

Abstract
Circadian and sleep defects are well documented in Huntington's disease (HD). Modulation of the autophagy pathway has been shown to mitigate toxic effects of mutant Huntingtin (HTT) protein. However, it is not clear whether autophagy induction can also rescue circadian and sleep defects. Using a genetic approach, we expressed human mutant HTT protein in a subset of Drosophila circadian neurons and sleep center neurons. In this context, we examined the contribution of autophagy in mitigating toxicity caused by mutant HTT protein. We found that targeted overexpression of an autophagy gene, Atg8a in male flies, induces autophagy pathway and partially rescues several HTT-induced behavioral defects, including sleep fragmentation, a key hallmark of many neurodegenerative disorders. Using cellular markers and genetic approaches, we demonstrate that indeed the autophagy pathway is involved in behavioral rescue. Surprisingly, despite behavioral rescue and evidence for the involvement of the autophagy pathway, the large visible aggregates of mutant HTT protein were not eliminated. We show that the rescue in behavior is associated with increased mutant protein aggregation and possibly enhanced output from the targeted neurons, resulting in the strengthening of downstream circuits. Overall, our study suggests that, in the presence of mutant HTT protein, Atg8a induces autophagy and improves the functioning of circadian and sleep circuits.
SIGNIFICANCE STATEMENT Defects in sleep and circadian rhythms are well documented in Huntington's disease. Recent literature suggests that circadian and sleep disturbances can exacerbate neurodegenerative phenotypes. Hence, identifying potential modifiers that can improve the functioning of these circuits could greatly improve disease management. We used a genetic approach to enhance cellular proteostasis and found that overexpression of a crucial autophagy gene, Atg8a, induces the autophagy pathway in the Drosophila circadian and sleep neurons and rescues sleep and activity rhythm. We demonstrate that the Atg8a improves synaptic function of these circuits by possibly enhancing the aggregation of the mutant protein in neurons. Further, our results suggest that differences in basal levels of protein homeostatic pathways is a factor that determines selective susceptibility of neurons.
Keywords: Atg8a, autophagy, Drosophila circadian circuit, Huntington's disease, pigment dispersing factor, sleep
Introduction
Huntington's disease (HD) is a poly-glutamine (poly-Q) disorder and is characterized by motor, cognitive, and sleep defects (Roos, 2010). Mutation in the Huntingtin gene (Htt) causes an increase in poly-Q repeats in the exon 1 of the protein (MacDonald et al., 1993; Brandt et al., 1996; Gusella and MacDonald, 2006). Expanded poly-Q repeats lead to structural and functional changes in the protein. Mutant huntingtin protein (mHTT) forms nuclear and cytoplasmic aggregates (Davies et al., 1997; Chen et al., 2002) and affects synaptic transport, causes high ROS levels, and leads to transcriptional changes (Schulte and Littleton, 2011). Perturbation in circadian and sleep-wake cycles is also seen in HD patients (Goodman et al., 2011; Musiek and Holtzman, 2016; Leng et al., 2019; Colwell, 2021). Animal models of HD also show changes in the firing pattern of clock neurons, reduction in expression of clock genes, clock output neuropeptides, decrease in total sleep, and increased sleep fragmentation (Morton et al., 2005; Maywood et al., 2010; Kudo et al., 2011; Bellosta Diago et al., 2017; Kuljis et al., 2018).
For sustained neuronal health, the proper functioning of autophagy pathway appears to be crucial. Brain - specific KO of autophagy related genes Atg5 or Atg7 in mouse models leads to the accumulation of ubiquitinylated protein aggregates and neuronal loss (Hara et al., 2006; Komatsu et al., 2006; Nishiyama et al., 2007). Multiple steps of the autophagy pathway are defective in neurodegenerative disorders (Yamamoto and Yue, 2014; Nah et al., 2015). Specifically, in HD patient samples and animal models, cargo recognition and loading steps are defective (Martinez-Vicente et al., 2010). However, other studies have shown that the expression of critical autophagy genes is not altered in other mouse models of HD (Baldo et al., 2013; Kumar et al., 2021), suggesting that this pathway can be targeted for therapeutic intervention.
Pharmacological modulation of the autophagy pathway has been shown to mitigate the toxic effect of mHTT in many model organisms (Ravikumar et al., 2004; Sarkar et al., 2005; Spilman et al., 2010; Koga et al., 2011). Modulators such as rapamycin, glucose-6-phosphate, lithium, etc., have been shown to boost the clearance of mHTT in the neurons (Sarkar et al., 2005; Spilman et al., 2010). However, the role of autophagy modulation in circadian and sleep centers in the background of mHTT is unclear. Proper functioning of circadian and sleep centers is critical for organisms, and defects in these centers have been hypothesized to aggravate neurodegenerative phenotypes (Musiek and Holtzman, 2016; Leng et al., 2019). Hence, we asked whether autophagy induction can ameliorate the toxicity caused by mHTT in these circuits using Drosophila, a well-established model system in the field of neurodegeneration (Chan and Bonini, 2000; Allocca et al., 2018). We took a genetic approach and asked whether overexpression of key autophagy genes mitigates the toxic effect of mHTT in Drosophila circadian and sleep neurons. We found rescue of the behavioral rhythm and sleep parameters and further investigated its underlying cellular basis.
Previous studies from our group and others have shown that expression of HTT protein containing 128 poly-Q repeats in a critical subset of the Drosophila circadian circuit, the ventral lateral neurons (LNvs), recapitulates various features of the disease, including the formation of protein aggregates, breakdown of molecular clock oscillations, and activity-rest rhythm (Sheeba et al., 2008; Prakash et al., 2017; F. Xu et al., 2019). Our targeted screen conducted for potential modifiers that can rescue behavioral phenotypes caused by the expression of mutant HTT-Q128 protein in the lateral ventral neurons suggested Atg8a, a key autophagy gene, to be a strong candidate for further investigations (Prakash et al., 2022).
Given that the role of Atg8a in neurons is not well understood (Ratliff et al., 2015), we conducted further studies to examine the cellular processes that enabled behavioral rescue. Here we show that overexpression of Atg8a in the presence of mHTT rescues locomotor activity rhythms and sleep phenotypes. Although Atg8a-mediated rescue was dependent on the autophagy pathway, we observed enhanced aggregation of the mutant protein in the targeted circuits. We further showed that the presence of mHTT in the circadian neurons hampers the expression of Cathepsin-D (an important lysosomal enzyme), a phenotype rescued in a subset of the targeted neurons on Atg8a overexpression. Finally, we show that Atg8a behavioral rescue is possibly an outcome of improved communication of the targeted neurons to the downstream neurons. Overall, this study shows that genetic modulation of the autophagy pathway can largely mitigate the toxicity caused by mHTT in Drosophila circadian and sleep neurons.
Materials and Methods
Fly lines
Transgenic fly line with 548 aa of the human Huntingtin (Htt) gene either with pathogenic 128 poly-Q repeats (w1118; UAS-Htt128; +) or controls containing no poly-Q repeats (w1118; UAS-HttQ0; +) were a generous gift from Troy Littleton (Massachusetts Institute of Technology) (Lee et al., 2004). R23E10Gal4 line was a generous gift from Jeff Donlea's laboratory. PdfGAL4 driver line was obtained from Todd Holmes (University of California–Irvine). Other UAS lines: yw1118; +; UAS-GFP-Atg8a (BL 51656), UAS RNAi: yv; +; UAS Atg1RNAi (BL 26731), and w1118; +; + (BL 5905), lines were procured from Bloomington Stock Center. All fly lines were maintained on a standard cornmeal medium under LD12:12 at 25°C.
The UAS-HttQ128 and UAS-HttQ128; UAS-Atg8a lines were used to generate the w; PdfGal4/UAS-Q128 (mutant) and w; PdfGal4/UAS-Q128; UAS-Atg8a/+ (rescue) lines, which would express mutant HTT-Q128 protein or coexpress Atg8a and HTT-Q128 in the PDF neurons. To target sleep neurons, lines were generated using R23E10Gal4 driver. The UAS control lines are referred to as Q128; Atg8a and their driver control lines as Pdf/+. Their corresponding control lines with no poly-Q expansion is denoted Pdf>Q0, Atg8a.
Behavioral assays
Activity-rest rhythm for male flies was recorded by using Drosophila Activity Monitoring (DAM, Trikinetics) system. Two- to 3-d-old flies of the desired genotypes were loaded in glass tubes containing food at one end and a cotton plug at the other end. Circadian circuit experiments using PdfGal4 drivers were done in 7 mm glass tubes, and the locomotion data were recorded in 1 min bins continuously for 21 d (starting from day 3, after eclosion) under constant darkness (DD, 25°C). Experiments for sleep neurons were performed in 5 mm glass tubes, and the locomotion data were recorded in 1 min bins continuously for 10 d (starting from day 5, after eclosion) under LD 12:12 at 25°C. Experiments were performed in incubators manufactured by Sanyo or Percival or in light-tight boxes placed in temperature-controlled cubicles.
Activity-rest data analysis
For activity rhythm analysis, raw activity count data were scanned and binned in 15 min. Data were analyzed with CLOCKLAB software (Actimetrics) using χ2 periodogram with a cutoff of p < 0.05 to determine whether the flies are rhythmic and the amplitude of the periodogram. A fly was considered rhythmic if the periodogram amplitude was above the cutoff and was also validated by visual inspection of its actogram. For circadian circuit data, activity rhythm features, such as percentage rhythmicity and robustness of rhythm, were calculated over three 7 d windows to track progressive changes. The three temporal windows (age windows [AWs]) spanning 21 d are designated as AW1 (age 3-9 d), AW2 (age 10-16 d), and AW3 (age 17-23 d). To obtain a measure of changes in activity consolidation across days, we calculated “r” for each day of individual flies using a custom-made MATLAB code as described by Prakash et al. (2022). For experiments with R23E10Gal4 driver, sleep analysis was done using Pysolo software for three AWs comprising 3 d each (Gilestro and Cirelli, 2009).
Immunocytochemistry
Adult male fly brains were dissected at specified ages in ice-cold PBS and fixed with 4% PFA at room temperature for 45 min; 10% horse serum in 0.5% PBT was used as blocking solution. Overnight blocking was done at 4°C. After blocking, samples were incubated in primary antibodies for 48 h at 4°C. Incubation with secondary antibodies was done for 24 h at 4°C. Whole brains were mounted on slides using 70% glycerol in PBS. Primary antibodies used were anti-Huntingtin – mouse (1:500) (Millipore MAB2166), anti-PDF – rabbit (1:30,000) (a gift from Michael Nitabach, Yale University), anti-PDF – mouse (1:5000) (DSHB PDF C7), anti-HSP70 – mouse (Fisher Scientific), anti-PDF – rat (1:3000) (a gift from Jae Park), anti-PER – rabbit (1:20 000) (a gift from Jeffrey C. Hall, Brandeis University), anti-GFP – chicken (1:2000) (Invitrogen), anti-Cathepsin-D (1:250) (CST, 2284), and anti-Ref(2)P – rabbit (1:500) (Abcam, ab56416). Secondary antibodies AlexaFluor (Invitrogen) (1:3000) anti-rabbit-488, -546, and -647, anti-mouse-546 and -647, and anti-chicken-488 and anti-rat-647 were used.
Image acquisition and quantification
Brain samples were imaged using Zeiss microscope (LSM 880) at 20×, 40× (oil-immersion) or 63× (oil immersion) objective with the same zoom, laser power, gain, and other settings for a given experiment. While the specific settings varied for different experiments, they were kept constant for all the control and experimental genotypes within an experiment. All image analyses were conducted on raw image files. PDF+ small and large neurons were distinguished based on their position and size. Quantification (cell number) of both the cell types was done based on anti-PDF and -HTT staining by going through each stack of the captured images for all the genotypes.
Aggregate quantification was done using ImageJ. Maximum intensity projection (MIP) images were used for quantification of both aggregated and nonaggregated HTT-Q128 staining. Quantification was done for each brain hemisphere separately by marking the area of the PDF neurons and some initial part of the projection. Thresholding of the MIP images was done keeping consistent parameters for both genotypes. The analyze particles tool with size specification of 0.5 to ∞ (for large neurons) and 0.6 to ∞ (for small neurons) was used to obtain measures of inclusion number and size of inclusion. The quantification method did not distinguish between HTT inclusions and spots, resulting in spots being included in the inclusion number and size quantification. Colocalized Ref(2)P-HTT or Cath-D-HTT levels were quantified from the area marking the PDF neurons using the colocalization tool in ImageJ. In small neurons, the number of colocalization events was quantified. However, in large neurons, intensity of colocalization was quantified and the same is plotted in Results.
The signal intensity of mutant HTT, PERIOD protein, and PDF neuropeptide was quantified using MIP images. The raw intensity of the respective proteins/neuropeptide was quantified by marking the area of the projections or the cell bodies. Further, using the same area, background staining intensity was also quantified. Before plotting and analysis, background intensity was subtracted from raw values.
In case of Figures 2–9 where representative images are provided, to enable better visualization, adjustments for brightness and contrast have been applied for individual channels separately.
Figure 2.

Atg8a overexpression in PDF+ lateral ventral neurons does not improve the levels of PDF neuropeptide in the small LNvs (s-LNv). A, Representative MIP images (day 1, adult brain) of PDF neurons (arrow indicates large neurons; dotted circles represent small neurons) depicting staining of GFP-Atg8a (green), mutant HTT-Q128 (red), and PDF (magenta). Right top diagram represents the right half of the Drosophila brain depicting the position of PDF neurons in the brain. Scale bar, 20 µm. (+) and (–) in the bar graphs represent the presence or absence, respectively, of the gene in the fly. B, Plot represents quantification of HTT protein intensity from small and large neurons when only HTT-Q0 protein or HTT-Q0 and Atg8a protein were coexpressed. No significant decrease in the intensity of HTT protein was observed on the expression of two UAS constructs in the PDF neurons. n > 16 brain hemispheres. *p < 0.05. C, Plot represents % rhythmicity values for control and experimental genotypes. Control flies were rhythmic; however, no significant improvement in % rhythmicity was observed when mutant HTT-Q128 protein was coexpressed with UAS-GFP. n > 17 flies/genotype. *p < 0.05. D, Plot represents quantification of the number of PDF+ small neurons. Based on PDF neuropeptide and HTT-Q128 staining, quantification was done at three different ages (days 1, 5, and 10). Control genotype showed ∼4 PDF+ small neurons per hemisphere. Compared with the control genotype, mutant HTT-Q128 protein expression led to a significant reduction in the number of small neurons. A small improvement in the number of small neurons was observed on Atg8a overexpression. n > 12 brain hemispheres (for control genotype), and n > 16 brain hemispheres (for experimental genotype). *p < 0.05. E, Plot represents quantification of the number of PDF+ large neurons. Based on PDF neuropeptide and HTT-Q128 staining, quantification was done at three different ages (days 1, 5, and 10). Control genotype showed ∼4 PDF+ large neurons per hemisphere. Compared with the control genotype, mutant HTT-Q128 protein expression in the large neurons does not lead to any significant reduction in the number of neurons. Neither any change was observed on Atg8a overexpression in the presence of mutant HTT-Q128 protein. n > 12 brain hemispheres (for control genotype), and n > 16 brain hemispheres (for both experimental genotypes). *p < 0.05. Asterisk on individual genotypes indicates that the genotype is significantly different from all other plotted genotypes.
