

Abstract
Aims:
Glucokinase (GK) is expressed in the glucose-sensing cells of the islets of Langerhans and plays a critical role in glucose homeostasis. Here, we tested the hypothesis that genetic actvation of GK in a small subset of β-cells is sufficient to change the glucose set-point of the whole islet.
Material and Methods:
Mouse models of cell-type specific GK deficiency (GKKO) and genetic enzyme activation (GKKI) in a subset of β-cells were obtained by crossing the αGSU (gonadotropin alpha subunit)-Cre transgene with the appropriate GK mutant alleles. Metabolic analyses consisted of glucose tolerance tests, perifusion of isolated islets and intracellular calcium measurements.
Key Findings:
The αGSU-Cre transgene produced genetically mosaic islets, as Cre was active in 15 ± 1.2% of β-cells. While mice deficient for GK in a subset of islet cells were normal, unexpectedly, GKKI mice were chronically hypoglycemic, glucose intolerant, and had a lower threshold for glucose stimulated insulin secretion. GKKI mice exhibited an average fasting blood glucose level of 3.5 mM. GKKI islets responded with intracellular calcium signals already at that spread through the whole islets at 1 mM, and secreted insulin at 3 mM glucose.
Significance:
Genetic activation of GK in a minority of β-cells is sufficient to change the glucose threshold for insulin secretion in the entire islet and thereby glucose homeostasis in the whole animal. These data support the model in which β-cells with higher GK activity function as ‘hub’ or ‘trigger’ cells and thus control insulin secretion by the β-cell collective within the islet.
Keywords: Islet, beta cell, glucokinase, hub cell, trigger cell
1. Introduction:
Glucokinase (GK), unique among hexokinases in displaying a high Km for glucose, is a central enzyme involved in maintaining glucose homeostasis. Blood glucose levels must be maintained within the physiological range (4–6 mM), as deviations from these concentration in either direction cause hypoglycemia or hyperglycemia, respectively, both conditions that are not tolerated long-term. Glucose metabolism is primarily regulated by the pancreatic islets of Langerhans, where β-cells use GK to directly respond to rising glucose levels to induce glucose stimulated insulin secretion (GSIS) (Matschinsky and Wilson, 2019). In α-cells, GK is emerging as an important regulator of glucose induced glucagon suppression (Basco et al. , 2018, Moede et al. , 2020), and recent data from our laboratories demonstrate that changing the Km of GK for glucose specifically in α-cells alters glucose suppression of glucagon secretion proportionally (Bahl et al. , 2021). The secretion of both hormones from pancreatic islets is responsible for regulating blood sugar levels, which is essential for organismal survival.
Insulin secretion is coupled to glycolytic flux through multiople intermediary steps, translating biochemical signals in the form of an increased ATP/ADP ratio into electrical signals. This is accomplished via the ATP-dependent potassium channel on the β-cell plasma membrane, which closes when ATP/ADP ratios increase (Wilson et al. , 2017), causing membrane depolarization which results in the opening of voltage gated calcium channels. The rise in intracellular calcium is the final trigger for the fusion of predocked insulin granules with the plasma membrane and insulin release (Kaestner et al. , 2021). In rodent islets, the glucose response in the form of a calcium signal is first observed in a leader cell, which is followed by a synchronous calcium signal in neighboring cells that is dependent at least in part on electrical coupling via connexin 36 gap junctions (Benninger et al. , 2008, MacDonald and Rorsman, 2006). There are several explanations for this phenomenon: leader cells may have a different gene or protein expression profile than other cells (Johnston et al. , 2016), or stochastic gene expression and protein activity of glucose homeostasis genes generates a small fraction of cells which are more sensitive to glucose. More specifically, it is possible that within an islet, there exists a Gaussian distribution of GK expression and/or activity between beta cells. This would naturally bring about cells that are slightly more glucose responsive than average, as first observed by Pipeleers (Pipeleers, 1992), and thus behave as trigger cells. We recently developed a conditional GK mutant allele renders the enzyme half-saturated with glucose at 1 mM instead of 7 mM for the native enzyme (Tornovsky-Babeay et al. , 2021). We reasoned that this allele could be used to directly test if beta cells could be forced to become ‘trigger’ cells by simply increasing their glycolytic flux. To this end we took advantage of our serrendipidous discovery that the αGSU-Cre transgene, active in gonadotrophs of the pituitary, is active in a small subset of pancreatic β-cells. We derived mice with a glucokinase activating mutation present in ~15% of β-cells and tested their metabolic phenotype, calcium signaling and insulin secretory response. As a control, we employed mice in which GK was ablated in a minority of β-cells using the same αGSU-Cre. The phenotypes described below demonstrate the control strength that glucokinase exerts within β-cells to regulate glucose homeostasis and suggest that β-cells with high glycolytic flux can serve as ‘hub’ or ‘trigger’ cells to determine the secretory behavior of the β-cell collective.
2. Materials and Methods
2.1. Mice
All animal procedures were approved by the IACUC of the University of Pennsylvania. Mice with a conditional null allele for glucokinase (Postic et al. , 1999), conditional glucokinase ins454A mice (Tornovsky-Babeay et al., 2021), and Sun1-GFP (Mo et al. , 2015) were crossed with αGSU-Cre (Singh et al. , 2009) mice to generate the experimental cohorts. Animals were housed in cages containing maximally 5 mice, fed a standard chow diet, and maintained on a 12-hour light/dark cycle. Animal studies were performed on experimental and control mice of 8 weeks to 24 weeks of age. No sex-based differences were observed in any experimental measurements except body weight.
2.2. Histology and Immunofluorescence.
Pituitary glands or pancreata were isolated from mice and fixed with 4% paraformaldehyde. Fixed tissues were embedded in paraffin and sectioned by the Radioimmunoassay and Biomarkers Core of the Penn DRC. Slides were treated with a dewax and rehydration scheme: Xylenes, 100%, 95%, 80%, 70% ethanol, and ddH2O. Antigen retrieval was performed on slides using citric acid buffer under high temperature and pressure for 90 minutes. PBST was used for permeabilization and washes. Cas block buffer was used for blocking and diluting antibodies. Antibodies and corresponding dilutions used for immunostaining are listed in Supplemental Table 1. DAPI at a 10,000:1 dilution in H2O was used to stain nuclei before image acquisition. Islets were imaged at 40X magnification. Cells were counted manually using the ImageJ counting tool. The percentage of GFP+ cells for each cell type was calculated as GFP and insulin/glucagon/somatostatin double positive cells over the total number of insulin/glucagon/somatostatin positive cells for β-, α-, and δ- cells, respectively.
