

Abstract
Background
Alcohol-associated liver disease (ALD) and non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH) are two of the most common etiologies of chronic liver disease worldwide. Changes in intestinal permeability and increased gut microbial translocation have been posited as important contributors to inflammation in both ALD and NAFLD. However, gut microbial translocation has not been compared between the two etiologies and can lead to better understanding of the differences in their pathogenesis to liver disease.
Methods
We compared serum and liver markers in the following five models of liver disease to understand the differences in the role of gut microbial translocation on liver disease progression caused by ethanol versus Western diet: 1) 8-week chronic ethanol feeding model. 2) 2-week chronic-plus-binge (National Institute on Alcohol Abuse and Alcoholism (NIAAA)) ethanol feeding model. 3) 2-week chronic-plus-binge (NIAAA) ethanol feeding model in microbiota humanized gnotobiotic mice colonized with stool from patients with alcohol-associated hepatitis. 4) 20-week Western-diet feeding model of NASH. 5) 20-week Western-diet feeding model in microbiota humanized gnotobiotic mice colonized with stool from NASH patients.
Results
Translocation of bacterial lipopolysaccharide to the peripheral circulation was seen in both ethanol-induced and diet-induced liver disease, but translocation of bacteria itself was restricted to only ethanol-induced liver disease. Moreover, the diet-induced steatohepatitis models developed more significant liver injury, inflammation, and fibrosis compared with ethanol-induced liver disease models, and this positively correlated with the level of lipopolysaccharide translocation.
Conclusions
More significant liver injury, inflammation, and fibrosis are seen in diet-induced steatohepatitis, which positively correlates with translocation of bacterial components, but not intact bacteria.
Keywords: alcohol-associated liver disease, non-alcoholic fatty liver disease, steatohepatitis, bacterial translocation, gut-liver-axis, microbiome
INTRODUCTION
Alcohol-associated liver disease (ALD) and non-alcoholic fatty liver disease (NAFLD) are two of the leading causes of cirrhosis and cirrhosis-related mortality in the United States and worldwide1–5. ALD and NAFLD share disease progression through a similar spectrum from hepatic steatosis to steatohepatitis (alcohol-associated hepatitis (AH) and non-alcoholic steatohepatitis (NASH), respectively), development of fibrosis, and liver cirrhosis6. Clinically distinguishing NAFLD from ALD remains challenging, with increasing prevalence of both alcohol use and obesity leading to a larger overlap of these two major risk factors7. Yet, the distinction is important, as modest alcohol use in NAFLD patients is associated with less improvement in steatosis and aspartate transaminase levels, as well as lower odds of NASH resolution, compared with no use of alcohol8. Further, increased investigation into the metabolic-associated fatty liver disease (MAFLD) diagnosis has found that while it shares a similar clinical profile and long-term outcomes with NAFLD, the primary driver of increased liver-related mortality is driven by ALD rather than insulin resistance9, suggesting fundamental differences in the mechanisms driving these two diseases.
Many mechanistic pathways contribute to the development of liver injury in alcohol and non-alcoholic liver disease, including lipotoxicity, generation of reactive oxygen species, activation of immune receptors, and immune-mediated inflammation. Alterations in intestinal permeability and increased translocation of intestinal bacteria and gut-derived pathogen-associated molecular patterns across a disrupted gut barrier into the liver and systemic circulation via the portal vein have been implicated in the pathogenesis of both ALD and NASH10,11. The translocation of bacteria and bacterial endotoxins such as lipopolysaccharide (LPS) into the liver stimulates Kupffer cells to release pro-inflammatory cytokines through pattern-recognition receptors such as toll-like receptors (TLRs), initiating a cascade of hepatocellular injury and fibrosis. Direct comparison of bacterial translocation between ALD and NASH injury models, which may elucidate differences in the pathogenesis of the two disease mechanisms, has not yet been performed. Here, we identify key differences in the bacterial translocation between alcohol and their effects on downstream hepatic inflammation and fibrosis.
METHODS
Mouse models.
8-week chronic ethanol feeding model
Wild type (WT) C57BL/6 female mice (age, 9–12 weeks) bred at University of California San Diego were placed on Lieber DeCarli diet for 8 weeks, as previously described12. The caloric intake from ethanol was 0% on day 1, 10% on days 2 and 3, 20% on days 4 and 5, 30% from day 6 until the end of 6 weeks, and 36% for the last 2 weeks. On the last day, mice were gavaged with a single dose of ethanol (5 g/kg body weight) in the early morning and sacrificed 9 h later. Pair-fed control mice received a diet with an isocaloric substitution of dextrose.
2-week chronic-plus-binge (NIAAA) ethanol feeding model
WT C57BL/6 male mice (age, 9–12 weeks) bred at University of California San Diego were placed on a chronic–binge ethanol diet (NIAAA model), as previously described12,13. Mice were fed with the Lieber–DeCarli diet and the caloric intake from ethanol was 0% on days 1–5 and 36% from day 6 until the end of the study period. At day 16, mice were gavaged with a single dose of ethanol (5 g/kg body weight) in the early morning and sacrificed 9 h later. Pair-fed control mice received a diet with an isocaloric substitution of dextrose.
2-week chronic-plus-binge (NIAAA) ethanol feeding model in microbiota humanized gnotobiotic mice colonized with stool from patients with alcohol-associated hepatitis (AH)
C57BL/6 germ-free mice (male and female) were bred at UCSD and fecal transplantation was performed using stool samples from patients with alcoholic hepatitis, as previously described14. Mice were gavaged with 100 μl of stool samples (1 g stool dissolved in 30 ml Luria–Bertani (LB) medium containing 15% glycerol under anaerobic conditions), starting at an age of 5–6 weeks and repeated 2 weeks later. Two weeks after the second gavage, mice were placed on the ethanol or control (isocaloric) diet as described above.
20-week Western-diet feeding model of NASH
WT C57BL/6 mice were purchased from Charles River. 8- to 9-week-old male WT mice were started on a Western diet (AIN-76A, TestDiet, St. Louis, MO, with a fat content of 20% and energy from fat was 40%), together with glucose (18.9 g/L) and fructose (23.1 g/L) in the drinking water for 20 weeks, as previously described15. Control mice received standard chow diet. Mice had free access to food and water and all tissues were harvested from nonfasted mice.
20-week Western-diet feeding model in microbiota humanized gnotobiotic mice colonized with stool from NASH patients
Male germ-free C57BL/6 mice were bred at the University of California San Diego. Fecal microbiota transplantation with samples from 2 patients with NASH was performed at the age of 5–6 weeks and repeated 2 weeks later, as previously described16. Briefly, mice were gavaged 100 μL of a stool suspension from a single donor, which was prepared by dissolving 1 g stool in 30 mL Luria-Bertani (LB) medium containing 15% glycerol under anaerobic conditions. Two weeks after the second gavage, mice were placed for 20 weeks on an irradiated Western-style fast-food diet (“Western diet”) (TD.200289; containing 41.9% kcal from fat, 43.0% kcal from carbohydrate, 15.2% from protein, 4.6 kcal/g). As a control, mice were fed for 20 weeks with an irradiated low-fat control diet (“chow diet”) (TD.110637; containing 13.0% kcal from fat, 67.9% kcal from carbohydrate, 19.1% kcal from protein, 3.6 kcal/g). Diets were manufactured by Teklad Diets, Madison, WI. Mice were randomly assigned to the different groups at the beginning of the study. Animals were maintained on a 12h:12h light–dark cycle in Sentry SPP systems (Allentown, NJ) under gnotobiotic conditions. All manipulations were performed during the light cycle. Control mice received standard chow diet. Mice had free access to food and water and all tissues were harvested from nonfasted mice.
