

Abstract
Background
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm that harbors the Philadelphia chromosome. Tyrosine kinase inhibitor (TKI) therapy has dramatically improved the survival of patients with CML. Nevertheless, 20%–40% of CML patients require changes in TKI therapy due to intolerance or drug resistance. A total of 30%–60% of resistant cases result from kinase domain (KD) mutations. There is currently no published data on CML KD mutations in South Africa.
Methods
This retrospective, descriptive study collected data from 206 CML patients attending the King Edward Hospital Hematology clinic. Patient‐based and mutation‐based factors were analyzed using descriptive statistical analysis and Kaplan–Meier curves for survival analysis.
Results
KD mutations were detected in 29.1% (n = 60 of 206). A total of 40 different KD mutations were detected, with unknown responses to TKI therapy in 65% (n = 26 of 40). A total of 57.7% (n = 15 of 26) of mutations with an unknown response, showed a response to specific TKIs in our study. Four patients had A399T mutations, of which two showed good responses to Nilotinib. Patients with I293N and V280M mutations showed good responses to Imatinib. G250E was most frequently detected. Despite M351T being one of six most commonly reported KD mutations globally, this mutation was not detected in our patient cohort. A total of 20.9% (n = 43 of 206) human immunodeficiency virus (HIV) positive patients were identified, of which 25.6% (n = 11 of 43) had KD mutations. HIV status showed no significant effect on mutational status or overall survival.
Conclusion
The predicted response to TKI therapy was unknown in more than half of the KD mutations detected in our patient population. Additionally, eight patients with mutations with known responses to TKIs showed responses discordant to that expected. HIV status and KD mutations had no statistically significant effect on overall survival. Although some data were comparable to international publications, few notable differences warrant further investigation.
Keywords: chronic myeloid leukemia, Imatinib, kinase domain, mutations, resistance
1. INTRODUCTION
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm that accounts for approximately 15% of all leukemias. 1 , 2 , 3 It is characterized by the presence of the Philadelphia chromosome (Ph+), which is found in approximately 95% of CML patients. 3 , 4 The Ph+ is produced by the fusion of the Abelson Leukemia Virus (ABL) gene found on chromosome 9, with the Breakpoint Cluster Region (BCR) gene found on chromosome 22. 1 , 4 This occurs as a result of a reciprocal translocation of genetic material between both chromosomes, termed t(9;22)(q34.1;q11.2). 1 , 2 , 3 , 4 , 5 The fusion gene codes for a 210‐kilo Dalton fusion protein (p210) which plays a role in the initial development of CML. 4 The growth, survival and proliferation signaling pathways seen in CML are driven by the expression of this oncoprotein, the BCR::ABL1 fusion gene. 4 , 6 The detection of the BCR::ABL1 fusion gene by molecular studies, confirms the diagnosis of CML. 4
In 2001, the Food and Drug Administration approved a tyrosine kinase inhibitor (TKI), Imatinib Mesylate (Gleevec) for first‐line therapy of CML. 1 , 6 Other TKIs have subsequently been approved for the treatment of CML. The clinician's choice of TKI therapy is dependent upon drug cost, availability, toxicity, efficacy, and tolerability, as well as patient‐related factors. 7 , 8 The use of targeted drug therapy has improved the 10‐year overall survival (OS) rate from 20% to 80%–90%, with some patients going on to reach a life expectancy close to that of the general population. 1 , 9
The response to TKI therapy is determined by the assessment of hematologic, cytogenetic and molecular parameters. 3 , 7 A complete hematologic response (CHR) requires the normalization of peripheral blood counts (absolute leucocyte count <10 × 109/L, platelet count <450 × 109/L, and the lack of immature granulocytes or blasts in the peripheral blood), no signs or symptoms of the disease, and the resolution of splenomegaly. 3 , 7 The absence of Ph+ metaphases defines complete cytogenetic response (CCyR). 3 , 7 Major molecular response (MMR) is achieved when BCR::ABL1 International Standardized is ≤0.1% or ≥3‐log reduction in BCR::ABL1 mRNA on quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT‐PCR). 3 , 7
Despite high rates of hematologic and cytogenetic responses to Imatinib, a significant proportion of patients (20%–40%) require a change in treatment. 10 , 11 This may be as a result of clinical or molecular resistance, or due to intolerance to Imatinib. 10 , 11 Kinase Domain (KD) mutations are detected in 30%–60% of patients with Imatinib resistance, with more than a hundred KD mutations having been reported to date. 2 , 11 , 12 , 13 , 14 These mutations typically arise in or within proximity to one of four regions of the KD which includes the phosphate‐binding (P) loop, Src Homology 3 contact, SH2 contact, and activation (A) loop. 12 , 15
