

Abstract
Excessive reactive oxygen species production by mitochondria (mtROS) is a key contributor to age-related vascular endothelial dysfunction. We recently showed in a crossover design, placebo-controlled clinical trial in older adults that 6 wk of treatment with the mitochondria-targeted antioxidant (MitoQ) improved endothelial function, as measured by nitric oxide (NO)-mediated endothelium-dependent dilation (EDD), by lowering mtROS and was associated with reduced circulating levels of oxidized low-density lipoprotein (oxLDL). Here, we conducted an ancillary analysis using plasma samples from our clinical trial to determine if MitoQ treatment-mediated changes in the “circulating milieu” (plasma) contribute to improvements in endothelial function and the mechanisms involved. With the use of an ex vivo model of endothelial function, acetylcholine-stimulated NO production was quantified in human aortic endothelial cells (HAECs) exposed to plasma collected after chronic MitoQ and placebo supplementation in 19 older adults (67 ± 1 yr; 11 females). We also assessed the influence of plasma on endothelial cell (EC) mtROS bioactivity and the role of lower circulating oxLDL in plasma-mediated changes. NO production was ∼25% higher (P = 0.0002) and mtROS bioactivity was ∼25% lower (P = 0.003) in HAECs exposed to plasma collected from subjects after MitoQ treatment versus placebo. Improvements in NO production ex vivo and NO-mediated EDD in vivo with MitoQ were correlated (r = 0.4683; P = 0.0431). Increasing oxLDL in plasma collected after MitoQ to placebo levels abolished MitoQ treatment effects on NO production and mtROS bioactivity, whereas inhibition of endogenous oxLDL binding to its lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1) prevented these effects. These findings provide novel insight into the mechanisms by which MitoQ treatment improves endothelial function in older adults.
NEW & NOTEWORTHY Chronic supplementation with a mitochondria-targeted antioxidant (MitoQ) improves vascular endothelial function in older adults, but the mechanisms of action are incompletely understood. Here, we show that MitoQ supplementation leads to changes in the circulating milieu (plasma), including reductions in oxidized low-density lipoprotein that enhance nitric oxide production and reduce mitochondrial oxidative stress in endothelial cells. These findings provide new information regarding the mechanisms by which MitoQ improves age-related endothelial dysfunction.
Keywords: aging, nitric oxide, plasma, postmenopausal women, reactive oxygen species
INTRODUCTION
Cardiovascular disease (CVD) is the leading cause of death in the United States and advancing age is the strongest independent risk factor for CVD (1, 2). The increase in CVD risk with aging is largely mediated by reductions in vascular function, including endothelial dysfunction (2). A key event underlying endothelial dysfunction with aging is a decrease in bioavailability of the vasodilatory molecule nitric oxide (NO) secondary to increased reactive oxygen species (ROS)-related oxidative stress (3). Excessive production of ROS by mitochondria (mtROS) is a contributor to vascular oxidative stress that promotes age-associated endothelial dysfunction (4–6).
MitoQ is a mitochondria-targeted antioxidant that accumulates in the mitochondria where it is optimally positioned to reduce mtROS (7). We recently showed in a randomized, placebo-controlled, crossover-design pilot clinical trial in older men and postmenopausal women (8) (PMW) that 6 wk of MitoQ supplementation improved NO-mediated endothelium-dependent dilation (EDD), as measured by brachial artery flow-mediated dilation (FMDBA) (9). Complementary assessments suggested that the improvement in endothelial function likely was mediated by reduced mtROS-associated suppression of EDD (9). However, a more in-depth interrogation of the mechanisms involved was not possible based on the in vivo measurements performed in that study.
Using an innovative ex vivo model, we have recently shown that some interventions that improve EDD in vivo are associated with changes to the circulating milieu that improve endothelial cell (EC) function (10, 11). In this model, ECs in culture are exposed to plasma collected from subjects after treatment or placebo and acetylcholine (ACh)-stimulated NO production is quantified as an index of EC function (10, 11). We can also assess the effect of plasma exposure on EC mtROS bioactivity (11). However, whether chronic MitoQ supplementation alters the circulating milieu to improve EC function and whether reductions in EC mtROS bioactivity are mechanistically involved are unknown.
Excessive mtROS leads to oxidation of circulating lipids (12), which further promote endothelial dysfunction (13, 14). Oxidized low-density lipoprotein (oxLDL) is an oxidized lipid species that increases with aging and is strongly related to endothelial dysfunction (15). When bound to its lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1), oxLDL initiates a signaling cascade in ECs that increases mtROS (16) to reduce NO bioavailability. In our pilot trial, MitoQ treatment in older adults reduced circulating levels of oxLDL (9). However, whether lower oxLDL with chronic MitoQ treatment contributes to plasma treatment-mediated improvements in EC function and reductions in mtROS bioactivity has not been assessed.
To address these research gaps, we conducted an ancillary analysis using plasma collected in our pilot crossover clinical trial assessing the efficacy of chronic MitoQ supplementation for improving endothelial function in older adults (9). We used our laboratory’s ex vivo model to assess the effect of subject plasma exposure on ACh-stimulated NO production (10, 11) and mtROS bioactivity in ECs ex vivo and the mechanistic role of changes in oxLDL with MitoQ on these outcomes. We also assessed the relation between improvements in EC NO production ex vivo with the improvements in NO-mediated EDD in vivo with MitoQ supplementation. We hypothesized that 1) chronic MitoQ supplementation in older adults would favorably modulate the circulating milieu by lowering plasma oxLDL levels to result in higher EC NO production and lower mtROS bioactivity and 2) improvements in EC NO production ex vivo and NO-mediated EDD in vivo with chronic MitoQ supplementation would be related.