DNA extraction from sterile liver
The right lobes of the murine livers were carefully dissected in a sterile fashion and then genomic DNA was isolated to quantify bacterial 16S as previously described with slight modifications17. Briefly, the tissue was weighed and homogenized using 1.0mm Zirconia/Silica beads in PBST supplemented with Proteinase K, then digested with RNaseA and 10% SDS. The suspensions were then homogenized with UltraPure Buffer-Saturated Phenol (Thermo Fisher Scientific) and lysate was extracted twice with UltraPure Phenol:Chloroform:isoamyl alcohol (Thermo Fisher Scientific) and once with chloroform and sodium acetate buffer. DNA was then precipitated, washed with ethanol, and resuspended in sterile water. The 16S ribosomal RNA gene was amplified using established primers detailed below and the gene expression was normalized to host 18S.
Real-time quantitative PCR.
RNA was extracted from mouse livers and cDNA was generated. Primer sequences for mouse genes were obtained from the NIH qPrimerDepot. All primers used in this study are as follows, and gene expression was determined with Sybr Green (Bio-Rad Laboratories) using the ABI StepOnePlus real-time PCR system. The qPCR value was normalized to 18S.
| Gene | Primer | Nucleotide Sequence |
|---|---|---|
| Mouse 18S | F | 5’-AGTCCCTGCCCTTTGTACACA-3’ |
| R | 5’-CGATCCGAGGGCCTCACTA-3’ | |
| Mouse Acta2 | F | 5’-GTTCAGTGGTGCCTCTGTCA-3’ |
| R | 5’-ACTGGGACGACATGGAAAAG-3’ | |
| Mouse Col1a1 | F | 5’-TAGGCCATTGTGTATGCAGC-3’ |
| R | 5’-ACATGTTCAGCTTTGTGGACC-3’ | |
| Mouse Cxcl1 | F | 5’-TGCACCCAAACCGAAGTC-3’ |
| R | 5’-GTCAGAAGCCAGCGTTCACC-3’ | |
| Mouse Ccl2 | F | 5’-ATTGGGATCATCTTGCTGGT-3’ |
| R | 5’-CCTGCTGTTCACAGTTGCC-3’ | |
| Mouse Il1b | F | 5’-GGTCAAAGGTTTGGAAGCAG-3’ |
| R | 5’-TGTGAAATGCCACCTTTTGA-3’ | |
| Mouse Tgfb1 | F | 5’-GGAGAGCCCTGGATACCAAC-3’ |
| R | 5’-CAACCCAGGTCCTTCCTAAA-3’ | |
| Mouse Tlr2 | F | 5’-CATCACCGGTCAGAAAACAA-3’ |
| R | 5’-ACCAAGATCCAGAAGAGCCA-3’ | |
| Mouse Tlr4 | F | 5’-ATGGCATGGCTTACACCACC-3’ |
| R | 5’-GAGGCCAATTTTGTCTCCACA-3’ | |
| Mouse Tnfa | F | 5’-AGGGTCTGGGCCATAGAACT-3’ |
| R | 5’-CCACCACGCTCTTCTGTCTAC-3’ | |
| Universal bacterial 16S | F | 5’-GTGSTGCAYGGYTGTCGTCA-3’ |
| R | 5’-ACGTCRTCCMCACCTTCCTC-3’ | |
| Bacteriodetes 16S | F | 5’-GGCGACCGGCGCACGGG-3’ |
| R | 5’-GRCCTTCCTCTCAGAACCC-3’ | |
| Firmicutes 16S | F | 5’-GGAGYATGTGGTTTAATTCGAAGCA-3’ |
| R | 5’-AGCTGACGACAACCATGCAC-3’ | |
| Proteobacteria 16S | F | 5’-TCGTCAGCTCGTGTYGTGA-3’ |
| R | 5’-CGTAAGGGCCATGATG-3’ |
Biochemical analysis.
LPS was measured in mouse serum extracted from the inferior vena cava using the Mouse Lipopolysaccharides ELISA Kit (Cusabio). The albumin content in mouse feces was measured by ELISA (Bethyl Laboratories). Feces were diluted in dilution buffer (100mg/ml) and analyzed following the manufacturer’s instructions. Serum levels of alanine aminotransferase (ALT) were determined using a previously described protocol which quantitates the activity of ALT via rate of NADH oxidation18. Hepatic triglyceride levels were measured using Triglyceride Liquid Reagents kit (Pointe Scientific).
Staining procedures.
To determine liver fibrosis, formalin-fixed tissue samples were embedded in paraffin and stained with Sirius red.
Statistical analysis.
Results are expressed as mean ± s.e.m. (except when otherwise stated). Significance of two groups or multiple groups were evaluated using two-sided unpaired Student’s t-test, or one-way analysis of variance (ANOVA) with Tukey’s post-hoc test, respectively.
RESULTS
Translocation of bacteria is restricted to ethanol-induced liver injury
First, we assessed translocation of bacterial LPS into the peripheral circulation of conventional mice treated subjected to ethanol- and Western diet-induced liver disease models. WT mice subjected to 2-week chronic-plus-binge ethanol, 8-week chronic ethanol, or Western diet all had significantly higher levels of serum LPS compared with controls, but Western diet resulted in significantly more LPS translocation than chronic ethanol (Figure 1a). Next, to model the effects of human microbiota from liver disease patients on liver disease progression, germ-free mice were humanized with AH and NASH microbiota from two different patients each. The microbiota composition between AH and NASH patients were different, with AH patients harboring higher abundance of Akkermansia and Veillonella species (Supplementary Figure 1). AH-microbiota humanized mice exhibited higher baseline serum LPS levels, with a trend towards higher levels in chronic-plus-binge ethanol-fed mice. NASH-microbiota humanized mice fed a Western diet also exhibited significantly higher levels of serum LPS as compared with their controls. Compared with ethanol-induced liver disease models, treatment with Western diet in both traditional and microbiota-humanized mouse models resulted in significantly higher levels of bacterial LPS translocation (Figure 1a).
Figure 1. Gut permeability and bacterial translocation are affected differently in ethanol versus diet-induced liver disease.