Point mutations are the most frequent type of KD mutation within the ABL1 KD to cause resistance to Imatinib. 10 , 14 , 15 These point mutations impair or prevent the binding of certain TKIs to their binding sites. 12 , 15
Screening for KD mutations at the time of diagnosis has been recommended in patients who are in the accelerated or blastic phase of disease. 12 , 15 Mutational analysis may also be performed at any time that loss of CHR, MMR, or CCyR is detected; or when additional clonal chromosomal abnormalities in the Ph+ clone are detected by chromosome banding analysis. 12
Despite its limited sensitivity, Sanger sequencing remains the gold standard for the detection of ABL1 KD mutations. 16 The advent of Next Generation Sequencing has provided an alternate method for the detection of mutations. 2 , 15 , 16
There is a scarcity of published data concerning CML in South Africa, with most available data documented in small studies. These studies have shown the median age at diagnosis of CML to be younger than that reported internationally. 17 , 18
Despite the high incidence of human immunodeficiency virus (HIV) in South Africa, there is very little South African literature on people living with HIV (PLWH) and CML concurrently. A South African publication, by Moosa et al. in 2012, reported a study of 18 PLWH and CML. These patients made up 7.5% of all patients with CML, had a median age of 37 years, and had more aggressive disease at presentation. 18 International studies have shown the co‐existence of these two disease entities to be coincidental. 17 , 18
To date, there is no published data describing the frequency and types of KD mutations detected in the South African population.
Thus, the aim of this study was to define and determine the frequency of KD mutations in CML patients in our local setting, both in patients with and without HIV. We also aimed to determine the response to changes in treatment, disease progression, and the OS of these patients.
2. MATERIALS AND METHODS
A retrospective chart review of CML patients who attended the Hematology Clinic at King Edward VIII Hospital in Kwa‐Zulu Natal between January 1, 2011 and December 31, 2021, was performed. King Edward Vlll Hospital is a tertiary, academic hospital in the province of KwaZulu‐Natal, South Africa. Of the 247 patients with CML who attended the clinic during this period, 41 patients were excluded (Table 1). Reasons for exclusion were that attendance at the clinic did not fall within the study period, the detection of mutations predated the study period, and/or an insufficient data. Data were collected for 206 patients meeting the inclusion criteria, which included patients ≥12 years of age with BCR‐ABL1 positivity, and who were commenced on Imatinib within the stipulated time period (Table 1).
Table 1.
Summary of the study population.
| Total number | n = 247 | ||
|---|---|---|---|
| (Diagnosed with CML, attended KEH hematology between 01/01/2011 and 31/12/2021) | |||
| Patients excluded | n = 41 | ||
| (Attendance at the clinic which did not fall within the study period, detection of mutations which predated the study period, and/or an insufficiency of data) | |||
| Patients included | n = 206 | ||
| (≥12 years of age, BCR‐ABL1 positive, commenced on Imatinib) | |||
| Patients with mutations | n = 60 (29.1%) | ||
| HIV status | Positive | Negative | Unknown |
| n = 11 (18.3%) | n = 40 (66.7%) | n = 9 (15.0%) | |
| Age | ≥50 years | <50 years | |
| n = 20 (33.3%) | n = 40 (66.7%) | ||
Abbreviations: CML, chronic myeloid leukemia; KEH, King Edward VIII Hospital.
All mutational analysis was performed by Sanger Sequencing at one of two referral centers, using Applied Biosystems 3500 or 3730 genetic analyzers, which are supplied by Thermofisher Life Technologies.
Both paper‐based patient records and the National Health Laboratory Service Laboratory Information System were used to source the required information. Patient names were not recorded, and each patient was assigned a unique number to protect patient confidentiality. The relevant information was extracted and documented on a data collection sheet, which was then captured electronically in password‐protected devices. Coding of variables was used to exclude subjectivity and ensure consistent interpretation.
Parameters recorded on the data collection sheet included patient age at diagnosis, HIV status, phase of disease at presentation, reason for changing TKI, result of mutational analysis, sensitivity/resistance to TKIs, choice of second TKI, and clinical and molecular response. Molecular responses were assessed according to the latest European LeukemiaNet and National Comprehensive Cancer Network defined response criteria—with the same response milestones as used for first‐line treatment. 3 , 7 qRT‐PCR results were reviewed to assess intervals of 1‐log shifts in BCR::ABL1 transcript levels, reported in IS units.
