

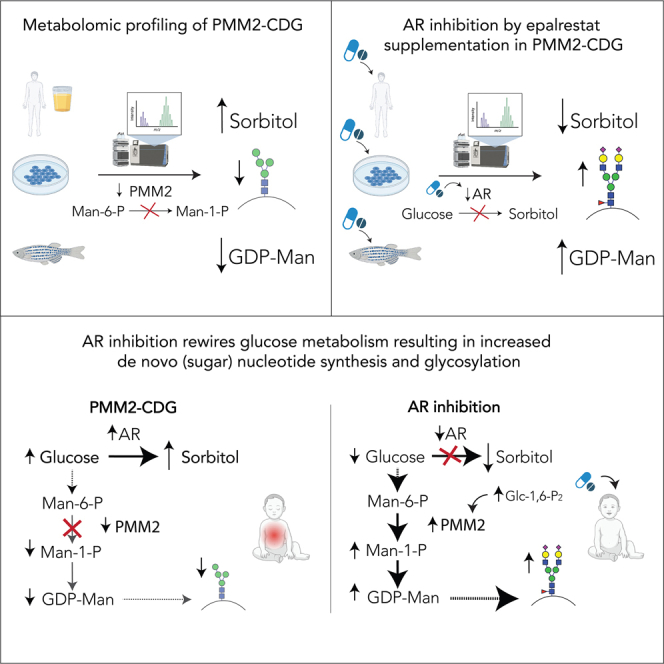
Summary
Abnormal polyol metabolism is predominantly associated with diabetes, where excess glucose is converted to sorbitol by aldose reductase (AR). Recently, abnormal polyol metabolism has been implicated in phosphomannomutase 2 congenital disorder of glycosylation (PMM2-CDG) and an AR inhibitor, epalrestat, proposed as a potential therapy. Considering that the PMM2 enzyme is not directly involved in polyol metabolism, the increased polyol production and epalrestat’s therapeutic mechanism in PMM2-CDG remained elusive. PMM2-CDG, caused by PMM2 deficiency, presents with depleted GDP-mannose and abnormal glycosylation. Here, we show that, apart from glycosylation abnormalities, PMM2 deficiency affects intracellular glucose flux, resulting in polyol increase. Targeting AR with epalrestat decreases polyols and increases GDP-mannose both in patient-derived fibroblasts and in pmm2 mutant zebrafish. Using tracer studies, we demonstrate that AR inhibition diverts glucose flux away from polyol production toward the synthesis of sugar nucleotides, and ultimately glycosylation. Finally, PMM2-CDG individuals treated with epalrestat show a clinical and biochemical improvement.
Keywords: phoshomannomutase-2 deficiency, polyol metabolism, aldose reductase, aldose reductase inhibition, glycosylation, congenital disorder of glycosylation
Graphical abstract
Highlights
-
•
Metabolomic profiling of different PMM2-CDG models shows abnormal polyol metabolism
-
•
AR inhibitor epalrestat decreases polyols and increases GDP-mannose
-
•
Tracer studies show AR inhibition promotes sugar nucleotide synthesis and glycosylation
-
•
Individuals with PMM2-CDG treated with epalrestat note clinical improvement
Radenkovic et al. discover that aldose reductase (AR) and its product sorbitol are upregulated in PMM2-CDG, while GDP-mannose, an essential glycosylation precursor, is depleted. Inhibition of AR decreases sorbitol, increases GDP-mannose, and ultimately improves glycosylation. Individuals with PMM2-CDG undergoing AR inhibition by oral epalrestat therapy show clinical improvement.
Introduction
Glycosylation is one of the most biologically significant post-translational modifications. Glycan synthesis begins in the cytosol, where sugar nucleotides, essential glycan building blocks, are produced. Then, they are used as sugar donors in the endoplasmic reticulum and in the Golgi apparatus to make chains of sugars called glycans, which are attached to nascent proteins and ultimately transported to the cell surface. Glycosylation is essential for protein stability, cell integrity, cell signaling, etc.1
Congenital disorders of glycosylation (CDGs) are a rapidly growing group of inherited metabolic disorders caused by pathogenic variants in genes coding for the enzymes and transporters affecting glycosylation. There are more than 160 CDGs described to date.2,3,4 The most common CDG is phosphomannomutase-2 (PMM2)-CDG, with more than 1,000 reported patients and an estimated prevalence of 1:18,745 (non-Finnish Europeans) to 1:366,999 (South Asian population).5 PMM2-CDG individuals present with a multisystem phenotype with the main clinical findings being neurologic involvement (ataxia, speech delay, and seizures), global developmental delay, intellectual disability, coagulation abnormalities, and liver abnormalities.6 PMM2-CDG is caused by a deficiency in the phosphomannomutase-2 (PMM2) enzyme,7 which leads to the depletion of mannose-1-P (Man-1-P) and its downstream metabolite GDP-mannose,8 an essential sugar nucleotide involved in glycosylation. Finally, depletion of GDP-mannose in PMM2-CDG results in immature and incomplete glycan chains.9
There is no cure for PMM2-CDG,6 and there is a critical unmet medical need in identifying and developing possible therapeutic options for this ultra-rare disorder.
One of the promising therapeutic candidates is epalrestat,10 a non-competitive aldose reductase (AR) inhibitor.11 AR is involved in polyol metabolism and converts sugars (e.g., glucose) to their respective sugar alcohols (e.g., sorbitol). AR inhibitors, such as epalrestat, are used to treat diabetes,11,12,13 where excess glucose is converted to sorbitol and can lead to secondary diabetic complications.14,15,16,17,18 A recent high-throughput drug screen, showed epalrestat increased PMM enzymatic activity in pmm-2 hypomorphic mutant CRISPR-Cas9 worms and PMM2-CDG patients’ fibroblasts.10
Apart from increasing PMM enzymatic activity,10 epalrestat improved glycosylation in vitro in patient fibroblasts.19 Considering polyol metabolism is not closely associated with PMM enzyme or glycosylation, the ability of epalrestat to increase PMM enzymatic activity and improve glycosylation was puzzling. To probe whether polyol metabolism could be affected in PMM2-CDG, we assessed the urine polyol levels in 24 PMM2-CDG individuals and found elevated urine sorbitol in the majority of them.19 Additionally, urine sorbitol levels correlated with disease severity and neurological involvement in our PMM2-CDG cohort.19 This was crucial, as high sorbitol levels are associated with peripheral neural damage20,21,22 and could contribute to peripheral neuropathy often observed in PMM2-CDG.6 Finally, we performed a safety and efficacy study in a pediatric PMM2-CDG individual using oral epalrestat. We found that epalrestat was safe, decreased urine sorbitol levels, and resulted in normalization of carbohydrate deficient transferrin (CDT; a clinical glycosylation marker).19
Despite promising clinical results, the mechanism of increased urine polyols and the therapeutic effect of epalrestat in PMM2-CDG remained elusive. Unlike individuals with diabetes, PMM2-CDG individuals do not present with blood hyperglycemia,6 which could explain the elevated polyol production, nor has AR ever been implicated in the regulation of glycosylation. Therefore, there is a significant knowledge gap in the link between polyol metabolism and glycosylation.
To address this knowledge gap, we hypothesized that PMM2 deficiency causes a change in glucose flux away from Man-1-P, GDP-mannose, and glycosylation toward polyol production, and that targeting AR with epalrestat would result in metabolic rewiring and improved protein glycosylation.
To test these hypotheses, we performed extensive metabolic investigations in PMM2-CDG patient-derived fibroblasts, pmm2 mutant zebrafish, and patient samples (blood and urine). To assess the clinical significance of AR inhibition in PMM2-CDG, three pediatric PMM2-CDG patients underwent long-term (1–2.5 years) treatment with epalrestat.
Results
Increase of urine polyols is common in PMM2-CDG
In our previous study, we identified elevated sorbitol levels in 24 patients with PMM2-deficient congenital disorder of glycosylation.19 In this study, we assessed urine polyol levels (sorbitol, mannitol, galactitol) in an additional 26 PMM2-CDG individuals, in one of the biggest PMM2-CDG cohorts to date (a total of 50 patients who are currently enrolled in our CDG natural history study NCT03173300) (Table S1). While no increased levels of urine galactitol was observed, 86% (43 out of 50) of PMM2-CDG individuals had elevated urine sorbitol (mean, 16.3 mmol/mol creatinine; control reference range, <5 mmol/mol creatinine), while 46% (23 out of 50) had elevated urine mannitol (mean, 50.18 mmol/mol creatinine; control reference range, <20 mmol/mol creatinine) (Figure 1A). These results confirm that abnormal polyol metabolism is common in PMM2-CDG.
Figure 1.

Disturbed polyol metabolism is a hallmark of PMM2-CDG
(A) Urine sorbitol and mannitol are increased in PMM2-CDG G. Urine sorbitol and mannitol were measured in PMM2-CDG patients (n = 50). Higher limit of the control reference range (n = 533; sorbitol <5 mmol/mol creatine; mannitol 20 mmol/mol creatine) is given on the left.
(B) Heatmap of the 25 top discriminating intracellular metabolites. Metabolomics analysis showed PMM2-CDG fibroblasts clustered separately from healthy control due to significant differences in relative abundances of multiple metabolites in PMM2 deficient fibroblasts compared with healthy controls. Clustering result shown as heatmap (distance measured using Euclidean), and clustering algorithm using Ward.
(C) Mannose-1-P and GDP-mannose are depleted in PMM2-CDG fibroblasts, while intracellular polyols (sorbitol and mannitol) are increased. GDP-mannose and polyol pool (total pool of sorbitol, mannitol, galactitol) measured by LC/MS. Mannose-1-P, sorbitol, and mannitol were measured separately by gas chromatography-mass spectrometry (GC/MS). CTR, healthy control.
(D) Zebrafish model of PMM2-CDG shows similar metabolic changes to PMM2-CDG fibroblasts. GDP-mannose is significantly lower in pmm2 mutant zebrafish compared with the wild-type (WT) controls, while polyols are increased. PMM enzymatic activity measured in pmm2 mutant zebrafish (n = 3). Average pmm activity of WT is given on the left. Each technical replicate contained 10 zebrafish.
(E) PMM2-CDG affects multiple biochemical pathways. Representation of main findings identified by metabolomic profiling of PMM2-CDG fibroblasts and zebrafish. All metabolite abundances are represented as relative compared with the control samples. The number of biological (n) and technical (t) replicates is PMM2-CDG (n = 6, t = 1–8) and CTR fibroblasts (n = 5, t = 1–6) (B and C), pmm2 mutant (n = 5, t = 1), WT (n = 4, t = 1) (D). Means are represented with SD. Student’s t test was performed. Polyol refers to six-carbon-sugar alcohol (galactitol, mannitol, and sorbitol) pool measured by LC/MS. Hexose refers to six-carbon-sugar (glucose, galactose, fructose, and mannose) pool measured by LC/MS. UDP-hexose (UDP-hex) refers to the pool of UDP-galactose and UDP-glucose. UDP-hexNAc refers to the pool of UDP-glcNac, UDP-galNac, and UDP-manNac. C1-5, control 1–5; P, phosphate; P1–6, patient 1–6; ref range, reference range.
Global metabolic changes in PMM2-CDG result in increased intracellular polyols
Since PMM2-CDG does not commonly present with hyperglycemia in blood,6 we hypothesized that PMM2 deficiency resulted in altered glucose metabolism leading to an intracellular increase in polyols. To assess global metabolic changes elicited by PMM2 deficiency, we metabolically profiled PMM2-CDG patient fibroblasts (P1–P6) with confirmed decreases in PMM enzymatic activity.19 Using high-throughput liquid chromatography-mass spectrometry (LC/MS) metabolomics, we simultaneously monitored over 45 metabolites covering major metabolic pathways (glycolysis, polyol metabolism, pentose phosphate pathway [PPP], nucleotide metabolism, hexosamine biosynthesis, tricarboxylic acid [TCA] cycle, etc.) in both PMM2-CDG and healthy fibroblasts, cultured in the presence of 5.5 mM (physiological) glucose.
Metabolomics analysis showed that PMM2-CDG fibroblasts clustered separately from healthy controls (Figure 1B). Man-1-P and GDP-mannose were depleted in PMM2-CDG (Figures 1B and 1C), corroborating the PMM2 deficiency. However, hexoses and polyol pool (sorbitol, mannitol, galactitol), specifically sorbitol and mannitol, but not galactitol or galactose, were increased (Figures 1B, 1C, and S1A). As there was no significant difference in the abundance of uridine diphosphate (UDP)-hexoses (UDP-glucose and UDP-galactose), which are involved in galactose metabolism (Figure S2A), galactose metabolism is probably not affected in PMM2-CDG. Therefore, these results strongly indicate intracellular accumulation of glucose in PMM2 deficiency, resulting in increased sorbitol and mannitol production. Furthermore, we noted changes in other pathways (glycolysis, TCA cycle) (Figure 1B) and found that the nucleotide-phosphate pools (NXPs; NMP, NDP, NTP) were also depleted in PMM2-CDG fibroblasts (Figure 1B). These findings suggest that PMM2 deficiency results in broad metabolic rearrangements.
Fibroblasts are valuable but limited in vitro model systems to the study of PMM2-CDG, a disease that predominantly affects metabolically active organs such as liver, brain, and heart. Therefore, we validated our in vitro findings in the genetically stable pmm2m/m (from here on referred to as pmm2 mutant) zebrafish. The pmm2 mutant zebrafish harbors a mutation in an essential splice site (pmm2sa1050), which results in a hypomorphic pmm2 allele.23 By 6 days post fertilization (dpf), PMM activity is significantly decreased in pmm2 mutant zebrafish embryos (Figure 1D),23 with levels comparable with the enzymatic activity measured in PMM2-CDG patient fibroblasts.19 Like the in vitro PMM2-CDG fibroblast model, GDP-mannose levels were reduced in pmm2 mutant embryos compared with wild type (Figure 1D). Polyols were also increased in pmm2 mutant embryos, corroborating our findings from PMM2-CDG patient fibroblasts (Figure 1D). Given that zebrafish lysate contains both circulating (blood) and intracellular (tissue) metabolites, the observed metabolites pools from the zebrafish experiments reflect both the intracellular and extracellular compartments.
In summary, our findings suggested that PMM2-CDG affects multiple biochemical pathways, resulting in a decrease of GDP-mannose and an increase of intracellular polyols (Figure 1E).
Intracellular polyols are elevated in PMM2-CDG regardless of the environment
Since polyols were increased both intracellularly in PMM2-CDG fibroblasts and in the urine of PMM2-CDG patients (Figure 1), we asked whether the increased polyols are the result of environmental factors (e.g., diet, medication, medium used for culture of fibroblasts) or rather the direct consequence of the intracellular biochemical changes caused by PMM2 deficiency. To eliminate the possibility that increased intracellular polyols are caused by the presence and consequent uptake of polyols in the cell culture medium, we compared the abundance of polyols in the standard medium containing non-dialyzed fetal bovine serum (FBS) with the medium containing dialyzed FBS. We found that non-dialyzed FBS contained 50–80 times more sorbitol and mannitol compared with the dialyzed FBS (Figure S1B). Therefore, the availability of extracellular polyols in the non-dialyzed FBS could have an effect on the intracellular polyols observed in the PMM2-CDG fibroblasts (Figure 1). To eliminate the uptake of polyols from the medium, we used dialyzed FBS in all subsequent experiments. Polyols remained significantly increased in all six patient cell lines studied (Figures 2A and 2B), indicating increased uptake of polyols from the medium did not cause increased intracellular polyol levels in PMM2-CDG.
