

Abstract
Crouzon syndrome with acanthosis nigricans is an autosomal dominant disease, with typical features of classic Crouzon craniosynostosis, verrucous hyperplasia, and hyperpigmentation of the skin. While several mutations in FGFR2 cause classic Crouzon syndrome, Crouzon syndrome with acanthosis nigricans results from a point mutation in the fibroblast growth factor receptor 3 gene (FGFR3). We report the case of an 8-year-old Vietnamese girl diagnosed with Crouzon syndrome with acanthosis nigricans, showing typical clinical features, including a crouzonoid face and dark plaques on the skin. Genetic testing showed a missense variation in FGFR3, associated with Crouzon syndrome with acanthosis nigricans. Following diagnosis, we treated acanthosis nigricans with 10% urea cream. This case study and literature review discuss the cutaneous manifestations and dermatological treatments while demonstrating the importance of clinical examination and evaluation of the patient’s medical history during diagnosis. Our findings contribute to the global pool of data, providing practical insights into the manifestations of Crouzon syndrome.
Key words: Crouzon syndrome, acanthosis nigricans, FGFR3 mutation
Introduction
Crouzon syndrome (CS), first described in 1912 by the French neurosurgeon Octave Crouzon, is a rare genetic disorder caused by a mutation in the fibroblast growth factor receptor 2 (FGFR2) gene.1 Craniosynostosis is the premature fusion of the skull bones. It is the main cause of the prominent characteristics of CS, such as midfacial and maxillary hypoplasia, shallow orbit, mandibular prognathism, overcrowding of upper teeth, V-shaped maxillary dental arch, and upper airway obstruction.1,2 However, acanthosis nigricans (AN) is a cutaneous disorder characterized by symmetric, thick, dark plaques which commonly appear on the neck, axillae, inframammary fold, and groin area. The disease may be related to metabolic or genetic disorders, autoimmune conditions, malignant diseases, or the use of certain medications.3,4
In 1968, Curth described the association between CS and AN.5 Since then, several similar cases have been reported. Mutations in the FGFR3 gene, such as a heterozygous A391E missense mutation, are mainly responsible for this disorder.6 The variation in clinical manifestations and gene mutations strongly indicate that this disorder is distinct from classic Crouzon syndrome.7 The name Crouzonodermoskeletal syndrome was proposed for Crouzon syndrome with acanthosis nigricans (CSAN) to delineate its four phenotypical characteristics: the crouzonoid phenotype (Crouzono), acanthosis nigricans (dermo), jaw cementomas, and vertebral alterations (skeletal).8 Mutations in FGFR3 cause inadequate stimulation of fibroblast growth factor receptors in keratinocytes and fibroblasts, resulting in hyperkeratotic plaques on the flexural area of the skin.9 In CSAN, AN mostly occurs on the neck. However, it also occurs on the face, axillae, abdomen, upper and lower extremities, and lower back.10 Besides AN, other cutaneous features, such as melanotic nevi, hypopigmented scars, sacral pits, and verrucae vulgaris have also been described in patients with CSAN.10
Data on the cutaneous characteristics of CSAN are extremely limited. Mir et al. described 35 cases of CSAN, focusing on the manifestations of AN.10 Since then, several cases of CSAN have been reported. Herein, we describe the case of an 8-year-old Vietnamese girl diagnosed with CSAN based on clinical presentation and genetic testing. In addition, we reviewed published literature to provide further insights into the pathogenesis, cutaneous manifestations, and management of AN in CSAN.
Case Report
An 8-year-old Vietnamese girl was referred to our hospital for examination of dark, thick plaques covering most of her body. She was the third child of a family with clinically healthy parents and siblings. Since birth, she had surgical intervention for hydrocephalus and surgery to treat craniosynostosis at 22 months of age. At 3 months old, her parents noticed dark plaques on her neck and elbows. No treatment was pursued because of the absence of other symptoms. In the past year, these plaques spread across other areas such as the face, chest, back, abdomen, and thighs; and they had become thicker, darker, and itchy. Examination revealed craniofacial deformities, such as exophthalmos, hypertelorism, beaked nose, and mandibular prognathism. The patient found it difficult to breathe through her nose, so she breathed through her mouth. Furthermore, skin examination revealed marked verrucous hyperkeratosis on the groin, abdomen, back, axillae, chest, neck, and face, especially around the eyes and mouth. The patient also experienced pruritus (Figure 1).
