

Abstract

The aim of this work was to bring forth some new hybrid molecules having pharmacologically potent indole and 1,3,4-oxadiazole heterocyclic moieties unified with a propanamide entity. The synthetic methodology was initiated by esterification of 2-(1H-indol-3-yl)acetic acid (1) in a catalytic amount of sulfuric acid and ethanol in excess, to form ethyl 2-(1H-indol-3-yl)acetate (2), which was converted to 2-(1H-indol-3-yl)acetohydrazide (3) and further transformed to 5-(1H-indole-3-yl-methyl)-1,3,4-oxadiazole-2-thiol (4). 3-Bromopropanoyl chloride (5) was reacted with various amines (6a–s) in aqueous alkaline medium to generate a series of electrophiles, 3-bromo-N-(substituted)propanamides (7a–s), and these were further reacted with nucleophile 4 in DMF and NaH base to yield the targeted N-(substituted)-3-{(5-(1H-indol-3-ylmethyl)-1,3,4-oxadiazol-2-yl)sulfanyl}propanamides (8a–s). The chemical structures of these biheterocyclic propanamides were confirmed by IR, 1H NMR, 13C NMR, and EI-MS spectral techniques. These compounds were evaluated for their enzyme inhibitory potentials against the α-glucosidase enzyme, where the compound 8l showed promising enzyme inhibitory potential with an IC50 value less than that of the standard acarbose. Molecular docking results of these molecules were coherent with the results of their enzyme inhibitory potentials. Cytotoxicity was assessed by the percentage of hemolytic activity method, and these compounds generally exhibited very low values as compared to the reference standard, Triton-X. Hence, some of these biheterocyclic propanamides might be considered as salient therapeutic agents in further stages of antidiabetic drug development.
1. Introduction
The identification of α-glucosidase inhibitors has been dynamically pursued with the aim of developing therapeutics for the treatment of diabetes mellitus type 2 (D2M).1 Diabetes mellitus is a group of metabolic dysfunctions of carbohydrate metabolism characterized by hyperglycemia (high blood glucose levels), resulting from defects in insulin action, insulin secretion, or both.2 α-Glucosidase is a membrane-bound enzyme at the epithelium of the small intestine and hydrolyzes terminal nonreducing 1–4 linked α-glucose residues to release monomeric glucose molecules, which is mainly responsible for causing hyperglycemia.3 The inhibition of α-glucosidase can delay the carbohydrate absorption and has been used as one of the therapeutic approaches for the treatment of diabetes.3,4 Therefore, design and synthesis of small hybrid molecules as α-glucosidase inhibitors is an important research area in medicinal chemistry.5
Many of the biologically active synthetic compounds have a five-membered nitrogen-containing heterocyclic ring in their structures. It has been established from the structure–activity relationship of synthetic compounds that almost half of the therapeutic agents consist of heterocyclic moieties. Indole is an aromatic heterocyclic organic compound. It has a bicyclic structure, consisting of a six-membered benzene ring fused to a five-membered nitrogen-containing pyrrole ring. Indole is a popular component of fragrances and the precursor to many pharmaceuticals. Notably, the indolic amino acid tryptophan is the precursor of the neurotransmitter serotonin. In the past few years, it was reported that indole, its bioisosters, and derivatives have antimicrobial activity against Gram-negative and Gram-positive bacteria and yeast. Other properties of indole-containing drug molecules include antihypertensive, antidepressant, antipsychotic agents, antiemetic, analgesic, antiasthmatic, antiviral, antiarrhythmic activities, involvement in nonsteroidal anti-inflammatory drugs (NSAIDS), B-blocker drug toxins, inhibitors of RNA polymerase-11, agonists for the cannabinoid receptors, non-nucleoside reverse transcriptase inhibitors, opioid agonists, and in sexual dysfunction.5 In recent years, the oxadiazole chemistry has also been studied extensively, and some of the drugs comprising oxadiazole in association with other heterocyclic rings are in clinical practice. Literature survey revealed that 1,3,4-oxadiazoles are related to a wide range of pharmacological activities.5,6 The 2,5-substituted oxadiazole derivatives have been reported to possess anticonvulsant activity and antifungal activity.7 Compounds containing an oxadiazole moiety have been reported to possess several biological properties such as anticancer,8,9 antimicrobial,10−12 anti-inflammatory,13 anticonvulsant,14 antioxidant,15 and anti-HIV.16 The mechanical stability of the membrane of red blood cells (RBCs) is a good indicator to evaluate the in vitro effects of various compounds while screening for cytotoxicity.17,18 Treating cells with a cytotoxic compound (including drug molecules) can cause different damages to red blood cells in animals and human beings. The cells may undergo a loss of membrane integrity and die rapidly because of cell lysis.19
The main drawbacks of the currently used α-glucosidase inhibitors, such as acarbose, are the side effects such as abdominal distention, flatulence, meteorism, and possible diarrhea.20 In continuation of our previous efforts on indole-oxadiazole-bearing acetamides as α-glucosidase inhibitors,4,21,22 the present investigation was aimed to explore the antidiabetic potential of newly synthesized indole-oxadiazole-bearing propanamides. Moreover, the hemolytic profile of these molecules was ascertained, and in silico molecular docking studies were also carried out to find out the best binding conformational pose of these synthesized ligands against the α-glucosidase enzyme.
2. Results and Discussion
In the presented work, N-(substituted)-3-{(5-(1H-indol-3-ylmethyl)-1,3,4-oxadiazol-2-yl)sulfanyl}propanamides (8a–s) were synthesized, keeping in consideration the increasing demand for new antidiabetic agents. These compounds were explored for their enzyme inhibitory potential against α-glucosidase. The cytotoxicity of these molecules was evaluated by a percent hemolysis study. Moreover, their molecular docking studies were also carried out to delineate their binding affinity and binding mode of inhibition.
2.1. Chemistry
The synthetic route adopted is outlined in Scheme 1, and various substituents (−R groups) are listed in Table 1. The synthesis was initiated by the esterification of 2-(1H-indol-3-yl)acetic acid (1) with a catalytic amount of H2SO4 in ethanol, which was taken in excess to shift the equilibrium to the product side, i.e., ethyl 2-(1H-indol-3-yl)acetate (2), due to reversibility of the reaction. This reaction mixture was refluxed for 7–8 h, and on completion, 10% aqueous Na2CO3 was added to neutralize the catalyst and unreacted starting acid. In the second step, the ester 2 was reacted with hydrazine monohydrate in a methanol solvent and stirred for 10 h at room temperature. After complete conversion, methanol was distilled off, and the product was washed with cold n-hexane (soluble in water) to obtain corresponding 2-(1H-indol-3-yl)acetohydrazide (3) in a good yield. In the third step, intramolecular cyclization was carried out by refluxing 3, for 6–7 h, with CS2 and KOH in ethanol. After completion, reaction contents were acidified with dil. HCl keeping the pH at 5–6, and the precipitates thus obtained were filtered and washed with cold distilled water to afford a thiol-containing nuleophile, 5-(1H-indole-3-yl-methyl)-1,3,4-oxadiazole-2-thiol (4). In the parallel sequence of reactions, 3-bromopropanoyl chloride (5) was reacted with respective cycloalkyl/aryl/aralkyl amines (6a–s) in the presence of 10% aqueous Na2CO3 to form 3-bromo-N-(substituted)propanamides (7a–s). Aromatic amines that had electron-donating groups were better reactive in these nucleophilic reactions than those ones having electron-withdrawing groups. One by one, the electrophiles, 7a–s, were reacted with 4 in a polar aprotic solvent, i.e., DMF, and NaH as an activator, to achieve the targeted N-(substituted)-3-{(5-(1H-indol-3-ylmethyl)-1,3,4-oxadiazol-2-yl)sulfanyl}propanamides (8a–s), and their structures were corroborated with spectral techniques.
Scheme 1. Outline for Synthesis of N-(Substituted)-3-{(5-(1H-indol-3-ylmethyl)-1,3,4-oxadiazol-2-yl)sulfanyl}propanamides (8a–s).
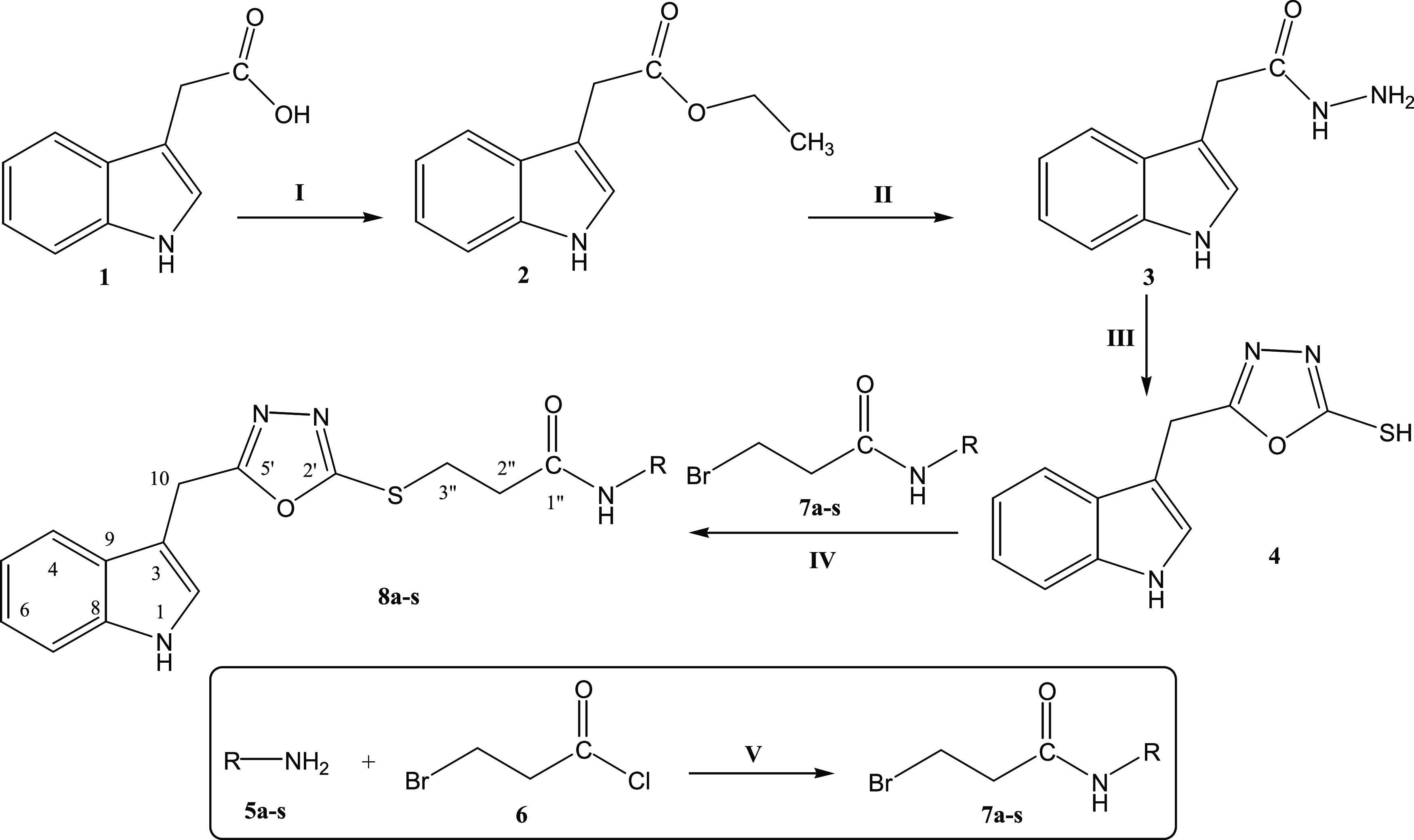
Reagents & conditions: (I) 2-(1H-indol-3-yl)acetic acid (1)/ethanol/sulfuric acid/ reflux/7–8 h. (II) Ethyl 2-(1H-indol-3-yl)acetate (2) methanol/hydrazine monohydrate/stirring/10 h. (III) 2-(1H-Indol-3-yl)acetohydrazide (3)/ethanol/CS2/KOH/reflux/7 h. (IV) Cycloalkyl/aralkyl/aryl amines (5a–s)/3-bromopropanoyl chloride (6)/10% Na2CO3/manual shaking/RT/25–30 min. (V) 5-(1H-Indol-3-yl-methyl)-1,3,4-oxadiazole-2-thiol (4)/3-bromo-N-(substituted)propanamides (7a–s)/NaH/DMF/stirring/7–8 h.
