

ABSTRACT
A comprehensive analysis of the molecular mechanism underlying colorectal tumor evaluated the development of colorectal cancer (CRC) and proposed targeting small molecular inhibitors. Nonetheless, the adoptive resistance of these therapies remains a challenge with respect to achieving an effective clinical response. Thus, identifying the molecular mechanisms guiding CRC growth is essential. The results of The Cancer Genome Atlas (TCGA) dataset analysis demonstrated a critical role of signal transducer and activator of transcription 3 (STAT3) pathway in tumor immune suppression via modulation of the recruitment of Treg cells and M2 type tumor-associated macrophages. The in vivo experiments elucidate that targeting STAT3 pathways markedly reduce the proportions of TAMs and Tregs by inhibiting tumor progression. These findings revealed crosstalk between Treg cells and M2 macrophages, proving a potential therapeutic strategy for CRC therapy. Combinatorial treatment with STAT3 inhibitor and programmed death 1 (PD-1) antibody therapy effectively prevents CRC tumor growth in a mouse model with high anti-tumor immunity. In summary, targeting STAT3 disrupts the interaction between Treg cells and M2 macrophages and improves the anti-tumor response in CRC, thereby offering a promising strategy to treat patients with CRC.
KEYWORDS: Tregs, M2 macrophages, Crosstalk, STAT3, CRC
Introduction
Colorectal cancer (CRC) is the third most common cancer and a major cause of cancer-related mortality worldwide1. Several mechanisms have been described with respect to cancer development2. The molecular pathways are activated through colorectal carcinogenesis, progress, and metastasis3. A comprehensive analysis of molecular mechanisms underlying colorectal tumors proposed several targeting small molecular inhibitors4. Although the development of small molecular inhibitors has significantly improved the outcome of CRC cancer patients, adoptive resistance to these therapies remains a challenge in achieving an effective clinical response5. Thus, it is crucial to excavate the molecular mechanisms guiding CRC growth and develop new practical strategies to enhance the therapeutical efficacy in these patients.
The tumor microenvironment is composed of tumor, stromal, and immune cells that modulate the growth and progression of tumors6,7. Reportedly, tumor immune microenvironment (TME) is closely related to CRC prognosis, influenced by the composition and activity of immune cells8. Several studies have focused on the immunosuppressive effect on immune cells such as T regulatory (Treg) cells, M2-type macrophages, and myeloid-derived suppressor cells (MDSCs)9–11. Moreover, the immune checkpoint expression and interaction among immune cells determined the immune response of TME12. In addition, various chemokines and cytokines in the TME modulate the proliferation and differentiation of immune cells, thereby contributing to the immune state of tumors13. The current studies have deciphered the interaction of immune cells with other cells14 . For instance, cancer associated fibroblasts assembled a physical barrier and restricted CD8+ T cells recruitment by secreting various chemokines and cytokines15. However, the correlation among suppressive immunes such as macrophages is yet to be elucidated in CRC.
Tumor-associated macrophages (TAMs) are the most prominent immune cells, exhibiting different subtypes, such as M2 macrophages, in regulating TME by various microenvironment signals16,17 and have been shown to regulate CRC tumor cells in tumor metastases18. Noteworthy, CRC cells derived IL-6 stimulated M2 macrophage polarization, in turn promoting the migration of CRC cells19. Interestingly, cancer cells undergo IL-4 excretion by extracellular matrix (ECM), which promotes macrophage proliferation, modulating the CRC metastasis20. The interaction between cancer cells and macrophages is modulated by the pathways, which in turn proposes a potential small molecular inhibitor in CRC therapies21. Moreover, cancer associated fibroblast (CAFs) also regulate the recruitment of macrophages and promote the differentiation into protumorigenic macrophages via various regulatory molecules, thereby inducing immune suppression in the TME22. Additionally, CAFs and M2 macrophages present a positive interaction through SDF-1/CXCR4 axis for progression23. Thus, an in-depth understanding between TAMs and other suppressive immune cells would explain the mechanisms of macrophages and further reveal the potential molecular targets for CRC therapy.
Given the importance of TAMs in the immune suppressive state and CRC progression, we speculated that the crosstalk between M2 macrophages and Treg cells modulates immune suppression and promotes tumor growth. In this study, we found that M2 macrophages were significantly correlated with tumor infiltrating Treg cells in CRC. The TCGA datasets demonstrated that the STAT3 pathway plays a critical role in tumor immune suppression by modulating the recruitment of Treg cells and M2 macrophages. The in vivo experiments elucidate that targeting STAT3 pathways markedly reduced the proportions of TAMs and Tregs by inhibiting tumor progression. These findings revealed a crosstalk between Treg cells and M2 macrophages, proving a potential therapeutic strategy for CRC therapy.
Materials and methods
Materials and regents
Antibodies for immunohistochemical staining: anti-CD8α (D4W2Z, 98941), Foxp3 (D6O8R, 12653S), F4/80 (D2S9R, 70076S), CD206 (E6T5J, 24595S) were obtained from Cell Signaling Technology (Danvers, MA). TUNEL Reagent Test Kit (11684795910) was purchased from Roche (Shanghai, China).
Antibodies for Flow Analysis: CD8α-PerCP-Cy5.5 (53–6.7, 561109), CD25-BV510 (PC61, 563037), CD279 (PD-1)-BV605 (RMP1–30, 748267), CD3-BV650 (145-2C11, 564378), IL-10-BV711 (JES5-16E3, 564081), IL-17A-BV786 (TC11-18H10, 564171), CD45-BUV395 (30-F11, 565967), CD366 (Tim-3)-PE (5D12, 566346), STAT3-PE-CF594 (4/P-STAT3, 562673), CD4-PE-Cy7 (GK1.5, 563933), CD206-Alexa Fluor® 647 (MR5D3,565250), F4/80-BV510 (T45-2342,743280) were all obtained from BD Bioscience (San Jose, CA, USA). Foxp3-Alexa Fluor® 647 (150D, 320014), TGF-β1-FITC (TW7-16B4, 141414), Granzyme B-BV421(QA18A28, 396414) were purchased from BioLegend (San Diego, CA, USA). Fixable viability Dye eFluorTM 780 (65-0865-14) were purchased from eBioscienceTM (San Diego, CA, USA).
Cell culture
CRC cells CT26 were purchased from ATCC (Virginia, USA) and incubated in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Waltham, USA) with 10% fetal bovine serum (FBS) (Gibco, Waltham, USA) and 1% penicillin/streptomycin (Gibco, Waltham, USA).
Correlation of CRC infiltrated Tregs and M2 macrophages
Colon adenocarcinoma (COAD) (n = 458) and rectum adenocarcinoma (READ) (n = 166) data were downloaded from The Cancer Genome Atlas (TCGA) database using Tumor Immune Estimation Resource (TIMER2.0) (http://timer.cistrome.org). The correlation between tumor infiltrated Tregs and M2 macrophages) was estimated by their coding genes. The correlation of the Treg coding gene FOXP3 with M2 macrophages was analyzed in both COAD and READ tumors. Similarly, M2 macrophages coding genes (CD163 and MRC1) were correlated with tumor infiltrated Treg cells in CRC patient cohorts. Moreover, the expression of cytokines including IL-10 and TGF-β1 has been estimated both on tumor infiltrated Tregs and M2 macrophages in COAD and READ by using Gene Expression Profiling Interactive Analysis (GEPIA) online (http://gepia.cancer-pku.cn).