Figure 9.

Atg8a overexpression in the dorsal fan-shaped body neurons (dFB) rescues sleep defects. A, Sleep profiles of control and experimental genotypes for 3 AWs each comprising 3 d of data. Plots represent data from two independent replicate experiments. Expression of mHTT leads to reduction in predominantly nighttime sleep in the flies. Atg8a overexpression rescues the sleep defects in the flies. B, Plot represents quantification of mean total sleep quantified for a single replicate experiment for 3 AWs. Control genotypes show good sleep levels; however, the expression of mHTT leads to a significant decrease in total sleep in the initial two AWs (red bar). Expression of Atg8a with mutant protein rescues sleep defects, and the values are comparable to the control genotypes. n > 17 flies/genotype. *p < 0.05. C, Plot represents quantification of mean day sleep quantified for a single replicate experiment for 3 AWs. Control genotypes show good sleep levels; however, the expression of mHTT leads to a significant decrease in daytime sleep in the initial two AWs (red bar). Expression of Atg8a with mutant protein rescues daytime sleep, and the values are comparable to the control genotypes (green bar). n > 17 flies/genotype. *p < 0.05. D, Plot represents quantification of mean nighttime sleep quantified for a single replicate experiment for 3 AWs. Control genotypes show good sleep levels; however, the expression of mHTT leads to a significant decrease in nighttime sleep in the initial two AWs (red bar). Expression of Atg8a with mutant protein rescues nighttime sleep. n > 17 flies/genotype. *p < 0.05. E, Plot represents quantification of mean length of daytime sleep bouts quantified for a single replicate experiment for 3 AWs. Expression of mHTT leads to a significant decrease in daytime sleep bout length (red bar). Expression of Atg8a with mutant protein rescues length of daytime sleep episode. n > 17 flies/genotype. *p < 0.05. F, Plot represents quantification of mean length of nighttime sleep bouts quantified for a single replicate experiment for 3 AWs. Expression of mHTT leads to a significant decrease in nighttime sleep bout length (red bar). Expression of Atg8a with mutant protein rescues length of nighttime sleep episode. n > 17 flies/genotype. *p < 0.05. (+) and (–) in the bar graphs represent the presence or absence, respectively, of the gene in the fly. G, Plot represents quantification of nonaggregates and aggregated mHTT. Expression of mHTT in dFB leads to accumulation of nonaggregated mHTT in the axons (left, red bar). Atg8a overexpression in the presence of mHTT significantly reduces the nonaggregated mHTT from the projections (left, green bar). However, Atg8a overexpression does not lead to any significant change in the mutant aggregate number in the targeted neurons (right). n > 10 brains/genotype. *p < 0.05. H, Representative MIP images (day 3, adult brain) of dFB neurons depicting staining of HTT (red). Dotted area represents the projections from the neurons. Arow points to the neuropil region. Scale bar, 40 µm. Asterisk on individual genotypes indicates that the genotype is significantly different from all other plotted genotypes.
Experimental design and statistical analysis
Each assay or experiment included all possible experimental genotypes with age-matched individuals. All statistical analysis was done using Statistica 7. Data for fraction of rhythmic individuals and Cathepsin-D-positive cells were compared using the χ2 test. m × n and 2 × 2 Fisher Exact test were done using iCalcu (https://www.icalcu.com/stat/chisqtest.html) and vassarstats (http://vassarstats.net/newcs.html). For pairwise comparisons, p value was adjusted through Bonferroni corrections. Amplitude of periodogram, period, and r values were compared using repeated-measures ANOVA, keeping genotype as a fixed factor. Sleep parameters, mHTT aggregates, and size analysis, large neurons Ref(2)P-HTT-Q128 colocalization, PDF neuropeptide levels were compared using one-way ANOVA, keeping genotype as a fixed factor. Post hoc multiple comparisons in all the cases were conducted using Tukey's Honest Significant Difference test with α = 0.05. For PDF+ cell number, PERIOD protein levels, Cath-D-HTT-Q128 colocalization, and PER+ DN cells, quantification compared using Kruskal–Wallis test was done keeping genotype as a fixed factor. HSP70+ small and large neuron numbers, nonaggregated HTT protein intensity, small neurons Ref(2)P-HTT-Q128 colocalization were compared using Mann–Whitney U test, keeping genotype as a fixed factor. Statistics and p value until four decimal places are mentioned in Results. When the values were so small as to have >4 zeroes after decimal, they are represented as p ≪ 0.05.
Results
Atg8a overexpression rescues activity rest rhythm under DD
An initial screen in our laboratory revealed that Atg8a is a potential modifier of behavioral defects caused by mHTT (Ganguly, 2015; Prakash et al., 2022). Using a similar approach, we first quantified the fraction of individuals that show rhythmic locomotor activity when Atg8a is coexpressed with HTT-Q128 in a subset of circadian pacemaker neurons. As expected, almost all control individuals were rhythmic for at least 3 weeks (Fig. 1A). To examine age-dependent changes in rhythmicity, we analyzed features of the rhythm in three consecutive AWs, each comprising 7 d (Fig. 1B–D). HTT-Q128-expressing flies showed a significant reduction in rhythmicity, even as early as AW1 (χ(df=4) = 63.5210, p = 1e−12, Pdf/+ vs Pdf>Q128, p = 2.2138e−7, Q128; Atg8a/+ vs Pdf>Q128, p = 2.9382e−7, Pdf>Q0; Atg8a vs Pdf>Q128, p = 2.2138e−7) (Fig. 1B, red bar). Atg8a overexpression in the presence of the mHTT led to a significant improvement in the fraction of rhythmic individuals (AW1 – χ(df=4) = 63.5210, p = 1e−12, Pdf>Q128; Atg8a vs Pdf>Q128, p ≪ 0.05; AW2 – χ(df=4) = 93.2304, p = 0, Pdf>Q128; Atg8a vs Pdf>Q128, p = 5.6452e−7; AW3 – χ(df=4) = 57.1185, p = 1.1e−11, Pdf>Q128; Atg8a vs Pdf>Q128, p = 0.0003) (Fig. 1B, green bar). However, when we examined robustness of the rhythm using the amplitude of the periodogram (χ2 periodogram analysis), we find that Atg8a-overexpressing flies exhibited poorer quality of rhythms (repeated-measures ANOVA: main effect of genotype only, F(3,12) = 12.795, p = 0.0005, Pdf>Q128; Atg8a vs Pdf/+, p = 0.0490, Pdf>Q128; Atg8a vs Q128; Atg8a/+, p = 0.0458, Pdf>Q128; Atg8a vs Pdf>Q0; Atg8a, p = 0.0004) (Fig. 1C, green bar). We also tabulated the circadian period values of the control and experimental genotypes. Overexpression of Atg8a or coexpression of nonpathogenic HTT-Q0 with Atg8a in the PDF neurons leads to an increase in circadian period in the later AWs (repeated-measures ANOVA (AW × genotype interaction, F(10,24) = 4.3, p = 0.0015) (Fig. 1D). The few flies that remained rhythmic on expression of mutant HTT-Q128 protein in the PDF neurons exhibited periodicity close to 24 h and do not appear to be different from parental controls (visual observation; no statistical analysis because of low sample size of Pdf>Q128 flies). Further, in AW1, flies coexpressing the mutant HTT-Q128 protein with Atg8a showed period values comparable to the control genotypes. However, at later AWs, period values decrease (Fig. 1D), possibly because of the presence of mutant HTT-Q128 protein. To obtain greater temporal resolution for the quality of activity rhythm, we calculated r, an estimator of consolidation of activity (Prakash et al., 2017, 2022). Control flies (Pdf>Q0; Atg8a) showed a higher r value (of ∼0.5), which was consistent across days. HTT-Q128 expression led to a significant reduction in r value (of ∼0.2) (repeated-measures ANOVA: genotype, F(4,15) = 26.478, p ≪ 0.05, Pdf/+ vs Pdf>Q128, p = 0.0001, Q128; Atg8a/+ vs Pdf>Q128, p = 0.0001, Pdf>Q0; Atg8a vs Pdf>Q128, p = 0.0001). Coexpression of Atg8a with HTT-Q128 led to a significant improvement in the r value and was sustained across days (repeated-measures ANOVA: genotype, F(4,15) = 26.478, p ≪ 0.05, Pdf>Q128; Atg8a vs Pdf>Q128, p = 0.0003) (Fig. 1E, green line).
Figure 1.
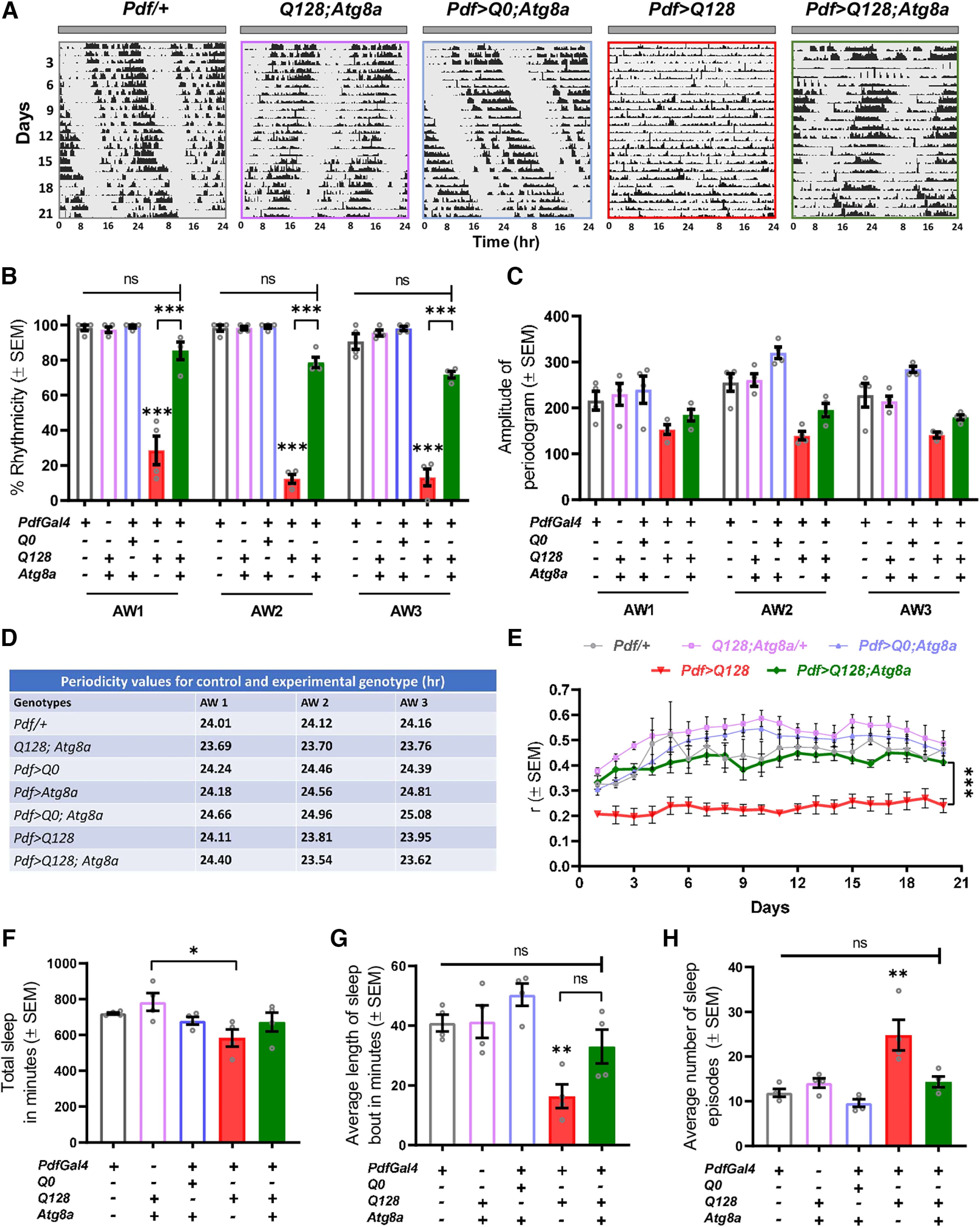
Atg8a overexpression in PDF+ lateral ventral neurons improves activity-rest rhythm in the presence of mutant HTT-Q128 protein. A, Representative double-plotted actograms for control (Pdf/+, Q128; Atg8a, Pdf>Q0; Atg8a) and experimental (Pdf>Q128, Pdf>Q128; Atg8a) genotypes, showing activity data for 21 d in DD at 25°C. (+) and (–) in the bar graphs represent the presence or absence, respectively, of the gene in the fly. B, Plot represents quantification of rhythmicity values from four independent replicate experiments for control and experimental genotypes (red bar, green bar) plotted across 3 AWs (AW1-AW3; 7 d each). Compared with the control genotypes, a significant decrease in percentage rhythmicity was observed in flies expressing mutant HTT-Q128 protein in PDF neurons. Further overexpression of Atg8a in the presence of mutant HTT-Q128 protein improves the number of rhythmic individuals in all 3 AWs, and the values were comparable to the control genotype. n > 16 flies for all the genotypes/replicate experiment. *p < 0.05. C, Plots represent mean amplitude values from four independent experiments plotted across 3 AWs (AW1-AW3; 7 d each) for all the genotypes. A significant difference was observed in the amplitude values of the control and Pdf>Q128; Atg8a (rescue) flies. Pdf>Q128 (mutant) genotype was not included in the analysis because of low rhythmic individuals. n > 16 flies for all the genotypes/replicate experiment. *p < 0.05. D, Table representing mean period values from three independent replicate experiments for control and experimental genotypes (red bar, green bar) for 3 AWs (AW1-AW3; 7 d each). Compared with control flies, overexpression of Atg8a in PDF neurons leads to a significant increase in clock period. Further, we observed an additive effect in the clock period value when both HTT-Q0 and Atg8a protein were expressed in the PDF neurons. However, no significant change was observed in the flies coexpressing mutant HTT-Q128 protein and Atg8a, and the values were comparable to the control flies. n > 16 flies for all genotypes/replicate experiment. E, Plot represents mean r values from four independent experiments plotted across 20 d for all the genotypes. Compared with control genotypes, a significant decrease was observed in the r values of mutant HTT-Q128 protein-expressing flies. A significant improvement in r value was observed on Atg8a overexpression in the presence of mutant HTT-Q128 protein, and the values were comparable to all control genotypes (except Q128; Atg8a). n > 16 flies for all the genotypes/replicate experiment. *p < 0.05. F, Plot represents mean values of total sleep from four independent experiments obtained by averaging total sleep across the first 4 d of the run for all the genotypes. Mutant HTT-Q128 expression and Atg8a coexpression in the PDF neurons does not affect the total sleep of the flies, and the values were comparable to all the control genotypes (except Q128; Atg8a). n > 16 flies for all the genotypes/replicate experiment. *p < 0.05. G, Plot represents mean values of length of sleep bout from four independent experiments obtained by averaging values across the first 4 d of the run for all the genotypes. Compared with control genotypes, mutant HTT-Q128 protein expression in PDF neurons led to a significant reduction in the length of sleep bouts of flies. Atg8a overexpression in the presence of mutant HTT-Q128 protein led to improvement in the length of sleep bouts; however, the values were not comparable to the control genotypes (except Pdf>Q0; Atg8a). n > 16 flies for all the genotypes/replicate experiment. *p < 0.05. H, Plot represents mean values of the number of sleep episodes from four independent experiments obtained by averaging the number of sleep episodes across the first 4 d of the run for all the genotypes. Compared with control genotypes, mutant HTT-Q128 protein expression in PDF neurons leads to a significant increase in the number of sleep episodes of flies. Atg8a overexpression in the presence of mutant HTT-Q128 protein significantly reduces the number of sleep episodes, and the values were comparable to all the control genotypes. n > 16 flies for all the genotypes/replicate experiment. *p < 0.05. Asterisk on individual genotypes indicates that the genotype is significantly different from all other plotted genotypes.