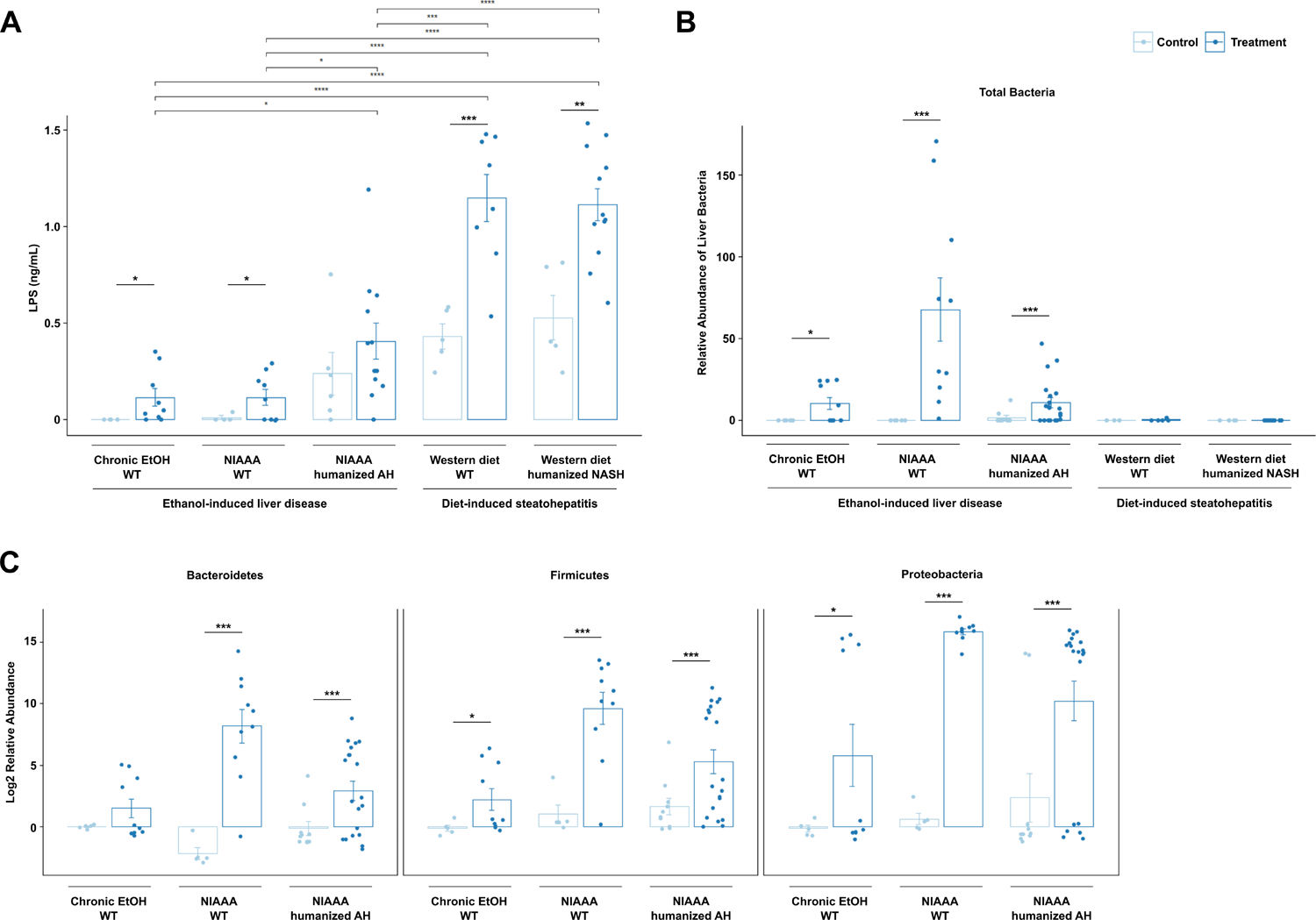
(a) Bacterial LPS concentrations in sterile harvested serum, (b) relative abundance of total bacterial DNA isolated from mouse livers dissected in a sterile fashion normalized to host 18S gene expression, and (c) hepatic DNA levels of Bacteroidetes, Firmicutes, and Proteobacteria, in the five models of liver disease, normalized to total amount of liver using mouse 18S primers. Results are expressed as mean ± s.e.m. (a–c). P values between mice in the control versus treatment groups were determined by two-sided Student t-test and p values among mice in different treatment groups were determined by one-way ANOVA with Tukeýs post-hoc test. *P < 0.05, **P < 0.01, ***P < 0.001.
In contrast, when we assessed translocation of bacteria to the liver using 16S qPCR, we were only able to detect bacteria in the sterilely procured livers of mice subjected to ethanol treatment (Figure 1b). Ethanol-induced liver disease was associated with translocation of bacteria from both the gram-positive Firmicutes phylum and gram-negative Bacteroidetes and Proteobacteria phyla to the liver (Figure 1c).
To assess the effect of ethanol versus Western diet on intestinal permeability, fecal albumin levels were measured19 (Supplementary Figure 2). Similar to the effect seen in LPS translocation, fecal albumin levels were higher in treatment groups than control groups, indicating increased intestinal permeability in response to ethanol or Western diet treatment. However, in contrast to serum LPS levels, there was no significant difference in fecal albumin between Western diet and ethanol treatment in conventional mice; only NASH-microbiota humanized mice fed a Western diet demonstrated increased intestinal permeability compared with other treatments. Differences in the microbiota between conventional mice and microbiota humanized groups might contribute to changes in permeability.
Liver injury, inflammation, and fibrosis are correlated with translocation of LPS but not translocation of bacteria
We next compared the level of liver injury and inflammation in the five mouse models. While significant injury was induced by both Western diet and alcohol, Western diet induced more severe liver injury than chronic alcohol use, as demonstrated by serum ALT (Figure 2a). Further, serum ALT levels were significantly correlated with LPS translocation (Figure 2b). Gene expression of Ccl2 (C-C Motif Chemokine Ligand 2), an inflammatory chemokine produced primarily by injured hepatocytes and activated Kupffer cells and responsible for the recruitment of monocytes and lymphocytes, was also significantly elevated in livers subjected to Western diet compared with ethanol-induced liver injury (Figure 3a), whereas Cxcl1 (C-X-C Motif Chemokine Ligand 1), a hepatocyte-derived chemokine responsible for neutrophil recruitment, was only modestly elevated in Western diet-induced liver injury as compared with ethanol-induced injury (Figure 3c). Tnf (tumor necrosis factor) and Il1b (Interleukin 1 beta) showed similar patterns of significant elevation in both Western diet injury models as well as in AH-microbiota humanized mice fed ethanol (Figure 3e, g). Interestingly, though the gene expression of each of these inflammatory mediators was significantly correlated with LPS translocation (Supplementary Figure 3), when correlation was assessed on data stratified by ethanol versus diet-mediated injury, significant positive correlation was only seen for diet-induced steatohepatitis models (Figure 3b, d, f, h).
Figure 2. Diet-induced steatohepatitis is associated with more liver injury.