Differential expressed gene in the tumor tissues
To reveal the interaction between Tregs and M2 macrophages in CRC, we compared the differential expressed genes in the development of CRC tumors from TCGA datasets (GSE146587). Genes presented according fold change > 1 and padj < 0.05 in the CRC tumors. Cytokines and chemokines were also analyzed in CRC tumor compared to normal tissues. Moreover, gene set enrichment analysis of cancer pathways was presented in normal tissues versus tumors using KEGG analysis.
Mouse model
All animal experiments were performed in accordance with the guidelines approved by the institutional Animal Care and Use Committee of Tongji University.
Efficacy of targeting STAT3 in vivo of CRC mouse model
Six-week-old BALB/c mice were purchased from Shanghai SLAC Animal Laboratory Co. Ltd. (Shanghai, China) and fed at the Animal Center of Tongji University. CT26 cells (1 × 106 cells) were administered into the right limbs of BALB/c mice. Tumor size was determined on the indicated days using the formula (width)2 × length/2. After the tumor grew into 100 mm3, mice were treated with different strategies and segregated into four groups according to the tumor therapy received: phosphate-buffer saline (PBS), STAT3 inhibitor (5 µM, MedChemExpress, New Jersey, USA), PD-1 mAb (10 µg/mL, Bioxcell, Lebanon, NH, USA) or combinatorial treatment with STAT3 inhibitor (5 µM) and PD-1 mAb (10 µg/mL), a replicate for each treatment group for each day for intraperitoneal injection. At the endpoint of experiments, all mice were euthanized, and organs were harvested for flow cytometry and histopathological analysis.
Flow cytometry for peripheral blood
After treatment, mouse blood was obtained and treated with red blood cell (RBC) lysis buffer. Peripheral blood mononuclear cells (PBMCs) were cultured in RPMI 1640 medium and then labeled with different antibodies at 4°C for extracellular and intercellular staining. Fixable viability Dye eFluorTM 780 (Invitrogen, Waltham, MA, USA) was used to exclude dead cells from subsequent flow cytometric analysis. Cells were resuspended in staining buffer and blocked with anti-mouse CD16/32 Fc block (BioLegend, clone 2.4G2,1:50) at 4°C for 15 min. For extracellular staining, single cells were incubated with anti-mouse CD8α-PerCP-Cy5.5, CD25-BV510, PD-1-BV605, CD3-BV650, CD45-BUV395, Tim-3-PE, CD4-PE-Cy7, F4/80-BV510 with staining buffer at 4°C in the dark for 30 min. Then, the samples were fixed and permeabilized with a fix-perm buffer for 45 min, followed by intracellular staining (IL-10-BV711, IL-17A-BV786, STAT3-PE-CF594, Foxp3-Alexa Fluor® 647, TGF-β1-FITC, Granzyme B-BV421 and CD206-Alexa Fluor® 647). All buffer solutions were used in accordance with the manufacturer’s instructions (BD Bioscience). The CD4+ T cells, cytotoxic CD8+ T cells, Tregs and macrophages were analyzed via BD LSRFortessa.
Tumor single cell preparation and staining
Tumor specimens from BALB/c mice were minced and enzymatically digested in DMEM supplemented with Collagenase D (2 mg/mL, Sigma) and DNase I (0.5 mg/mL, Sigma) at 37°C for 45 min with sharking. The cell lysates were strained through a 70-μm cell strainer and resuspended in a staining buffer. For mice spleen, samples were minced and prepared through a 70-μm cell strainer. Then, fixable viability Dye eFluorTM 780 was used for flow cytometric analysis to exclude the debris. The cells were resuspended in a staining buffer and blocked with anti-mouse CD16/32 Fc block at 4°C for 15 min. The staining suspensions of blood samples were prepared as described above.
Histopathological analysis
Tissue samples from colon tumors and other organs of mice were fixed with paraformaldehyde and embedded in paraffin. Samples were cut into 5-µm sections and stained with hematoxylin and eosin (H&E). Tumor-associated immune cells were analyzed with histopathological and immunohistochemical staining (IHC). The histopathological toxicity was assessed in the main organs, including the liver, heart, spleen, kidney, and lung. Tumor H&E and TdT-mediated dUTP Nick-End Labeling (TUNEL) staining were performed to determine the different treatments’ efficacies. Immunohistochemical staining including CD8, Foxp3, F4/80, and CD206 were performed for all CRC tissues. Images were obtained using NanoZoomer S210 (Hamamatsu, Hamamatsu, Japan) under a 10X objective lens. The positive cells were quantitated and analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Digitally scanned slices were imaged under 5× and 10× objective lenses. The statistical differences were calculated using ImageJ software after four treatments.
Statistical analysis
GraphPad Prism v.9.3 software (San Diego, CA, USA) was performed to analyze the data. Two-tailed paired or unpaired Student’s t-test was used to estimate the difference between the two groups. Multiple-group comparisons were analyzed by one- or two-way ANOVA with multiple comparisons. Statistical significance is indicated as * p < .05, ** p < .01, *** p < .001, **** p < .0001.
Results
Positive correlation between tumor-infiltrated Tregs and M2 macrophages in human CRC cohorts
The tumor immune microenvironment (TIME) composes innate and adaptive immune cells with the tumor progression. The proportion of immunosuppressive cells such as Treg cells, MDSCs, and M2-type macrophages were significantly increased in tumor development. The evaluation of COAD-TCGA dataset found that FOXP3 gene expression is correlated with tumor-associated M2 macrophages (Figure 1(a)). Similarly, a positive correlation was established between M2 macrophages cell genes (CD163 and MRC1) with tumor-associated Tregs (Figure 1(a)). A similar trend was estimated in the READ cohorts, suggesting that Tregs might regulate M2 macrophage infiltration into CRC tumors. Notably, anti-tumor immune response was determined by the composition of immune cells, as well as the chemokines and cytokines expression and interaction within the associated matrix24. In Figure 1b, we observed that cytokines IL-10 coding gene expression of IL-10 and TGF-β (TGF-β1) correlated with tumor infiltrated Tregs both in COAD and READ, revealing that the cytokines are crucial for Tregs infiltration (Figure 1(b)). Moreover, we also found that M2 macrophages demonstrated a high positive correlation with different cytokines IL-10 and TGF-β in human CRC cohorts (Figure 1(b)). These results indicated that Tregs and M2 macrophages have strong connection in colorectal cancer.
Figure 1.