LNv neurons are also important modulators of sleep (Sheeba et al., 2008; Potdar and Sheeba, 2018), and expression of HTT-Q128 protein leads to sleep defects (Faragó et al., 2019). We asked whether Atg8a overexpression can mitigate sleep defects. We quantified total sleep, length, and the number of sleep episodes for the first 4 d under DD (age 5-8 d). Total sleep of HTT-Q128-expressing flies and those coexpressing Atg8a were not different from control genotypes (one-way ANOVA: genotype, F(4,15) = 2.777, p = 0.0656) (Fig. 1F). More detailed characterization showed that sleep quality in terms of bout length and number were affected by HTT-Q128 expression (bout length – one-way ANOVA: genotype, F(4,15) = 8.2632, p = 0.0009, Pdf/+ vs Pdf>Q128, p = 0.0106, Q128; Atg8a/+ vs Pdf>Q128, p = 0.0092, Pdf>Q0; Atg8a vs Pdf>Q128, p = 0.0007; bout number – genotype, F(4,15) = 10.6704, p = 0.0002, Pdf/+ vs Pdf>Q128, p = 0.0011, Q128; Atg8a/+ vs Pdf>Q128, p = 0.0055, Pdf>Q0; Atg8a vs Pdf>Q128, p = 0.0003) (Fig. 1G,H, red bar). We find that Atg8a overexpression led to changes in both; however, significant improvement was only observed in the number of sleep bouts (bout length – one-way ANOVA: genotype, F(4,15) = 8.2632, p = 0.0656, Pdf>Q128; Atg8a vs Pdf>Q128, p = 0.1103; bout number – genotype, F(4,15) = 10.6704, p = 0.0002, Pdf>Q128; Atg8a vs Pdf>Q128, p = 0.0067) (Fig. 1G,H, green bar). Thus, our results show that Atg8a overexpression can rescue the behavioral phenotypes of arrhythmic locomotion and sleep disruptions caused by HTT-Q128 expression in circadian pacemaker neurons.
Behavioral rescue is not accompanied by improvement in PDF neuropeptide levels
PDF is an important signaling molecule only released by the lateral ventral neurons. Expression of mutant HTT-Q128 protein has been shown to affect the levels of PDF neuropeptide in a subset of lateral ventral neurons named small lateral ventral neurons: s-LNv and another subset named large lateral ventral neurons (l-LNv) remain unperturbed (Prakash et al., 2017). However, before correlating the behavioral improvement with cellular changes, we first asked whether the changes in the behavior are because of the expression of two UAS constructs leading to dilution effects. To assess this, we quantified the levels of HTT-Q0 protein from both subsets of neurons in flies only expressing HTT-Q0 protein or coexpressing HTT-Q0 with Atg8a in the PDF neurons. Coexpression of HTT-Q0 protein and Atg8a does not lead to any decrease in the levels of HTT-Q0 protein in both the subsets of neurons (small neurons – Mann–Whitney U test – Z(1,25) = −4.72778, p ≪ 0.05; large neurons – one-way ANOVA, F(1,30) = 4.2034, p = 0.04917) (Fig. 2B). We also recorded activity-rest rhythm for flies only expressing HTT-Q128 protein or coexpressing HTT-Q128 and UAS-GFP (another UAS transgene in place of UAS-Atg8a, which is expected to be benign but could potentially result in dilution effects) in the PDF neurons. It was observed that coexpression of both UAS constructs in PDF neurons does not lead to any significant improvement in the activity-rest rhythm compared with flies only expressing HTT-Q128 protein (χ(df=2) = 34.6180, p = 3.0394e−8, Pdf>Q0 vs Pdf>Q128, p ≪ 0.05; Pdf>Q0 vs Pdf>Q128; GFP, p ≪ 0.05; Pdf>Q128 vs Pdf>Q128; GFP, p = 1) (Fig. 2C). Overall, both immunocytochemistry and behavioral experiments point out that the expression of two UAS constructs does not decrease the expression of the HTT protein or lead to any improvement in the activity rhythm, suggesting that the behavioral rescue on Atg8a overexpression is possibly not an outcome of dilution effects.
Now to relate behavioral improvements to cellular changes, we asked whether Atg8a overexpression rescues PDF neuropeptide levels in the targeted neurons. To access, we quantified the number of detectable LNvs in both the experimental genotypes (Pdf> HttQ128 and Pdf> HttQ128; Atg8a) based on PDF staining. Coexpression of nontoxic Huntingtin protein HTT-Q0 with Atg8a showed ≈ 4 small and large LNvs (Fig. 2D,E, blue unfilled bar). We observe that HTT-Q128 expression in the LNv neurons causes a sharp decline in the number of small neurons (Fig. 2D, red bar); and with age, the number further declines (day 1 – Kruskal–Wallis test – H(2,52) = 25.7229, p ≪ 0.05, Pdf>Q0; Atg8a vs Pdf>Q128, p < 0.05; day 5 – H(2,49) = 20.7545, p ≪ 0.05, Pdf>Q0; Atg8a vs Pdf>Q128, p ≪ 0.05; day 10 – H(2,47) = 27.6967, p ≪ 0.05, Pdf>Q0; Atg8a vs Pdf>Q128, p ≪ 0.05). Coexpression of Atg8a with mutant HTT-Q128 protein showed a slight increase in the small neurons number, but the improvement was not significant (day 1 – Kruskal–Wallis test – H(2,52) = 25.7229, p ≪ 0.05, Pdf>Q128; Atg8a vs Pdf>Q128, p = 0.0636; day 5 – H(2,49) = 20.7545, p ≪ 0.05, Pdf>Q128; Atg8a vs Pdf>Q128, p = 0.5357; day 10 – H(2,47) = 27.6967, p = 0, Pdf>Q128; Atg8a vs Pdf>Q128, p = 0.6473) (Fig. 2D, green bar). Furthermore, as expected, no change in the number of large neurons was observed in any of the experimental genotypes, and the numbers were comparable to the control genotype (day 1 – Kruskal–Wallis test – H(2,52) = 2.8307, p = 0.2428; day 5 – H(2,49) = 0.9214, p = 0.6308; day 10 – H(2,47) = 0.2760, p = 0.8711) (Fig. 2E, red and green bars).
Since no changes were observed in the large neurons number or PDF levels, we asked whether their ability to tolerate stressful conditions is better than the small neurons. To examine this, adult fly brains were dissected at two different ages (day 1 and 18) and were coimmunostained against PDF and Heat Shock Protein 70 (HSP70, a stress marker) (Kim et al., 2020). No HSP70 staining was observed in the small and large neurons of the control genotypes (Fig. 3A, top). On day 1, all the small neurons of HTT-Q128-expressing flies showed HSP70 staining, while only a few large neurons were HSP70+ (Fig. 3B,C, red bar). Coexpression of Atg8a with the mutant HTT-Q128 protein did not lead to any significant decrease in the number of HSP70+ small or large neurons (day 1 (small neurons) – Mann–Whitney U test – Z(1,34) = 0.9163, p = 0.3594; large neurons – Z(1,34) = 1.8377, p = 0.0660; day 18 (small neurons) – Z(1,45) = −1.3108, p = 0.1899; large neurons – Z(1,45) = 0.9963, p = 0.3190) (Fig. 3B,C, green bar). When compared between day 1 and day 18, no significant difference was observed in the number of HSP70+ large neurons between experimental genotypes; however, a significant reduction in the number of HSP70+ small neurons was observed in both the genotypes (Pdf>Q128 – day 1 vs day 18 – Mann–Whitney U test – Z(1,37) = 2.3718, p = 0.0176; Pdf>Q128; Atg8a – day 1 vs day 18 – Z(1,42) = 2.8427, p = 0.0044) (Fig. 3B). Together, PDF-based cell quantification and HSP70 staining data suggest that Atg8a overexpression does not lead to any significant improvement in the number of small neurons. Moreover, HSP70 staining suggests that the large neurons are more resistant to stress, and the gradual decline in the number of HSP70+ small neurons possibly points toward the loss of these neurons with age.
Figure 3.
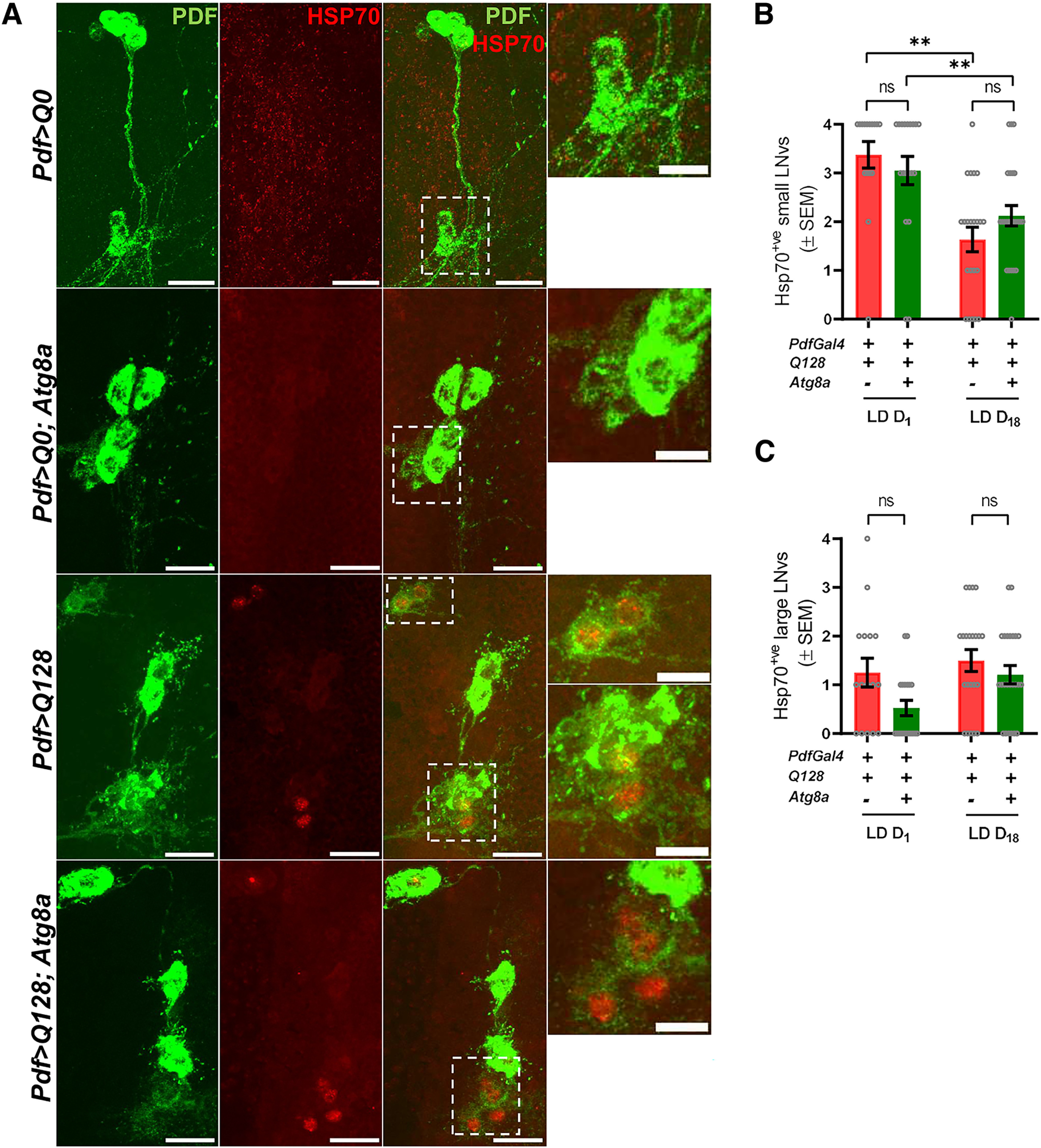
Mutant HTT-Q128 protein induces HSP70 expression in the PDF+ lateral ventral neurons. A, Representative MIP images (day 1, adult brain) of PDF neurons (large neurons and small neurons) depicting staining of PDF neuropeptide (green), and HSP70 (red). Scale bar, 20 µm. Zoomed in panel shows small neurons. Scale bar, 10 µm. (+) and (–) in the bar graphs represent the presence or absence, respectively, of the gene in the fly. B, Plot represents quantification of the number of HSP70+ small neurons. Based on PDF neuropeptide and HSP70 staining, quantification was done at two different ages (day 1 and day 18). Mutant HTT-Q128 protein expression induces HSP70 protein expression in mainly small neurons at an early age. Compared with flies only expressing mutant HTT-Q128 protein, no significant change was observed in the number HSP70+ small neurons on Atg8a overexpression at both ages. n > 16 brain hemispheres (for both genotypes). *p < 0.05. C, Plot represents quantification of the number of HSP70+ large lateral ventral neurons. Based on PDF neuropeptide and HSP70 staining, quantification was done at two different ages (day 1 and day 18). Mutant HTT-Q128 expression led to the induction of HSP70 protein in large neurons. Compared with flies only expressing mutant HTT-Q128, no significant change was observed in the number of HSP70+ large neurons on Atg8a overexpression. n > 16 brain hemispheres (for both experimental genotypes). *p < 0.05. Asterisk on individual genotypes indicates that the genotype is significantly different from all other plotted genotypes.