2.3. In vivo Metabolic Assays.
Longitudinal physiological measurements on mice for body weight and non-fasting blood glucose were taken every two weeks on the same day and at the same time. Blood was drawn through tail tip amputation and measured using an AlphaTrack glucometer. Glucose tolerance tests were conducted on 18 week old mice fasted for 16 hours in fasting cages containing ALPHA-dri bedding and access to water. Mice were injected intraperitoneally with 2 mg/kg body weight of 10% dextrose. Blood was drawn by tail tip amputation. Blood glucose was measured by an AlphaTrack glucometer pre-injection and 15, 30, 60, 90, and 120 minutes post-injection.
2.4. Islet Studies.
Islets were isolated using collagenase (EC 3.4.24.3 Serva, 17449) digestion in Hanks buffer followed by separation of islets from exocrine tissue in a Ficoll (Sigma, F-9378) gradient as previously described (Doliba et al. , 2012). Islets were cultured for 3–4 days in RPMI 1640 medium (Sigma) containing 10% fetal bovine serum, 10 ml/l penicillin-streptomycin-amphotericin B solution (GIBCO BRL) and 10 mM glucose. For perifusion studies, islets (200–500 islets) were placed on a nylon filter in a plastic perifusion chamber (Millipore, Bedford, MA) and perfused with a flow rate of 1 ml per min. Islets from one mouse or pooled islets from three mice matched for sex, age, and genotype were used for each experiment. The perifusion apparatus consisted of a computer-controlled low-pressure chromatography system (BIO-RAD Econo system) that allowed programmable rates of flow and glucose concentration in the perfusate, a water bath (37°C), and fraction collector (BIO-RAD, model 2128). The perifusate was a Krebs buffer (pH 7.4) containing: 114 mmol/l NaCl, 5 mmol/l KCl, 24 mmol/l NaHCO3, 1 mmol/l MgCl2 6H2O, 2.2 mmol/l Ca2+, 1 mM Pi, 10 mmol/l HEPES (pH 7.4), 0.25% of BSA equilibrated with 95% O2 and 5% CO2. Treatments consisted of: a physiological 4 mM Amino Acid Mixture (AAM), 1, 3, 8, 16.7 mM glucose, 0.1 mM IBMX, and 30 mM KCl. Insulin levels were determined via radioimmune assay by Radioimmunoassay and Biomarkers Core of the Penn DRC.
2.5. Intracellular Ca2+ Measurements.
Mouse islets were loaded with fura-2AM during a 40 min pretreatment at 37°C in 2 ml KRBB supplemented with 5 mmol/l fura-2 acetoxymethylester (Molecular Probes, Eugene, OR). The loaded islets were transferred to the perifusion chamber and placed on the homeothermic platform of an inverted Zeiss microscope. Islets were perfused with KRBB at 37°C at a flow rate 1 ml/min, while 1, 3, 9, 27 mM glucose, and 30 mM KCl were applied. Intracellular Ca2+ was measured by dual wavelength fluorescence microscopy using a Zeiss AxioVision system as described previously (Doliba et al. , 2010, Li et al. , 2013).
2.6. Single cell calcium measurements by confocal Imaging.
Islets from GKKI and control mice were left to recover overnight in Islet Medium (RPMI 1640, 11mM Glucose, 10% FBS, with Penicillin-Streptomycin 100 IU-μg/ml). On the day of the experiment, 8 islets per group were transferred to a Petri dish with Krebs-Ringer Bicarbonate buffered medium + 10 mM HEPES (KRBH) at pH 7.40, supplemented with + 0.1 % Bovine Serum Albumin and 1 mM glucose. The cell permeant calcium indicator Cal-590 AM (AAT Bioquest, Inc.) was added to the dish at a concentration of 4 μM, and the dish was set on a shaker at room temperature for 30 minutes. Next, the islets were transferred to a 4-chamber glass bottom dish (D35C4–20-1.5-N, Cellvis) containing KRBH with 0.1 % BSA and 1 mM glucose, and the dish was placed in a stage-top incubator set at 37°C, 5% CO2, 100% humidity for the duration of the experiment. Imaging was performed using a Zeiss LSM880 microscope with a Plan-Apochromat 20x/0.8 M27objective, with the following settings: 561 nm laser excitation, 570–695 nm bandpass filter, pixel size 1.661 μm, pixel dwell time 33.0 μs. After each glucose concentration was added, we waited 6 minutes before starting the 5-minute time-course with one image every 5 seconds. For the study of calcium waves across islets, we employed a pixel size of 1.371 μm and a pixel dwell time of 3.45 μs to acquire images every 42 ms.
3. Results
3.1. The αGSU-Cre transgene is active in small subpopulations of alpha, beta, and delta cells of the islet.
To characterize the activity of the αGSU-Cre transgene in the pituitary gland and pancreatic islets, we crossed αGSU-Cre mice to the Sun1-GFP reporter mouse, where Cre expression excises the stop codon of Sun1-GFP, resulting in expression of the Sun1-GFP green fluorescent fusion protein localized to the nuclear envelope (Mo et al., 2015). Anterior pituitary glands of Sun1-GFPloxP/LoxP; αGSU-Cre+ mice were analyzed by immunofluorescence using antibodies against luteinizing hormone, which marks gonadotrophs, and GFP. A fraction of anterior pituitary cells displayed GFP expression in the nuclear envelope as expected, and a subset of those cells co-expressed luteinizing hormone (Figure 1, A).
Figure 1: Expression of Cre recombinase in luteinizing hormone expressing cells in the pituitary and, unexpectedly, in the β-, α-, and δ-cells of pancreatic islets.