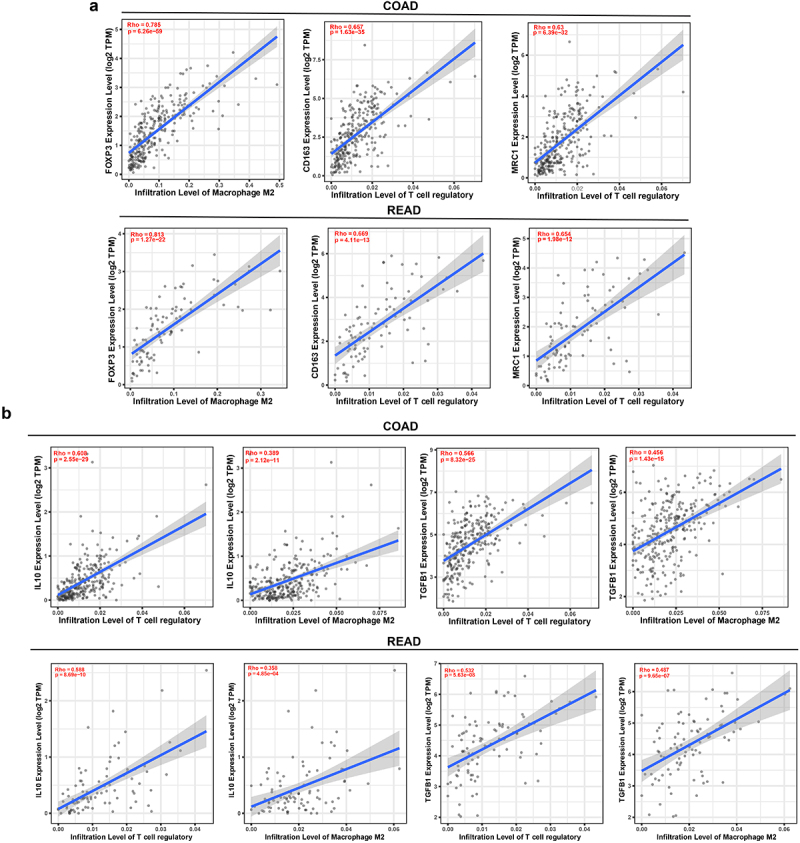
Correlations between Tregs and M2 macrophages in colorectal cancer. (a) A positive correlation between Treg coding gene FOXP3 with M2 macrophage. M2 macrophage coding genes CD163 and MRC1 presented high positive correlations with tumor infiltrated Tregs in both COAD (n=458) and READ (n=166). (b) Cytokines IL-10 coding gene IL-10 and TGF-β coding gene TGF-β1 demonstrated a high positive correlation neither in Tregs or M2 macrophages in CRC patients’ cohorts (COAD and READ).
Crosstalk between tumor infiltrated Tregs and M2 macrophages
To reveal the interaction between Tregs and M2 macrophages in CRC, we compared the differential expressed genes in the development of CRC tumors from TCGA datasets. As shown in Figure 2a, Tregs coding foxp3 and macrophage coding gene CD163 were up-regulated in tumor tissues compared to normal tissues. Treg and Macrophages related cytokines and chemokines such as TGF-β1, IL-6, IL-1β, CCL3 and CXCL2 were also enhanced in the development of colorectal cancer (Figure 2(a)). Moreover, we found that cytokine and cytokine receptor and JAK-STAT3 signaling pathways regulated the TME in tumor progression (Figure 2(b)). Subsequently, we observed that tumor-associated Tregs are correlated with STAT3 expression in COAD and READ cohorts with coefficients of 0.525 and 0.615, respectively (Figure 2(c)). M2 macrophages also demonstrated a positive correlation with STAT3 levels in the CRC TME (Figure 2(c)). Since TGF-β and IL-10 cytokines were secreted both in Tregs and M2 Macrophages, we aimed to explore the regulation of STAT3 in cytokine secretion. Strikingly, STAT3 emerges as a regulator of TGF-β and IL-10 expression via a positive interaction of marker genes TGF-β1 and IL-10 in COAD TME (Figure 2d). On the other hand, a significant connection was detected between TGF-β1 and IL-10 with immune suppressive Treg cells in READ patients (Figure 2d). These results indicated that STAT3 serves as a crosstalk between Tregs and M2 macrophages in CRC progression.
Figure 2.
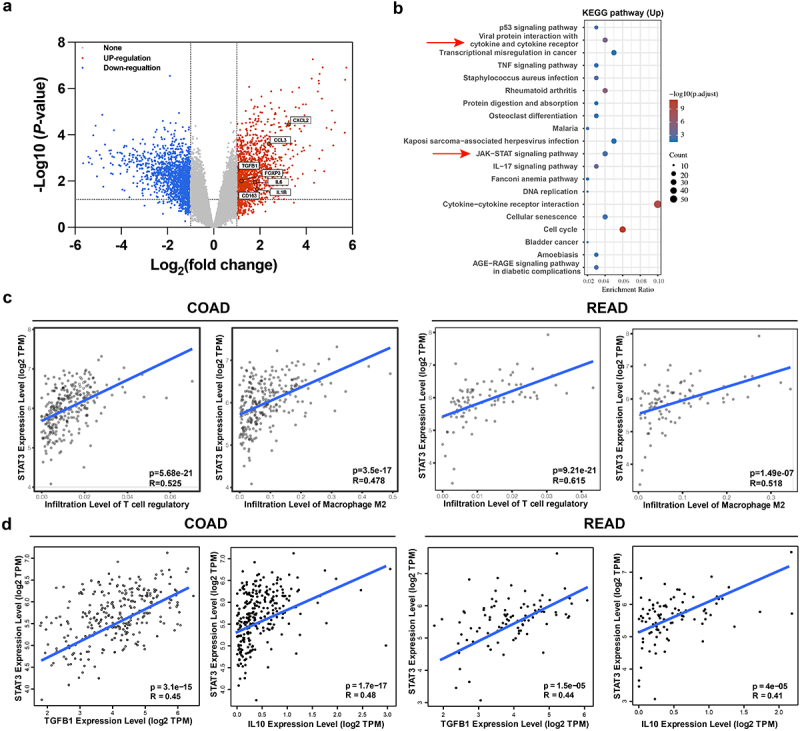
Crosstalk between Tregs and M2 macrophages in colorectal cancer. (a) Volcano plot of differentially expressed genes of human RNA-sequencing data from TCGA dataset (GSE146587). Genes presented according fold change >1 and padj <0.05 in the CRC tumors. Treg coding gene FOXP3 and M2 macrophage coding gene CD163 were significantly upregulated in the tumors. Cytokines IL-6, TGFβ1 and IL1β and chemokines CXCL2 and CCL3 that were markedly increased in CRC tumor. (b) Gene set enrichment analysis of cancer pathways were presented in normal tissues versus tumors using KEGG analysis. Cytokine and cytokine receptor and JAK-STAT3 signaling pathways regulated the TME in tumor progression. (c) Positive correlation between STAT3 with Treg or M2 macrophage both in COAD and READ cohorts. (d) Cytokines IL-10 and TGF-β coding gene IL-10 and TGF-β1 presented high positive correlations with tumor infiltrated Tregs and M2 macrophage in COAD and READ by Gene Expression Profiling Interactive Analysis (GEPIA).
