

SUMMARY
Male mice lacking the androgen receptor (AR) in pancreatic b cells exhibit blunted glucose-stimulated insulin secretion (GSIS), leading to hyperglycemia. Testosterone activates an extranuclear AR in β cells to amplify glucagon-like peptide-1 (GLP-1) insulinotropic action. Here, we examined the architecture of AR targets that regulate GLP-1 insulinotropic action in male b cells. Testosterone cooperates with GLP-1 to enhance cAMP production at the plasma membrane and endosomes via: (1) increased mitochondrial production of CO2, activating the HCO3−-sensitive soluble adenylate cyclase; and (2) increased Gαs recruitment to GLP-1 receptor and AR complexes, activating transmembrane adenylate cyclase. Additionally, testosterone enhances GSIS in human islets via a focal adhesion kinase/SRC/phosphatidylinositol 3-kinase/mammalian target of rapamycin complex 2 actin remodeling cascade. We describe the testosterone-stimulated AR interactome, transcriptome, proteome, and metabolome that contribute to these effects. This study identifies AR genomic and non-genomic actions that enhance GLP-1-stimulated insulin exocytosis in male β cells.
In brief
Xu et al. show that androgen receptor activation in male β cells increases mitochondrial CO2 production, activating soluble adenylate cyclase; testosterone increases Gαs recruitment to GLP-1 and androgen receptors, activating transmembrane adenylate cyclase; and testosterone also engages an actin remodeling cascade. These potentiate GLP-1-mediated cAMP production in microdomains and enhance insulin exocytosis.
Graphical Abstract
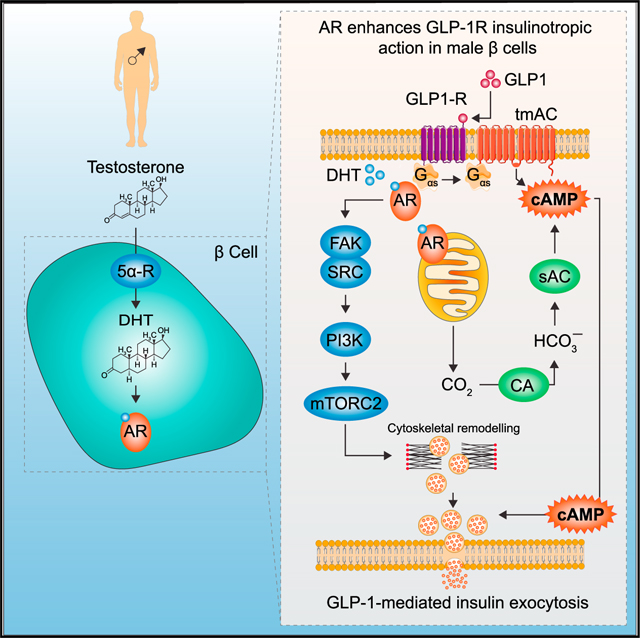
INTRODUCTION
Testosterone deficiency predisposes men to type 2 diabetes (T2D),1–5 and testosterone treatment prevents or reverses T2D in androgen-deficient men.6,7, Testosterone deficiency produces insulin resistance that is instrumental in its diabetogenic effect. However, the role of testosterone deficiency in the dysfunction of insulin-producing b cells as a factor predisposing to T2D, and the importance of testosterone in improving β cell function and reversing T2D in androgen-deficient men, remain unexplored.8, This lack of knowledge is surprising, since it is established that insulin resistance cannot induce hyperglycemia without β cell failure to compensate.9–11 Male mice lacking the androgen receptor (AR) in β cells (βARKO) exhibit decreased glucose-stimulated insulin secretion (GSIS), leading to glucose intolerance,12, and their islets display dysregulated genes involved in inflammation and insulin secretion.13, When exposed to a Western diet, male βARKO mice develop β cell failure, which leads to inadequate compensation for insulin resistance and hyperglycemia.12, Thus, in the absence of AR in β cells, testosterone cannot maintain normoglycemia. In human β cells, testosterone is converted to the potent AR agonist dihydrotestosterone (DHT) by the enzyme 5α-reductase, which is necessary for GSIS.14, In rodent and human b cells, DHT activates an extranuclear AR that amplifies the insulinotropic action of glucagon-like peptide-1 (GLP-1).12 Here, we used a multidisciplinary approach to study the architecture of AR pathways enhancing GLP-1 insulinotropic action. We show that DHT activation of AR: (1) increases mitochondrial production of CO2, which activates the bicarbonate-sensitive soluble adenylate cyclase (AC); and (2) increases the small GTPase Gαs recruitment to the GLP-1 receptor and AR, further activating transmembrane adenylate cyclase (tmAC). This potentiates GLP-1-mediated cAMP production at the plasma membrane and endosomes, thereby sensitizing pools of insulin granules for exocytosis. In parallel, DHT and GLP-1 collaborate to promote AR interactions with actin remodeling proteins via a focal adhesion kinase (FAK)/SRC/phosphatidylinositol 3-kinase (PI3K)/ mammalian target of rapamycin complex 2 (mTORC2) cascade that enhance GSIS in male human β cells.
RESULTS
Male βARKOMIP mice exhibit blunted GSIS following intraperitoneal, but not oral, glucose administration
On a Western diet, male β cell-specific ARKO (βARKOMIP) mice developed fed hyperglycemia (Figures 1A and 1B) and hypoinsulinemia (Figures 1C and 1D) compared with controls. DHT amplifies the insulinotropic effect of GLP-1 in cultured mouse and human islets.12, To explore the relevance of these findings in vivo, we compared the effect of intraperitoneal-injected glucose (IP-GTT, to partially bypass gut GLP-1 release) versus orally administered glucose (O-GTT, to stimulate gut GLP-1 release) in male control and βARKOMIP mice. Following IP-GTT, βARKOMIP mice exhibited impaired GSIS (Figure 1E), and glucose intolerance (Figure 1F). The defect was selective to glucose, as βARKOMIP showed no alteration in arginine-stimulated insulin secretion (Figure 1G). In contrast, during O-GTT, βARKOMIP mice exhibited similar glucose tolerance (Figure 1H) and β cell function (insulin/glucose ratio at 30 min) as controls (Figure 1I). Thus, loss of β cell AR may impair the insulinotropic action of islet-derived GLP-1 but does not seem to alter gut-derived GLP-1 insulinotropic actions.
Figure 1. DHT activation of AR in β cell amplifies the insulinotropic effect of islet-derived GLP-1 via GLP-1R.

(A–D) Data are from male βARKOMIP and ARlox MIP-CreERT (control) mice fed a western diet since weaning. (A) Random-fed blood glucose and corresponding area under the curve (AUC) from 9 to 29 weeks. (B and C) Random-fed blood glucose and insulin measured at 25 weeks. (D) Insulin/glucose index of insulin deficiency from (B) and (C).
(E) Intraperitoneal glucose-stimulated insulin secretion (GSIS) (3 g/kg) with corresponding insulin AUC.
(F) Intraperitoneal glucose tolerance test (GTT) (2 g/kg) with corresponding glucose AUC.
(G) Intraperitoneal arginine-stimulated insulin secretion (ASIS) (1 g/kg) with corresponding insulin AUC.
(H) Oral GTT (2 g/kg) with corresponding glucose AUC.
(I) Insulin/glucose ratio at 30 min into the oral GTT. Mice were studied at 23–35 weeks of age (n = 10–15).
(J and K) GSIS measured in static incubation in chow-fed RIP-Cre (control), (J) βGLP-1RKORIP islets and (K) βARKORIP islets treated with DHT (10 nM) and GLP-1 (10 nM) for 40 min. Values represent the mean ± SE of n = 2 mice/group measured in triplicate.
(L) Dynamic insulin secretion measured via perifusion in male human islets challenged with 2.8 mM glucose, 16.7 mM glucose, and 20 mM KCl + 16.7 mM glucose. Islets were cultured overnight in vehicle or DHT (10 nM). During perifusion, islets were treated with vehicle, DHT, Exendin9–39 (100 nM), or DHT + Exendin9–39.
(M) AUC for insulin secretion during 16.7 mM glucose (10–30 min) from (L).
(N) AUC for insulin secretion during second KCl + glucose (45–60 min) from (L).
In (L) to (N), data represent a mean ± SE. of two chambers/donor using n = 3 donors. Values represent the mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001.
Activation of β cell AR amplifies the insulinotropic action of GLP-1 via GLP-1R
We assessed GSIS in cultured islets from male control and β cell-specific GLP-1R knockout (βGLP1RKORIP) mice. We used the RIP-Cre transgenic mouse to generate βGLP1RKORIP and βARKORIP mice. Consistent with results observed in vivo, DHT increased GSIS and amplified the insulinotropic action of GLP-1 in control islets (Figure 1J). In contrast, DHT had no effect on GSIS and did not amplify the insulinotropic action of GLP-1 in βGLP1RKORIP islets (Figure 1J). Thus, DHT amplifies the insulinotropic action of islet-derived and exogenous GLP-1, and this effect requires the presence of the β cell GLP-1R. In parallel, we assessed GLP-1 insulinotropic action using male βARKORIP islets. As expected, DHT alone enhanced GSIS and amplified the insulinotropic action of GLP-1 in control islets (Figure 1K). In contrast, DHT alone or in the presence of GLP-1 failed to enhance GSIS in βARKORIP islets (Figure 1K).
We examined the effect of DHT and the requirement of the GLP-1R on dynamic insulin secretion in male human islets in a perifusion system where GSIS is characterized by a biphasic pattern. At 16.7 mM glucose, DHT enhanced second-phase insulin secretion compared with vehicle (Figures 1L and 1M). Moreover, DHT further enhanced GSIS under conditions of maximum membrane depolarization (16.7 mM glucose with 30 mM KCl), suggesting sensitization of exocytosis of the readily releasable pool of insulin granules. In contrast, the stimulatory effect of DHT on GSIS and during membrane depolarization was not observed in the presence of the specific GLP1-R antagonist exendin(9–39)15, (Figures 1L, 1N, and S1F), thus confirming the importance of GLP-1R in DHT second-phase GSIS in human β cells.
Testosterone requires conversion to DHT in target tissues via action of the enzyme 5α-reductase (5α-R) to activate AR.16, βARKOMIP mice exhibit decreased intraperitoneal-injected GSIS and fed hyperglycemia (Figure 1E), suggesting that testosterone is converted to DHT in male mouse islets via 5α-R to increase GSIS, as in human β cells.14, Here, we confirmed that male mouse islets convert testosterone to DHT via 5α-R activity, a process essential to enhancing GSIS (Figures S1A and S1B).
Since GLP-1 corrected the insulin secretory defect of cultured βARKORIP islets (Figure 1K), we explored whether preventing GLP-1 degradation in vivo following treatment with linagliptin, a dipeptidyl peptidase-4 (DPP-4) inhibitor,17, would restore the b cell defect of male βARKOMIP mice. As expected, linagliptin improved b cell response to a glucose load in βARKOMIP mice (Figures S1C–S1E).
DHT amplifies the insulinotropic action of GLP-1 selectively
Since DHT amplifies the insulinotropic action of GLP-1 via GLP-1R in male islets (Figure 1 and Navarro et al.12,), we sought to determine whether DHT could similarly amplify the insulinotropic action of glucose-dependent insulinotropic polypeptide (GIP) and glucagon, which also bind G-protein-coupled receptors (GPCRs) coupled to Gαs.18, We examined the effect of DHT on cAMP production in INS1 832/3 cells, an incretin-responsive β cell model,19, using a modified FRET sensor based upon Epac2-camps.20, Using this approach, GLP-1, GIP, glucagon, and forskolin (FSK; a pharmacological activator of tmAC) all elicited a rapid and sustained rise in cAMP production (Figures 2A–2F). DHT alone elicited a small amplitude rise in cAMP (Figures 2A–2F). Notably, DHT elicited a further increase in GLP-1-induced cAMP production (Figures 2A and 2F). The ability of DHT to amplify GLP-1 production of cAMP was also observed with the GLP-1R agonist exendin 4 (Figure S2A and S2B). In contrast, DHT did not increase glucagon-, GIP-, or FSK-induced cAMP production (Figures 2B–2F and S2C–S2H). We assessed the effect of DHT on GLP-1-, GIP-, glucagon-, and FSK-stimulated GSIS in INS1 832/3 cells. In these cells, DHT alone did not significantly increase GSIS. However, consistent with cAMP production data (Figures 2A–2F), DHT amplified the insulinotropic effect of GLP-1 (Figure 2G) but failed to amplify the insulinotropic effect of GIP, glucagon, or FSK (Figures 2H–2J). Similar results were obtained using wild-type male islets (Figures S2I–S2L).
Figure 2. DHT amplifies GLP-1-stimulated cAMP production at the plasma membrane and endosomes.

(A–E) 832/3 cells were infected with adenovirus harboring the FRET Epac2 camps probe and treated with DHT (10 nM) in the presence of (A) GLP-1 (10 nM), (B) GIP (100 nM), (C) glucagon (20 nM), and (D) forskolin (FSK, 10 μM) starting at the indicated time (Tx arrow, 5 min). cAMP production was monitored in real time from live cells at single-cell resolution. The same control trace is shown in (A) to (D) for all experiments. Traces represent mean ± SE from all cells imaged over six independent replicates (i.e., 180–240 cells per state). (E) Summary graph showing amplitude of cAMP responses from (A) to (C) of six independent experiments, each with 30–40 cells/treatment imaged at single-cell resolution.
(F) Representative pictures from (A) to (E). Scale bar, 20 μm.
(G–J) GSIS in static incubation in 832/3 cells treated with vehicle, DHT (10 nM) in the presence of (G) GLP-1 (10 nM), (H) GIP (100 nM), (I) glucagon (20 nM), and (J) forskolin (100 nM) for 40 min. Values represent the mean ± SE of n = 3 independent wells and 3–8 independent experiments.
(K) INS1 832/3 cells were transfected with GLP-1R-GFP (green) and FLAG-AR (red) and treated with vehicle, DHT (10 nM), GLP-1 (10 nM), and DHT + GLP-1 for 15 min. Receptor localization was assessed by immunofluorescence.
(L–Q) Chinese hamster ovarian (CHO-K1) cells were transfected with cAMP biosensors, (L) PM-TEpacVV (plasma membrane localized) or (O) Endo-TEpacVV (endosomally localized), and images were captured by confocal microscopy 24 h after transfection. Note peripheral distribution for the plasma membrane biosensor and punctate pattern for the endosomal biosensor. Scale bars, 10 μm. (M, N, P, and Q) 832/3 cells were transfected with PM-TEpacVV or Endo-TEpacVV plasmids and treated with DHT (10 nM), GLP-1 (10 nM), and DHT + GLP-1 (n = 4 independent replicates, 55–103 cells). (M and P) cAMP production was monitored in real time from live cells imaged at single-cell resolution, with treatment starting at the indicated time (Tx arrow, 5 min). (N and Q) AUC of the cAMP peak between 5 and 12 min.
(R and S) GSIS was measured in static incubation in male human islet donors (10 islet equivalents [IEQ]/condition measured in 3–6 replicates and two human donors) treated with vehicle, DHT (10 nM) for 40 min in the presence or absence of (R) the PKA inhibitor H89 (10 mM) or (S) EPAC inhibitor ESI-09 (10 μM).
Values represent the mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
DHT amplifies GLP-1 production of cAMP at the plasma membrane and endosomes
We assessed whether ligand-activated AR and GLP-1R interact within the same cell compartment, as suggested by the selective effect of DHT on GLP-1-induced but not glucagon-or GIP-induced cAMP increases. We transfected INS1 832/3 cells with GLP-1R-GFP and FLAG-AR plasmids and studied receptor localization. At basal (vehicle), GLP-1R-GFP was localized to the plasma membrane (PM) and likely the Golgi compartments (due to receptor overexpression from a plasmid cytomegalovirus promoter). FLAG-AR was localized close to the PM and throughout the cytosol (Figure 2K). DHT treatment did not alter the location of FLAG-AR or GLP-1R-GFP. Following GLP-1 stimulation alone or in combination with DHT, a fraction of internalized GLP-1R-GFP came in proximity to an FLAG-AR fraction (Figures 2K and S3A). Following staining of endosomes with transferrin, we determined that stimulation with GLP-1 alone or combined with DHT triggered GLP-1R-GFP internalization to endosomes (Figure S3B) but did not noticeably co-localize with AR (Figure S3C).