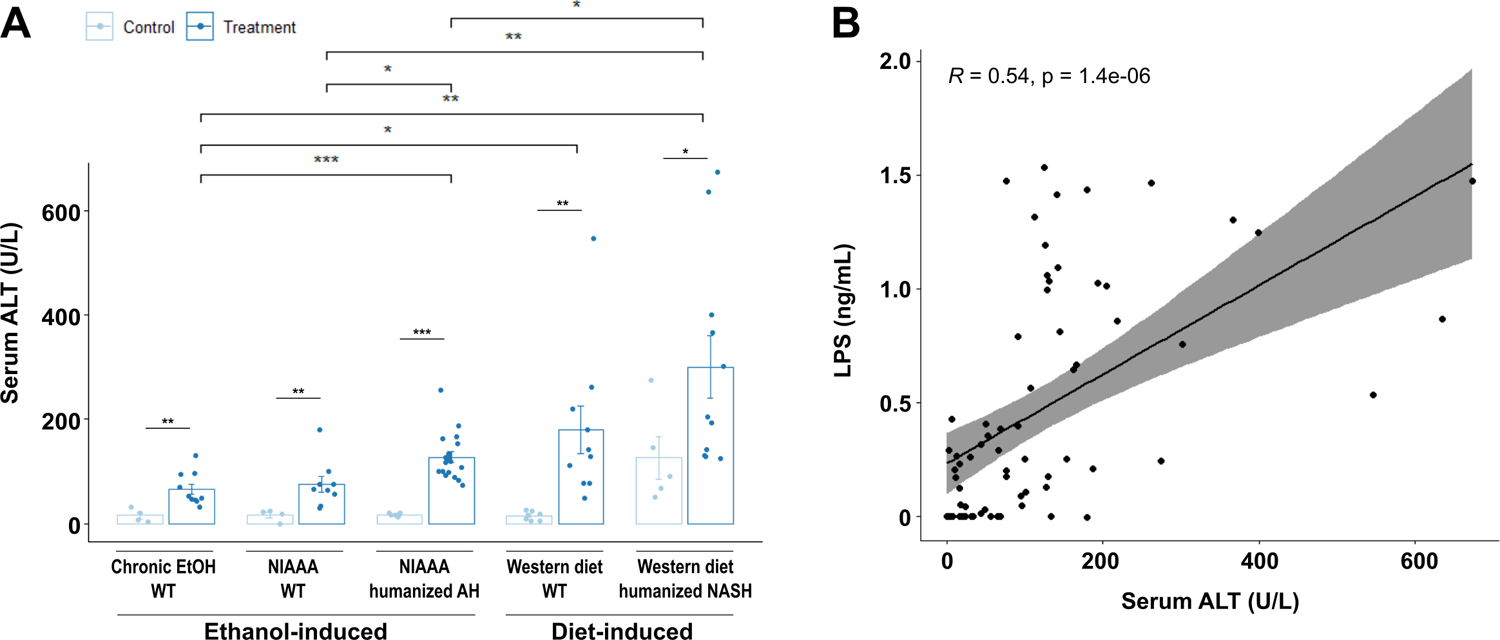
(a) Serum levels of ALT in the five models of liver disease. (b) Pearson’s correlation of serum ALT and serum LPS concentrations in mice from all groups. P values between mice in the control versus treatment groups were determined by two-sided Student t-test and p values among mice in different treatment groups were determined by one-way ANOVA with Tukeýs post-hoc test. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 3. Diet-induced steatohepatitis is associated with more liver inflammation.
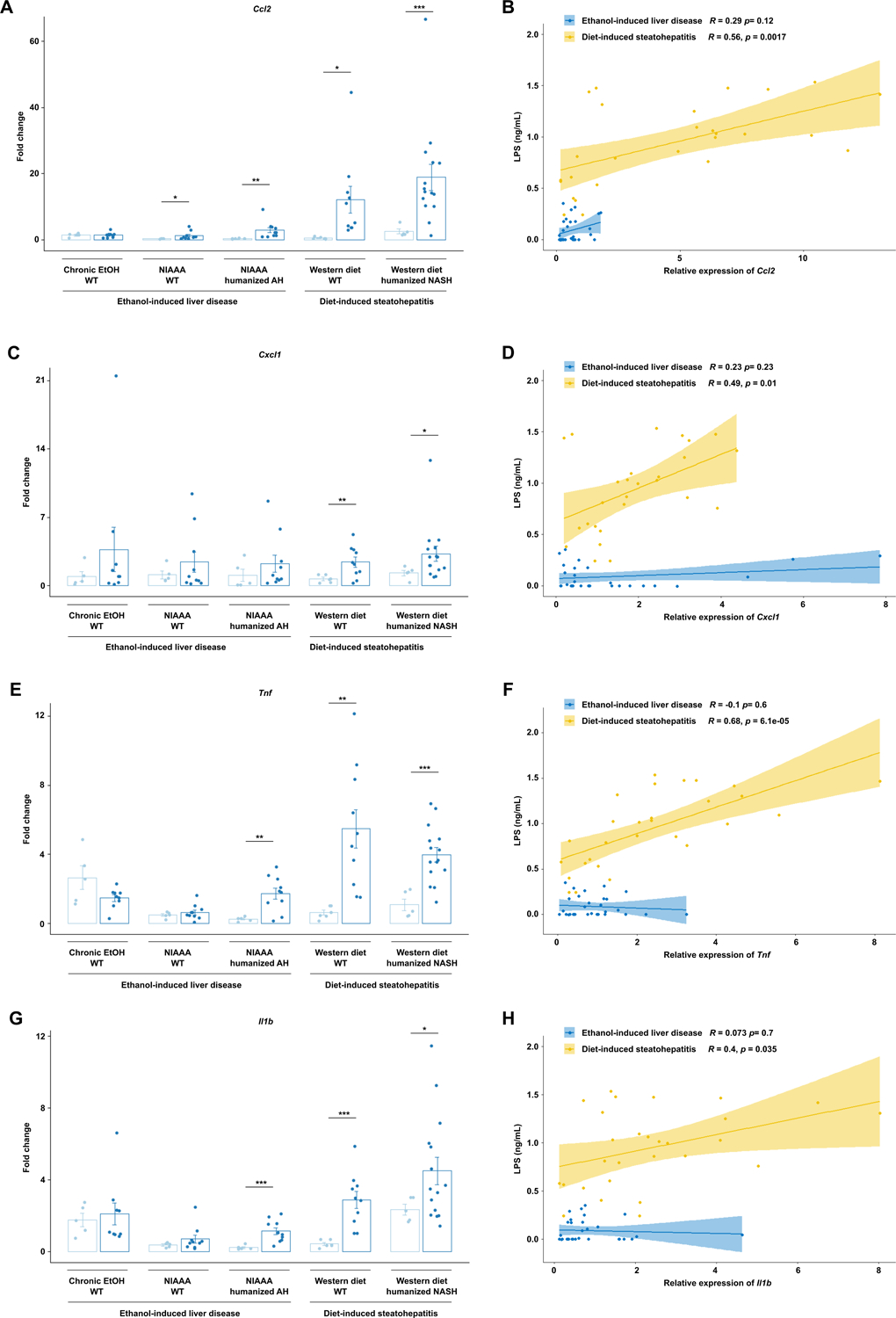
(a) Hepatic levels of (a) Ccl2, (c) Cxcl1, (e) Tnf, and (g) Il1b mRNAs in the five models of liver disease. Pearson’s correlation of (b) Ccl2, (d) Cxcl1, (f) Tnf, and (h) Il1b mRNAs and serum LPS concentrations in mice from all groups, stratified by ethanol versus diet-induced liver injury. P values between mice in the control versus treatment groups were determined by two-sided Student t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
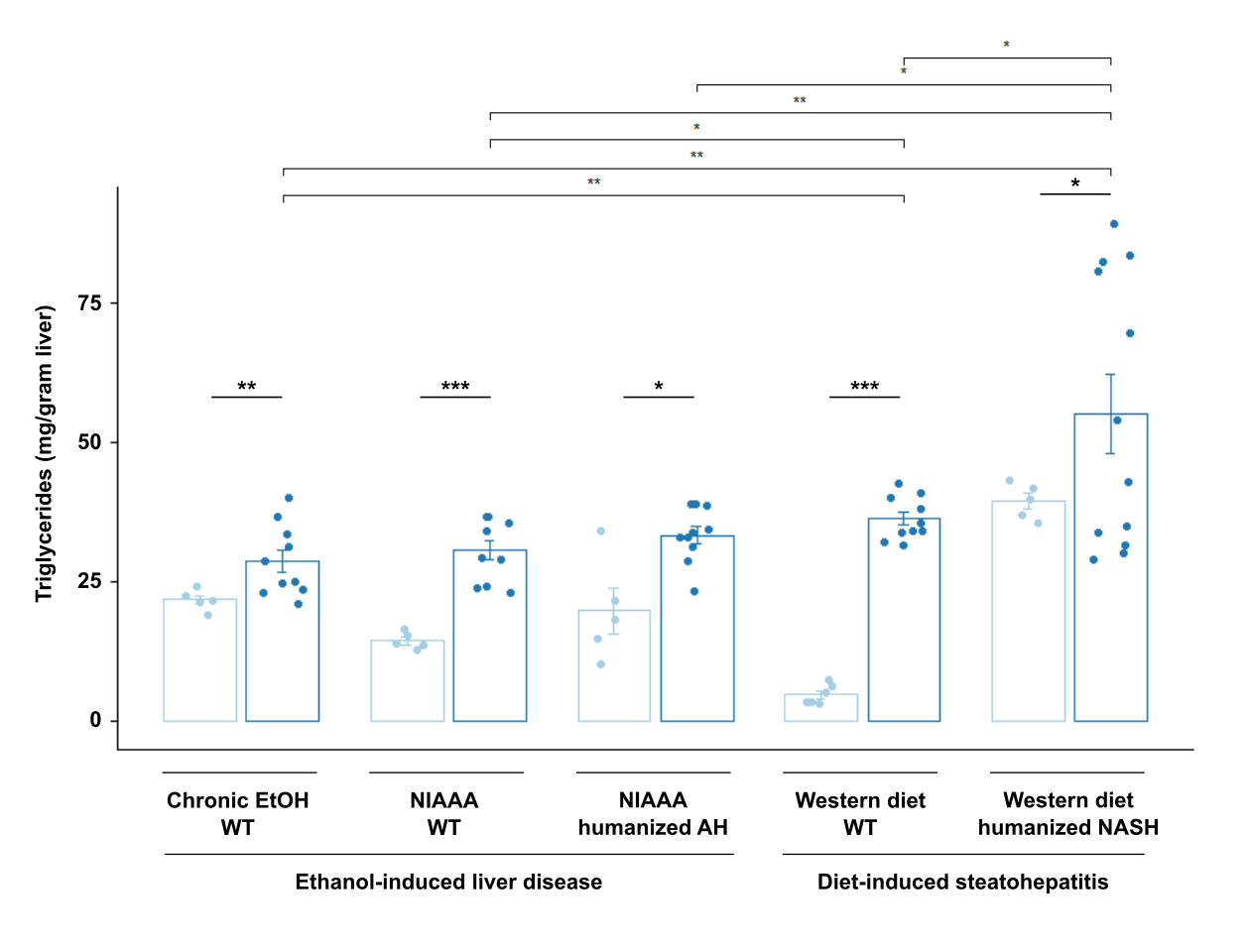
Similar to the patterns of liver inflammation, expression of genes associated with hepatic fibrosis such as Col1a1 (collagen type I alpha 1), Tgbf (transforming growth factor-beta), and Acta2 (actin alpha 2) were also most significantly elevated in livers subjected to Western