Targeting STAT3 reduced the recruitment of Tregs and M2 macrophages in CRC mouse model
Herein, we established a CRC mouse model to elucidate the therapeutic effect on STAT3 in vivo. While using a STAT3 inhibitor Stattic, we found that the proportion of Treg cells both in spleen and tumor samples significantly decreased by analyzing flow TSNE data (Figure 3(a–b)). Moreover, spleen associated CD8+ T cells were enhanced in STAT3 inhibitor treatment group (Figure 3(a)), and abundant cells were recruited into tumors after STAT3 pathway inhibition (Figure 3(b)), implying an excellent anti-tumor anti-immunity in CRC mouse tumors. To characterize the infiltration of immune cells and the efficacy of STAT3 inhibitor (Stattic), we performed tumor H&E and IHC (Figure 3(c–d)). Briefly, after STAT3 treatment, tumor cell necrosis and apoptosis accumulated in CRC tumors (Figure 3(c)). To map the immune hallmarks in the tumor, IHC results were analyzed in the inhibition of STAT3 pathway. Strikingly, a larger number of CD8+ T cells were infiltrated into tumor nests and exerted anti-tumor immunity via STAT3 inhibitor (Figure 3(c–d)). Consistent with flow cytometry results, tumor-associated Foxp3 Treg cells were depleted after the suppressed of STAT3 pathway in the CRC tumor mouse model (Figure 3(c–d)). Importantly, not only the target Treg cells but also M2 types of tumor-associated macrophages were depleted by Stattic. In the crosstalk of Treg cells and macrophages, STAT3 regulates the recruitment of the two immune cells. The inhibition of STAT3 pathway decreased the level of M2 macrophage marker CD206 with no alteration of F4/80 expression (Figure 3(c–d)). These results concluded that inhibition of STAT3 reshapes the immune TME and further ameliorates the anti-tumor immunity of CRC mouse model.
Figure 3.
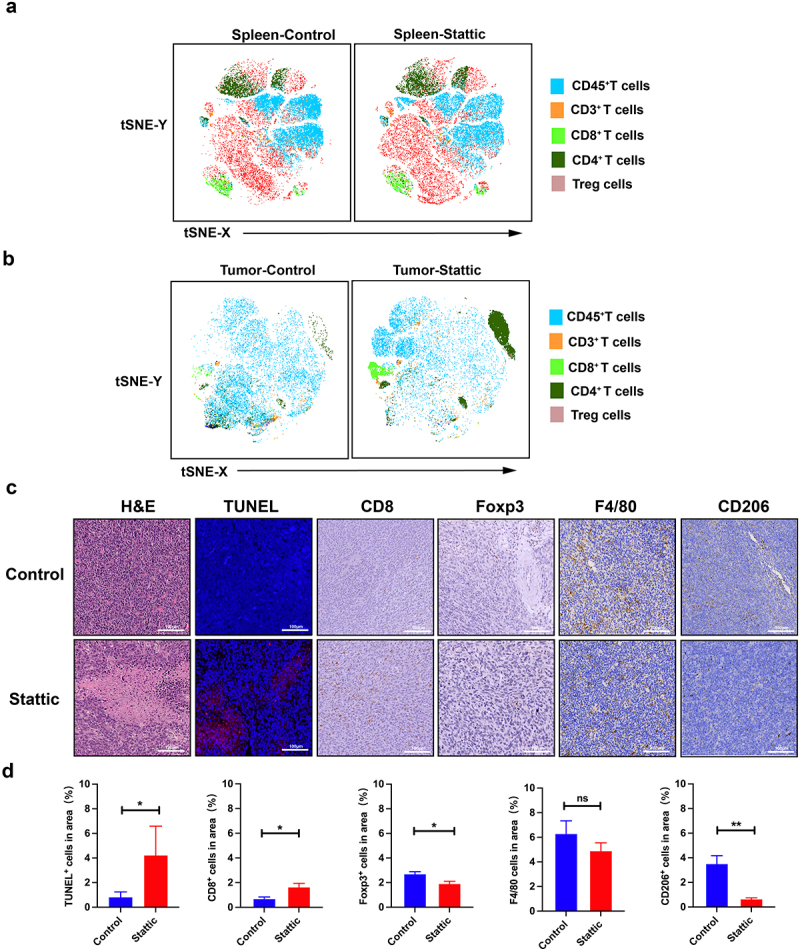
Anti-tumor immunity of STAT3 inhibitor in the CT26 mouse model. (a) tSNE map of spleen associated CD45+T cells, CD3+T cells, CD4+T cells, CD8+T cells and Treg cells after Stattic therapy. Spleen associated CD8+ T cells were enhanced in STAT3 inhibitor treatment group. (b) Tumor tSNE map of the analyzed tumor-associated CD45+T cells, CD3+T cells, CD4+T cells, CD8+T cells and Treg cells after Stattic treatment. (c) Tumor necrosis in tumor sections, as indicated through H&E staining. Apoptosis in tumor sections was examined through TUNEL staining. Immunohistochemical staining for CD8, Foxp3, F4/80 and CD206 in the tumor after STAT3 inhibitor therapy. A larger number of CD8+ T cells were infiltrated into tumor nests and exerted anti-tumor immunity via STAT3 inhibitor. (d) Quantification of positive TUNEL cells, CD8+, Foxp3+, F4/80+ and CD206+ cells in the CRC mouse model after STAT3 inhibitor treatment. Tumor-associated Foxp3 Treg cells were depleted after the suppressed of STAT3 pathway.
Targeting STAT3 enhanced anti-PD-1 therapeutic efficacy in the CRC mouse model
Previous studies highlighted that only 15% of MSI-H CRC patients responded adequately to PD-1 mAb with an improved survival rate25. However, a larger number of CRC patients gained only a few benefits from anti-PD-1 treatment26. Thereby, it is crucial to develop new practical strategies to enhance ICB efficacy in CRC patients. To address this issue, we assessed the characteristics of a combination Stattic with PD-1 mAb in CRC mouse models (Figure 4(a)). First, we analyzed the impact of STAT3 inhibitor combined ICB on tumor growth in CRC (Figure 4(b)). Compared to the IgG control group, anti-PD-1 with or without Stattic inhibited tumor growth, but the combined therapy had a stronger inhibitory effect compared with the monotherapy (Figure 4(b–c)). Consistently, the combination therapy suppressed tumor regression and increased the mice response to STAT3 inhibitor by markedly decreasing the relative tumor size compared to IgG control or anti-PD-1 monotherapy (Figure 4(d)). Finally, we performed H&E and TUNEL staining to confirm the combination effect of Stattic and PD-1 mAb in mouse cancer tissues (Figure 4(e)). In agreement with tumor volume, tumor cell necrosis and apoptosis were markedly increased in combined STAT3 inhibitor and PD-1 mAb treatment compared to the anti-PD-1 sets, suggesting an excellent therapeutic effect on CRC tumors (Figure 4(f)). These data established a promising anti-cancer strategy by combination of Stattic with PD-1 mAb in CRC mice.
Figure 4.