Table 1. Different −R Groups in Compounds 5a–s, 7a–s, and 8a–s of Scheme 1.

The structural characterization of one of the biheterocyclic propanamides is given hereby in detail for the benefit of the readers. The compound 8r was purified as a light amorphous solid with 83% yield having a melting point of 110–112 °C. Its molecular formula, C22H22N4O2S, was established through its molecular ion peak in its EI-MS at m/z 406. The mass fragmentation pattern also supported this molecular formula. Moreover, counting the number of protons in its 1H NMR spectrum and the carbon resonances in its 13C NMR spectrum also augmented the assignment of the molecular formula. The vibrational studies of the molecules were examined using Fourier transform infrared (FT-IR) spectroscopy to affirm various functionalities. Various absorption bands appeared at υ 3340 (N–H), 3088 (Ar C–H), 1663 (C=N), 1653 (C=O str.), 1558 (Ar C=C), 1061 (C–O–C), and 632 (C–S) cm–1. The 1H NMR spectrum of this compound is given in Figures S1 and S2. Two singlets in the most downfield region at 11.05 (s, 1H, NH-1) and 9.86 (s, 1H, −CONH) ppm were assigned to heteroatom protons of the indole-3-ylmethyl group. Two ortho-coupled doublets with integration of one proton each at 7.51 (d, J = 7.8 Hz, 1H, H-4) and 7.38 (d, J = 7.2 Hz, 1H, H-7) ppm belonged to two methine protons of the phenyl ring of the indole-3-ylmethyl moiety. The remaining two protons of this ring resonated as two diortho-coupled triplets at 7.10 (t, J = 7.2 Hz, 1H, H-5) and 7.00 (t, J = 7.6 Hz, 1H, H-6) ppm. The methine proton of the pyrrole ring of the indole-3-ylmethyl moiety appeared as a broad singlet at 7.34 (br. s, 1H, H-2) ppm. Methylene protons of the indole-3-ylmethyl group appeared at 4.33 (s, 2H, CH2-10) ppm, as a singlet in the aliphatic region. Additionally, two broad singlets appeared with two- and one-proton integrations for the substituted 3,5-dimethylphenyl ring at 7.8 (br. s, 2H, H-2‴ & H-6‴) and 6.69 (br. s, 1H, H-4‴) ppm, respectively, in the aromatic region. The aliphatic region presented one intense singlet for six protons of two methyl groups at 2.22 (s, 6H, CH3-7‴ & CH3-8‴) ppm and two triplets for two propanoylmethylenes at 3.44 (t, J = 6.6 Hz, 2H, CH2-3″) and 2.84 (t, J = 6.6 Hz, 2H, CH3-2″) ppm. The 13C NMR spectrum of 8r was analyzed to confirm the carbon skeleton of the organic molecule (Figures S3 and S4). The spectrum presented 19 signals for 22 carbons, including nine quaternary at 168.53 (C-5′), 167.10 (C-1″), 163.21 (C-2′), 138.75 (C-1‴), 137.62 (C-3‴ & C-5‴), 136.15 (C-8), 126.57 (C-9), and 106.59 (C-3) ppm, eight methine signals at 124.75 (C-4‴), 124.16 (C-2), 121.30 (C-6), 118.75 (C-5), 118.15 (C-4), 116.86 (C-2‴ & C-6‴), and 111.58 (C-7) ppm, three methylene signals at 35.72 (C-2″), 27.74 (C-3″), and 21.42 (C-10) ppm, and two magnetically equivalent methyl carbons resonating as one signal at 21.05 (C-7‴ & C-8‴) ppm. Because of the symmetry in the 3,5-dimethylphenyl ring, three signals are depicted less from the total number of carbons contained in this entity. The EI-MS spectrum of 8r and its proposed fragmentation pattern are sketched in Figures S5 and S6, respectively. The base peak appeared at m/z 130 for the indole-3-ylmethyl cation, while the two peaks at m/z 176 and 231 were also very diagnostic to deduce its molecular structure. So, on the basis of aforementioned evidence, the structure of 8r was confirmed, and it was named as N-(3,5-dimethylphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide. The structures of all other derivatives in the series were also characterized in a similar manner.
2.2. α-Glucosidase Inhibitory, Hemolytic, and Molecular Docking Studies
All these biheterocyclic amides were subjected to in vitro evaluation for their α-glucosidase inhibitory potential, and it was inferred from the results that these molecules exhibited moderate to good inhibitory potentials as shown by their IC50 values (Table 2). A promising inhibitory potential was shown by compound 8l with an IC50 value of 25.78 ± 0.05 μM, which was lower and better than the standard acarbose (38.25 ± 0.12 μM). This good activity may be attributed to the presence of the N-phenylpropanamide group, due to which this molecule through its in silico study has shown the best conformational fit to the binding pocket of this enzyme. So, this molecule might prove to be a suitable inhibitor of α-glucosidase in further drug development investigations. Compounds 8p (2,6-dimethylphenyl), 8q (3,4-dimethylphenyl), 8r (3,5-dimethylphenyl), and 8f (3-methylphenyl) with IC50 values of 47.47 ± 0.13, 49.81 ± 0.17, 48.96 ± 0.13, and 50.15 ± 0.11 μM, respectively, also displayed considerable inhibitory potentials against the α-glucosidase enzyme. Synthesized compounds can be arranged in the following row according to their inhibitory activity: 8l > 8p > 8r > 8q > 8f > 8k > 8g > 8j > 8a > 8i > 8h > 8b > 8m > 8s > 8e > 8o > 8n > 8d > 8c (see Table 2 for IC50 values).
Table 2. Percent Inhibition at 0.5 mM and IC50 Values of Biheterocyclic Propanamides, 8a–s, for α-Glucosidasea.
| α-glucosidase |
||||
|---|---|---|---|---|
| compound | (%) inhibition | IC50 (μM) | hemolysis (%) | binding affinity (kcal/mol) |
| 8a | 95.25 ± 0.23 | 141.39 ± 0.12 | 2.66 ± 0.02 | –8.1 |
| 8b | 97.12 ± 0.38 | 171.46 ± 0.14 | 8.16 ± 0.03 | –8.5 |
| 8c | 87.76 ± 0.49 | 311.75 ± 0.16 | 57.08 ± 0.03 | –8.5 |
| 8d | 93.38 ± 0.38 | 232.75 ± 0.17 | 0.05 ± 0.01 | –8.9 |
| 8e | 98.56 ± 0.43 | 217.23 ± 0.12 | 3.85 ± 0.01 | –8.7 |
| 8f | 98.54 ± 0.39 | 50.15 ± 0.11 | 3.83 ± 0.02 | –8.6 |
| 8g | 98.14 ± 0.41 | 115.96 ± 0.15 | 1.57 ± 0.01 | –8.5 |
| 8h | 98.21 ± 0.31 | 159.8 ± 0.14 | 0.83 ± 0.01 | –9.3 |
| 8i | 84.25 ± 0.21 | 157.61 ± 0.11 | 4.16 ± 0.02 | –8.8 |
| 8j | 99.83 ± 0.36 | 128.62 ± 0.18 | 12.34 ± 0.03 | –9.4 |
| 8k | 96.72 ± 0.29 | 88.52 ± 0.13 | 2.66 ± 0.02 | –7.9 |
| 8l | 97.56 ± 0.57 | 25.78 ± 0.05 | 8.16 ± 0.02 | –9.7 |
| 8m | 97.56 ± 0.58 | 180.95 ± 0.11 | 57.08 ± 0.03 | –8.2 |
| 8n | 99.53 ± 0.42 | 229.75 ± 0.15 | 0.05 ± 0.01 | –8.7 |
| 8o | 97.11 ± 0.32 | 217.83 ± 0.26 | 3.85 ± 0.02 | –9.1 |
| 8p | 93.88 ± 0.21 | 47.47 ± 0.13 | 3.83 ± 0.02 | –9.4 |
| 8q | 98.51 ± 0.25 | 49.81 ± 0.17 | 1.57 ± 0.01 | –9.4 |
| 8r | 95.75 ± 0.29 | 48.96 ± 0.13 | 0.83 ± 0.01 | –8.2 |
| 8s | 95.83 ± 0.27 | 189.59 ± 0.48 | 4.16 ± 0.02 | –9.9 |
| acarbose | 92.23 ± 0.14 | 38.25 ± 0.12 | ||
| Triton-X | 95.32 ± 0.01 | |||
Note: IC50 values (concentration at which there is 50% enzyme inhibition) of compounds were calculated from the inhibition data obtained after doing assays at high dilutions of the compounds as given in the assay method, and data were computed using EZ-Fit Enzyme kinetics software (Perrella Scientific, Inc., Amherst, USA). Data are the mean of three values (mean ± S.E.M., n = 3), with S.E.M. denoting the standard error of the mean. Note: PBS (% hemolysis) = 2.45 ± 0.01%. Hemolytic activity and docking energy (kcal/mol) values are also given.
All the compounds were evaluated for their cytotoxicity profile in terms of the percentage of hemolytic activity. Most of the compounds showed (Table 2) very mild hemolytic activity values, where compound 8d having substitution of the 2-phenethyl group exhibited the lowest hemolytic activity (0.05 ± 0.01%) and the highest hemolytic activity was displayed by 8c (57.08 ± 0.03%) having substitution of the benzyl group. Anyhow, the overall values were much lower than Triton-X (95.32 ± 0.01%), which was used as a positive control. Phosphate-buffered saline (PBS: 2.45 ± 0.01% hemolysis value) was used as a negative control.
α-Glucosidase (EC no. 3.2.1.106) belongs to the class of hydrolase proteins and is considered as a receptor molecule against diabetes. The α-glucosidase comprises 811 amino acids with the secondary structure composed of 35% helices, 25% β-sheets, and 38% coils. The X-ray diffraction studies of α-glucosidase confirmed its resolutions of 2.04 Å. The α-glucosidase Ramachandran plot indicated that 97.6% of residues were present in favored regions. These selected Ramachandran graph values showed the good accuracy of phi (φ) and psi (ψ) angles among the coordinates of target proteins. The Ramachandran graph of α-glucosidase is shown in Figure S7.