Endocrine laboratory tests, including those for follicle stimulating hormone, luteinizing hormone, estradiol, testosterone, and prolactin levels, were conducted; all values were in the normal range except for cortisol, the level of which was lower than normal. Computed tomography scans revealed midface hypoplasia with exophthalmos and fibrous dysplasia of the maxillary sinus that matched the characteristics of a crouzonoid face (Figure 2). Plain radiographs of the pelvis and lumbar spine showed slight posterior scalloping of the vertebral body, which may have been caused by the previous occurrence of hydrocephalus. The nasendoscopy revealed a narrow nasal cavity and enlarged grade III tonsils, which would explain the patient’s breathlessness. No anomaly was found on the abdominal or cardiac ultrasonography.
Analysis of target genes FGFR2 (exon 2, 3, 7 and 8) and FGFR3 (exon 9) revealed a point mutation, 1172C>A in exon 9 of FGFR3. This transversion caused a missense mutation, resulting in the substitution of alanine with glutamic acid (Ala391Glu), which has been reported in other CSAN cases (Figure 3).
Discussion
Our patient had typical characteristics of CSAN, including a crouzonoid face, which exhibited craniosynostosis, proptosis, hypertelorism, beaked nose, mandibular prognathism, upper airway obstruction, and AN on the face and flexural areas, such as the neck, axillae, inframammary fold, and groin (Table 1).7,10-16 These findings were in agreement with those of Mir et al., who examined 35 cases of CSAN, including genetic confirmation, and concluded that the neck was the most common area of AN, followed by the axillae, face, chest, abdomen, groin, upper extremities, lower extremities, and lower back.10 Melanotic nevi and hypopigmented scars were also common in their study, but we did not find these in our case. In general, the characteristics of AN in CSAN are not different from those of AN in other conditions; however, AN in CSAN is usually severe and widespread.10 CSAN manifestation has also been shown to differ by age. Sharda et al. reported a 10-hour-old male neonate with CSAN, diagnosed through clinical examination including genetic testing.11 Interestingly, skin examination showed slight coarseness and hyperpigmentation on the ear lobule, axillae, areolae, scrotum, and perioral and perineal region. Skin biopsy showed hyperkeratotic and papillomatotic epidermis with mild acanthosis. This study by Sharda et al. showed that AN in CSAN may be atypical in several ways;11 thus, AN should not be the primary symptom used to diagnose CSAN as it is not specific. It is important to consider other characteristics such as the crouzonoid face before genetic testing for FGFR3 mutation.
Figure 1.
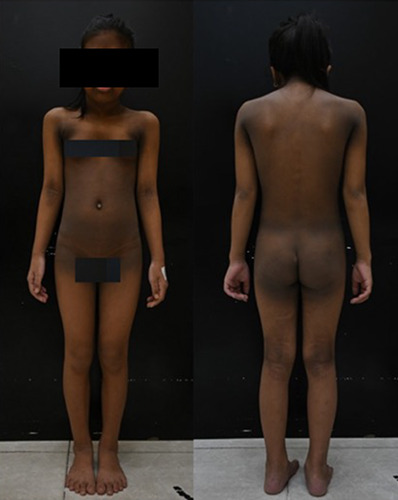
Clinical manifestations of the patient. Acanthosis nigricans can be observed on her face (around her eyes and mouth), neck, axillary, elbow, groin, around the umbilicus, and spreading to her abdomen and back.
Figure 2.
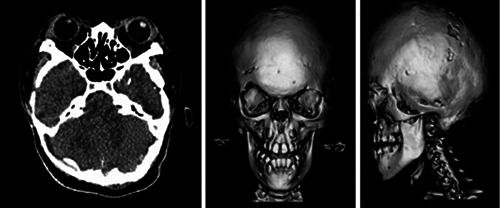
Computed tomography scans showing exophthalmos, mild midface hypoplasia, and mandibular prognathism.
Figure 3.