HTT aggregates persist in LNv despite behavioral rescue
The presence of mutant protein aggregates is thought to be toxic to neurons (Davies et al., 1997; DiFiglia et al., 1997). However, other studies have suggested that the soluble form is more toxic (Takahashi et al., 2008; Lajoie and Snapp, 2010). We asked whether the rescue in locomotor activity rhythm on Atg8a overexpression is an outcome of clearance of mutant protein aggregates from the LNvs. To assess this, we quantified the number of mutant protein aggregates (present both in the cell bodies and projections of both the subsets of neurons) and aggregate size from the targeted neurons. Quantification from small neurons was done at the third instar larval stage where mutant HTT-Q128 protein aggregates are present, but no loss in the small neurons was observed. For large neurons, quantification was done at days 1 and 10 (after eclosion). Expression of mutant HTT-Q128 protein led to the formation of protein aggregates in both groups of LNvs, while no aggregates were observed in flies expressing nontoxic HTT-Q0 construct (Figs. 2A, 4A). At the larval stage (quantified at L3), coexpressing HTT-Q128 and Atg8a resulted in a significant increase in both the number and size of mutant HTT aggregates in small neurons (one-way ANOVA: F(1,56) = 12.4975, p = 0.0010) (Fig. 4B,C). At the adult stage, where the large neurons can be visualized, mutant HTT-Q128 protein (which does not appear to be aggregated) is detected both in the neurons on day 1 (Fig. 4A, right). Further, no change was observed in its intensity on Atg8a overexpression (one-way ANOVA: F(1,66) = 0.3955, p = 0.5324) (Fig. 4D, green bar). The number of large neurons with either the diffuse or aggregated forms of mutant HTT-Q128 protein was also not altered (Fig. 4E). Additionally, in the large neurons Atg8a overexpression does not lead to any significant change in the number of mutant protein aggregates or size (quantified at D1 and D10) (aggregate number: day 1 – one-way ANOVA, F(1,35) = 0.4255, p = 0.5186; day 10 – F(1,31) = 0.069, p = 0.7943; aggregate size: day 1 – one-way ANOVA, F(1,35) = 0.4658, p = 0.4993; day 10 – F(1,31) = 0.2783, p = 0.6016) (Fig. 4F,G, green bar). Overall, these results show that Atg8a overexpression in the presence of mutant HTT-Q128 protein led to a significant increase in the number and size of the mutant protein aggregates in the small neurons.
Figure 4.
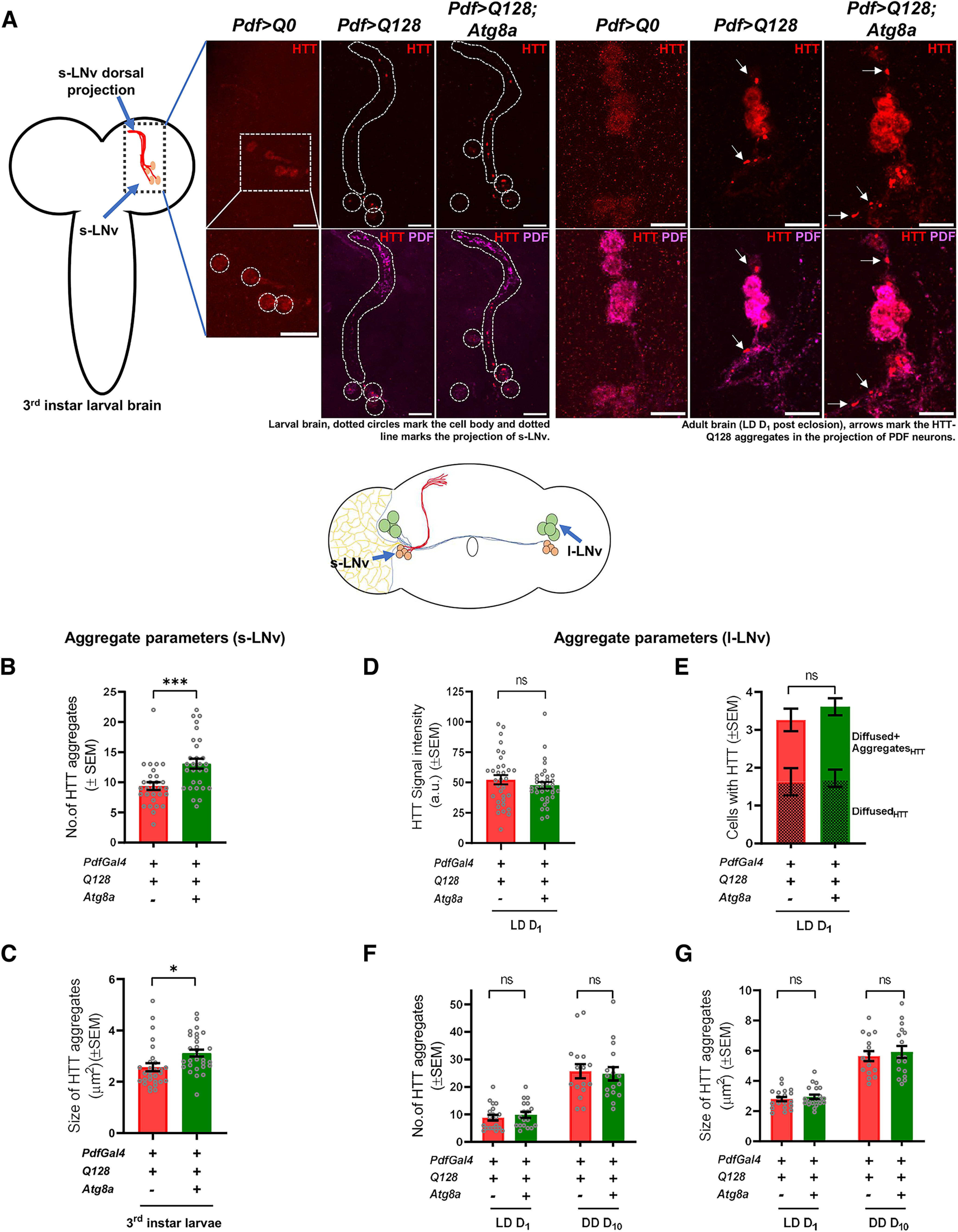
Atg8a overexpression increases mutant HTT-Q128 protein aggregates in the PDF+ small lateral ventral neurons. A, Representative MIP images of PDF neurons (small – (larva brain; left) and large neurons – (adult brain; right) depicting staining of mutant HTT-Q128 protein (red) & PDF neuropeptide (magenta). Left diagram represents the larval Drosophila brain depicting the position of small PDF neurons. Dotted circles represent small neuron cell bodies. Dotted areas represent small neuron projections. Scale bar, 20 µm. (+) and (–) in the bar graphs represent the presence or absence, respectively, of the gene in the fly. B, Plot represents quantification of the number of mutant protein aggregates, quantified from the small neurons (at L3 stage). Mutant HTT-Q128 protein expression led to the formation of protein aggregates in the small neurons. Compared with flies only expressing mutant HTT-Q128 protein, a significant increase in the number of mutant HTT-Q128 protein aggregates was observed on Atg8a overexpression. n > 24 brain hemispheres (for both experimental genotypes). *p < 0.05. C, Plot represents quantification of the size of mutant protein aggregates, quantified from the small neurons (at L3 stage). Compared with flies only expressing mutant HTT-Q128 protein, a significant increase in the size of mHTT aggregates was observed on Atg8a overexpression. n > 24 brain hemispheres (for both experimental genotypes). *p < 0.05. D, Plot represents quantification of signal intensity of nonaggregated mutant HTT-Q128 protein, quantified from the large neurons (on day 1, after eclosion). Compared with flies only expressing mutant HTT-Q128 protein, Atg8a overexpression does not lead to any significant change in the intensity of nonaggregated mutant HTT-Q128 protein. n > 30 neurons (for both experimental genotypes). *p < 0.05. E, Plot represents quantification of the cell numbers positive for either only mutant HTT-Q128 protein aggregates or have both aggregated and nonaggregated mutant HTT-Q128 protein. Quantification was done from the large neurons (on day 1, after eclosion). Compared with mutant HTT-Q128-expressing flies, no significant change was observed in both the quantifications on Atg8a overexpression. n > 30 neurons (for both experimental genotypes). *p < 0.05. F, Plot represents quantification of the number of mutant protein aggregates, quantified from the large neuron cell bodies and some part of the projections at two different ages (days 1 and 10). As observed in small neurons, mutant HTT-Q128 protein expression in large neurons led to the formation of protein aggregates. Compared with flies only expressing mutant HTT-Q128 protein, at both ages, no significant change was observed in the number of mutant HTT-Q128 protein aggregates on Atg8a overexpression. n > 18 brain hemispheres (day 1, for both experimental genotype), and n > 16 brain hemispheres (day 10, for both experimental genotypes). *p < 0.05. G, Plot represents quantification of the size of mutant protein aggregates, quantified from the large neuron cell bodies and some part of the projections at two different ages (days 1 and 10). Compared with flies only expressing mutant HTT-Q128 protein, at both ages, no significant change was observed in the size of mutant HTT-Q128 aggregates on Atg8a overexpression. n > 18 brain hemispheres (day 1, for both the experimental genotypes), and n > 16 brain hemispheres (day 10, for both experimental genotypes). *p < 0.05. Asterisk on individual genotypes indicates that the genotype is significantly different from all other plotted genotypes.
Autophagy pathway is involved in Atg8a-mediated behavioral rescue
Although Atg8a is crucial for the autophagy pathway (Nguyen et al., 2016), to the best of our knowledge, it is not known whether its overexpression induces autophagy in neuronal cells. To confirm that indeed autophagy pathway upregulation is the basis of the Atg8a-mediated rescue, we first quantified the punctate form of Ref(2)P, a key autophagy adaptor protein (Lippai and Low, 2014), and hypothesized that changes in Ref(2)P levels would reflect the status of the autophagy. We detected colocalized Ref(2)P in both the experimental genotypes (Fig. 5). In small neurons (third instar), we observed that coexpression of Atg8a with mutant HTT-Q128 protein led to a significant increase in colocalized Ref(2)P levels (Mann–Whitney U test, Z(df=1) = −3.2463, p = 0.00082) (Fig. 5A, green bar). No significant change was observed in the large neurons in adult flies on Atg8a overexpression (day 1) (one-way ANOVA, F(1,33) = 0.7471, p = 0.3937). However, at a later stage (day 10), a small but significant increase in the Ref(2)P-HTT-Q128 (one-way ANOVA, F(1,31) = 6.1704, p = 0.0187) colocalization was observed (Fig. 5B, green bar). Further, comparison between days 1 and 10 also revealed a significant increase in Ref(2)P-HTT-Q128 colocalization on Atg8a overexpression (one-way ANOVA, F(1,33) = 16.6173, p = 0.0003); however, such an increase was not observed in flies only expressing HTT-Q128. Overall, increased Ref(2)P colocalization to the HTT-Q128 aggregates suggests the involvement of the autophagy pathway in Atg8a-mediated rescue.
Figure 5.
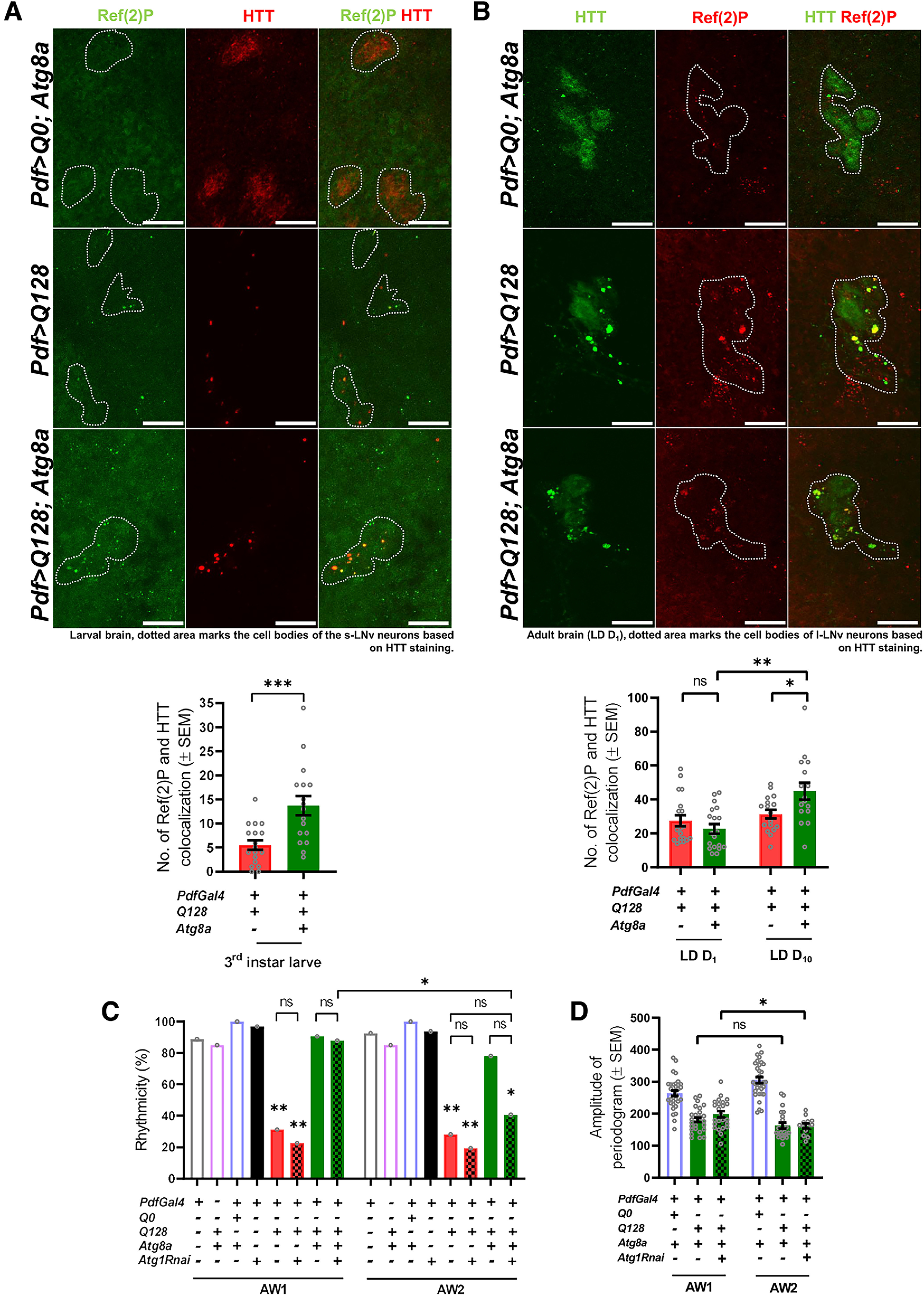
Increased Ref(2)P levels in the rescue genotype suggest that Atg8a-mediated rescue in activity rhythm is autophagy-dependent. A, (+) and (–) in the bar graphs represent the presence or absence, respectively, of the gene in the fly. Representative MIP images of small PDF neurons (from L3 stage, larval brain) depicting staining for Ref(2)P protein (green), control (HTT-Q0), and mutant HTT-Q128 (red). Dotted area represents the cell bodies of the small neurons. Scale bar, 20 µm. Images for HTT-Q0 were acquired separately and not subjected to quantification. Plot represents quantification of the Ref(2)P-HTT colocalization events in small neurons for both the experimental genotypes. A mean Ref(2)P-HTT colocalization event of ∼5 was observed in flies expressing mutant HTT-Q128 protein (red bar). Compared with the flies only expressing mutant HTT-Q128 protein, a significant increase in Ref(2)P-HTT colocalization event (∼15) was observed on Atg8a overexpression. n > 16 brain hemispheres (for both genotypes). *p < 0.05. B, Representative MIP images (LD day 1, adult brain) of PDF neurons depicting staining for Ref(2)P protein (red), control (HTT-Q0), and mutant HTT-Q128 (green). Dotted area represents the cell bodies of the small neurons. Scale bar, 20 µm. Plot represents quantification of the number of Ref(2)P-HTT colocalization events quantified across two different ages (days 1 and 10) for both the experimental genotypes from large neurons. A mean Ref(2)P-HTT colocalization of ∼30 was observed in flies expressing mutant HTT-Q128 protein. Additionally, no significant change was observed in the values with age for the mutant genotype (red bar). Compared with the flies only expressing mutant HTT-Q128 protein, Atg8a coexpression does not lead to a significant increase in the number of Ref(2)P-HTT colocalization events on day 1. However, a significant increase in the colocalization was observed on day 10 compared with the values obtained from the same genotype at an earlier age (day 1) and flies only expressing mutant HTT-Q128 protein on day 10. n > 16 brain hemispheres (for both genotypes). *p < 0.05. C, Plot represents quantification of rhythmicity values quantified across 2 AWs (8 d each) for all the genotypes. Consistent with the previous results, expression of mutant HTT-Q128 protein in PDF neurons results in a significant decrease in percentage rhythmicity compared with the control genotypes (red bar). Further, coexpression of Atg8a with mutant HTT-Q128 improves the number of rhythmic individuals in both the AWs (green bar). In the first AW, downregulation of Atg1 in the rescue genotype (Pdf>Q128; Atg8a) does not lead to any significant decrease in the rhythmicity, and the values were comparable to both control and rescue genotypes. However, compared with the first AW, a significant decrease in the number of rhythmic individuals was observed in the second AW (green-filled bars). n > 16 flies/genotype. *p < 0.05. D, Plot represents rhythm amplitude values for Pdf>Q0; Atg8a, Pdf>Q128; Atg8a, and Pdf>Q128; Atg8a, Atg1Rnai flies. A significant decrease in rhythm amplitude was only observed on Atg1 downregulation in the Pdf>Q128; Atg8a background. n > 16 flies/genotypes. *p < 0.05. Asterisk on individual genotypes indicates that the genotype is significantly different from all other plotted genotypes.