Informed consent was not required due to the retrospective, observational nature of the study. The study protocol was approved by the Biomedical Research Committee (BREC00003764/2022) and by each participating center according to the Declaration of Helsinki.
2.1. Statistical analysis
Descriptive statistics were used to summarize the data. Ā total of 2 × 2 χ 2 tests were used to examine the association of KD mutations with age groups and HIV status. Frequencies and percentages were reported for demographic variables, name of KD mutations and response to therapy.
Time from the first presentation to the date last seen or date of death (OS) was computed and Kaplan–Meier curves were drawn for patients with and without KD mutations, and by HIV status. A log‐rank test was then used to compare the equality of the survivor function. Five‐year survival rates and 95% confidence intervals are reported for both groups. A p < 0.05 was considered statistically significant. Stata v 17 statistical software was used for statistical analysis.
3. RESULTS
3.1. Patient‐based analysis
The 206 patients included in this study had a median age of 41.5 years at diagnosis (12–76 years) and a male‐to‐female ratio of 0.9. The white cell counts at presentation ranged from 23 to 874 × 109/L. 20.9% (n = 43) of patients were PLWH. However, the HIV status of 14.6% (n = 30) patients was documented or could not be traced. Additional patient‐based data can be found in Figure 1.
Figure 1.
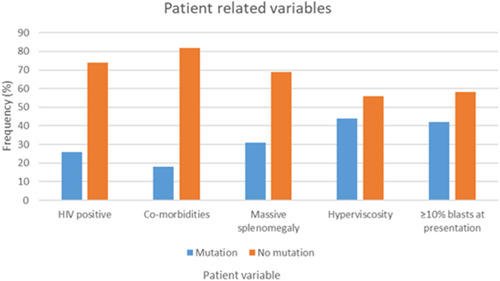
Patient‐related variables.
The majority of patients presented in the chronic phase of disease (93.2%, n = 192), followed by accelerated phase (4.4%, n = 9), and blastic phase (1.5%, n = 3). Two patients did not have sufficient data available for the classification of the phase of disease.
All patients were commenced on first‐line therapy with Imatinib, except one patient who was diagnosed at another facility before being transferred to our facility. This patient had been commenced on Nilotinib.
A total of 45.6% (n = 94) patients required a change in TKI. A total of 63.8% (n = 60) of these patients had a detectable KD mutation, however, 14 of the 60 patients had mutations sensitive to Imatinib therapy. Those patients were counseled on compliance and continued on Imatinib therapy, with or without dose adjustments. Other reasons for change in TKI included intolerance to, or cytopenias secondary to a specific TKI, suboptimal clinical or molecular responses (with negative mutational studies), disease progression, and drug availability at the facility. A total of 38.8% of patients (n = 80) had a poor molecular response, with no detected mutations.
The median time to resistance was 36 months, and the median time for a change in TKI was 63 months (4–207 months).
3.2. Mutation‐based analysis
A total of 29.1% (n = 60 of 206) of patients in this cohort developed a KD mutation during the study period.
Only 25.6% (n = 11 of 43) of PLWH had detectable mutations compared to 30.1% (n = 49 of 163) of HIV‐negative patients with KD mutations detected.
KD mutations were slightly more common in the patients diagnosed with CML who were younger than 50 years of age (30.3% vs. 27.0%, p = 0.6), however, this correlation was not statistically significant.
Forty‐six patients had one detectable KD mutation, while two and three different KD mutations were detected in nine and five patients respectively. It could not be determined from this retrospective study whether these were compound or polyclonal mutations.
Forty different KD mutations were detected in this cohort (Table 2). Of the 40 types of KD mutations detected, 14 are known to have complete or partial responses to specific TKI's, while the response to TKI therapy is not known in 26 of the 40 KD mutations (Table 2). A total of 57.7% (n = 15 of 26) of KD mutations without a documented effect on response to TKIs, showed a response to a specific TKI in this study.
Table 2.