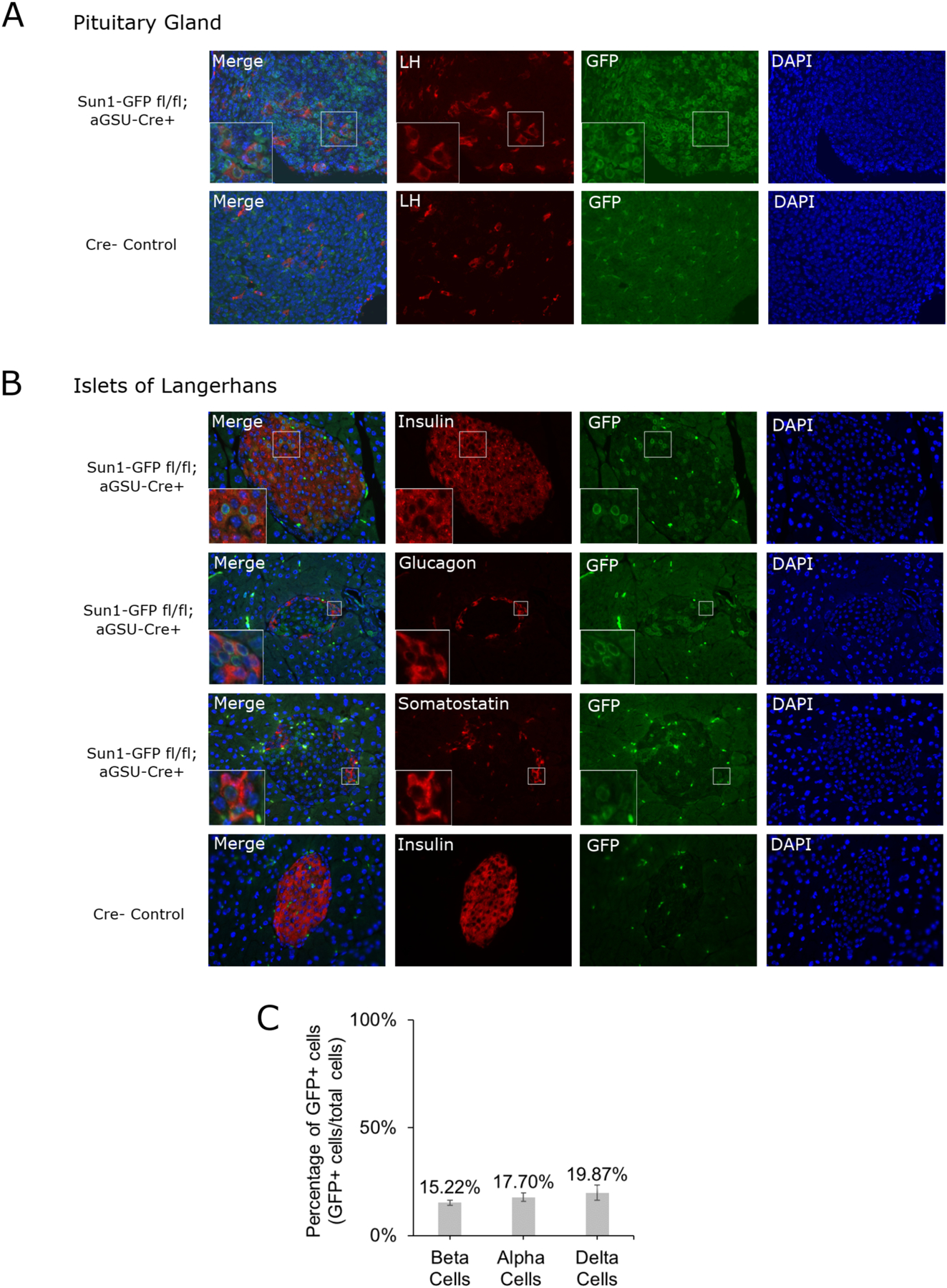
(A) Representative immunofluorescence images of pituitary sections and (B) islets of Langerhans from αGSU-Cre+;Sun1-GFP reporter mice indicating active Cre recombination in co-immunolabeled cell types. Perinuclear GFP localization of Sun1-GFP is detectable in αGSU-Cre+ mice, but not Cre- mice. (C) Islets stained for insulin (n=53), glucagon (n=26), and somatostatin (n=27) were quantified for GFP and hormone colocalization as a percentage of total GFP+ cells of each cell type.
Because of an unexpected hypoglycemic phenotype observed in the GKins454A;αGSU-Cre+ mice, (see below) pancreata from the Sun1-GFPloxP/LoxP;αGSU-Cre+ reporter mouse were isolated and analyzed by immunofluorescence with antibodies against insulin, glucagon, somatostatin, and GFP, to test if Cre expression was also present in the major endocrine cell types of the islets of Langerhans. The imaging results and quantification revealed Sun1-GFP expression in 15.22 ± 1.22% of insulin expressing β-cells, 17.70 ± 1.93% of glucagon expressing α-cells, and 19.87 ± 3.46% of somatostatin expressing δ-cells (Figure 1, B and C). Therefore, αGSU-Cre expression in these pancreatic endocrine cells in our GKKO and GKKI mouse models leads to GK depletion and activation, respectively, and could thus alter glucose-sensing function. In this study we further investigated this unexpected phenotype.
3.2. Deletion of GK in 15% of β-cells does not affect glucose metabolism.
First, we analyzed mice deficient for GK in a subset of islet endocrine cells in the GKKO model. To this end, the αGSU-Cre mouse was crossed with the GK floxed conditional null mouse, originally characterized by Postic and colleagues (Postic et al., 1999), to derive GKKO mice. In this model, Cre recombination excises exons 9 and 10 of GK, generating a null allele. In all experiments, controls for GKKO mice were Cre-negative mice homozygous or heterozygous for the GK loxP allele. We tracked body weight and non-fasting blood glucose levels in cohorts of GKKO mice and control mice over a period of 8 weeks and found no significant differences between these groups for both sexes (Fig. 2A; Suppl. Fig. S1). In addition, random and fasting glucose levels of GKKO mice were not different from control mice (Fig. 2B, C [t=0], Suppl. Fig. S2). Glucose tolerance tests (GTT) after 16 hours of fasting likewise were unremarkable (Fig 2, C).
Figure 2: Metabolic Characterization of GKKO mice and GKKO islets.