METHODS
This study is an ancillary analysis using plasma samples collected during our completed randomized, double-blind, placebo-controlled, crossover-design clinical trial investigating the efficacy of MitoQ for improving vascular function in healthy older adults (9) (NCT02597023). Twenty older men and PMW aged 60–79 yr without overt CVD, but with subclinical endothelial dysfunction at baseline, were studied (9). All subjects underwent both MitoQ and placebo supplementation. Specifically, subjects were randomized to either 6 wk of supplementation with MitoQ or placebo followed by 6 wk of the alternate condition (i.e., MitoQ and then placebo, or placebo and then MitoQ) in a counterbalanced manner. The two supplementation phases were separated by a 2-wk washout period. Remaining plasma samples were available from 19 of the 20 subjects studied in the original trial. A detailed description of the study design, intervention, and outcome assessments can be found as previously published (9). All procedures were reviewed and approved by the Institutional Review Board at the University of Colorado Boulder. The nature, benefits, and risks of all study procedures were explained to volunteers, and their written informed consents were obtained before participation in the study.
Plasma was collected from venous blood samples from subjects after each 6-wk treatment phase (MitoQ and placebo). All blood samples were obtained >24 h after the last dose of MitoQ or placebo (9), when circulating MitoQ levels were below levels that exert effects on endothelial function (see results). Human aortic ECs (HAECs) (PromoCell; used after 4–6 passages) were cultured in 96-well glass bottom plates (CellVis) under standard conditions (37.5°C, 100% relative humidity, 5% CO2) in basal media [Endothelial Cell Growth Medium-2 (EGM-2) BulletKit + 1% penicillin-streptomycin (pen-strep); PromoCell] supplemented with 10% subject plasma (17). To determine endothelial function ex vivo, NO production was quantified in HAECs following exposure to subject plasma for 24 h. To assess the effects of subject plasma on EC mtROS bioactivity, HAECs were exposed to subject plasma for 2 h. All experiments were performed in triplicate. Plasma exposure durations were determined from preliminary experiments to identify the optimal time point to quantify each outcome measure (data not shown).
Following subject plasma exposure, HAECs were washed three times with Hank’s balanced salt solution (HBSS; Thermo Fisher) before fluorescent probe staining. HAECs were incubated with the fluorescent probe Hoechst (nuclei stain; Thermo Fisher) and either 10 µM diaminorhodamine-4M AM (DAR-4M AM) (to quantify NO production; Sigma-Aldrich) for 45 min or 5 µM MitoSOX (to quantify mtROS bioactivity; Thermo Fisher) for 15 min. Following staining, HAECs were washed three times with HBSS. A final 100 µL of HBSS was added, and HAECs were allowed to rest for 15 min before imaging.
Live HAECs were imaged at ×20 using wide-field fluorescence microscopy (EVOS M7000 Imaging System; Thermo Fisher) under standard incubation conditions using the EVOS Onstage Incubator. HAECs stained with DAR-4M AM were imaged before and 6 min after addition of 100 µM ACh (Sigma) to stimulate NO production. Six minutes was determined from preliminary experiments to identify the time to maximum ACh-stimulated NO production (Supplemental Fig. S1A; all Supplemental material is available at https://doi.org/10.6084/m9.figshare.23220539). The ACh concentration of 100 µM represents the concentration used by our laboratory to stimulate maximal endothelium-dependent dilation of mouse carotid arteries ex vivo (13).
Images were quantified using Celleste 5.0 Image Analysis Software (Thermo Fisher) (17). To quantify ACh-stimulated changes in NO production, integrated fluorescence intensity (IFI) was determined before and after administration of ACh. To calculate fold change in ACh-stimulated NO production, IFI measured post-ACh administration was normalized to IFI pre-ACh in each well and averaged across three replicates. To quantify mtROS bioactivity, IFI was normalized to total cell count in each well and averaged across three replicates. Responses to plasma exposure for each subject were normalized to the overall average of the placebo condition for ACh-stimulated NO production and mtROS bioactivity.
Under the experimental conditions described earlier, we confirmed that DAR-4M AM is specific for NO as the ACh-stimulated increase in signal was abolished by a NO synthase inhibitor [NG-nitro-l-arginine methyl ester (l-NAME); 100 µM (13), Sigma-Aldrich], and the signal was increased with the exogenous NO donor sodium nitroprusside [SNP; 10 µM (13), Santa Cruz Biotechnology] in HAECs with and without plasma treatment (4% FBS + EGM-2 + 1% pen-strep) (Supplemental Fig. S1B). Moreover, we confirmed that MitoSOX detects mtROS as the signal was enhanced by rotenone [100 nM (18, 19), Sigma-Aldrich], an inducer of mtROS production at mitochondrial complex I, and reduced by MitoQ [1 µM (13), Antipodean Pharmaceuticals], a mitochondrial-targeted antioxidant, in HAECs with and without plasma treatment (Supplemental Fig. S1C). These results supporting the specificity of DAR-4M AM for NO and MitoSOX for mtROS are in agreement with previous work in ECs (20, 21).