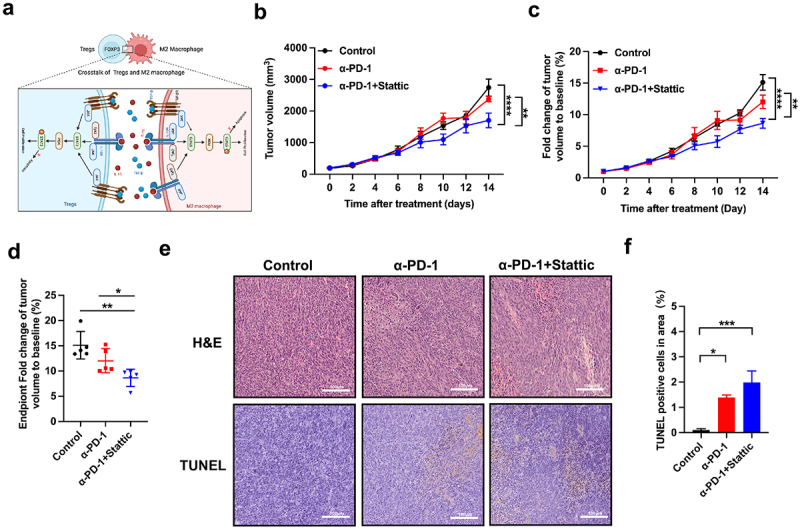
Anti-tumor response of Stattic and PD-1 mAb in the CT26 mouse model. (a) Schematical mechanism of the interaction of tumor-associated Treg and macrophages in the CRC tumor. IL-10 activating its receptor leads to STAT3 phosphorylation, promoting cell proliferation both in Treg and macrophage. TGF-β interacts with its receptor with subsequent PI3K and STAT3 activation. These two pathways promote proliferation and activation of Tregs and macrophages indicate alternative target upon inhibition of STAT3 in the CRC treatment. (b) Tumor growth volume curve of CT26 tumors after treatment with PD-1 mAb, and combinatorial Stattic and PD-1 mAb therapy. The combined therapy had a stronger anti-tumor effect compared with the monotherapy (n = 5; **** indicated p < .0001, ** indicated p < .01). (c) Fold changes of tumor volume at different therapies in CRC tumors (n = 5; **** indicated p < .0001, ** indicated p < .01). The combination therapy suppressed tumor regression and increased the mice response. (d) Fold changes of tumor volume at experiment endpoint after combination therapy with Stattic and PD-1 mAb compared with control or PD-1 mAb in CT26 mouse model (n = 5; **indicated p < .01, * indicated p < .05). (e,f) Tumor necrosis in tumor sections, as indicated through H&E staining. Apoptosis in tumor sections was examined through TUNEL staining. Quantification of apoptotic cells were presented in the combination therapy with Stattic and PD-1 mAb (n = 5; *** indicated p < .001, * indicated p < .05).
Combination Stattic with anti-PD-1 reshaped CRC tumor immune microenvironment
To further explore the TME of CRC after combination therapy, we analyzed the tumor infiltrated immune cells by flow cytometry and found that combining Sttatic with anti-PD-1 treatment increased the frequency of tumor infiltrated CD8+ and CD4+ T cells compared with single anti-PD-1 therapy (Figure 5(a–b)). A markedly enhanced proportion of tumor infiltrated CD8+ T cells were also found in combination group than IgG control ones (Figure 5(a)). Similarly, CD4+ T cells showed a slightly increased in combination with no significance (Figure 5(b)). Meanwhile, a higher CD8+/CD4+ ratio was presented in colorectal cancer after combined Sttatic with anti-PD-1 treatment (Figure 5(c)). Expectedly, tumor infiltrated Treg cells were markedly decreased in the combination therapy when compared to control group (Figure 5(d)). Interestingly, combined Stattic and anti-PD-1 therapy did not change the infiltration of F4/80+ cells (Figure 5(e)). While, M2 macrophages marker CD206 on F4/80+CD11b+ cells significantly decreased in the Sttatic with anti-PD-1 treatment (Figure 5(f)). Thus, targeting STAT3 regulates the crosstalk between Treg cells and M2 macrophages and reactive tumor immunity and enhances anti-PD-1 therapy of CRC tumors.
Figure 5.
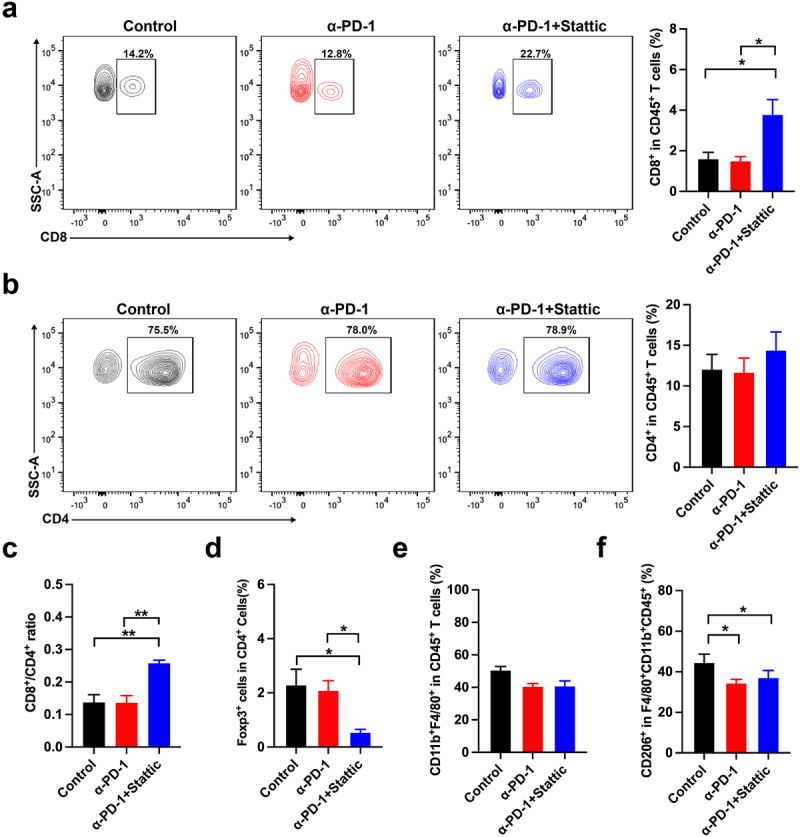
Stattic enhanced the immune response of PD-1 mAb therapy in the CT-26 mouse model. (a) Contour images and quantification of tumor infiltrated CD8+T cells in the CRC mouse model. (b) Tumor-associated CD4+ T cells were presented and quantified by flow analysis. (c) Analysis of the ratio of CD8+T cells to CD4+T cells in the tumors after combinatorial Stattic and PD-1 mAb therapy. (d) Percentages of CD4+Foxp3+ cells from CT26 tumors upon combinatorial Stattic and PD-1 mAb therapy compared to control groups. (e) CD11b+F4/80+ cells in the tumor after combinatorial Stattic and PD-1 mAb therapy. (f) Quantification of M2 macrophages marker CD206 positive cells from tumors after different therapies. Significance is indicated as * p < .05 or ** p < .01.
To better understand the therapeutic effect in vivo, we analyzed the data using flow TSNE analysis. When using a STAT3 inhibitor Stattic with anti-PD-1 therapy, we found that the proportion of CD8+ T cells and CD4+ T cells in tumor infiltrated T cells map were significantly increased of flow TSNE data (Figure 6(a)). A slight decrease of Treg cells was also found in the combination group (Figure 6(a)). These results were consistent with above data (Figure 5(d)). A further comparison of molecular characteristics of immune cells was revealed in the three groups. The MFI level of TIM-3+CD8+T cells was decreased in the TME after combination therapy, indicating active CD8+T cells (Figure 6(b)). Given the critical role of IL-10 and TGF-β in tumor immune suppression by activated Treg and M2 macrophages, we analyzed the cytokine production in the TME. Strikingly, the production of IL-10 and TGF-β on Treg cells was reduced, and MFI expression of TGF-β on CD206 M2 macrophages was observed after combination therapy, suggesting an increased abundance of activated anti-tumor immunity (Figure 6(d–e)). Furthermore, systemic toxicity was conducted in different therapeutic groups, wherein we did not find any significant damage in the main organs, including the heart, liver, spleen, lung and kidney (Figure 6(f)). Finally, we concluded that combined PD-1 mAb and STAT3 inhibitor decreased the number of suppressive immune cells in CRC tumors associated with increased anti-PD-1 efficacy.