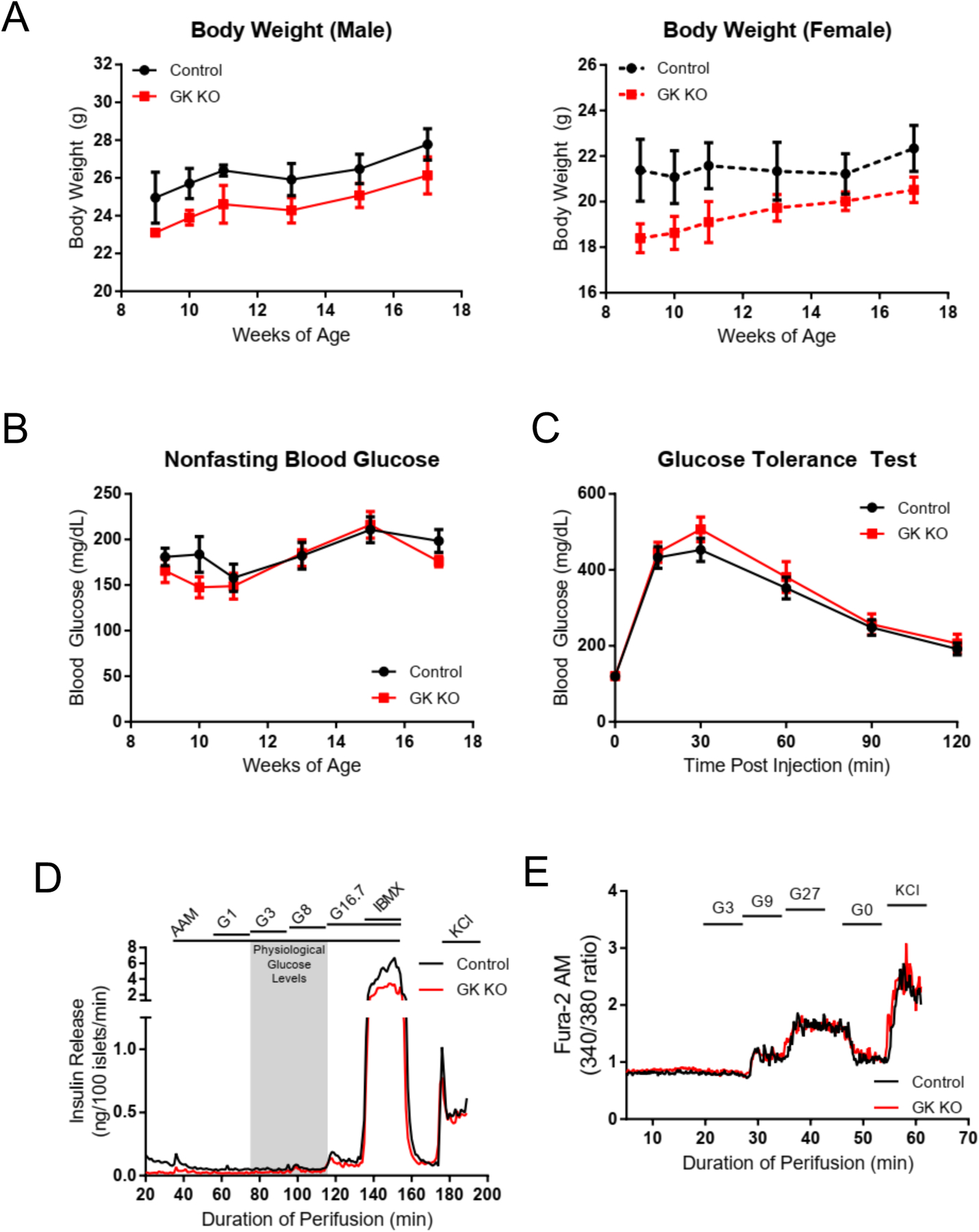
(A) Body weights of male GKKO (n=5) and control (n=6), and female GKKO (n=6) and control (n=5) mice were recorded over 8 weeks, with no statistically significant differences between control and GKKO mice. (B) Nonfasting blood glucose in the same cohort of mice over the same 8-week period (GKKO, n=11. Control, n=12). (C) Glucose tolerance as assessed by IPGTT after a 16-hour fast in 18 week old GKKO mice (n=11) compared to 18 week old control mice (n=11). Note that fasting glucose levels. (t=0) are not different between the two groups. (D) Pooled islets from three age, sex, and genotype matched mice were isolated (n=3 experiments) and perifused with the regime indicated with no difference in GSIS. A physiological 4 mM amino acid mixture was used to establish baseline secretion before the addition of the indicated glucose regimen or IBMX to increase intracellular cAMP levels. (E) Islets isolated from GKKO (n=2) and control (n=2) mice were perifused with a physiological 4 mM amino acid mixture before addition of the indicated glucose regime and Fura2-AM fluorescence was measured to determine intracellular calcium levels. Data shown in A,B and C are presented as mean +/− SEM.
To assess insulin secretion while removing any possible non-islet effects related to possible ectopic expression of αGSU in other tissues which were not evaluated, islets were isolated and perifused with increasing glucose concentrations. Islets from GKKO and control mice did not differ in their insulin secretion profile (Fig 2, D). In agreement with perifusion data, calcium imaging of the GKKO islets showed a normal increase in intracellular calcium at glucose levels of 9 mM and 27 mM, and no signal at 3 mM glucose (Fig 2, E). Thus, GKKO mice were similar to controls, despite 15% of their β-cells lacking GK. These findings are consistent with a previous study by Piston and Magnuson that demonstrated that deleting GK in 30% of β-cells does not significantly alter whole islet physiology (Piston et al. , 1999).
3.3. GK activation in 15% of β-cells causes hypoglycemia, impaired glucose tolerance, and a lower threshold for glucose stimulated insulin secretion.
Next, we crossed the αGSU-Cre mouse with the GK activating mutation mouse (Tornovsky-Babeay et al., 2021) to generate GKKI mice, in order to assess whether rendering a small subset of β-cells hyper-responsive to glucose would be sufficient to alter the islet secretory response and glucose homeostasis. In the GKKI mouse, Cre recombination removes the endogenous exon 10 and replaces it with an ins454A mutant exon 10 cassette, resulting in a GK activating mutation. The same mutation was originally observed in the human GCK gene in a patient with hyperinsulinemic hypoglycemia (Sayed et al. , 2009). Mutant GKins454A displays a significantly increased affinity for glucose, with a half-saturation point of 1.1 mM glucose compared to 7 mM for wild type GK. Controls for GKKI mice were Cre-negative with homozygous or heterozygous conditional activation alleles. The same metabolic tests conducted for the GKKO mice were performed on GKKI mice, and both homozygous GKKI and heterozygous GKKI mice were included. Body weights of male and female GKKI mice were not significantly different from controls except at the 22 week time-point in female mice (Fig 3, A). We discovered a striking difference in non-fasting blood glucose levels between control, GKKI heterozygous, and GKKI homozygous mice with average values of 124.2 ± 5.1 mg/dl for control, 94.7 ± 9.5 mg/dl for heterozygous GKKI, and 70.5 ± 8.2 mg/dl for homozygous GKKI, which was maintained over an 8-week timeframe (Fig 3, B). There were no observable sex differences and Cre- GKins454A homozygous and heterozygous control mice behaved similarly. These results were surprising given that only 15% of islet β-cells contain GKins454A, and suggest that enzyme activation in a minority of β-cells is sufficient to lower the glycemic set point of the whole animal.