Figure 2.

AR inhibition with epalrestat decreases polyol levels and elicits global metabolic changes in both PMM2-CDG and healthy controls
(A) Epalrestat, unlike glucose-1,6-P2, does not stabilize PMM2 protein. Heat-induced melting profiles of WT human PMM2 (top panels) and the mutant (F119L HOM variant, bottom panels) recorded by thermal shift. T0.5 (50% unfolded protein) is indicated by dashed line. As previously reported, mutant protein shows lower T0.5, indicating it is less stable than WT. WT PMM2 and the F119L variant with varying concentrations of the known activator glc-1,6-P2, showing increased T0.5 with increasing ligand concentration (left panels) indicating protein stabilization. WT PMM2 and the F119L variant with epalrestat, showing less than 1.0°C change in T0.5 (right panels), indicating no effect of epalrestat on PMM2 stability. T0.5 values are provided in Table S1.
(B) Epalrestat decreases intracellular polyol levels. Polyol pathway with major enzymes and cofactors is shown (top). PMM2-CDG and healthy fibroblasts were treated with 10 μM epalrestat. LC/MS analysis showed a significant effect of epalrestat on overall polyol pool (sorbitol, mannitol, galactitol). GC/MS analysis identified sorbitol as the main polyol being decreased by epalrestat, while mannitol was fluctuating (bottom).
(C) Metabolite abundances linked to PMM2 enzyme are increased upon epalrestat supplementation. GDP-mannose synthesis pathway with major enzymes (PMM2) and metabolites is shown (top). Epalrestat had a significant effect on the abundances of Man-1-P, Man-6-P, GDP-mannose, and Glc-1,6-P2 in patient fibroblasts (bottom).
(D) Metabolic effect of epalrestat is corroborated in vivo.Pmm2 mutant and wild-type zebrafish were treated with 10 or 40 μM epalrestat for 24 h. Asterisk (∗) indicates that each zebrafish sample contains 10 zebrafish. All metabolite abundances are represented as relative to the untreated control samples. The number of biological (n) and technical (t) replicates is PMM2-CDG n = 6, t = 2–4, CTR n = 5, t = 1–4 (B and C), and Pmm2 mutant (n = 12–16∗, t = 1) and wild-type (n = 14–19∗,t = 1) (D). Means are represented with SD. Two-way ANOVA or mixed-effects model with repeated measures was used for statistical analysis where applicable. For additional metabolites, see Figure S2. CTR, control; MDH, mannitol dehydrogenase; NAD(H), nicotinamide adenine dinucleotide (hydrogen); NADP(H), nicotinamide adenine dinucleotide phosphate (hydrogen); PMM2, phosphomannomutase-2; P, patient; SORD, sorbitol dehydrogenase; g, p value reflecting the effect of genotype; t, p value reflecting the effect of the treatment; i, p value reflecting the interaction between treatment and genotype as calculated by statistical analysis.
To rule out potential dietary causes behind increased urine polyol levels in PMM2-CDG, we evaluated the influence of dietary fructose on the urine polyol levels. Fructose has become one of the major sources of energy in the Western diet and has been associated with numerous disorders, including diabetes, hypertension, and obesity.24 Peripheral fasting fructose levels range between 0.01 and 0.07 mM and can increase 5-fold after consumption of a fructose-rich meal.25 Fructose is present in the normal diet of PMM2-CDG patients who do not follow any special dietary regimen, and increased fructose intake could result in increased urine polyol levels. We therefore tested the possibility of fructose contributing to the polyol increase seen in PMM2-CDG.
First, we performed a fructose tolerance test in a PMM2-CDG individual (P5) and healthy controls. While we saw no significant increase in the urine sorbitol or mannitol after fructose loading in healthy controls, the urine sorbitol in PMM2-CDG increased approximately seven times after the fructose tolerance test, while mannitol increased three times (Figure S1C), indicating that dietary fructose could increase urine polyols in PMM2-CDG.
Then, to investigate the contribution of exogenous fructose to the intracellular polyol accumulation, we incubated patient and control fibroblasts in the presence of 13C6-fructose (Figure S1D; see STAR Methods). Addition of 0.15 mM exogenous fructose (average blood fructose concentration upon fructose-rich meal) resulted in an increase in the polyol and hexose pool abundances (Figure S1D). Moreover, fractional contribution (FC) from 13C6-fructose in the hexose pool was found in both PMM2-CDG and controls, while no significant labeling from 13C6-fructose was found in the polyol pool (Figure S1D). Specifically, intracellular mannitol and fructose were increased in PMM2-CDG in the presence of exogenous fructose (Figure S1E), while fructose did not have a significant effect on intracellular glucose and sorbitol, which were elevated in the patient cells regardless of the fructose availability (Figure S1E). It is well known that fructose stimulates glucose metabolism.26,27,28 No significant labeling from 13C6-fructose was found in the polyol pool. However, the polyol abundance (mannitol) was increased in the presence of fructose, suggesting availability of fructose indirectly increases polyol production by, e.g., regulating glucose metabolism or increasing the overall concentrations of available sugars in the cell. Further, these results indicated increased intracellular sorbitol and glucose levels are not caused by environmental factors such as fructose but are intrinsic to the intracellular biochemical changes caused by PMM2 deficiency.
Finally, as mannitol is produced from fructose by mannitol dehydrogenase (MDH), an enzyme not targeted by epalrestat, the availability of exogenous fructose in PMM2-CDG could hence contribute to the additional accumulation of intracellular fructose and polyols (specifically mannitol) and might have implications for the dietary regimen of PMM2-CDG individuals. Therefore, the role of fructose in PMM2-CDG should further be explored.
AR inhibition with epalrestat decreases polyol levels and elicits global metabolic changes
Because we found that intracellular sorbitol and glucose were increased in PMM2-CDG regardless of the environment, we wondered whether the AR itself is upregulated in PMM2-CDG. We found that AR (AKR1B1) gene expression was increased in PMM2-CDG even in the presence of 5.5 mM (physiological) glucose (Figure S2A), suggesting that AR inhibitors (e.g., epalrestat) might be therapeutic in PMM2-CDG by directly inhibiting AR. To investigate this further, we performed AR enzymatic activity assays in PMM2-CDG and healthy control fibroblasts grown in 5.5 mM glucose + dialyzed serum and treated with 10 μM epalrestat. Like AKR1B1 expression, AR enzymatic activity was increased in PMM2-CDG fibroblasts, and epalrestat was able to decrease it (Figure S2B).
We have already shown that the epalrestat increased PMM enzymatic activity (Table S1)19 and improved glycosylation in vitro and in vivo in PMM2-CDG.19 As epalrestat could increase PMM enzymatic activity by directly binding to PMM2 protein, we performed thermal shift binding assay in WT and F119L HOM PMM2 protein variants. As a positive control, we used glucose-1,6-P2, which, alongside Man-1,6-P2, is a known activator of the PMM enzyme.29,30 Our results confirmed that mutant PMM2 protein is less stable than the WT31 and that glucose-1,6-P2, is able to stabilize it29 (Figure 2A top panels; Table S1). Moreover, the addition of 10 μM epalrestat did not result in a significant stabilization of either WT or mutant PMM2 protein (T0.5 < 1°C) (Figure 2A bottom panels; Table S1).
Considering the increase in PMM activity could not be explained by the direct binding of epalrestat to PMM2 protein, we hypothesized AR inhibition by epalrestat rewires the metabolism by redirecting glucose away from the excess polyol production toward GDP-mannose synthesis, ultimately improving glycosylation.19
To test our hypothesis, we assessed the global metabolic effect of epalrestat by analyzing the metabolome of PMM2-CDG and control samples treated with 10 μM epalrestat. The concentration of epalrestat was previously established based on the pharmacological profile of epalrestat in the patient treated with 0.8 mg/kg/day three times a day (TID).19
Metabolomics showed that epalrestat reduced the overall polyol pool in both patient and control cell lines (Figure 2B). Specifically, sorbitol was significantly reduced following epalrestat treatment (Figure 2B). Mannitol was also reduced in the majority of patient fibroblasts, although its abundance fluctuated on epalrestat (Figure 2B). We also found that the NADPH/NADP ratio was increased in epalrestat-treated cells, consistent with the inhibition of AR, which oxidizes NADPH to NADP (Figures 2B and S2B).
Moreover, epalrestat increased PMM2-related metabolites mannose-6-P (Man-6-P), Man-1-P, and GDP-mannose (Figure 2C). This is crucial, as PMM2-CDG individuals are unable to produce sufficient Man-1-P and consequently GDP-mannose, which is necessary for correct glycosylation. These results also corroborated our previous findings of epalrestat-mediated increased glycosylation in PMM2-CDG fibroblasts.19
Furthermore, the majority of patient fibroblasts showed decreased levels of glucose-1,6-P2 (Glc-1,6-P2) compared with the controls, and epalrestat increased the overall abundance of Glc-1,6-P2 in PMM2-CDG (Figure 2C). As Glc-1,6-P2 can activate PMM229,30 by directly binding to PMM2 protein (Figure 2A), and no interaction between epalrestat and PMM2 protein was observed (Figure 2A), our data indicate that the increase in PMM enzymatic activity measured in patient fibroblasts (Table S1)10,19 is likely mediated by the Glc-1,6-P2 increase.
Finally, epalrestat treatment altered metabolite abundances across other biochemical pathways (e.g., glycogen metabolism, the hexosamine biosynthesis pathway, and the PPP) (Figure S2C), further supporting our hypothesis that AR inhibition results in global metabolic changes and shifts glucose metabolism away from polyol production toward (sugar) nucleotide production, which is beneficial in PMM2-CDG.
Effects of aldose inhibition are corroborated in vivo in pmm2 mutant zebrafish
To corroborate these findings in vivo, 6-dpf wild-type and pmm2 mutant zebrafish were treated with a single dose of either 10 or 40 μM epalrestat for 24 h. We found a significant effect of epalrestat on both GDP-mannose and polyol abundance, like the ones seen in fibroblasts (Figure 2D). Specifically, there was a significant increase in GDP-mannose abundance in pmm2 mutant zebrafish treated with 10 μM epalrestat (Figure 2D).
Then, using an automated behavioral tracking system (Figures 3A and 3B), we asked if epalrestat improved the previously characterized defects in pmm2 zebrafish swim behavior.23 We chose 6-dpf embryos, as the pmm2 mutant zebrafish first exhibit differences in swim behavior at 5–6 dpf. These differences become more pronounced as the animals age, resulting in death between 10 and 13 dpf.23 Therefore, we analyzed epalrestat’s effect at 6–10 dpf. This time window provides the best chance to evaluate epalrestat’s ability to improve swim behavior without the complicating effects of global system death. Choosing this time window is further supported by the fact that the pmm2 mutant zebrafish have other tissue defects (beyond altered swim behavior)23 that may also contribute to their early mortality. Specifically, development of craniofacial cartilage is disrupted in pmm2 mutants, which raises the possibility that feeding problems also contribute to system failure in these animals. Therefore, analyzing the swim behavior at the early time point eliminates the chance that feeding problems make the effects of epalrestat difficult to measure at later stages.
Figure 3.

Effects of aldose inhibition are corroborated in vivo in pmm2 mutant zebrafish
(A) Schematic representation of imaging and treatment of zebrafish. Briefly, 5-dpf wild-type and pmm2 embryos were placed one per well in a 12-well culture dish and treated daily with either 20 μM epalrestat or vehicle, refreshing the media daily. After 15 min of sensory deprivation, locomotor activity is monitored for 10-min intervals each day (6–10 dpf) using the Zebrabox system.
(B) Track of swim paths of wild-type and pmm2 zebrafish with and without epalrestat treatment on 6 and 8 dpf. Slow speed is indicated in green, while fast speed is indicated in red.
(C) Epalrestat increases swimming distance (mm). Epalrestat significantly increased swimming distance at 6 and 7 dpf in pmm2 mutant zebrafish.
(D) Epalrestat increases number of initiated swim events. Epalrestat significantly increased the number of initiated swim events. Mixed-effect analysis with repeated measures was used to statistically evaluate data. Means are represented with SD. e, p value reflecting the effect of epalrestat treatment; lt, p value reflecting the length of treatment; g, p value reflecting the effect of genotype calculated by statistical analysis. The number of biological (n) and technical (t) replicates was wild-type, n = 42–46, t = 1; pmm2, n = 22–51, t = 1.
We first treated 6-dpf WT and pmm2 mutant zebrafish with 10 μM daily epalrestat treatment, which resulted in no improvement in swimming behavior (data not shown). Then, we treated both WT and pmm2 mutant zebrafish with 20 μM daily epalrestat (5–10 dpf), which increased the distance swum and the number of swim events initiated in the pmm2 mutant zebrafish (Figures 3C and 3D). The improvement was most significant at 6 and 7 dpf (Figures 3C D). Moreover, we found no significant difference in the viability between treated and untreated pmm2 mutant zebrafish (data not shown).
Treatment with nucleosides, specifically guanosine, increases GDP-mannose abundance in PMM2-CDG to the levels seen in healthy controls
Although we noticed an overall increase in sugar nucleotides after epalrestat treatment, GDP-mannose levels in patient fibroblasts never reached the ones seen in controls. As nucleotide levels in PMM2-CDG were also depleted (Figure 1), we tested whether the addition of nucleosides, specifically uridine or guanosine (30 μM each), could restore sugar nucleotide pools. While guanosine can be directly used for GDP-mannose biosynthesis, uridine can be used to make UDP, which is a part of several nucleotide sugars essential for glycosylation (UDP-glucose, UDP-galactose, UDP-glcNac, UDP-galNac). By increasing nucleotide sugar pools, glycosylation could also be increased.32 Further, uridine is safe and has frequently been used to treat other genetic metabolic disorders, including different CDGs.33,34
GDP-mannose abundance further increased after the combination of epalrestat and nucleosides or uridine (Figures 3A and 3B). However, the combination of guanosine and epalrestat restored GDP-mannose and GDP-fructose abundance comparable with levels seen in healthy controls (Figure S3C). Therefore, PMM2-CDG patients could additionally benefit from a combination therapy of epalrestat and nucleosides, specifically guanosine.
Tracer studies reveal AR inhibition rewires glucose metabolism, resulting in increased de novo (sugar) nucleotide synthesis and glycosylation
To elucidate the link between AR and glycosylation and confirm that the therapeutic effect of epalrestat is through the inhibition of the AR and not an off-target effect, we performed glucose tracer experiments following the small interfering RNA (siRNA) inhibition of AKR1B1 (AR gene) (Figure 4A). Although pharmacological inhibition of AR with epalrestat, a non-competitive and reversible inhibitor of AR,11 cannot be directly compared with the AKR1B1 knockdown (KD), we hypothesized that the siRNA AKR1B1 KD would result in a similar metabolic effect, confirming the beneficial mechanism of epalrestat in PMM2-CDG.
Figure 4.