The ligand-protein docked complexes were analyzed on the basis of minimum energy values and binding interaction (hydrogen/hydrophobic) patterns. All the synthesized compounds were bound in the active region of the target protein with different conformational poses. Based on in vitro results, compound 8l was the most active one having a good IC50 value (25.78 ± 0.05 μM). The decent activity of 8l and 8s was also justified by the docking results (−9.70 and −9.90 kcal/mol) whereby both bind inside the active region of the target protein. Our docking outcomes disclosed very good correlations with the in vitro experimental results. The binding energy values of all compounds are listed in Table 2.
Binding pocket analysis showed that compounds bind within the active region of the target protein (Figure 1). All the ligands were confined in the same positions having different binding poses. The common binding pattern showed the significance of the ligand interacting with α-glucosidase. In the target protein, chains B and C are the most active parts, and most of the interacting residues were located in these domains.
Figure 1.

Binding pocket of the target protein 4J5T.
The structure–activity relationship (SAR) analysis showed that 8l binds with α-glucosidase in the active region of the target protein. The active binding residues of α-glucosidase that are functionally participating in the inhibition and signaling pathways were justified from literature studies. It has been observed that Glu771 and Asp568 were considered as key functional players in the inhibition kinetics and downstream signaling pathways.23 Moreover, other studies also proposed that the binding pocket residues such as Asp568, Trp381, Trp710, Trp715, and Trp789 were are also present in the same binding domain of α-glucosidase.24,25 Our docking results showed that 8l binds with the active binding pocket of the target protein. The ligand 8l formed four hydrogen bonds with Asp568, Glu707, Glu771, and Arg445 residues with bond lengths of 3.46, 3.20, 2.96, and 2.10 Å, respectively (Figure 2A,B). The 2D depiction of all docking interaction results is shown in Figures S8–S25.
Figure 2.

Docking of 8l with α-glucosidase. (A) The docking complex of α-glucosidase is shown in purple color in surface format to depict the binding pocket of 8l within the active region of the target protein. (B) The binding complex is shown in which the basic skeleton of 8l is depicted in green color while heteroatoms like oxygen, sulfur, and nitrogen are labeled with red, yellow, and dark-blue colors, respectively. The blue color was used to show the interacting residues of the target protein, while the receptor (α-glucosidase) is shown in ribbon format. The red color-labeled residues actively participated in hydrogen bonding, and their distances are shown in angstrom (Å).
Acarbose and 8l docked complexes were superimposed to check the binding interaction pattern. Results showed that both acarbose and 8l bind at the same position at little different conformation positions. However, the interacting residues were approximately the same in both complexes. Glu707 was a common residue, which is involved in hydrogen bonding. Moreover, Trp715, Tyr709, and Trp789 were also common in both complexes (Figure 3). The common interaction pattern confers that our synthetic compound has the same binding behavior as the standard structure.
Figure 3.

Docking complex acarbose and 8l in a superimposed form.
3. Conclusions
The designed biheterocyclic hybrids amalgamated with propanamides were synthesized in a successful manner, and their structures were corroborated authentically with spectral analysis. The screening of these amides against α-glucosidase explored that the molecule, 8l, exhibited very promising inhibitory potential with an IC50 value smaller than that of the standard acarbose. This compound also showed mild cytotoxicity. Moreover, it binds within the active region of the target protein with a good binding affinity. So, based upon the in vitro enzyme inhibitory and in silico molecular docking results, it can be summated that 8l might be a potential lead molecule in further drug development studies for the treatment of D2M.
4. Experimental Section
4.1. General
All the chemicals, along with analytical-grade solvents, were purchased from Sigma Aldrich, Alfa Aesar (Germany), or Merck through local suppliers. Precoated silica gel Al plates were used for TLC with ethyl acetate and n-hexane as a solvent system. Spots were detected by UV254. A Gallenkamp apparatus was used to detect melting points in capillary tubes. IR spectra (ν, cm–1) were recorded by the KBr pellet method in a Jasco-320-A spectrometer. 1H NMR spectra (δ, ppm) were recorded at 600 MHz (13C NMR spectra, at 150 MHz) in DMSO-d6 using a Bruker Advance III 600 Ascend spectrometer using a BBO probe. The electron ionization mass (EI-MS) spectra were measured on a JEOL JMS-600H instrument with a data processing system.
4.2. Synthesis of Ethyl 2-(1H-Indol-3-yl)acetate (2)
2-(1H-Indol-3-yl)acetic acid (0.2 mol; 1) dissolved in absolute ethanol (70 mL) and a catalytic amount of concentrated sulfuric acid (20 mL) were taken in a 500 mL round-bottom (RB) flask and refluxed for 7–8 h until the maximum completion of the reaction, supervised through TLC. Some glass chips were added to avoid bumping of reaction contents. At the end, the reaction mixture was neutralized with 10% aqueous sodium carbonate (40 mL). The product was isolated by solvent extraction by chloroform (50–60 mL × 3). The solvent was distilled off, and the compound 2 was obtained as a reddish-brown liquid, which became solid at refrigeration. Brownish liquid; mol. formula: C12H13N2O; mol. weight: 203 g/mol; IR (KBr, υ, cm–1): 3415 (N–H), 3037 (Ar C–H), 1733 (C=O), 1534 (Ar C=C); 1H NMR (400 MHz, DMSO-d6, δ, ppm): 10.9 (s, 1H, NH-1), 7.48 (br. d, J = 8.0 Hz, 1H, H-4), 7.34 (br. d, J = 8.0 Hz, 1H, H-7), 7.23 (br. s, 1H, H-2), 7.06 (t, J = 7.6 Hz, 1H, H-5), 6.97 (t, J = 7.6 Hz, 1H, H-6), 4.16 (q, J = 7.2 Hz, 2H, CH2-1′), 3.71 (s, 2H, CH2-10), 1.17 (t, J = 7.2 Hz, 3H, CH3-2′); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 157.2 (C-1′), 136.1 (C-8), 130.0 (C-9), 124.3 (C-2), 122.3 (C-6), 121.2 (C-4), 119.6 (C-5), 114.2 (C-7), 108.7 (C-3), 63.6 (C-2′), 32.4 (C-10), 16.2 (C-3′); EI-MS (m/z): 203 (C12H13N2O)·+ (M)·+, 158 (C10H8NO)+, 130 (C9H8N)+, 73 (C3H5O2)+.
4.3. Synthesis of 2-(1H-Indol-3-yl)acetohydrazide (3)
Ethyl 2-(1H-indol-3-yl)acetate (0.15 mol; 2) in 60 mL of methanol and hydrazine monohydrate (80%; 25 mL) were taken in a 500 mL round-bottom flask. The reaction mixture was stirred for 10 h. After absolute conversion, the acid hydrazide was obtained by distilling methanol off from the reaction mixture. The precipitates were filtered, washed with cold n-hexane, and air-dried to get pure 2-(1H-indol-3-yl)acetohydrazide (3). Brownish crystals; yield: 89%; m.p. 113 °C; mol. formula: C10H11N3O; mol. weight: 189 g/mol; IR (KBr, υ, cm–1): 3431 (N–H), 3032 (C–H Ar), 1630 (C=O), 1529 (Ar C=C); 1H NMR (400 MHz, DMSO-d6, δ, ppm): 10.8 (s, 1H, NH-1), 9.08 (s, 1H, NHNH2), 7.55 (br. d, J = 7.6 Hz, 1H, H-4), 7.31 (br. d, J = 8.0 Hz, 1H, H-7), 7.16 (br. s, 1H, H-2), 7.04 (t, J = 7.2 Hz, 1H, H-5), 6.95 (t, J = 7.6 Hz, 1H, H-6), 4.16 (br. s, 2H, NHNH2), 3.43 (s, 2H, CH2-10); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 159.1 (C-11), 137.4 (C-8), 129.1 (C-9), 125.3 (C-2), 123.6 (C-6), 120.2 (C-4), 118.5 (C-5), 113.4 (C-7), 108.5 (C-3), 32.3 (C-10); EI-MS (m/z): 189 (C10H11N3O)·+ (M)·+, 158 (C10H8NO)+, 130 (C9H8N)+, 116 (C8H6N)+, 59 (CHN2O)+.
4.4. Synthesis of 2-(1H-Indol-3-ylmethyl)-1,3,4-oxadiazol-5-thiol (4)
2-(1H-Indol-3-yl)acetohydrazide (20.0 g; 0.11 mol; 3) in absolute ethanol (100 mL) was added to a 500 mL RB flask. Potassium hydroxide (6.3 g; 0.11 mol) was added to the solution followed by the addition of carbon disulfide (14.0 mL; 0.22 mol), and the mixture was refluxed with stirring for 7 h. Progress of the reaction was monitored by TLC, and on completion, it was diluted with distilled water and acidified with dilute hydrochloric acid to pH 5–6. The precipitates formed were filtered, washed with water, and recrystallized from ethanol to obtain pure 5-(1H-indole-3-yl-methyl)-1,3,4-oxadiazole-2-thiol (4) in good yield. Dark-brown powder; yield: 76%; m.p. 125 °C; mol. formula: C11H9N3OS; mol. weight: 231 g/mol; IR (KBr, υ, cm–1): 3337 (N–H), 3085 (Ar C–H), 1562 (Ar C=C), 1666 (C=N), 1065 (C–O–C), 638 (C–S); 1H NMR (400 MHz, DMSO-d6, δ, ppm): δ 11.0 (s, 1H, NH-1), 7.49 (br. d, J = 7.6 Hz, 1H, H-4), 7.37 (br. d, J = 8.0 Hz, 1H, H-7), 7.34 (br. s, 1H, H-2), 7.09 (t, J = 7.6 Hz, 1H, H-5), 7.00 (t, J = 7.6 Hz, 1H, H-6), 4.20 (s, 2H, CH2-10); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 167.6 (C-5′), 162.8 (C-2′), 136.1 (C-8), 132.2 (C-9), 124.1 (C-2), 121.3 (C-6), 118.7 (C-5), 118.0 (C-4), 111.5 (C-7), 106.5 (C-3), 21.4 (C-10); EI-MS (m/z): 233 (C11H9N3OS + 2)·+ (M + 2)+, 231 (C11H9N3OS)·+ (M)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 130 (C9H8N)+, 103 (C2HN2OS + 2)+, 101 (C2HN2OS)+.
4.5. Synthesis of 3-Bromo-N-(substituted)propanamides (7a–s)
3-Bromo-N-(substituted)propanamides (7a–s) were synthesized by reacting various cycloalkyl/aryl/aralkyl amines (0.02 mol; 5a–s) with 3-bromopropanoyl chloride (6) in equimolar quantities (0.001 m) and shaking manually in 10% aqueous Na2CO3. Solid precipitates were formed after 25–30 min, filtered, and washed with cold distilled water to obtain the desired electrophiles (7a–s).
4.6. Synthesis of N-(Substituted)-3-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamides (8a–s)
5-(1H-Indole-3-yl-methyl)-1,3,4-oxadiazol-2-thiol (0.001 mol; 4), N,N-dimethylformamide (DMF, 7 mL) as a solvent, and NaH (0.002 mol) as an activator were taken in a 50 mL RB flask and stirred for half an hour. Equimolar quantities of 3-bromo-N-(substituted)propanamides (7a–s) as electrophiles were then added into the reaction mixture. The reaction mixture was stirred for 7–8 h at 35 °C. After absolute conversion, the reaction mixture was poured on crushed ice; precipitates thus formed were filtered, washed with distilled water, and dried to afford pure N-(substituted)-3-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamides (8a–s) in good yields.