Figure 3: Metabolic Characterization of GKKI mice and GKKI islets.
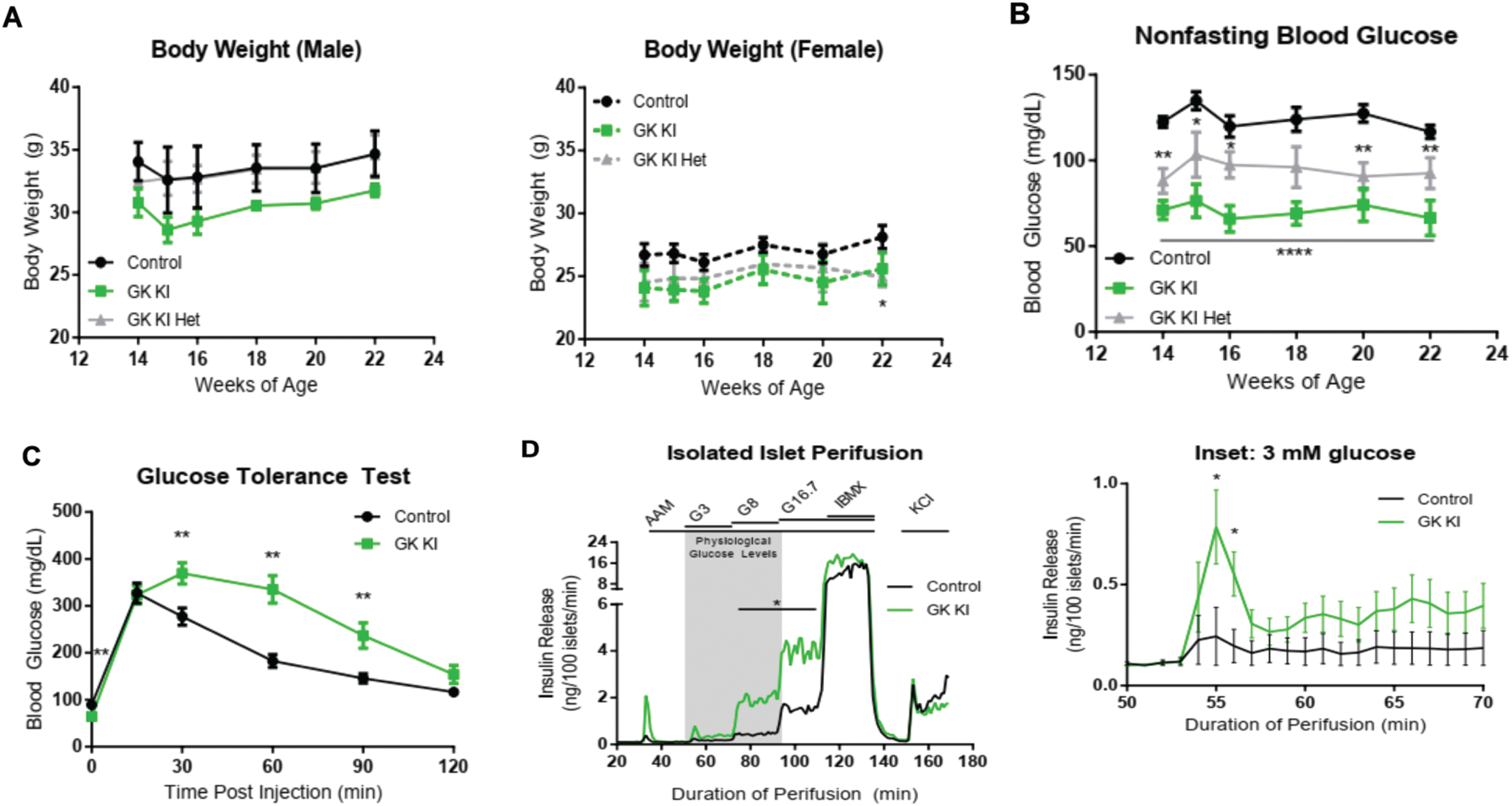
(A) Body weights of male GK KI (n=6), GK KI Het (n=4), and Control (n=7) mice and female GKKI (n=4), GK KI Het (n=4), and Control (n=7) mice were recorded over 8 weeks with no statistically significant difference between KI and control mice except for one week between female control and GK KI Het mice. (B) Nonfasting blood glucose levels of the same cohort of mice were recorded over the same 8-week period (GK KI, n=10; GK KI Het, n=8; Control, n=14). The GK ins454A allele had a gene-dosage dependent effect on lowering basal blood glucose levels. (C) IPGTT tests after a 16-hour fast in control (n=16) and GKKI (n=11) mice showed lower fasting blood glucose levels (t = 0) and decreased glucose tolerance over 2 hours post injection in GK KI mice. (D) Perifusion assay of Islets isolated from GKKI (n=8) and Control (n=8) mice. A physiological 4 mM amino acid mixture was used to establish baseline secretion before the addition of the indicated glucose regimen or IBMX to increase intracellular cAMP levels. The inset shows the response of the GK KI islets to 3 mM glucose, which is driven by the activating mutation in glucokinase in a subset of beta cells. Data shown in A,B and C are presented as mean +/− SEM. *. P<
During an intraperitoneal glucose tolerance test following a 16-hour fast, GKKI mice had significantly lower average fasting blood glucose levels (65.2 ± 6.5 mg/dl) compared to control mice (89.3 ± 4.8 mg/dl; p <0.01) (Fig. 3C). Counterintuitively, GKKI mice displayed impaired glucose tolerance after glucose injection during the GTT compared to control mice (Fig 3, C). A similar impairment in glucose tolerance had been described by Tornovsky-Babeay and colleagues in mice with heterozygous GKins454A expression in all β-cells (Tornovsky-Babeay et al., 2021), an effect that was attributed to glucotoxicity-induced β-cell death. We propose a different model for this paradoxical result in the Discussion section below.