Tracer studies with 13C6 glucose upon AKR1B1 siRNA inhibition reveal metabolic rewiring and increased de novo sugar nucleotide synthesis
(A) Methodology. To assess the changes in glucose flux in AKR1B1 KD cells, cells were incubated with either siRNA targeting the AR gene AKR1B1 or non-targeting (negative) siRNA for 48 h. Then, medium was changed and the medium containing either 13C6-glucose (tracer glucose) or 12C6-glucose was added. Tracer was then metabolized throughout subsequent biochemical pathways. Metabolites were extracted for metabolomics analysis. Furthermore, membrane-bound sialic acid, the end sugar of glycan chains, was isolated and subjected to metabolomics analysis.
(B) AKR1B1 KD results in a decrease in polyols and increase in metabolites related to glycosylation. Relative polyol abundances and FC of 13C6 glucose in polyol were decreased following AR KD, while UDP-hexose and CMP-sialic acid abundances were increased.
(C) Deconvolution of positional labeling of 13C6 glucose in CMP-sialic acid. Increase in m11, m14, and m16 after AR inhibition and incubation with 13C6 glucose in CMP-sialic acid indicates multiple pathways (PPP, nucleotide biosynthesis, glucosamine biosynthesis, and glycolysis) are simultaneously upregulated upon AR inhibition.
(D) Increased glucose flux is observed in membrane-derived sialic acid, terminal glycan sugar. Increase in abundance and FC of 13C6-glucose in cytosolic sialic acid is observed in cells treated with siRNA targeting AKR1B1 (see Figure S4B). These changes ultimately lead to increase in abundance and FC of 13C6-glucose in CMP-sialic acid and sialic acid derived from membranes, suggesting that sialylation and overall glycosylation are improved upon AR inhibition. Relative metabolite abundances were calculated based on the average of CTR treated with non-targeting (negative) siRNA. FC of 13C6 glucose was calculated for each metabolite based on the isotopologue distribution and corrected for naturally occurring 13C isotopes (see STAR Methods). Two-way repeated-measures ANOVA or mixed-effect analysis with repeated-measures analysis were performed. For the additional metabolites, see Figure S4B. Specific relative metabolite abundances in PMM2-CDG and CTR and %FC can be found in Figure S4B. The number of biological (n) and technical (t) replicates: PMM2-CDG n = 2, t = 3; healthy control n = 3, t = 1–3 (B and C); PMM2-CDG n = 2, t = 2; healthy control n = 2, t = 2 (D). The artworks are a visual representation of the results, where the size of the pies represents the arbitrary abundance of represented metabolites across both CTR and PMM2-CDG, while the color of the pie represents the average %FC of 13C6-glucose in the specific metabolite. FC, fractional contribution; negat siRNA, negative/non-targeting siRNA; AKR1B1 siRNA, siRNA targeting ARK1B1 gene; CTR, control; P, patient; g, p value reflecting effect of genotype; s, p value reflecting the effect of the siRNA targeting AKR1B1; i, p value reflecting the interaction between AKR1B1 KD and positional labeling.
Following AKR1B1 siRNA KD—confirmed by RT-qPCR and western blot (WB) (Figure S4A)—we observed significant changes in abundance and FC of 13C6-glucose in several metabolites, implying that glucose flux was significantly altered upon AR inhibition in both patient and control fibroblasts (Figures 4A and S4B). The changes in abundances of several metabolites were comparable between the two approaches used to inhibit AR (KD vs. epalrestat).
Crucially, the abundance and the FC of 13C6-glucose in polyols were significantly decreased after AKR1B1 KD (Figure 4B), verifying that glucose conversion to sorbitol was drastically inhibited (Figure 4B). Besides the reduction in polyol synthesis metabolism, AKR1B1 KD resulted in a significant increase in the FC of 13C6-glucose and metabolite abundance of hex-P (which includes mannose-6-P and mannose-1-P), hexosamine-P (Figure S4B), sialic acid (Figure S4B), and (sugar) nucleotides (Figures 4B and S4B), which suggested de novo (sugar) nucleotide synthesis was promoted upon AR inhibition. Since de novo nucleotide synthesis occurs during intensive proliferation,35 we assessed the effect of AR KD on proliferation. We found that AR inhibition increased proliferation in both control and patient fibroblasts (Figure S4C), corroborating our tracer experiment findings.
Unfortunately, we were not able to assess the abundance and labeling in GDP-mannose, as the m/z of m2-m16 of GDP-mannose overlaps with m/z of m0-m14 of UDP-hexNAc (see STAR Methods). However, CMP-sialic acid is synthesized from UDP-hexNAc and offers further insights into the pathway activities related to glycosylation. Like GDP-mannose and UDP-hexNAc, CMP-sialic acid is an important glycosylation building block that provides sialic acid to the terminus of the correctly assembled glycan chains (Figure 4A). As CMP-sialic acid (Figure 4C) is synthetized through the combination of several moieties coming from different biochemical pathways including the five-carbon ribose (from PPP), six-carbon hexose (from hexosamine biosynthesis), two-carbon acetyl group (from acetyl-CoA), and a three-carbon group (from phospho-enolpyruvate [PEP]).1,36 Therefore, the positional labeling—the percentage of labeled carbons ranging from m0 (no carbons labeled) to m20 (all carbons labeled)—can provide information about the activities of the pathways providing those moieties. A similar approach has previously been described using UDP-glcNAc.37,38 Upon AKR1B1 KD, we noted an increase in m11, m14, and m16 of CMP-sialic acid, while m5, m6, and m8 decreased (Figure 4C). These data imply an increased flux of glucose going toward glycolysis (m3-labeled moiety from PEP), acetyl-coenzyme A (CoA) production (m2-labeled moiety), and de novo nucleotide synthesis (m11 is an m5-labeled moiety of ribose + m6-labeled moiety of hexose) (Figure 4C).
The increase of CMP-sialic acid abundance and FC of 13C6-glucose indicated that AR inhibition promoted de novo synthesis of sugar nucleotides, sugar donors of glycans. These data, however, were not sufficient to conclude that AKR1B1 KD results in increased glycosylation, which we previously observed in epalrestat-treated cells.19 Therefore, we checked whether the observed changes in glucose flux upon AR inhibition would also be reflected in improved glycosylation by isolating the cell membranes containing mature glycans and enzymatically removing the terminal sialic acid upon incubation with sialidase (neuraminidase). We then specifically assessed the abundance and the isotopologues of glycan-derived sialic acid in analogy with intracellular sialic acid and CMP-sialic acid. Here, the abundance and the overall FC of 13C6-glucose of the sialic acid generated from membrane bound glycans suggested that AR inhibition led to an increase in sialylation (Figure 4D), corroborating our previous finding that AR inhibition leads to overall glycosylation improvement as assessed by glycoproteomics in PMM2-CDG fibroblasts treated with epalrestat.19
Off-label epalrestat treatment improves clinical outcome measures in PMM2-CDG patients
Epalrestat was trialed in three pediatric PMM2-CDG patients (P5–7) to assess long-term treatment safety and efficacy. No side effects were observed. Escalating doses of epalrestat were well tolerated.
Due to the multisystem PMM2-CDG phenotype (liver function abnormalities, coagulation abnormalities, developmental delay, and neurologic involvement such as ataxia), the beneficial effects of epalrestat were assessed in relation to several aspects of the PMM2-CDG phenotype (see STAR Methods). Safety parameters (complete blood cell count, serum transaminases, bilirubin, alkaline phosphatase, prothrombin time, international normalized ratio), growth, and vital signs were monitored throughout the treatment.
Treatment with epalrestat decreased urine polyol levels, which remained mostly stable during treatment (Table 1). We noted fluctuation in mannitol in P5, which underwent a fructose loading test (Figure S1B). Furthermore, we observed improvement in biochemical markers that are commonly abnormal in PMM2-CDG, such as serum transaminase levels, coagulation parameters (ATIII), and CDT. At baseline, CDT was abnormal to a different degree in all three patients. Normalization of CDT was seen in P5. P6 showed significant improvement in CDT with a steady trend toward normalization at 18 months of therapy and escalating doses of epalrestat. Baseline CDT results of P7 were not available, and the first result was obtained after 3 months of therapy with epalrestat. However, at the 1-year mark, a slight improvement was seen in CDT. Moreover, the declining modified Brief Ataxia Rating Scale (BARS)39 scores indicated improvement in ataxia (P5 and P6). Although P7 was not yet eligible for the BARS assessment given her age, improvement in lower body strength and speech was observed on epalrestat. Phenotype severity was assessed by Nijmegen Pediatric CDG Rating Scale (NPCRS),40 and improvement was noted in two out of three patients (P5 and P7). Additionally, all patients self-reported clinical improvement and quality-of-life improvement during epalrestat treatment as assessed by goal attainment scale (GAS) (Table 1; see STAR Methods).41
Table 1.
Off-label epalrestat treatment improves clinical outcome measures in PMM2-CDG patients
| Patient | P5 | P6 | P7 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Agea/gender | 9/F | 8/M | 3/F | ||||||
| PMM2 pathogenic variants | c.422G>A | c.415G>A | – | – | c.422G>A | c.548T>C | – | c.422G>A | c.647A>T |
| Parameter/time point (months) | baseline | 12 | 24 | 28 | baseline | 12 | 18 | baseline | 12 |
| Epalrestat dose (mg/kg/day) | 0.8 | 1.5 | 2 | 3 | 0.8 | 1.5 | 3 | 0.5; 0.8d | 1.5 |
| Total NPCRS (maximum 78) | 24 | 20 | 18 | 18 | 19 | 19 | 18 | 18 | 15 |
| Modified BARS (maximum 54) | 39 | 29 | 33 | 30 | 45 | 29 | 27 | N/A | N/A |
| GAS (scale −2 to +2) | |||||||||
| Goal 1b | −2 | −1 | −1 | N/A | −2 | −1 | N/A | −2 | −2 |
| Goal 2b | −2 | −1 | −1 | N/A | −1 | −1 | N/A | −2 | −1 |
| Goal 3b | −2 | −2 | −2 | N/A | −2 | −2 | N/A | −2 | −1 |
| Laboratory investigations | |||||||||
| AST U/L (C <50 UL) | 48 | 37 | 34 | 40 | 58 | 40 | 38 | 548 | 80 |
| ALT U/L (C < 45 U/L) | 23 | 24 | 25 | 25 | 67 | 32 | 49 | 997 | 105 |
| AT III (C 0.80–1.30 IU/mL) | 0.88 | 0.99 | 0.97 | 1.03 | 0.25 | – | 0.39 | 0.21 | 0.37 |
| CDT mono-oligo/di-oligo ratio (C < 0.06) | 0.09 | 0.06 | 0.09 | 0.07 | 0.69 | 0.43 | 0.47 | 1.60c | 1.58 |
| CDT a-oligo/di-oligo ratio (C < 0.011) | 0.005 | 0.003 | 0.010 | 0.08 | 0.196 | 0.111 | 0.105 | 0.919c | 1.033 |
| Urine sorbitol (<5 mmol/mol Cr) | 19.9 | 10.7 | 7.5 | 7.86 | 50.80c | 12.81 | 9 | 9.85 | 0.03 |
| Urine mannitol (<20 mmol/mol Cr) | 648.6 | 32.6 | 45.3 | 93.2 | 23.90c | 14.77 | 47 | 10.37 | 1.89 |
Clinical and biochemical findings of patients at baseline and intermittent results during treatment with epalrestat. Abnormal values are marked in bold. ALT, alanine transaminase AST, aspartate transaminase; ATIII, antithrombin III; BARS, Brief Ataxia Rating Scale; CDT, carbohydrate-deficient transferrin; Cr, creatinine; C, control; NPCRS, Nijmegen Pediatric CDG Rating Scale; N/A, not applicable. Dash (–) indicates value not reported.
Age at the time of publication (years).
See STAR Methods for goals description.
First available results 3 months after starting treatment.
Patient was first started on 0.5 mg/kg/day, and after a month switched to 0.8 mg/kg/d.
Discussion
AR is a key enzyme of the polyol pathway, converting glucose to sorbitol.42 It is strongly implicated in diabetes, where excess glucose (hyperglycemia) is converted to sorbitol.15 The accumulation of sorbitol is considered toxic and is associated with neuropathy, nephropathy, and retinopathy in diabetic patients.15,17,18,21,22 AR has also been linked to other pathologic conditions such as cardiac disease, inflammatory disorders, asthma, sepsis, and cancer.43 Consequently, most research around AR inhibition has been related to diabetes.44,45,46 AR inhibition ameliorates diabetes-related complications by regulating the glucose flux and prevents the toxic accumulation of sorbitol.12,13,44,46 Recently, increased intracellular sorbitol levels in fibroblasts were reported in a form of hereditary neuropathy, sorbitol dehydrogenase (SORD) deficiency.20 SORD enzyme participates in the second step of the polyol pathway, at which sorbitol is converted to fructose, and its deficiency results in the accumulation of sorbitol.20 The authors showed that inhibition of AR with epalrestat had therapeutic effects and resulted in decreased sorbitol levels in vitro in SORD-deficient fibroblasts and in vivo in a SORD-deficient Drosophila model.
We have recently reported on abnormal polyol metabolism in PMM2-CDG. Specifically, we found elevated urine sorbitol levels in a smaller cohort of PMM2-CDG individuals.19 Similar to diabetes, urine sorbitol levels correlated with the degree of neuropathy in PMM2-CDG.19 Moreover, urine polyol levels decreased and clinical glycosylation markers improved in the pediatric PMM2-CDG individual treated with epalrestat,19 suggesting an association between the abnormal glycosylation and polyol metabolism.
In this study, to explore the mechanistic link between AR and glycosylation and the beneficial effects of epalrestat in PMM2-CDG, we first confirmed that increased urine sorbitol was a hallmark of PMM2-CDG by assessing urine polyol levels in a cohort of 50 PMM2-CDG patients. Our results showed that 86% of PMM2-CDG patients had increased urine sorbitol and 46% increased urine mannitol (Figure 1). Increased urine galactitol was not observed in our cohort, suggesting galactitol does not have the same clinical relevance as sorbitol and mannitol. To evaluate the intracellular levels of polyols and metabolic changes due to PMM2 deficiency, we then investigated metabolite levels in two models of PMM2-CDGs: patient-derived fibroblasts and pmm2 mutant zebrafish. While fibroblasts enabled us to study intracellular metabolic changes directly in patient-derived cells, we leveraged a zebrafish model to corroborate our findings in vivo. Increased polyols were present in both PMM2-CDG fibroblasts and pmm2 mutant zebrafish embryos, while GDP-mannose, a downstream metabolite of PMM2 and a major glycosylation building block, was depleted (Figure 1). Additionally, we found that glycolysis, TCA cycle, and nucleotide synthesis pathway metabolites were affected in patient fibroblasts (Figure 1), which might have further therapeutic implications in PMM2-CDG. To test if the increase in polyols was the consequence of PMM2 deficiency, we first ruled out the effect of environment on polyol abundance. Intracellular polyols, specifically sorbitol, were increased in PMM2-CDG, regardless of the presence of extracellular polyols or fructose (Figure S1). Then, we investigated whether AR itself is upregulated in PMM2-CDG and found both AKR1B1 expression (Figure S2) and AR enzymatic activity increased, which was then decreased by epalrestat (Figure S2B).