4.6.1. N-(Cyclohexyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8a)
Dark-brown amorphous powder; yield: 85%; m.p. 95–97 °C; mol. formula C20H24N4O2S; mol. weight: 384 g/mol; IR (KBr, υ, cm–1): 3346 (N–H), 3081 (Ar C–H), 1668 (C=N), 1650 (C=O str.), 1558 (Ar C=C), 1062 (C–O–C), 635 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.15 (s, 1H, NH-1), 7.84 (d, J = 7.5 Hz, 1H, −CONH), 7.51 (br. d, J = 7.9 Hz, 1H, H-4), 7.38 (br. d, J = 8.1 Hz, 1H, H-7), 7.34 (br. s, 1H, H-2), 7.10 (t, J = 7.3 Hz, 1H, H-5), 7.00 (t, J = 7.3 Hz, 1H, H-6), 4.33 (s, 2H, CH2-10), 3.42 (br. s, 1H, H-1‴), 3.34 (t, J = 6.78 Hz, 2H, CH2-3″), 2.56 (t, J = 6.78 Hz, 2H, CH2-2″), 1.71–1.07 (m, 10H, CH2-2‴ to CH2-6‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.8 (C-5′), 167.7 (C-1″), 163.6 (C-2′), 136.2 (C-8), 131.4 (C-9), 124.1 (C-2), 121.6 (C-6), 118.8 (C-5), 118.6 (C-4), 111.3 (C-7), 106.8 (C-3), 49.8 (C-1‴), 36.5 (C-2″), 29.4 (C-2‴ & 6‴), 28.2 (C-3″), 22.4 (C-10), 22.1 (C-3‴ & 5‴), 21.8 (C-4‴); EI-MS (m/z): 386 (C20H24N4O2S + 2)·+ (M + 2)+, 384 (C20H24N4O2S)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 154 (C9H16NO)+, 130 (C9H8N)+, 98 (C7H12N)+, 83 (C6H11)+.
4.6.2. N-(Phenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2yl}sulfanyl)propanamide (8b)
Light-brown amorphous powder; yield: 81%; m.p. 134–135 °C; mol. formula: C20H18N4O2S; mol. weight: 378 g/mol; IR (KBr, υ, cm–1): 3336 (N–H), 3084 (Ar C–H), 1669 (C=N), 1062 (C–O–C), 1654 (C=O str.), 1558 (Ar C=C), 633 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.05 (s, 1H, NH-1), 10.02 (s, 1H, −CONH), 7.57 (br. d, J = 8.5 Hz, 2H, H-2‴ & H-6‴), 7.52 (d, J = 7.9 Hz, 1H, H-4), 7.37 (br. d, J = 8.1 Hz, 1H, H-7), 7.34 (br. s, 1H, H-2), 7.29 (br. t, 2H, J = 8.4 Hz, H-3‴ & H-5‴), 7.10 (t, J = 7.0 Hz, 1H, H-5), 7.04 (br. t, J = 7.38 Hz, 1H, H-4‴), 7.00 (t, J = 7.0 Hz, 1H, H-6), 4.33 (s, 1H, CH2-10), 3.44 (t, J = 6.78 Hz, 2H, CH2-3″), 2.87 (t J = 7.08 Hz, 2H, CH2-2″); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.7 (C-5′), 167.6 (C-1″), 163.8 (C-2′), 137.5 (C-1‴), 136.4 (C-8), 128.9 (C-9), 128.7 (C-3‴ & C-5‴), 126.5 (C-4‴), 124.1 (C-2), 121.6 (C-6), 118.5 (C-5), 118.2 (C-4), 118.0 (C-2‴ & C-6‴), 111.5 (C-7), 106.5 (C-3), 36.4 (C-2″), 28.2 (C-3″), 21.4 (C-10); EI-MS (m/z): 380 (C20H18N4O2S)·+ (M + 2)+, 378 (C8H16N4O2S)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 148 (C9H10NO)+, 147 (C9H9NO)+, 130 (C9H8N)+, 92 (C6H6N)+, 77 (C6H5)+.
4.6.3. N-(Benzyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8c)
Dark-brown amorphous powder; yield: 88%; m.p. 85–87 °C; mol. formula: C21H20N4O2S; mol. weight: 392 g/mol; IR (KBr, υ, cm–1): 3340 (N–H), 3080 (Ar C–H), 1669 (C=N), 1651 (C=O str.), 1560 (Ar C=C), 1060 (C–O–C), 636 (C–S); 1H NMR(600 MHz, DMSO-d6, δ, ppm): 10.87 (s, 1H, NH-1), 10.31 (t, J = 5.6 Hz, 1H, −CONH), 7.86 (br. d, J = 8.5 Hz, 2H, H-2‴ & H-6‴), 7.64 (d, J = 9.3 Hz, 1H, H-4), 7.60–7.46 (m, 3H, H-3‴, H-4‴ & H-5‴), 7.34 (br. d, J = 9.0 Hz, 1H, H-7), 7.28 (br. s, 1H, H-2), 7.07 (t, J = 8.4 Hz, 1H, H-5), 6.98 (t, J = 8.4 Hz, 1H, H-6), 4.31 (s, 2H, CH2-10), 3.44 (t, J = 7.9 Hz, 2H, CH2-3″), 3.96 (t, J = 8.7 Hz, 2H, CH2-2″), 3.01 (br. s, 2H, CH2-7‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.5 (C-5′), 167.1 (C-1″), 163.7 (C-2′), 138.5 (C-1‴), 136.2 (C-8), 130.5 (C-9), 129.6 (C-3‴ & C-5‴), 127.8 (C-4‴), 124.1 (C-2), 121.6 (C-6), 18.7 (C-2‴ & C-6‴), 118.3 (C-5), 118.1 (C-4), 111.5 (C-7), 106.5 (C-3), 38.4 (C-7‴), 35.6 (C-2″), 27.1 (C-3″), 22.3 (C-10); EI-MS (m/z): 394 (C21H20N4O2S + 2)·+ (M + 2)+, 392 (C21H20N4O2S)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 162 (C10H12NO)+, 161 (C10H11NO)+, 130 (C9H8N)+, 106 C7H8NO)+, 91 (C7H7)+.
4.6.4. N-(2-Phenethyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8d)
Dark-brown colored amorphous powder; yield: 86%; m.p. 276–278 °C; mol. formula: C22H22N4SO2; mol. weight: 406 g/mol; IR (KBr, υ, cm–1): 3332 (N–H), 3086 (Ar C–H), 1668 (C=N), 1650 (C=O str.), 1560 (Ar C=C), 1063 (C–O–C), 633 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.06 (s, 1H, NH-1), 8.04 (t, J = 5.6 Hz, 1H, −CONH), 7.52 (br. d, J = 7.8 Hz, 1H, H-4), 7.38 (br. d, J = 8.1 Hz, 1H, H-7), 7.35 (br. s, 1H, H-2), 7.24 (d, J = 7.4 Hz, 2H, H-2‴ & H-6‴), 7.8–7.15 (m, 3H, H-3‴ to H-5‴), 7.10 (t, J = 7.9 Hz, 1H, H-5), 7.02 (t, J = 7.0 Hz, 1H, H-6), 4.34 (s, 2H, CH2-10), 3.37–3.25 (m, signals merged with DMSO-d6, 4H, CH2-3″ & CH2-2″), 2.69 (t, 2H, J = 7.4 Hz, CH2-8‴), 2.57 (t, J = 6.7 Hz, 2H, CH2-7‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.1 (C-5′), 166.9 (C-1″), 163.8 (C-2′), 138.3 (C-1‴), 136.1 (C-8), 130.1 (C-9), 128.2 (C-2‴ & C-6‴), 127.8 (C-3‴ & C-5‴), 125.4 (C-4‴), 124.1 (C-2), 121.9 (C-6), 118.7 (C-5), 118.1 (C-4), 111.1 (C-7), 106.2 (C-3), 35.7 (C-2″), 33.2 (C-8‴), 28.3 (C-7‴), 28.1 (C-3″), 21.5 (C-10); EI-MS (m/z): 408 (C21H20N4SO2 + 2)·+ (M + 2)+, 406 (C21H20N4SO2)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 176 (C11H14NO)+, 175 (C11H13NO)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 130 (C9H8N)+, 120 (C7H8N)+, 105 (C8H9)+, 77 (C6H6)+.
4.6.5. N-(2-Methylphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8e)
Dark-brown colored amorphous powder; yield: 86%; m.p. 127–128 °C; mol. formula: C21H20N4O2S; mol. weight: 392 g/mol; IR (KBr, υ, cm–1): 3341 (N–H), 3079 (Ar C–H), 1663 (C=N), 1653 (C=O str.), 1569 (Ar C=C), 1064 (C–O–C), 633 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.08 (s, 1H, NH-1), 9.42 (s, 1H, −CONH), 7.53 (d, J = 7.8 Hz, 1H, H-4), 7.39 (br. d, J = 8.0 Hz, 1H, H-6‴), 7.37 (d, J = 7.6 Hz, 1H, H-7), 7.35 (br. s, 1H, H-2), 7.20 (d, J = 7.3 Hz, 1H, H-3‴), 7.15 (t, J = 7.2 Hz, 1H, H-5), 7.11–7.07 (m, 2H, H-4‴ & H-5‴), 7.01 (t, J = 7.4 Hz, 1H, H-6), 4.34 (s, 1H, CH2-10), 3.46 (t, J = 6.6 Hz, 2H, CH2-3″), 2.88 (t, J = 6.6 Hz, 2H, CH2-2″), 2.16 (s, 3H, CH3-7‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.2 (C-5′), 167.1 (C-1″), 163.3 (C-2′), 137.1 (C-1‴), 136.4 (C-3‴), 136.2 (C-8), 131.0 (C-9), 129.1 (C-5‴), 126.2 (C-4‴), 124.1 (C-2), 121.4 (C-6), 18.2 (C-2‴), 118.7 (C-5), 118.4 (C-6‴), 118.1 (C-4), 111.5 (C-7), 106.5 (C-3), 36.7 (C-2″), 28.8 (C-3″), 21.4 (C-10), 18.4 (C-7‴); EI-MS (m/z): 394 (C20H18N4SO2 + 2)·+ (M + 2)+, 392 (C20H18N4SO2)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 162 (C10H12NO)+, 161 (C10H11NO)+, 130 (C9H8N)+, 106, (C7H8N)+, 91 (C7H7)+.