We reasoned that, if Atg8a overexpression was indeed bringing about the rescue via the autophagy pathway, then the downregulation of key autophagy genes should prevent the observed rescue. We downregulated an upstream autophagy gene, Atg1 in flies coexpressing HTT-Q128 and Atg8a and recorded activity rhythms for two AWs. Control flies showed ∼90% rhythmicity in both AWs (Fig. 5C). Downregulation of Atg1 alone in the LNvs did not lead to any decrease in the fraction of rhythmic individuals (χ(df=7) = 114.3759, p ≪ 0.05, Pdf/+ vs Pdf>Atg1Rnai, p = 0.3232, Q128; Atg8a/+ vs Pdf>Atg1Rnai, p = 0.2975, Pdf>Q0; Atg8a vs Pdf>Atg1Rnai, p = 0.9999) (Fig. 5C, black bar). As expected, flies coexpressing HTT-Q128 and Atg8a were rhythmic (χ(df=7) = 114.3759, p ≪ 0.05, Pdf>Q128; Atg8a vs Pdf>Q128, p ≪ 0.05) (Fig. 5C, green bar). Downregulation of Atg1 in the rescue background (Pdf>Q128; Atg8a, Atg1RNAi) did not lead to any significant reduction in the rhythmicity in the first AW (AW1) (χ(df=7) = 114.3759, p ≪ 0.05, Pdf>Q128; Atg8a vs Pdf>Q128; Atg8a, Atg1Rnai, p = 1). However, by the second AW, these flies showed significantly reduced rhythmicity (χ(df=7) = 107.2783, p = 1e−12, Pdf>Q128; Atg8a, Atg1Rnai [AW1] vs Pdf>Q128; Atg8a, Atg1Rnai [AW2], p ≪ 0.05) (Fig. 5C, filled green bar). We also observed a significant drop in rhythm amplitude on Atg1 downregulation in the rescue background (one-way ANOVA: Pdf> Q128 Atg8a AW comparison – F(1,47) = 2.2637, p = 0.1392; Pdf> Q128 Atg8a, Atg1Rnai, AW comparison – F(1,35) = 6.9472, p = 0.0124) (Fig. 5D, filled green bar). Although a significant decrease in the percentage rhythmicity and amplitude was observed on Atg1 downregulation, changes in the expression level of multiple UAS transgenes through a single Gal4 driver can also contribute to the behavioral phenotype. Overall, compromised rescue at later stages and increased Ref(2)P-HTT colocalization in small neurons indicates that the autophagy pathway is being harnessed in mitigating the toxicity of HTT-Q128.
HTT-Q128 hampers Cathepsin-D expression
Lysosomes are a key component of the autophagy pathway. Studies have shown that the activity of lysosomal enzymes involved in the degradation of cargo increases on autophagy induction (Settembre et al., 2011, 2013; Stransky and Forgac, 2015). We examined the status of lysosomes starting at the third instar larval stages when small neurons already show aggregates of HTT-Q128. We stained for Cathepsin-D (Cath-D), a key enzyme involved in lysosome function (Benes et al., 2008; Sevlever et al., 2008). Cath-D was not observed in the small neurons of control or flies expressing mutant HTT-Q128 protein. However, flies coexpressing HTT-Q128 and Atg8a showed punctate staining for the Cath-D in the soma of small neurons (Pdf>Q128; Atg8a vs Pdf>Q128 χ(df=1) = 0.8, p = 0.0052) (Fig. 6A, right top). We further checked whether mutant HTT-Q128 protein aggregates colocalize with Cath-D staining; and indeed, we observed colocalization of Cath-D-HTT-Q128 in flies coexpressing both mutant HTT-Q128 protein and Atg8a (Kruskal–Wallis test – H(2,54) = 20.2135, p ≪ 0.05, Pdf>Q0; Atg8a vs Pdf>Q128, p = 1; Pdf>Q0; Atg8a vs Pdf>Q128; Atg8a, p = 0.02478; Pdf>Q128; Atg8a vs Pdf>Q128, p = 0.0315) (Fig. 6A, right bottom).
Figure 6.
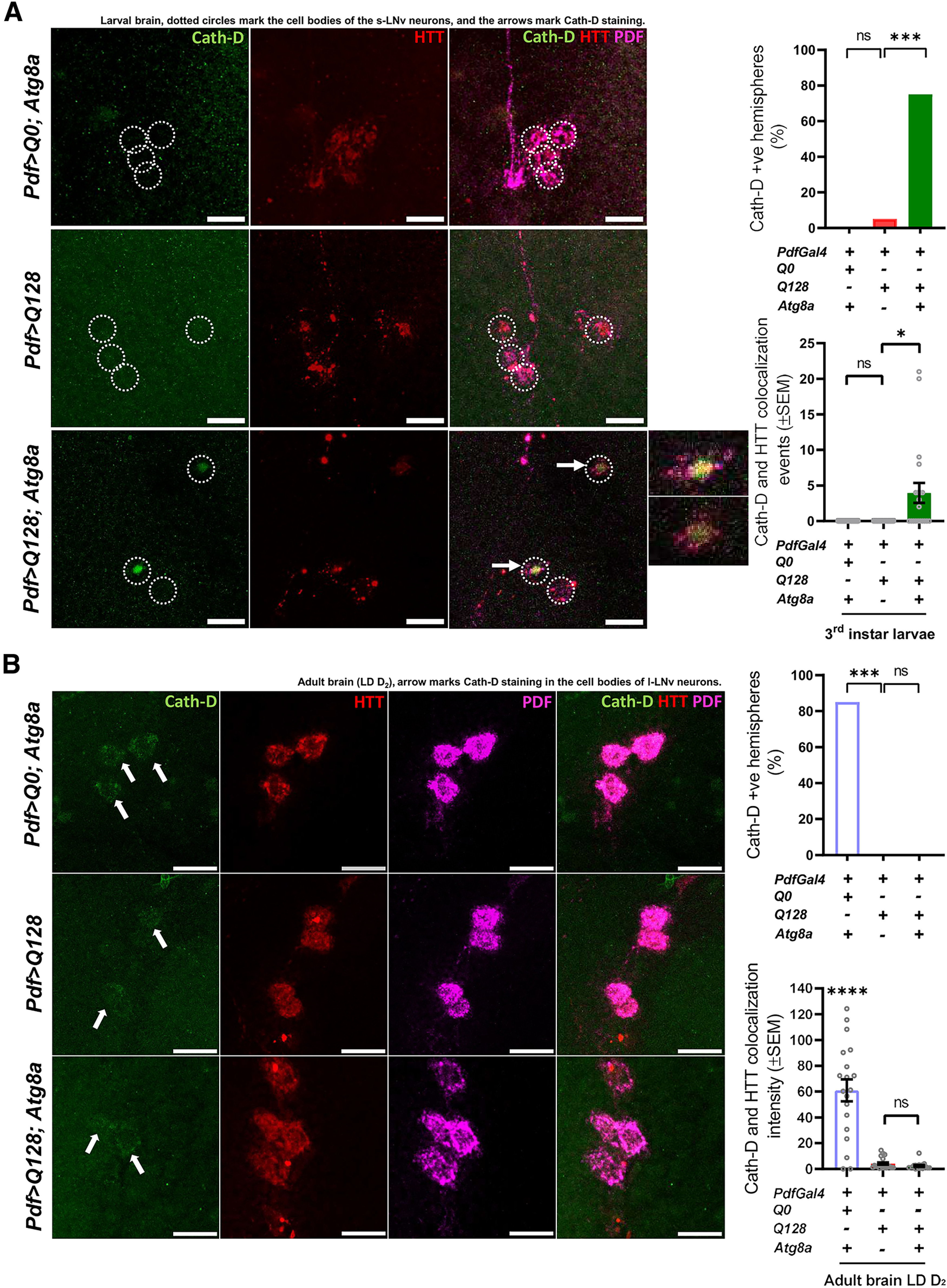
Flies overexpressing Atg8a shows Cathepsin-D staining (possibly lysosome functioning) in the small neurons. (+) and (–) in the bar graphs represent the presence or absence, respectively, of the gene in the fly. A, Representative MIP images (at L3 stage, larval brain) of small PDF+ neurons, depicting staining of Cath-D (green), control (HTT-Q0) and HTT-Q128 (red), and PDF neuropeptide (magenta). Scale bar, 20 µm. Top right, Quantification showing that, in both the control genotype and flies expressing only mutant HTT-Q128 protein, no staining for Cath-D was observed in the cell bodies. However, big spot-like staining for Cath-D was observed in flies overexpressing Atg8a in the presence of mHTT. n > 14 brain hemispheres/genotype. *p < 0.05. Bottom, Quantification of Cath-D-HTT-Q128 colocalization events in control and experimental genotypes. Compared with other control and experimental genotypes, Atg8a-overexpressing flies show a significantly high number of Cath-D-HTT colocalization events. n > 14 brain hemispheres/genotype. *p < 0.05. B, Representative MIP images (LD day 1, adult brain) of large PDF+ neurons, depicting staining of Cath-D (green), control (HTT-Q0) and HTT-Q128 (red), and PDF neuropeptide (far-red). Arrow indicates the Cath-D staining in the large neurons for all genotypes. Scale bar, 20 µm. Top right, Quantification showing that staining for Cath-D (both diffuse and punctate) can be observed in the large neurons for the control genotype. No such staining pattern was observed in flies expressing either mutant HTT-Q128 protein or coexpressing Atg8a with the mutant protein. n > 16 brain hemispheres/genotype. *p < 0.05. Bottom, Quantification of Cath-D-HTT-Q128 colocalization intensity in control and experimental genotypes. Control flies show a significantly high level of Cath-D-HTT colocalization intensity compared with other experimental genotypes. n > 16 brain hemispheres/genotype. *p < 0.05. Asterisk on individual genotypes indicates that the genotype is significantly different from all other plotted genotypes.
To examine the large neurons which appear only during pupal stages, we imaged adult brains (on day 2, after eclosion). The control genotype showed diffuse and punctate Cath-D staining in the soma. However, very faint or no staining was observed in flies either expressing only HTT-Q128 or coexpressing Atg8a with the mutant HTT-Q128 protein (χ(df=2) = 9.7901, p = 0.0074, Pdf>Q128; Atg8a vs Pdf>Q0; Atg8a, p = 0.00402; Pdf>Q128; Atg8a vs Pdf>Q128, p = 1 & Pdf>Q0; Atg8a vs Pdf>Q128, p = 0.0033) (Fig. 6B, right top). Colocalization quantification further supports these results, wherein significantly high colocalization intensity of Cath-D-HTT-Q128 was observed in the control genotype compared with the experimental genotypes (Kruskal–Wallis test – H(2,56) = 23.1158, p ≪ 0.05, Pdf>Q0; Atg8a vs Pdf>Q128, p = 0.0006; Pdf>Q0; Atg8a vs Pdf>Q128; Atg8a, p = 0.0002; Pdf>Q128; Atg8a vs Pdf>Q128, p = 1) (Fig. 6B, right bottom). Overall, improved Cath-D staining and colocalization of mutnat HTT protein to Cath-D in the small neurons further strengthens the idea of autophagy-mediated rescue by Atg8a overexpression.
Behavioral rescue is not dependent on PERIOD protein oscillation
Daily oscillation of core circadian clock component-PERIOD (PER) in the small LNv is crucial for the maintenance of locomotor rhythm under DD (Yang and Sehgal, 2001; Peng et al., 2003). Previous studies showed that expression of mutant HTT-Q128 in LNvs disrupts the level and dampens the oscillation of PER protein in both LNv subtypes (Prakash et al., 2017, 2022). To assess whether Atg8a-mediated behavioral rescue in the activity-rest rhythm is an outcome of improvement in PER protein levels, we examined four different circadian time points on DD day 3 (CT22, CT2, CT11, and CT15). The signal intensity of PER protein was quantified from small, large, and PDF- fifth s-LNv (as nontargeted control). Control flies showed circadian oscillation of PER protein in all three sets of neurons (Kruskal–Wallis test: Pdf>Q0; Atg8a – H(3,60) = 53.9578, p ≪ 0.05, CT22 vs CT2, p = 0.0348, CT22 vs CT11, p ≪ 0.05, CT22 vs CT15, p ≪ 0.05; Pdf>Q128 – H(3,69) = 58.0768, p ≪ 0.05, CT22 vs CT2, p = 0.0039, CT22 vs CT11, p ≪ 0.05, CT22 vs CT15, p = 0.0061; Pdf>Q128; Atg8a – H(3,74) = 56.5582, p ≪ 0.05, CT22 vs CT2, p = 0.2662, CT22 vs CT11, p ≪ 0.05, CT22 vs CT15, p = 0.0033) (Fig. 7A,B). In both the experimental genotypes, a clear time-dependent oscillation of PER protein was observed in fifth s-LNv (Fig. 7B, red and green curve, top). Expression of mutant HTT-Q128 protein led to a significant reduction in both the levels and oscillation of PER protein in both small and large neurons (Mann–Whitney U test: large neurons – CT22 – Z(df=1) = 8.5027, p ≪ 0.05; small neurons – CT22 – Z(df=1) = 8.0976, p ≪ 0.05) (Fig. 7B, red curve, bottom). Interestingly, coexpression of Atg8a with mutant HTT-Q128 protein failed to improve levels or oscillation of PER protein in both neuronal subsets (Mann–Whitney U test: large neurons – CT22 – Z(df=1) = 9.5070, p ≪ 0.05; small neurons – Z(df=1) = 8.6602, p ≪ 0.05) (Fig. 7B, green curve, bottom). Overall, this result suggests that the observed behavioral rescue is not dependent on PERIOD protein oscillation.