Since we previously measured an increase in PMM enzymatic activity in epalrestat-treated PMM2-CDG fibroblasts (Table S1),19 we investigated the possibility that epalrestat increases PMM activity by directly binding to PMM2 protein by performing thermal shift assay in WT and mutant (F119L HOM) protein (Figure 2). There was no significant difference in either WT or mutant PMM2 protein stability in the presence of epalrestat, while glc-1,6- P2, a known PMM2 activator,29 stabilized both WT and the mutant (Figure 2A).
Based on these results, we hypothesized that epalrestat-mediated AR inhibition rewires the cellular metabolism by redirecting glucose from the production of polyols toward other biochemical pathways, ultimately improving glycosylation.19 To test this, we assessed the metabolomes of PMM2-CDG and healthy fibroblasts treated with epalrestat. We found Man-1-P, GDP-mannose, and glc-1,6-P2, which were depleted in PMM2-CDG, increased in the presence of epalrestat (Figure 2C). The increase in Glc-1,6-P2 was of specific interest, as glc-1,6-P2 is able to activate PMM enzyme29 (Figure 2A). Hence, these data imply that the increase in intracellular concentration of glc-1,6-P2 likely plays a role in the increased PMM enzyme activity on epalrestat (Table S1).19 Alongside the increase in metabolites related to the PMM enzyme, AR inhibition resulted in changes across pathways of hexosamine biosynthesis, PPP, and (sugar) nucleotide synthesis in both patient and control fibroblasts (Figure S2), suggesting a global epalrestat-induced metabolic rewiring.
As fibroblasts are not the most metabolically active tissue, we further investigated the beneficial effects of epalrestat in vivo in pmm2 mutant zebrafish. We found that 24 h of epalrestat treatment was sufficient to significantly decrease polyol levels and increase GDP-mannose (Figure 2). Daily supplementation with epalrestat also resulted in improvement in swimming behavior in pmm2 mutant zebrafish (Figure 3), further supporting beneficial effects of epalrestat in PMM2-CDG.
The mechanistic link between AR inhibition and glycosylation was further established by targeting AKR1B1 (AR gene) with siRNA. Although pharmacological inhibition of AR with epalrestat differs kinetically from the siRNA approach and cannot be directly compared, these two methods led to comparable global metabolic changes in PMM2-CDG and healthy controls (Figures 4 and S4). Furthermore, tracer studies subsequently provided insight into the changes in pathway activities upon AR KD and demonstrated that glucose was redirected away from polyol production toward sugar nucleotide synthesis and protein glycosylation in both patient and healthy control fibroblasts (Figures 4 and S4), corroborating our previous results.19
Finally, the beneficial effect of AR inhibition by epalrestat was assessed in the three PMM2-CDG individuals treated with oral epalrestat (1–2.5 years). Epalrestat substantially decreases urine sorbitol and mannitol (Table 1). However, mannitol fluctuations following long-term use of epalrestat (>1 year, P5 and P6) were observed, similar to the ones in fibroblasts (Figure 2). These results are expected, as mannitol is produced by MDH enzyme, which is not directly targeted by epalrestat (Figure 2). Nevertheless, oral epalrestat treatment resulted in a clinical improvement in all three PMM2-CDG individuals (Table 1). Specifically, phenotype (NPCRS) and ataxia (BARS) severity decreased, while serum transaminases and coagulation parameters improved. Critically, CDT also improved (Table 1).
Although AR inhibition showed promising therapeutic effects in PMM2-CDG, there are additional therapeutic considerations. For example, PMM2-CDG individuals might additionally benefit from dietary fructose restriction and/or nucleoside supplementation. Specifically, we found the presence of fructose resulted in an increase in mannitol even in the presence of epalrestat (Figure S1), which might influence the effects of the treatment and result in a lessened clinical improvement. We also showed that the addition of nucleosides, e.g., guanosine, to the epalrestat treatment resulted in the full restoration of GDP-mannose levels (Figure S3), which was not seen on epalrestat alone (Figure 2). Further studies using combined epalrestat + guanosine supplementation in pmm2 mutant zebrafish might help elucidate the potential benefits of guanosine in treating PMM2-CDG. Moreover, AR inhibition by epalrestat resulted in an increased availability of nicotinamide-related cofactors (NADPH and NAD) after epalrestat treatment and changes in NADPH/NADP and NAD/NADH ratios (Figure S2). Considering NAD supplementation has been proposed as a treatment for other inborn metabolic disorders,47,48 these changes could also contribute to the overall beneficial effects of epalrestat and should be further considered. Next, we measured increased proliferation in fibroblasts treated with siRNA targeting AKR1B1. The effect of AR inhibition in PMM2-CDG has not been explored prior to this study, and AR inhibition was shown to decrease proliferation in cancer when excess growth factors or glutathione synthesis products are present.49,50,51,52 Although both PMM2-CDG and cancer affect metabolism, their metabolic adaptations are vastly different, and therefore treatments targeting AR might have different effects across these conditions. Still, the potential proliferation regulation role of AR should be further explored. We also have to note that, although inhibition of AR with either epalrestat or AKR1B1 KD strongly suggests improved protein glycosylation, the mechanisms of action could be different. As both AKR1B1 gene and AR enzymatic activity seem to be increased in PMM2-CDG, the possible role of PMM2 on the regulation of AR on gene and protein level should be explored.
Altogether, this study sheds light on the role of AR and polyol metabolism in the regulation of glycosylation and offers a deeper understanding of the therapeutic mechanism of AR inhibition by epalrestat in PMM2-CDG.
While increased AR activity results in an increase in sorbitol production, AR inhibition results in an increase of the availability of sugar nucleotides and protein glycosylation. In PMM2-CDG, this is therapeutic, as AR inhibition decreases toxic intracellular polyol production caused by PMM2 deficiency, boosts key metabolites Man-1-P and GDP-mannose, and restores glycosylation.19 Based on our in vivo and in vitro mechanistic studies establishing the role of AR inhibition in improving glycosylation, the efficacy of epalrestat is currently undergoing further assessment in a double-blind, crossover, placebo-controlled, phase III clinical trial (Oral Epalrestat Therapy in Pediatric Subjects with PMM2-CDG; ClinicalTrials.gov NCT04925960). Finally, we propose that polyol metabolism should be assessed in patients with other CDGs and AR inhibition therapy considered, especially for the CDGs affecting early glycosylation (e.g., ALG3-CDG) or the ones presenting with defects in sialic acid metabolism (e.g., NANS-CDG).
Limitations of the study
The limitations of our study involve the available disease models of PMM2-CDG and the MS technology-related limitations. Fibroblasts are an insufficient model to study metabolic disorders, such as CDG, which preferentially affect metabolically active organs such as the heart, liver, muscle, and brain. Therefore, metabolic changes in the fibroblasts might not fully mirror the metabolic rewiring occurring in the metabolically active, disease-relevant tissues. To overcome this, we studied a pmm2 mutant zebrafish model capturing both metabolic and morphological changes. While epalrestat treatment improved several metabolic parameters, it did not fully restore swimming behavior in PMM2-deficient zebrafish. However, pmm2 mutants have multiple phenotypes beyond swimming defects, including craniofacial abnormalities, liver deficiency, and neurological impairments. The impact of epalrestat on these systems, particularly the CNS, is currently unclear, which makes it difficult to determine why epalrestat only partially reversed the behavioral pathology. Finally, the limitations of MS technology did not allow us to assess the changes in the FC of 13C6-glucose after AKR1B1 KD of metabolites, including GDP-mannose, because of the spectra overlap with labeled UDP-hexNAc. While we could trace glucose flux changes through the de novo nucleotide synthesis metabolites and glycans, showing glucose flux through GDP-mannose is also increased after AR KD would directly corroborate the metabolomic findings observed after epalrestat-mediated AR inhibition.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Donkey anti-Mouse DyLight 800 secondary antibody | Invitrogen | RRID:AB_2556752 |
| Donkey anti-Rabbit DyLight 680 secondary antibody | Invitrogen | RRID: AB_2556622 |
| Mouse anti-AKR1B1 | Merck | SAB1409018 |
| Rabbit anti-ACTB | ABClonal | RRID: AB_2768234 |
| Bacterial and virus strains | ||
| E. coli BL21(DE3)pLysS - Novagen | Millipore Sigma | 69451 |
| Biological samples | ||
| Human blood | ClinicalTrials.gov Identifier: NCT04199000 | N/A |
| Human urine | ClinicalTrials.gov Identifier: NCT04199000 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Acetonitrile HPLC grade | Merck | 75-05-8 |
| Ammonium Bicarbonate | Sigma | A6141 |
| Ampicillin sodium salt | Thermo Fisher | AAJ6380709 |
| BSA | Sigma | A2153 |
| CaCl2 | Merck | 233-140-8 |
| Chloramphenicol | Thermo Fisher | 56-75-7 |
| Chloroform | VRW | 67-66-3 |
| cOmplete™ Protease inhibitor cocktail | Roche | 11697498001 |
| D-Fructose-6-P | Sigma | F1502 |
| D-Galactitol | Sigma | D0256 |
| D-Galactose-1-P | Sigma | G0380 |
| D-Glucose | Sigma | G8270 |
| D-GLUCOSE (1,2,3,4,5,6,6-D7, 97-98%) | Cambridge Isotope Laboratories |
DLM-2062-0.5 |
| D-Glucose (U-13C6, 98%) | Cambridge Isotopes | CLM-1396 |
| D-Glucose 1,6-diphosphate potassium salt hydrate | Sigma | G6893 |
| D-Glucose-1-P | Sigma | G1259 |
| D-Glucose-6-P | Sigma | G7250 |
| D-Mannitol | Sigma | M4125 |
| D-Mannose-1-P | Sigma | M1755 |
| D-Mannose-6-P | Sigma | M6876 |
| D-Ribose 5-phosphate barium salt hexahydrate | Sigma | 83870 |
| D-Sorbitol | Sigma | S1876 |
| Dichlormethane | Sigma | 650463 |
| Dimethylsulfoxide | Merck | D8418 |
| Dimethylsulfoxide | Thermo Fisher | 06-802-561 |
| Dynabeads streptatividin T1 | invitrogen | 65601 |
| EmbryoMax ® nucleosides (100x) | Sigma | ES-008-D |
| Epalrestat | Thermo Fisher | 82159-09-09 |
| Epalrestat HPLC grade | Selleck’s chem | S2035 |
| Ethanol (EtOH) 99% | Acros Organics BVBA | E/0650DF/15 |
| EZ-Link-Sulfo-NHS-LC-Biotin | Thermo Fisher | 21335 |
| Glycerol | Thermo Fisher | G33-4 |
| Glycerol | VWR chemicals | 56-81-5 |
| Guanosine, suitable for cell culture | Sigma | G6264-1G |
| HEPES | Thermo Fisher | BP310-1 |
| HEPES | Sigma | H3375 |
| HEPES sodium salt, 99% | ACROS organics | 215000010 |
| IGEPAL®- CA-630 | Sigma | 18896 |
| Imidazole | Thermo Fisher | O3196 |
| Intercept® (PBS) Protein-Free Blocking Buffer | Licor | 927-90001 |
| Isopropyl β-D-1-thiogalactopyranoside | Thermo Fisher | 15529019 |
| KCl | Thermo Fisher | P217 |
| Kinedak (Epalrestat) tablets 50mg | Ono Pharmaceuticals | NF611 |
| L-Arginine: HCl (D7, 98%; 15N4, 98%) | Cambridge Isotope Laboratories |
DNLM-7543-0.25 |
| L-Glutamic acid (13C5, 97-99%; D5, 97-99%; 15N, 97-99%) | Cambridge Isotope Laboratories |
CDNLM-6804-0.25 |
| L-Glutamine | Sigma | 49419 |
| Lipofectamine 2000 | Invitrogen | 11668027 |
| Luria Broth | Thermo Fisher | 12780052 |
| Methanol (MeOH) HPLC grade | VRW | 67-56-1 |
| Methoxyamine (MOX) hydrochloride for GC derivatisation 97,5-102,5% | Sigma Aldrich | 89803 |
| MgCl2 | Merck | 7791-18-6 |
| Myristic-d27 (98%) acid | Sigma | 366889 |
| n-Heptane ≥99% | VRW | 142-82-5 |
| N,O-bis (trimethylsilyl) trifluoroacetamide (BSTFA) | Sigma | 15222 |
| Na-azide | Sigma | S2002 |
| Na2HPO4x12H20 | Sigma | 10039-32-4 |
| NaCl | Sigma | S9888 |
| NaH2PO4x2H2O | Sigma | 13472-35-0 |
| NaOH | Sigma | 37576 |
| Neuraminidase from A. ureafaciens | Roche | 10269611001 |
| Page Ruler prestained protein ladder | Thermo Scientific | 404-266-16 |
| PhosSTOP™ Phosphatase inhibitor | Roche | 4906837001 |
| PNGase-F | New England Biolabs | P0704L |
| RIPA | Sigma | R0278 |
| Silencer™ Negative Control No. 1 siRNA | Ambion | AM4611 |
| siRNA targeting AKR1B1 | Ambion | AM16704 |
| Sodium chloride | Thermo Fisher | S271 |
| TaqMan probe for RT-qPCR AKR1B1 | Thermo Fisher | Hs01091553_g |
| TaqMan probe for RT-qPCR ACTB | Thermo Fisher | hs99999903_m1 |
| Tris base, Tris(hydroxymethyl)aminomethane | Roche | 77-86-1 |
| Tris(2-carboxyethyl)phosphine | Hampton Research | HR2-801 |
| Triton™ X-100 | Sigma | X100 |
| TRIzol reagent | Fisher Scientific | 12034977 |
| Tryptone | Thermo Fisher | BP1421 |
| Tween 20 | Millipore Sigma | P9416 |
| Tween® 20 | Sigma | P1379 |
| Uridine, suitable for cell culture | Sigma | U3003 |
| Yeast extract | Thermo Fisher | H26769.36 |
| α-D-Glucose 1,6-bisphosphate potassium salt hydrate | Sigma | 49225 |
| β-Nicotinamide adenine dinucleotide phosphate disodium salt | Sigma | 10128031001 |
| Critical commercial assays | ||
| 500 RXN FAST UNIVERSAL PCR MASTER MIX (2X), NO PCR Reagent, Real-Time TaqMan Fast Universal PCR Master Mix (2X), No AmpErase | Fisher Scientific | 10311135 |
| Aldose reductase activity assay (colorimetric) | AbCam | ab273276 |
| Applied Biosystems Protein Thermal Shift Dye Kit | ThermoFisher | 4461146 |
| Pierce BCA Protein Assay Kit | ThermoFisher | 23225 |
| PKH26 Red Fluorescent Cell Linker Mini Kit for General Cell Membrane Labeling | Sigma | MINI26-1KT |
| RNAeasy Midi kit (50) | Quiagen | 75144 |
| RNAeasy Mini plus kit (50) | Quiagen | 74134 |
| SuperScript Reverse transcriptase kit | Invitrogen | 18064014 |
| SuperScript™ III First-Strand Synthesis System | Invitrogen | 18080051 |
| SYBR™ Green PCR Master Mix | Applied Biosystems | 4309155 |
| TaqMan Fast Advanced Master Mix | LifeTechnologies | 4444963 |
| Experimental models: Cell lines | ||
| GM05381 Healthy control fibroblasts | Coriell | RRID:CVCL_7419 |
| GM05400 Healthy control fibroblasts | Coriell | RRID:CVCL_7426 |
| GM05757 Healthy control fibroblasts | Coriell | RRID:CVCL_7437 |
| GM08399 Healthy control fibroblasts | Coriell | RRID:CVCL_7482 |
| Human fibroblasts | See methods | N/A |
| Deposited data | ||
| Zebrafish metabolomics data | NMDR | ST002561 |
| Fibroblasts metabolomics data | NMDR | ST002566 |
| Fibroblasts metabolomics data fructose experiments | NMDR | ST002565 |
| Fibroblasts metabolomics data epalrestat experiments | NMDR | ST002562 |
| Fibroblasts metabolomics data siRNA experiments | NMDR | ST002563 |
| Fibroblasts metabolomics data neuraminidase experiment | NDMR | ST002564 |
| Fibroblasts GC/MS sugar quantification | NDMR | ST002575 |
| Experimental models: Organisms/strains | ||
| Zebrafish (Danio rerio) pmm2 mutant | Zebrafish International Resource Center | ZDB-ALT-130411-138 |
| Zebrafish (Danio rerio) wt | Zebrafish International Resource Center | N/A |
| Oligonucleotides | ||
| AKR1B1 Forward primer 5’-GGTGATCCCCAAGTCTGTGA-3’ | IDT | N/A |
| AKR1B1 Reverse primer 5’-AGGTGGTATCCTGGCTG-3’ | IDT | N/A |
| ACTB Forward primer 5’-AGAGCTACGAGCTGCCTGAC-3’ | IDT | N/A |
| ACTB Reverse primer 5’-AGCACTGTGTTGGCGTACAG-3’ | IDT | N/A |
| Recombinant DNA | ||
| Plasmid: WT human PMM2 | This paper | N/A |
| Plasmid: F119L variant | This paper | N/A |
| Software and algorithms | ||
| Adobe Illustrator for MacBook | Adobe | RRID:SCR_010279 |
| Biorender | Biorender.com | RRID:SCR_018361 |
| El-Maven Polly | Elucidata | RRID:SCR_022159 |
| GraphPad Prism v 9.00 MacBook | GraphPad software | RRID:SCR_000306 |
| Mass Hunter Workstation software with the Quantitative Analysis Version B.06.00/Build 6.0.388.0 | Agilent | RRID:SCR_015040 |
| Metaboanalyst v 5.0 | https://www.metaboanalyst.ca | RRID:SCR_015539 |
| National Metabolomics Data Repository (NMDR) Metabolomics Workbench |
NIH | RRID: SCR_013794 |
| Thermo Xcalibur software | ThermoFisher Scientific | RRID:SCR_014593 |
| Other | ||
| 10X Bolt™ Sample Reducing Agent | Invitrogen | B0009 |
| 20X Bolt™ MES SDS Running Buff | Invitrogen | B0002 |
| 4X Bolt™ LDS Sample Buffer | Invitrogen | B0008 |
| Anti-Anti | Gibco | 15240062 |
| Bolt™ 10%, Bis-Tris, 1.0 mm, Mini Protein Gels | Invitrogen | NW00100BOX |
| Bolt™ Transfer Buffer (20X) | Invitrogen | BT00061 |
| DMEM low glucose | Gibco | 11885084 |
| DMEM no glucose, no glutamine, no phenol red | Gibco | A1443001 |
| Fetal bovine serum (FBS) | Sigma | F7524 |
| Fetal bovine serum dialyzed | Gibco | 26400044 |
| Nitrocellulose membrane | BioRad | 1620115 |
| OptiMEM- Reduced Serum Medium | Gibco | 31985062 |
| PBS | Gibco | 10010023 |
| Trypsin-EDTA 10x 100ml | VWR | X0930-100 |
| HisPur Ni-NTA resin | Thermo Fisher | PI88222 |
| Superdex 200 10/300 GL size exclusion column | Cytiva | 10309283 |
Resource availability
Lead contact
Further information and requests for resources and reagents can be directed to the lead contact.
