalyst. Beilstein J. Org. Chem 2014, 10, 1272–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nikitas NF; Gkizis PL; Kokotos CG Thioxanthone: A Powerful Photocatalyst for Organic Reactions. Org. Biomol. Chem 2021, 19, 5237–5253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.