diet compared with ethanol diet (Figure 4a–c). Further, significant positive correlation between LPS translocation and gene expression of Col1a1, Tgfb, and Acta2 was seen only in diet-induced steatohepatitis models (Figure 4b, d, f, Supplementary Figure 4a–c). Sirius red staining also demonstrates more severe fibrosis in Western diet-induced liver disease compared with ethanol-induced liver injury models (Figure 3g). No significant fibrosis was seen in the two-week alcohol models in both wild type and AH-microbiota humanized mice. Hepatic steatosis, as measured by hepatic triglyceride levels, is also increased in Western diet-induced compared with ethanol-induced liver disease (Supplementary Figure 5).
Figure 4. Diet-induced steatohepatitis is associated with more liver fibrosis.
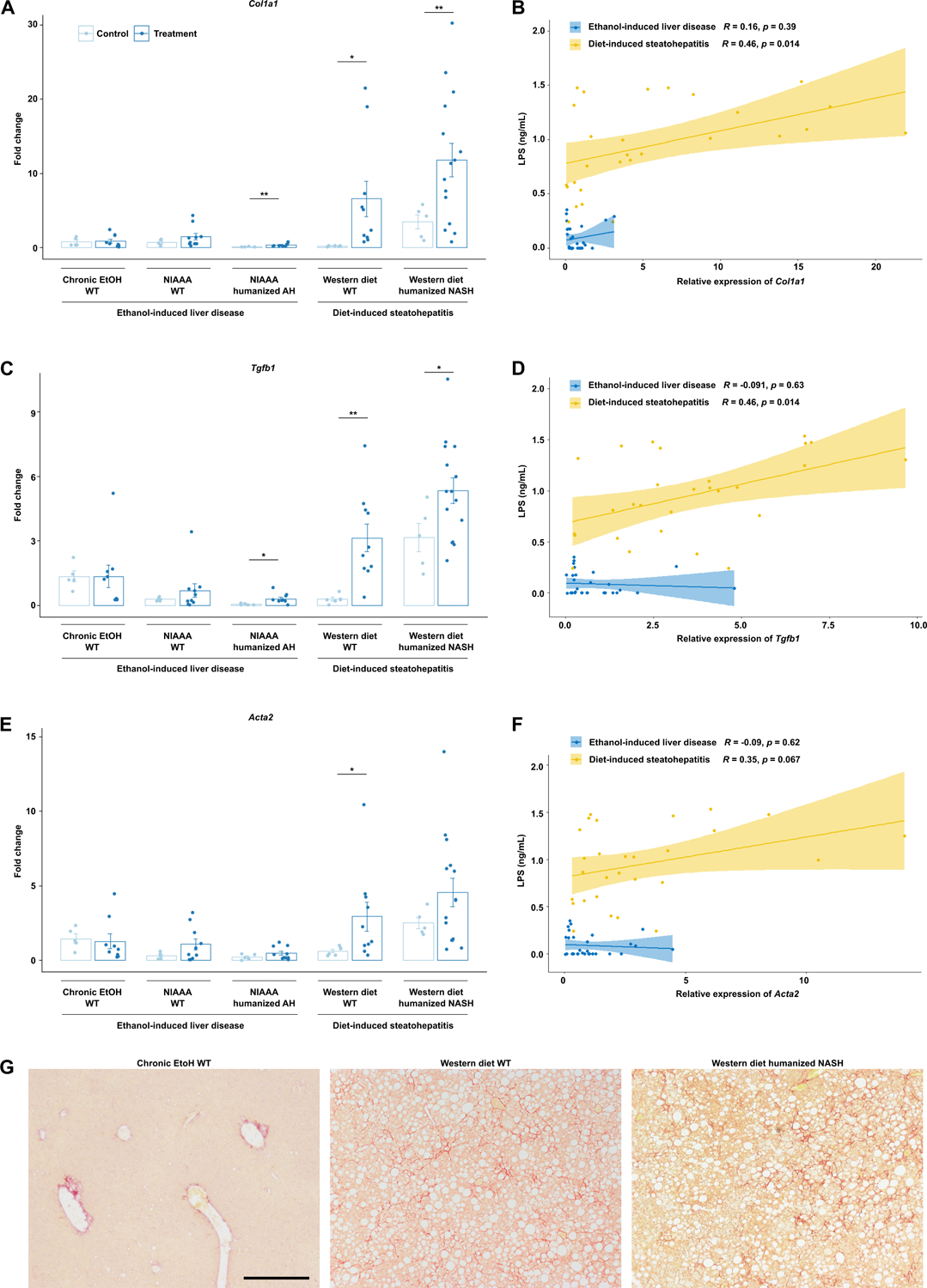
Hepatic levels of (a) Col1a1, (c) Tgfb, and (e) Acta2 mRNAs in the five models of liver disease. Pearson’s correlation of (b) Col1a1, (d) Tgfb, and (f) Acta2 mRNAs and serum LPS concentrations in mice from all groups, stratified by ethanol versus diet-induced liver injury. (g) Representative Sirius red-stained liver sections. P values between mice in the control versus treatment groups were determined by two-sided Student t-test. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bar indicates 200 um.