Mutations detected, frequency and response to TKI therapy.
| Name of mutation | Number detected | Imatinib | Nilotinib | Dasatinib |
|---|---|---|---|---|
| G250E | 11 | |||
| T315I | 6 | |||
| F359I | 5 | |||
| F359V | 4 | |||
| A399T | 4 | |||
| M244V | 4 | |||
| E255K | 4 | |||
| H396R | 2 | |||
| E255V | 2 | |||
| F317L | 2 | |||
| D363_R386del | 2 | |||
| V260A | 1 | |||
| V260G | 1 | |||
| V260M | 1 | |||
| K219R | 1 | |||
| A412S | 1 | |||
| H396Y | 1 | |||
| E275K | 1 | |||
| G259D | 1 | |||
| Y253F | 1 | |||
| Y253H | 1 | |||
| I242V | 1 | |||
| I293N | 1 | |||
| E282V | 1 | |||
| V280M | 1 | |||
| E279K | 1 | |||
| V339M | 1 | |||
| G254R | 1 | |||
| Y312C | 1 | |||
| R307N | 1 | |||
| V299L | 1 | |||
| F311I | 1 | |||
| C330R | 1 | |||
| L248V | 1 | |||
| V225F | 1 | |||
| P367Q | 1 | |||
| V304RfsTer14 | 1 | |||
| L248_L247del | 1 | |||
| N331D | 1 | |||
| Q300* | 1 |
Note: Full/partial resistance  , Full/partial sensitivity
, Full/partial sensitivity  , Unknown
, Unknown  .
.
The response to TKI therapy for patients with KD mutations is documented in Table 3 (unknown effect of mutation on TKI therapy) and Table 4 (known effect of mutation on TKI therapy).
Table 3.
Mutations with unknown effect on TKI therapy, choice of TKI, and molecular response observed.
| Patient number | Mutation detected | TKI choice | Molecular responsea |
|---|---|---|---|
| 2 | Q300* | Imatinib | Good |
| 7 | D363_R386del | Nilotinib | Poor |
| 15 | V260G | Imatinib | Undetermined/static |
| 24 | V304RfsTer14 | Imatinib | Undetermined |
| 30 | K219R | Nilotinib | Good |
| 32 | A412S | Imatinib | Good |
| 44 | E275Kb | Nilotinib | Poor |
| Dasatinib | Good | ||
| 70 | G259D | Imatinib | Undetermined |
| 81 | A399T | Nilotinib | Good |
| 89 | I242V | Imatinib | Poor |
| 91 | A399T | Imatinib | Undetermined |
| 93 | I293N | Imatinib | Good |
| N331D | Imatinib | Good | |
| 94 | V260M | Nilotinib | Good |
| E282V | Nilotinib | Good | |
| 96 | V280M | Imatinib | Good |
| 99 | H396Y | Imatinib | Good |
| 100 | A399T | Imatinib | Poor |
| 102 | V339M | Imatinib | Undetermined |
| 105 | G254R | Dasatinib | Good |
| 113 | Y312C | Nilotinib | Good |
| 142 | R307N | Imatinib | Good |
| 170 | C330R | Nilotinib | Poor |
| 195 | L248Vc | Dasatinib | Undetermined |
| L248_L247del | Dasatinib | Undetermined | |
| 198 | V225F | Imatinib | Good |
| 199 | P367Q | Dasatinib | Undetermined |
| 204 | A399T | Nilotinib | Good |
| 205 | V260Ad | Nilotinib | Poor |
Abbreviation: TKI, tyrosine kinase inhibitor.
Predicted responses:
Molecular responses were assessed according to the current European LeukemiaNet and National Comprehensive Cancer Network defined response criteria—using the same response milestones as for first‐line treatment. 3 , 7
Imatinib—resistant, Nilotinib—sensitive, Dasatinib—unknown.
Imatinib—resistant, Nilotinib—sensitive, Dasatinib—unknown.
Imatinib—resistant, Nilotinib—unknown, Dasatinib—unknown.
Table 4.
Mutations with known effect on TKI, choice of TKI, and response observed.