To determine if levels of circulating MitoQ observed in subjects following chronic MitoQ supplementation modulate NO production and mtROS bioactivity, we exposed HAECs to 0.3 pmol of MitoQ (9). NO production and mtROS bioactivity were measured, in triplicate, as described earlier and compared with control (4% FBS + EGM-2 + 1% pen-strep) (n = 6/condition).
To determine if physiological levels of oxLDL impair NO production and increase mtROS bioactivity, we exposed HAECs to levels of oxLDL found in plasma from subjects after placebo treatment (90 ng/mL oxLDL + EGM-2 + 1% pen-strep). NO production and mtROS bioactivity were measured, in triplicate, as described earlier and compared with control (n = 6/condition).
To elucidate the role of oxLDL in circulating milieu-related changes following MitoQ supplementation, we normalized oxLDL (L34357; Invitrogen; Thermo Fisher) levels between placebo and MitoQ conditions by adding the average reduction of oxLDL with MitoQ treatment (14 ng/mL) to plasma from subjects after MitoQ treatment. NO and mtROS were then measured, in triplicate, and normalized as described earlier (n = 19/group).
To determine if lower endogenous oxLDL imparts favorable plasma-mediated changes to NO production and mtROS bioactivity through changes in interaction with its LOX-1 receptor, LOX-1 blocking antibodies (R&D Systems; Goat IgG) were used (10 µg/mL), as previously described (22), in a representative subset of subjects (n = 8 or 9/condition). LOX-1 antibodies were administered to HAECs in standard cell culture medium for 1 h before plasma exposure, per manufacturer recommendation. NO and mtROS were then measured, in triplicate, and normalized as described earlier.
Fluorescence intensity was normalized within a plate to the placebo plasma condition on a given plate. The placebo plasma condition on all plates was normalized to 1 to allow comparison across plates. As such, the data from the MitoQ plasma + oxLDL condition and the LOX-1 blockade experiments are presented and compared statistically with the effects of placebo plasma and MitoQ plasma using the data presented in Fig. 1.
Figure 1.

A and B: acetylcholine (ACh)-stimulated nitric oxide (NO) production (A; ***P < 0.001) and mitochondrial reactive oxygen species (mtROS) bioactivity (B; **P < 0.01) measured in cultured human aortic endothelial cells following exposure to plasma collected from subjects after mitochondria-targeted antioxidant (MitoQ) or placebo supplementation (n = 19/condition). Individual subject responses and group means are shown. C–F: NO and mtROS responses in older men (C and D; n = 8) and postmenopausal women (E and F; n = 11) (*P < 0.05). Paired t tests.
Statistical Analyses
Data analyses were performed using GraphPad Prism v9.5.0. Paired t tests and a two-way repeated-measures ANOVA were used to assess the specificity of DAR-4M AM and MitoSOX to NO and mtROS, respectively. A one-way repeated-measures ANOVA was used to assess the optimal time point to quantify NO production. The impact of subject plasma, collected from the same subjects after completion of both the MitoQ and placebo phases of the crossover pilot trial, on outcome variables was evaluated using paired t tests. A two-way repeated-measures ANOVA was used to compare absolute levels of NO production before and after ACh stimulation. Unpaired t tests were used to compare conditions in experiments determining the effect of circulating MitoQ and physiological levels of oxLDL on outcome measures. A Shapiro–Wilk test for normality (P = 0.0364) followed by a Spearman correlation test was used to determine the relation between in vivo NO-mediated EDD and ex vivo NO production. To assess the effect of oxLDL and LOX-1 on outcome variables, one-way repeated-measures ANOVAs were used. The Grubbs test was used to identify outliers (α = 0.05). An outlier was identified in mtROS bioactivity in Fig. 3 within “placebo − LOX-1” and “MitoQ + oxLDL − LOX-1” groups and another identified within “absolute NO production” and differences in “ACh-stimulated NO production” in Supplemental Fig. S2, A and B. Significance was P < 0.05 for all tests.
Figure 3.