Upon ligand binding, endosomal trafficking of GLP-1R promotes cAMP generation from tmAC, which is important for insulin exocytosis.21, The biased agonist exendin-phe1, which promotes PM retention of the GLP-1R, also increases cAMP production and GSIS.22, Therefore, both GLP-1R PM and endosomal cAMP production are important for GSIS. To determine whether DHT enhances GLP-1-induced cAMP production by promoting GLP-1R PM retention, we studied the effect of DHT in MIN6B1 cells stably expressing SNAP-tagged GLP-1R following treatment with either exendin-4 (which promotes GLP-1R internalization) or exendin-phe1 (which promotes GLP-1R PM retention).22, We did not detect any effect of DHT on GLP-1R trafficking with either exendin-4 or exendin-phe1 treatment (Figure S4A), suggesting that DHT does not alter GLP-1R PM trafficking but rather enhances GLP-1R signaling from PM or endosomes.
To determine whether DHT enhances cAMP production in these compartments, we generated cAMP sensors specific to the PM (TEpac2VV-PM), endosomes (TEpac2VV-Endo), or cytoplasm (control, Epac2-camps-VV). Using these sensors, we studied GLP-1 and DHT-induced subcellular production of cAMP in INS1 832/3 cells (Figures 2L–2Q and S4B). Compared with vehicle, both GLP-1 and DHT increased cAMP production at the PM (Figures 2M and 2N), endosomes (Figures 2P and 2Q), and cytoplasm (Figure S4B). We reasoned that if DHT amplifies GLP-1-induced cAMP at the PM and the endosomes, DHT should sensitize cAMP-dependent effectors in these compartments, namely, the protein kinase A (PKA) and the exchange protein activated by cAMP islet 2 (Epac2).23, Accordingly, DHT amplification of GSIS in cultured islets from male wild-type mouse (Figure S4C) and human donors (Figures 2R and 2S) was not observed following pharmacological inhibition of PKA or Epac2.
DHT amplifies GLP-1 production of cAMP and GSIS via soluble adenylate cyclase
Two distinct forms of AC generate cAMP: a family of tmACs and the soluble AC (ADCY10, sAC).24, Following GLP-1R activation, coupling with the stimulatory G protein, Gαs, activates tmAC, leading to rapid cAMP production. In contrast, sAC lacks membrane-spanning domains and is distributed throughout the cytoplasm, PM, and endosomes, leading to spatial distribution of cAMP.25, Unlike the tmAC-induced rapid rise in cAMP, sAC responds with a slow and sustained cAMP production in response to prolonged cues, such as bicarbonates. Glucose elicits a slow rise in cAMP via sAC, which participates in GSIS.26, We investigated the contribution of sAC to DHT and GLP-1 enhancement of cAMP production in INS1 832/3 cells using PM-and endosomal-specific cAMP probes. Consistent with and complementing data shown in Figure 2, at the PM, DHT prolonged GLP-1-stimulated cAMP production leading to a second cAMP peak (Figures 3A and 3B) without altering overall cAMP production (Figure S5A). At the endosomes, DHT triggered an earlier peak of GLP-1-stimulated cAMP compared with GLP-1 alone (Figures 3C and 3D) without altering overall cAMP production (Figure S5C). Thus, DHT prolongs GLP-1 action at the PM and promotes earlier onset of GLP-1 action at the endosomes. Prior DHT exposure decreased FSK-stimulated cAMP production at the PM and endosomes (Figures 3A, 3C, S5B, and S5D). Similarly, prior exposure to GLP-1 blunted FSK-stimulated cAMP on top of the steady-state cAMP response, consistent with tmAC desensitization by prior GLP-1 exposure (compare Figures 3A and 3C with Figures S5B and S5D). Surprisingly, the sAC-selective allosteric inhibitor LRE127, produced an early peak of cAMP in response to vehicle and DHT at the PM and endosomes (Figures 3E–3H). We interpret this finding as an off-target effect of LRE1, which also activates tmAC,27, in the context of sAC inhibition. Consistent with this possibility, LRE1 also promoted an early peak of GLP-1-mediated cAMP production at the PM and endosomes (Figures 3E–3H). Most importantly, at the PM, LRE1 decreased the late cAMP responses to DHT + GLP-1 (Figures 3I, 3J, and S5E). In addition, at the endosomes, LRE1 blunted the early cAMP response to DHT + GLP-1 (Figures 3K and 3L). Thus, the amplifying effects of DHT upon GLP-1-stimulated cAMP generation at the PM and endosomes are mediated via sAC. We examined the consequence of sAC inhibition on DHT-enhanced GSIS in cultured male human islets using pharmacological inhibitors. KH-728, and LRE1 did not impair GSIS (Figures 3M and 3N). However, DHT potentiation of GSIS was not observed in the presence of KH7 or LRE1 (Figures 3M and 3N). Finally, we used the highly selective sAC inhibitor TDI-10229, which does not affect tmAC activity.29, TDI-10229 used at a concentration of 10 μM inhibited GSIS (Figure S5J). However, at 5 μM, TDI-10229 showed no effect on GSIS but inhibited DHT potentiation of GSIS (Figure 3O). Thus, DHT stimulates cAMP production and enhances islet GSIS from human β cells via sAC.
Figure 3. DHT amplifies GLP-1-stimulated cAMP production at the plasma membrane and endosomes via sAC.

(A–L) 832/3 cells were transfected with PM-TEpacVV or Endo-TEpacVV plasmids and treated with DHT (10 nM), GLP-1 (10 nM), and DHT + GLP-1 in the presence or absence of sAC inhibitor LRE-1 (5 μM). Forskolin (FSK, 10 μM) was added at 15 min. Data represent n = 4 independent replicates of 55–103 cells. (A, E, and I) cAMP production was monitored in real time at the PM or ENDO (C, G, and K) from live cells imaged at single-cell resolution, with treatment starting at the indicated time (Tx arrow, 5 min). (B and J) AUC of second GLP-1 peak of cAMP at PM from (A) and (I). (F and H) AUC of LRE1 early peak of cAMP at PM and ENDO from (E) and (G). (D and L) AUC of GLP-1 early peak of cAMP at ENDO from (C) and (K).
(M–O) Male human islets treated with vehicle, DHT (10 nM) overnight and again for the duration of the experiment in the presence of absence of the sAC inhibitors KH7 (1 μM) (M), LRE-1 (10 μM) (N), and TDI-10229 (5 μM) (O). Data represent 10 IEQ/condition measured in triplicate of n = 3–4 donors.
Values represent the mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
DHT activates sAC via glucose metabolism-induced bicarbonate production
Since sAC is activated by bicarbonates (HCO3−), we examined the possibility that DHT activates sAC by increasing the production of HCO3−. The product of glucose metabolism via glycolysis and the tricarboxylic acid (TCA) cycle is CO2, which is instantly converted to HCO3− by carbonic anhydrase (CA). We hypothesized that if DHT increases GSIS via sAC, it must promote CO2 conversion to HCO3− by CA. Consistent with this possibility, in cultured male human islets the CA inhibitor acetazolamide (AZZ) blunted DHT enhancement of GSIS (Figure 4A). The family of Na+/HCO3− co-transporters (NBCs) and the cystic fibrosis transmembrane regulator (CFTR), an anion channel, play fundamental roles in intracellular pH homeostasis by controlling HCO3− absorption and secretion.30, We reasoned that if DHT stimulates the production of HCO3−, it may require NBCs and CFTR to maintain pH homeostasis. Cultured human islets incubated with either a selective CFTR channel blocker (CFTRinh-172)31, or a selective NBC inhibitor (S0859)32, exhibited no significant alteration in GSIS (Figures 4B and 4C). However, in the presence of either S0859 or CFTRinh-172, DHT failed to enhance GSIS, suggesting that DHT activation of sAC requires NBCs and CFTR.
Figure 4. DHT increases glucose metabolism and bicarbonate production.

(A–C) GSIS was measured via static incubation in male human islets (n = 3–7 donors, 10 IEQ/condition measured in triplicate) cultured for 24 h with vehicle or DHT (10 nM) and then for 40 min in the presence or absence of (A) a carbonic anhydrase inhibitor AZZ (50 μM), (B) CFTR inhibitor CFTRi (500 μM), or (C) NA+/HCO3− co-transporter inhibitor S0859 (2 μM).
(D) Schematic representation of bioenergetics, metabolomics, proteomics, and transcriptomics workflow.
(E–K) Oxygen consumption rate (OCR) was measured in male C57/BL6 mouse islets treated with vehicle or DHT (10 nM) in a Seahorse analyzer during a mitochondrial stress test using oligomycin, FCCP, and antimycin A/rotenone at the indicated times. Islets were pooled from 10 mice with 150 islets per well in 10 wells for each condition and n = 3 experiments. (E) Line graph of OCR showing the key parameters of mitochondrial function measured in (F) to (K).
(L–P) Extracellular acidification rate (ECAR) was measured in a Seahorse analyzer during (L–N) a glycolytic stress test using 2-DG and (O and P) a mitochondrial stress test using antimycin A/rotenone at the indicated times. Islets were pooled from 5 mice with 150 islets per well in 5 wells for each condition and n = 2 experiments.
(Q) Human islets (n = 3 donors, 1,200 IEQ/condition) were treated with DHT for 5 min, 30 min, and 18 h, compared with control untreated islets, and studied by reverse-phase protein array (RPPA). Normalized values from the three individual donors were averaged to generate an average Z score and map proteins in the heatmap. *p < 0.05 (t test) and #p < 0.05 (Fisher’s LSD test) compared with control.
(R) Human islets (n = 3 donors, 500 IEQ/condition) were treated with DHT at 16.7 mM glucose for 30 min in the presence of [13C]glucose, and metabolites were measured by LC-MS. Results represent the mean ± SE of the ratio 13C/12C metabolite abundance.
(S) Schematic representation of the pathway, enzymes, and metabolites described in (A) to (R).
Values represent the mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To elucidate the mechanisms by which DHT enhances HCO3− production, we combined mouse islet bioenergetics, human islet proteomics, metabolomics, and single-cell transcriptomics (scRNA-seq) (Figure 4D). In male wild-type mouse islets, DHT did not increase basal, glucose-stimulated, or maximal respiration (Figures 4E–4H) and produced no significant effect on ATP-mediated respiration after oligomycin addition (an inhibitor of ATP synthase, complex V of the electron transport chain [ETC]) (Figures 4E and 4I). However, after addition of rotenone/antimycin A (inhibitors of complex I and III, blocking ETC-mediated respiration), DHT-treated mouse islets retained a higher oxygen consumption rate than vehicle-treated islets, suggesting that DHT increases respiration via an ETC-independent mechanism (Figures 4E and 4J). DHT produced a trend toward decreased coupling efficiency (Figure 4K). Extracellular acidification rate (ECAR) reflects the combined H+ acidification from lactate (glycolysis) and CO2 (TCA cycle). DHT produced a greater acidification than vehicle at high glucose (Figures 4L and 4M). Notably, compared with vehicle, DHT-treated mouse islets retained higher non-glycolytic acidification following addition of 2-deoxyglucose (2-DG), an inhibitor of glycolysis (Figures 4L and 4N), and produced a trend toward a greater fall in ECAR following rotenone/antimycin A, suggesting increased mitochondrial acidification (Figures 4O and 4P). In a similar experiment, DHT-treated human islets exhibited increased non-glycolytic (Figures S5K–S5M) and mitochondrial acidification (Figures S5N and S5O), both of which were blunted in the presence of AZZ, confirming that DHT-induced mitochondrial acidification requires conversion of CO2 to HCO3−. Thus, DHT increases islet glucose metabolism, leading to increased mitochondrial production of H+ (and HCO3−).
We examined DHT-stimulated glucose metabolism in male human islets by functional proteomics using a reverse-phase protein array (RPPA). Under these conditions, DHT increased the expression of enolase, a glycolytic enzyme, and mitochondrial pyruvate dehydrogenase (PDH), which converts pyruvate to acetyl-CoA, producing CO2, concomitant to a decrease in lactate dehydrogenase (Figure 4Q). DHT decreased cytochrome c oxidase (ETC complex IV) and produced a late increase in ATP synthase (ETC complex V). To assess fluxes through glycolysis and the TCA cycle, we examined the fate of DHT-stimulated [13C]glucose in human islets. Consistent with proteomics results, DHT increased the 13C/12C ratio for glycolytic intermediates fructose 6-phosphate (F6-P), F1,6-P, and phosphoglycerate, as well as acetyl-CoA, without changes in lactate (Figure 4R). DHT decreased the 13C/12C ratio in citrate and increased it in glutamate, indicating increased citrate conversion to α-ketoglutarate (AKG) followed by AKG conversion to glutamate. We observed no change in 13C incorporation into succinate or malate or in ATP levels. Thus, DHT increases islet glycolysis and mitochondrial TCA production of CO2 (likely via PDH), which is converted to HCO3-by CA to activate sAC. Surprisingly, DHT does not increase ETC-dependent respiration or ATP production (Figure 4S).
We analyzed DHT’s effects on the human islet cell transcriptome using scRNA-seq. We identified the four main endocrine cell subtypes, namely α, β, γ, and pancreatic polypeptide (PPY) cells (Figure 5A). Eliminated doublet-cell clusters were annotated based on cell-specific markers (Figures 5C and S6A–S6D). We focused on α and β cells, the two prominent human islet cell types. Unbiased clustering of insulin-positive and glucagon-positive cells revealed the presence of two main b cell subpopulations (β1 and β2), two main α cell subpopulations (α1 and α2), and a population of transendocrine cells (co-expressing low insulin and glucagon) (Figures 5A and 5C). Among islet cells, AR was mostly expressed in α and β cells as well as transendocrine cells (Figure 5D). Notably, AR and GLP-1R were co-expressed in β1, α1, and transendocrine cells (Figures 5B and 5D). In β cells, and consistent with data presented in Figure 4, DHT increased expression of glycolytic genes (GK, TPI1, ENO2, PGK1, and PKM) and decreased expression of mitochondrial genes of the ETC complexes I, III, IV, and V (Figures 5E and 5G). Gene ontology (GO), used to evaluate the function of differentially expressed genes (DEGs), revealed that in β cells, DHT upregulates genes enriched in pathways involved in translation, glycolysis, and response to hypoxia and downregulates genes enriched in pathways involved in ETC (Figures 5F and 5H). In α cells, analysis of GO or DEGs also revealed DHT increasing or decreasing expression of mitochondrial ETC genes (Figures 5I and 5L).
Figure 5. DHT-stimulated human islet a and b cell transcriptome involved in glycolysis and TCA cycle.

(A) Human islets (750 IEQ/condition) were cultured for 24 h with vehicle and DHT and single-cell RNA sequencing. Data represent a multiple-donor integrated multidimensional uniform manifold approximation projection (UMAP) plot. The plot shows 28,809 single cells where each dot represents the transcriptome of a single cell. Clusters of characteristic cell types on the basis of their transcriptional footprint are color coded and labeled.
(B) UMAP plot of data classified in (A) showing the expression of GLP-1R transcripts across individual islet cells.
(C) Dot plot showing the percentage of cells across each cluster expressing cell-type-specific genes.
(D) UMAP plot of data classified in (A) showing the expression of AR transcripts across individual islet cells.
(E) Volcano plot showing differentially expressed genes (DEGs) for DHT vs. vehicle treatment in the Beta 1 cluster. Blue, genes with FDR < 0.05 and log2 fold change < −0.137; red, genes with FDR < 0.05 and log2 fold change > 0.137; black, genes below both thresholds.