Figure 7.
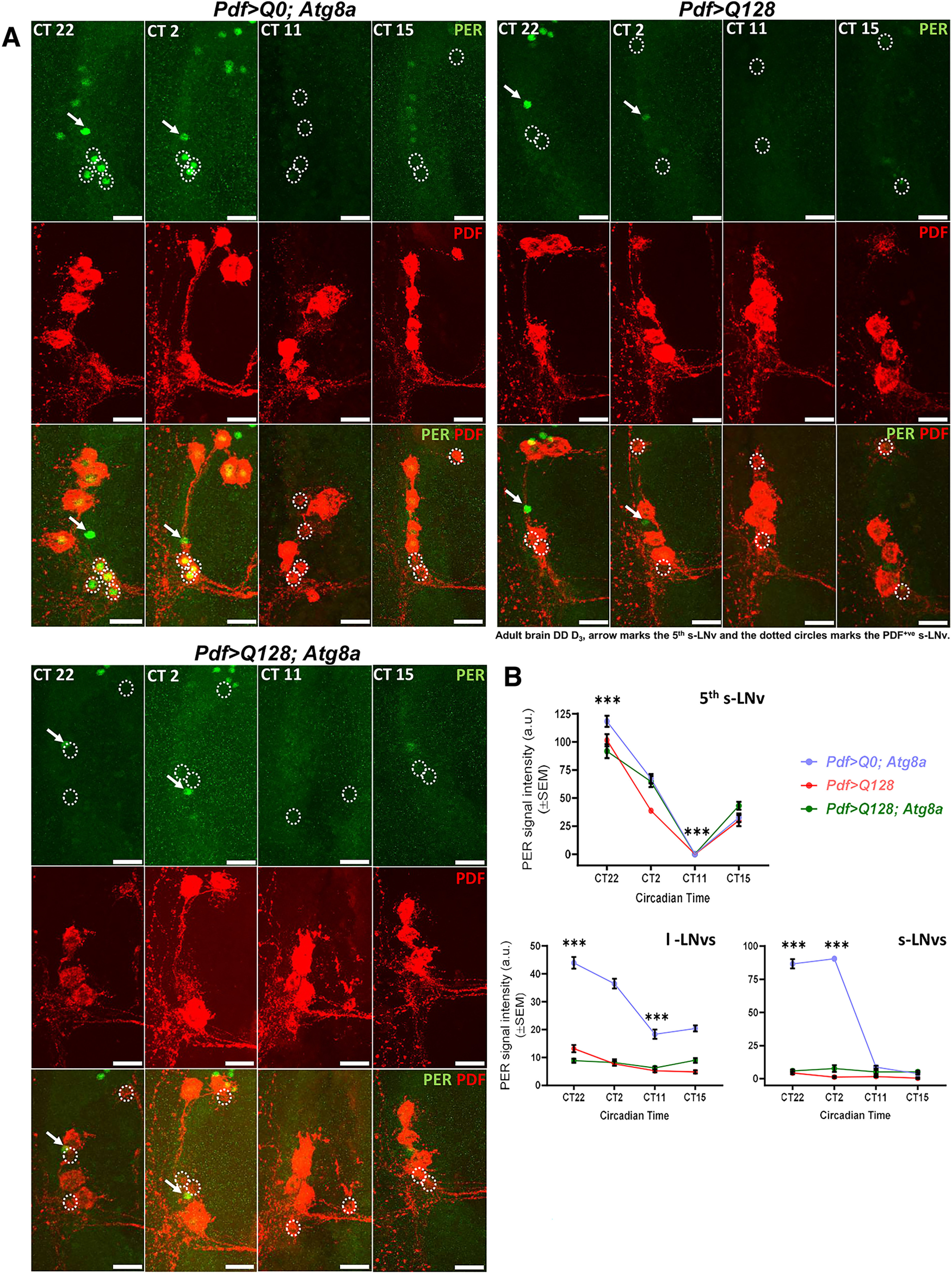
Atg8a overexpression does not improve PERIOD (PER) protein oscillations in the PDF+ lateral ventral neurons. A, Representative MIP images (DD day 3, adult brain) of PDF neurons and fifth small lateral ventral neuron (do not express PDF neuropeptide and served as our control for the PER staining) depicting staining for PER protein (green) and PDF neuropeptide (red) for four circadian time points (CT22, CT2, CT11, and CT15). Arrow indicates fifth s-LNv. Dotted circles represent small PDF neurons. Scale bar, 20 µm. B, Plots represent quantification of the mean signal intensity of PER protein, quantified on DD day 3 from fifth small lateral ventral neuron (left), PDF+ small neurons (middle), and large neurons (right) at four different circadian time points. Fifth small ventral neuron shows time-dependent oscillation of PER protein in all the genotypes. Compared with the control genotype, expression of mutant HTT-Q128 protein in both small and large neurons significantly hampers the PER protein levels and oscillation in both small and large neurons (red curve). No significant improvement was observed in PER protein levels or oscillation on Atg8a overexpression in the presence of mutant HTT-Q128 protein in the PDF neurons (green curve). n > 16 brain hemispheres/time point (for control genotype), and n > brain 18 hemispheres/time point (for experimental genotypes). *p < 0.05. Asterisk on individual genotypes indicates that the genotype is significantly different from all other plotted genotypes and time points.
Atg8a overexpression improves output from the PDF neurons
The small neurons exhibit a circadian rhythm in the level of PDF neuropeptide in the dorsally located terminal projections, and its release at the dorsal projections is critical for synchronizing core clock protein oscillation in the downstream circadian neuronal groups (Renn et al., 1999; Peng et al., 2003). Previously, our laboratory has reported that the expression of mutant HTT-Q128 protein in PDF neurons does not lead to a breakdown in PDF oscillations in the dorsal projections (Prakash et al., 2017). Here we also report that oscillation of PDF neuropeptide occurs in the dorsal projections for all the tested genotypes, including flies expressing mutant HTT-Q128 protein (Fig. 8A,B). However, based on our sampling of four time points across a day, we find that the oscillation of PDF neuropeptide in mutant HTT-Q128 protein-expressing flies did not show the gradual rise and fall as seen in the controls (Fig. 8B, red bars). Interestingly, on coexpression of Atg8a, along with HTT-Q128, a gradual rise and fall in the PDF levels were now restored in the dorsal projections (Fig. 8B, green bars).
Figure 8.
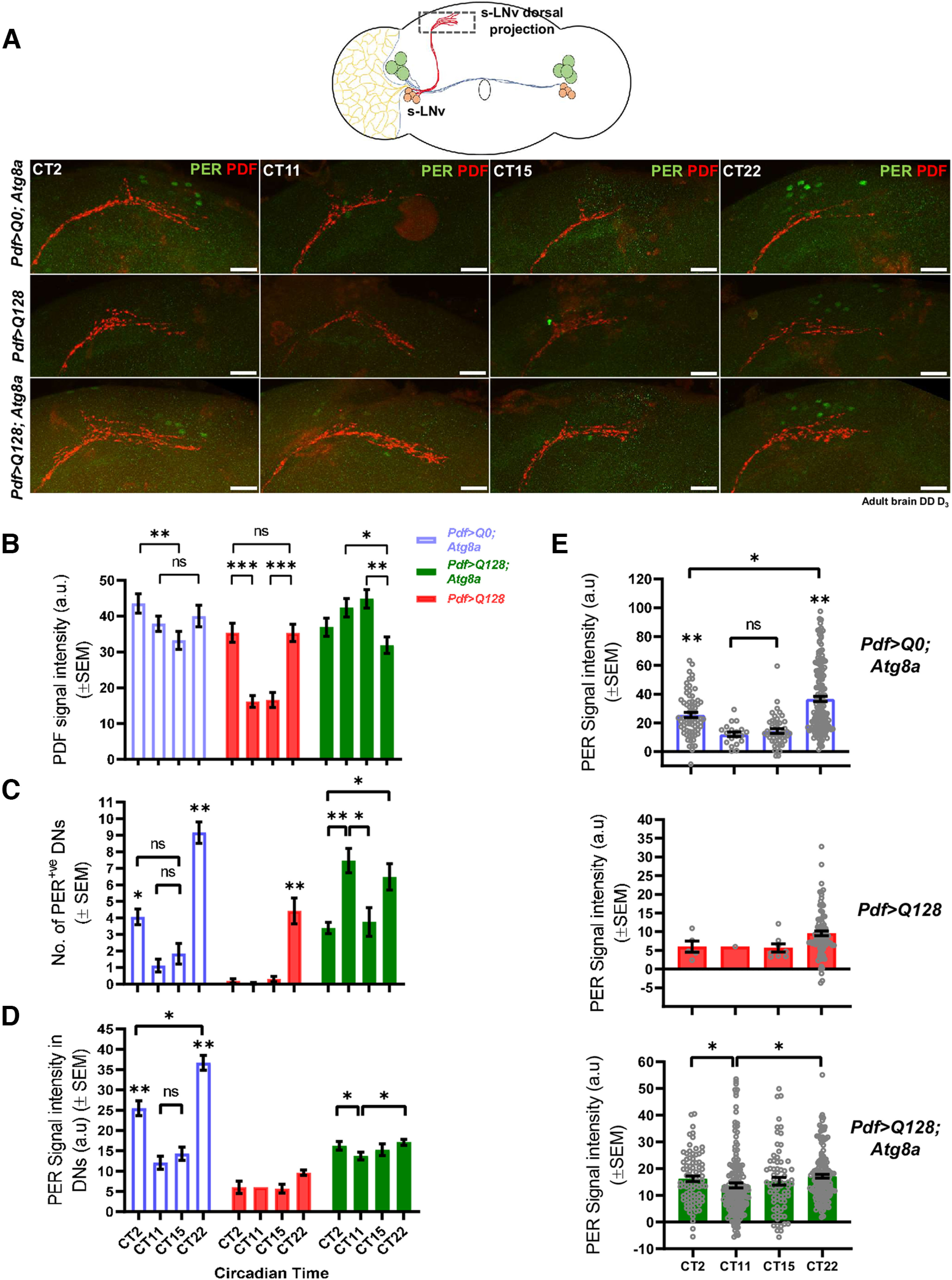
Atg8a overexpression improves the output from the PDF+ small neurons to the downstream neurons. A, Representative MIP images (DD day 3, adult brain) of dorsal projections (part of small lateral ventral neurons) and DNs, depicting staining for PER protein (green) and PDF neuropeptide (red) for four circadian time points (CT22, CT2, CT11, and CT15). Scale bar, 20 µm. B, Plots represent quantification of the mean signal intensity of PDF neuropeptide, quantified from the dorsal projections of small neurons at four different circadian time points on DD day 3. Time-dependent oscillation of PDF neuropeptide was observed in the control genotype (blue bars). Expression of mutant HTT-Q128 in the PDF neurons does not hamper the oscillation of PDF neuropeptide in the dorsal projections, but like the control genotype, no gradual decrease or increase was observed in the intensity (red bars). Atg8a-overexpressing flies in the presence of mutant HTT-Q128 protein also show PDF oscillation (green bar), but the change in the intensity is distinctly different from what was observed in flies only expressing mutant HTT-Q128 protein. n > 16 brain hemispheres/time point (for control genotype), and n > 18 brain hemispheres/time point (for experimental genotype). *p < 0.05. C, Plot represents quantification of the number of PER+ DN1 and DN2 (together mentioned as DNs) neurons quantified across four different circadian time points. Nice time-dependent oscillation was observed in the number of PER+ DNs for the control genotype. Expression of mutant HTT-Q128 protein in the PDF neurons hampers PER expression in the DNs, and hardly any cells were visualized at CT2, CT11, and CT15 time points. Overexpression of Atg8a in the PDF neurons improves PER expression in the DNs; however, the oscillation in the cell number was not the same as observed in the control genotype. n > 16 brain hemisphere/time point (for control genotype), and n > 18 brain hemisphere/time point (for experimental genotype). *p < 0.05. D, Plots represent quantification of the mean signal intensity of PER protein, quantified from DNs at four different circadian time points. The control genotype showed a nice time-dependent oscillation of PER protein in the DNs (blue bars). Expression of mutant HTT-Q128 protein in the PDF neurons hampers PER oscillation in the DNs (red bars). Atg8a overexpression in the PDF neurons results in a low amplitude PER oscillation in downstream DNs (green bars). n > 16 brain hemispheres/time point (for control genotype), and n > 18 brain hemispheres/time point (for experimental genotype). *p < 0.05. E, Plots (same as in D) represent individual values of PER protein intensity quantified from the DNs. *p < 0.05. Asterisk on individual genotypes indicates that the genotype is significantly different from all other plotted genotypes.
Since PDF oscillations appear to have been modified by Atg8a overexpression, we asked whether the downstream circuits have also been affected. We quantified PER protein oscillation in a subset of circadian clock neurons called dorsal neurons (DNs- DN1, DN2, and DN3), which express PDF-receptor and are known to receive input from the s-LNv (Mertens et al., 2005; Lear et al., 2009). DN1s receive input from the PDF+ s-LNv and communicate with downstream motor centers (Cavanaugh et al., 2014). Since we were not always able to reliably distinguish the DN1 and DN2 subtypes, we consider them as one entity in our analysis. In control flies, the number of PER+ DNs showed the expected oscillation with a significant peak at CT22 and low values at CT 11 and 15 (Kruskal–Wallis test: H(3,77) =43.9153, p ≪ 0.05, CT22 vs CT11, p ≪ 0.05, CT11 vs CT15, p = 1.0000) (Fig. 8C, blue bars). Expression of mutant HTT-Q128 protein in PDF neurons led to an overall reduction in PER+ DNs and a few neurons showed PER staining at CT22 (Fig. 8C, red bars). Atg8a overexpression in the PDF neurons improved PER expression in the DNs, although the distribution of cell numbers was different from controls with ∼3 or 4 cells being detected even at CT15 and there being relatively larger numbers even at CT 11 (Kruskal–Wallis test: H(3,92) = 18.7867, p = 0.0003, CT22 vs CT2, p = 0.0254, CT2 vs CT11, p = 0.0019, CT11 vs CT15, p = 0.0123, CT15 vs CT22, p = 0.1078) (Fig. 8C, green bars). Further, we quantified the PER protein intensity and found the expected oscillation in control flies (Kruskal–Wallis test: H(3,302) =66.2835, p ≪ 0.05, CT22 vs CT11, p = 0.0004) (Fig. 8D, blue bars; Fig. 8E, top). The presence of mutant HTT-Q128 protein in PDF neurons caused an overall reduction in the PER protein in DNs (Fig. 8D, red bar; Fig. 8E, middle). Coexpression of Atg8a with HTT-Q128 protein in PDF neurons led to a significant improvement in PER protein level in the DNs, although only a low-amplitude PER protein oscillation was detected (Kruskal–Wallis test: H(3,481) = 17.7307, p = 0.0005, CT22 vs CT11, p = 0.0004, CT2 vs CT11, p = 0.0257) (Fig. 8D, green bar; Fig. 8E, bottom). Overall, in the presence of mutant HTT-Q128 protein, overexpression of Atg8a in the PDF neurons improves output from the small neurons.