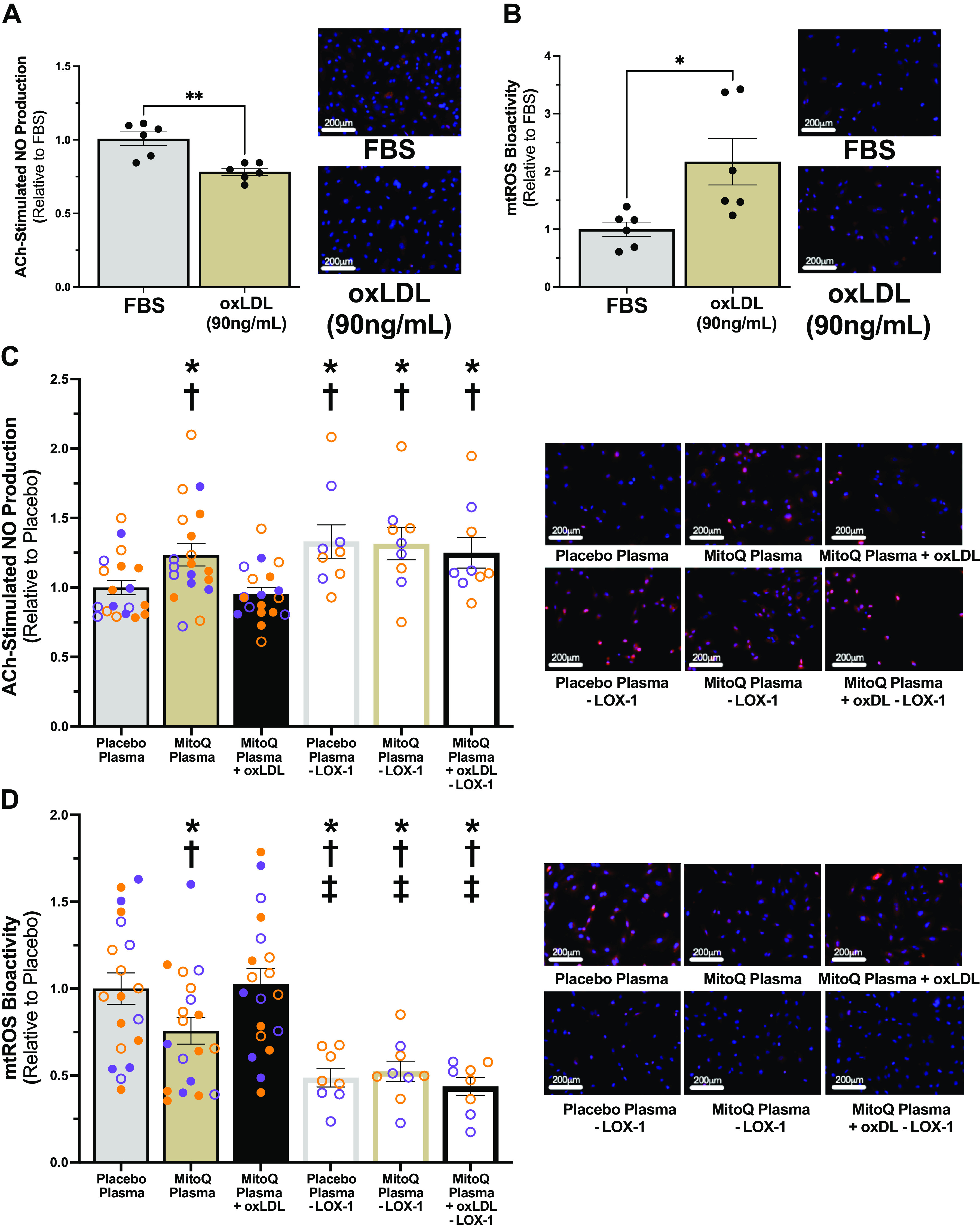
A and B: acetylcholine (ACh)-stimulated nitric oxide (NO) production (A; **P < 0.01) and mitochondrial reactive oxygen species (mtROS) bioactivity (B; *P < 0.05) in response to levels of oxidized low-density lipoprotein (oxLDL) measured in plasma from subjects after placebo supplementation (90 ng/mL) or fetal bovine serum (FBS) (n = 6/condition). Independent t tests. C and D: ACh-stimulated NO production (C) and mtROS bioactivity (D) following exposure to plasma with oxLDL normalization (n = 19/condition) and/or lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1) antibody blockade (n = 8 or 9/condition; open circles). Data and representative images presented for the placebo plasma and mitochondria-targeted antioxidant (MitoQ) plasma conditions in C and D are the same as those presented in Fig. 1, A and B, respectively, to facilitate comparison of the magnitude of effect of oxLDL addback and LOX-1 blockade. Purple, older men (n = 8); orange, postmenopausal women (n = 11). P < 0.05 compared with *placebo; †MitoQ + oxLDL; and ‡MitoQ. One-way repeated-measures ANOVA. Data are means ± SE.
RESULTS
Plasma collected from 19 older adults (8 men; 11 PMW) after completion of the MitoQ and placebo phases of our pilot crossover trial (9) was used. Following chronic MitoQ supplementation, endothelial function (FMDBA) was higher and plasma oxLDL levels were lower compared with after placebo; other subject characteristics were unaltered (9) (Table 1).
Table 1.
Participant characteristics
| Characteristics | Placebo | MitoQ |
|---|---|---|
| Sex, men/women | 8/11 | |
| Age, yr | 67 ± 0.9 | |
| Years since menopause | 14.7 ± 1.4 | |
| Brachial artery flow-mediated dilation | 3.5 ± 0.4 | 5.0 ± 0.5* |
| Body mass index, kg/m2 | 23.5 ± 0.8 | 23.3 ± 0.8 |
| Systolic blood pressure, mmHg | 113 ± 4 | 112 ± 3 |
| Diastolic blood pressure, mmHg | 67 ± 2 | 68 ± 1 |
| Resting heart rate, beats/min | 61 ± 2 | 62 ± 2 |
| Physical activity caloric expenditure, kcal/wk | 5,279 ± 802.1 | 5,222 ± 548.9 |
| Total cholesterol, mg/dL | 195.0 ± 8.6 | 194.3 ± 9.0 |
| High-density lipoprotein cholesterol, mg/dL | 53.4 ± 3.0 | 53.2 ± 3.2 |
| Low-density lipoprotein cholesterol, mg/dL | 120.4 ± 7.6 | 122.4 ± 7.6 |
| Triglycerides, mg/dL | 108.3 ± 12.2 | 103.8 ± 18.5 |
| Fasting glucose, mg/dL | 83.0 ± 1.5 | 84.4 ± 1.6 |
| Oxidized low-density lipoprotein, ng/mL | 89.9 ± 5.7 | 75.6 ± 5.5* |
Values are means ± SE. MitoQ, mitochondria-targeted antioxidant.
Paired t test, *P < 0.05.