Materials availability
All reagents are commercially available. This study did not result in any new reagents. Recombinant WT and F119 PMM2 protein generated in this study are available on demand.
Experimental model and subject details
Ethics
Informed research content was obtained from all patients included in the study. Samples from PMM2-CDG affected individuals were collected during the Natural History Study (ClinicalTrials.gov Identifier: NCT04199000) in accordance with Mayo Clinic IRB study (IRB: 19-005187). Fibroblasts collected from the patients were analyzed in accordance with ethics application number S58358 and S60206 (‘‘Retrospective metabolomic analysis of archived fibroblasts’’) and Mayo Clinic IRB study (IRB: 16-004682). Additional healthy fibroblasts were obtained from Coriell institute (C7-C9, GM07575, GM05381, GM08399). Handling and euthanasia of fish for all experiments were in compliance with policies of the Greenwood Genetic Center, as approved by the Institutional Animal Care and Use Committee (permit #A2019 01-003-Y3-A2).
Human subjects
Fifty individuals with PMM2-CDG were included in this study (Table S1). Clinical data was collected both retrospectively and prospectively from the individuals recruited by Frontiers for Congenital Disorders of Glycosylation Consortium (FCDGC) at Mayo Clinic according to the IRB, within the scope of the standard of care (see19). Urine samples (P1-P50) and blood samples (P5-P7) were collected.
Cell culture
Patient (P1-P6) and control fibroblasts were obtained by a skin punch biopsy or purchased from Coriell (see key resources). The detailed clinical information of the six patients is given in Table S1. Fibroblasts were maintained in low glucose DMEM (Gibco) supplemented with 10% FBS in an incubator at 37°C, 5% CO2, no longer than two months or until they reached passage 15 to avoid senescence. Routine mycoplasma testing was performed.
E.coli culture
We used E.coli BL21(DE3)pLysS cells (Millipore Sigma) for recombinant expression of His-tagged human PMM2 proteins for thermal shift assays. The cells were cultured using standard practices.
Zebrafish strains, maintenance, and husbandry.
Animals were maintained according to standard protocols. The zebrafish strains were originally obtained from the Zebrafish International Resource Center (ZIRC, Eugene, OR)23. Staging was done according to established criteria.53 Pmm2sa10150 mutant zebrafish was established as previously described.23
Method details
Outcomes
The primary objective of the study was to assess the efficacy and safety of epalrestat treatment in 3 PMM2-CDG pediatric subjects prior to a planned Phase III clinical trial (Oral epalrestat therapy in Pediatric Subjects with PMM2-CDG, NCT04925960). The secondary objective was to assess the ability of epalrestat to improve clinically relevant aspects of PMM2-CDG such as increased urine polyols (sorbitol, mannitol), abnormal glycosylation (carbohydrate deficient transferrin), abnormal coagulation (ATIII-antithrombin III), impaired liver function (ALT- Alanine Transaminase, AST- Aspartate Transaminase), neurologic function (Brief Ataxia Rating Scale - BARS),39,54 severity of phenotype (Nijmegen Pediatric CDG Rating Scale - NPCRS).40 We monitored the ability of epalrestat to inhibit polyol production by evaluating urine polyol levels (sorbitol, mannitol), which are elevated in PMM2-CDG. Next, carbohydrate deficient transferrin, which is abnormal in PMM2-CDG, was evaluated to assess improvement of glycosylation following epalrestat treatment. We monitored changes in the liver enzymes AST, ALT, which are frequently elevated in PMM2-CDG, both as part of a safety study and as part of monitoring clinical improvement in coagulation. Neurologic improvement was assessed by modified BARS scores in 2 patients (3rd patient was not eligible for the test due to age at the start of the trial). NPCRS was assessed at every visit to assess changes in the severity of the patients’ phenotype. Additionally, we monitored the frequency of adverse events, changes in vital signs, complete blood count, and changes in BMI.
Procedures
Two patients were started on epalrestat on single patient investigational new drug (IND) (P5 IRB: 19-010017; IND #145262; P6 Protocol PMM2-CDG-001/A). Epalrestat was initiated on the parents’ own initiative in the third patient (P7). Two patients are currently in their second year of epalrestat treatment, while the third, previously reported patient19 is in the 3rd year of treatment. Physical, neurological, and biochemical examinations were performed in 3 PMM2-CDG patients at baseline, and at every three months after the start of the epalrestat treatment. Patients underwent NPCRS phenotype severity assessment and biological sampling at every visit. Epalrestat (Ono Pharmaceuticals, Osaka, Japan) was taken orally 3 times per day (TID) before meals in a divided dose, with dosage escalation throughout the treatment. The pharmacological profile of epalrestat was previously established.19 The starting dosage in P5 and P6 was 0.8 mg/kg/day, while P7 was started on 0.5 mg/kg/day due to her young age. P7 dosage was increased to 0.8 mg/day after a month, once safety was established. The complete overview of the dosage escalation is given in Table 1. Blood and urine samples were collected routinely. Urine polyols, carbohydrate deficient transferrin (CDT), anti-thrombin III (ATIII), liver enzymes (ALT, AST), and complete blood count (CBC) were performed. Modified Brief Ataxia Rating Scale39 was used to assess ataxia in the 2 patients (P5, P6; P7 was not eligible for the test due to age). The modified BARS scale included Archimedes spiral test, but not the abnormalities of the ocular pursuit system and the scale range was 0-54. Goal attainment scale (GAS) was administered as described per study protocol (ClinicalTrials.gov Identifier: NCT04199000). Quantification of goal attainment levels is done on a 5-point scale ranging from -2 to +2, with 0 indicating the expected level of goal attainment.41 Three individual goals were established by each patient (parent) at baseline and the progress was annually assessed. Goals included: walking without assistance (Goal #1 P5, P6), communicating with strangers (Goal #2 P5, P6), independent toilet training (Goal #3 P5), to be able to have a healthy and independent life (Goal #3 P6), to be able to equally play with peers (Goal #1 P7), normalization of laboratory results (liver enzymes) (Goal #2 P7), and improved balance to stand and walk (Goal #3 P7).
Fructose tolerance test
Fructose tolerance test was performed twice in patient and healthy controls according to the standard clinical protocol at fasting state with 1 g/kg oral fructose, and the first voided urine was collected. Urine collected while fasting prior to the fructose tolerance test was used as baseline.
Human sample analysis
Urine from 50 PMM2-CDG individuals was analyzed for the presence of polyols (sorbitol, mannitol, etc.) as previously described (Ligezka et al., 2021). Control reference range was established from n = 533 individuals out of which male = 226; female = 267; pediatric = 324 (<18 years old); adult = 209 (>18 years old). Collected blood samples were used for CDT analysis, ApoCIII analysis, CBC, liver enzymes, and coagulation factor analysis. Three patients (P1-P3) had urine and blood tests performed at UZ Leuven. All tests performed in blood are CLIA certified and routinely performed at Mayo Clinic.
PMM enzymatic activity assay
PMM enzymatic activity assay was previously performed in patient fibroblasts by CLIA certified PMM enzymatic activity assay.19 This method was adapted from.7 PMM and phosphomannoisomeraase (MPI) enzymatic activity was then measured in parallel by providing substrates for PMM and MPI (Man-1-P and Man-6-P respectively) and subsequently fluorometrically measuring the difference in generated NADPH. In this reaction, the activity of PGM is not measured, as the substrate for PGM (Glucose-1-P, Glucose-6-P, Ribose-1-P, Ribose-5-P) are not provided. Briefly, patient fibroblasts were grown in the presence or absence of 10 μM epalrestat in standard culture medium containing 5.5 mM glucose and 10 % FBS. Medium was removed and cells washed in PBS. Cell pellet was collected by scraping in PBS. Then, 1mg/ml of protein pellet was homogenized by sonication in 200 μL enzymatic buffer (25 mmol/L HEPES buffer, pH 7.1 + 25 mmol/L KCl + 0.02% (w/v) Na-azide) and incubated overnight. Total protein is determined by a protein assay (BioRad). Next, 50ul of patient and control lysate are pipetted into separate wells of a 96-cell microtiter plate followed by addition of 190ul of PMM reaction mixture (HEPES Reaction Buffer, NADP, 5% Inactivated BSA, Mannose-1,6-P2 (enzyme activator, produced at Mayo clinic Biochemical Genetics Laboratory), and intermediate enzymes [MPI, phosphoglucoseisomerase- PGI, glucose-6-phosphate dehydrogenase -G6PD]). 17.6 mmol/L Man-1-Phosphate is added at time 0. Man-1-P is metabolized to Glucose-6-Phosphate (Glc-6-P) by exogenous MPI and PGI which is then converted to 6-P-Glucono-delta-lactone by the NADP dependent reaction catalyzed by exogenous G6PD (PMM is the rate limiting step in this reversible pathway). Absorbance is read at 340nm at 30 minutes and 40 minutes by a BMG FLUOstar plate reader pre-equilibrated to 37°C. PMM2 activity is then determined by the change in absorbance from 30min to 40min and calculated in nmol/hr/mg total protein.
PMM enzymatic activity in the zebrafish was assessed in the same manner as decribed above with following adjustments: 10 embryos per sample were homogenized by sonication in 200 μL enzymatic buffer (25 mmol/L HEPES (Sigma), 25 mmol/L KCl (Fisher), pH 7.1; 0.02 % (w/v) Na-azide (Sigma)) and stored at -80 °C.
All reagents are from Sigma unless otherwise noted.
Analysis of AKR1B1 gene expression by RT-qPCR
To determine AKR1B1 gene expression RT-qPCR was performed as previously described55. Briefly, patient (P2, P3, P4, P5) and healthy fibroblasts (GM5381, GM5400, GM5757) were grown in DMEM (Gibco) containing 5.5 mM (physiological) glucose in 2 T75 flasks. Cell pellet was collected by scraping in PBS when cells were 80-90% confluent. RNA was then isolated from the samples using the RNeasy Mini Plus kit (Qiagen). RNA concentration and purity were determined using a NanoDrop spectrophotometer (ThermoFisher). Superscript III kit (Invitrogen) was used to prepare cDNA from isolated RNA as described above. Primer mix was prepared for the gene of interest (AKR1B1, IDT) and housekeeping gene (ACTB, IDT). 1 μL of forward and reverse primer, 5 μL SYBR universal PCR master mix buffer (Applied biosciences), and 2 μL of RNase-free water was added to 1 μL of previously prepared cDNA. The mixture was then pipetted to a 324-well PCR plate, and the plate was sealed and briefly centrifuged before being placed in a Lightcycler real time PCR system (Roche). The machine built-in protocol was used to perform melt curve analysis. Next, Ct values were exported from the program and analyzed. The 2ct method was used to analyze the relative changes in gene expression normalized against house-keeping gene mRNA expression56.
Aldose reductase (AR) enzymatic activity assay
AR enzymatic activity was assessed by AR activity assay kit (AbCam ab273276) according to the manufacturer’s protocol. The assay measures the change in NADPH in the presence of AR substrate to estimate the activity of AR. Briefly, PMM2-CDG (P2, P3, P5, P6) and healthy cells (GM5381, GM5400, GM8399) were prepared in T75 flasks as described above (see methods epalrestat supplementation). Cells were washed with PBS twice, scraped in PBS and pelleted in centrifuge at 2,000 rpm, 10 min, 4 °C. Next, fresh pellets were lysed on ice in 50 μL of AR extraction buffer and centrifuged at 12,000 rpm, 10 min, 4 °C. The supernatant was collected, 20 μL of the supernatant was used for the assay, while the rest was used for protein concentration estimation (Pierce BCA kit, ThermoFisher). NADPH standards, negative control, positive controls, samples, NADPH and AR substrate were added to the plate in duplicates to the UV 96-well plate according to the manufacturer’s instructions. Oddysey plate (Omega) reader is used to measure absorbance at 340 nm for one hour. NADPH concentrations in the samples was estimated based on the standard curve. AR activity was calculated by subtracting the final NADPH concentration from the initial one and normalizing it to the protein content. Relative enzymatic activity was calculated based on the average of untreated healthy control samples.