(F) Bar plot showing enriched gene ontology (GO) pathway terms for the DEGs shown in (E).
(G–L) (G) Volcano plot showing DEGs for DHT vs. vehicle treatment in the Beta 2 cluster, as in (E). (H) Bar plot showing enriched GO pathway terms for the DEGs shown in (G). (I) Volcano plot showing DEGs for DHT vs. vehicle treatment in the Alpha 1 cluster, as in (E). (J) Bar plot showing enriched GO pathway terms for the DEGs shown in (I). (K) Volcano plot showing DEGs for DHT vs. vehicle treatment in the Alpha 2 cluster, as in (E). (L) Bar plot showing enriched GO pathway terms for the DEGs shown in (K).
(M) Schematic representation of the effect of DHT on glycolysis and TCA cycle genes and pathways described in (E) to (L).
Data represent n = 3 donors. Values represent the mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To identify DHT-stimulated AR-interacting proteins, we performed immunoprecipitation (IP)-based proteomics in INS1 832/3 cells (Figure 6A). Consistent with omics data reported above (Figures 4O and 4P), GO enrichment analysis using Enrichr revealed DHT-induced AR interactions with mitochondrial proteins (Figure 6B). We integrated the GO analysis with Ingenuity Pathway Analysis (IPA) and a protein-protein interaction network enrichment prediction via STRING and constructed a DHT-stimulated AR interactome (Figure 6C). DHT promoted AR interaction with mitochondrial protein complexes involved in TCA cycling including PDH, isocitrate dehydrogenase (which catalyzes the decarboxylation of isocitrate, producing AKG and CO2), and NADH dehydrogenase (ETC complex I).
Figure 6. DHT and GLP-1 collaborate to promote GLP-1R and AR recruitment of Gαs.

(A) Schematic representation of immunoprecipitation-based proteomics analysis workflow.
(B–E) IP-based proteomics analysis was performed in INS1 832/3 cells treated with DHT (10 nM) or vehicle overnight followed by DHT in the presence or absence of GLP-1 (10 nM) for 5 min. (B) GO enrichment analysis of DHT-treated cells using Enrichr. Bar plot shows DHT-stimulated AR interactions with proteins over- or under-represented in the indicated pathways. (C) DHT-stimulated AR interactome (left) and corresponding heatmap (right) were constructed by integrating the GO, IPA, and STRING analyses. (D) GO enrichment analysis of DHT + GLP-1-treated cells using Enrichr. Bar plots shows DHT-and GLP-1-stimulated AR interactions with proteins over-or under-represented in the indicated pathways. (E) DHT -and GLP-1-stimulated AR interactome (left) and corresponding heatmap (right) were constructed as in (C).
(F) Schematic representation of the concept of SmBiT and LgBiT interactions resulting in luminescence.
(G) Luminescence of INS1 832/3 cells transfected with plasmids for SmBiT-AR, LgBiT-GLP-1R, or empty vector SmBiT-Empty and treated with DHT (10 nM) GLP-1 (10 nM), or both, for 20 min.
(H) INS1 832/3 cells were transfected with SmBiT-GLP-1R and LgBiT-Gαs. After basal measurements for 6 min, cells were treated (Tx) with DHT, GLP-1, or both for 22 min. Measurements were taken every 2 min. Line graph shows Gαs recruitment to GLP-1R.
(I) AUC for (H).
(J) INS1 832/3 cells were transfected with SmBiT-AR and LgBiT-Gαs and treated with DHT (10 nM) or GLP-1 (10 nM) as in (H). Line graph shows Gαs recruitment to GLP-1R.
(K) AUC for (J).
(L) GSIS was measured in static incubation in male human islets (5 donors, 10 IEQ/condition measured in triplicate) treated with vehicle, DHT (10 nM) for 40 min in the presence of absence of the tmAC inhibitor ddAdo (1 μM).
In (G) to (K), AR and GLP-1R interaction assays were performed in n = 12 biological replicates. Gαs recruitment assays were performed in n = 6 biological replicates. Values represent the mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Thus, experiments described in Figures 4–6 suggest that DHT programs an increase in islet glycolysis and mitochondrial TCA production of CO2 (likely via PDH and isocitrate dehydrogenase [IDH]), which is converted to HCO3− by CA to activate sAC (Figures 4S and 5M).
DHT and GLP-1 collaborate to promote GLP-1R and AR recruitment of Gαs
DHT promoted AR interaction with protein complexes involved in cAMP signaling, including Gα proteins, and cAMP-dependent PKA (Figure 6C). In addition, GLP-1 promoted a fraction of GLP-1R to internalize to endosomes to come into proximity with a pool of DHT-activated AR (Figures 2J and S3). We used the NanoBiT split-luciferase system to investigate GLP-1R and AR interactions. Since ligand-activated GLP-1R couples with the stimulatory G protein, Gαs, to activate tmAC, we also assessed GLP-1R and AR association with Gαs. GLP-1R and AR were cloned into LgBiT and SmBiT constructs, respectively and expressed in INS1 832/3 cells. Under these conditions, if the SmBiT-AR and LgBiT-GLP-1R come into proximity, either via direct interaction or as part of a larger protein complex, the two parts of NanoBiT assemble, which increases the luciferase signal (Figure 6F). Co-expression of SmBiT-AR with LgBiT-GLP-1R increased the luminescence compared with co-expression of LgBiT-GLP-1R SmBiT-Empty (used as a control), confirming that GLP-1 and AR are in proximity. DHT did not increase SmBiT-AR and LgBiT-GLP-1R proximity but GLP-1 (with or without DHT) increased proximity for SmBiT-AR with LgBiT-GLP-1R compared with vehicle and DHT alone, suggesting that GLP-1 enhances AR and GLP-1R interaction (directly or as part of a complex) (Figure 6G). To test Gαs recruitment to GLP-1R or AR, we cloned GLP-1R and AR into SmBiT and Gαs into LgBiT. As expected, upon GLP-1 stimulation, in the presence or absence of DHT, LgBiT-Gαs was recruited to SmBiT-GLP-1R (Figures 6H and 6I). DHT progressively increased LgBiT-Gαs recruitment to SmBiT-GLP-1R, although it did not reach significance. Notably, DHT and GLP-1 alone or in combination increased LgBiT-Gαs recruitment to SmBiT-AR (Figures 6J and 6K). In addition, GLP-1 and DHT seemed to further amplify this recruitment. No significant LgBiT-Gαs recruitment to SmBiT-Empty control was observed (Figures S7B and S7C). Thus, GLP-1 stimulation increases GLP-1R and AR proximity. In addition, DHT and GLP-1 collaborate to enhance Gαs recruitment to GLP-1R and AR complexes.
Since Gαs recruitment to GPCRs activates tmAC, we assessed the contribution of tmAC to DHT-enhanced GSIS in cultured male human islets using the tmAC-selective inhibitor 2′,5′-dideoxyadenosine (ddAdo). When used alone, ddAdo did not impair GSIS (Figure 6L). However, DHT potentiation of GSIS was not observed in the presence of ddAdo, suggesting that DHT insulinotropic action also requires tmAC.
DHT-stimulated AR pathways promoting glucose-stimulated actin remodeling and exocytosis
The analysis of DHT-stimulated human β cell transcriptome described above revealed DEGs in pathways related to secretory granules, focal adhesion, vesicles, exocytosis, cytoskeletal trafficking, microtubules, and protein localization to membrane (Figures 5F and 5H), suggesting that DHT promotes cytoskeleton remodeling in relation to insulin granule exocytosis. Consistent with this possibility, the IP-based proteomics analysis in INS-1 cells revealed DHT-stimulated AR interactions with gelsolin, twinfilin-1, and F-actin-capping proteins, all promoting actin depolymerization33, (Figures 6B and 6C). DHT stimulated AR interactions with proteins involved in insulin granule maturation, including the lysosomal vacuolar-type ATPase (V-ATPase), which promotes insulin granule acidification34, and the prohormone convertase 2 (PC2) that converts proinsulin to insulin (Figure 6C). Notably, in the presence of GLP-1, DHT promoted AR interaction with proteins involved in vesicular trafficking, including microtubule motor proteins (myosin 1E, 1C, Va, and KIF5)35, and small GTPases involved in actin remodeling and second-phase GSIS36, such as guanine nucleotide exchange factors (GEFs) RhoGEF1, RhoGEF2, RhoGEF4, and EPAC2, GTPase-activating proteins (GAPs) RasGAP1, RhoGAP17, and USP6NL, and the GTPase dynamin2 (Figures 6D and 6E). Thus, DHT alone promotes AR interaction with proteins promoting glucose-stimulated actin depolymerization, whereas in the presence of GLP-1, DHT stimulates AR interaction with proteins involved in latter steps of glucose-stimulated insulin granule trafficking. DHT alone or in the presence of GLP-1 promoted AR dissociation from proteins favoring actin or microtubule polymerization (Figure S7A). Descriptions of all proteins are provided in Data S1.
Consistent with GLP-1 and DHT promoting AR interaction with tyrosine kinases (Figure 6E), the analysis of DHT-stimulated phosphoproteins in male human islets by ontologies revealed enrichment in tyrosine kinase activation pathways (Figure S8A). At the individual phosphoprotein level, DHT rapidly increased the tyrosine phosphorylation of FAK, the protein-tyrosine kinase 2β (PYK2), and SRC (Figures 7A and S8C). We assessed the role of FAK and PYK2 in DHT amplification of GSIS in male human islets following pharmacological inhibition using two FAK family kinase selective inhibitors, PF43139637, and PF562271,38, which inhibit both FAK and PYK2. While the inhibitors used alone had no effect on GSIS, inhibition of FAK/PYK2 blunted the effect of DHT (Figure 7B). Notably, the potent and FAK-selective inhibitor PF57322839, (Figure 7C), but not the PYK2-selective inhibitor PF461843337, (Figure 7D), abolished the stimulatory effect of DHT on GSIS. Thus, FAK is instrumental in DHT insulinotropic actions in human islets. DHT stimulation of SRC phosphorylation was confirmed by western blotting, and, accordingly, the SRC-selective inhibitor PP240, blunted DHT enhancement of GSIS (Figures 7E and S8D). In prostate cells, androgen activation of SRC activates PI3K,41, and we observed DHT-induced phosphorylation of phosphoinositide-dependent kinase-1 (PDK-1), a PI3K target (Figures 7A and S8C). We assessed the consequence of PI3K inhibition on DHT-induced GSIS in male human islets using two highly selective class IA PI3K inhibitors, GDC-094142, and BMK-120.43, Although GDC-0941 and BMK-120 used alone decreased GSIS, the ability of DHT to amplify GSIS was further decreased by both PI3K inhibitors, suggesting that PI3K is involved in DHT insulinotropic action in human islets (Figure 7F).
Figure 7. AR-activated pathway promoting glucose-stimulated actin remodeling and exocytosis.

(A) Human islets (n = 3 donors, 10 IEQ/condition) were treated with DHT for 5 min, 30 min, and 18 h, compared with control untreated islets, and studied by RPPA. Normalized values from the three individual donors were averaged to generate an average Z score and map phosphoproteins in the heatmap. *p < 0.05 (t test). (B–H) GSIS was measured in static incubation in male human islets (10 IEQ/condition measured in triplicate) cultured for 24 h with vehicle or DHT (10 nM) followed by 40 min with DHT in the presence or absence of the following inhibitors: (B) dual FAK and PYK2 inhibitors PF431396 (15 nM) and PF562271 (15 nM) (n = 3–5 donors), (C) selective FAK inhibitor PF573228 (10 nM) (n = 3–7 donors), (D) selective PYK2 inhibitor PF4618433 (1 μM) (n = 3–7 donors), (E) SRC kinase inhibitor PP2 (1 μM) (n = 3–5 donors), (F) PI3K inhibitors BKM120 (1 μM) and GDC-0941 (1 μM) (n = 3–4 donors), and (H) mTORC2 inhibitor JR-AB2–011 (5 μM) (n = 5 donors). (G) GSIS was measured in static incubation in islets treated with vehicle or DHT (10 nM) for 40 min from male control (RIP-Cre), βRAHET, and βRAKO mice (n = 7 mice, with each condition measured in triplicate, n = 10 islets per replicate),.
(I) Representative transmission electron micrographs of human islet β cell treated with vehicle (left) and DHT (right). Insets highlight insulin granules, where red arrows show multigranules. Scale bar, 1 μm.
(J) (Top) Quantification of insulin granule size. (Bottom) Quantification of percentage of insulin multigranules over total insulin granules/cell across n = 3 donors (I and J) and 15 cells.
(K) Plasma membrane content of VAMP2 normalized for STX4 in INS-1 832/3 cells treated with DHT (10 nM).
(L and M) Insulin exocytosis measured by change in capacitance (fF) from human islet β cells with DHT (10 nM) applied via patch pipette in the presence of cAMP (100 μM) (n = 22 cells per treatment). Changes in fF are normalized to cell size (pF).
(N and O) Calcium current measured in human islet b cells with DHT (10 nM) applied via patch pipette in the presence of cAMP (100 μM) (n = 22 cells per treatment). Calcium current (ICa), calcium influx (QCa), and calcium influx (pC) are normalized to cell size (pF).
(P) Schematic representation of proposed mechanism. (1 and 2) DHT-activated AR pools in the PM vicinity, mitochondria (mito), and nucleus program glycolysis and TCA cycle, increasing CO2 production, which is converted to HCO3− via carbonic anhydrase (CA). HCO3− activates the sAC at (3) the PM and (4) endo while CFTR and NBC promote HCO3− efflux to maintain pH homeostasis. In parallel, DHT-activated AR pools at the (3) PM and (4) endo collaborate with ligand-activated GLP-1R to promote Gαs recruitment to AR and GLP-1R complexes and activate tmAC. Together, this results in DHT enhancing GLP-1-mediated cAMP production at the PM and endo, which (5) activates cAMP-dependent effectors PKA and EPAC2 to promote insulin granule exocytosis. (6) DHT-activated AR in the PM vicinity activates a signaling cascade including FAK/SRC/PI3K/mTORC2 that further enhances insulin granule exocytosis. AR-DHT may also promote actin remodeling via gelsolin (GSN).
Values represent mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The mammalian target of rapamycin (mTOR) exists in two distinct complexes: mTORC1, containing the regulatory associated protein of mTOR (raptor), and mTORC2, containing the rapamycin-insensitive companion of mTOR (rictor) (Figures 7A and S8C). FAK44, and PI3K promote the activation of mTORC2, and we observed phosphorylation of rictor and its targets, protein kinase C (PKC) and Akt (also a PI3K target) (Figures 7A and S8C). We also observed decreased phosphorylation of the mTORC1 targets, ribosomal protein S6 kinase (p70S6K) and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1), together suggesting that DHT differentially activates mTORC1 and mTORC2. In male human islets, the ability of DHT to enhance GSIS was blocked by rapamycin, an mTOR inhibitor (Figure S8E). In a parallel experiment, we used cultured islets from male transgenic mice overexpressing one copy of a kinase-dead mTOR (KD-mTOR) exclusively in b cells that exhibit a deficit in GSIS.45, DHT failed to rescue the deficient GSIS in KD-mTOR islets (Figure S8F). Islets from male mice with heterozygous deletion of raptor (βRAHET) exhibited a blunted GSIS in static incubation, consistent with the known importance of mTORC1 in GSIS (Figure 7G). However, DHT not only restored GSIS in cultured male βRAHET islets but also amplified GSIS compared with vehicle. In contrast, and consistent with the RPPA phosphorylation pattern, islets from male mice harboring a homozygous deletion of rictor (βRIKO) showed no abnormal GSIS at baseline, and DHT failed to amplify GSIS in βRIKO islets compared with controls (Figure 7G). Consistent with results in the mouse, DHT failed to amplify GSIS in male human islets exposed to JR-AB2–011, a small-molecule-selective inhibitor of mTORC246, (Figure 7H). Thus, in male mouse and human islets, DHT enhances GSIS via mTORC2. Consistent with DHT increasing cAMP and the requirement of PKA in DHT-stimulated GSIS, we also observed DHT-induced phosphorylation of cAMP-response element binding protein, a PKA target (Figures 7A and S8C). Additional protein expression altered by DHT is shown on Figure S8B.