LPS induces hepatic inflammation via the TLR4 signaling pathway20, which has been implicated in the pathogenesis of both NASH and ALD, but direct comparison of the two modalities of injury has not yet been performed21. We found that in Western diet-induced liver injury models, the expression of Tlr4 was significantly increased compared with controls, similar to serum LPS levels (Figure 5a), while there was no significant difference with ethanol-induced injury models. Tlr2, which recognizes gram-positive bacterial cell wall components and has also been shown to promote liver inflammation and fibrosis in NASH and ALD, was also only significantly overexpressed in the livers of mice on Western diet and not in mice subjected to ethanol (Figure 5c). Increased LPS translocation was significant positively correlated with expression of Tlr2 and Tlr4 only in diet-induced steatohepatitis models (Figure 5b, d, Supplementary Figure 4d, e). Tlr2 and Tlr4 is expressed on parenchymal and non-parenchymal liver cells22.
Figure 5. Diet-induced steatohepatitis is associated with upregulation in the TLR signaling pathway.
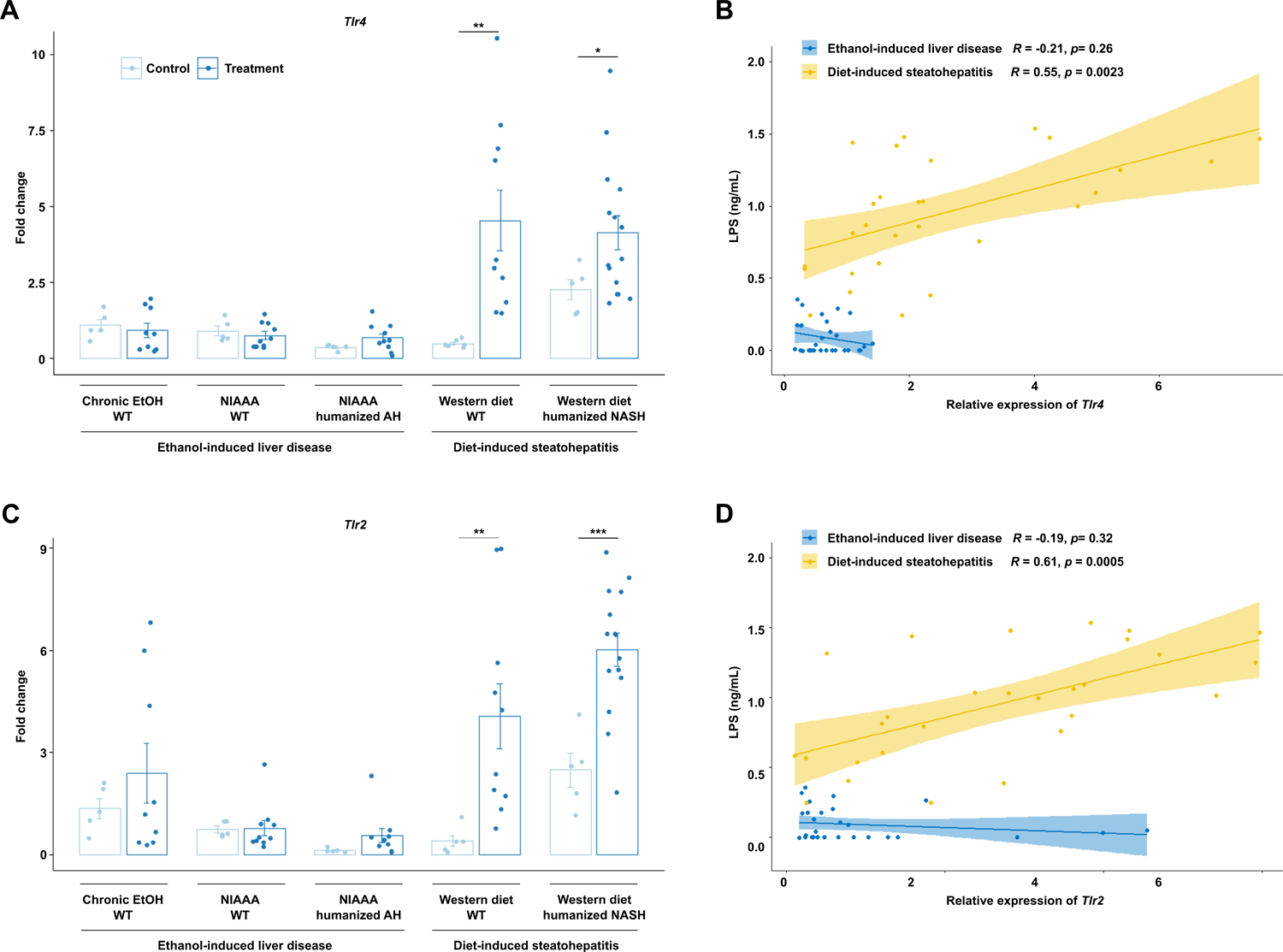
Hepatic levels of (a) Tlr4 and (c) Tlr2 mRNAs in the five models of liver disease. Pearson’s correlation of (b) Tlr4 and (d) Tlr2 mRNAs and serum LPS concentrations in mice from all groups, stratified by ethanol versus diet-induced liver injury. P values between mice in the control versus treatment groups were determined by two-sided Student t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
DISCUSSION
In both alcohol-associated liver disease and nonalcoholic steatohepatitis, several mechanisms are disrupted at the interface between the gut microbiota and the intestinal epithelium, including increased intestinal permeability, dysregulation of bile acid metabolism, and decreased protective microbial metabolite production. Bacterial translocation is an important contributor to the liver inflammation and fibrosis seen in both ALD and NASH, but the role that it plays in disease pathogenesis in the two disease etiologies is not well understood. Most existing studies assess bacterial translocation via proxy measures such as quantifying bacterial components in the blood or inflammatory markers in the liver or serum11 or via in vitro assays23, and few studies have quantified bacterial abundance in the liver. In this study, we assessed bacterial translocation using two different methods: 1) we quantify LPS using an enzyme-linked immunoassay with antibodies purified against an E. coli LPS polymer in sterile harvested serum and confirmed these findings by measuring fecal albumin, and 2) we measured 16S rDNA quantitatively by PCR from bacterial DNA isolated directly from mouse livers dissected in a sterile fashion. We found that while there is increased intestinal permeability and translocation of bacterial products in all models, translocation of bacteria is restricted to ethanol-induced liver disease. Further, liver injury, inflammation, and fibrosis were positively associated with the severity of LPS translocation only in diet-induced steatohepatitis and not in ethanol-induced liver injury.
The differences we see in translocation of viable bacteria versus bacterial components such as LPS in ethanol-induced versus diet-induced models of liver injury are consistent with existing literature. For example, mice deficient in Reg3g, an antimicrobial molecule that prevents translocation of viable bacteria but has no effect on LPS translocation, have increased susceptibility to ethanol-induced liver disease, but not to Western diet‐induced steatohepatitis17,24. Intestinal Reg3b and Reg3g are downregulated in mice fed an ethanol diet25, while they are not significantly changed in the intestine of mice fed a high fat diet26. This may explain the differences we see in bacterial abundance in the liver between these two etiologies of liver disease. We also see significant variation in bacterial translocation and intestinal permeability between individual mice in both ALD and NASH models, as has been reported in pre-cirrhotic ALD and NASH patients11,27, and the reason for this is not yet understood.