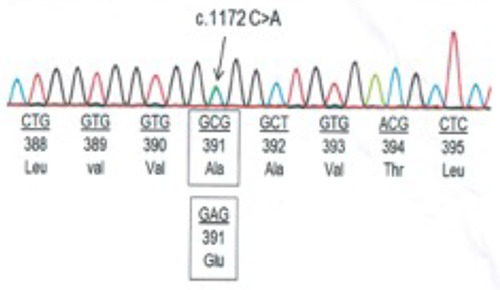
Figure 3. Genetic test showing C to A mutation at nucleotide 1172 on exon 9 in the FGFR3 gene.
In our case study, AN onset in the flexural areas of the patient occurred at 3 months of age, indicating that AN has an early onset. Arnaud- Lopez et al. described 35 cases of CSAN and found that AN occurred in 80% of the patients within the first decade of life,12 while Mir et al. reported 4.5 years as the mean age of onset.10 The earliest age of onset was described by Sharda et al., where AN was observed in a 10-hour-old male neonate.11
Previous studies have shown that, in addition to AN, up to 25% of patients have other skin anomalies, such as melanotic nevi and hypopigmented scars.12 However, pruritus and mild xerosis were the only symptoms observed in our case.
In our case study, genetic testing revealed an A391E mutation in FGFR3, which confirmed CSAN. CSAN and classic CS, in which mutations are mainly located on the FGFR2 gene, share similar characteristics such as cranial synostosis. Interestingly, FGFR2 and FGFR3 are located on two distinct chromosomes; thus, it is possible that other factors such as proteoglycans, which induce dimerization and activation of FGFRs, regulate the interaction between FGFR mutations from either gene.7 Nagase et al. reported both FGFR2 and FGFR3 mutations in a family in which the elder sister had an FGFR2 mutation and the younger sister had an FGFR3 mutation.17 Rymer et al. described a case of Pfeiffer syndrome type 3 that had an FGFR3 mutation associated with CSAN.18 Although further studies are needed to clarify the relationship between FGFR gene locations, the present findings reveal that phenotypical differences may be related to specific deleterious variants.7
Treating the underlying disease is the best approach for resolving AN; however, in cases where AN is related to genetic disorders such as CSAN, relieving the symptoms seems to be the only treatment option. Although there are no guidelines on the first-line therapy for AN, many topical and systemic agents have been used. In our case, topical tacrolimus helped reduce itching, and 10% urea cream decreased the thickness and darkness of the AN plaques. Among the topical agents, retinoids are the preferred choice of treatment because their efficacy is well-proven. Lahiri et al. reported the clinical and histological efficacy of 0.05% tretinoin cream in 30 patients with idiopathic AN in whom the condition was recalcitrant to conventional treatments.19 Treesirichod et al. conducted a randomized trial of 10% urea and 0.025% tretinoin creams for the treatment of AN.20 Although both medications improved hyperpigmentation, the efficacy of 0.025% tretinoin cream was significantly better than that of the 10% urea creams. After considering its side effects, we applied the 10% urea cream to a significant area of the skin. After 2 months of treatment, the results were unsatisfactory; however, no side effects were observed. In addition, we used tacrolimus to control the pruritus. Inflammation is not a prominent factor in the pathogenesis of AN, and evidence supporting the use of tacrolimus in AN is also limited; however, this agent may be considered safe to relieve itching. Moreover, melanocytes express FGFR3. The activating A391E mutation increases signals downstream of FGFR3, where serine/threonine-protein kinase B-raf (BRAF) and MAPK/ERK kinase (MEK) are located. Thus, the topical application of these kinase inhibitors should be considered as a therapy for CSAN.6 Recently, the efficacy of FGFR3 inhibitors in the management of carcinomas has been described and provides a promising therapy for CSAN.
Evidence related to the prognosis of CSAN is extremely limited. Recent studies revealed that the mutation Ala391Glu in FGFR3 is associated with severe disease presentation and requires surgical treatment.21 However, reported cases of CSAN indicate that this disease may have a good prognosis if it is diagnosed and treated early.
Table 1.