DHT enhances GLP-1-stimulated insulin granule docking to the PM and exocytosis
We finally assessed the ability of DHT to enhance GLP-1-stimulated insulin granule docking to the PM and to promote insulin granule exocytosis. We first examined the effect of DHT on human β cell insulin granule ultrastructure. DHT increased the size of insulin granules as well as the frequency of multivesicular granules containing several intracellular structures (Figures 7I and 7J). This suggests that DHT promotes multivesicular exocytosis, which increases the release of insulin granule equivalents.47, Upon glucose stimulation, VAMP2-bound insulin granules translocate to the PM, where VAMP2 fuses with the PM-localized protein STX4.48, In INS-1 cells, compared with GLP-1 alone DHT enhanced GLP-1-mediated VAMP2 accumulation in PM fractions, consistent with DHT enhancing GLP-1-stimulated insulin granule docking to the PM (Figure 7K). We measured the effect of DHT on the capacitance of dispersed primary human male β cells. When intracellular cAMP was clamped, DHT increased depolarization-induced granule exocytosis (Figures 7L and 7M), independent of calcium influx (Figures 7N and 7O), confirming that DHT potentiates cAMP-stimulated insulin granule exocytosis.
Together, experiments described in Figures 5, 6, and 7 suggest that (1) in the presence of GLP-1, DHT promotes AR interaction with protein complexes involved in glucose-stimulated actin remodeling and insulin granule trafficking and increases GLP-1-mediated insulin granule docking to the PM, and (2) DHT engages an FAK-SRC-PI3K-mTORC2 cascade to enhance GSIS and potentiates cAMP-stimulated insulin granule exocytosis in human β cells.
DISCUSSION
This study provides an architecture of AR targets enhancing GLP-1R insulinotropic action in male rodent and human β cells. We identified several interdependent pathways summarized in Figure 7P.
Activation of sAC and tmAC
DHT activation of the β cell AR enhances cAMP production and GSIS, which requires GLP-1 activation of the GLP-1R. DHT prolongs GLP-1-mediated cAMP generation at the PM and accelerates GLP-1-mediated cAMP generation at the endosomes. Since agonist-mediated GLP-1R retention at the PM increases cAMP production and GSIS,22, and internalization of the GLP-1R in endosomes allows the cAMP signal to be prolonged at the endosomes,21, the combined actions of DHT and GLP-1 in these microdomains likely sensitizes the pool of insulin granules for exocytosis. First, GLP-1 increases GLP-1R and AR proximity, and the combination of DHT and GLP-1 promotes Gαs recruitment to GLP-1R and AR. Thus, activation of a pool of AR in the vicinity of the PM may directly recruit Gαs 49, or enhance GLP-1R recruitment of Gαs, both of which are expected to activate tmAC and cAMP production at the PM and endosomes. Accordingly, DHT insulinotropic action in human islets is absent following tmAC inhibition. Second, DHT potentiation of GLP-1-mediated cAMP production at the PM and endosomes, and enhancement of GSIS from human islets, require sAC activation. Our multidisciplinary approach reveals that DHT programs glucose metabolism toward mitochondrial CO2 production, which is converted to HCO3− by CA and is sensed by sAC at the PM and endosomes to produce cAMP. In the presence of GLP-1, DHT promoted AR interaction with protein complexes involved in cAMP compartmentalization, including phosphodies-terases that restrict the cAMP signal to the PM and endosomes, the cAMP effector EPAC2, and AKAP that organizes the PKA signalosome to specific domains.50 Pharmacological inhibition of EPAC2 and PKA abolished DHT insulinotropic action in mouse and human islets.
The mechanism by which DHT enhances glucose oxidative flux may involve transcriptional and translational regulation of the glycolytic and TCA cycle machinery via different pools of membrane-associated, mitochondrial, and nuclear ARs. For example, DHT rapidly increases PDH recruitment to AR complexes and PDH expression, likely increasing PDH activity, as suggested by DHT-increasing [13C]glucose metabolism in [13C] acetyl-CoA. Surprisingly, DHT increases glucose metabolism to acetyl-CoA without increasing the activity of the mitochondrial ETC. A possible explanation is that to maintain the TCA cycle CO2 production (via PDH and IDH activity) without overwhelming the ETC with a surplus of electrons, DHT action on a mitochondrial AR diverts a portion of the TCA cycle carbons away from full oxidation via AKG conversion to glutamate.51
DHT insulinotropic action in human islets requires NBCs and CFTR. Notably, NBCs and CFTR are activated by cAMP,52, and CFTR requires activation by sAC.53, Thus, DHT, by stimulating HCO3− production, may require NBCs and CFTR activation via sAC as a feedback control of local pH homeostasis. Activation of sAC is also essential for the localization of the V-ATPase to lysosomes and their acidification.54, In β cells, V-ATPase promotes insulin granule acidification, which increases PC2 activity.34 Notably, V-ATPase and PC2 are enriched in the DHT-stimulated AR interactome, suggesting that DHT activation of sAC promotes insulin granule maturation, which is consistent with DHT supporting the formation of larger and multivesicular insulin granules in human β cells.
AR activation influences GLP-1-stimulated cytoskeleton remodeling and insulin exocytosis
Activation of AR in β cells enhances glucose-stimulated first-and second-phase insulin release from male mouse12, and human islets. The first phase of insulin secretion corresponds to the rapid fusion of a readily releasable pool (RRP) of granules docked at the PM; the second phase occurs by replenishment of the RRP by an intracellular pool of granules.33 Filamentous actin (F-actin) acts as a polymerized web preventing the reserve pool of granules access to the RRP. Glucose induces a localized conversion of F-actin to monomeric globular actin (G-actin), allowing granule movement through the actin network and their recruitment to the PM.
DHT binding to AR is involved in glucose-stimulated actin remodeling. First, in INS1 cells, DHT promoted AR interaction with F-actin-depolymerizing proteins, including gelsolin. Gelsolin associates with syntaxin 4 to promote insulin granule exocytosis,55, and mice lacking gelsolin exhibit impaired actin remodeling and GSIS.56, DHT-induced AR interaction with gelsolin and capping proteins may prime the actin cytoskeleton to enable GLP-1-mediated insulin granule exocytosis. Second, ontologies used to evaluate the function of DEGs in human β cells revealed that DHT promotes enrichment in focal adhesion, cytoskeletal trafficking, and exocytosis pathways. Finally, in human islets we identified a signaling cascade whereby DHT activation of focal adhesions and culmination in mTORC2 activation is instrumental to GSIS. In β cells, glucose activation of FAK and SRC promotes actin remodeling, allowing docking of insulin granules to the PM.55,57,58, In various cells, FAK and SRC activate mTORC2 to promote cytoskeleton remodeling.44,59, Notably, cAMP activates mTORC2,60,61, and mTORC2 activity is localized at the plasma membrane and endosomes,62, where DHT increases cAMP. Thus, in the presence of islet-derived GLP-1, both FAK/SRC and sAC-generated cAMP pathways may be required for DHT to activate mTORC2, thus amplifying glucose-stimulated actin remodeling toward insulin granule exocytosis. Accordingly, in INS1 cells, in the presence of GLP-1, DHT promotes AR interaction with small GTPases GαPs and GEFs involved in actin remodeling and second-phase GSIS,63,64, as well as myosin and kinesin motor proteins that favor vesicular transport.35 These findings suggest that DHT influences GLP-1-and glucose-stimulated cytoskeleton remodeling and exocytosis in β cells. Indeed, in INS-1 cells, DHT enhanced GLP-1-mediated VAMP2 accumulation in PM fractions, consistent with DHT enhancing GLP-1-stimulated insulin granules docking to the PM.
Testosterone deficiency increases T2D risk in older men. This study warrants new clinical trials to assess the benefits of combining testosterone and GLP-1 receptor agonists in androgen-deficient men with T2D. The AR is a well-characterized drug target, and selective androgen receptor modulators (SARMs) are in development to prevent age-related frailty by providing androgen anabolic activity in muscle and bone but without androgen stimulation of the prostate.65 The design of SARMs with AR agonistic action in β cells represents a therapeutic avenue to prevent T2D in older men and those who are undergoing androgen depletion for prostate cancer.
Limitations of the study
The analysis of AR-interacting proteins in IP-based proteomics was not designed to provide causality in pathways studied. Thus, the AR interactome was not confirmed by co-IP and does not inform on the functional significance of AR-interacting proteins to DHT-enhanced GSIS. The time course of DHT and GLP-1-stimulated AR association with Gαs in the IP-based proteomics (limited to 5 min) may underestimate the actual kinetics of their interactions observed in the NanoBiT split-luciferase assay (ranging from 0 to 30 min). The RPPA was designed to investigate cancer biology and lacks antibodies to multiple pathways that are key to islet biology. In addition, given the heterogeneity of human islets, the small sample size used in the RPPA (n = 3) limits the power of our analysis.
In conclusion, this study identifies AR targets that enhance GLP-1-stimulated insulin exocytosis in male human and rodent β cells through (1) enhanced cAMP production at the PM and endosomes via activation of the bicarbonate sensor sAC, Gαs recruitment to GLP-1R, and AR to activate tmAC, and (2) cooperation with effectors of cytoskeleton remodeling and the vesicular transport machinery.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Franck Mauvais-Jarvis (fmauvais@tulane.edu).
Materials availability
This study did not generate any new materials.
Data and code availability
Single cell RNA sequencing data has been deposited in the National Center for Biotechnology Information (NCBI) gene expression omnibus (GEO) with the accession number GEO: GSE201256, mass spectrometry data at EMBL-EBI Proteomics Identification database (PRIDE) with accession number PRIDE: PXD041592 and, metabolomics data at UCSD metabolomics Workbench/National Metabolomics Data Repository (NMDR) with accession number NMDR: PR001654. All data reported in this paper will be shared by the lead contact upon request.
A description of coding environments required to reproduce scRNAseq analysis in this paper are outlined in: https://github.com/FMJLabTulane/AR-DHT.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
To generate βARKOMIP mice we crossed mice carrying the AR gene with floxed exon 2 on their X chromosome (ARlox) with the Ins1-Cre/ERT (MIP-Cre+/−) transgenic mouse (The Jackson Laboratory). Generation and characterization of ARlox −/− have been described.66 We induced tamoxifen (tam; Sigma) inactivation of AR after puberty and following 4 weeks of tam treatment in silastic tubing, and all metabolic measures were taken after a 4-week waiting period. 10mm silastic laboratory tubing (Dow Corning) was filled with 15mg tam, capped with wooden applicator sticks, and sealed with silastic medical adhesive (Dow Corning). Because the MIP-CreERT transgenic mouse exhibits transgene-driven expression of human growth hormone,67 we validated the ARlox MIP-CreERT mice (without Tam injection) as controls of βARKOMIP.12 The βARKORIP mouse was generated by crossing ARlox with transgenic mice overexpressing the Cre recombinase under control of the RIP promoter (Ins2-creTg(Ins2−cre)25Mgn/J, Jackson Laboratory). The βGLP-1KORIP mouse was generated by crossing GLP-1Rlox (a kind gift from Dr. David A. D’Alessio of Duke University)68 with RIP-Cre. Since RIP-Cre transgenic mice exhibit mild impaired GSIS,12,69 we used the RIP-Cre as the control group. The generation of β-cell-specific kinase-dead mTOR (KD-mTOR) was described previously.45 To generate mice with heterozygous deletion of raptor (βRAHET)70 and mice with homozygous deletion of rictor (βRIKO),71 raptorfl/fl and rictortm1.1Mnh mice were crossed with Ins2-creTg(Ins2-cre)23Herr transgenic mice, which express Cre recombinase under the control of a rat insulin (Ins2) promoter (Rip-Cre). Littermates negative for the Rip-Cre transgene were used as controls. All mice were generated on a C57/BL6 background. All studies were performed with the approval of the Tulane University Animal Care and Use Committee in accordance with the NIH Guidelines.
Cell lines and cell culture
INS-1 832/3 cells, were cultured in phenol containing (and 24hours prior to an experiment) phenol red free RPMI medium (Gibco) containing 11mM glucose, supplemented with charcoal stripped FBS (10% v/v; Invitrogen), Sodium Pyruvate (1mM; Gibco), HEPES buffer (10mM; Invitrogen), GlutaMAX (2mM; Gibco), β-mercaptoethanol (50μM; Invitrogen) and, Penicillin-Streptomycin (1x; Gibco). MIN6B1 cells were cultured in DMEM (Invitrogen) containing glucose (4.5 g/L), supplemented with FBS (15% v/v; Invitrogen), β-mercaptoethanol (50μM; Invitrogen) and penicillin/streptomycin (1% v/v; Invitrogen). Cells were seeded in 24-well plates with glass coverslips using medium containing charcoal-stripped FBS (10% v/v). Both cell types were cultured at 37°C in a humidified incubator containing 5% CO2.
Human pancreatic islets
De-identified human pancreatic islets from thirty-five male donors were obtained from PRODO Laboratories Inc, and the Integrated Islet Distribution Program (IIDP). Donor information are provided on Table S2. Islets were left in culture at 37°C in a humidified incubator containing 5% CO2 overnight before any experiments were performed. Islets were cultured in phenol red free RPMI medium (Gibco) containing 11mM glucose, supplemented with 10% Charcoal Stripped FBS (Invitrogen), HEPES (10mM; Gibco), Sodium Pyruvate (1mM; Gibco), β-mercaptoethanol (50μM; Invitrogen), GlutaMAX (2mM; Gibco) and Penicillin-Streptomycin (1x; Gibco).
Studies involving human cadaveric tissue
Samples used in this study originate from de-identified cadaveric donors and are institutional review board exempt.
METHOD DETAILS
In vivo studies
Mice aged 9 to 35 weeks (see Figure 1 legend for details) were weaned onto a customized diet designed to be high in saturated fat and simple sugars (sucrose and fructose) to mimic a western diet (30% AMF; 14.9% Kcal protein, 33.2% Kcal carbohydrates, 51.9% Kcal fat; Harlan Teklad) for 9 weeks. Mice were housed and experimented upon with approval by the Tulane department of comparative medicine and institutional animal care and use committee (IACUC) authorization.
Mice were injected with a glucose solution for glucose tolerance test (IP-GTT, 2 g/kg) and for glucose stimulated insulin secretion (IP-GSIS, 3 g/kg). Mice were fasted overnight before glucose injection. Blood glucose was measured from tail vein blood using True Metrix (Trividia Health). Insulin was measured in plasma by ELISA kit (Millipore).
Mouse islet isolation and measurement of insulin secretion in static incubation
Mouse islet isolation was performed following pancreatic duct injection with collagenase (Sigma) as described.72 For measurement of insulin secretion, mouse or human islets were hand-picked under a dissection microscope in batches of three replicates of 10 islets per condition and treated with DHT (10nM; Steraloids) for 24 h prior to GSIS. Islets were then treated with vehicle (95% ethanol), DHT, GLP-1 (10nM; Indiana University Bloomington), GIP (100nM; Tocris), glucagon (20nM; Sigma), forskolin (10μM; Sigma), H89 (10μM; CST), ESI-09 (10μM; Sigma), Exendin9–39 (100nM; Sigma), KH7 (1μM; Sigma Aldrich), LRE1 (10μM; Sigma Aldrich), TDI-10229 (2μM, 5μM, 10μM; Cornell), Acetazolamide/AZZ (50μM; MedChemExpress), CFTRinh-172 (500μM; Selleckchem), S0859 (2μM; MedChemExpress), 2′,5′-Dideoxyadenosine/ddADO (1μM), PF431396 (15nM; Selleckchem), PF562271 (15nM; Selleckchem) PF573228 (10nM; MedChemExpress), PF4618433 (1μM; MedChemExpress), PP2 (1μM; Sigma Aldrich), BKM120 (1μM; Selleckchem), GDC-0941 (1μM; Sigma Aldrich), Rapamycin (27.4nM; Sigma Aldrich), JR-AB2–011 (5μM; TargetMol), Linagliptin (83 μg/ kg; Boehringer Ingelheim), Finasteride (100nM; Sigma Aldrich), Dutasteride (100nM; Sigma Aldrich), Exendin4 (100nM), ExendinPhe1 (100nM; Imperial College London) at 2.8mM and then 16.7mM glucose for 40 min sequentially. Insulin release from islets was measured as described with Rat/Mouse Insulin ELISA kit (Millipore Sigma).