Further studies are also needed to understand why translocation of bacteria and LPS are discordant in diet-induced models and whether this contributes to the increased severity of liver injury, inflammation, and fibrosis. While increased intestinal permeability in treatment conditions (ethanol or Western-diet) mirrors the trends seen in LPS translocation, differences in intestinal permeability alone do not explain translocation of whole bacteria. One possibility is that phagocytic receptors on Kupffer cells, which are important for promoting hepatic clearance of translocated bacteria and found to be decreased in ALD12, may be affected differently in NASH. Another possibility is that the bacterial translocation that occurs in ethanol-induced liver injury promotes hepatic immune activation and clearance of LPS such that there is less detectable LPS in the peripheral circulation. An interesting recent study demonstrated a liver microbiome distinct from the gut microbiome and found that the recruitment of inflammatory cells to the liver and the level of intrahepatic leukocytic proliferation was dependent on the liver microbiome28. They found the liver microbiome was enriched for Proteobacteria, similar to our findings, and the levels of hepatic immune activation were specifically correlated to the relative abundance of Bacteroidetes in the liver. This would suggest that the increased serum LPS levels we see in diet-induced liver disease models may be secondary to a deficiency in immune activation and LPS clearance, which may also result in increased liver injury.
There may be additional implications for increased translocation of viable bacteria in ALD compared with NASH as liver disease progresses. For example, spontaneous bacterial peritonitis, a serious complication of cirrhosis associated with significant morbidity and mortality, is disproportionately seen in patients with cirrhosis secondary to ALD compared with NAFLD/NASH29,30. Additionally, increased translocation of viable bacteria in ALD may explain why liver-related mortality in MAFLD patients is driven by ALD9. As the prevalence of both ALD and NASH continues to rise, better understanding of the differences in liver injury driven by alcohol versus non-alcoholic means may help us understand the additive effects of injury when the two etiologies overlap.
Supplementary Material
Supplementary Figure 1. Bacterial composition of AH and NASH patient microbiota used to colonize germ-free mice. Mean relative abundance of the top 21 most abundant bacterial genera; 0–1 corresponds to 0%−100% abundance.
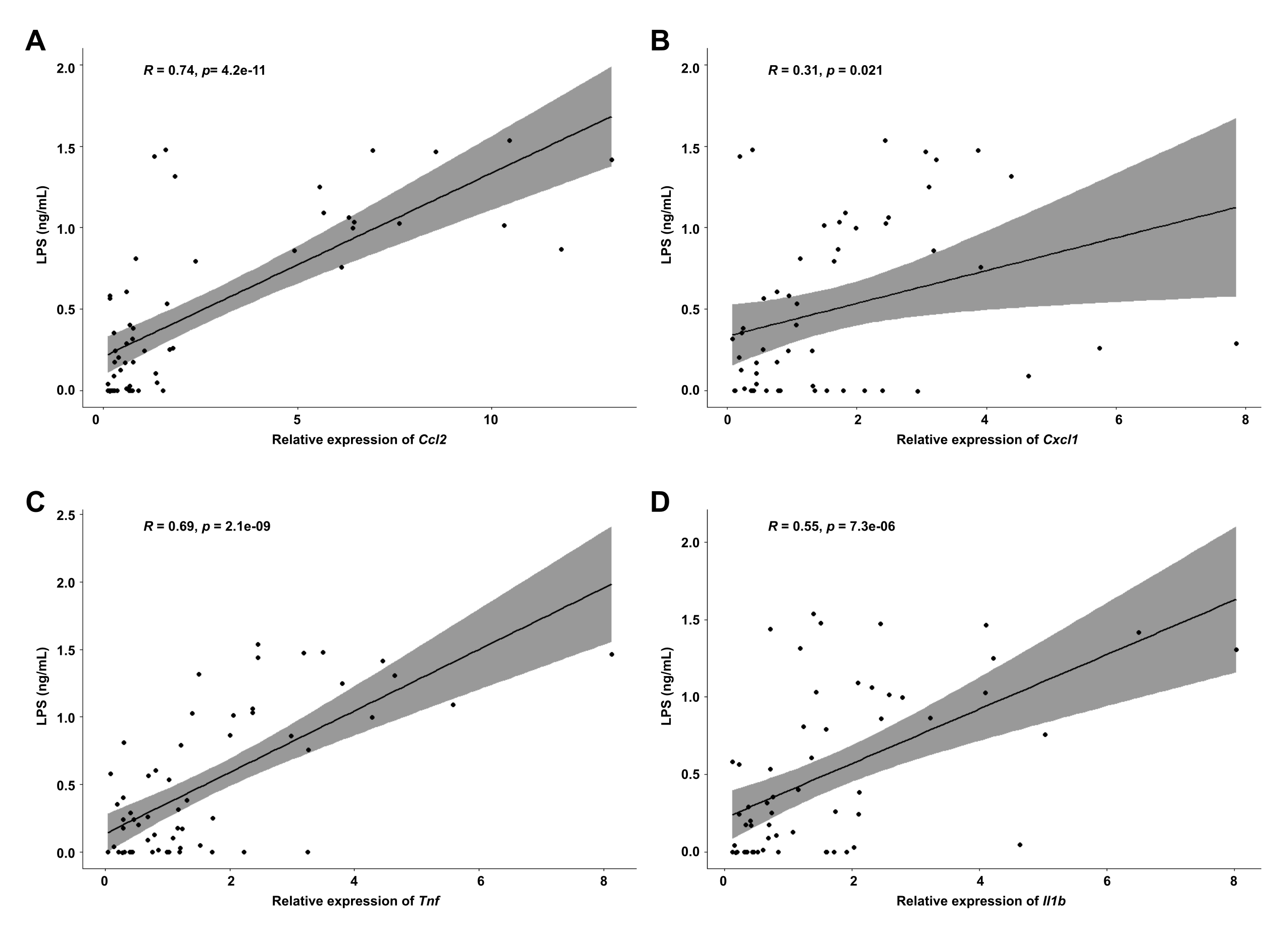
Supplementary Figure 3. Hepatic inflammation is positively correlated with LPS translocation. Pearson’s correlation of (a) Ccl2, (b) Cxcl1, (c) Tnf, and (d) Il1b mRNAs and serum LPS concentrations in mice from all groups.
{kind=link}
Supplementary Figure 4. Hepatic fibrosis and TLR signalling is positively correlated with LPS translocation. Pearson’s correlation of (a) Col1a1, (b) Tgfb, (c) Acta2, (d) Tlr4, and (e) Tlr2 mRNAs and serum LPS concentrations in mice from all groups.
{kind=link}
Supplementary Figure 5. Diet-induced steatohepatitis is associated with increased steatosis. Hepatic triglyceride content in the five models of liver disease. P values between mice in the control versus treatment groups were determined by two-sided Student t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
{kind=link}
Supplementary Figure 2. Intestinal permeability in ethanol and diet-induced liver disease models. Fecal albumin levels in the five different models of liver disease.