4.6.6. N-(3-Methylphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8f)
Brown-color amorphous; yield: 78%; m.p. 136–138 °C; mol. formula: C21H20N4O2S; mol. weight: 392 g/mol; IR (KBr, υ, cm–1): 3338 (N–H), 3087 (Ar C–H), 1659 (C=O str.), 1661 (C=N), 1568 (Ar C=C), 1060 (C–O–C), 634 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.03 (s, 1H, NH-1), 9.97 (s, 1H, −CONH), 7.51 (d, J = 9.4 Hz, 1H, H-4), 7.41 (br. s, 1H, H-2‴), 7.37 (br. d, J = 9.7 Hz, 1H, H-7), 7.34–7.33 (m, 2H, H-6‴ & H-2), 7.16 (t, J = 9.3 Hz, 1H, H-5‴), 7.09 (t, J = 8.6 Hz, 1H, H-5), 7.00 (t, J = 8.5 Hz, 1H, H-6), 6.86 (br. d, J = 8.9 Hz, 1H, H-4‴), 4.33 (s, 1H, CH2-10), 3.43 (t, J = 7.9 Hz, 2H, CH2-3″), 2.85 (t, J = 7.9 Hz, 2H, CH2-2″), 2.26 (s, 3H, CH3-7‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.3 (C-5′), 167.4 (C-1″), 163.1 (C-2′), 137.5 (C-1‴), 136.1 (C-8), 131.0 (C-9), 129.1 (C-3‴ & 5‴), 126.5 (C-4‴), 124.1 (C-2), 121.3 (C-6), 18.1 (C-2‴ & C-6‴), 118.7 (C-5), 118.1 (C-4), 111.5 (C-7), 106.5 (C-3), 36.7 (C-2″), 28.1 (C-3″), 21.4 (C-10), 18.3 (C-7‴); EI-MS (m/z): 380 (C20H18N4SO2 + 2)·+ (M + 2)+, 392 (C20H18N4SO2)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 162 (C10H12NO)+, 161 (C10H11NO)+, 130 (C9H8N)+, 106, (C7H8N)+, 91 (C7H7)+.
4.6.7. N-(4-Methylphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8g)
Light-brown amorphous solid; yield: 81%; m.p. 111–113 °C; mol. formula: C21H20N4O2S; mol. weight: 392 g/mol; IR (KBr, υ, cm–1): 3336 (N–H), 3081 (Ar C–H), 1664 (C=N), 1062 (C–O–C), 1650 (C=O str.), 1560 (Ar C=C), 633 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.03 (s, 1H, NH-1), 10.24 (s, 1H, −CONH), 7.50 (d, J = 8.4 Hz, 1H, H-4), 7.48 (d, J = 8.4 Hz, 2H, H-2‴ & H-6‴), 7.37 (br. d, J = 7.8 Hz, 1H, H-7), 7.34 (br. s, 1H, H-2), 7.14 (d, J = 7.2 Hz, 2H, H-3‴ & H-5‴), 7.09 (t, J = 7.5 Hz, 1H, H-5), 6.99 (t, J = 6.9 Hz, 1H, H-6), 4.34 (s, 1H, CH2-10), 3.44 (t, J = 7.9 Hz, 2H, CH2-3″), 2.87 (t, J = 7.9 Hz, 2H, CH2-2″), 2.24 (s, 3H, CH2-7‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.5 (C-5′), 167.8 (C-1″), 163.3 (C-2′), 138.1 (C-1‴), 136.1 (C-8), 130.3 (C-9), 128.2 (C-3‴ & 5‴), 126.3 (C-4‴), 124.1 (C-2), 121.6 (C-6), 18.2 (C-2‴ & C-6‴), 118.7 (C-5), 118.1 (C-4), 111.5 (C-7), 106.5 (C-3), 36.7 (C-2″), 27.1 (C-3″), 21.5 (C-10), 18.3 (C-7‴); EI-MS (m/z): 380 (C20H18N4SO2 + 2)·+ (M + 2)+, 392 (C20H18N4SO2)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 162 (C10H12NO)+, 161 (C10H11NO)+, 130 (C9H8N)+, 106, (C7H8N)+, 91 (C7H7)+.
4.6.8. N-(2-Ethylphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanmide (8h)
Brown amorphous solid; yield: 83%; m.p. 135–137 °C; mol. formula: C22H22N4O2S; mol. weight: 406 g/mol; IR (KBr, υ, cm–1): 3334 (N–H), 3084 (Ar C–H), 1665 (C=N), 1656 (C=O str.), 1562 (Ar C=C), 1066 (C–O–C), 632 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.05 (s, 1H, NH-1), 9.40 (s, 1H, −CONH), 7.52 (d, J = 7.8 Hz, 1H, H-4), 7.38 (br. d, J = 8.0 Hz, 1H, H-7), 7.35 (s, 1H, H-2), 7.32 (d, J = 7.3 Hz, 1H, H-6‴), 7.22 (d, J = 7.8 Hz, 1H, H-3‴), 7.17–7.12 (m, 2H, H-4‴, H-5‴), 7.10 (t, J = 7.2 Hz, 1H, H-5), 7.01 (t, J = 7.2 Hz, 1H, H-6), 4.35 (s, 2H, CH2-10), 3.45 (t, J = 6.6 Hz, 2H, CH2-3″), 2.88 (t, J = 6.8 Hz, 2H, CH2-2″), 2.53 (q, J = 7.5 Hz, 2H, CH2-7‴), 1.07 (t, J = 7.5 Hz, 3H, CH3-8‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 167.9 (C-5′), 166.8 (C-1″), 163.5 (C-2′), 137.8 (C-1‴), 136.3 (C-8), 130.6 (C-9), 128.8 (C-3‴), 128.1 (C-5‴), 127.4 (C-2‴), 125.5 (C-4‴), 124.1 (C-2), 122.5 (C-6), 118.7 (C-5), 118.1 (C-6‴), 117.7 (C-4), 111.2 (C-7), 106.6 (C-3), 35.7 (C-2 28.2 (C-3″), 26.8 (C-7‴), 21.2 (C-10), 17.7 (C-8‴); EI-MS (m/z): 408 (C22H22N4O2S)·+ (M + 2)+, 406 (C21H20N4O2S)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 176 (C11H14NO)+, 175 (C11H13NO)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 130 (C9H8N)+, 120 (C8H10N)+, 105 (C8H9)+.
4.6.9. N-(4-Ethylphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8i)
Light-brown amorphous solid; yield: 79%; m.p. 130–132 °C; mol. formula: C22H22N4O2S; mol. weight: 406 g/mol; IR (KBr, υ, cm–1): 3332 (N–H), 3083 (Ar C–H), 1667 (C=N), 1656 (C=O str.), 1559 (Ar C=C), 1065 (C–O–C), 630 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.05 (s, 1H, NH-1), 9.94 (s, 1H, −CONH), 7.52 (br. d, J = 7.8 Hz,1H, H-4), 7.46 (d, J = 8.4 Hz, 2H, H-2‴ & H-6‴), 7.38 (br. d, J = 8.4 Hz, 1H, H-7), 7.34 (br. s, 1H, H-2), 7.13 (d, J = 8.4 Hz, 2H, H-3‴ & H-5‴), 7.10 (t, J = 7.8 Hz, 1H, H-5), 7.00 (t, J = 7.2 Hz, 1H, H-6), 4.33 (s, 1H, CH2-10), 3.44 (t, J = 6.6 Hz, 2H, CH2-3″), 2.84 (t, J = 6.6 Hz, 2H, CH2-2″), 2.55 (q, J = 7.5 Hz, 2H, CH2-7‴), 1.16 (t, J = 6.4 Hz, 3H, CH3-8‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.9 (C-5′), 167.6 (C-1″), 163.6 (C-2′), 189.0 (C-4‴), 137.1 (C-1‴), 136.6 (C-8), 128.3 (C-9), 127.0 (C-3‴ & C-5‴), 124.6 (C-2), 121.8 (C-6), 18.6 (C-2‴ & C-6‴), 118.2 (C-5), 118.0 (C-4), 111.0 (C-7), 107.0 (C-3), 36.1 (C-2″), 28.2 (C-3″), 28.0 (C-7‴), 21.9 (C-10), 16.1 (C-8‴); EI-MS (m/z): 408 (C22H22N4O2S)·+ (M + 2)+, 406 (C21H20N4O2S)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 176 (C11H14NO)+, 175 (C11H13NO)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 130 (C9H8N)+, 120 (C8H10N)+, 105 (C8H9)+.
4.6.10. N-(2-Ethoxyphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8j)
Brown-colored amorphous powder; yield: 76%; m.p. 124–125 °C; mol. formula: C22H22N4O3S; mol. weight: 422 g/mol; IR (KBr, υ, cm–1): 3336 (N–H), 3081 (Ar C–H), 1654 (C=O str.), 1664 (C=N), 1565 (Ar C=C), 1065 (C–O–C), 630 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.01 (s, 1H, NH-1), 9.07 (s, 1H, −CONH), 7.86 (d, J = 9.0 Hz, 1H, H-6‴), 7.49 (d, J = 9.0 Hz, 1H, H-4), 7.35 (br. d, J = 9.6 Hz, 1H, H-7), 7.31 (br. s, 1H, H-2), 7.07 (t, J = 8.4 Hz, 1H, H-5), 7.01–6.96 (m, 3H, H-3‴ to H-5‴), 6.85 (t, J = 7.6 Hz, 1H, H-6), 4.31 (s, 2H, CH2-10), 4.14 (q, J = 7.6 Hz, 2H, CH2-7‴), 3.41 (t, J = 7.8 Hz, 2H, CH2-3″), 2.90 (t, J = 7.8 Hz, 2H, CH2-2″), 1.04 (t, J = 7.4 Hz, 3H, CH3-8‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.5 (C-5′), 167.0 (C-1″), 163.2 (C-2′), 142.5 (2‴), 136.5 (C-8), 130.6 (C-9), 128.5 (C-1‴), 126.9 (C-4‴), 126.5 (C-6‴), 124.3 (C-2), 122.3 (C-6), 18.8 (C-5‴), 118.7 (C-5), 117.8 (C-4), 112.6 (C-3‴), 111.1 (C-7), 106.1 (C-3), 64.1 (C-7‴), 36.6 (C-2″), 28.4 (C-3″), 21.4 (C-10), 14.8 (C-8‴); EI-MS (m/z): 424 (C22H22N4O3S)·+ (M + 2)+, 422 (C22H22N4O3S)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 82 (C11H14NO2)+, 81 (C11H13NO2)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 136 (C9H10NO)+, 130 (C9H8O)+, 121 (C8H9O)+.
4.6.11. N-(4-Ethoxyphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8k)
Brown amorphous solid; yield: 84%; m.p. 128–129 °C; mol. formula: C22H22N4O3S; mol. weight: 422 g/mol; IR (KBr, υ, cm–1): 3337 (N–H), 3080 (Ar C–H), 1667 (C=N), 1657 (C=O str.), 1561 (Ar C=C), 1062 (C–O–C), 637 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.03 (s, 1H, NH-1), 9.15 (s,1H, −CONH), 7.52 (d, J = 7.8 Hz, 1H, H-4), 7.45 (d, J = 7.2 Hz, 2H, H-2‴ & H-6‴), 7.38 (br. d, J = 8.6 Hz, 1H, H-7), 7.33 (br. s, 1H, H-2), 7.09 (t, J = 8.0 Hz, 1H, H-5), 7.04 (d, J = 7.0 Hz, 2H, H-3‴ & H-5‴), 7.00 (t, J = 7.2 Hz, 1H, H-6), 4.34 (s, 2H, CH2-10), 4.15 (q, J = 7.2 Hz, 2H, CH2-7‴), 3.40 (t, J = 7.2 Hz, 2H, CH2-3″), 2.94 (t, J = 7.8 Hz, 2H, CH2-2″), 1.03 (t, J = 7.4 Hz, 3H, CH3-8‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.7 (C-5′), 167.5 (C-1″), 163.9 (C-2′), 154.7 (C-4‴), 136.8 (C-1‴), 136.1 (C-8), 131.0 (C-9), 124.3 (C-2), 122.3 (C-6), 121.1 (C-2‴ & C-6‴), 118.7 (C-5), 118.2 (C-4), 115.2 (C-3‴ & C-5‴), 111.2 (C-7), 106.3 (C-3), 64.2 (C-7‴), 36.5 (C-2 27.6 (C-3″), 21.4 (C-10), 16.9 (C-8‴); EI-MS (m/z): 424 (C22H22N4O3S)·+ (M + 2)+, 422 (C22H22N4O3S)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 82 (C11H14NO2)+, 81 (C11H13NO2)+, 158 (C10H8NO)+ 156 (C10H8N2)+, 136 (C9H10NO)+, 130 (C9H8O)+, 121 (C8H9O)+.