Atg8a improves the functioning of Drosophila sleep homeostatic circuit
One of the common characteristic features of HD is sleep defects (Morton et al., 2005; Maywood et al., 2010; Goodman et al., 2011). Lack of proper sleep has been shown to negatively impact the functioning of individuals, and recently it has been hypothesized that sleep defects can be a factor that exacerbates neurodegenerative phenotypes (Musiek and Holtzman, 2016; Leng et al., 2019; Voysey et al., 2021). Since sleep is strongly influenced by sleep centers distinct from circadian circuits, we targeted the dorsal fan-shaped body-dFB (one of the well-recognized sleep homeostatic circuits) and asked whether Atg8a overexpression rescues sleep phenotypes. Initial AW showed a significant decrease in total, daytime, and nighttime sleep on mutant HTT-Q128 protein expression (one-way ANOVA: total sleep – 1-3 day – F(3,104) = 23.901, p ≪ 0.05, R23E10>Q128 vs R23E10> Q0; Atg8a, p ≪ 0.05, R23E10>Q128 vs R23E10/+, p ≪ 0.05; 4-6 d – F(3,104) = 13.249, p ≪ 0.05, R23E10>Q128 vs R23E10> Q0; Atg8a, p ≪ 0.05, R23E10>Q128 vs R23E10/+, p ≪ 0.05; day sleep – 1-3 d – F(3,104) = 19.784, p ≪ 0.05, R23E10>Q128 vs R23E10> Q0; Atg8a, p = 0.0001, R23E10>Q128 vs R23E10/+, p = 0.0045; night sleep – 1-3 d – F(3,104) = 19.663, p ≪ 0.05, R23E10>Q128 vs R23E10> Q0; Atg8a, p = 0.0001, R23E10>Q128 vs R23E10/+, p = 0.0001; 4-6 d – F(3,104) = 15.877, p ≪ 0.05, R23E10>Q128 vs R23E10> Q0; Atg8a, p = 0.0001, R23E10>Q128 vs R23E10/+, p = 0.0001) (Fig. 9A–D, red bar). Further, detailed characterization revealed that the mutant protein also leads to a decrease in the length of daytime and nighttime sleep bout length (one-way ANOVA: length of daytime sleep episodes – 1-3 d – F(3,104) = 6.7622, p = 0.0003, R23E10>Q128 vs R23E10> Q0; Atg8a, p = 0.0016, R23E10>Q128 vs R23E10/+, p = 0.0011; length of nighttime sleep episodes – 1-3 d – F(3,104) = 11.6154, p ≪ 0.05, R23E10>Q128 vs R23E10> Q0; Atg8a, p = 0.0001, R23E10>Q128 vs R23E10/+, p = 0.0011, 4-6 d – F(3,104) = 7.9562, p ≪ 0.05, R23E10>Q128 vs R23E10> Q0; Atg8a, p = 0.0003, R23E10>Q128 vs R23E10/+, p = 0.0005) (Fig. 9E,F, red bar). In addition to these defects, we also observed that, with time, defects in sleep levels were reversed and comparable to control genotypes, possibly as a compensatory response from other sleep circuits. Atg8a overexpression in dFB rescues the defect in sleep level, and all the parameters were comparable to control flies (one-way ANOVA: total sleep – 1-3 d – F(3,104) = 23.901, p ≪ 0.05, R23E10>Q128 vs R23E10> Q128; Atg8a, p ≪ 0.05; 4-6 d – F(3,104) = 13.249, p ≪ 0.05, R23E10>Q128 vs R23E10> Q128; Atg8a, p ≪ 0.05; daytime sleep – 1-3 d – F(3,104) = 19.784, p ≪ 0.05, R23E10>Q128 vs R23E10> Q128; Atg8a, p = 0.0001; nighttime sleep – 1-3 d – F(3,104) = 19.663, p ≪ 0.05, R23E10>Q128 vs 23E10> Q128; Atg8a, p = 0.0001, 4-6 d – F(3,104) = 15.877, p ≪ 0.05, R23E10>Q128 vs R23E10> Q128; Atg8a, p = 0.0001) (Fig. 9A–D, green bar). Further, changes in total, daytime, and nighttime sleep on Atg8a overexpression, improvement in the length of daytime and nighttime sleep episodes was also observed on Atg8a overexpression (one-way ANOVA: length of daytime sleep episodes – 1-3 d – F(3,104) = 6.7622, p = 0.0003, R23E10>Q128 vs R23E10> Q128; Atg8a, p = 0.0069; 4-6 d – F(1,52) = 7.0615, p = 0.0104; length of nighttime sleep – 1-3 d – F(3,104) = 11.6154, p ≪ 0.05, R23E10>Q128 vs R23E10> Q128; Atg8a, p = 0.0006; 4-6 d – F(1,52) = 14.4124, p = 0.0003) (Fig. 9E,F).
We further asked what cellular changes result in behavioral rescue. We dissected adult brains of the relevant genotypes at day 3 (after eclosion) and stained for HTT-Q128 protein. In both experimental genotypes, we observed mutant HTT-Q128 aggregates. Further, we observed an accumulation of nonaggregated mutant HTT-Q128 protein in the axonal projections of the circuit, a phenotype not observed in Atg8a-overexpressing flies (Fig. 9G,H). The same was verified by quantifying the nonaggregated mutant form in the axonal projections, and we observed that Atg8a overexpression leads to a significant reduction in the same (one-way ANOVA: F(1,20) = 21.4120, p = 0.0001, R23E10>Q128 vs R23E10>Q128; Atg8a, p = 0.0003) (Fig. 9G, left). We further quantified the mutant protein aggregates and observed an increase in the aggregate number in the whole circuit; however, the increase was not significantly different from the flies only expressing mutant HTT-Q128 protein (one-way ANOVA: F(1,20) = 1.4271, p = 0.2471) (Fig. 9G, right). Overall, these results suggest that Atg8a overexpression improves the functioning of the dFB circuit and improves sleep phenotypes in the presence of mutant HTT-Q128 protein.
Discussion
Circadian and sleep defects are well documented in both patient and animal models of HD. While there is no direct evidence, recent studies have highlighted the potential of circadian therapies in neurodegenerative models (Pallier et al., 2007; Wang et al., 2017; Whittaker et al., 2018). Here, using a genetic approach, we targeted a subset of Drosophila circadian neurons whose functioning is directly correlated with self-sustained locomotor rhythms, and sleep-center neurons that are known to promote sleep and asked whether genetic modulation of the autophagy pathway within these cells can mitigate the toxicity caused by mHTT.
Our study shows that overexpression of a key autophagy gene, Atg8a in the presence of mutant HTT-Q128 protein, led to sustained (up to 3 weeks as adults), although partial (low amplitude) rescue of rhythmic locomotion. In addition to the improvement in activity rhythm, Atg8a overexpression also ameliorated sleep defects in the flies. Interestingly, despite the clear behavioral rescue, some cellular phenotypes revealed novel and unexpected patterns. Staining for a molecular clock protein PERIOD revealed that Atg8a-mediated behavioral rescue is not dependent on improvement in the oscillation of the PERIOD protein (Fig. 7). The maintenance of activity rhythms in the absence of PERIOD protein in the small neurons indicates that the protein within these neurons might be dispensable for rhythm sustenance. Two very recent studies support this reasoning and show that PERIOD protein in LNvs is not necessary for the persistence of activity rhythms under DD but is vital for rhythm strength (Delventhal et al., 2019; Schlichting et al., 2019). Overall, these results point out that autophagy modulations can mitigate the toxicity caused by the mHTT in circadian and sleep neurons.
We show that Atg8a overexpression results in increased Ref(2)P-HTT-Q128 and Cath-D -HTT-Q128 colocalization in the targeted neurons. Further, Atg1 downregulation attenuated the Atg8a-mediated rescue. These results strongly point toward the involvement of Atg8a in regulating the autophagy pathway in neurons. Two recent studies lend support to this observation, wherein they show that Atg8a positively regulates the autophagy pathway (Jacomin et al., 2020; Hwang et al., 2022).
Basal level of autophagy may vary among neuronal subtypes, and they may clear cargo at different rates (Tsvetkov et al., 2013). Our finding of low level of Ref(2)P colocalization in small neurons (Fig. 5) also points toward possible differences in the level of autophagy between the two subsets of neurons. Cathepsin-D staining (Fig. 6) also supports this notion, as small neurons failed to show any visible Cathepsin-D staining in control flies while large neurons showed both diffused and punctate staining. These inherent differences between large and small neurons can possibly explain why small neurons are more susceptible to aggregate stress. The lack of Cathepsin-D staining in large lateral ventral neurons on Atg8a overexpression along with lower HSP70 (Figs. 3, 6B) suggest that large neurons have greater ability to tolerate stress. This is also supported by the RNAseq studies, which indicate that large neurons are well equipped with pathways required for tackling stressful conditions (Kula-Eversole et al., 2010; Ma et al., 2021). A recent study from our laboratory also shows that overexpression of HSP70 protein can decrease toxicity of the mutant HTT-Q128 protein in the small neurons leading to improvement in activity-rest rhythm (Prakash et al., 2022). The lack of change in aggregate number in the large neurons also points toward lack of autophagy induction on Atg8a overexpression. Given these differences, it will be interesting to examine stage specificity of Atg8a-mediated autophagy induction in the large neurons and whether inherent differences in protein homeostatic pathways are one of the reasons for the selective susceptibility of small neurons.
Despite the involvement of the autophagy pathway in Atg8a-mediated rescue of behavior, we did not observe any decrease in mHTT aggregates in the circadian and sleep neurons. Interestingly, we observed an increase in the levels of HTT aggregates on Atg8a overexpression in both circuits. It is possible that increased aggregation in the targeted neurons is an outcome of defects at either the cargo recognition and loading or fusion steps of the autophagy pathway, which result in the accumulation of aggregates in these neurons (Martinez-Vicente et al., 2010). Studies have also shown that soluble forms of mutant HTT is more toxic to neurons and increased aggregation helps neurons to better deal with toxicity (Arrasate et al., 2004; Miller et al., 2010). This could be another possible reason for the increase of aggregates on Atg8a overexpression. The prolonged survival of large neurons despite the presence of a high aggregate load is not surprising. Similar observations have been reported for mammalian cortical neurons, where high aggregate load is not correlated with cell death (Gutekunst et al., 1999; Kuemmerle et al., 1999). It further suggests that the large neurons are more competent in tackling stress, as no major defects were observed in the large neurons despite the accumulation of mutant HTT aggregates.
Synaptic transmission of neuropeptides and neurotransmitters is critical for proper behavioral and physiological outputs. In neurodegenerative disorders including HD, defects in axonal transport, vesicular fusion, and release of neurotransmitters or neuropeptides are seen (Li et al., 2001; Q. Xu et al., 2013). Here we show that the presence of mHTT in LNvs hampers the expression of PDF neuropeptide in the soma of small neurons. However, the axonal projections of these neurons show strong PDF staining and oscillation. The presence of PDF staining in axons suggests that the vesicular transport of PDF mRNA or peptides in the axons is not significantly affected by HTT. Loss of PDF, PERIOD, and Cathepsin-D staining in the cell bodies of the small neurons further suggests that HTT-mediated defects are more prominent in cell bodies. Further, the oscillation of PDF neuropeptide in the neuronal processes hints toward contribution from other cells, including glia in maintaining the PDF oscillations. This observation is further supported by a recent study, which shows the involvement of glial cells in regulating PDF oscillations in the projections of the small neurons (Damulewicz et al., 2022). Lack of PERIOD protein expression and oscillation in the downstream DNs on mutant HTT expression in the small neurons point toward defective synaptic communication between the small and DNs, which might be the case with the sleep neurons as well. Atg8a overexpression in the small neurons led to an improvement in PERIOD protein levels in DNs (Fig. 8), suggesting that the behavioral rescue is possibly an outcome of improvement in synaptic communication between the small neurons and downstream DNs. Further, improved sleep is also associated with decrease in nonaggregated mHTT from the neuronal processes of sleep neurons, again pointing out that the rescue is possibly an outcome of improved synaptic communication. We speculate that this improved synaptic communication is an outcome of increased aggregations leading to decreased toxicity of mutant HTT in the neurons, or that Atg8a being a vesicular protein improves PDF release from the small neurons.
In conclusion, we present evidence for compromised autophagy in circadian pacemaker circuit when mHTT is expressed, and demonstrate that genetic upregulation of Atg8a enables the circadian pacemakers and sleep circuits to drive behavioral rhythmicity in locomotion and restore the quality of sleep. Further, our studies suggest that this occurs through improvement in synaptic output of the circuits. We propose that Atg8a in the targeted neurons enhances autophagy, which in turn increases the aggregation of mHTT and rescues the strength of connections with downstream neurons in the circadian circuit and sleep circuit, thus enabling overall rescue of circadian and sleep phenotypes.
Footnotes
This work was supported by Jawaharlal Nehru Centre for Advanced Scientific Research and Department of Biotechnology BT/INF/22/SP27679/2018 to V.S.; and Science and Engineering Research Board Core Research Grant CRG/2019/006802. We thank all laboratory members for valuable discussion and critical inputs during the study and manuscript preparation.
The authors declare no competing financial interests.