Acknowledgment of grant support:
C.H. is supported by T32 DK007202. This study was supported in part by the Southern California Research Center for ALPD and Cirrhosis funded by the National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health P50AA011999 (to C.L.H.), NIH grants K12 HD85036, University of California San Diego Altman Clinical and Translational Research Institute (ACTRI)/NIH grant KL2TR001444, Pinnacle Research Award in Liver Diseases Grant #PNC22–159963 from the American Association for the Study of Liver Diseases Foundation (to P.H.), NIH grants R01 AA24726, R37 AA020703, U01 AA026939, U01 AA026939–04S1, by Award Number BX004594 from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development, and a Biocodex Microbiota Foundation Grant (to B.S.) and services provided by NIH centers P50 AA011999 and the San Diego Digestive Diseases Research Center (SDDRC) P30 DK120515.
Conflicts of interest:
B.S. has been consulting for Ambys Medicines, Ferring Research Institute, Gelesis, HOST Therabiomics, Intercept Pharmaceuticals, Mabwell Therapeutics, Patara Pharmaceuticals and Takeda. B.S.’s institution UC San Diego has received research support from Axial Biotherapeutics, BiomX, CymaBay Therapeutics, NGM Biopharmaceuticals, Prodigy Biotech and Synlogic Operating Company. B.S. is founder of Nterica Bio. UC San Diego has filed several patents with Y.D., C.L. and B.S. as inventors related to this work.
REFERENCES
- 1.Dang K, Hirode G, Singal AK, Sundaram V, Wong RJ. Alcoholic Liver Disease Epidemiology in the United States: A Retrospective Analysis of 3 US Databases. Am J Gastroenterol. Jan 2020;115(1):96–104. [DOI] [PubMed] [Google Scholar]
- 2.Mitra S, De A, Chowdhury A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl Gastroenterol Hepatol. 2020;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong T, Dang K, Ladhani S, Singal AK, Wong RJ. Prevalence of Alcoholic Fatty Liver Disease Among Adults in the United States, 2001–2016. JAMA. May 7 2019;321(17):1723–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. Jul 2016;64(1):73–84. [DOI] [PubMed] [Google Scholar]
- 5.Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. Nov 10 2018;392(10159):1789–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tannapfel A, Denk H, Dienes HP, et al. Histopathological diagnosis of non-alcoholic and alcoholic fatty liver disease. Virchows Arch. May 2011;458(5):511–523. [DOI] [PubMed] [Google Scholar]
- 7.Staufer K, Huber-Schonauer U, Strebinger G, et al. Ethyl glucuronide in hair detects a high rate of harmful alcohol consumption in presumed non-alcoholic fatty liver disease. J Hepatol. May 20 2022. [DOI] [PubMed] [Google Scholar]
- 8.Ajmera V, Belt P, Wilson LA, et al. Among Patients With Nonalcoholic Fatty Liver Disease, Modest Alcohol Use Is Associated With Less Improvement in Histologic Steatosis and Steatohepatitis. Clin Gastroenterol Hepatol. Sep 2018;16(9):1511–1520 e1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Younossi ZM, Paik JM, Al Shabeeb R, Golabi P, Younossi I, Henry L. Are there outcome differences between NAFLD and metabolic-associated fatty liver disease? Hepatology. Apr 1 2022. [DOI] [PubMed] [Google Scholar]
- 10.Trebicka J, Macnaughtan J, Schnabl B, Shawcross DL, Bajaj JS. The microbiota in cirrhosis and its role in hepatic decompensation. J Hepatol. Jul 2021;75 Suppl 1:S67–S81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luther J, Garber JJ, Khalili H, et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol Gastroenterol Hepatol. Mar 2015;1(2):222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duan Y, Chu H, Brandl K, et al. CRIg on liver macrophages clears pathobionts and protects against alcoholic liver disease. Nat Commun. Dec 9 2021;12(1):7172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc. Mar 2013;8(3):627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duan Y, Llorente C, Lang S, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. Nov 2019;575(7783):505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou R, Llorente C, Cao J, et al. Intestinal alpha1-2-Fucosylation Contributes to Obesity and Steatohepatitis in Mice. Cell Mol Gastroenterol Hepatol. 2021;12(1):293–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Demir M, Lang S, Hartmann P, et al. The fecal mycobiome in non-alcoholic fatty liver disease. J Hepatol. Apr 2022;76(4):788–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bluemel S, Wang L, Martino C, et al. The Role of Intestinal C-type Regenerating Islet Derived-3 Lectins for Nonalcoholic Steatohepatitis. Hepatol Commun. Apr 2018;2(4):393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartmann P, Schnabl B. Inexpensive, Accurate, and Stable Method to Quantitate Blood Alanine Aminotransferase (ALT) Levels. Methods Protoc. Oct 14 2022;5(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartmann P, Chen P, Wang HJ, et al. Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology. Jul 2013;58(1):108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roh YS, Seki E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J Gastroenterol Hepatol. Aug 2013;28 Suppl 1:38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiziltas S. Toll-like receptors in pathophysiology of liver diseases. World J Hepatol. Nov 18 2016;8(32):1354–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun L, Dai JJ, Hu WF, Wang J. Expression of toll-like receptors in hepatic cirrhosis and hepatocellular carcinoma. Genet Mol Res. Jul 14 2016;15(2). [DOI] [PubMed] [Google Scholar]
- 23.Mouries J, Brescia P, Silvestri A, et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J Hepatol. Dec 2019;71(6):1216–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, Fouts DE, Starkel P, et al. Intestinal REG3 Lectins Protect against Alcoholic Steatohepatitis by Reducing Mucosa-Associated Microbiota and Preventing Bacterial Translocation. Cell Host Microbe. Feb 10 2016;19(2):227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan AW, Fouts DE, Brandl J, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology. Jan 2011;53(1):96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartmann P, Seebauer CT, Mazagova M, et al. Deficiency of intestinal mucin-2 protects mice from diet-induced fatty liver disease and obesity. Am J Physiol Gastrointest Liver Physiol. Mar 1 2016;310(5):G310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leclercq S, Matamoros S, Cani PD, et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc Natl Acad Sci U S A. Oct 21 2014;111(42):E4485–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leinwand JC, Paul B, Chen R, et al. Intrahepatic microbes govern liver immunity by programming NKT cells. J Clin Invest. Apr 15 2022;132(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niu B, Kim B, Limketkai BN, et al. Mortality from Spontaneous Bacterial Peritonitis Among Hospitalized Patients in the USA. Dig Dis Sci. May 2018;63(5):1327–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim KH, Potts JR, Chetwood J, Goubet S, Verma S. Long-term outcomes after hospitalization with spontaneous bacterial peritonitis. J Dig Dis. Apr 2015;16(4):228–240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Bacterial composition of AH and NASH patient microbiota used to colonize germ-free mice. Mean relative abundance of the top 21 most abundant bacterial genera; 0–1 corresponds to 0%−100% abundance.
Supplementary Figure 3. Hepatic inflammation is positively correlated with LPS translocation. Pearson’s correlation of (a) Ccl2, (b) Cxcl1, (c) Tnf, and (d) Il1b mRNAs and serum LPS concentrations in mice from all groups.
Supplementary Figure 4. Hepatic fibrosis and TLR signalling is positively correlated with LPS translocation. Pearson’s correlation of (a) Col1a1, (b) Tgfb, (c) Acta2, (d) Tlr4, and (e) Tlr2 mRNAs and serum LPS concentrations in mice from all groups.
Supplementary Figure 5. Diet-induced steatohepatitis is associated with increased steatosis. Hepatic triglyceride content in the five models of liver disease. P values between mice in the control versus treatment groups were determined by two-sided Student t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
Supplementary Figure 2. Intestinal permeability in ethanol and diet-induced liver disease models. Fecal albumin levels in the five different models of liver disease.