Linagliptin treatment
Linagliptin was formulated into the western diet at concentration of 83 μg/kg βARKOMIP and control mice received either western diet without linagliptin or linagliptin diet at week 18. Metabolic tests were performed after 4 weeks of linagliptin treatment.
Mouse islet steroid conversion assays
Mouse islets were isolated from 10 wild-type male mice and recovered overnight in complete medium: RPMI-1640 (Gibco) supplemented with 10% charcoal-stripped FBS and Pen/Strep (100 U/ml, 100 μg/mL). Approximately 250 islets were in each replicate and each condition was run in triplicate. Islets were treated with T (100nM; Sigma), the 5α-R inhibitors finasteride (100nM; Sigma) and dutasteride (100nM; Sigma), or vehicle (ethanol and DMSO). Other control conditions included culture medium without FBS and complete medium with finasteride and dutasteride. Culture medium and islets were harvested for further analysis after a 24-h incubation period.
Steroid quantification by ultra-high-performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS)
For the measurement of androgens, 500 μL of culture medium or external standard mix were combined with an internal standard mixture and extracted by liquid-liquid extraction with tert-butyl methyl ether (Acros Organics). Chromatographic separation and steroid quantification were performed using an ACQUITY ultra performance liquid chromatography system (Waters) coupled to a XE-VO™ TQ-XS triple quadrupole mass spectrometer (Waters). Mass-to-charge transitions monitored in multiple reaction monitoring used for quantification are summarized in Table S2. Peak area ratios of analyte and internal standard, 1/x weighting and linear least squares regression were used to produce the standard curves for quantification. Limits of quantifications were 0.24 nM for T and 0.24 nM for 5α-dihydrotestosterone (DHT).
Measurement of insulin secretion in perifusion
Perifusion experiments were performed in Krebs buffer containing 125 mM NaCl, 5.9 mM KCl, 1.28 mM CaCl2, 1.2 mM MgCl2, 25 mM HEPES, and 0.1% bovine serum albumin at 37°C using a PERI4–02 machine (Biorep Technologies). Fifty hand-picked human islets were loaded in Perspex microcolumns between two layers of acrylamide-based microbead slurry (Bio-Gel P-4, Bio-Rad Laboratories). Cells were challenged with either low or high glucose (2.8 mM; 16.7 mM) or potassium chloride (KCl = 20 mM) at a rate of 100 μL/min. After 60 min of stabilization in 2.8mM glucose, cells were stimulated with the following sequence: 10 min at 2.8mM glucose, 20 min at 16.7mM glucose, 10 min at 20mM KCl +16.7mM glucose, and 10 min 2.8mM glucose. Samples were collected every minute on a plate kept at <4°C, while the perifusion solutions and islets were maintained at 37°C in a built-in temperature controlled chamber. Insulin concentrations were determined using commercially available ELISA kits (Mercodia). Total insulin release was normalized per total insulin content using a human insulin ELISA kit (Mercodia). See Table S1 for donor information.
Generation of organelle-targeted FRET cAMP biosensors
Organelle-targeted cAMP FRET biosensors were generated from the untargeted Epac-and mTurquoise-based TEpacVV sensor (a gift from Professor Kees Jalink, The Netherlands Cancer Institute)73 as follows: Endo-TEpacVV was generated by PCR cloning of the 2xFYVE domain (endosomal targeting sequence) of the bPAC-Endo construct (a kind gift from Prof Mark Von Zastrow, UC San Francisco)74 to the N-terminus end of TEpacVV using HindIII and NotI restriction sites. PM-TEpacVV was generated by PCR cloning of the lipid raft-binding domain of Lyn kinase (plasma membrane targeting sequence) of the bPAC-PM construct to the N-terminus end of TEpacVV using the same restriction sites as above. bPAC-Endo and bPAC-PM construct were a gift from Prof Mark Von Zastrow, UC San Francisco.74
Immunofluorescence analysis of GLP-1R and AR co-localization
INS1 832/3 cells were plated in 24-well plates and the following day, transfected with GLP-1R-GFP and FLAG-AR plasmids (0.5μg of each plasmid/well plus 2μL Lipofectamine 2000). After 24h, the cells were trypsinized and plated in 24-well plates with glass coverslips using phenol-red-free medium containing charcoal-stripped FBS (RPMI 1640 no phenol red, 10% v/v charcoal-stripped FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, 10mM HEPES, and 1mM sodium pyruvate). 48h post-transfection, the cells were treated with transferrin-555 for 30 min to label the endosomal pathway. Subsequently, treatments with control medium, GLP-1 100nM, DHT 10nM, or GLP-1 + DHT for 5 or 10 min were performed. After a wash with ice-cold PBS, the cells were fixed in 4% PFA for 20 min at 4C. After two washes with PBS, 0.1% Triton X-100 was added for 10 min to allow permeabilization. Cells were washed with PBS twice and blocking buffer (3% w/v BSA, 1% v/v goat serum, 0.1% v/v Tween 20 in PBS) was added for 30 min. The blocking buffer was discarded, and primary anti-FLAG antibody (mouse monoclonal antibody, F3165 Sigma) diluted 1:500 in blocking buffer was added overnight at 4C. After three 5-min washes in PBS +0.1% v/v Tween 20 (PBST), secondary antibody anti-mouse Alexa Fluor 647 (1:1,000 in blocking buffer) was incubated for 30 min at room temperature. Following three 5-min washes in PBST, the coverslips were mounted onto slides using ProLong Diamond Antifade Mountant with DAPI (ThermoFisher Scientific). Images were captured using a Zeiss LSM-780 inverted confocal microscope with a 63x/1.4numerical aperture oil-immersion objective from the Facility for Imaging by Light Microscopy (FILM) at Imperial College London and analyzed in Fiji ImageJ.
Dynamic measurement of cAMP production
INS-1 832/3 cells were cultured in complete medium composed of RPMI 1640 medium supplemented with 10% fetal calf serum, 100IU/ml penicillin, 100 mg/L streptomycin, 10mM HEPES, 2mM L-glutamine, 1mM sodium pyruvate, and 50μM beta-mercaptoethanol. Cells were split twice a week and cultured in an incubator at 37°C supplemented with 5% CO2. For imaging, INS-1 832/3 cells were grown on glass coverslips. Adenoviral infection with the FRET Epac2-camps probe (a kind gift from Professor Dermot Cooper, University of Cambridge) was performed 48h prior to imaging. Transfection of INS-1 832/3 cells with cAMP FRET biosensors targeted to the plasma membrane (PM-TEpacVV), cytoplasm (TEpacVV) or endosomes (Endo-TEpacVV) was performed using Lipofectamine-2000 with 1–3 μg DNA 48h prior to imaging. Imaging of cAMP was conducted using a Crest X-Light spinning disk system in widefield mode, coupled to a Nikon Ti-E, SPECTRA X light engine, 20× objective and 16-bit Photometrics Evolve Delta EM-CCD, as described.20 Excitation was delivered at λ = 430–450 nm, with emitted signals detected at λ = 460–500 nm and 520–550 nm for Cerulean and Citrine, respectively. HEPES-bicarbonate buffer was used, containing (in mM): 120 NaCl, 4.8 KCl, 24 NaHCO3, 0.5 Na2HPO4, 5 HEPES, 2.5 CaCl2, 1.2 MgCl2 and 16.7 D-glucose. FRET responses were calculated as the fluorescence ratio of Cerulean/Citrine and normalized as F/Fmin.
Confocal analysis of SNAP-GLP-1R internalization
MIN6B1 cells (a gift from Prof Philippe Halban, University of Geneva) stably expressing human SNAP-GLP-1R (‘‘MIN6B1-SNAP-GLP-1R’’)22 were generated by transfection of an SNAP-GLP-1R vector (Cisbio) followed by G418 selection and maintained in DMEM with 15% FBS, 50 μM β-mercaptoethanol and 1% penicillin/streptomycin. Cells were seeded in 24-well plates with glass coverslips using medium containing charcoal-stripped FBS (10% v/v). 24h later, cells were labeled at 37C for 30 min with 1 μM SNAP-Surface 549 to label surface receptors, treated for 30 min with 100 nM Exendin-4 or the biased agonist Exendin-Phe122 in the presence or absence of 10 nM DHT. After a wash with ice-cold PBS, the cells were fixed in 4% PFA for 20 min at 4C, washed in PBS, mounted onto slides using ProLong Diamond Antifade Mounting Medium with DAPI (ThermoFisher Scientific). Images were captured using a Zeiss LSM-780 inverted confocal microscope with a 63x/1.4numerical aperture oil-immersion objective from the Facility for Imaging by Light Microscopy (FILM) at Imperial College London and analyzed in Fiji ImageJ.
Transmission electron microscopy
For ultrastructural analyses, tissue sections were deparaffinized in three changes of xylene followed by rehydration in a graded series of ethanol. The tissue samples were then refixed in 2% paraformaldehyde/2.5% glutaraldehyde (Ted Pella Inc.) in 100 mM sodium cacodylate buffer, pH 7.2 for 2 h at room temperature. Samples were washed in sodium cacodylate buffer and postfixed in 1% osmium tetroxide (Polysciences Inc.) for 1 h. Samples were then rinsed extensively in dH20 prior to en bloc staining with 1% aqueous uranyl acetate (Ted Pella Inc., Redding, CA) for 1 h. Following several rinses in dH20, samples were dehydrated in a graded series of ethanol and embedded in Eponate 12 resin (Ted Pella Inc.). Sections of 95 nm were cut with a Leica Ultracut UCT ultramicrotome (Leica Microsystems Inc., Bannockburn, IL), stained with uranyl acetate and lead citrate, and viewed on a JEOL 1200 EX transmission electron microscope (JEOL USA Inc) equipped with an AMT 8-megapixel digital camera and AMT Image Capture Engine V602 software (Advanced Microscopy Techniques).
Bioenergetics
Islets isolated from male C57/BL6N mice were washed once with assay buffer (made from Agilent Seahorse XF Base Medium supplemented with 3mM glucose and 1% charcoal striped FBS). Around 150 islets were transferred to each well of Seahorse XF24 Islet Capture Microplate (Agilent) and were incubated in assay buffer at 37°C for 60 min before being transferred to Agilent Seahorse XFe24 Analyzer. Islets were maintained in the assay medium throughout the experiment, while oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured at basal (3 mM), glucose-stimulated level (20 mM) and after addition of oligomycin, carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), rotenone/antimycin or 2-deoxy-gluxose (2-DG) according to manufacturer’s instructions.
Metabolomics
Human islets were treated overnight with 10nM DHT or EtOH in a humidified incubator containing 4% CO2 at 37°C and then batched in groups of 100 islets in microcentrifuge tubes and treated for either 30 min with media containing either 2.8mM or 16.7 mM 13C6 glucose; the same DHT or EtOH concentrations were maintained. Following the incubation period, tubes were briefly centrifuged at low speed and media was withdrawn. 150mM ammonium acetate in water were added to rinse residual media (150mM ammonium acetate is an osmolarity-matching, mass spectrometry compatible rinse buffer with near-neutral pH). Tubes were centrifuged, supernatant was withdrawn, and they were frozen by immersion in liquid nitrogen and stored at −80C prior to extraction. On the day of metabolomics analysis, islets were extracted in 200μL of ice-cold 8:1:1 methanol:water:chloroform, with tissue disruption aided by immediate probe sonication for 20 s with a Branson 450 Sonifier set to output level 2, duty cycle 20%. The disrupted samples were allowed to incubate on ice for 10 min and were then centrifuged for 5 min at 14,000 xg to pellet cell debris. 180μL of supernatant were dried under a gentle stream of nitrogen Gαs at room temperature, and were then reconstituted in 45μL of 4:1 water:methanol and transferred to autosampler vials for analysis.
LC-MS analysis. Samples were analyzed on an Agilent 1290 Infinity II coupled to a 6530 Q-ToF MS system with a Dual JetStream Ionization (ESI) source. The separation method was ion paring-chromatography (IPC-MS) performed using an Agilent Zorbax Extend C18 1.8 μ RRHD column, 2.1 × 150mm ID with a matched guard column. Mobile phase A was 97% water, 3% methanol. Mobile phase B was 100% methanol. Both mobile phases contained 15mM tributylamine and 10 mM acetic acid. Mobile phase C was 100% acetonitrile. Elution was carried out using a linear gradient followed by a multi-step column wash including automated (valve-controlled) backflushing (see Table S2). Column temperature was 35°C and injection volume was 5 μL. MS acquisition was performed in negative ion mode, scan rate 2 spectra/sec, mass range 50–1200 m/z. Source parameters were: drying gas temperature 250°C, drying gas flow rate 13 L/min, nebulizer pressure 35 psig, sheath gas temp 325°C and flow 12 L/min, capillary voltage 3500V, internal reference mass correction enabled. Data analysis was performed using Agilent Profinder 10.0 software in batch isotopologue extraction mode with automated natural isotope abundance correction enabled. Compound identification was performed by matching accurate mass and retention time to those of authentic standards analyzed using the same method.
Immunoprecipitation-based proteomics analysis of AR interactome
832/3 cells were treated with vehicle, DHT (10−8 M) (Steraloids) or DHT and GLP-1 (10−8 M) all treatments were added 5 min before harvesting, DHT was added the day before as well. Cell monolayers were rinsed with phosphate buffered saline and then scraped with nondenaturing IP buffer containing phosphatase inhibitors (PhosSTOP, Sigma-Aldrich) and protease inhibitor cocktail (cOmplete, Sigma-Aldrich). The cell suspension was passed through a 20-gauge needle 10 times. The whole-cell extract was purified by centrifugation at 12000 g for 10 min at 4°C. Cell extracts (1 mg total protein) were incubated with 2μg of anti-AR antibody (Abcam) or rabbit IgG control (Cell Signaling) overnight at 4°C under rotation. Protein A-conjugated agarose beads (Cell Signaling) were added to protein-antibody mixture the following day for 2 h. The beads were pelleted at 13,000 g for 3 min at 4°C. The supernatant was removed, and beads were washed 5 times with ice-cold 1X IP buffer. The IP antibody and immunoprecipitated proteins were eluted from the beads into 4X SDS loading buffer containing BME by boiling the samples for 10 min at 98°C. Samples were flicked during the heat step to ensure efficient elution. The purified samples were briefly centrifuged, and proteins were separated by SDS-PAGE. The gel was stained, and the protein bands were cut from the gel following PierceTM Silver Stain Kit for Mass Spectrometry (ThermoFisher) protocol. The gel bands were cut into 1 mm3 pieces and destained with 50% acetonitrile in 25 mM ammonium bicarbonate. The reduction was performed by adding 25 mM Dithiothreitol in 25 NH4HCO3 and incubating at 37°C for 1 h, followed by alkylation with 55 mM Iodoacetamide in 25 mM NH4HCO3 for 1 h in the dark at room temperature. Gel pieces were dehydrated by acetonitrile wash, dried by SpeedVac for 2 min, and submitted to trypsin digestion with a 1:50 ratio of protein vs. enzyme solution at 37°C overnight. Digested peptides were extracted using 1% formic acid and an additional extraction with 70% acetonitrile and 5% formic acid. The final tryptic peptides were dried to concentrate the peptides and reconstituted in 2% acetonitrile with 0.1% formic acid for LC-MSMS analysis. Proteomics analysis was performed on a Q Exacitive Orbitrap hybrid mass spectrometer with an Ultimate 3000 nano-LC and nanoelectrospray ionization. Peptides were separated with an nC18 analytical column (C18 Pepmap 100, 3 μm particle, 100 Å pore, 75 μm i.d. ×150 mm) using 100 min buffer gradient a low flow rate at 300 nL/min. Data-dependent acquisition in positive mode was performed for data collection. Acquired data was searched with Proteome Discoverer 2.4 using the SEQUEST search engine with label-free quantification workflow against the UniProt database of Rattus norvegicus (http://www.uniprot.org; Proteome ID: UP000002494). Search parameters were trypsin cleavage sites with two missed cleavage site allowance; precursor and fragment mass tolerance were set at ±10ppm and 0.02 Da. Carbamidomethyl of cysteine was set as a fixed modification and oxidation of methionine as a variable modification. As most proteins identified in IP experiments are non-specific binders, we compared proteins identified in the AR vs. IgG interactome and eliminated contaminant proteins that were enriched in both. Proteins enriched in the AR immunoprecipitate by 2-fold up or down between treatments were selected. Enriched proteins were analyzed and integrated using Enrichr (https://maayanlab.cloud/Enrichr/), Ingenuity Pathways Analysis (Ingenuity Systems, www.ingenuity.com ) and STRING version 11.5 (https://string-db.org/). Proteins of interest were selected for heatmap representation, where proteins were normalized via a Z-test in R using customized scripts and plotted using the ggplot2 and heatmap.2 packages.