Figure 6.
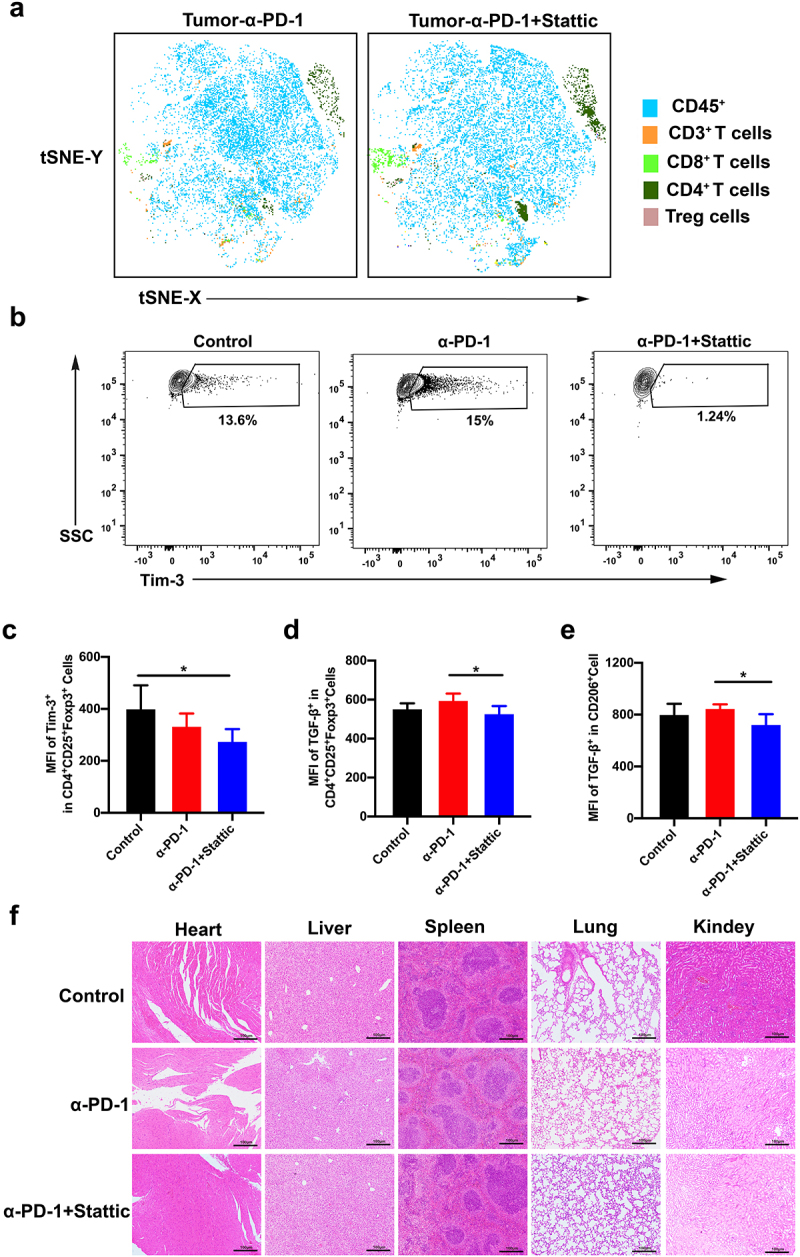
Immune response of tumor microenvironment after combinatorial STAT3 inhibitor and PD-1 mAb therapy in the CT26 mouse model. (a) tSNE analysis for CD45+ T cells, CD3+ T cells, CD4+ T cells, CD8+ T cells and Treg cells in the tumor after combinatorial Stattic and PD-1 mAb therapy. (b-c) Represented contour images (b) and quantification of Tim-3 (c) in Treg cells after combinatorial Stattic and PD-1 mAb therapy in CRC tumor. (d-e) Quantification of MFI for TGF-β both in Tregs and M2 macrophages in CRC mouse model after different treatment. (f) No significant organ toxicity was assessed through H&E staining of samples of the heart, liver, spleen, lung, and kidneys in mouse after treatment.
Discussion
Recently, progress has been made in understanding the cancer cell intrinsic mechanisms in cancer development27. Numerous publications have described how malignant cells in the TME regulate tumor progression and metastasis by interactions between cancer cells and other cells8. Researchers have focused on the cellular and molecular characteristics of the interaction of cells in the CRC TME28. Cancer associated fibroblasts assembled a physical barrier and restricted CD8+ T cells recruitment by secreting different chemokines and cytokines15. Presently, the TAMs regulate CRC tumor cells in tumor metastases18. Noteworthy, CRC cells derived IL-6 stimulated M2 macrophage polarization, in turn promoting the migration of CRC cells19. However, the correlation among suppressive immune cells, such as macrophages, is poorly known in CRC. An in-depth understanding between TAMs and other suppressive immune cells elucidates the mechanisms effectuated by macrophages and further reveals the potential molecular targets for CRC therapy. In the present study, a positive correlation was established between CRC infiltrated Tregs and M2 macrophages. Next, we investigated the efficacy of STAT3 inhibitor and the altered tumor-infiltrated Tregs and M2 macrophages by targeting STAT3 in a CRC mouse model. The current results demonstrated that targeting STAT3 enhances the anti-tumor efficacy of anti-PD-1 by decreasing Tregs and M2 macrophages infiltration, which increased anti-tumor immunity.
Notably, macrophages are suppressive immune cells in the TME and regulate CRC tumor cells in tumor metastases18. Cancer associated fibroblasts produce M-CSF, IL-6, and CCL2 promoting macrophage polarization into M2 macrophages with higher CD163 expression22. Therefore, the suppressive immune cells Tregs and M2 macrophages might interact in tumor progression. Human datasets provided a positive correlation between Tregs and M2 macrophages in both COAD and READ. Given the key role of cytokines in cell–cell interaction, we also screened the expression of IL-10 and TGF-β genes. As expected, IL-10 and TGF-β presented a high correlation with tumor-infiltrated Tregs and M2 macrophages in human CRC cohorts. As key cytokines to Treg cell proliferation and differentiation, IL-10 is complicated by tumor-genesis and progression in multiple cancers21. While, we did not estimate the changes of tumor infiltrated Tregs and M2 macrophages after IL-10 and TGF-β blockade. Previous studies demonstrated that increased expression of IL-10 activates the PI3K and STAT3 pathway of Tregs in PD-1-resistant and radioresistant cancer29,30. IL-10 also regulated the polarization and proliferation of macrophages in the development of TME by suppressing immunity31. Based on the previous and current data, we proposed that STAT3 regulates the inaction between Tregs and macrophages in CRC.