Effect of Changes in the Circulating Milieu with MitoQ on EC Function
To determine if chronic MitoQ supplementation in older adults alters the circulating milieu to improve EC function, we used our ex vivo model of endothelial function. ECs exposed to subject plasma collected after MitoQ supplementation had ∼25% higher ACh-stimulated NO production [placebo, 1.0 ± 0.05 arbitrary units (AU); MitoQ, 1.24 ± 0.08 AU; P = 0.0002] (Fig. 1A) and ∼25% lower mtROS bioactivity (placebo, 1.0 ± 0.09 AU; MitoQ, 0.76 ± 0.08 AU; P = 0.003) (Fig. 1B) compared with placebo. Favorable plasma-mediated changes on NO production and mtROS bioactivity were observed to a similar extent in both older men (Fig. 1, C and D) and PMW (Fig. 1, E and F). Similar results were obtained when the data were analyzed as absolute values. Specifically, although basal EC NO production (before ACh stimulation) was not different between conditions (placebo, 3.0 × 106 ± 3.6 × 105 AU; MitoQ, 2.9 × 106 ± 3.8 × 105 AU; P = 0.55) (Supplemental Fig. S2A), there was a greater increase in NO in response to ACh in ECs treated with plasma from subjects after MitoQ supplementation (Post-ACh – Pre-ACh: placebo, 1.8 × 106 ± 3.3 × 105 AU; MitoQ, 3.1 × 106 ± 7.5 × 105 AU; P = 0.018) (Supplemental Fig. S2, A and B). Moreover, mtROS bioactivity (expressed as absolute values) was lower in ECs treated with plasma from subjects following MitoQ supplementation (placebo, 4.5 × 104 ± 4.0 × 103 AU; MitoQ, 3.3 × 104 ± 3.4 × 103 AU; P = 0.003) (Supplemental Fig. S2C).
We then investigated if chronic MitoQ supplementation-mediated improvements in EC NO production ex vivo and NO-mediated EDD in vivo, as assessed by FMDBA, were correlated. We found a moderate positive relation (r = 0.4684; P = 0.0431) between improvements in in vivo and ex vivo measures of endothelial function with MitoQ supplementation (Fig. 2). These observations provide initial evidence that increases in NO production assessed in EC culture after exposure to plasma following chronic MitoQ treatment may be associated with corresponding improvements in endothelial function measured in vivo.
Figure 2.

Spearman correlation between improvements in nitric oxide (NO)-mediated endothelium-dependent dilation in vivo, as assessed by brachial artery flow-mediated dilation, and acetylcholine (ACh)-stimulated NO production ex vivo with chronic mitochondria-targeted antioxidant (MitoQ) treatment [difference in percent change (%change) between placebo and MitoQ] (n = 19).
Circulating levels of MitoQ were measured at trace amounts in plasma samples obtained 24 h after the last dose following chronic MitoQ supplementation compared with placebo (9). These low levels of MitoQ have not been shown to exert biological effects (22). To confirm this in the present study, we compared the addition of 0.3 pmol of MitoQ to FBS control and found no differences in NO production (FBS, 1.00 ± 0.05 AU; MitoQ, 0.92 ± 0.04 AU; P = 0.2) or mtROS bioactivity (FBS, 1.00 ± 0.12 AU; MitoQ, 1.07 ± 0.10 AU; P = 0.67). This suggests that plasma levels of MitoQ following chronic MitoQ supplementation in older adults do not contribute to the plasma treatment effects in NO production or mtROS bioactivity.
Effect of Lower OxLDL with MitoQ on Higher EC Function
Circulating oxLDL was lower in subject plasma following chronic MitoQ supplementation compared with placebo (9). Previous research investigating the role of oxLDL in EC dysfunction and increases in mtROS ex vivo have used supraphysiological concentrations of oxLDL, ultimately limiting the physiological relevance of their findings (4, 5, 23, 24). Therefore, we first determined if oxLDL levels measured following the placebo phase (90 ng/mL) of our study (9) could cause EC dysfunction and increase mtROS ex vivo. ECs exposed to 90 ng/mL of oxLDL had lower NO production (FBS, 1.00 ± 0.05 AU; oxLDL, 0.78 ± 0.02 AU; P = 0.002) (Fig. 3A) and higher mtROS bioactivity (FBS, 1.00 ± 0.12 AU; oxLDL, 2.19 ± 0.40 AU; P = 0.02) (Fig. 3B) compared with control. These results suggest that physiological levels of oxLDL observed in older adults can cause EC dysfunction and increase mtROS bioactivity.
We next sought to determine if the reduction in circulating oxLDL following MitoQ supplementation contributes to plasma treatment-driven differences in NO production and mtROS bioactivity. To do so, we replaced the average reduction of oxLDL with MitoQ supplementation (14 ng/mL) (9) in plasma collected following the MitoQ phase of the trial to normalize oxLDL levels between conditions. Normalizing oxLDL levels between conditions abolished group differences in NO production (placebo, 1.00 ± 0.05 AU; MitoQ + oxLDL, 0.95 ± 0.04 AU; P = 0.70) (Fig. 3C) and mtROS bioactivity (placebo, 1.00 ± 0.09 AU; MitoQ + oxLDL, 1.03 ± 0.09 AU; P = 0.99) (Fig. 3D). These findings suggest that lower plasma oxLDL with MitoQ supplementation in older adults contributes to the plasma treatment-stimulated improvement in EC function, which is likely a result of decreased mtROS bioactivity.