| Patient number | Name of mutation | Reported effect on TKI | Choice of TKI | Patient response |
|---|---|---|---|---|
| 8 | F359I | S – D, R – N, PR ‐ I | Dasatinib | Undetermined |
| 10 | F359I | S – D, R – N, PR ‐ I | Dasatinib | Poora |
| 16 | G250E | R – I and N, S ‐ D | Nilotinib | Poor |
| 40 | G250E | R – I and N, S ‐ D | Nilotinib | Poor |
| F359V | S – D, R – I and N | Dasatinib | Poora | |
| 43 | H396R | S – D and N, PR ‐ I | Nilotinib | Good |
| 74 | F359I | S – D, R – N, PR ‐ I | Dasatinib | Good |
| 82 | M244V | S – D and N, PR ‐ I | Nilotinib | Undetermined |
| 85 | T315I | R – D, N, I | Nilotinib | Gooda |
| 86 | F359V | S – D, R – I and N | Dasatinib | Good |
| 87 | G250E | R – I and N, S ‐ D | Dasatinib | Poora |
| Y253F | S – D, R – I, PR ‐ N | Dasatinib | Poora | |
| F359V | S – D, R – I and N | Dasatinib | Poora | |
| 88 | E255K | S – D, R – I, PR ‐ N | Dasatinib | Good |
| 90 | M244V | S – D and N, PR ‐ I | Dasatinib | Poora |
| F359I | S – D, R – N, PR ‐ I | Dasatinib | Poora | |
| 92 | T315I | R – D, N, I | Nilotinib | Undetermined |
| 95 | E255K | S – D, R – I, PR ‐ N | Dasatinib | Undetermined |
| 97 | F359I | S – D, R – N, PR ‐ I | Dasatinib | Undetermined |
| 98 | E279K | S – D and N, PR ‐ I | Nilotinib | Undetermined |
| 100 | E255K | S – D, R – I, PR ‐ N | Nilotinib | Poor |
| Dasatinib | Undetermined | |||
| 101 | F359V | S – D, R – I and N | Dasatinib | Poora |
| 104 | G250E | R – I and N, S ‐ D | Nilotinib | Undetermined |
| 105 | G250E | R – I and N, S ‐ D | Nilotinib | Poor |
| 106 | Not documented | S – D and N, PR ‐ I | Nilotinib | Poor |
| T315I | R – D, N, I | Dasatinib | Poor | |
| 107 | H396R | S – D and N, PR ‐ I | Dasatinib | Undetermined |
| 124 | M244V | S – D and N, PR ‐ I | Imatinib | Undetermined |
| 125 | T315I | R – D, N, I | Hydroxyurea | Undetermined |
| 136 | E255V | R – I, PR – D and N | Nilotinib | Poor |
| 137 | G250E | R – I and N, S ‐ D | Nilotinib | Undetermined |
| 140 | G250E | R – I and N, S ‐ D | Nilotinib | Poor |
| T315I | R – D, N, I | Nilotinib | Poor | |
| 143 | G250E | R – I and N, S ‐ D | Dasatinib | Poora |
| Nilotinib | Undetermined | |||
| V299L | S – I and N, PR ‐ D | Nilotinib | Undetermined | |
| 145 | T315I | R – D, N, I | Nilotinib | Poor |
| 154 | F311I | R – I and D, PR ‐ N | Nilotinib | Undetermined |
| 159 | Not documented | S – D and N, PR ‐ I | Dasatinib | Good |
| 195 | Y253H | S – D, R – I, PR ‐ N | Dasatinib | Undetermined |
| 199 | G250E | R – I and N, S ‐ D | Nilotinib | Poor |
| 200 | E225K | R – I and N, PR ‐ D | Dasatinib | Poor |
| 201 | F317L | S – N, PR – I and D | Nilotinib | Poora |
| 203 | M244V | S – N and D, PR ‐ I | Imatinib | Undetermined |
| F317L | S – N, PR – I and D | Imatinib | Undetermined | |
| 205 | G250E | R – I and N, S ‐ D | Dasatinib | Poora |
| E225V | S – D, R – I and N | Dasatinib | Poora | |
| 206 | G250E | R – I and N, S ‐ D | Imatinib | Undetermined |
Abbreviation: D, Dasatinib; I, Imatinib; N, Nilotinib; PR, partial resistance; R, resistant; S, sensitive; TKI, tyrosine kinase inhibitor.
Discordant response to TKI.
Of the detected 26 KD mutations which are known to have an unknown response to TKI therapy, our study showed patients with Q300*, A412S, I293N, N331D, V280M, H396Y, A399T, R307N, and V225F mutations had responded to Imatinib; patients with K219R, A399T, V260M, E282V, and Y312C mutations responded to Nilotinib; and Dasatinib was effective in patients with E275R and G254R mutations (Table 3).
3.3. Survival analysis
Survival analysis showed an OS of 97.1% (n = 149, CI 93.2–98.8%) and 95.6% (n = 80, CI 90.9–97.9%) at 2 and 5 years respectively. Patients without KD mutations had a 5‐year OS of 95.8% (n = 47, CI 90.1–98.2%). There was no significant difference in OS in patients with KD mutations (5‐year OS 95.7%, n = 33, CI 84.0%–98.9%, p = 0.7) or the PLWH (5‐year OS 94.1%, n = 12, 95% CI 65.0%–99.2%, p = 0.6) (Figure 2).