To assess the glucose dependency of insulin release, while removing any possible islet independent effect of possible ectopic aGSU expression in other cell types, we isolated islets and performed perifusion assays, measuring insulin secretion at different glucose concentrations. GKKI islets had a decreased threshold for GSIS (Fig 3, D and inset), with insulin secretion induced already at 3 mM glucose. GKKI islets secreted significantly more insulin at 8 mM glucose than controls (GKKI: 1.94 ± 0.40 ng insulin/100 islets/min. Control: 0.47 ± 0.27 ng insulin/100 islets/min) and 16.7 mM glucose (GKKI: 4.04 ± 0.74 ng insulin/100 islets/min. Control: 1.59 ± 0.58 ng insulin/100 islets/min). Maximal capacity of GKKI islets was not altered as seen when islets were treated with IBMX to increase intracellular cAMP levels. GK activation in a subset of β-cells is thus able to sensitize the entire islet to glucose, and is the likely cause of hypoglycemia in GKKI mice.
Finally, we employed calcium imaging to further characterize the glucose sensitivity of GKKI islets. We perifused GKKI islets with increasing glucose concentrations and detected a rise in intracellular calcium at 3 mM glucose (GKKI: 1.59 ± 0.18. Control: 0.92 ± 0.09). At 9 mM glucose, both control and GKKI islets showed increased calcium activity, but the GKKI response was greater in magnitude (GKKI: 1.84 ± 0.18. Control: 1.25 ± 0.08). Of note, even 1 mM glucose levels increased intracellular calcium in GKKI islets (Fig 4, A and inset).
Figure 4: Analysis of intracellular calcium confirms left shift in glucose response of GKKI islets.
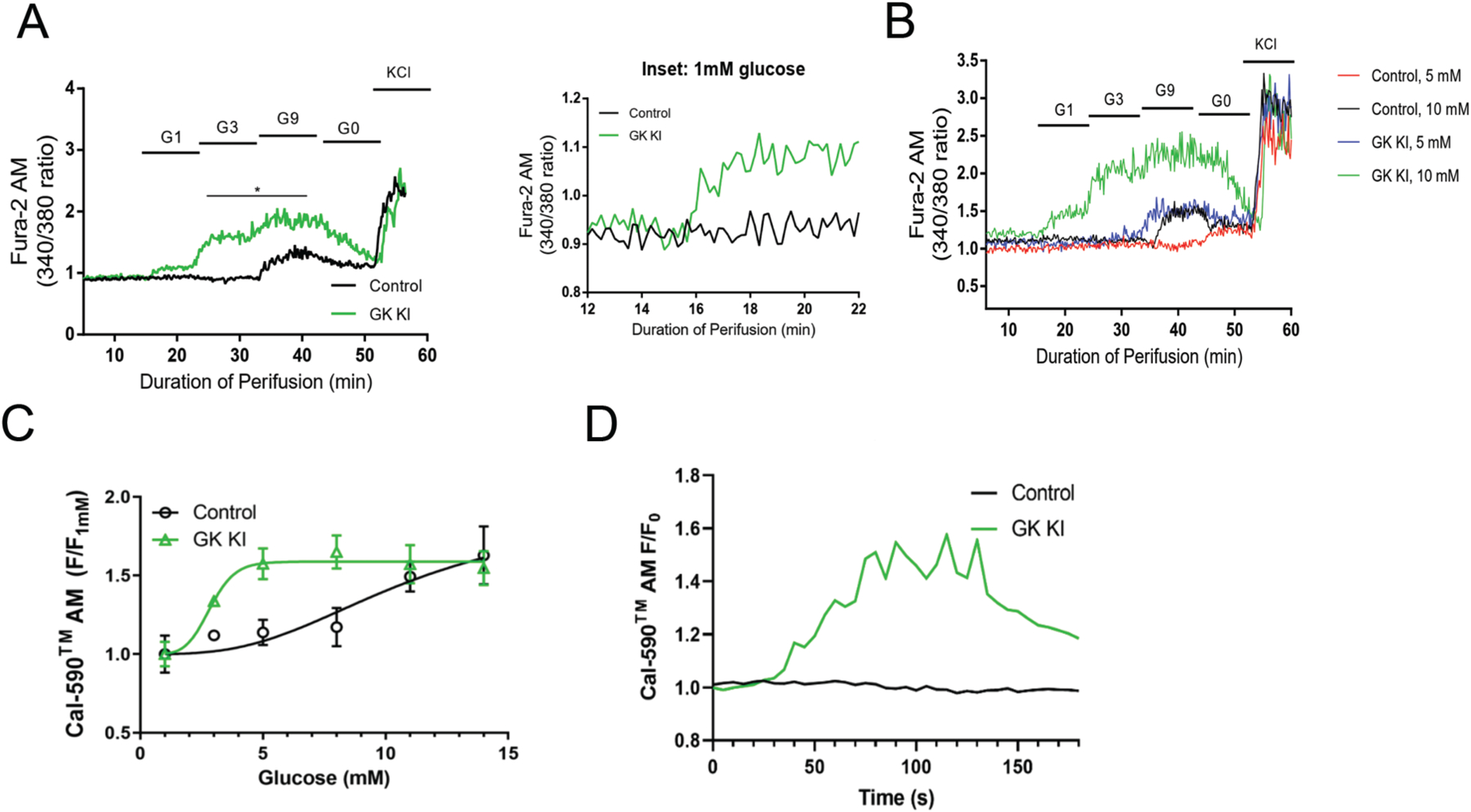
(A) Isolated islets from GKKI (n=4) and control (n=4) mice were perifused with the indicated glucose regime and Fura2-AM fluorescence was measured to determine calcium activity. (B) Intracellular calcium was measured in control (n=2) and GKKI (n=2) islets cultured at either 5 mM glucose or 10 mM glucose, demonstrating that glucose sensitivity is lost in control but retained in GKKI islets cultured at 5 mM glucose. (C) Intracellular calcium was measured in islets from GKKI and control mice at six glucose concentrations; the GKKI islets showed a dose-response profile shifted towards lower glucose concentrations. (D) Representative calcium traces for GKKI and Control islets at 1 mM glucose. Data shown are presented as mean +/− SEM. *, p < .05.