Aberrant JAK/STAT3 signaling has been identified as a crucial factor in tumor immunity involving tumor growth and metastasis32. JAK family proteins regulate various cellular cytokines receptors, such as the IL-6, IL-10, and interferon-γ (IFN-γ), by STAT3 phosphorylation and activation.33 In melanoma, STAT3 phosphorylation activates the proliferation of tumor infiltrated Treg cells.30 Moreover, TAMs promote tumor progression and drug resistance by activating the JAK-STAT3 pathway34. Thus, potent STAT3 inhibitors have been developed and employed in multiple cancers. Based on the analysis of the correlation of STAT3 with tumor-infiltrated Treg cells and M2 macrophages, we hypothesized STAT3 as a crosstalk regulating the interaction between suppressive immune cells. Using STAT3 inhibitor, a high anti-tumor immunity was estimated by high infiltrated CD8+ T cells. Moreover, the proportion of CRC tumor-associated M2 macrophages and Treg cells were reduced after using Stattic which resulted in high tumor therapeutic efficacy. These results showed that inhibition STAT3 remodeled the tumor immune microenvironment and further ameliorated the anti-tumor immunity in the CRC.
As only colorectal cancer responded worse to the treatment of PD-1 mAb, a strategy by enhancing ICB efficacy was needed. Combined STAT3 inhibitor with PD-1 mAb, tumor growth was significantly inhibited and rescued the suppressed immunologic tumor microenvironment. In our study, we found that combinatorial therapy significantly enhanced the tumor infiltrated CD8+ T cells and decreased the suppressive cells such as Tregs and macrophages. Moreover, the checkpoint Tim-3 expression on CD8+ T cells was significantly suppressed after using combined treatment. Decreased promotion of Tim-3+CD8+ T cells indicated activated CD8+T cells in CRC tumor. These findings facilitated STAT3 as a promising target in cancer immunotherapy, while the influence of STAT3 in other immune cells, such DCs, and MDSCs, has not been identified. Moreover, the combination of STAT3 inhibitors with other therapies, such as a vaccine, oncolytic adenovirus, and other ICB, could be explored further.
Taken together, the preset study showed that treatment with STAT3 inhibitor and anti-PD-1 therapy prevents tumor growth of CRC in a mouse model with high anti-tumor immunity. However, underlying mechanisms need to be elucidated in future studies. Also, the potential synergism of the STAT3 inhibitor with other ICBs or immune agents need to be investigated. In summary, targeting STAT3 disrupts the interaction between Treg cells and M2 macrophages and improves the anti-tumor response in CRC, thereby providing a promising strategy to treat patients with CRC.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No.: 82172735, 81803090 and 81902896); Shanghai Pujiang Program (No.: 21PJD055); and Pujiang Fostering Program of Shanghai Tenth Peoples’ Hospital under grant (No.: 040118024 and 2021YPDRC016); The Shanghai Program (No.: 19YF1438300).
Biographies
Lili Huang received a PHD degree in Oncology at Tongji University, Shanghai China in 2020.
Yu Zhao received a PHD degree in Oncology at Tongji University, Shanghai China in 2016.
Mengying Shan received a Master degree in Oncology at Tongji University in 2019.
Sitong Wang received a PHD degree in Oncology at Tongji University, Shanghai China in 2020.
Jianhua Chen received a PHD degree in Oncology at Tongji University, Shanghai China in 2017.
Zhuqing Liu received a PHD degree in Oncology at Tongji University, Shanghai China in 2017.
Qing Xu received a PHD degree in Oncology at Tongji University, Shanghai China in 2017.
Funding Statement
This work was supported by grants from Fundamental Research Funds for the Central Universities (No. 15012150092, 22120220656), National Natural Science Foundation of China Foster Program of Shanghai Tenth People’s Hospital (No. 04.03.21.022), Shanghai Shenkang Hospital Development Group Medical and Enterprise Integration and Innovation Cooperation Special Project (No. SHDC2022CRT009).
Author contribution
Lili Huang: Conceptualization, Software, Writing-Original draft preparation; Yu Zhao: Methodology, Data curation; Mengying Shan: Data curation; Sitong Wang: Data curation; Jianhua Chen: Writing and Reviewing; Zhuqing Liu: Visualization, Supervision and Editing; Qing Xu: Supervision, Validation.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Ethical approval statement
All animal experiments were performed in accordance with the guidelines approved by the institutional Animal Care and Use Committee of Tongji University (Project number: SHDSYY-2020-4309, data approval: 2020-08-15).
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Schreuders EH, Ruco A, Rabeneck L, Schoen RE, Sung JJ, Young GP, Kuipers EJ.. Colorectal cancer screening: a global overview of existing programmes. Gut. 2015;64(10):1637–12. doi: 10.1136/gutjnl-2014-309086. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen LH, Goel A, Chung DC. Pathways of colorectal carcinogenesis. Gastroenterology. 2020;158(2):291–302. doi: 10.1053/j.gastro.2019.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malki A, ElRuz RA, Gupta I, Allouch A, Vranic S, Al Moustafa AE. Molecular mechanisms of colon cancer progression and metastasis: recent insights and advancements. Int J Mol Sci. 2020;22(1):22. doi: 10.3390/ijms22010130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inoue A, Robinson FS, Minelli R, Tomihara H, Rizi BS, Rose JL, Kodama T, Srinivasan S, Harris AL, Zuniga AM, et al. Sequential administration of XPO1 and ATR inhibitors enhances therapeutic response in TP53-mutated colorectal cancer. Gastroenterology. 2021;161(1):196–210. doi: 10.1053/j.gastro.2021.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itatani Y, Kawada K, Yamamoto T, Sakai Y. Resistance to anti-angiogenic therapy in cancer—alterations to anti-VEGF pathway. Int J Mol Sci. 2018;19(4):1232. doi: 10.3390/ijms19041232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizuno R, Kawada K, Sakai Y. Prostaglandin E2/EP signaling in the tumor microenvironment of colorectal cancer. Int J Mol Sci. 2019;20(24):6254. doi: 10.3390/ijms20246254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Y, Bian S, Zhou X, Cui Y, Wang W, Wen L, Guo L, Fu W, Tang F. Single-cell multiomics sequencing reveals prevalent genomic alterations in tumor stromal cells of human colorectal cancer. Cancer Cell. 2020;38(6):818–828 e815. doi: 10.1016/j.ccell.2020.09.015. [DOI] [PubMed] [Google Scholar]
- 8.Goc J, Lv M, Bessman NJ, Flamar AL, Sahota S, Suzuki H, Teng F, Putzel GG, Bank JRILC, Eberl G, et al. Dysregulation of ILC3s unleashes progression and immunotherapy resistance in colon cancer. Cell. 2021;184(19):5015–5030 e5016. doi: 10.1016/j.cell.2021.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Jin XH, Luo J, Duan JL, Cai MY, Chen JW, Feng ZH, Guo AM, Wang FW, Xie D. ITLN1 inhibits tumor neovascularization and myeloid derived suppressor cells accumulation in colorectal carcinoma. Oncogene. 2021;40(40):5925–5937. doi: 10.1038/s41388-021-01965-5. [DOI] [PubMed] [Google Scholar]