Figure 2.
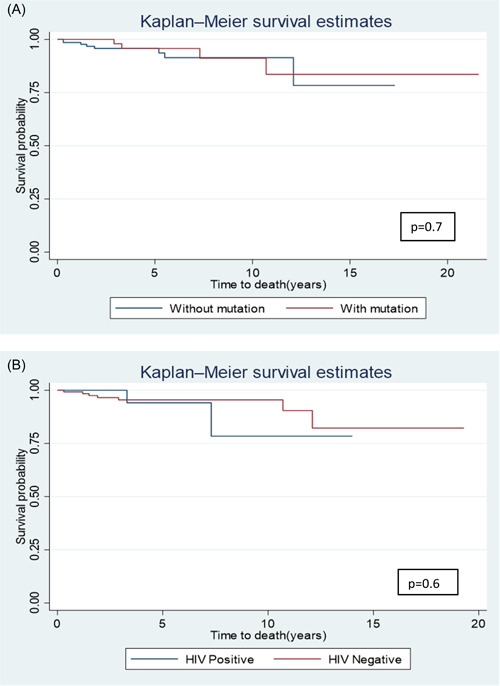
Survival analysis curves (A) Survival probability curves for patients with mutations and those without mutations. (B) Survival probability curves for patients living with human immunodeficiency virus (HIV) and those without HIV.
A total of 5.8% (n = 12 of 206) of patients in this cohort had documentation of death related to the disease or a complication thereof. A total of 58% of these deaths (n = 7) occurred within 5 years of diagnosis. A total of 33% of these patients (n = 4) had detectable KD mutations, two of which had more than one KD mutation.
4. DISCUSSION
The World Health Organization and other publications report CML as having a male‐to‐female ratio of 1.2–1.7, with a median age at diagnosis of 50–60 years of age. 4 , 7 , 19 , 20 Our patient group had a male‐to‐female ratio of 0.9, and a much younger median age of 41.5 years of age at diagnosis. The youngest patient diagnosed with CML was 12 years old. 64.1% patients (n = 132 of 206) were below the age of 50 years at the time of diagnosis. These results are consistent with the limited data available in South Africa, which has shown a younger age of diagnosis of patients with CML in this population. 17 , 18 , 21
Our results also showed that almost 50% of patients required a change in TKI after treatment with Imatinib, which is slightly higher than current international data (20%–40%). 10 , 11 Almost half of the patients who required a change in TKI, had a detectable KD mutation by Sanger Sequencing, which is compatible with global findings of 30%–60%. 2 , 11 , 12 , 13
A subset of patients was confirmed to be HIV positive (21%, n = 43 of 206). To our knowledge, this is the first South African study reporting on KD mutations in PLWH. Although the cohort is small there was no substantial correlation between the presence of a KD mutation and being HIV positive (p = 0.6). Additionally, there were no notable differences in the types of KD mutations found in PLWH and patients without HIV.
Globally, the six most frequently reported KD mutations are documented to be T315I, G250E, E255K, M244V, M351T, and Y253F. 14 , 15 The most frequently found KD mutations in our cohort were G250E (18.3%, n = 11 of 60), T315I (10%, n = 6 of 60), F359I (8.3%, n = 5 of 60), and A399T, M244V, E255K, and F359V each detected in 6.7% (n = 4 of 60) of patients. M351T was not detected in our study population, and this differs from data published from other African countries. 22 , 23
The G250E KD mutation constitutes a substitution of glycine for glutamic acid at amino acid 250, positioned at the p‐loop. The G250E mutation confers variable levels of resistance to most TKI's, but is sensitive to Dasatinib. 3 , 24 This mutation was the most frequently detected mutation (n = 11 of 60) in our cohort. More than half the patients with G250E mutation had more than one mutation detected (n = 7 of 11, 63.6%). All 11 of our patients showed poor clinical or molecular response to the TKI drugs available at the facility, including Dasatinib.
More than half of the mutations with an unknown response responded to a specific TKI.
A399T KD mutation was detected in four patients, and the response to TKI therapy is unknown. Two of the four patients with this mutation were changed to Nilotinib and showed a good molecular and clinical response. Two patients were continued on Imatinib, one having a poor molecular response and the response of the other was not yet determined at the time of the study.