Reverse phase protein array (RPPA)
Human islets (1200 IEQ/condition) were cultured overnight and treated with DHT (10−8 M) (steraloids) for 5 min, 30 min or 18 h (control islets received no treatment) and stored at −80°C until shipment to MD-Anderson Cancer Center Functional Proteomics RPPA Core Facility. Cell lysates were serially diluted two-fold for 5 dilutions (undiluted, 1:2, 1:4, 1:8; 1:16) and arrayed on nitrocellulose-coated slides in a 11×11 format to produce sample spots. Sample spots were then probed with 485 unique antibodies by a tyramide-based signal amplification approach and visualized by DAB colorimetric reaction to produce stained slides. Stained slides were scanned on a Huron TissueScope scanner to produce 16-bit tiff images. Sample spots in tiff images were identified and their densities quantified by Array-Pro Analyzer. In each sample, relative protein levels were determined by interpolating each dilution curve produced from the densities of the 5-dilution sample spots using a “standard curve” (SuperCurve) for each slide (antibody). All relative protein level data points were normalized for protein loading and transformed to linear values. Protein levels were further normalized by GαPDH and β-actin. Enrichment (fold-change) between treatments and control group were reported for all phosphoproteins of interest. For heatmap representation proteins were normalized via a Z-test in R using customized scripts and plotted using the ggplot2 and heatmap.2 packages.
Single cell RNA sequencing
Human islets (750 IEQ per condition) were treated with either 10nM DHT or EtOH overnight in a humidified incubator containing 4% CO2 at 37°C. Islet cells were then dispersed using TrypLE (Thermofischer), and immediately evaluated for viability (89.88 ± 1.77%) by Cellometer Automated Cell Counter (Nexcelom Bioscience) prior to single cell RNAseq library preparation. For 10x single cell RNAseq library preparation, 5000 individual live cells per sample were targeted by using 10x Single Cell 3′ RNAseq technology provided by 10x Genomics (10X Genomics Inc). Briefly, viable single cell suspensions were partitioned into nanoliter-scale Gel Beads-In-EMulsion (GEMs). Full-length barcoded cDNAs were then generated and amplified by PCR to obtain sufficient mass for library construction. Following enzymatic fragmentation, end-repair, A-tailing, and adaptor ligation, single cell 3′ libraries comprising standard Illumina P5 and P7 paired-end constructs were generated. Library quality controls were performed by using Agilent High Sensitive DNA kit with Agilent 2100 Bioanalyzer (Agilent) and quantified by Qubit 2.0 fluorometer (ThermoFisher). Pooled libraries at a final concentration of 750pM were sequenced with paired end single index configuration by Illumina NextSeq 2000 (Illumina).
Computational architecture
A complete compendium of the coding software, coding environment and data-analysis pipeline is available online as an R script repository on Github: https://github.com/FMJLabTulane/AR-DHT. These scripts replicate the data analysis outlined in this manuscript.
1. Alignment and Generation of Filtered Gene counts Using CellRanger:
For all datasets, we utilized CellRanger v4.0.0 software using the [-mkfastq] command to de-multiplex FASTQ data. Reads were mapped and aligned to the human genome (10X genomics pre-built GRCh38–2020-A Homo sapiens reference transcriptome assembly) with STAR (95.83 ± 0.54% of reads confidently mapped to the human genome).75 Subsequently, final digital gene expression matrices and c-loupe files were generated for downstream multimodal analysis. Cellranger identified 31,198 correctly allocated barcodes (cells) (98.18% ± 0.14% valid barcodes), having 20,625 ± 5634 reads/cell (99.65 ± 0.39% valid UMIs) and 3848 ± 560 genes/cell.
2. Multimodal single-cell Transcriptomic Analysis using Seurat
We deployed Seurat v4 (https://github.com/satijalab/seurat/releases/tag/v4.1.0) scripts to perform merging, thresholding, normalization, principal component analysis (linear dimensionality reduction), clustering analysis (non-linear multidimensional reduction), visualization and differential gene expression analysis. Cells having total mitochondrial RNA contribution beyond 20% were eliminated from the analysis, along with cells expressing less than 200 or greater than 8000 total genes (doublet elimination). At this point we were left with 28,809 cells (4802 ± 1101), having 22,259 ± 6944 reads/cell and 4037 ± 805 genes/cell. This denotes that 92.3% of cells were sequenced within acceptable parameters representing high quality scRNA sequencing data. After this, we performed global normalization (using the [SCTransform] function in Seurat). SCTransform is a mathematical model which derives Pearson’s residuals from regularized negative bionomial regression values, calculated across gene expression count-data derived from each cell. This method removes technical effects yet maintains biological heterogeneity.76 [SCTransform] also calculates and stores data pertaining to the 3000 most variable genes (SCTransform completely removes the necessity of performing [NormalizeData], [ScaleData] and [FindVariableFeatures]). Since we did not want to give any one sample more weight over the other, we performed an unbiased cross-sample dataset reciprocal PCA integration as outlined and recommended by Seurat developers (https://satijalab.org/seurat/articles/integration_rpca.html ) to create an integrated dataset.
We then performed a PCA analysis on the integrated dataset. Using Seurat’s non-linear dimensional reduction algorithm [RunU-MAP], we performed Uniform Manifold Approximation and Projection (UMAP) to cluster cells based on variable genes identified previously. Differential gene expression analysis was performed using the [FindAllMarkers] function in Seurat. We used a Wilcoxon ranksum test to perform the analysis and confirmed with another algorithm: Model-based Analysis of Single-cell Transcriptomics (MAST) using the [FindAllMarkers] function on default settings. In our analysis.
A differentially expressed gene is defined as any gene which is expressed in at least 10% cells, has a p value <0.001 and >1.1x average fold change from all other clusters being tested.
Cell clusters were defined to have at least 20 differentially expressed genes for which at least 10 were unique when compared to other clusters.
Dimensional reduction was within recommended parameters outlined by linear dimensionality testing using the Seurat functions [RunPCA], [JackStraw] and [PCElbowPlot]. In order to test for cluster separation, and to insure our dimensional reduction was within acceptable parameters, we performed cluster analysis using [clustree],77 an algorithm that models multiple dimensionality reductions (dimensionality reductions control for the degree of clustering in the data) and allows the user to visualize cluster stability as higher resolutions are used for dimensionality reduction. This mathematical modeling allows for optimal clustering of single cell data, where only stable clusters (maintaining their characteristics over a spectrum of resolutions) are used to cluster data. This allows for a more accurate representation of clusters, without over or under-clustering.
3. Pathway Analysis using Enrichr and GOPlot
Gene ontology analysis was performed using Enrichr.78 GO Biological process output comma-delimited (CSV) files were saved, and all those pathways having an adjusted p value <0.01 were selected in the analysis. We selected commonly reoccurring and highly ranked GO pathways, highly enriched in genes derived from the differential gene expression analysis. Bar plots of GO pathways were generated using Graphpad Prism v6.01, p-adjusted values were normalized using the following formula: -Log(p value).
4. Graphical visualization of scRNAseq data
Seurat has a comprehensive palette of integrated graphical visualization tools derived in part from the R function [ggplot2]. We used the [DimPlot] function to visualize UMAP plots, using metadata files to color-code various cells and variable genes to assist in cell clustering. Using the [VlnPlot] function, we generated Violin plots to look at gene expression across clusters, and [DoHeatmap] to generate heatmaps of topmost differentially expressed genes.
NanoBiT assays
NanoBiT assays were performed using methods adapted from published studies describing Gα recruitment assays.79 Cells were plated in six-well plates and transfected 24 h later using lipofectamine 2000 (Invitrogen) with: 250 ng LgBiT-GLP-1R and 250 ng SmBiT-AR or 250 ng SmBiT-Empty for AR and GLP-1R interaction assays; and 125 ng SmBiT-AR, SmBiT-GLP1R or SmBiT-Empty with 250 ng LgBiT-Gαs for G-protein recruitment assays. Following 48 h, cells were harvested in fresh media, and each well seeded across 8 wells of a 96-well plate (each well was seeded at a density of 40,000 cells/well in a volume of 40 μL). Approximately 4 h later, each well was loaded with 40 μL NanoLuc substrate (Promega) and luminescence baseline signal read on a Pherastar FSX plate reader (BMGLabtech) at 37°C for four cycles (equivalent to 6 min). Vehicle and agonists (DHT, GLP-1, combined DHT+GLP-1, each at 10–8 M) were prepared in Hank’s Balanced Saline Solution (HBSS, Sigma Aldrich) at 10× concentration and added to wells once baselines were stable and responses recorded immediately following agonist addition every 2 min for a further 22 min. All responses were normalized by subtracting an averaged vehicle luminescence from all measurements and expressed relative to luminescence at 0 min. For Gαs recruitment assays all values were plotted, while the maximal response was plotted for AR and GLP-1R interaction assays using GraphPad Prism. AR and GLP-1R interaction assays were performed in n = 12 biological replicates. G-protein recruitment assays were performed in n = 6 biological replicates.
Plasma membrane preparation for granule docking measurements
In brief, INS-1 832/3 were treated with DHT (10 nM) followed by a change to low glucose (2.5mM)/low serum (2.5%) overnight. For GLP-1 agonism, cells were exposed to KRBH for 1h, followed by the addition of either high glucose (11.0 mM) plus GLP-1 (10 nM), or high glucose (11.0 mM) plus GLP-1 (10 nM) and DHT (10 nM) for 15 min. After 15 min, the cells were harvested, and the plasma membrane (PM) fractions were isolated by differential centrifugation using a sucrose gradient as described.48 Eight micrograms of protein from experimental conditions were resolved via 15% freshly hand-poured SDS-PAGE and VAMP2 content in plasma membrane fractions detected by immunoblotting for VAMP2 (Synaptic Systems, Cat #104211) and normalized for plasma membrane content using constitutively present plasma membrane marker protein STX4.80
Human β-cell capacitance measurements of exocytosis
Human islets from male donors (1500 IEq) were washed briefly with VERSENE solution (ThermoFisher 15040066) and dissociated using TrypLE Select Enzyme solution (ThermoFisher 12563011) at 37°C, with occasional trituration. Dissociated β-cells were purified as described previously using mouse anti-human NTPDase3 antibodies (Ectonucleotidases-ab) and CELLection Pan Mouse IgG kit (ThermoFisher 11531D).81 Cells were plated on sterilized glass shards and cultured overnight at 37°C before experiments. The base media was glucose-free RPMI-1640 supplemented with 8 mM glucose, 10% (v/v) fetal bovine serum (ThermoFisher A31605), 10,000 units/mL penicillin and 10,000 mg/mL streptomycin (Fisher Scientific). Staining for insulin and glucagon were performed to control for the purity of sorted β-cells. Rat anti-insulin antibodies were obtained from DSHB (GN-ID4) and rabbit anti-glucagon antibodies from Cell Signaling (#2760). Alexa Fluor 555 conjugated anti-rat IgG (#4417) and Alexa Fluor 488 conjugated anti-rabbit IgG (#4412) were obtained from Cell Signaling. β-cell Ca2+ current and exocytosis were measured as in82 with minor changes. Briefly, a Sutter MP-225 micromanipulator was used together with a HEKA EPC10 patch-clamp amplifier (Heka Instruments, Bellmore, NY) in the whole cell patch-clamp configuration to record Ca2+ current from intact islets perfused with standard external solution (above) at 33°C. Pipette tips were filled with an internal solution (in mM: 125 Cs-glutamate, 10 CsCl, 10 NaCl, 1 MgCl2$6H2O, 0.05 EGTA, 5 HEPES, 0.1 cAMP, 3 MgATP; pH 7.15 with CsOH) and 10 nM DHT or vehicle (0.1% DMSO) was added to this as indicated for dialysis into the cell. The extra cellular bath solution contained (in mM): 118 NaCl, 5.6 KCl, 1.2 MgCl2, 2.6 CaCl2, 5 Glucose, 20 TEA-Cl, 5 HEPES, pH 7.4, adjusted with NaOH. 30 s after membrane rupture, Ca2+ current was quantified from a 15 ms depolarization from 70 to 0 mV using a P/4 leak subtraction protocol. After an additional 1.5 min, exocytosis was stimulated by activating Ca2+ influx with a series of ten 250 ms membrane depolarizations from −70 to 0 mV. Capacitance responses (fF) and Ca2+ currents (pA) were normalized to initial cell size (pF).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed with GraphPad Prism v8 and R v4.2. Results are presented as mean ± SEM as specified in figures. When results showed a Gaussian distribution, statistical analyses were performed using the unpaired Student’s t test, Wilcoxon test or ANOVA (with Bonferroni, Dunnett or Fisher’s LSD post hoc test). A p value less than 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| RPPA antibodies | MD-Anderson Cancer Center | https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core/antibody-information-and-protocols.html |
| Anti-FLAG | Sigma Aldrich | Cat# F3165; RRID: AB_259529 |
| Anti-AR | Abcam | Cat# Ab133273; RRID: AB_11156085 |
| Anti-AR | Millipore Sigma | Cat# 06-680; RRID: AB_310214 |
| Anti-VAMP2 | Synaptic Systems | Cat# 104 211; RRID: AB_887811 |
| Anti-NTPDase3 | Ectonucleotidases-ab | Cat# hN3-B3S and hN3-H10S |
| Anti-STX4 | Custom Designed | N/A |
| Ant-rat-Insulin | DSHB | Cat# GN-ID4; RRID: AB_2255626 |
| Anti-Glucagon | Cell signaling | Cat# 2760; RRID: AB_659831 |
| Anti-mouse Alexa Fluor 647 | Thermofischer Scientific | Cat# A-31571; RRID: AB_162542 |
| Anti-rat Alexa Fluor 555 | Cell signaling | Cat# 4417; RRID: AB_10696896 |
| Anti-rabbit Alexa Fluor 488 | Cell signaling | Cat# 4412; RRID: AB_1904025 |
|
| ||
| Biological samples | ||
|
| ||
| De-identified human pancreatic islets | PRODO Laboratories Inc | https://prodolabs.com/ |