4.6.12. N-((2-Methoxycarbonyl)phenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8l)
Brown amorphous solid; yield: 86%; m.p. 233–234 °C; mol. formula: C22H20N4O4S; mol. weight: 436 g/mol; IR (KBr, υ, cm–1): 3340 (N–H), 3081 (Ar C–H), 1669 (C=N), 1651 (C=O str.), 1564 (Ar C=C), 1063 (C–O–C), 634 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.09 (s, 1H, NH-1), 10.57 (s, 1H, −CONH), 8.10 (d, J = 8.4 Hz, 1H, H-3‴), 7.90 (d, J = 7.5 Hz, 1H, H-6‴), 7.60 (t, J = 7.2 Hz, 1H, H-4‴), 7.48 (d, J = 7.8 Hz, 1H, H-4), 7.38 (d, J = 8.1 Hz, 1H, H-7), 7.31 (br. s, 1H, H-2′), 7.22 (t, J = 7.2 Hz, 1H, H-5‴), 7.08 (t, J = 7.8 Hz, 1H, H-5), 6.92 (t, J = 7.4 Hz, 1H, H-6), 4.32 (s, 2H, CH2-10), 3.79 (s, 3H, CH3-8‴), 3.28 (t, J = 7.2 Hz, 2H, CH2-3″), 2.84 (t, J = 7.8 Hz, 2H, CH2-2″); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 167.9 (C-5′), 167.0 (C-1″), 163.5 (C-2′), 156.2 (C-7‴), 138.3 (C-1‴), 136.1 (C-8), 135.5 (C-5‴), 132.8 (C-3‴), 131.6 (C-9), 124.1 (C-2), 121.3 (C-6), 121.0 (C-4‴), 18.2 (C-6‴), 118.5 (C-5), 117.7 (C-4), 114.7 (C-2‴), 111.1 (C-7), 106.1 (C-3), 56.2 (C-8‴), 36.6 (C-2″), 27.4 (C-3″), 21.4 (C-10); EI-MS (m/z): 438 (C21H18N4O4S)·+ (M + 2)+, 436 (C21H18N4O4S + 2)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 206 (C11H12NO3)+, 205 (C11H11NO3)+, 158 (C10H8NO)+ 156 (C10H8N2)+, 150 (C8H8NO2)+, 135 (C8H7O2)+, 130 (C9H8N)+, 104 (C7H6N)+.
4.6.13. N-(2,3-Dimethylphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8m)
Brown-colored amorphous solid; yield: 90%; m.p. 103–105 °C; mol. formula: C22H22N4O2S; mol. weight: 406 g/mol; IR (KBr, υ, cm–1): 3346 (N–H), 3079 (Ar C–H), 1663 (C=N), 1653 (C=O str.), 1566 (Ar C=C), 1062 (C–O–C), 634 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.05 (s, 1H, NH-1), 9.47 (s, 1H, −CONH), 7.52 (br. d, J = 7.8 Hz, 1H, H-4), 7.38 (br. d, J = 8.4 Hz, 1H, H-7), 7.35 (br. s, 1H, H-2), 7.11–7.09 (m, 2H, H-6‴ & H-5), 7.04–6.99 (m, 3H, H-6, H-4‴ & H-5‴), 4.34 (s, 2H, CH2-10), 3.45 (t, J = 6.7 Hz, 2H, CH2-3″), 3.86 (t, J = 6.7 Hz, 2H, CH2-2″), 2.24 (s, 3H, CH3-7‴), 2.02 (s, 3H, CH3-8‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.6 (C-5′), 167.3 (C-1″), 163.4 (C-2′), 136.3 (C-3‴), 136.2 (C-1‴), 136.1 (C-8), 131.3 (C-9), 128.2 (C-2‴), 126.8 (C-4‴), 129.6 (C-5‴), 124.1 (C-2), 122.4 (C-6‴), 121.3 (C-6), 118.7 (C-5), 118.4 (C-4), 111.5 (C-7), 106.5 (C-3), 36.7 (C-2″), 28.4 (C-3″), 21.4 (C-10), 18.5 (C-7‴), 18.3 (C-8‴); EI-MS (m/z): 408 (C22H22N4O2S)·+ (M + 2)+, 406 (C21H20N4O2S)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 176 (C11H14NO)+, 175 (C11H13NO)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 130 (C9H8N)+, 120 (C8H10N)+, 105 (C8H9)+, 65 (C5H5)+.
4.6.14. N-(2,4-Dimethylphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8n)
Brown amorphous solid; yield: 87%; m.p. 140–141 °C; mol. formula: C22H22N4O2S; mol. weight: 406 g/mol; IR (KBr, υ, cm–1): 3331 (N–H), 3085 (Ar C–H), 1668 (C=N), 1655 (C=O str.), 1564 (Ar C=C), 1062 (C–O–C), 632 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.10 (s, 1H, NH-1), 9.35 (s, 1H, −CONH), 7.52 (br. d, J = 7.8 Hz, 1H, H-4), 7.38 (br. d, J = 7.8 Hz, 1H, H-7), 7.35 (br. s, 1H, H-2), 7.21 (br. d, J = 9.2 Hz, 1H, H-6‴), 7.10 (t, J = 7.2 Hz, 1H, H-5), 7.02–6.99 (m, 2H, H-3‴ & H-6), 6.92 (br. d, J = 8.1 Hz, 1H, H-5‴), 4.34 (s, 2H, CH2-10), 3.44 (t, J = 6.6 Hz, 2H, CH2-3″), 3.85 (t, J = 6.6 Hz, 2H, CH2-2″), 2.24 (s, 3H, CH3-7‴), 2.11 (s, 3H, CH3-8‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.6 (C-5′), 167.3 (C-1″), 163.3 (C-2′), 138.3 (C-1‴), 136.1 (C-8), 133.3 (C-2‴), 132.3 (C-4‴), 130.3 (C-9), 129.3 (C-5‴), 125.5 (C-3‴), 118.1 (C-6‴), 124.1 (C-2), 121.3 (C-6), 118.7 (C-5), 118.1 (C-4), 111.5 (C-7), 106.5 (C-3), 35.9 (C-2″), 27.6 (C-3″), 21.4 (C-10), 18.7 (C-7‴), 18.2 (C-8‴); EI-MS (m/z): 408 (C22H22N4O2S)·+ (M + 2)+, 406 (C21H20N4O2S)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 176 (C11H14NO)+, 175 (C11H13NO)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 130 (C9H8N)+, 120 (C8H10N)+, 105 (C8H9)+, 65 (C5H5)+.
4.6.15. N-(2,5-Dimethylphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8o)
Dark-brown amorphous solid; yield: 86%; m.p. 142–144 °C; mol. formula: C22H22N4O2S; mol. weight: 406 g/mol; IR (KBr, υ, cm–1): 3338 (N–H), 3081 (Ar C–H), 1669 (C=N), 1651 (C=O str.), 1559 (Ar C=C), 1062 (C–O–C), 634 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.05 (s, 1H, NH-1), 9.34 (s, 1H, −CONH), 7.52 (d, J = 7.8 Hz, 1H, H-4), 7.38 (br. d, J = 8.1 Hz, 1H, H-7), 7.35 (br. s, 1H, H-2), 7.18 (br. s, 1H, H-6‴), 7.10 (t, J = 7.2 Hz, 1H, H-5), 7.07 (br. d, J = 7.8 Hz, 1H, H-3‴), 7.01 (t, J = 7.8 Hz, 1H, H-6), 6.90 (br. d, J = 7.2 Hz, 1H, H-4‴), 4.34 (s, 1H, CH2-10), 3.45 (t, J = 6.6 Hz, 2H, CH2-3″), 2.86 (t, J = 6.6 Hz, 2H, CH2-2″), 2.23 (s, 3H, CH3-7‴), 2.11 (s, 3H, CH3-8‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.6 (C-5′), 167.3 (C-1″), 163.2 (C-2′), 138.4 (C-1‴), 136.1 (C-8), 133.7 (C-5‴), 132.5 (C-2‴), 126.3 (C-9), 126.5 (C-4‴), 125.5 (C-3‴), 124.1 (C-2), 123.4 (C-6‴), 121.3 (C-6), 118.7 (C-5), 118.1 (C-4), 111.5 (C-7), 106.5 (C-3), 35.6 (C-2″), 27.6 (C-3″), 21.4 (C-10), 18.6 (C-7‴), 18.8 (C-8‴); EI-MS (m/z): 408 (C22H22N4O2S)·+ (M + 2)+, 406 (C21H20N4O2S)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 176 (C11H14NO)+, 175 (C11H13NO)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 130 (C9H8N)+, 120 (C8H10N)+, 105 (C8H9)+, 65 (C5H5)+.
4.6.16. N-(2,6-Dimethylphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8p)
Brown amorphous solid; yield: 89%; m.p. 143–145 °C; mol. formula: C22H22N4O2S; mol. weight: 406 g/mol; IR (KBr, υ, cm–1): 3339 (N–H), 3085 (Ar C–H), 1652 (C=O str.), 1665 (C=N), 1561 (Ar C=C), 1062 (C–O–C), 635 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.02 (s, 1H, NH-1), 9.98 (s, 1H, −CONH), 7.51 (d, J = 8.1 Hz, 1H, H-4), 7.38 (br. d, J = 8.4 Hz, 1H, H-7), 7.35 (br. s, 1H, H-2), 7.10 (t, J = 7.8 Hz, 1H, H-5), 7.06–7.02 (m, 3H, H-3‴ to H-5‴), 7.00 (t, J = 7.2 Hz, 1H, H-6), 4.34 (s, 2H, CH2-10), 3.44 (t, J = 6.6 Hz, 2H, CH2-3″), 2.85 (t, J = 6.6 Hz, 2H, CH2-2″), 2.15 (s, 6H, CH3-7‴ & CH3-8‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.5 (C-5′), 167.1 (C-1″), 163.2 (C-2′), 138.6 (C-1‴), 136.2 (C-8), 136.0 (C-2‴ & C-6‴), 130.3 (C-9), 128.4 (C-3‴ & C-5‴), 126.4 (C-4‴), 124.1 (C-2), 121.3 (C-6), 118.7 (C-5), 116.2 (C-4), 111.5 (C-7), 106.5 (C-3), 35.7 (C-2″), 28.2 (C-3″), 21.4 (C-10), 18.6 (C-7‴ & C-8‴); EI-MS (m/z): 408 (C22H22N4O2S)·+ (M + 2)+, 406 (C21H20N4O2S)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 176 (C11H14NO)+, 175 (C11H13NO)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 130 (C9H8N)+, 120 (C8H10N)+, 105 (C8H9)+, 65 (C5H5)+.