Two patients with KD mutations of unknown effect on TKI therapy (I293N and V280M), were continued on Imatinib and went on to achieve a sustained deep molecular remission, of 51 and 30 months respectively.
Of note, eight patients with KD mutations of known TKI effect also showed conflicting response to therapy, which can likely be attributed to the presence of more than one KD mutation, or other confounding factors such as compliance to medication dosages and scheduled appointments (Table 3).
The limitations of our study should be acknowledged. The accurate assessment of patient outcome was prevented by the high incidence of repeated defaulting of appointments and medication (n = 45), as well as some patients being lost to follow‐up (n = 36 of 45). Thirty‐six patients were lost to follow up, of which fourteen had detected mutations. It was not possible to establish the outcome of those patients.
The reasons for patients defaulting appointments and treatment were largely socioeconomic, including lack of finances and transport, as well as poor understanding regarding the importance for compliance of treatment, and following dosages and times as prescribed.
Furthermore, it should be noted that Imatinib was first available to select patients in this institution from 2001 on a trial basis. Before this, patients were treated with Hydroxyurea and Interferon, until Imatinib became accessible. Few patients who were diagnosed before the study period were only commenced on Imatinib in the course of 2011 and 2012.
5. CONCLUSION
This study has identified that just under a third of CML patients in our population had KD mutations; which is consistent with global data. The age of the patients, as well as their HIV status showed no statistically significant correlation with the incidence of KD mutations.
Our study showed that patients with mutations with unknown responses to TKI therapy, in some cases did actually respond to specific TKI therapy.
Further studies are warranted to gain more insight into our CML patient population. This includes the investigation of factors that may contribute to the younger age of diagnosis, the prevalence of each BCR‐ABL1 fusion transcript in this setting and its impact on disease evolution. A prospective study of the KD mutations found in our population, together with monitoring of these patients will be beneficial to the assessment of response to specific TKI therapy in patients with KD mutations, the impact on disease evolution, and OS as compared to published global data.
AUHTOR CONTRIBUTION
Caryn Benjamin: Project administration; conceptualization (lead); methodology (lead); investigation (lead); visualization (equal); writing—original draft preparation (lead); writing—review and editing (equal). Stephanie Murugan: Conceptualization (supporting); methodology (supporting); investigation (supporting); visualization (equal); writing—review and editing (equal). Siddeeq Hoosen: Conceptualization (supporting); writing—review and editing (equal). Nadine Rapiti: Conceptualization (supporting); writing—review and editing (equal).
CONFLICT OF INTEREST STATEMENT
The authors declare there are no conflicts of interest.
ETHICS STATEMENT
This study was approved by the Biomedical Research Committee (BREC00003764/2022) and by each participating center according to the Declaration of Helsinki. There was no study‐specific consent due to the retrospective nature of the study.
TRANSPARENCY STATEMENT
The lead author Benjamin affirms that this manuscript is an honest, accurate, and transparent account of the study being reported; that no important aspects of the study have been omitted; and that any discrepancies from the study as planned (and, if relevant, registered) have been explained.
ACKNOWLEDGMENTS
The authors would like to thank Saffiya Ibrahim for providing information about the analyzers used at the testing facilities, and Catherine Connolly, biostatistician at the South African Research Council, for assistance with the statistical analysis.