Expression and purification of recombinant human PMM2
The gene for human PMM2 (UniProt ID H3BT06) was commercially synthesized (GenScript) with codon optimization for bacterial expression and inserted into a pET-14b vector with an N-terminal His6-affinity tag and tobacco etch virus protease site (tag sequence MHHHHHHENLYFQG). The F119L variant was prepared via commercial site-directed mutagenesis of the wild-type vector (GenScript). For protein expression, the respective vectors were transformed into E. coli BL21(DE3)pLysS cells. Bacterial cultures were initiated in 10 ml LB media with 100 μg mL-1 ampicillin and 34 μg mL-1 chloramphenicol and grown for 12 h in a shaking incubator at 37 °C. The 10 mL culture was used to inoculate 1 liter of terrific broth supplemented with the same antibiotics and grown at 37°C to an A600 of 0.8. The culture was induced with isopropyl 1-thio-b-D-galactopyranoside (final concentration 0.5 mM) and grown overnight (16 hr) at 19°C. Cells were harvested by centrifugation, and the cell pellets flash frozen in liquid N2 and stored at -80°C.
Protein purification was carried out at 4 °C. Pellets were resuspended in buffer A [50 mM Hepes, pH 7.5, 500 mM NaCl, 5% glycerol, and 0.5 mM tris(2-carboxyethyl) phosphine (TCEP)] supplemented with 20 mM imidazole and 1% Tween-20 and disrupted by sonication. The lysate was clarified by centrifugation at 16,500 rpm for 1 h. The supernatant was passed through a 0.45 μm filter and loaded onto a Ni2+ column. Ten column volumes of buffer A plus 20 mM imidazole were used to wash the column. The protein was eluted with four column volumes of buffer A plus 250 mM imidazole. For the F119L variant, the protein was further purified by size-exclusion chromatography on a Superdex 200 10/300 column using a buffer containing 20 mM Hepes, pH 7.5, 150 mM NaCl, and 5 mM MgCl2. The purified proteins were dialyzed into 50 mM Hepes, pH 7.5, 150 mM NaCl, 1 mM MgCl2 and 0.5 mM TCEP, concentrated to 10 mg/mL, flash-frozen in liquid nitrogen, and stored at -80°C. We note that higher concentrations of epalrestat could not be tested due to solubility limitations in aqueous solution.
Thermal shift assays
Recombinant human PMM2 (wild-type and F119L variant) samples were diluted to 1.0 mg/mL (33.6 mM) in 50 mM Hepes, pH 7.4, and 50 mM NaCl in the presence and absence of various ligands. Protein samples with the known activator glucose 1,6-bisphosphate (Millipore Sigma) were prepared at concentrations of ligand ranging from 0.5 to 5 mM. Samples with epalrestat (Thermo Fisher) were prepared at 10 mM ligand to match experimental conditions in other assays. The final concentration of DMSO in all samples was <0.5 %. Samples were incubated with SYPRO Orange dye from the Applied Biosystems Protein Thermal Shift kit per manufacturer’s recommendation for 1 hour at 4°C. A Quant Studio 3 Real-Time PCR System (ThermoFisher Scientific) was used to ramp from 20°C to 99° C in 0.1°C increments with 10 second holds between ramping steps. Fluorescence values were normalized as in31 and T0.5 was calculated as the midpoint of the normalized fluorescence response. Samples were run in duplicate.
Metabolomic profiling of fibroblasts
Metabolomic profiling of fibroblasts was done as previously reported32. Briefly, 15,000 fibroblasts were plated in 1.5 mL DMEM (Gibco, 5.5 mM glucose, 2 mM glutamine, 10 % FBS) per well in two 6-well plates. The medium change was performed on day 2 and day 4. On day 6, the medium was removed, cells were washed with ice-cold 0.9 % NaCl (Sigma) (physiological solution) and 250 μL of extraction buffer (80 % MeOH, IS) was added to the cells. Next, cells were scraped and the cell extracts transferred to 1.5 mL Eppendorf tubes and placed overnight at -80 °C. Next, samples were centrifuged at 15,000 rpm, 4 °C, 20 min. The supernatant was transferred to a vial for direct analysis by LC/MS, while 100 μL of 200 mM NaOH was added to the (protein) pellet. Pellets were incubated at 95 °C for 30min after which they were centrifuged at 5,000 rpm, 4 °C, 10 min. Supernatant was used to determine protein concentration by BCA protein assay kit (Pierce).
Metabolomic profiling of zebrafish
10 embryos per sample were homogenized by sonication in extraction buffer (80% MeOH, IS). The metabolites were then precipitated at -80 °C overnight, after which the samples were centrifuged at 15,000 rpm, 20 min, 4 °C. Next, the supernatant was transferred to a fresh Eppendorf tube. The samples were centrifuged again at 15,000 rpm, 20 min, 4 °C to remove any leftover impurities and transferred to a fresh MS vial. Finally, the samples were analyzed by LC/MS (see below).
Epalrestat supplementation
Fibroblasts were plated in 6-well plates for LC/MS (15.000 cells per well) and T75 flasks (112.500 cells per flask) for GC/MS analysis and aldose reductase activity assay (see methods below). After 24 h, medium was removed and cells washed with PBS. Then, DMEM containing 5.5 mM glucose, 2 mM glutamine, and 10 % dialyzed FBS was added. Epalrestat was freshly prepared and fibroblasts were supplemented with 10 μM epalrestat daily as previously described10,19. After 48 hours media was changed, and 48 h after that, metabolites were extracted and measured by both LC/MS and GC/MS (see below). Zebrafish embryos were genotyped at 2 days post fertilization (dpf) as previously described23 and treated as described in the results, by adding the epalrestat (solubilized in DMSO) directly into the embryo media at 5 dpf. Control embryos were similarly treated with DMSO alone. Embryos were harvested for metabolic analyses 24 h post-treatment at 6 dpf. The final concentration of DMSO was equivalent between control and treatment, never exceeding 0.5 %.
Nucleoside supplementation experiments
The experiments were performed as described above (epalrestat experiments) with following changes. Cells were plated in 6-well plates. 24 h after plating, medium was changed to either A) DMEM medium containing 5.5 mM glucose, 2 mM glutamine, 10 % dialyzed FBS supplemented with EmbryoMax nucleosides (100x) to the final concentration 0.03 mM per nucleotide (Sigma), or B) DMEM medium containing 5.5 mM glucose, 2 mM glutamine, and 10 % dialyzed FBS. Epalrestat was given to the patient and control fibroblasts as described above. Zebrafish were treated either with combination of 1mM guanosine (Sigma) and 40 uM epalrestat (dissolved in DMSO), or DMSO alone. The final concentration of DMSO was equivalent between control and treatment, never exceeding 0.5 %. Embryos were harvested 24-hour post-treatment at 6dpf and homogenized in the extraction buffer as described above.
Uridine/guanosine supplementation experiments
The experiments were performed as described above with following changes. Cells were plated in 6-well plates. 24 h after plating medium was changed to either A) DMEM medium containing 0.03 mM guanosine or 0.03 mM uridine, 5.5 mM glucose, 2 mM glutamine, and 10 % dialyzed FBS, or B) DMEM medium containing 5.5 mM glucose, 2 mM glutamine, and 10 % dialyzed FBS. Epalrestat was given to the patient and control fibroblasts as described above. Zebrafish was supplemented by 1mM guanosine and epalrestat dissolved in DMSO for 24h.
Zebrabox motility assay
Progeny of a pmm2sa1050/+ (pmm2m/+) mating were collected and raised in the light following standard protocols. 5dpf embryos were placed one per well in a 12-well culture dish and treated daily with either 20 μM epalrestat in DMSO or DMSO only, refreshing the embryo media daily. Embryos were also fed a small number of rotifers daily once behavioral readings were completed. Locomotor activity (swim paths, speeds) was monitored for ten-minute intervals from 6-10 dpf using the Zebrabox System (ViewPoint Inc., Toronto, Canada) as previously described57. Data was analyzed and graphed using GraphPad Prism.
Inhibition of AKR1B1 by siRNA
Due to limited experimental capacity, we selected two representative patient cell lines (P2 most severe, P5 mild presentation) for our subsequent experiments. We first performed a titration of AKR1B1 siRNA (0 nM, 2.5 nM, 5nM, 10 nM, 20 nM, and 40 nM) in both patient and control fibroblasts to determine the optimal concentration for our experiments. We found that 5 nM siRNA concentration was the optimal dose, resulting in significant KD of the AKR1B1 and the minimal cell death (data not shown).
For metabolite extraction, membrane bound sialic acid isolation and quantification, and RNA and protein extraction, 15,000 cells were plated in quadruplicates in 6-well plates in 1.5 mL of DMEM (Gibco) containing 5.5 mM glucose, 2 mM glutamine, and 10% FBS. The following day, medium was removed and the cells were washed with 1 mL PBS before adding 1.2 mL of DMEM containing 5.5 mM glucose, 2 mM glutamine and 10% dialyzed serum. Then, 5 nM siRNA targeting AKR1B1 (Ambion, cat AM16704) and non-targeting (negative) siRNA (Ambion, cat AM4611) was prepared using Lipofectamine 2000 (Invitrogen) and OptiMEM (Gibco) according to the manufacturer’s protocol (final volume of 300 μL). This siRNA mix was then added to the cells. After 48 h, the medium was removed, and cells were washed with PBS, and DMEM medium containing 2 mM glutamine, 10% dialyzed FBS and either A) 5.5 mM 13C6-glucose or B) 5.5 mM 12C6-glucose was added. Cells were incubated for another 48h followed by membrane sialic acid isolation, and RNA, protein, and metabolite extraction. Extracted metabolites were subjected to LC/MS. RNA was used to generate cDNA and validate the AKR1B1 KD with RT-qPCR, while protein was used for Western Blotting (see below).
Membrane sialic acid isolation and quantification
Control and PMM2-CDG fibroblasts were plated in 6-well plates in DMEM containing 5.5 mM glucose, 2 mM glutamine, and 10% FBS, followed by inhibition of AKR1B1 by siRNA (described above). Briefly, medium was removed from the cells and the cells were washed 3 times with 1 mL Dulbecco PBS containing 0.901 mM CaCl2 (Merck) and 0.492 mM MgCl2 (Merck). Next, cells were incubated with 1 mg/100 mL EZ-Link-Sulfo-NHS-LC-Biotin (Thermo) in Dulbecco for 30 min, RT, shaking. Cells were then washed twice with 1 mL Dulbecco, non-reacted biotin was blocked with 1 mL 20 mM glycine in Dulbecco for 15 min, and cells washed again with 1 mL Dulbecco. Dulbecco was then removed, cells scraped in 200 μL RIPA buffer (with protease inhibitors) and transferred to a fresh Eppendorf tube. Samples were lysed on ice with 3 consecutive freeze-thaw cycles. Further, 30 μL dynabeads streptavidin T1 (Invitrogen) was added to each sample and the samples were washed twice with 500 μL 10 mM ammonium bicarbonate and neuraminidase buffer (100 mM Sodium Acetate Buffer with 2 mM CaCl2 (Merck), pH 5.0). Neuraminidase buffer was removed and 500 μL PBS was added to the beads. 30 μL of prepared mix was added to each sample, and samples were incubated, shaking overnight at 4 °C. Samples were put on a dynabead rack (Invitrogen) and the supernatant was transferred to a new Eppendorf tube and used for protein concentration assay. Beads containing membrane fractions were washed 2 times with 500 μL lysis buffer (2 % IGEPAL (Sigma), 1% Triton X-100 (Sigma), and 10 % glycerol inPBS) and then washed with 1 mL PBS. Samples are centrifuged at 500 rcf, 5 min, 4 °C and PBS was removed. 100 μL neuraminidase buffer containing 0.05 U neuraminidase was added to each sample and samples were incubated overnight at 37 °C, shaking. Next, supernatant was transferred to a new Eppendorf tube and lyophilized at 4°C. Finally, pellets were resuspended in 100 μL of extraction buffer (80 % MeOH, IS). Sialic acid was measured by LC/MS as described below. El Maven Polly software was used to annotate sialic acid based on m/z ratio and elution time, and determine fractional contribution of glucose in sialic acid.
Confirmation of AKR1B1 knock-down
To confirm AKR1B1 siRNA experiments resulted in AKR1B1 KD, protein and RNA expression was assessed in fibroblasts incubated with negative siRNA or siRNA targeting AKR1B1. Briefly, 500μL of TRIZol (Fisher) was added to cells previously washed with PBS. The cells were scraped and transferred to a fresh 1.5 mL Eppendorf tube. After, the cells were kept overnight at -80°C. Next, 100 μL of chloroform was added and the sample briefly shaken. The samples were then centrifuged for 25 min, 12000 rpm, 4°C. The polar phase was transferred to a fresh Eppendorf tube and 250 L of 70% cold ethanol was added. RNA was then isolated from the sample using the RNeasy Mini kit (Qiagen). RNA concentration and purity were determined using a NanoDrop spectrophotometer (ThermoFisher). Superscript II kit (Invitrogen) was used to prepare cDNA from isolated RNA. Briefly, 0.5 μg of RNA was pipetted into a 0.2 mL PCR tube and RNAse free water was added to the final volume of 10.5 μL. The sample was incubated with 2 μL of Random Primers (1/20 diluted) in a thermocycler for 10 min at 70°C and then cooled to 4°C. Master mix containing 4 μL first strand buffer (5x undiluted), 2 μL DTT, 1 μL dNTP and 0.5 μL SuperScript II was added to each sample. The samples were returned to the thermocycler and the following program was applied: 10 min 25°C, 80 min 42°C, 15 min 70°C, and 4°C indef. After, the samples were stored at −80°C before performing RT-qPCR. Primer mix was prepared for the gene of interest (AKR1B1) and housekeeping gene (BACT). 0.5 μL of Taqman primer (ThermoFisher, AKR1B1-hs01091533_g1; ACTB hs99999903_m1), 0.5 μL TaqMan universal PCR mastermix buffer (2x undiluted) (Applied biosciences), and 2.5 μL of RNase-free water was added to 2 μL of previously prepared cDNA. The mixture was then pipetted to a 96-well PCR plate, and the plate was sealed and briefly centrifuged before being placed in a Lightcycler 96 real-time PCR system (Roche). The following protocol was run: preincubation (60s), amplification (95°C, 30s, 45cycles), acquisition 60°C, 30s. The machine built-in protocol was used to perform melt curve analysis. Next, Ct values were exported from the program and analyzed. The 2ct method was used to analyze the relative changes in gene expression normalized against house-keeping gene mRNA expression.56 For AKR1B1 protein expression analysis, cells were scraped in PBS, and centrifuged at 600 rpm, 4°C, 10 min. PBS was removed and the cell pellet was stored at −80°C overnight. RIPA buffer was added to the cell pellet and cells were lysed by 3 consecutive freeze-thaw cycles. Samples were centrifuged at 12,000 rpm, 4°C,10 min and the supernatant transferred to a new Eppendorf tube. Protein concentration was determined by BCA protein assay kit (Pierce). Total cell protein (20 μg) mixed with sample buffer (4x Bolt LDS sample buffer, 10% DTT) was denatured (30 min, 95°C) and loaded onto a 10% Bolt Bis-Tris gel (ThermoFisher). The proteins were then separated by PAGE (200 V, 1.5 h, RT). Next, protein transfer onto the nitrocellulose membrane (1620115 BioRad) was done using Bolt transfer buffer (Thermo Fisher) containing 10% methanol (MeOH) (3h, 35V, 4°C), followed by blocking of the membrane in Licor buffer (Oddysay) (1 h, RT). Incubation with primary antibodies (mouse anti-AKR1B1 1:200, rabbit anti-ACTB 1:5000 Cat#AC026 ABclonal, 4°C) in Intercept blocking buffer (Licor) was done overnight. Next, the membrane was washed 6 × 10 min with PBS-T (0.1% Tween 20) at RT after which the membrane was incubated with secondary antibodies (donkey anti-mouse 1:5000 Invitrogen Cat#SA510172 DyLight 800, donkey anti-rabbit 1:5000 Invitrogen Cat#SA5-10042 DyLight 680) (1 h, RT). Then, the membrane was again washed 6 × 10 min with PBS-T, 1 × 10 min PBS). Finally, Li-COR Oddysey Cx (700, 800channel) was used to visualize the membrane and quantify AKR1B1 and ACTB bands.