| De-identified human pancreatic islets | Integrated Islet Distribution Program (IIDP) | https://iidp.coh.org/ |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| 17β-Estradiol (E2) | Steraloids | Cat# E0950-000 |
| GLP-1 | Richard D. DiMarchi | N/A |
| Exendin 9–39 | Sigma Aldrich | Cat# E7269 |
| Forskolin | Sigma Aldrich | Cat# F3917 |
| GIP | Tocris | Cat# 2084 |
| Glucagon | Sigma Aldrich | Cat# G2044 |
| DHT | Steraloids | Cat# A2570-000 |
| H89 | CST | Cat# 9844 |
| ESI-09 | Sigma Aldrich | Cat# SML0814 |
| LRE1 | Sigma Aldrich | Cat# SML1857 |
| KH7 | Sigma Aldrich | Cat# K3394 |
| TDI-10229 | Cornell | N/A |
| Acetazolamide (AZZ) | MedChemExpress | Cat# HY-B0782 |
| CFTRinh-172 (CFTRi) | Selleckchem | Cat# S7139 |
| S0859 | MedChemExpress | Cat# HY-15529 |
| 2′,5′-Dideoxyadenosine | Sigma Aldrich | Cat# D7408 |
| PF431396 | Selleckchem | Cat# S7644 |
| PF562271 | Selleckchem | Cat# S2890 |
| PF573228 | MedChemExpress | Cat# HY-10461 |
| PF4618433 | MedChemExpress | Cat# HY-18312 |
| PP2 | Sigma Aldrich | Cat# P0042 |
| BKM120 | Selleckchem | Cat# S2247 |
| GDC-0941 | Sigma Aldrich | Cat# 5.09226 |
| Rapamycin | Sigma Aldrich | Cat# 37094 |
| JR-AB2-011 | TargetMol | Cat# T11728 |
| Linagliptin | Boehringer Ingelheim | N/A |
| Finasteride | Sigma Aldrich | Cat# F1293 |
| Dutasteride | Sigma Aldrich | Cat# SML1221 |
| SNAP-Surface 549 | NEB | Cat# S9112S |
| Exendin4 | Sigma Aldrich | Cat# E7144 |
| Exendin-Phe1 | Imperial College London | N/A |
| Streptozotocin | Sigma Aldrich | Cat# 18883-66-4 |
| D-(+)-Glucose | Sigma Aldrich | Cat# G7528 |
| PBS | Gibco | Cat# 10010023 |
| HBSS | Gibco | Cat# 14025092 |
| Phenol red free RPMI-1640 medium | Gibco | Cat# 11835030 |
| DMEN medium | Gibco | Cat# 11965126 |
| Phenol red free DMEN medium | Gibco | Cat# 21063029 |
| HEPES | Gibco | Cat# 15630080 |
| Sodium Pyruvate | Gibco | Cat# 11360070 |
| GlutaMAX | Gibco | Cat# 35050061 |
| Penicillin/streptomycin | Gibco | Cat# 15140063 |
| Collagenase | Sigma Aldrich | Cat# C9263 |
| Charcoal Stripped FBS | GemCell™ | Cat# 100–119 |
| FBS | GemCell™ | Cat# 100–500 |
| Glutaraldehyde | Ted Pella Inc. | Cat# 18436 |
| Sodium cacodylate buffer | Ted Pella Inc. | Cat# 18851 |
| Osmium tetroxide | Polysciences Inc. | Cat# 20816-12-0 |
| Uranyl acetate | Ted Pella Inc. | Cat# 19481 |
| Eponate 12 resin | Ted Pella Inc. | Cat# 18005 |
| Lead citrate | Sigma Aldrich | Cat# 15326 |
| Ammonium acetate | Sigma Aldrich | Cat# 73594 |
| Tributylamine | Sigma Aldrich | Cat# 8.08358 |
| Acetic acid | Sigma Aldrich | Cat# 27225 |
| Acetonitrile | Sigma Aldrich | Cat# 34851 |
| NanoLuc substrate | Promega | Cat# N1110 |
| Lipofectamine | Thermofischer Scientific | Cat# L3000001 |
| Tamoxifen | Sigma Aldrich | Cat# T5648 |
| TrypLE | Thermofischer Scientific | Cat# 12604013 |
| Ethanol | Sigma Aldrich | Cat# E7023 |
| Versene | ThermoFisher | Cat# 15040066 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat# 103010-100 |
| Human Insulin Elisa | Mercodia Inc. | Cat# 10-1113-01 |
| Rat/Mouse Insulin ELISA kit | Millipore | Cat# EZRMI-13K |
| Testosterone Elisa | IBL America | Cat# IB79106 |
| Luteinizing hormone Elisa | LSBio | Cat# LS-F22503-1 |
| 17β-estradiol Elisa | Calbiotech | Cat# ES380S |
| 10x Single Cell 3′ v3.1 RNAseq kit | 10X Genomics | Cat# PN-1000121 |
| Illumina NextSeq 2000 P3 100 kit | Illumina Biosciences | Cat# 20040559 |
| Illumina NextSeq 2000 P3 100 kit | Illumina Biosciences | Cat# 20040559 |
| CELLection Pan Mouse IgG kit | Thermofischer Scientific | Cat# 11531D |
|
| ||
| Deposited data | ||
|
| ||
| Single Cell RNAseq Data | This study. Deposited location: Gene Expression Omnibus - NCBI | GEO: GSE201256 |
| Proteomics Data | This study. Deposited location: Proteomics Identification Database - EMBL-EBI | PRIDE: PXD041592 |
| Metabolomics Data | This study. Deposited location: UCSD Metabolomics Workbench/National Metabolomics Data Repository | NMDR: PR001654 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| INS1 832/13 | Millipore Sigma | Cat# SCC207; RRID: CVCL_7226 |
| INS1 832/3 | Millipore Sigma | Cat# SCC208; RRID: CVCL_ZL55 |
| MIN6B | University of Geneva | N/A |
| MIN6B1-SNAP-GLP-1R | Imperial College London | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | The Jackson Laboratory | Strain#000664 |
| Mouse: MIP-CreERT | The Jackson Laboratory | Strain#:024709 |
| Mouse: ARfl/fl | Catholic University of Leuven | N/A |
| Mouse: Ins2-creTg(Ins2–cre)25Mgn/J | The Jackson Laboratory | Strain# 003573 |
| Mouse: Glp1rf/f | Dr. David D’Alessio | N/A |
| Mouse: Raptorfl/fl | The Jackson Laboratory | Strain# 013188 |
| Mouse: Rictortm1.1Mnh | The Jackson Laboratory | Strain# 020649 |
| Mouse: Ins2-creTg(Ins2–cre)23Herr | MMRC | Strain# 011155-MU |
| Mouse: KD-mTOR | Dr. Druie Cavender | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pTEpacVV | The Netherlands Cancer Institute) | N/A |
| pEndo-TEpacVV | University of Birmingham | N/A |
| pbPAC-Endo | UC San Francisco | N/A |
| pbPAC-PM | UC San Francisco | N/A |
| pPM-TEpacVV | University of Birmingham | N/A |
| pGLP-1R-GFP | Imperial College London | N/A |
| pFLAG-AR | Imperial College London | N/A |
| pEpac2-camps | University of Cambridge | N/A |
| LgBiT-GLP-1R | University of Birmingham | N/A |
| SmBiT-AR | University of Birmingham | N/A |
| SmBiT-Empty | University of Birmingham | N/A |
| SmBiT-GLP1R | University of Birmingham | N/A |
| LgBiT-Gαs | University of Birmingham | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Single cell analysis architecture (Github + Zenodo) | FMJ Lab | https://zenodo.org/record/7814544 https://github.com/FMJLabTulane/AR-DHT |
| Gen5 imager software | Agilent | https://www.biotek.com/products/software-robotics-software/gen5-microplate-reader-and-imager-software/ |
| Elements Advanced Research software | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements/nis-elements-advanced-research |
| Zeiss Zen Software | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html |
| Cellranger | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Seurat | The Satija Lab | https://satijalab.org/seurat/ |
| R | The R Consortium | https://www.r-project.org/ |
| RStudio v1.2.1335 (64x bit, for Windows) | The R Consortium | https://www.rstudio.com/products/ |
| Ingenuity Pathway Analysis (IPA) | Qiagen | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/?cmpid=QDI_GA_IPA&gclid=Cj0KCQrKQBhCNARIsACUEW_beTxh0IgfRAtfWcvbawY1cFpVLowb2G6pe8Qf7jWZtJY1s6KfuAGIaAuxEEALw_wcB |
| Proteome Discoverer 2.4 | ThermoFisher | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html |
| GraphPad Prism v8 | GraphPad | https://www.graphpad.com/ |
| STRING | v11.5 | https://string-db.org/ |
| EnrichR | Developed in the Ma’ayan Lab | https://maayanlab.cloud/Enrichr/ |
Highlights.
DHT enhances GLP-1-mediated cAMP production and insulin exocytosis in male β cells
DHT increases Gαs recruitment to GLP-1R and AR complexes, activating tmAC
DHT increases mitochondrial CO2 production and conversion to HCO3−, activating sAC
DHT enhances GSIS via an FAK/SRC/PI3K/mTORC2 actin remodeling signaling cascade
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants DK107444 and DK074970 (F.M.-J.), DK073716 (E.B.-M.), DK113103 and DK127637 (M.J.M.), DK102233 and DK067912 (D.C.T.), U.S. Department of Veterans Affairs Merit Awards BX003725 (F.M.-J.), BX002728 (E.B.-M.), and B005113 (M.J.M.), an Investigator-Initiated Study from Boehringer Ingelheim and Eli Lilly (F.M.-J.), MRC grants MR/N00275X/1 and MR/S025618/1 (D.J.H.), Diabetes UK grant 17/0005681 (D.J.H.), European Research Council/European Union’s Horizon 2020 research and innovation programme starting grant 715884 (D.J.H.), Academy of Medical Sciences Springboard Award SBF004|1034 supported by the British Heart Foundation, Diabetes UK, the Global Challenges Research Fund, the Government Department of Business, Energy and Industrial Strategy and the Wellcome Trust (CMG), Wellcome Trust Investigator Award 209492/Z/ 17/Z (W.A.), National Institute for Health Research Birmingham Biomedical Research Centre at the University Hospitals Birmingham NHS Foundation Trust, the University of Birmingham grant BRC-1215-20009 (W.A.), the A. Alfred Taubman Medical Research Institute, and the Tulane Center of Excellence in Sex-Based Biology & Medicine) (F.M.-J.). Human pancreatic islets were provided by the NIDDK-funded Integrated Islet Distribution Program (IIDP) (RRID:SCR _014387) at City of Hope (NIH grant 2UC4DK098085).
DECLARATION OF INTERESTS
F.M.-J. received an Investigator-Initiated Study award from Boehringer Ingelheim and Eli Lilly and consulting fees from Mithra Pharmaceutical, Inc. D.E.F. has received research funding from GTx, Inc and has familial relationships with Hummingbird Bioscience, Maia Biotechnology, Alms Therapeutics, Hinova Pharmaceuticals, and Barricade Therapeutics.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112529.
REFERENCES
- 1.Navarro G, Allard C, Xu W, and Mauvais-Jarvis F.(2015). The role of androgens in metabolism, obesity, and diabetes in males and females. Obesity 23, 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zitzmann M.(2009). Testosterone deficiency, insulin resistance and the metabolic syndrome. Nat. Rev. Endocrinol 5, 673–681. [DOI] [PubMed] [Google Scholar]
- 3.Traish AM (2014). Adverse health effects of testosterone deficiency (TD) in men. Steroids 88, 106–116. [DOI] [PubMed] [Google Scholar]
- 4.Kelly DM, and Jones TH (2013). Testosterone: a metabolic hormone in health and disease. J. Endocrinol 217, R25–R45. [DOI] [PubMed] [Google Scholar]
- 5.Saad F.(2017). Testosterone therapy and glucose homeostasis in men with testosterone deficiency (hypogonadism). Adv. Exp. Med. Biol 1043, 527–558. [DOI] [PubMed] [Google Scholar]
- 6.Wittert G, Bracken K, Robledo KP, Grossmann M, Yeap BB, Handelsman DJ, Stuckey B, Conway A, Inder W, McLachlan R, et al. (2021). Testosterone treatment to prevent or revert type 2 diabetes in men enrolled in a lifestyle programme (T4DM): a randomised, double-blind, placebo-controlled, 2-year, phase 3b trial. Lancet Diabetes Endocrinol. 9, 32–45. [DOI] [PubMed] [Google Scholar]
- 7.Yassin A, Haider A, Haider KS, Caliber M, Doros G, Saad F, and Garvey WT (2019). Testosterone therapy in men with hypogonadism prevents progression from prediabetes to type 2 diabetes: eight-year data from a registry study. Diabetes Care 42, 1104–1111. [DOI] [PubMed] [Google Scholar]
- 8.Xu W, Morford J, and Mauvais-Jarvis F.(2019). Emerging role of testosterone in pancreatic b-cell function and insulin secretion. J. Endocrinol 240, R97–R105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polonsky KS (1994). The beta-cell in diabetes: from molecular genetics to clinical research. Diabetes 44, 705–717. 1995. [DOI] [PubMed] [Google Scholar]
- 10.Prentki M, and Nolan CJ (2006). Islet beta cell failure in type 2 diabetes. J. Clin. Invest 116, 1802–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weyer C, Bogardus C, Mott DM, and Pratley RE (1999). The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Invest 104, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Navarro G, Xu W, Jacobson DA, Wicksteed B, Allard C, Zhang G, De Gendt K, Kim SH, Wu H, Zhang H, et al. (2016). Extranuclear actions of the androgen receptor enhance glucose-stimulated insulin secretion in the male. Cell Metabol. 23, 837–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu W, Niu T, Xu B, Navarro G, Schipma MJ, and Mauvais-Jarvis F.(2017). Androgen receptor-deficient islet beta-cells exhibit alteration in genetic markers of insulin secretion and inflammation. A transcriptome analysis in the male mouse. J. Diabet. Complicat 31, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu W, Schiffer L, Qadir MMF, Zhang Y, Hawley J, Mota De Sa P, Keevil BG, Wu H, Arlt W, and Mauvais-Jarvis F.(2020). Intracrine testosterone activation in human pancreatic b cells stimulates insulin secretion. Diabetes 69, 2392–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Göke R, Fehmann HC, Linn T, Schmidt H, Krause M, Eng J, and Göke B.(1993). Exendin-4 is a high potency agonist and truncated exendin-(9–39)-amide an antagonist at the glucagon-like peptide 1-(7–36)-amide receptor of insulin-secreting beta-cells. J. Biol. Chem 268, 19650–19655. [PubMed] [Google Scholar]
- 16.Wilson JD, Griffin JE, and Russell DW (1993). Steroid 5a-reductase 2 deficiency. Endocr. Rev 14, 577–593. [DOI] [PubMed] [Google Scholar]
- 17.Eckhardt M, Langkopf E, Mark M, Tadayyon M, Thomas L, Nar H, Pfrengle W, Guth B, Lotz R, Sieger P, et al. (2007). 8-(3-(R)-aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione (BI 1356), a highly potent, selective, long-acting, and orally bioavailable DPP-4 inhibitor for the treatment of type 2 diabetes. J. Med. Chem 50, 6450–6453. [DOI] [PubMed] [Google Scholar]
- 18.Pierce KL, Premont RT, and Lefkowitz RJ (2002). Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol 3, 639–650. [DOI] [PubMed] [Google Scholar]
- 19.Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, and Newgard CB (2000). Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424–430. [DOI] [PubMed] [Google Scholar]
- 20.Hodson DJ, Mitchell RK, Marselli L, Pullen TJ, Gimeno Brias S, Semplici F, Everett KL, Cooper DMF, Bugliani M, Marchetti P, et al. (2014). ADCY5 couples glucose to insulin secretion in human islets. Diabetes 63, 3009–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuna RS, Girada SB, Asalla S, Vallentyne J, Maddika S, Patterson JT, Smiley DL, DiMarchi RD, and Mitra P.(2013). Glucagon-like peptide-1 receptor-mediated endosomal cAMP generation promotes glucose-stimulated insulin secretion in pancreatic b-cells. Am. J. Physiol. Endocrinol. Metab 305, E161–E170. [DOI] [PubMed] [Google Scholar]