Benjamin C, Murugan S, Hoosen S, Rapiti N. Chronic myeloid leukemia kinase domain mutations: a retrospective descriptive study on the therapeutic and prognostic significance in patients at King Edward VIII Hospital, KwaZulu‐Natal, South Africa. Health Sci Rep. 2023;6:e1376. 10.1002/hsr2.1376
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2016 update on diagnosis, therapy, and monitoring. Am J Hematol. 2016;91(2):252‐265. [DOI] [PubMed] [Google Scholar]
- 2. Erbilgin Y, Eskazan AE, Hatirnaz Ng O, et al. Deep sequencing of BCR‐ABL1 kinase domain mutations in chronic myeloid leukemia patients with resistance to tyrosine kinase inhibitors. Leuk Lymphoma. 2019;60(1):200‐207. [DOI] [PubMed] [Google Scholar]
- 3. Deininger MW, Shah NP, Altman JK, et al. Chronic myeloid leukemia, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2020;18(10):1385‐1415. [DOI] [PubMed] [Google Scholar]
- 4. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. International Agency for Research on Cancer; 2017. [Google Scholar]
- 5. Schmidt M, Rinke J, Schäfer V, et al. Molecular‐defined clonal evolution in patients with chronic myeloid leukemia independent of the BCR‐ABL status. Leukemia. 2014;28(12):2292‐2299. [DOI] [PubMed] [Google Scholar]
- 6. Lim C, Miller GD, Bruno BJ. Resistant mutations in CML and Ph+ALL—role of ponatinib. Biologics. 2014;8:243‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hochhaus A, Baccarani M, Silver RT, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34(4):966‐984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saglio G, Jabbour E. First‐line therapy for chronic phase CML: selecting the optimal BCR‐ABL1‐targeted TKI. Leuk Lymphoma. 2018;59(7):1523‐1538. [DOI] [PubMed] [Google Scholar]
- 9. Patel AB, O'Hare T, Deininger MW. Mechanisms of resistance to ABL kinase inhibition in chronic myeloid leukemia and the development of next generation ABL kinase inhibitors. Hematol Oncol Clin North Am. 2017;31(4):589‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Khorashad JS, Kelley TW, Szankasi P, et al. BCR‐ABL1 compound mutations in tyrosine kinase inhibitor–resistant CML: frequency and clonal relationships. Blood. 2013;121(3):489‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Patkar N, Ghodke K, Joshi S, et al. Characteristics of BCR‐ABL kinase domain mutations in chronic myeloid leukemia from India: not just missense mutations but insertions and deletions are also associated with TKI resistance. Leuk Lymphoma. 2016;57(11):2653‐2660. [DOI] [PubMed] [Google Scholar]
- 12. Soverini S, Hochhaus A, Nicolini FE, et al. BCR‐ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118(5):1208‐1215. [DOI] [PubMed] [Google Scholar]
- 13. Kim H, Kim S, Kim H‐J, et al. Comparison of frequency and sensitivity of BCR‐ABL1 kinase domain mutations in asian and White patients with imatinib‐resistant chronic–phase chronic myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2018;18(10):e391‐e399. [DOI] [PubMed] [Google Scholar]
- 14. Bommannan K, Naseem S, Binota J, Varma N, Malhotra P, Varma S. Tyrosine kinase domain mutations in chronic myelogenous leukemia patients: a single center experience. J Postgrad Med. 2022;68(2):93‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Soverini S, Abruzzese E, Bocchia M, et al. Next‐generation sequencing for BCR‐ABL1 kinase domain mutation testing in patients with chronic myeloid leukemia: a position paper. J Hematol Oncol. 2019;12(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Romzova M, Smitalova D, Tom N, et al. Novel illumina‐based next generation sequencing approach with one‐round amplification provides early and reliable detection of BCR‐ABL1 kinase domain mutations in chronic myeloid leukemia. Br J Haematol. 2020;189(3):469‐474. [DOI] [PubMed] [Google Scholar]
- 17. Louw VJ. Chronic myeloid leukaemia in South Africa. Hematology. 2012;17(sup1):s75‐s78. [DOI] [PubMed] [Google Scholar]
- 18. Patel M. Human immunodeficiency virus infection and chronic myeloid leukemia: is there an association. J Leuk. 2014;02:e108. [DOI] [PubMed] [Google Scholar]
- 19. Lin Q, Mao L, Shao L, et al. Global, regional, and national burden of chronic myeloid leukemia, 1990–2017: a systematic analysis for the global burden of disease study 2017. Front Oncol. 2020;10:580759. 10.3389/fonc.2020.580759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2022 update on diagnosis, therapy, and monitoring. Am J Hematol. 2022;97(9):1236‐1256. [DOI] [PubMed] [Google Scholar]
- 21. Hodkinson KE, Bouwer N, Vaughan J. South African study of blast phase chronic myeloid leukaemia: a poor prognostic outlook. Afr J Lab Med. 2022;11(1):1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tadesse F, Asres G, Abubeker A, Gebremedhin A, Radich J. Spectrum of BCR‐ABL mutations and treatment outcomes in Ethiopian imatinib‐resistant patients with chronic myeloid leukemia. JCO Global Oncology. 2021;7:1187‐1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Khair H, Elderdery A, Nour B, et al. BCR‐ABL kinase domain mutations ‐E255K, Y253 H and M351T among sudanese population with CML. Pharmacophore. 2022;12(4):112‐118. 10.51847/MGa8PfuPAd [DOI] [Google Scholar]
- 24. Zabriskie MS, Eide CA, Tantravahi SK, et al. BCR‐ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome‐positive leukemia. Cancer Cell. 2014;26(3):428‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.