References
- Allocca M, Zola S, Bellosta P (2018) The fruit fly, Drosophila melanogaster: modeling of human diseases (Part II). In: Drosophila melanogaster: model for recent advances in genetics and therapeutics. London, United Kingdom: IntechOpen. [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810. 10.1038/nature02998 [DOI] [PubMed] [Google Scholar]
- Baldo B, Soylu R, Petersén Å (2013) Maintenance of basal levels of autophagy in Huntington's disease mouse models displaying metabolic dysfunction. PLoS One 8:e83050. 10.1371/journal.pone.0083050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellosta Diago E, Pérez Pérez J, Santos Lasaosa S, Viloria Alebesque A, Martínez Horta S, Kulisevsky J, López del Val J (2017) Circadian rhythm and autonomic dysfunction in presymptomatic and early Huntington's disease. Parkinsonism Relat Disord 44:95–100. 10.1016/j.parkreldis.2017.09.013 [DOI] [PubMed] [Google Scholar]
- Benes P, Vetvicka V, Fusek M (2008) Cathepsin D: many functions of one aspartic protease. Crit Rev Oncol Hematol 68:12–28. 10.1016/j.critrevonc.2008.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt J, Bylsma FW, Gross R, Stine OC, Ranen N, Ross CA (1996) Trinucleotide repeat length and clinical progression in Huntington's disease. Neurology 46:527–531. 10.1212/wnl.46.2.527 [DOI] [PubMed] [Google Scholar]
- Cavanaugh DJ, Geratowski JD, Wooltorton JR, Spaethling JM, Hector CE, Zheng X, Johnson EC, Eberwine JH, Sehgal A (2014) Identification of a circadian output circuit for rest:activity rhythms in Drosophila. Cell 157:689–701. 10.1016/j.cell.2014.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan HY, Bonini NM (2000) Drosophila models of human neurodegenerative disease. Cell Death Differ 7:1075–1080. 10.1038/sj.cdd.4400757 [DOI] [PubMed] [Google Scholar]
- Chen S, Ferrone FA, Wetzel R (2002) Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci USA 99:11884–11889. 10.1073/pnas.182276099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS (2021) Defining circadian disruption in neurodegenerative disorders. J Clin Invest 131:e148288. 10.1172/JCI148288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damulewicz M, Szypulski K, Pyza E (2022) Glia-neurons cross-talk regulated through autophagy. Front Physiol 13:886273. 10.3389/fphys.2022.886273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90:537–548. 10.1016/s0092-8674(00)80513-9 [DOI] [PubMed] [Google Scholar]
- Delventhal R, O'Connor RM, Pantalia MM, Ulgherait M, Kim HX, Basturk MK, Canman JC, Shirasu-Hiza M (2019) Dissection of central clock function in Drosophila through cell-specific CRISPR-mediated clock gene disruption. Elife 8:e48308. 10.7554/eLife.48308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990–1993. 10.1126/science.277.5334.1990 [DOI] [PubMed] [Google Scholar]
- Faragó A, Zsindely N, Bodai L (2019) Mutant huntingtin disturbs circadian clock gene expression and sleep patterns in Drosophila. Sci Rep 9:1–6. 10.1038/s41598-019-43612-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly P (2015) Effects of temporally controlled Huntingtin expression and autophagy upregulation in Drosophila melanogaster. MS thesis, Jawaharlal Nehru Centre for Advanced Scientific Research. Bangalore, India. Available at https://libjncir.jncasr.ac.in/jspui/browse?type=author&value=Ganguly%2C+Payel&value_lang=. [Google Scholar]
- Gilestro GF, Cirelli C (2009) pySolo: a complete suite for sleep analysis in Drosophila. Bioinformatics 25:1466–1467. 10.1093/bioinformatics/btp237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AO, Rogers L, Pilsworth S, McAllister CJ, Shneerson JM, Morton AJ, Barker RA (2011) Asymptomatic sleep abnormalities are a common early feature in patients with Huntington's disease. Curr Neurol Neurosci Rep 11:211–217. 10.1007/s11910-010-0163-x [DOI] [PubMed] [Google Scholar]
- Gusella JF, MacDonald ME (2006) Huntington's disease: seeing the pathogenic process through a genetic lens. Trends Biochem Sci 31:533–540. 10.1016/j.tibs.2006.06.009 [DOI] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ (1999) Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci 19:2522–2534. 10.1523/JNEUROSCI.19-07-02522.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889. 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- Hwang HJ, Ha H, Lee BS, Kim BH, Song HK, Kim YK (2022) LC3B is an RNA-binding protein to trigger rapid mRNA degradation during autophagy. Nat Commun 13:1436. 10.1038/s41467-022-29139-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacomin AC, Petridi S, di Monaco M, Bhujabal Z, Jain A, Mulakkal NC, Palara A, Powell EL, Chung B, Zampronio C, Jones A, Cameron A, Johansen T, Nezis IP (2020) Regulation of expression of autophagy genes by Atg8a-interacting partners Sequoia, YL-1, and Sir2 in Drosophila. Cell Rep 31:107695. 10.1016/j.celrep.2020.107695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Barua S, Huang MY, Park J, Yenari MA, Lee JE (2020) Heat shock protein 70 (HSP70) induction: chaperonotherapy for neuroprotection after brain injury. Cells 9:2020. 10.3390/cells9092020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga H, Martinez-Vicente M, Arias E, Kaushik S, Sulzer D, Cuervo AM (2011) Constitutive upregulation of chaperone-mediated autophagy in Huntington's disease. J Neurosci 31:18492–18505. 10.1523/JNEUROSCI.3219-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata JI, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884. 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- Kudo T, Schroeder A, Loh DH, Kuljis D, Jordan MC, Roos KP, Colwell CS (2011) Dysfunctions in circadian behavior and physiology in mouse models of Huntington's disease. Exp Neurol 228:80–90. 10.1016/j.expneurol.2010.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal F (1999) Huntingtin aggregates may not predict neuronal death in Huntington's disease. Ann Neurol 46:842–849. [PubMed] [Google Scholar]
- Kula-Eversole E, Nagoshi E, Shang Y, Rodriguez J, Allada R, Rosbash M (2010) Surprising gene expression patterns within and between PDF-containing circadian neurons in Drosophila. Proc Natl Acad Sci USA 107:13497–13502. 10.1073/pnas.1002081107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuljis D, Kudo T, Tahara Y, Ghiani CA, Colwell CS (2018) Pathophysiology in the suprachiasmatic nucleus in mouse models of Huntington's disease. J Neurosci Res 96:1862–1875. 10.1002/jnr.24320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar MJ, Shah D, Giridharan M, Yadav N, Manjithaya R, Clement JP (2021) Spatiotemporal analysis of soluble aggregates and autophagy markers in the R6/2 mouse model. Sci Rep 11:96. 10.1038/s41598-020-78850-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie P, Snapp EL (2010) Formation and toxicity of soluble polyglutamine oligomers in living cells. PLoS One 5:e15245. 10.1371/journal.pone.0015245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear BC, Zhang L, Allada R (2009) The neuropeptide PDF acts directly on evening pacemaker neurons to regulate multiple features of circadian behavior. PLoS Biol 7:e1000154. 10.1371/journal.pbio.1000154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Yoshihara M, Littleton JT (2004) Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington's disease. Proc Natl Acad Sci USA 101:3224–3229. 10.1073/pnas.0400243101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng Y, Musiek ES, Hu K, Cappuccio FP, Yaffe K (2019) Association between circadian rhythms and neurodegenerative diseases. Lancet Neurol 18:307–318. 10.1016/S1474-4422(18)30461-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li SH, Yu ZX, Shelbourne P, Li XJ (2001) Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. J Neurosci 21:8473–8481. 10.1523/JNEUROSCI.21-21-08473.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippai M, Low P (2014) The role of the selective adaptor Ref(2)P and ubiquitin-like proteins in autophagy. Biomed Res Int 2014:832704. 10.1155/2014/832704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Przybylski D, Abruzzi KC, Schlichting M, Li Q, Long X, Rosbash M (2021) A transcriptomic taxonomy of Drosophila circadian neurons around the clock. Elife 10:1–19. 10.7554/eLife.63056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ME, et al. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes: the Huntington's Disease Collaborative Research Group. Cell 72:971–983. 10.1016/0092-8674(93)90585-E [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM (2010) Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci 13:567–576. 10.1038/nn.2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maywood ES, Fraenkel E, McAllister CJ, Wood N, Reddy AB, Hastings MH, Morton AJ (2010) Disruption of peripheral circadian timekeeping in a mouse model of Huntington's disease and its restoration by temporally scheduled feeding. J Neurosci 30:10199–10204. 10.1523/JNEUROSCI.1694-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens I, Vandingenen A, Johnson EC, Shafer OT, Li W, Trigg JS, de Loof A, Schoofs L, Taghert PH (2005) PDF receptor signaling in Drosophila contributes to both circadian and geotactic behaviors. Neuron 48:213–219. 10.1016/j.neuron.2005.09.009 [DOI] [PubMed] [Google Scholar]
- Miller J, Arrasate M, Shaby BA, Mitra S, Masliah E, Finkbeiner S (2010) Quantitative relationships between Huntingtin levels, polyglutamine length, inclusion body formation, and neuronal death provide novel insight into Huntington's disease molecular pathogenesis. J Neurosci 30:10541–10550. 10.1523/JNEUROSCI.0146-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES (2005) Disintegration of the sleep-wake cycle and circadian timing in Huntington's disease. J Neurosci 25:157–163. 10.1523/JNEUROSCI.3842-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, Holtzman DM (2016) Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 354:1004–1008. 10.1126/science.aah4968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nah J, Yuan J, Jung YK (2015) Autophagy in neurodegenerative diseases: from mechanism to therapeutic approach. Mol Cells 38:381–389. 10.14348/molcells.2015.0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, Lazarou M (2016) Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol 215:857–874. 10.1083/jcb.201607039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama J, Miura E, Mizushima N, Watanabe M, Yuzaki M (2007) Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy 3:591–596. [DOI] [PubMed] [Google Scholar]
- Pallier PN, Maywood ES, Zheng Z, Chesham JE, Inyushkin AN, Dyball R, Hastings MH, Morton AJ (2007) Pharmacological imposition of sleep slows cognitive decline and reverses dysregulation of circadian gene expression in a transgenic mouse model of Huntington's disease. J Neurosci 27:7869–7878. 10.1523/JNEUROSCI.0649-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Stoleru D, Levine JD, Hall JC, Rosbash M (2003) Drosophila free-running rhythms require intercellular communication. PLoS Biol 1:e13. 10.1371/journal.pbio.0000013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potdar S, Sheeba V (2018) Wakefulness is promoted during daytime by PDFR signalling to dopaminergic neurons in Drosophila melanogaster. eNeuro 5:ENEURO.0129-18.2018-147. 10.1523/ENEURO.0129-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P, Nambiar A, Sheeba V (2017) Oscillating PDF in termini of circadian pacemaker neurons and synchronous molecular clocks in downstream neurons are not sufficient for sustenance of activity rhythms in constant darkness. PLoS One 12:e0175073. 10.1371/journal.pone.0175073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P, Pradhan AK, Sheeba V (2022) Hsp40 overexpression in pacemaker neurons protects against circadian dysfunction in a Drosophila model of Huntington's disease. Dis Model Mech 15:dmm049447. 10.1242/dmm.049447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratliff EP, Mauntz RE, Kotzebue RW, Gonzalez A, Achal M, Barekat A, Finley KA, Sparhawk JM, Robinson JE, Herr DR, Harris GL, Joiner WJ, Finley KD (2015) Aging and autophagic function influences the progressive decline of adult Drosophila behaviors. PLoS One 10:e0132768. 10.1371/journal.pone.0132768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36:585–595. 10.1038/ng1362 [DOI] [PubMed] [Google Scholar]
- Renn SC, Park JH, Rosbash M, Hall JC, Taghert PH (1999) A pdf neuropeptide gene mutation and ablation of PDF neurons each cause severe abnormalities of behavioral circadian rhythms in Drosophila. Cell 99:791–802. 10.1016/s0092-8674(00)81676-1 [DOI] [PubMed] [Google Scholar]
- Roos RA (2010) Huntington's disease: a clinical review. Orphanet J Rare Dis 5:40. 10.1186/1750-1172-5-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC (2005) Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 170:1101–1111. 10.1083/jcb.200504035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlichting M, Díaz MM, Xin J, Rosbash M (2019) Neuron-specific knockouts indicate the importance of network communication to Drosophila rhythmicity. Elife 8:e48301. 10.7554/eLife.48301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte J, Littleton JT (2011) The biological function of the Huntingtin protein and its relevance to Huntington's disease pathology. Curr Trends Neurol 5:65–78. [PMC free article] [PubMed] [Google Scholar]
- Settembre C, di Malta C, Polito VA, Arencibia MG, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433. 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, de Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Jürgen Klisch T, Wollenberg AC, di Bernardo D, Chan L, Irazoqui JE, Ballabio A (2013) TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 15:647–658. 10.1038/ncb2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevlever D, Jiang P, Yen SH (2008) Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 47:9678–9687. 10.1021/bi800699v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheeba V, Fogle KJ, Kaneko M, Rashid S, Chou YT, Sharma VK, Holmes TC (2008) Large ventral lateral neurons modulate arousal and sleep in Drosophila. Curr Biol 18:1537–1545. 10.1016/j.cub.2008.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V (2010) Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One 5:e9979. 10.1371/journal.pone.0009979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky LA, Forgac M (2015) Amino acid availability modulates vacuolar H+-ATPase assembly. J Biol Chem 290:27360–27369. 10.1074/jbc.M115.659128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O (2008) Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum Mol Genet 17:345–356. 10.1093/hmg/ddm311 [DOI] [PubMed] [Google Scholar]
- Tsvetkov AS, Arrasate M, Barmada S, Ando DM, Sharma P, Shaby BA, Finkbeiner S (2013) Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat Chem Biol 9:586–592. 10.1038/nchembio.1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voysey Z, Fazal SV, Lazar AS, Barker RA (2021) The sleep and circadian problems of Huntington's disease: when, why and their importance. J Neurol 268:2275–2283. 10.1007/s00415-020-10334-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HB, Whittaker DS, Truong D, Mulji AK, Ghiani CA, Loh DH, Colwell CS (2017) Blue light therapy improves circadian dysfunction as well as motor symptoms in two mouse models of Huntington's disease. Neurobiol Sleep Circadian Rhythms 2:39–52. 10.1016/j.nbscr.2016.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker DS, Loh DH, Wang HB, Tahara Y, Kuljis D, Cutler T, Ghiani CA, Shibata S, Block GD, Colwell CS (2018) Circadian-based treatment strategy effective in the BACHD mouse model of Huntington's disease. J Biol Rhythms 33:535–554. 10.1177/0748730418790401 [DOI] [PubMed] [Google Scholar]
- Xu F, Kula-Eversole E, Iwanaszko M, Hutchison AL, Dinner A, Allada R (2019) Circadian clocks function in concert with heat shock organizing protein to modulate mutant Huntingtin aggregation and toxicity. Cell Rep 27:59–70.e4. 10.1016/j.celrep.2019.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Huang S, Song M, Wang CE, Yan S, Liu X, Gaertig MA, Yu SP, Li H, Li S, Li XJ (2013) Synaptic mutant huntingtin inhibits synapsin-1 phosphorylation and causes neurological symptoms. J Cell Biol 202:1123–1138. 10.1083/jcb.201303146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Yue Z (2014) Autophagy and its normal and pathogenic states in the brain. Annu Rev Neurosci 37:55–78. 10.1146/annurev-neuro-071013-014149 [DOI] [PubMed] [Google Scholar]
- Yang Z, Sehgal A (2001) Role of molecular oscillations in generating behavioral rhythms in Drosophila. Neuron 29:453–467. 10.1016/s0896-6273(01)00218-5 [DOI] [PubMed] [Google Scholar]