Cutaneous features and treatments of mutation-confirmed cases of Crouzon syndrome with acanthosis nigricans.
| Source | Sex | Age of onset | Mutation gene | Clinical features of AN | Other cutaneous findings | Treatment of AN |
|---|---|---|---|---|---|---|
| Meyers et alP | F | lOy | FGFR3 | AN on neck, axillae, perioral, periorbital areas, chest, and abdomen | Nevi on face and trunk, hypopigmented scars, seborrheic dermatitis, acne vulgaris | NA |
| F | NA | FGFR3 | AN on neck, axillae, perioral, periorbital areas, chest, and abdomen | Nevi on face, trunk, and extremities | NA | |
| F | NA | FGFR3 | AN on neck, axillae, perioral, periorbital areas, chest, and abdomen | Nevi on face, trunk, and extremities | NA | |
| M | NA | FGFR3 | AN on neck, axillae, perioral, periorbital areas, chest, and abdomen | Nevi on face, trunk, and extremities | NA | |
| Wilkes et al.14 | M | 9y | FGFR3 | AN on axillae, neck, groin, and perioral and periorbital areas | Nevi on face and upper trunk, warty acanthoma | NA |
| F | 6y | FGFR3 | AN on neck, axillae, elbow flexure, and periorbital area and around umbilicus | NA | NA | |
| M | NA | FGFR3 | AN on neck, axillae, and groin | Warty acanthoma on back | NA | |
| Nagase et al? | M | 12y | FGFR3 | AN on neck, axillae, elbow flexure, lower eyelid, and perioral area | NA | NA |
| Schweitzer et al}5 | M | 16m | FGFR3 | AN on chest, abdomen, neck, lower extremities, axillae, and groin | Hypopigmented scar on abdomen | NA |
| F | <ly | FGFR3 | AN on neck, chest, and inguinal and axillary area | NA | NA | |
| F | 7y | FGFR3 | AN on neck, chest, groin, axillae, perioral and periorbital areas, umbilicus, abdominal surgical scar | Nevi and cafe-au-lait spot on the leg | NA | |
| Arnaud-Lopez et al.12 | F | 21 m | FGFR3 | AN on neck, flexural areas, nipples, abdomen, and perioral and chest regions | NA | NA |
| F | 4y | FGFR3 | AN on periorbital, paranasal, and perioral regions, neck, chest, abdomen, and flexural areas | Hypopigmented scar on neck and abdomen | NA | |
| Sharia et al.11 | M | 10h | FGFR3 | Coarseness of skin and hyperpigmentation on ear lobule, perioral area, axillae, areolae, and scrotum | NA | NA |
| Norgaardeia/.If | M | At birth | FGFR3 | No skin changes | NA | NA |
| Mir et al.w | F | 4y | FGFR3 | AN on neck, axillae, groin, and abdomen | Nevi on neck and trunk | NA |
| Our case | F | 3m | FGFR3 | AN on face, neck, axillae, groin and around umbilicus, chest, back, and flexure areas | Xerosis | Urea 10% + tacrolimus 0.03% |
M, male; F, female; AN, acanthosis nigricans; NA, not applicable; y, years; m, months, h, hours.
Conclusions
CSAN is a rare disorder with complex clinical manifestations. Further studies are needed to elucidate the pathogenesis of this disorder. Moreover, treatment modalities should be further evaluated to verify their safety and efficacy so that more information is available to assist in the development of treatment protocols. Based on our case study and literature review, we recommend genetic testing to confirm the diagnosis of CSAN, in addition to examining symptoms and assessing the patient’s medical history.
Acknowledgments
this work was supported by the Ho Chi Minh City Hospital of Dermato-Venereology. The authors thank the Department of Cosmetic Dermatology for helping with data collection.