A final test of hyperactivation of GK in our model employed long-term glucose starvation. As established decades ago, islets cultured at 5 mM behave similarly to starved islets and do not respond acutely to glucose with insulin secretion (Burch et al. , 1981, Liang et al. , 1990) (Fig 4B). For this study, islets from GKKI or control mice were cultured overnight at 5 mM glucose or the standard 10 mM glucose prior to calcium imaging. Control islets cultured at 5 mM glucose (red trace) lose all responsiveness to glucose as expected, compared to those pre-cultured at 10 mM glucose, which respond normally to 9 mM glucose (Figure 4B). In contrast, elevated GK activity in GKKI islets in the 5 mM glucose condition rescues this loss of glucose sensitivity to a significant degree, mimicking control islets cultured at 10 mM glucose (Figure 4B). When comparing calcium activity in islets from control and GKKI mice using Cal-590 instead of Fura-2, we again found a significant shift of the calcium response to increasing glucose concentrations (Fig 4, C). GKKI islets were active at lower glucose concentrations, and even 1 mM glucose was sufficient to elicit calcium activity in GKKI islets, but not in control islets (see representative calcium traces in Fig 4,D). This difference is also shown in the time-lapse videos taken on control and mutant islets incubated with 1 mM glucose. While control islets show no calcium activity, those with mosaic activation of GK in 15% of islet β-cells exhibit strong calcium waves at this very low glucose concentration (supplemental videos 1 and 2).
4. Discussion
We employed two genetic mouse models to investigate if manipulation of glucokinase activity in a small minority of pancreatic β-cells is sufficient to alter islet function and whole body glucose homeostasis, taking advantage of our serendipitous finding that the αGSU-Cre transgene is active in a small subset of islet cells. Expression of the αGSU-Cre transgene is expected to permanently label cells carrying the SUN1-GFP transgene even if αGSU-Cre expression is transient. The observation that only 15% of the adult β-cells are labeled suggests the αGSU expression identifies a descrete subset of beta cells and not stochastic expression in all cells at different times during growth development. The function and significance of this β-cell subpopulation is not known.
Using the αGSU-Cre transgene to ablate GK had no effect on glucose homeostasis or islet secretory response, demonstrating that 85% normally active β-cells are sufficient to maintain normal insulin secretion and that the sub-population of αGSU expressing β-cells does not play a critical, glucose-dependent role in net islet function. In sharp contrast, when GK was genetically activated in only 15% of β-cells, islets were sensitized to glucose levels as low as 1 mM as indicated by intracellular calcium imaging. Functionally, these islets secrete insulin at 3 mM glucose and significantly lower the glycemic setpoint of GKKI animals. Although the body weight of GKKI mice is not significantly different from that of control mice, these mice exhibit drastically lower non-fasting and fasting blood glucose levels. Strikingly, the heterozygous GKKI mice had an intermediate glycemic setpoint between control and homozygous GKKI animals, demonstrating a gene-dosage effect of GK, a result that parallels those from a study done with whole body glucokinase activating mutations in mice (Pino et al. , 2007).
Our ex vivo studies showed that GKKI islets have a significantly decreased threshold for GSIS at 3 mM glucose and have detectable calcium activity at 1 mM glucose, which extended over a large fraction of the islet. This observation is striking at the 1 mM glucose level, as the overactive β-cells appear to communicate with other islet cells to elicit a synchronous calcium signal within the whole islet (See supplemental videos). This finding is in keeping with the ‘hub’ or ‘trigger’ cell model for the islet reponse to glucose, where Johnston and coleagues had mapped the functional properties of mouse islets using a dual optogenetic and photopharmacological strategy, showing that ablation of ‘hub’ cells prevented the coordinate reponse of islet β-cells to glucose (Johnston et al., 2016). The fact that ablation of GK in the αGSU-Cre positive β-cells did not change islet function (Figure 2) suggests that these cells are not normally ‘hub’ or ‘trigger’ cells, but can be made to act as such by changing the Km for glucose in these cells from 7 to 1.1. mM.
Despite the increased sensitivity of GKKI islets to glucose, whole animal glucose tolerance tests paradoxically showed a decreased ability to lower blood glucose in GKKI animals. This finding recapitulates what occurs in mice in which the GKins454A mutation is present in all β-cells (Tornovsky-Babeay et al., 2021). Tornovsky and colleagues showed that this glucose intolerance is the result of loss of β-cell mass over time. For our current model, we propose an alternate explanation. Thus, we posit that the hypoglycemia (with blood glucose concentrations of 3.6 mM) that ensues in GKKI mice following the overnight fast that is necessary to perform the glucose tolerance test allows for activation of insulin secretion in response to the glucose bolus only in the small number of hypersensitive GKins454A β-cells, but not the majority wild type β-cells. This is plausible because the acute response of the β-cell to glucose is highly dependent on chronic glucose levels, as shown in rats that have been fed and fasted (Burch et al., 1981, Liang et al., 1990) and again demonstrated in Figure 4B above. The fact that islets from GKKI mice exposed to prolonged hypoglycemia showed only partial rescue of the calcium signal suggests that either the function of these induced trigger cells is partially impaired by the exposure to low glucose, or that the responsiveness of the remaining 85% of β-cells with normal GK function is impaired by the pretreatment. Future studies will aim to address this question.
Finally, our findings here have implications in medicine, namely, the treatment of diabetes using glucokinase activators (GKAs). GKAs emerged as promising candidates for the treatment of diabetes in the early 2000s, as these drugs have the potential to restore insulin secretion in β-cells. However, dosing regimens used in subsequent clinical trials for GKAs caused severe side effects (hypoglycemia) in patients, and many studies were cancelled (Meininger et al. , 2011). Recently, GKAs have reemerged as a promising drug based on a successfully completed phase 2 clinical trial (Zhu et al. , 2018). Likewise, improved glycemia, β-cell function, and glucose tolerance have been demonstrated with the mild GKA HMS5552 in diabetic rats (Wang et al. , 2017). In our mouse model, a small subset of β-cells expressing highly activated GK is sufficient to alter the whole islet response to glucose, demonstrating the power of GK in the context of β-cells.