To determine a mechanism by which lower circulating oxLDL with MitoQ supplementation improved NO production and reduced mtROS, we then inhibited endogenous oxLDL binding to its receptor using LOX-1 antibodies in plasma from a representative subset of subjects (n = 8 or 9; 5 PMW). Inhibition of LOX-1 increased NO production (placebo, 1.00 ± 0.05 AU; placebo − LOX-1, 1.33 ± 0.12 AU; MitoQ − LOX-1, 1.32 ± 0.12 AU; MitoQ + oxLDL − LOX-1, 1.25 ± 0.11 AU) (Fig. 3C) to levels comparable with the MitoQ condition and reduced mtROS bioactivity (placebo, 1.00 ± 0.09 AU; placebo − LOX-1, 0.49 ± 0.05 AU; MitoQ − LOX-1: 0.52 ± 0.06 AU; MitoQ + oxLDL − LOX-1, 0.44 ± 0.05 AU) (Fig. 3D) to levels lower than the MitoQ condition. These observations indicate that the reduction in circulating levels of endogenous oxLDL following MitoQ treatment leads to higher NO production and lower mtROS bioactivity through decreased oxLDL-LOX-1 interaction.
DISCUSSION
This study demonstrates that 6 wk of MitoQ supplementation in older adults changes the circulating milieu to improve endothelial cell function and decrease mitochondrial oxidative stress ex vivo by reducing oxLDL-LOX-1 signaling. These findings are the first to demonstrate that alterations in the circulating milieu are a mechanism by which chronic treatment with a mitochondria-targeted antioxidant improves endothelial function in older adults (9).
We recently showed that chronic MitoQ supplementation in older adults improves endothelial function in vivo, as assessed by FMDBA (9). We have also shown that interventions that improve endothelial function in older adults appear to do so, in part, by altering factors in circulation (10, 11). Here, we extend these observations to supplementation with a mitochondrial antioxidant and show that ECs exposed to plasma collected from subjects after chronic MitoQ supplementation exhibit higher NO production. Furthermore, improvements in EC function assessed ex vivo after chronic MitoQ supplementation were positively related to corresponding improvements in endothelial function measured in vivo.
Excess mtROS can react with NO to reduce bioavailability and impair endothelial function. Indeed, our previous findings suggest reductions in mtROS mediate improvements in EDD with MitoQ (9, 13). In the current study, we found that plasma from subjects after MitoQ supplementation reduced EC mtROS, implicating reductions in mtROS as a mechanism of circulating factor-related improvements in EC function. Importantly, improvements in EC NO production and reductions in mtROS were independent of circulating levels of MitoQ.
Higher NO production and lower mtROS bioactivity stimulated by MitoQ supplementation-mediated changes in the circulating milieu were observed in both older men and PMW. This is of particular interest as, in contrast to older men, aerobic exercise interventions have not been consistently found to improve endothelial function in PMW (25–28). In our pilot study, we observed similar efficacy of chronic MitoQ supplementation for improving endothelial function in vivo in older men and PMW (9). Here, we extend these observations to show that changes in the circulating milieu are a novel mechanism by which MitoQ treatment causes higher endothelial function in both older men and PMW.
OxLDL is thought to be mechanistically involved in endothelial dysfunction, in part by exacerbating mtROS. However, previous research investigating the deleterious effect of oxLDL on EC function and increases in mtROS bioactivity ex vivo has used supraphysiological concentrations of oxLDL (4, 5, 23, 24). Therefore, we first investigated if endogenous levels of oxLDL measured in healthy older adults elicit dysfunction in ECs and increase mtROS bioactivity before exploring the role of lower plasma levels of oxLDL for favorably modulating EC function and mtROS bioactivity in subject plasma. Indeed, we found that ECs exposed to oxLDL at levels measured in plasma from subjects after placebo caused EC dysfunction and increased mtROS bioactivity. To our knowledge, this is the first time that levels of circulating oxLDL measured in healthy older adults have been shown to reduce EC function and increase mtROS bioactivity ex vivo.
To determine the specific role of lower-circulating oxLDL with MitoQ supplementation on higher EC function and lower mtROS ex vivo, we normalized oxLDL levels between conditions. Normalization of oxLDL abolished differences in NO production and mtROS bioactivity, suggesting a contributing role of lower plasma levels of oxLDL with MitoQ supplementation. When oxLDL binds to its LOX-1 receptor, it promotes excessive mtROS by increasing mitochondria fission-related processes in ECs to lower NO production and impair endothelial function (16). Pharmacological blockade of endogenous oxLDL binding to LOX-1 in subject plasma ameliorated the oxLDL-LOX-1 signaling-mediated impairment of NO production and augmentation of mtROS bioactivity across conditions. Taken together, these experiments show that decreased oxLDL-LOX-1 binding is a mechanism by which reduced levels of circulating oxLDL following chronic MitoQ supplementation results in higher EC function and lower mtROS bioactivity.
We recognize a few limitations of our study. First, studies have shown that DAR-4M AM may not be entirely specific to NO, that its fluorescent signal may also reflect other reactive nitrogen species (29). Although we conducted rigorous positive and negative control experiments to assess the specificity of DAR-4M AM for NO, future studies should use an additional negative control, such as the NO scavenger c-PTIO, to confirm these findings. Similarly, we performed experiments to confirm the specificity of MitoSOX for mtROS; however, another positive control, such as antimycin, could be important to further verify that the changes in MitoSOX signal we observed are specific to mtROS. Finally, we showed that LOX-1 blockade eliminated group differences by increasing NO and decreasing mtROS in ECs through inhibition of oxLDL-LOX-1 signaling. It is important to consider that LOX-1 blockade may not only be inhibiting oxLDL signaling but also blocking the action of other unexplored/unidentified ligands. Future studies are warranted to identify other potential molecular transducers of the beneficial effect of MitoQ treatment.