References
- 1.Padmanabhan V, Hegde AM, Rai K. Crouzon's syndrome: a review of literature and case report. Contemp Clin Dent 2011;2:211-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hlongwa P. Early orthodontic management of Crouzon syndrome: a case report. J Maxillofac Oral Surg 2009;8:74-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popa ML, Popa AC, Tanase C, Gheorghisan-Galateanu AA. Acanthosis nigricans: to be or not to be afraid. Oncol Lett 2019;17:4133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Das A, Datta D, Kassir M, et al. Acanthosis nigricans: a review. J Cosmet Dermatol 2020;19:1857-65. [DOI] [PubMed] [Google Scholar]
- 5.Curth HO. The necessity of distinguishing four types of acanthosis nigricans. Congressus Internationalis Dermatologiae Munich. 1968;1:557-8. [Google Scholar]
- 6.Kariya H, Nakano H, Ishii N, et al. Crouzon syndrome with acanthosis nigricans and prominent diffuse hyperpigmentation associated with gain-of-function A391E mutation in FGFR3 gene. J Dermatol 2020;47:e451-2. [DOI] [PubMed] [Google Scholar]
- 7.Nagase T, Nagase M, Hirose S, Ohmori K. Crouzon syndrome with acanthosis nigricans: case report and mutational analysis. Cleft Palate Craniofac J 2000;37:78-82. [DOI] [PubMed] [Google Scholar]
- 8.Cohen Jr MM. Let's call it Crouzonodermoskeletal syndrome so we won't be prisoners of our own conventional terminology. Am J Med Genet 1999;84:74. [PubMed] [Google Scholar]
- 9.Herman TE, Sargar K, Siegel MJ. Crouzono-dermo-skeletal syndrome, crouzon syndrome with acanthosis nigricans syndrome. J Perinatol 2014;34:164-5. [DOI] [PubMed] [Google Scholar]
- 10.Mir A, Wu T, Orlow SJ. Cutaneous features of crouzon syndrome with acanthosis nigricans. JAMA Dermatol 2013;149:737-41. [DOI] [PubMed] [Google Scholar]
- 11.Sharda S, Panigrahi I, Gupta K, et al. A newborn with acanthosis nigricans: can it be crouzon syndrome with acanthosis nigricans? Pediatr Dermatol 2010;27:43-7. [DOI] [PubMed] [Google Scholar]
- 12.Arnaud-López L, Fragoso R, Mantilla-Capacho J, Barros-Núñez P. Crouzon with acanthosis nigricans. Further delineation of the syndrome. Clin Genet 2007;72:405-10. [DOI] [PubMed] [Google Scholar]
- 13.Meyers GA, Orlow SJ, Munro IR, et al. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in crouzon syndrome with acanthosis nigricans. Nat Genet 1995;11:462-4. [DOI] [PubMed] [Google Scholar]
- 14.Wilkes D, Rutland P, Pulleyn LJ, et al. A recurrent mutation, ala391glu, in the transmembrane region of FGFR3 causes crouzon syndrome and acanthosis nigricans. J Med Genet 1996;33:744-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schweitzer DN, Graham Jr JM, Lachman RS, et al. Subtle radiographic findings of achondroplasia in patients with crouzon syndrome with acanthosis nigricans due to an Ala391Glu substitution in FGFR3. Am J Med Genet 2001;98:75-91. [PubMed] [Google Scholar]
- 16.Nørgaard P, Hagen CP, Hove H, et al. Crouzon syndrome associated with acanthosis nigricans: prenatal 2D and 3D ultrasound findings and postnatal 3D CT findings. Acta Radiol Short Rep 2012;1:arsr.2012.110017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagase T, Nagase M, Hirose S, Ohmori K. Japanese sisters with pfeiffer syndrome and achondroplasia: a mutation analysis. J Craniofac Surg 1998;9:477-80. [DOI] [PubMed] [Google Scholar]
- 18.Rymer K, Shiang R, Hsiung A, et al. Expanding the phenotype for the recurrent p. Ala391Glu variant in FGFR3: beyond crouzon syndrome and acanthosis nigricans. Mol Genet Genomic Med 2019;7:e656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lahiri K, Malakar S. Topical tretinoin in acanthosis nigricans. Indian J Dermatol Venereol Leprol 1996;62:159-61. [PubMed] [Google Scholar]
- 20.Treesirichod A, Chaithirayanon S, Chaikul T, Chansakulporn S. The randomized trials of 10% urea cream and 0.025% tretinoin cream in the treatment of acanthosis nigricans. J Dermatol Treat 2021;32:837-42. [DOI] [PubMed] [Google Scholar]
- 21.De Planque CA, Wall SA, Dalton L, et al. Clinical signs, interventions, and treatment course of three different treatment protocols in patients with crouzon syndrome with acanthosis nigricans. J Neurosurg Pediatr 2021;28:425-31. [DOI] [PubMed] [Google Scholar]