- 10.Cagnoni AJ, Giribaldi ML, Blidner AG, Cutine AM, Gatto SG, Morales RM, Salatino M, Abba MC, Croci DO, Marino KV, et al. Galectin-1 fosters an immunosuppressive microenvironment in colorectal cancer by reprogramming CD8 + regulatory T cells. Proc Natl Acad Sci USA. 2021;118(21). doi: 10.1073/pnas.2102950118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang C, Dou R, Wei C, Liu K, Shi D, Zhang C, Liu Q, Wang S, Xiong B. Tumor-derived exosomal microRNA-106b-5p activates EMT-cancer cell and M2-subtype TAM interaction to facilitate CRC metastasis. Mol Ther. 2021;29:2088–2107. doi: 10.1016/j.ymthe.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yin Y, Liu B, Cao Y, Yao S, Liu Y, Jin G, Qin Y, Chen Y, Cui K, Zhou L, et al. Colorectal cancer-derived small extracellular vesicles promote tumor immune evasion by upregulating PD-L1 expression in tumor-associated macrophages. Adv Sci. 2022;9(9):2102620. doi: 10.1002/advs.202102620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, Li J, Wang G, Lan Z, Li J, et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell. 2019;35(4):559–572 e557. doi: 10.1016/j.ccell.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei C, Yang C, Wang S, Shi D, Zhang C, Lin X, Liu Q, Dou R, Xiong B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol Cancer. 2019;18(1):64. doi: 10.1186/s12943-019-0976-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang TX, Tan XY, Huang HS, Li YT, Liu BL, Liu KS, Chen X, Chen Z, Guan XY, Zou C, et al. Targeting cancer-associated fibroblast-secreted WNT2 restores dendritic cell-mediated antitumour immunity. Gut. 2022;71(2):333–344. doi: 10.1136/gutjnl-2020-322924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, Zhang W, Huo M, Wang P, Liu X, Wang Y, Li Y, Zhou Z, Xu N, Zhu H. XBP1 regulates the protumoral function of tumor-associated macrophages in human colorectal cancer. Signal Transduct Target Ther. 2021;6(1):357. doi: 10.1038/s41392-021-00761-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu L, Liu Y, Kong X, Wu R, Peng Q, Zhang Y, Zhou L, Duan L. Fusobacterium nucleatum facilitates M2 macrophage polarization and colorectal carcinoma progression by activating TLR4/NF-kappaB/S100A9 cascade. Front Immunol. 2021;12:658681. doi: 10.3389/fimmu.2021.658681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang C, Ou R, Chen X, Zhang Y, Li J, Liang Y, Zhu X, Liu L, Li M, Lin D, et al. Tumor cell-derived SPON2 promotes M2-polarized tumor-associated macrophage infiltration and cancer progression by activating PYK2 in CRC. J Exp Clin Cancer Res. 2021;40:304. doi: 10.1186/s13046-021-02108-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong Q, Fang Y, Lai Q, Wang S, He C, Li A, Liu S, Yan Q. CPEB3 inhibits epithelial-mesenchymal transition by disrupting the crosstalk between colorectal cancer cells and tumor-associated macrophages via IL-6R/STAT3 signaling. J Exp Clin Cancer Res. 2020;39(1):132. doi: 10.1186/s13046-020-01637-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin X, Wang S, Sun M, Zhang C, Wei C, Yang C, Dou R, Liu Q, Xiong B. MiR-195-5p/NOTCH2-mediated EMT modulates IL-4 secretion in colorectal cancer to affect M2-like TAM polarization. J Hematol Oncol. 2019;12(1):20. doi: 10.1186/s13045-019-0708-7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Liu Q, Yang C, Wang S, Shi D, Wei C, Song J, Lin X, Dou R, Bai J, Xiang Z, et al. Wnt5a-induced M2 polarization of tumor-associated macrophages via IL-10 promotes colorectal cancer progression. Cell Commun Signal. 2020;18(1):51. doi: 10.1186/s12964-020-00557-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stadler M, Pudelko K, Biermeier A, Walterskirchen N, Gaigneaux A, Weindorfer C, Harrer N, Klett H, Hengstschlager M, Schuler J, et al. Stromal fibroblasts shape the myeloid phenotype in normal colon and colorectal cancer and induce CD163 and CCL2 expression in macrophages. Cancer Lett. 2021;520:184–200. doi: 10.1016/j.canlet.2021.07.006. [DOI] [PubMed] [Google Scholar]
- 23.Teng F, Tian WY, Wang YM, Zhang YF, Guo F, Zhao J, Gao C, Xue FX. Cancer-associated fibroblasts promote the progression of endometrial cancer via the SDF-1/CXCR4 axis. J Hematol Oncol. 2016;9(1):8. doi: 10.1186/s13045-015-0231-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, McSkane M, Baba H, Lenz HJ. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - a target for novel cancer therapy. Cancer Treat Rev. 2018;63:40–47. doi: 10.1016/j.ctrv.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aguiar PN Jr., Tadokoro H, Forones NM, de Mello RA. MMR deficiency may lead to a high immunogenicity and then an improvement in anti-PD-1 efficacy for metastatic colorectal cancer. Immunotherapy. 2015;7(11):1133–1134. doi: 10.2217/imt.15.84. [DOI] [PubMed] [Google Scholar]
- 26.Damilakis E, Mavroudis D, Sfakianaki M, Souglakos J. Immunotherapy in metastatic colorectal cancer: could the latest developments hold the key to improving patient survival? Cancers Basel. 2020;12(4):12. doi: 10.3390/cancers12040889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang W, Kandimalla R, Huang H, Zhu L, Li Y, Gao F, Goel A, Wang X. Molecular subtyping of colorectal cancer: recent progress, new challenges and emerging opportunities. Semin Cancer Biol. 2019;55:37–52. doi: 10.1016/j.semcancer.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Ma X, Chakravarti D, Shalapour S, DePinho RA. Genetic and biological hallmarks of colorectal cancer. Genes Dev. 2021;35(11–12):787–820. doi: 10.1101/gad.348226.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oweida AJ, Darragh L, Phan A, Binder D, Bhatia S, Mueller A, Van Court B, Milner D, Raben D, Woessner R, et al. STAT3 modulation of regulatory T cells in response to radiation therapy in head and neck cancer. J Natl Cancer Inst. 2019;111(12):1339–1349. doi: 10.1093/jnci/djz036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang L, Xu Y, Fang J, Liu W, Chen J, Liu Z, Xu Q. Targeting STAT3 abrogates tim-3 upregulation of adaptive resistance to PD-1 blockade on regulatory T cells of melanoma. Front Immunol. 2021;12:654749. doi: 10.3389/fimmu.2021.654749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, Lindsey ML. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol. 2017;112(3):33. doi: 10.1007/s00395-017-0622-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen M, Xu Z, Xu W, Jiang K, Zhang F, Ding Q, Xu Z, Chen Y. Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J Exp Clin Cancer Res. 2019;38(1):149. doi: 10.1186/s13046-019-1161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li R, Huang Y, Lin J. Distinct effects of general anesthetics on lung metastasis mediated by IL-6/JAK/STAT3 pathway in mouse models. Nat Commun. 2020;11(1):642. doi: 10.1038/s41467-019-14065-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu XL, Duan W, Su CY, Mao FY, Lv YP, Teng YS, Yu PW, Zhuang Y, Zhao YL. Interleukin 6 induces M2 macrophage differentiation by STAT3 activation that correlates with gastric cancer progression. Cancer Immunol Immunother. 2017;66(12):1597–1608. doi: 10.1007/s00262-017-2052-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.