In sum, we have shown using a genetic model that rendering 15% of β-cells overactive in terms of glycolytic flux is sufficient to significantly lower blood glucose levels in mice. Our data support the model that a minority of hub or trigger β-cells, defined by higher than average glycolytic flux, are sufficient to dicate the insulin secretory response of the whole islet.
Supplementary Material
Acknowledgements
We thank Dr. Suzanne Shapira for careful editing of the manuscript We thank the following core facilities at the University of Pennsylvania Diabetes Research Center (supported by P30-DK19525) for their assistance: Pancreatic Islet Cell Biology Core and Radioimmunoassay and Biomarkers Core. This work was supported by UC4 DK116271 to KHK and an unresricted gift from HUA Medicine Shanghai Ltd. to FMM.
Footnotes
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
KHC designed experiments, collected data, analyzed data, drafted the article, and revised the article. ND, CLM, JR, AU, TT designed experiments and collected data. SR, DP and BG designed experiments and revised the article. KHK and FMM conceived the study, designed experiments, drafted and revised the article, and approved the final version for publication.
References
- Bahl V, Lee May C, Perez A, Glaser B, Kaestner KH. Genetic activation of alpha-cell glucokinase in mice causes enhanced glucose-suppression of glucagon secretion during normal and diabetic states. Mol Metab. 2021;49:101193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basco D, Zhang Q, Salehi A, Tarasov A, Dolci W, Herrera P, et al. alpha-cell glucokinase suppresses glucose-regulated glucagon secretion. Nat Commun. 2018;9:546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninger RK, Zhang M, Head WS, Satin LS, Piston DW. Gap junction coupling and calcium waves in the pancreatic islet. Biophys J. 2008;95:5048–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch PT, Trus MD, Berner DK, Leontire A, Zawalich KC, Matschinsky FM. Adaptation of glycolytic enzymes: glucose use and insulin release in rat pancreatic islets during fasting and refeeding. Diabetes. 1981;30:923–8. [DOI] [PubMed] [Google Scholar]
- Doliba NM, Qin W, Najafi H, Liu C, Buettger CW, Sotiris J, et al. Glucokinase activation repairs defective bioenergetics of islets of Langerhans isolated from type 2 diabetics. Am J Physiol Endocrinol Metab. 2012;302:E87–E102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doliba NM, Qin W, Vinogradov SA, Wilson DF, Matschinsky FM. Palmitic acid acutely inhibits acetylcholine- but not GLP-1-stimulated insulin secretion in mouse pancreatic islets. Am J Physiol Endocrinol Metab. 2010;299:E475–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston NR, Mitchell RK, Haythorne E, Pessoa MP, Semplici F, Ferrer J, et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab. 2016;24:389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner KH, Campbell-Thompson M, Dor Y, Gill RG, Glaser B, Kim SK, et al. What is a beta cell? - Chapter I in the Human Islet Research Network (HIRN) review series. Mol Metab. 2021;53:101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Liu C, Nissim I, Chen J, Chen P, Doliba N, et al. Regulation of glucagon secretion in normal and diabetic human islets by gamma-hydroxybutyrate and glycine. J Biol Chem. 2013;288:3938–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Najafi H, Matschinsky FM. Glucose regulates glucokinase activity in cultured islets from rat pancreas. J Biol Chem. 1990;265:16863–6. [PubMed] [Google Scholar]
- MacDonald PE, Rorsman P. Oscillations, intercellular coupling, and insulin secretion in pancreatic beta cells. PLoS Biol. 2006;4:e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matschinsky FM, Wilson DF. The Central Role of Glucokinase in Glucose Homeostasis: A Perspective 50 Years After Demonstrating the Presence of the Enzyme in Islets of Langerhans. Front Physiol. 2019;10:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meininger GE, Scott R, Alba M, Shentu Y, Luo E, Amin H, et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34:2560–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron. 2015;86:1369–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moede T, Leibiger B, Vaca Sanchez P, Dare E, Kohler M, Muhandiramlage TP, et al. Glucokinase intrinsically regulates glucose sensing and glucagon secretion in pancreatic alpha cells. Sci Rep. 2020;10:20145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pino MF, Kim KA, Shelton KD, Lindner J, Odili S, Li C, et al. Glucokinase thermolability and hepatic regulatory protein binding are essential factors for predicting the blood glucose phenotype of missense mutations. J Biol Chem. 2007;282:13906–16. [DOI] [PubMed] [Google Scholar]
- Pipeleers DG. Heterogeneity in pancreatic beta-cell population. Diabetes. 1992;41:777–81. [DOI] [PubMed] [Google Scholar]
- Piston DW, Knobel SM, Postic C, Shelton KD, Magnuson MA. Adenovirus-mediated knockout of a conditional glucokinase gene in isolated pancreatic islets reveals an essential role for proximal metabolic coupling events in glucose-stimulated insulin secretion. J Biol Chem. 1999;274:1000–4. [DOI] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–15. [DOI] [PubMed] [Google Scholar]
- Sayed S, Langdon DR, Odili S, Chen P, Buettger C, Schiffman AB, et al. Extremes of clinical and enzymatic phenotypes in children with hyperinsulinism caused by glucokinase activating mutations. Diabetes. 2009;58:1419–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SP, Wolfe A, Ng Y, DiVall SA, Buggs C, Levine JE, et al. Impaired estrogen feedback and infertility in female mice with pituitary-specific deletion of estrogen receptor alpha (ESR1). Biol Reprod. 2009;81:488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornovsky-Babeay S, Weinberg-Corem N, Ben-Haroush Schyr R, Avrahami D, Lavi J, Feleke E, et al. Biphasic dynamics of beta cell mass in a mouse model of congenital hyperinsulinism: implications for type 2 diabetes. Diabetologia. 2021;64:1133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Liu H, Chen L, Duan Y, Chen Q, Xi S. Effects of a Novel Glucokinase Activator, HMS5552, on Glucose Metabolism in a Rat Model of Type 2 Diabetes Mellitus. J Diabetes Res. 2017;2017:5812607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DF, Cember ATJ, Matschinsky FM. The thermodynamic basis of glucose-stimulated insulin release: a model of the core mechanism. Physiol Rep. 2017;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Gan S, Liu Y, Ma J, Dong X, Song W, et al. Dorzagliatin monotherapy in Chinese patients with type 2 diabetes: a dose-ranging, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Diabetes Endocrinol. 2018;6:627–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.