Proliferation assay
Cells were plated in 96-well plates in triplicates using the adapted protocol from the metabolomics siRNA experiments. PKH26 red fluorescent cell linker mini kit for general cell membrane labeling (Sigma) was used according to the manufacturer’s protocol. The next day, the medium was changed, and non-targeting (negative) and siRNA targeting AKR1B1 were added to the cells (see STAR Methods). The plate with the cells was then placed in the Incucyte live cell analysis system (Essen Bioscience) and the plate was read in 2 h intervals for 5 days. Four pictures per well were taken. Medium was changed on day 3. Cell confluence was measured using built-in software. The confluence rate of change was calculated as the difference between the confluence on the first day after plating and the final day of experiment, divided by the number of days cells were in culture.
Relative metabolite quantification by targeted LC/MS
Relative metabolite quantification by LC/MS was done as previously described.32 An ion-pairing liquid chromatography column (C18, Acquity UPLC-HSS) or Hilic (Agilent Afinity) was used to separate 10 μL of sample, after which the metabolites were resolved on a Thermo Fisher Q-Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (negative ion mode) or Thermo Fisher Q-Exactive Focuse (negative ion mode). Reversed phase (C18) chromatography method was as follows: solvent The gradient started with 5% of solvent B (100% methanol) and 95% solvent A (H20, 10mM tributyl-amine, 15mM acetic acid) and remained at 5% B until 2 min post injection. A linear gradient to 37% B was carried out until 7 min and increased to 41% until 14 min. Between 14 and 26 minutes the gradient increased to 95% of B and remained at 95% B for 4 minutes. At 30 min the gradient returned to 5% B. The chromatography was stopped at 40 min. Column temperature was 40°C. For Hilic chromatography method was as follows: A linear gradient was carried out starting with 90% solvent A (100% acetonitrile) and 10% solvent B (10 mM Na-acetate in mqH2O, pH 9.3). From 2 to 12 min the gradient changed to 60% B. The gradient was kept on 60% B for 3 minutes and followed by a decrease to 10% B. Column temperature was 25°C The chromatography was stopped at 25 min. Flow rate for both methods was 0.25ml/min. The ESI settings were as follows: 50 sheet gas flow rate, auxiliary gas flow rate 15, spray voltage of 4 kV, S-lens RF level of 60, and the capillary temperature at 350. A full scan (resolution 140,000 at 200 m/z, AGC at 3 e6, 512 ms ion fill time, and 70–850 m/z scan range) was performed. The metabolites were identified according to their elution time and m/z ratios by using in-house metabolite standard library and El-Maven v0. 12.0/Polly ™Labeled LC-MS Workflow or Thermo Fisher Xcalibur software. The correction of naturally occurring carbon isotopes was also performed (Elucidata, Polly, versions 2019-2023). Metabolite abundances were normalized to the internal standard and protein content. Relative metabolite abundances were calculated by comparing PMM2-CDG affected samples to control samples. Absolute quantification was not performed. Note: Due to their chemical properties and almost identical chemical structure, LC/MS method used cannot separate polyols (sorbitol, mannitol, galactitol), hexoses (glucose, mannose, fructose, galactose), hexose-P (glucose-1-P, glucose-6-P, mannose-6-P, etc.), UDP-hexose (UDP-glc, UDP-gal), UDP-hexNac (UDP-galNAc, UDP-glcNAc, UDP-manNAc) etc due to the identical m/z ratio and elution time. In addition, this method is not able to separate certain isotopologues in tracer experiments, such as GDP-Mannose and UDP-HexNac, which overlap (m0-m14 isotopologues of UDP-HexNAc overlap with m2-m16 of GDP-Mannose etc.) Targeted GC/MS, on the other hand, has a more powerful separating capability compared to LC/MS and can separate different hexose-P, such as glucose-1-P or glucose-6-P because, unlike in LC/MS they elute at different times. However, targeted GC/MS can only monitor certain metabolites at the same time and a new method has to be developed for each class of metabolites. Therefore, GC/MS and LC/MS are complimentary to each other and when used together give a comprehensive overview of the metabolism.
Relative sugar quantification by GC/MS
To separate metabolites that are not separable by LC/MS as described above, GC/MS measurements of hexose-P (glucose-1-P, glucose-6-P, mannose-6-P, etc.), hexose-bisphosphates (glucose-1,6-P2, etc.), hexose (glucose, mannose, fructose, galactose), and polyols (sorbitol, mannitol, galactitol) were performed. Briefly, fibroblasts were cultured in a similar fashion as for LC/MS with the following adjustments: 1) an appropriate number of fibroblasts was plated in T75 flasks instead of 6-well plates to ensure the optimal amount of metabolite concentration necessary for GC/MS; 2) metabolites were extracted from cells with 2 mL 80 % MeOH, IS and the supernatant was transferred to a fresh 2 mL Eppendorf tube. Standards containing metabolites of interest were prepared simultaneously and dried overnight together with cellular extracts by vacuum centrifugation at 4 °C. The following day, 20 μL of methoxyamine (MOX) (Sigma Aldrich) was added to the samples and they were incubated for 90 min at 37 °C. Next, 60 μL of N, O-bis(trimethylsilyl)trifluoroacetamide (TMS) (Sigma Aldrich) was added to the samples, which were then incubated at 60 °C for 30 min. Samples were kept overnight in a dry and cool place to allow further derivatization with TMS. The following day, samples were placed on GC/MS and metabolites measured by Agilent 7890A GC (Agilent Technologies) coupled with an HP-5 ms 5 % phenyl methyl silox capillary column (30 m, 0.25 mm, 0.25 mm, Agilent Technologies), interfaced with a QQQ MS (Agilent 7000B, Agilent Technologies) and operating under ionization by electron impact at 70 eV. The temperature of injection port, interface, and ion source was 230 °C. The quadrupole temperature was maintained at 150°C. 1 μL of sample was injected onto the machine at either 1:25 split ratio (polyol), 1:5 split ratio (hexoses) or in splitless mode (Hexose-P and Hexose-BIP). Constant helium flow was applied at 1 mL/min. The temperature gradient applied was as follows: 2 min 100°C, next 175 °C at Δ20 °C/min, followed by an increase to 230 °C at Δ4 °C/min, kept at 230 °C for 3 min, increased to 300 °C at Δ 40°C/min and lastly kept at 300 °C for 5 min. The column was further baked for another 3 min at 325°C after the gradient. Hexose-P (SIM 299 m/z), hexose-bisphosphates (SIM 387 m/z), hexoses (SIM 204, 205, 206, 207, 208), and polyols (319, 320, 321, 322, 333 m/z)58 (Hummel et al, 2007; Hummel et al, 2010) standards were used to determine the elution time of each metabolite. Data was then analyzed with the Mass Hunter Workstation software with the Quantitative Analysis Version B.06.00/Build 6.0.388.0. Metabolite abundances were normalized to protein content. Relative metabolite abundances were calculated by comparing PMM2-CDG affected samples to control samples. Absolute quantification was not performed. This method does not measure 13C6 labeled mannose-6-P and mannose-1-P, or other individual labeled hexose-P, due to the electron ionization used in the GC-MS setup, as a consequence of this ionization, metabolites fragment and in the case of the hexose-P species, solely the fragment carrying the phosphate moiety (thus not the carbons) is detectable.
Relative quantification of NADPH, NADP, NAD and NADH
Fibroblasts from three patient fibroblasts (P2, P4, P5) and two healthy controls were plated on day 1. The next day medium was changed. The cells were grown in the presence and absence of 10 μM epalrestat for another 24 h. The medium was then removed, cells were washed with cold saline solution (0.9 % NaCl) and 250 μL of extraction buffer (50% MeOH, 30% acetonitrile, 20% H2O Tris-HCl pH 7.6) was added to the cells. The metabolites were precipitated overnight at −80°C, samples were centrifugated at 20,000 rpm, 20 min, 4°C, and supernatant was transferred to a fresh vial. Pellets were used for determination of protein concentration. Supernatant containing metabolites was analyzed by LC/MS less than 24 h after the initial collection to avoid degradation of NADPH into NADP and NADH into NAD. Relative abundance of NADP and NADPH was obtained. NADP/NADPH ratio was determined by dividing abundances of NADP with the abundances of NADPH in the same sample. NAD/NADH ratio was determined by dividing the abundances of NAD and NADH in the same sample.
Measurement of sorbitol and mannitol in FBS
Briefly, 10 μL of dialyzed and non-dialyzed FBS was added to 990 μL of extraction buffer (80% MeOH, IS). The samples were further precipitated overnight at −80°C. Samples were centrifuged and supernatant transferred to a fresh Eppendorf tube. Next, samples were dried by vacuum centrifugation at 4°C. Derivatization with MOX and TMS as well as measurements of sorbitol and mannitol in FBS were done as described above (Sugar quantification by GC/MS).
Quantification and statistical analysis
Statistical analysis
All statistical analysis has been performed using GraphPad 9.0 prism for MacBook where applicable. Significant differences in metabolite abundances under native conditions (5.5 mM glucose, non-dialyzed FBS) between patients and control fibroblasts were calculated by t-test with Welch correction (Figures 1 and S2). Metaboanalyst59 was used to generate heatmaps (Figure 1). Unless otherwise stated, two-way repeated measures ANOVA and multiple comparisons with Sidak correction was used to determine the effects of treatment (presence of epalrestat/epalrestat and nucleosides/epalrestat and guanosine/epalrestat and uridine/presence of fructose/siRNA inhibition), genotype (PMM2-CDG vs. WT), and interaction on metabolite abundances in fibroblasts (Figures 2, 4, 1, and S2–S4). Mixed model analysis with repeated measures and multiple comparisons with Sidak correction was performed on zebrafish experiments (Figures 2, 3) and CMP-sialic acid positional labeling (Figure 4). Differences were considered statistically significant if p < 0.05. Number of samples and technical replicates are indicated under each figure. Error bars are shown with standard deviation SD unless otherwise indicated
Thermal shift analysis quantification and statistical analysis
Relative fluorescence units (RFU) were normalized to fraction unfolded using the minimum and maximum fluorescence reading for the well as in (Andreotti et al., 2015) with the formula Funfolded = (RFUi - RFUmin)/(RFUmax - RFUmin), where Funfolded is the relative fractional portion unfolded at a temperature point, RFUi is the fluorescence reading at a temperature point, RFUmin is the minimum reading for that sample, and RFUmax is the maximum reading for that sample. The point at which the protein is half unfolded (Funfolded = 0.50), designated T0.5, is used for comparison of samples. T0.5 > 1°C was considered significant.
Additional resources
Web-based databases
Ellucidata, Polly platform, Metabolomics workflow https://polly.elucidata.io.
National institute of standards and technology Chemistry webbook SRD 69 https://webbook.nist.gov/chemistry/
National Metabolomics Data Repository Metabolomics workbench
https://www.metabolomicsworkbench.org.
The human metabolome database https://hmdb.ca.
Illustration software
All artwork and graphs were made by using GraphPad prism for MacBook and Adobe Illustrator. The exception being icons of Petri dish with cells, zebrafish, patient, urine cup, and medication icon created by Biorender.com (through subscription). All colors are color blind-friendly.
Acknowledgments
We would like to thank patients and their families. We thank Juan Ji for assistance with the protein purification. This work was supported by 1U54NS115198-01 from the National Institute of Neurological Disorders and Stroke (NINDS), the National Center for Advancing Translational Sciences (NCATS), the National Institute of Child Health and Human Development (NICHD), and the Rare Disorders Consortium Disease Network (RDCRN). B.G. was supported by a KU Leuven internal C1 funding grant (EFF-D2860-C14/17/110). H.F.-S. is supported by CDG Care and P20 GM139769 from the National Institute of General Medical Sciences (NIGMS). L.J.B. is supported by TRIUMPH grant (University of Missouri School of Medicine). LFO is supported by Ryan Fellowship (Department of Biochemistry, University of Missouri). P.W. is partially funded by a senior clinical investigatorship from FWO Flanders, Belgium (18B4322N).
Author contributions
Study conceptualization, B.G., E.M., and S.R.; methodology, E.M., B.G., H.F.-S., L.J.B., and S.R.; validation, E.M., B.G., and S.R.; investigation, A.E., A.N.L., A.L., C.L., D.C., E.M., H.F.-S., K.D., L.D.-R., L.F.O., L.S., M.S., P.V., J.M., P.V., P.W., S.S.M., S.R., and S.S.M.; formal analysis, A.N.L., E.M., B.G., G.P., H.F.-S., L.J.B., S.R., and T.K.; resources, B.G., D.C., E.M., H.F.-S., L.J.B., P.V., and T.K.; data curation, B.G. and S.R.; writing – original draft, B.G., E.M., and S.R.; writing – review and editing, A.N.L., B.G., E.M., G.P., H.F.-S., K.D., L.J.B., M.S., P.V., P.W., S.S.M., S.R., and T.K.; visualization, H.F.-S., L.J.B., and S.R.; supervision, B.G., E.M., H.F.-S., L.J.B., P.W., and T.K.; project administration, B.G. and E.M.; funding acquisition, B.G., D.C., E.M., H.F.-S., L.J.B., P.V., and T.K. All authors have read and approved the manuscript.
Declaration of interests
Mayo Clinic and E.M. have a financial interest related to this research. This research has been reviewed by the Mayo Clinic Conflict of Interest Review Board and is being conducted in compliance with Mayo Clinic Conflict of Interest policies. E.M. has the following patents planned, issued, or pending: application title, “Methods and Materials for Treating Glycosylation Disorders”; application # 16/973,210; filing date, 12/08/2020; Mayo Case # 2018-132.
Inclusion and diversity
We worked to ensure sex balance in the selection of non-human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure diversity in experimental samples through the selection of the cell lines. One or more authors of this paper self-identifies as a member of the LGBTQIA+ community. One or more of the authors of this paper self-identifies as living with disability. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Published: May 30, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101056.