CONCLUSIONS
Here, we demonstrate that 6 wk of chronic MitoQ supplementation in older adults modulates the circulating milieu to improve endothelial function and decrease mitochondrial oxidative stress ex vivo by reducing oxLDL-LOX-1 signaling. These novel observations suggest that changes in the circulating milieu with MitoQ treatment are an important mechanism by which MitoQ improves endothelial function with aging.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Figs. S1 and S2: https://doi.org/10.6084/m9.figshare.23220539.
GRANTS
This work was supported by National Institutes of Health Grants R21AG049451 and R01AG066730 (to D.R.S.), 5T32DK007135 (to K.O.M.), K01DK115524 (to M.J.R.), and Colorado CTSA UL1 TR002535; American Heart Association Grants 23POST1025630 (to K.O.M.) (https://doi.org/10.58275/AHA.23POST1025630.pc.gr.161298) and 23CDA1056582 (to M.J.R.) (https://doi.org/10.58275/AHA.23CDA1056582.pc.gr.168037); and an industry contract with MitoQ Limited (provided MitoQ and some financial support).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.O.M., K.R.L., D.R.S., and M.J.R. conceived and designed research; K.O.M., K.R.L., and S.D. performed experiments; K.O.M., K.R.L., and S.D. analyzed data; K.O.M., D.R.S., and M.J.R. interpreted results of experiments; K.O.M. prepared figures; K.O.M. and M.J.R. drafted manuscript; K.O.M., K.R.L., S.D., M.E.C., D.R.S., and M.J.R. edited and revised manuscript; K.O.M., K.R.L., S.D., M.E.C., D.R.S., and M.J.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the staff of the University of Colorado Boulder Clinical Translational Research Center for technical assistance.
REFERENCES
- 1. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, , et al. Heart Disease and Stroke Statistics-2017 Update: a report from the American Heart Association. Circulation 135: e146–e603, 2017. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. I. Aging arteries: a ‘set up’ for vascular disease. Circulation 107: 139–146, 2003. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- 3. Seals DR, Jablonski KL, Donato AJ. Aging and vascular endothelial function in humans. Clin Sci (Lond) 120: 357–375, 2011. doi: 10.1042/CS20100476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zmijewski JW, Moellering DR, Le Goffe C, Landar A, Ramachandran A, Darley-Usmar VM. Oxidized LDL induces mitochondrially associated reactive oxygen/nitrogen species formation in endothelial cells. Am J Physiol Heart Circ Physiol 289: H852–H861, 2005. doi: 10.1152/ajpheart.00015.2005. [DOI] [PubMed] [Google Scholar]
- 5. Roy Chowdhury SK, Sangle GV, Xie X, Stelmack GL, Halayko AJ, Shen GX. Effects of extensively oxidized low-density lipoprotein on mitochondrial function and reactive oxygen species in porcine aortic endothelial cells. Am J Physiol Endocrinol Metab 298: E89–E98, 2010. doi: 10.1152/ajpendo.00433.2009. [DOI] [PubMed] [Google Scholar]
- 6. Rossman MJ, Gioscia-Ryan RA, Clayton ZS, Murphy MP, Seals DR. Targeting mitochondrial fitness as a strategy for healthy vascular aging. Clin Sci (Lond) 134: 1491–1519, 2020. doi: 10.1042/CS20190559. [DOI] [PubMed] [Google Scholar]
- 7. Smith RAJ, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci 1201: 96–103, 2010. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- 8. Soules MR, Sherman S, Parrott E, Rebar R, Santoro N, Utian W, Woods N. Stages of Reproductive Aging Workshop (STRAW). J Womens Health Gend Based Med 10: 843–848, 2001. doi: 10.1089/152460901753285732. [DOI] [PubMed] [Google Scholar]
- 9. Rossman MJ, Santos-Parker JR, Steward CAC, Bispham NZ, Cuevas LM, Rosenberg HL, Woodward KA, Chonchol M, Gioscia-Ryan RA, Murphy MP, Seals DR. Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension 71: 1056–1063, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Craighead DH, Heinbockel TC, Freeberg KA, Rossman MJ, Jackman RA, Jankowski LR, Hamilton MN, Ziemba BP, Reisz JA, D'Alessandro A, Brewster LM, DeSouza CA, You Z, Chonchol M, Bailey EF, Seals DR. Time-efficient inspiratory muscle strength training lowers blood pressure and improves endothelial function, no bioavailability, and oxidative stress in midlife/older adults with above-normal blood pressure. J Am Heart Assoc 10: e020980, 2021. doi: 10.1161/JAHA.121.020980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rossman MJ, Gioscia-Ryan RA, Santos-Parker JR, Ziemba BP, Lubieniecki KL, Johnson LC, Poliektov NE, Bispham NZ, Woodward KA, Nagy EE, Bryan NS, Reisz JA, D'Alessandro A, Chonchol M, Sindler AL, Seals DR. Inorganic nitrite supplementation improves endothelial function with aging. Hypertension 77: 1212–1222, 2021. doi: 10.1161/HYPERTENSIONAHA.120.16175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mabile L, Meilhac O, Escargueil-Blanc I, Troly M, Pieraggi MT, Salvayre R, Nègre-Salvayre A. Mitochondrial function is involved in LDL oxidation mediated by human cultured endothelial cells. Arterioscler Thromb Vasc Biol 17: 1575–1582, 1997. doi: 10.1161/01.atv.17.8.1575. [DOI] [PubMed] [Google Scholar]