4.6.17. N-(3,4-Dimethylphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8q)
Brown amorphous solid; yield: 85%; m.p. 152–153 °C; mol. formula: C22H22N4O2S; mol. weight: 406 g/mol; IR (KBr, υ, cm–1): 3337 (N–H), 3082 (Ar C–H), 1669 (C=N), 1656 (C=O str.), 1551 (Ar C=C), 1063 (C–O–C), 632 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.05 (s, 1H, NH-1), 9.86 (s, 1H, −CONH), 7.52 (br. d, J = 7.8 Hz, 1H, H-4), 7.38 (br. d, J = 8.1 Hz, 1H, H-7), 7.34 (br. s, 2H, H-2 & H-2‴), 7.28 (dd, J = 1.9, 8.1 Hz, 1H, H-6‴), 7.10 (t, J = 7.0 Hz, 1H, H-5), 7.04 (br. d, J = 8.1 Hz, 1H, H-5‴), 7.00 (t, J = 7.8 Hz, 1H, H-6), 4.33 (s, 2H, CH2-10), 3.43 (t, J = 6.6 Hz, 2H, CH2-3″), 2.83 (t, J = 6.6 Hz, 2H, CH2-2″), 2.17 (s, 3H, CH3-7‴), 2.15 (s, 3H, CH3-8‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 167.7 (C-5′), 164.3 (C-1″), 162.8 (C-2′), 136.3 (C-3‴), 136.3 (C-1‴), 136.1 (C-8), 131.3 (C-9), 129.6 (C-4‴), 126.5 (C-5‴), 124.3 (C-2‴), 124.1 (C-2), 121.3 (C-6), 118.7 (C-5), 118.1 (C-6‴), 117.6 (C-4), 111.5 (C-7), 106.5 (C-3), 35.7 (C-2″), 28.2 (C-3″), 21.3 (C-10), 18.9 (C-7‴), 18.4 (C-8‴); EI-MS (m/z): 408 (C22H22N4O2S)·+ (M + 2)+, 406 (C21H20N4O2S)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 176 (C11H14NO)+, 175 (C11H13NO)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 130 (C9H8N)+, 120 (C8H10N)+, 105 (C8H9)+, 65 (C5H5)+.
4.6.18. N-(3,5-Dimethylphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8r)
Light amorphous solid; yield: 83%; m.p. 110–112 °C; mol. formula: C22H22N4O2S; mol. weight: 406 g/mol; IR (KBr, υ, cm–1): 3340 (N–H), 3088 (Ar C–H), 1663 (C=N), 1653 (C=O str.), 1558 (Ar C=C), 1061 (C–O–C), 632 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.05 (s, 1H, NH-1), 9.86 (s, 1H, −CONH), 7.51 (d, J = 7.8 Hz, 1H, H-4), 7.38 (d, J = 7.2 Hz, 1H, H-7), 7.34 (br. s, 1H, H-2), 7.8 (br. s, 2H, H-2‴ & H-6‴), 7.10 (t, J = 7.2 Hz, 1H, H-5), 7.00 (t, J = 7.6 Hz, 1H, H-6), 6.69 (br. s, 1H, H-4‴), 4.33 (s, 2H, CH2-10), 3.44 (t, J = 6.6 Hz, 2H, CH2-3″), 2.84 (t, J = 6.6 Hz, 2H, CH2-2″), 2.22 (s, 6H, CH3-7‴ & CH3-8‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.5 (C-5′), 167.1 (C-1″), 163.2 (C-2′), 138.7 (C-1‴), 137.6 (C-3‴ & C-5‴), 136.1 (C-8), 126.5 (C-9), 124.7 (C-4‴), 124.1 (C-2), 121.3 (C-6), 118.7 (C-5), 118.1 (C-4), 116.8 (C-2‴ & C-6‴), 111.5 (C-7), 106.5 (C-3), 35.7 (C-2″), 27.7 (C-3″), 21.4 (C-10), 21.0 (C-7‴ & C-8‴); EI-MS (m/z): 408 (C22H22N4O2S)·+ (M + 2)+, 406 (C21H20N4O2S)·+ (M)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 176 (C11H14NO)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 130 (C9H8N)+, 120 (C8H10N)+, 105 (C8H9)+.
4.6.19. N-(2-Ethyl-6-methylphenyl)-2-({5-((1H-indol-3-yl)methyl)-1,3,4-oxadiazol-2-yl}sulfanyl)propanamide (8s)
Dark-brown amorphous solid; yield: 81%; m.p. 126–128 °C; mol. formula: C23H24N4O3S; mol. weight: 436 g/mol; IR (KBr, υ, cm–1): 3334 (N–H), 3080 (Ar C–H), 1668 (C=N), 1651 (C=O str.), 1566 (Ar C=C), 1063 (C–O–C), 631 (C–S); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 11.04 (s, 1H, NH-1), 9.69 (s, 1H, −CONH), 7.51 (br. d, J = 7.8 Hz, 1H, H-4), 7.38 (br. d, J = 8.1 Hz, 1H, H-7), 7.35 (br. s, 1H, H-2), 7.10 (t, J = 6.9 Hz, 1H, H-5), 7.07–7.03 (m, 3H, H-3‴ to H-5‴), 7.00 (t, J = 7.8 Hz, 1H, H-6), 4.34 (s, 2H, CH2-10), 3.43 (t, J = 6.9 Hz, 2H, CH2-3″), 2.89 (t, J = 6.6 Hz, 2H, CH2-2″), 2.45 (q, merged with DMSO-d6, J = 7.4 Hz, 2H, CH2-7‴), 2.08 (s, 3H, CH3-9‴), 1.02 (t, J = 6.9 Hz, 3H, CH3-8‴); 13C NMR (150 MHz, DMSO-d6, δ, ppm): 168.4 (C-5′), 167.1 (C-1″), 163.2 (C-2′), 140.8 (C-1‴), 136.8 (C-2‴), 136.1 (C-8), 134.0 (C-6‴), 131.2 (C-9), 127.5 (C-4‴), 126.6 (C-5‴), 124.1 (C-2), 121.3 (C-6), 118.7 (C-5), 118.1 (C-4), 111.5 (C-7), 106.5 (C-3), 35.5 (C-2″), 27.8 (C-3″), 24.1 (C-7‴), 21.4 (C-10), 17.8 (C-9‴), 14.7 (C-8‴); EI-MS (m/z): 438 (C23H24N4O2S + 2)·+ (M + 2)+, 436 (C23H24N4O2S)·+ (M)+, 288 (C14H12N3O2S + 2)+, 286 (C14H12N3O2S)+, 260 (C1H12N3OS + 2)+, 258 (C13H12N3OS)+, 233 (C11H9N3OS + 2)+, 231 (C11H9N3OS)+, 80 (C12H16NO)+, 189 (C12H15NO)+, 158 (C10H8NO)+, 156 (C10H8N2)+, 134 (C9H12N)+, 130 (C9H8O)+, 18 (C9H11)+.
4.7. α-Glucosidase Inhibition Assay
The α-glucosidase inhibitory activity was performed according to an established method.26 A reaction mixture having 70 μL (50 mM) of phosphate-buffered saline at pH 6.8, 10 μL (0.5 mM) of the test compound, and 10 μL (0.057 units) of the enzyme was prepared. The contents were mixed and preincubated for 10 min at 37 °C, and absorbance was measured at 400 nm. Ten microliters of 0.5 mM substrate (p-nitrophenylglucopyranoside) was added to initiate the reaction. Acarbose was used as a positive control for the comparison of activity of tested compounds. After 30 min of incubation at 37 °C, absorbance was measured at 400 nm using a Synergy HT microplate reader for the final 100 μL sample. All experiments were carried out in triplicates. The percentage inhibition (%) was calculated by the formula given below
where control is the total enzyme activity without the inhibitor and test is the activity in the presence of a test compound.
4.8. Statistical Analysis
All assays were carried out in triplicate. The results are presented as means ± SEM with 85–95% CL. Statistical analysis was performed by Microsoft Excel 2010. IC50 values (concentration at which there is 50% enzyme inhibition) of compounds were calculated using EZ-Fit Enzyme kinetics software (Perella Scientific, Inc., Amherst, USA).
4.9. Hemolytic Activity
The hemolytic activity of the compound was studied as per the reported method.27,28 Freshly obtained heparinized bovine blood (3 mL) was collected from the Department of Clinical Medicine and Surgery, University of Agriculture, Faisalabad, Pakistan. Blood was centrifuged for 5 min at 1000g, plasma was discarded, and cells were washed three times with 5 mL of chilled (4 °C) sterile isotonic phosphate-buffered saline (PBS) at pH 7.4. Erythrocytes were maintained 108 cells per mL for each assay. Hundred microliters of each compound was mixed with human cells (108 cells/mL) separately. Samples were incubated for 35 min at 37 °C and agitated after 10 min. Immediately after incubation, the samples were placed on ice for 5 min and then centrifuged for 5 min at 1000g. Supernatant 100 μL samples were taken from each tube and diluted 10 times with chilled (4 °C) PBS. Triton X-100 (0.1% v/v) was taken as a positive control, and phosphate-buffered saline (PBS) was taken as a negative control and passed through the same process. The absorbance was noted at 576 nm using μQuant (BioTek, USA). The % RBC lysis for each sample was calculated. The study protocol was approved by the director of graduate studies (Institutional Ethical Committee) vide notification no. DGS/8786-89 dated 09-03-2015, University of Agriculture, Faisalabad, Pakistan29 and was conducted in accordance with the 1964 Declaration of Helsinki and its later amendments.29
4.10. Computational Methodologies
4.10.1. Selection of Target Proteins from PDB
The crystal structure of α-glucosidase with PDB ID 4J5T was accessed from the Protein Data Bank (PDB) (www.rcsb.org). The selected target protein structure was minimized with the Amber force field by employing the conjugate gradient algorithm in UCSF Chimera 1.10.1.30 The overall protein architecture and statistical percentage values of helices, β-sheets, coils, and turns were retrieved from the online server VADAR 1.8.31 Discovery Studio 4.1 Client,32 a visualizing tool, was used to generate the hydrophobicity graph and graphical depiction of target proteins.
4.10.2. Designing of Ligands and Molecular Docking
The structures of synthesized ligands were drawn in the ACD/ChemSketch tool and minimized with UCSF Chimera 1.10.1. All the synthesized ligands were sketched in the ACD/ChemSketch tool and accessed in mol format. Furthermore, the UCSF Chimera 1.10.1 tool was employed for energy minimization of each ligand separately having default parameters such as steepest descent steps of 100 with a step size of 0.02 (Å) and conjugate gradient steps of 100 with a step size of 0.02 (Å), and the update interval was fixed at 10. Finally, Gasteiger charges were added using Dock Prep in ligand structure to obtain the good structure conformation. A molecular docking experiment was employed on all the synthesized ligands against α-glucosidase by using the PyRx virtual screening tool with the AutoDock Vina Wizard approach.33 The grid box parameter values in the Vina search space for α-glucosidase were adjusted as center_x = −18.44, center_y = −20.91, center_z = 8.22 while size_x = 77.93, size_y = 68.98, and size_z = 103.65 in angstrom (Å), respectively. We have adjusted the grid box size to be sufficient enough to allow the ligand to move freely in the search space around the binding pocket residues. The default exhaustiveness value of 8 was adjusted in both dockings to maximize the binding conformational analysis. In all docked complexes, the ligand conformational poses were keenly observed to produce the best docking results. The docked complexes were evaluated based on the lowest binding energy (kcal/mol) values and structure–activity relationships. The graphical depictions of all the docking complexes were carried out using Discovery Studio (2.1.0).