- 22.Jones B, Buenaventura T, Kanda N, Chabosseau P, Owen BM, Scott R, Goldin R, Angkathunyakul N, Correâ IR Jr., Bosco D, et al. (2018). Targeting GLP-1 receptor trafficking to improve agonist efficacy. Nat. Commun 9, 1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burgers PP, Ma Y, Margarucci L, Mackey M, van der Heyden MAG, Ellisman M, Scholten A, Taylor SS, and Heck AJR (2012). A small novel A-kinase anchoring protein (AKAP) that localizes specifically protein kinase A-regulatory subunit I (PKA-RI) to the plasma membrane. J. Biol. Chem 287, 43789–43797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamenetsky M, Middelhaufe S, Bank EM, Levin LR, Buck J, and Steegborn C.(2006). Molecular details of cAMP generation in mammalian cells: a tale of two systems. J. Mol. Biol 362, 623–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zippin JH, Farrell J, Huron D, Kamenetsky M, Hess KC, Fischman DA, Levin LR, and Buck J.(2004). Bicarbonate-responsive “soluble” adenylyl cyclase defines a nuclear cAMP microdomain. J. Cell Biol 164, 527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zippin JH, Chen Y, Straub SG, Hess KC, Diaz A, Lee D, Tso P, Holz GG, Sharp GWG, Levin LR, and Buck J.(2013). CO2/HCO3(−)- and calcium-regulated soluble adenylyl cyclase as a physiological ATP sensor. J. Biol. Chem 288, 33283–33291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramos-Espiritu L, Kleinboelting S, Navarrete FA, Alvau A, Visconti PE, Valsecchi F, Starkov A, Manfredi G, Buck H, Adura C, et al. (2016). Discovery of LRE1 as a specific and allosteric inhibitor of soluble adenylyl cyclase. Nat. Chem. Biol 12, 838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bitterman JL, Ramos-Espiritu L, Diaz A, Levin LR, and Buck J.(2013). J. Pharmacological distinction between soluble and transmembrane adenylyl cyclases. J. Pharmacol. Exp. Therapeut 347, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fushimi M, Buck H, Balbach M, Gorovyy A, Ferreira J, Rossetti T, Kaur N, Levin LR, Buck J, Quast J, et al. (2021). Discovery of TDI-10229: a potent and orally bioavailable inhibitor of soluble adenylyl cyclase (sAC, ADCY10). ACS Med. Chem. Lett 12, 1283–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steward MC, Ishiguro H, and Case RM (2005). Mechanisms of bicarbonate secretion in the pancreatic duct. Annu. Rev. Physiol 67, 377–409. [DOI] [PubMed] [Google Scholar]
- 31.Taddei A, Folli C, Zegarra-Moran O, Fanen P, Verkman AS, and Galietta LJV (2004). Altered channel Gαting mechanism for CFTR inhibition by a high-affinity thiazolidinone blocker. FEBS Lett. 558, 52–56. [DOI] [PubMed] [Google Scholar]
- 32.Ch’En FFT, Villafuerte FC, Swietach P, Cobden PM, and Vaughan-Jones RD (2008). S0859, an N-cyanosulphonamide inhibitor of sodium-bicarbonate cotransport in the heart. Br. J. Pharmacol 153, 972–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalwat MA, and Thurmond DC (2013). Signaling mechanisms of glucose-induced F-actin remodeling in pancreatic islet β cells. Exp. Mol. Med 45, e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stiernet P, Guiot Y, Gilon P, and Henquin JC (2006). Glucose acutely decreases pH of secretory granules in mouse pancreatic islets. Mechanisms and influence on insulin secretion. J. Biol. Chem 281, 22142– 22151. [DOI] [PubMed] [Google Scholar]
- 35.Papadopulos A, Tomatis VM, Kasula R, and Meunier FA (2013). The cortical acto-Myosin network: from diffusion barrier to functional Gαteway in the transport of neurosecretory vesicles to the plasma membrane. Front. Endocrinol 4, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Veluthakal R, and Thurmond DC (2021). Emerging roles of small GTPases in islet b-cell function. Cells 10, 1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han S, Mistry A, Chang JS, Cunningham D, Griffor M, Bonnette PC, Wang H, Chrunyk BA, Aspnes GE, Walker DP, et al. (2009). Structural characterization of proline-rich tyrosine kinase 2 (PYK2) reveals a unique (DFG-out) conformation and enables inhibitor design. J. Biol. Chem 284, 13193–13201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crompton BD, Carlton AL, Thorner AR, Christie AL, Du J, Calicchio ML, Rivera MN, Fleming MD, Kohl NE, Kung AL, and Stegmaier K.(2013). High-throughput tyrosine kinase activity profiling identifies FAK as a candidate therapeutic target in Ewing sarcoma. Cancer Res. 73, 2873–2883. [DOI] [PubMed] [Google Scholar]
- 39.Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, Luzzio MJ, Cooper B, Kath JC, Roberts WG, and Parsons JT (2007). Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem 282, 14845–14852. [DOI] [PubMed] [Google Scholar]
- 40.Wong WP, Tiano JP, Liu S, Hewitt SC, Le May C, Dalle S, Katzenellenbogen JA, Katzenellenbogen BS, Korach KS, and Mauvais-Jarvis F.(2010). Extranuclear estrogen receptor-alpha stimulates NeuroD1 binding to the insulin promoter and favors insulin synthesis. Proc Natl Acad Sci USA 107, 13057–13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, Barone MV, Ametrano D, Zannini MS, Abbondanza C, and Auricchio F.(2000). Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 19, 5406–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, et al. (2008). The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem 51, 5522–5532. [DOI] [PubMed] [Google Scholar]
- 43.Burger MT, Pecchi S, Wagman A, Ni ZJ, Knapp M, Hendrickson T, Atallah G, Pfister K, Zhang Y, Bartulis S, et al. (2011). Identification of NVP-BKM120 as a potent, selective, orally bioavailable class I PI3 ki-nase inhibitor for treating cancer. ACS Med. Chem. Lett 2, 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thompson WR, Guilluy C, Xie Z, Sen B, Brobst KE, Yen SS, Uzer G, Styner M, Case N, Burridge K, and Rubin J.(2013). Mechanically activated Fyn utilizes mTORC2 to regulate RhoA and adipogenesis in mesenchymal stem cells. Stem Cell. 31, 2528–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alejandro EU, Bozadjieva N, Blandino-Rosano M, Wasan MA, Elghazi L, Vadrevu S, Satin L, and Bernal-Mizrachi E.(2017). Overexpression of kinase-dead mTOR impairs glucose homeostasis by regulating insulin secretion and not beta-cell mass. Diabetes 66, 2150–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benavides-Serrato A, Lee J, Holmes B, Landon KA, Bashir T, Jung ME, Lichtenstein A, and Gera J.(2017). Specific blockade of Rictor-mTOR association inhibits mTORC2 activity and is cytotoxic in glioblastoma. PLoS One 12, e0176599. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Hoppa MB, Jones E, Karanauskaite J, Ramracheya R, Braun M, Collins SC, Zhang Q, Clark A, Eliasson L, Genoud C, et al. (2012). Multivesicular exocytosis in rat pancreatic beta cells. Diabetologia 55, 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spurlin BA, and Thurmond DC (2006). Syntaxin 4 facilitates biphasic glucose-stimulated insulin secretion from pancreatic beta-cells. Mol. Endocrinol 20, 183–193. [DOI] [PubMed] [Google Scholar]
- 49.Mauvais-Jarvis F, Lange CA, and Levin ER (2022). Membrane-initiated estrogen, androgen and progesterone receptor signaling in health and disease. Endocr. Rev 43, 720–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnstone TB, Agarwal SR, Harvey RD, and Ostrom RS (2018). cAMP signaling compartmentation: adenylyl cyclases as anchors of dynamic signaling complexes. Mol. Pharmacol 93, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pavlova NN, Zhu J, and Thompson CB (2022). The hallmarks of cancer metabolism: still emerging. Cell Metab. 34, 355–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berger HA, Anderson MP, Gregory RJ, Thompson S, Howard PW, Maurer RA, Mulligan R, Smith AE, and Welsh MJ (1991). Identification and regulation of the cystic fibrosis transmembrane conductance regulator-generated chloride channel. J. Clin. Invest 88, 1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, Lam CS, Wu F, Wang W, Duan Y, and Huang P.(2005). Regulation of CFTR channels by HCO(3)–sensitive soluble adenylyl cyclase in human airway epithelial cells. Am. J. Physiol. Cell Physiol 289, C1145–C1151. [DOI] [PubMed] [Google Scholar]
- 54.Rahman N, Ramos-Espiritu L, Milner TA, Buck J, and Levin LR (2016). Soluble adenylyl cyclase is essential for proper lysosomal acidification. J. Gen. Physiol 148, 325–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalwat MA, Wiseman DA, Luo W, Wang Z, and Thurmond DC (2012). Gelsolin associates with the N terminus of syntaxin 4 to regulate insulin granule exocytosis. Mol. Endocrinol 26, 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tomas A, Yermen B, Min L, Pessin JE, and Halban PA (2006). Regulation of pancreatic beta-cell insulin secretion by actin cytoskeleton remodelling: role of gelsolin and cooperation with the MAPK signalling pathway. J. Cell Sci 119, 2156–2167. [DOI] [PubMed] [Google Scholar]
- 57.Rondas D, Tomas A, Soto-Ribeiro M, Wehrle-Haller B, and Halban PA (2012). Novel mechanistic link between focal adhesion remodeling and glucose-stimulated insulin secretion. J. Biol. Chem 287, 2423–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chatterjee Bhowmick D, Aslamy A, Bhattacharya S, Oh E, Ahn M, and Thurmond DC (2022). DOC2b enhances b-cell function via a novel tyrosine phosphorylation-dependent mechanism. Diabetes 71, 1246–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu L, Das S, Losert W, and Parent CA (2010). mTORC2 regulates neutrophil chemotaxis in a cAMP-and RhoA-dependent fashion. Dev. Cell 19, 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Olsen JM, Sato M, Dallner OS, Sandströ m AL, Pisani DF, Chambard JC, Amri EZ, Hutchinson DS, and Bengtsson T.(2014). Glucose uptake in brown fat cells is dependent on mTOR complex 2-promoted GLUT1 translocation. J. Cell Biol 207, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sato M, Dehvari N, Oberg AI, Dallner OS, Sandström AL, Olsen JM, Csikasz RI, Summers RJ, Hutchinson DS, and Bengtsson T.(2014). Improving type 2 diabetes through a distinct adrenergic signaling pathway involving mTORC2 that mediates glucose uptake in skeletal muscle. Diabetes 63, 4115–4129. [DOI] [PubMed] [Google Scholar]
- 62.Ebner M, Sinkovics B, Szczygie1 M, Ribeiro DW, and Yudushkin I.(2017). Localization of mTORC2 activity inside cells. J. Cell Biol 216, 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bishop AL, and Hall A.(2000). Rho GTPases and their effector proteins. Biochem. J 348, 241–255. [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Z, and Thurmond DC (2009). Mechanisms of biphasic insulingranule exocytosis -roles of the cytoskeleton, small GTPases and SNARE proteins. J. Cell Sci 122, 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mohler ML, Bohl CE, Jones A, Coss CC, Narayanan R, He Y, Hwang DJ, Dalton JT, and Miller DD (2009). Nonsteroidal selective androgen receptor modulators (SARMs): dissociating the anabolic and androgenic activities of the androgen receptor for therapeutic benefit. J. Med. Chem 52, 3597–3617. [DOI] [PubMed] [Google Scholar]
- 66.De Gendt K, Swinnen JV, Saunders PTK, Schoonjans L, Dewerchin M, Devos A, Tan K, Atanassova N, Claessens F, Lećureuil C, et al. (2004). A Sertoli cell-selective knockout of the androgen receptor causes spermatogenic arrest in meiosis. Proc. Natl. Acad. Sci. USA 101, 1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oropeza D, Jouvet N, Budry L, Campbell JE, Bouyakdan K, Lacombe J, Perron G, Bergeron V, Neuman JC, Brar HK, et al. (2015). Phenotypic characterization of MIP-CreERT1Lphi mice with transgene-driven islet expression of human growth hormone. Diabetes 64, 3798–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chambers AP, Sorrell JE, Haller A, Roelofs K, Hutch CR, Kim KS, Gutierrez-Aguilar R, Li B, Drucker DJ, D’Alessio DA, et al. (2017). The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metabol. 25, 927–934.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee JY, Ristow M, Lin X, White MF, Magnuson MA, and Hennighausen L.(2006). RIP-Cre revisited, evidence for impairments of pancreatic b-cell function. J. Biol. Chem 281, 2649–2653. [DOI] [PubMed] [Google Scholar]
- 70.Blandino-Rosano M, Barbaresso R, Jimenez-Palomares M, Bozadjieva N, Werneck-de-Castro JP, Hatanaka M, Mirmira RG, Sonenberg N, Liu M, Ru€egg MA, et al. (2017). Loss of mTORC1 signalling impairs b-cell homeostasis and insulin processing. Nat. Commun 8, 16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bentzinger CF, Romanino K, Cloëtta D, Lin S, Mascarenhas JB, Oliveri F, Xia J, Casanova E, Costa CF, Brink M, et al. (2008). Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metabol. 8, 411–424. [DOI] [PubMed] [Google Scholar]
- 72.Tiano JP, Delghingaro-Augusto V, Le May C, Liu S, Kaw MK, Khuder SS, Latour MG, Bhatt SA, Korach KS, Najjar SM, et al. (2011). Estrogen receptor activation reduces lipid synthesis in pancreatic islets and prevents beta cell failure in rodent models of type 2 diabetes. J. Clin. Invest 121, 3331–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Klarenbeek JB, Goedhart J, Hink MA, Gαdella TWJ, and Jalink K.(2011). A mTurquoise-based cAMP sensor for both FLIM and ratiometric read-out has improved dynamic range. PLoS One 6, e19170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsvetanova NG, and von Zastrow M.(2014). Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat. Chem. Biol 10, 1061–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hafemeister C, and Satija R.(2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zappia L, and Oshlack A.(2018). Clustering trees: a visualization for evaluating clusterings at multiple resolutions. GigaScience 7, giy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Inoue A, Raimondi F, Kadji FMN, Singh G, Kishi T, Uwamizu A, Ono Y, Shinjo Y, Ishida S, Arang N, et al. (2019). Illuminating G-protein-coupling selectivity of GPCRs. Cell 177, 1933–1947.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wiseman DA, Kalwat MA, and Thurmond DC (2011). Stimulusinduced S-nitrosylation of Syntaxin 4 impacts insulin granule exocytosis. J. Biol. Chem 286, 16344–16354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zaborska KE, Dadi PK, Dickerson MT, Nakhe AY, Thorson AS, Schaub CM, Graff SM, Stanley JE, Kondapavuluru RS, Denton JS, and Jacobson DA (2020). Lactate activation of α-cell K(ATP) channels inhibits glucagon secretion by hyperpolarizing the membrane potential and reducing Ca(2+) entry. Mol. Metabol 42, 101056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Merrins MJ, and Stuenkel EL (2008). Kinetics of Rab27a-dependent actions on vesicle docking and priming in pancreatic beta-cells. J. Physiol 586, 5367–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single cell RNA sequencing data has been deposited in the National Center for Biotechnology Information (NCBI) gene expression omnibus (GEO) with the accession number GEO: GSE201256, mass spectrometry data at EMBL-EBI Proteomics Identification database (PRIDE) with accession number PRIDE: PXD041592 and, metabolomics data at UCSD metabolomics Workbench/National Metabolomics Data Repository (NMDR) with accession number NMDR: PR001654. All data reported in this paper will be shared by the lead contact upon request.
A description of coding environments required to reproduce scRNAseq analysis in this paper are outlined in: https://github.com/FMJLabTulane/AR-DHT.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.