- 13. Gioscia-Ryan RA, LaRocca TJ, Sindler AL, Zigler MC, Murphy MP, Seals DR. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J Physiol 592: 2549–2561, 2014. doi: 10.1113/jphysiol.2013.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. López-Armada MJ, Riveiro-Naveira RR, Vaamonde-García C, Valcárcel-Ares MN. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 13: 106–118, 2013. doi: 10.1016/j.mito.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 15. Eskurza I, Kahn ZD, Seals DR. Xanthine oxidase does not contribute to impaired peripheral conduit artery endothelium-dependent dilatation with ageing. J Physiol 571: 661–668, 2006. doi: 10.1113/jphysiol.2005.102566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, Duess M-A, Levit A, Kim B, Hartman M-L, Joseph L, Shirihai OS, Vita JA. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 124: 444–453, 2011. doi: 10.1161/CIRCULATIONAHA.110.014506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Murray KO, Berryman-Maciel M, Darvish S, Coppock ME, You Z, Chonchol M, Seals DR, Rossman MJ. Mitochondrial-targeted antioxidant supplementation for improving age-related vascular dysfunction in humans: a study protocol. Front Physiol 13: 980783, 2022. doi: 10.3389/fphys.2022.980783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem 278: 8516–8525, 2003. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- 19. Hu H, Doll DN, Sun J, Lewis SE, Wimsatt JH, Kessler MJ, Simpkins JW, Ren X. Mitochondrial impairment in cerebrovascular endothelial cells is involved in the correlation between body temperature and stroke severity. Aging Dis 7: 14–27, 2016. doi: 10.14336/AD.2015.0906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murthy S, Koval OM, Ramiro Diaz JM, Kumar S, Nuno D, Scott JA, Allamargot C, Zhu LJ, Broadhurst K, Santhana V, Kutschke WJ, Irani K, Lamping KG, Grumbach IM. Endothelial CaMKII as a regulator of eNOS activity and NO-mediated vasoreactivity. PLoS One 12: e0186311, 2017. doi: 10.1371/journal.pone.0186311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nazarewicz RR, Dikalova AE, Bikineyeva A, Dikalov SI. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am J Physiol Heart Circ Physiol 305: H1131–H1140, 2013. doi: 10.1152/ajpheart.00063.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Imanishi T, Hano T, Sawamura T, Nishio I. Oxidized low-density lipoprotein induces endothelial progenitor cell senescence, leading to cellular dysfunction. Clin Exp Pharmacol Physiol 31: 407–413, 2004. doi: 10.1111/j.1440-1681.2004.04022.x. [DOI] [PubMed] [Google Scholar]
- 23. Chavakis E, Dernbach E, Hermann C, Mondorf UF, Zeiher AM, Dimmeler S. Oxidized LDL inhibits vascular endothelial growth factor-induced endothelial cell migration by an inhibitory effect on the akt/endothelial nitric oxide synthase pathway. Circulation 103: 2102–2107, 2001. doi: 10.1161/01.cir.103.16.2102. [DOI] [PubMed] [Google Scholar]
- 24. Kinumi T, Ogawa Y, Kimata J, Saito Y, Yoshida Y, Niki E. Proteomic characterization of oxidative dysfunction in human umbilical vein endothelial cells (HUVEC) induced by exposure to oxidized LDL. Free Radic Res 39: 1335–1344, 2005. doi: 10.1080/10715760500306695. [DOI] [PubMed] [Google Scholar]
- 25. Lew LA, Ethier TS, Pyke KE. The impact of exercise training on endothelial function in postmenopausal women: a systematic review. Exp Physiol 107: 1388–1421, 2022. doi: 10.1113/EP090702. [DOI] [PubMed] [Google Scholar]
- 26. Pierce GL, Eskurza I, Walker AE, Fay TN, Seals DR. Sex-specific effects of habitual aerobic exercise on brachial artery flow-mediated dilation in middle-aged and older adults. Clin Sci (Lond) 120: 13–23, 2011. doi: 10.1042/CS20100174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moreau KL, Stauffer BL, Kohrt WM, Seals DR. Essential role of estrogen for improvements in vascular endothelial function with endurance exercise in postmenopausal women. J Clin Endocrinol Metab 98: 4507–4515, 2013. doi: 10.1210/jc.2013-2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murray KO, Mahoney SA, Venkatasubramanian R, Seals DR, Clayton ZS. Aging, aerobic exercise, and cardiovascular health: barriers, alternative strategies and future directions. Exp Gerontol 173: 112105, 2023. doi: 10.1016/j.exger.2023.112105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lacza Z, Horváth EM, Pankotai E, Csordás A, Kollai M, Szabó C, Busija DW. The novel red-fluorescent probe DAR-4M measures reactive nitrogen species rather than NO. J Pharmacol Toxicol Methods 52: 335–340, 2005. doi: 10.1016/j.vascn.2005.06.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1 and S2: https://doi.org/10.6084/m9.figshare.23220539.
Data Availability Statement
Data will be made available upon reasonable request.