Acknowledgments
M.H. acknowledges the Ohio State University for providing the “President’s Postdoctoral Scholars Program (PPSP)” award and for financial support to complete this computational research. A.K. acknowledges financial support from NIH Grants R01 GM127701 and R01HG012117.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c01882.
NMR results and 2D docking complexes of all compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hyun T. K.; Eom S. H.; Kim J.-S. Molecular docking studies for discovery of plant-derived a-glucosidase inhibitors. Plant Omics 2014, 7, 166–170. [Google Scholar]
- Dabhi A. S.; Bhatt N. R.; Shah M. J. Voglibose: an alpha glucosidase inhibitor. J. Clin. Diagn. Res. 2013, 7, 3023–3027. 10.7860/JCDR/2013/6373.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidit D.; Frommer W.; Junge B.; Muller L.; Wingender W.; Truscheit E.; Schafer D. α-Glucosidase inhibitors. Naturwissenschaften 1977, 64, 535–536. 10.1007/BF00483561. [DOI] [PubMed] [Google Scholar]
- Rubab K.; Abbasi M. A.; Siddiqui S. Z.; Ashraf M.; Shaukat A.; Ahmad I.; Lodhi M. A.; Khan F. A.; Shahid M.; Akhtar M. N. Convergent synthesis of new N-substituted 2-{[5-(1H-indol-3-ylmethyl)-1, 3, 4-oxadiazol-2-yl] sulfanyl} acetamides as suitable therapeutic agents. Braz. J. Pharm. Sci. 2015, 51, 931–947. 10.1590/S1984-82502015000400019. [DOI] [Google Scholar]
- Naureen S.; Chaudhry F.; Munawar M. A.; Ashraf M.; Hamid S.; Khan M. A. Biological evaluation of new imidazole derivatives tethered with indole moiety as potent α-glucosidase inhibitors. Bioorg. Chem. 2018, 76, 365–369. 10.1016/j.bioorg.2017.12.014. [DOI] [PubMed] [Google Scholar]
- Kavitha S.; Kannan K.; Gnanavel S. Synthesis, characterization and biological evaluation of novel 2, 5 substituted-1, 3, 4 oxadiazole derivatives. Saudi Pharm. J. 2017, 25, 337–345. 10.1016/j.jsps.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.-Z.; Mulholland N.; Beattie D.; Irwin D.; Gu Y.-C.; Chen Q.; Yang G.-F.; Clough J. Synthesis and antifungal activity of 3-(1, 3, 4-oxadiazol-5-yl)-indoles and 3-(1, 3, 4-oxadiazol-5-yl) methyl-indoles. Eur. J. Med. Chem. 2013, 63, 22–32. 10.1016/j.ejmech.2013.01.038. [DOI] [PubMed] [Google Scholar]
- Bondock S.; Adel S.; Etman H. A.; Badria F. A. Synthesis and antitumor evaluation of some new 1, 3, 4-oxadiazole-based heterocycles. Eur. J. Med. Chem. 2012, 48, 192–199. 10.1016/j.ejmech.2011.12.013. [DOI] [PubMed] [Google Scholar]
- Shaharyar M.; Mazumder A.; Ahsan M. J. Synthesis, characterization and anticancer evaluation of 2-(naphthalen-1-ylmethyl/naphthalen-2-yloxymethyl)-1-[5-(substituted phenyl)-[1, 3, 4] oxadiazol-2-ylmethyl]-1H-benzimidazole. Arabian J. Chem. 2014, 7, 418–424. 10.1016/j.arabjc.2013.02.001. [DOI] [Google Scholar]
- Abbasi M. A.; Shahzad B.; Nafeesa K.; Rasool S.; Ashraf M.; Ejaz S. A.; Ismail H.; Mirza B. Synthesis, spectral characterization and bioactivity studies of some S-substituted derivatives of 5-(4-chlorophenyl)-1, 3, 4-Oxadiazol-2-thiol. World J. Pharm. Sci. 2014, 32–40. 10.3906/kim-2008-44. [DOI] [Google Scholar]
- Desai N. C.; Bhatt N.; Somani H.; Trivedi A. Synthesis, antimicrobial and cytotoxic activities of some novel thiazole clubbed 1, 3, 4-oxadiazoles. Eur. J. Med. Chem. 2013, 67, 54–59. 10.1016/j.ejmech.2013.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malladi S.; Isloor A. M.; Peethambar S. K.; Fun H. K. Synthesis and biological evaluation of newer analogues of 2, 5-disubstituted 1, 3, 4-oxadiazole containing pyrazole moiety as antimicrobial agents. Arabian J. Chem. 2014, 7, 1185–1191. 10.1016/j.arabjc.2013.12.020. [DOI] [Google Scholar]
- Gadegoni H.; Manda S. Synthesis and screening of some novel substituted indoles contained 1, 3, 4-oxadiazole and 1, 2, 4-triazole moiety. Chin. Chem. Lett. 2013, 24, 127–130. 10.1016/j.cclet.2013.01.001. [DOI] [Google Scholar]
- Harish K. P.; Mohana K. N.; Mallesha L.; Prasanna kumar B. N. Synthesis of novel 1-[5-(4-methoxy-phenyl)-[1, 3, 4] oxadiazol-2-yl]-piperazine derivatives and evaluation of their in vivo anticonvulsant activity. Eur. J. Med. Chem. 2013, 65, 276–283. 10.1016/j.ejmech.2013.04.054. [DOI] [PubMed] [Google Scholar]
- Musad E. A.; Mohamed R.; Saeed B. A.; Vishwanath B. S.; Lokanatha Rai K. M. Synthesis and evaluation of antioxidant and antibacterial activities of new substituted bis (1, 3, 4-oxadiazoles), 3, 5-bis (substituted) pyrazoles and isoxazoles. Bioorg. Med. Chem. Lett. 2011, 21, 3536–3540. 10.1016/j.bmcl.2011.04.142. [DOI] [PubMed] [Google Scholar]
- Hajimahdi Z.; Zarghi A.; Zabihollahi R.; Aghasadeghi M. R. Synthesis, biological evaluation, and molecular modeling studies of new 1, 3, 4-oxadiazole-and 1, 3, 4-thiadiazole-substituted 4-oxo-4 H-pyrido [1, 2-a] pyrimidines as anti-HIV-1 agents. Med. Chem. Res. 2013, 22, 2467–2475. 10.1007/s00044-012-0241-5. [DOI] [Google Scholar]
- Sharma P.; Sharma J. D. In vitro hemolysis of human erythrocytes—by plant extracts with antiplasmodial activity. J. Ethnopharmacol. 2001, 74, 239–243. 10.1016/S0378-8741(00)00370-6. [DOI] [PubMed] [Google Scholar]
- Baillie J.; Thompson A.; Irving J.; Bates M.; Sutherland A.; Macnee W.; Maxwell S.; Webb D. Oral antioxidant supplementation does not prevent acute mountain sickness: double blind, randomized placebo-controlled trial. QJM 2009, 102, 341–348. 10.1093/qjmed/hcp026. [DOI] [PubMed] [Google Scholar]
- Tiwari P.; Kumar B.; Kaur M.; Kaur G.; Kaur H. Phytochemical screening and Extraction: A Review. Int. Pharm. Sci. 2011, 1, 98–106. [Google Scholar]
- Al-Zuhair S.; Dowaidar A.; Kamal H. Inhibitory effect of dates-extract on α-Amylase and α-glucosidase enzymes relevant to non-insulin dependent diabetes mellitus. J. Biochem. Technol. 2010, 2, 158–160. [Google Scholar]
- Nazir M.; Abbasi M. A.; Aziz-Ur-Rehman; Siddiqui S. Z.; Khan K. M.; Salar U.; Shahid M.; Ashraf M.; Lodhi M. A.; Khan F. A. New indole based hybrid oxadiazole scaffolds with N-substituted acetamides: As potent anti-diabetic agents. Bioorg. Chem. 2018, 81, 253–263. 10.1016/j.bioorg.2018.08.010. [DOI] [PubMed] [Google Scholar]
- Abbas Q.; Hassan M.; Raza H.; Kim S. J.; Chung K.-W.; Kim G.-H.; Seo S.-Y. In vitro, in vivo and in silico anti-hyperglycemic inhibition by sinigrin. Asian Pac. J. Trop. Med. 2017, 10, 372–379. 10.1016/j.apjtm.2017.03.019. [DOI] [PubMed] [Google Scholar]
- Hu W.-p.; Cao G.-d.; Zhu J.-h.; Li J.-z.; Liu X.-h. Naturally occurring Batatasins and their derivatives as α-glucosidase inhibitors. RSC Adv. 2015, 5, 82153–82158. 10.1039/C5RA15328J. [DOI] [Google Scholar]
- Barker M. K.; Rose D. R. Specificity of processing α-glucosidase I is guided by the substrate conformation: crystallographic and in silico studies. J. Biol. Chem. 2013, 288, 13563–13574. 10.1074/jbc.M113.460436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapdelaine P.; Tremblay R. R.; Dubé J. P-Nitrophenol-alpha-D-glucopyranoside as substrate for measurement of maltase activity in human semen. Clin. Chem. 1978, 24, 208–211. 10.1093/clinchem/24.2.208. [DOI] [PubMed] [Google Scholar]
- Shahid M.; Bukhari S. A.; Gul Y.; Munir H.; Anjum F.; Zuber M.; Jamil T.; Zia K. M. Graft polymerization of guar gum with acryl amide irradiated by microwaves for colonic drug delivery. Int. J. Biol. Macromol. 2013, 62, 172–179. 10.1016/j.ijbiomac.2013.08.018. [DOI] [PubMed] [Google Scholar]
- Zuber M.; Tabasum S.; Jamil T.; Shahid M.; Hussain R.; Feras K. S.; Bhatti K. P. Biocompatibility and microscopic evaluation of polyurethane–poly (methyl methacrylate)–titnanium dioxide based composites for dental applications. J. Appl. Polym. Sci. 2014, 131, 3. 10.1002/app.39806. [DOI] [Google Scholar]
- Abbasi M. A.; Islam M.; Rehman A.-u.; Rasool S.; Rubab K.; Hussain G.; Ahmad I.; Ashraf M.; Shahid M.; Shah S. A. A. Synthesis, characterization, antibacterial, α-glucosidase inhibition and hemolytic studies on some new N-(2, 3-dimethylphenyl) benzenesulfonamide derivatives. Trop. J. Pharm. Res. 2016, 15, 591–598. 10.4314/tjpr.v15i3.22. [DOI] [Google Scholar]
- World Medical Association Declaration of Helsinki. Jama 2013, 310, 2191–2194. 10.1001/jama.2013.281053. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Willard L.; Ranjan A.; Zhang H.; Monzavi H.; Boyko R. F.; Sykes B. D.; Wishart D. S. VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 2003, 31, 3316–3319. 10.1093/nar/gkg565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studio D., Discovery studio. Accelrys [2.1]; Studio D; 2008. [Google Scholar]
- Dallakyan S.; Olson A. J. Small-molecule library screening by docking with PyRx. Chem. Biol. 2015, 1263, 243–250. 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.