Contributor Information
Silvia Radenkovic, Email: radenkovic.silvia@mayo.edu.
Eva Morava, Email: morava-kozicz.eva@mayo.edu.
Supplemental information
Data and code availability
-
•
Deidentified metabolomics data have been deposited at National Metabolomics Data Repository (NMDR) Metabolomics workbench, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This study does not report a novel code.
-
•
Any additional information required to reanalyze the data reported in this paper such as values and complete statistical analysis used to create graphs in this paper as well as original western blot images are available from the lead contact upon request.
References
- 1.Varki A., Cummings R.D., Esko J.D. Cold Spring Harbor; 2015. Essentials of Glycobiology; pp. 2015–2017. [DOI] [PubMed] [Google Scholar]
- 2.Ferreira C.R., Rahman S., Keller M., Zschocke J., ICIMD Advisory Group An international classification of inherited metabolic disorders (ICIMD) J. Inherit. Metab. Dis. 2021;44:164–177. doi: 10.1002/jimd.12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freeze H.H., Jaeken J., Matthijs G. CDG or not CDG. J. Inherit. Metab. Dis. 2022;45:383–385. doi: 10.1002/jimd.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyer S.W., Johnsen C., Morava E. Nutrition interventions in congenital disorders of glycosylation. Trends Mol. Med. 2022;28:463–481. doi: 10.1016/j.molmed.2022.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pajusalu S., Vals M.A., Mihkla L., Šamarina U., Kahre T., Õunap K. The estimated prevalence of N-linked congenital disorders of glycosylation across various populations based on allele frequencies in general population databases. Front. Genet. 2021;12:719437. doi: 10.3389/fgene.2021.719437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altassan R., Péanne R., Jaeken J., Barone R., Bidet M., Borgel D., Brasil S., Cassiman D., Cechova A., Coman D., et al. International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: diagnosis, treatment and follow up. J. Inherit. Metab. Dis. 2019;42:5–28. doi: 10.1002/jimd.12024. [DOI] [PubMed] [Google Scholar]
- 7.Schaftingen E.V., Jaeken J. 1995. Phosphomannomutase Deficiency Is a Cause of Carbohydrate-Deficient Glycoprotein Syndrome Type I. [DOI] [PubMed] [Google Scholar]
- 8.Körner C., Lehle L., Von Figura K. 1998. Abnormal synthesis of mannose 1-phosphate derived carbohydrates in carbohydrate-deficient glycoprotein syndrome type I fibroblasts with phosphomannomutase deficiency. [DOI] [PubMed] [Google Scholar]
- 9.Freeze H.H. Towards a therapy for phosphomannomutase 2 deficiency, the defect in CDG-Ia patients. Biochim. Biophys. Acta. 2009;1792:835–840. doi: 10.1016/j.bbadis.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyer S., Sam F.S., DiPrimio N., Preston G., Verheijen J., Murthy K., Parton Z., Tsang H., Lao J., Morava E., Perlstein E.O. Repurposing the aldose reductase inhibitor and diabetic neuropathy drug epalrestat for the congenital disorder of glycosylation PMM2-CDG. Dis. Model. Mech. 2019;12 doi: 10.1242/dmm.040584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma S.R., Sharma N. Epalrestat, an aldose reductase inhibitor, in diabetic neuropathy: An Indian perspective. Ann. Indian. Acad. Neurol. 2008;11:231–235. doi: 10.4103/0972-2327.44558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Q.R., Wang Z., Zhou W., Fan S.R., Ma R., Xue L., Yang L., Li Y.S., Tan H.L., Shao Q.H., Yang H.Y. Epalrestat protects against diabetic peripheral neuropathy by alleviating oxidative stress and inhibiting polyol pathway. Neural Regen. Res. 2016;11:345–351. doi: 10.4103/1673-5374.177745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramirez M.A., Borja N.L. Epalrestat: an aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy. 2008;28:646–655. doi: 10.1592/phco.28.5.646. [DOI] [PubMed] [Google Scholar]
- 14.Dunlop M. Direct effects of high glucose aldose reductase and the role of the polyol pathway in diabetic nephropathy. Kidney Int. Suppl. 2000;77:S3–S12. doi: 10.1046/j.1523-1755.2000.07702.x. [DOI] [PubMed] [Google Scholar]
- 15.Heaf D.J., Galton D.J. Diabetes Mellitus. 1975. Sorbitol and other polyols in lens, adipose tissue and urine. [DOI] [PubMed] [Google Scholar]
- 16.Lee A.Y.W., Chung S.K., Chung S.S.M., Kan Y.W. ). Demonstration that polyol accumulation is responsible for diabetic cataract by the use of transgenic mice expressing the aldose reductase gene in the lens. Proc. Natl. Acad. Sci. USA. 1995;92:2780–2784. doi: 10.1073/pnas.92.7.2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorenzi M. The polyol pathway as a mechanism for diabetic retinopathy: attractive, elusive, and resilient. Exp. Diabetes Res. 2007;2007:61038. doi: 10.1155/2007/61038. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.VAN HEYNINGEN R. Formation of polyols by the lens of the rat with ‘sugar’ cataract. Nature. 1959;184:194–195. doi: 10.1038/184194b0. [DOI] [Google Scholar]
- 19.Ligezka A.N., Radenkovic S., Saraswat M., Garapati K., Ranatunga W., Krzysciak W., Yanaihara H., Preston G., Brucker W., McGovern R.M., et al. Sorbitol is a severity biomarker for PMM2-CDG with therapeutic implications. Ann. Neurol. 2021;90:887–900. doi: 10.1002/ana.26245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cortese A., Zhu Y., Rebelo A.P., Negri S., Courel S., Abreu L., Bacon C.J., Bai Y., Bis-Brewer D.M., Bugiardini E., et al. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat. Genet. 2020;52:473–481. doi: 10.1038/s41588-020-0615-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dyck P.J., Zimmerman B.R., Vilen T.H., Minnerath S.R., Karnes J.L., Yao J.K., Poduslo J.F. Nerve glucose, fructose, sorbitol, myo -inositol, and fiber degeneration and regeneration in diabetic neuropathy. N. Engl. J. Med. 1988;319:542–548. doi: 10.1056/NEJM198809013190904. [DOI] [PubMed] [Google Scholar]
- 22.Bril V., Ono Y., Buchanan R.A. Sural nerve sorbitol in patients with diabetic sensorimotor polyneuropathy. Diabetes Care. 2004;27:1160–1163. doi: 10.2337/diacare.27.5.1160. [DOI] [PubMed] [Google Scholar]
- 23.Klaver E.J., Dukes-Rimsky L., Kumar B., Xia Z.J., Dang T., Lehrman M.A., Angel P., Drake R.R., Freeze H.H., Steet R., Flanagan-Steet H. Protease-dependent defects in N-cadherin processing drive PMM2-CDG pathogenesis. JCI Insight. 2021;6 doi: 10.1172/jci.insight.153474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson R.J., Segal M.S., Sautin Y., Nakagawa T., Feig D.I., Kang D.-H., Gersch M.S., Benner S., Sánchez-Lozada L.G. Potential Role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr. 2007;86:899–906. doi: 10.1093/ajcn/86.4.899. [DOI] [PubMed] [Google Scholar]
- 25.Douard V., Ferraris R.P. The role of fructose transporters in diseases linked to excessive fructose intake. J. Physiol. 2013;591:401–414. doi: 10.1113/jphysiol.2011.215731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu L., Li T., Liao Y., Wang Y., Gao Y., Hu H., Huang H., Wu F., Chen Y.G., Xu S., Fu S. Triose kinase controls the lipogenic potential of fructose and dietary tolerance. Cell Metabol. 2020;32:605–618.e7. doi: 10.1016/j.cmet.2020.07.018. [DOI] [PubMed] [Google Scholar]
- 27.Agius L., Peak M. Intracellular binding of glucokinase in hepatocytes and translocation by glucose, fructose and insulin. Biochem. J. 1993;296:785–796. doi: 10.1042/bj2960785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laughlin M.R. Normal roles for dietary fructose in carbohydrate metabolism. Nutrients. 2014;6:3117–3129. doi: 10.3390/nu6083117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monticelli M., Liguori L., Allocca M., Andreotti G., Cubellis M.V. β-Glucose-1,6-Bisphosphate stabilizes pathological phophomannomutase2 mutants in vitro and represents a lead compound to develop pharmacological chaperones for the most common disorder of glycosylation, PMM2-CDG. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20174164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Citro V., Cimmaruta C., Liguori L., Viscido G., Cubellis M.V., Andreotti G. A mutant of phosphomannomutase1 retains full enzymatic activity, but is not activated by IMP: possible implications for the disease PMM2-CDG. PLoS One. 2017;12:e0189629. doi: 10.1371/journal.pone.0189629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andreotti G., Monti M.C., Citro V., Cubellis M.V. Heterodimerization of two pathological mutants enhances the activity of human phosphomannomutase2. PLoS One. 2015;10:e0139882. doi: 10.1371/journal.pone.0139882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radenkovic S., Bird M.J., Emmerzaal T.L., Wong S.Y., Felgueira C., Stiers K.M., Sabbagh L., Himmelreich N., Poschet G., Windmolders P., et al. The metabolic map into the pathomechanism and treatment of PGM1-CDG. Am. J. Hum. Genet. 2019;104:835–846. doi: 10.1016/j.ajhg.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koch J., Mayr J.A., Alhaddad B., Rauscher C., Bierau J., Kovacs-Nagy R., Coene K.L.M., Bader I., Holzhacker M., Prokisch H., et al. CAD mutations and uridine-responsive epileptic encephalopathy. Brain. 2017;140:279–286. doi: 10.1093/brain/aww300. [DOI] [PubMed] [Google Scholar]
- 34.Nolting K., Park J.H., Tegtmeyer L.C., Zühlsdorf A., Grüneberg M., Rust S., Reunert J., Du Chesne I., Debus V., Schulze-Bahr E., et al. Limitations of galactose therapy in phosphoglucomutase 1 deficiency. Mol. Genet. Metab. Rep. 2017;13:33–40. doi: 10.1016/j.ymgmr.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lane A.N., Fan T.W.M. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015;43:2466–2485. doi: 10.1093/nar/gkv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y., Chen X. Sialic acid metabolism and sialyltransferases: natural functions and applications. Appl. Microbiol. Biotechnol. 2012;94:887–905. doi: 10.1007/s00253-012-4040-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moseley H.N.B., Lane A.N., Belshoff A.C., Higashi R.M., Fan T.W.M. A novel deconvolution method for modeling UDP-N-acetyl-D-glucosamine biosynthetic pathways based on 13C mass isotopologue profiles under non-steady-state conditions. BMC Biol. 2011;9:37. doi: 10.1186/1741-7007-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verdegem D., Moseley H.N.B., Vermaelen W., Sanchez A.A., Ghesquière B. MAIMS: a software tool for sensitive metabolic tracer analysis through the deconvolution of 13C mass isotopologue profiles of large composite metabolites. Metabolomics. 2017;13 doi: 10.1007/s11306-017-1250-7. [DOI] [Google Scholar]
- 39.Schmahmann J.D., Gardner R., MacMore J., Vangel M.G. Development of a brief ataxia rating scale (BARS) based on a modified form of the ICARS. Mov. Disord. 2009;24:1820–1828. doi: 10.1002/mds.22681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Achouitar S., Mohamed M., Gardeitchik T., Wortmann S.B., Sykut-Cegielska J., Ensenauer R., De Baulny H.O., Õunap K., Martinelli D., De Vries M., et al. Nijmegen paediatric CDG rating scale: a novel tool to assess disease progression. J. Inherit. Metab. Dis. 2011;34:923–927. doi: 10.1007/s10545-011-9325-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaasterland C.M.W., Van Der Weide M.C.J., Roes K.C.B., Van Der Lee J.H. Goal attainment scaling as an outcome measure in rare disease trials: a conceptual proposal for validation. BMC Med. Res. Methodol. 2019;19:227. doi: 10.1186/s12874-019-0866-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hers H.G. 1956. I. ∗ Associ∼ du FNRS Effects of irradiation in v/vo on bacterial deoxyribonucleic acid∗. [Google Scholar]
- 43.Singh M., Kapoor A., Bhatnagar A. Physiological and pathological roles of aldose reductase. Metabolites. 2021;11 doi: 10.3390/metabo11100655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He J., Gao H.X., Yang N., Zhu X.D., Sun R.B., Xie Y., Zeng C.H., Zhang J.W., Wang J.K., Ding F., et al. The aldose reductase inhibitor epalrestat exerts nephritic protection on diabetic nephropathy in db/db mice through metabolic modulation. Acta Pharmacol. Sin. 2019;40:86–97. doi: 10.1038/s41401-018-0043-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chalk C., Benstead T.J., Moore F. Aldose reductase inhibitors for the treatment of diabetic polyneuropathy. Cochrane Database Syst. Rev. 2007;2007:CD004572. doi: 10.1002/14651858.CD004572.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hotta N., Akanuma Y., Kawamori R., Matsuoka K., Oka Y., Shichiri M., Toyota T., Nakashima M., Yoshimura I., Sakamoto N., Shigeta Y. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: the 3-year, multicenter, comparative aldose reductase inhibitor-diabetes complications trial. Diabetes Care. 2006;29:1538–1544. doi: 10.2337/dc05-2370. [DOI] [PubMed] [Google Scholar]
- 47.Brakedal B., Dölle C., Riemer F., Ma Y., Nido G.S., Skeie G.O., Craven A.R., Schwarzlmüller T., Brekke N., Diab J., et al. The NADPARK study: a randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metabol. 2022;34:396–407.e6. doi: 10.1016/j.cmet.2022.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Pirinen E., Auranen M., Khan N.A., Brilhante V., Urho N., Pessia A., Hakkarainen A., Kuula J., Heinonen U., Schmidt M.S., et al. Niacin cures systemic NAD+ deficiency and improves muscle performance in adult-onset mitochondrial myopathy. Cell Metabol. 2020;31:1078–1090.e5. doi: 10.1016/j.cmet.2020.04.008. [DOI] [PubMed] [Google Scholar]
- 49.Tammali R., Ramana K.V., Srivastava S.K. Aldose reductase regulates TNF-α-induced PGE2 production in human colon cancer cells. Cancer Lett. 2008;252:299–306. doi: 10.1016/j.canlet.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tammali R., Reddy A.B.M., Ramana K.V., Petrash J.M., Srivastava S.K. Aldose reductase deficiency in mice prevents azoxymethane-induced colonic preneoplastic aberrant crypt foci formation. Carcinogenesis. 2009;30:799–807. doi: 10.1093/carcin/bgn246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tammali R., Ramana K.V., Singhal S.S., Awasthi S., Srivastava S.K. Aldose reductase regulates growth factor-induced cyclooxygenase-2 expression and prostaglandin E2 production in human colon cancer cells. Cancer Res. 2006;66:9705–9713. doi: 10.1158/0008-5472.CAN-06-2105. [DOI] [PubMed] [Google Scholar]
- 52.Zhang K.-R., Zhang Y.-F., Lei H.-M., Tang Y.-B., Ma C.-S., Lv Q.-M., Wang S.-Y., Lu L.-M., Shen Y., Chen H.-Z., et al. 2021. Targeting AKR1B1 inhibits glutathione de novo synthesis to overcome acquired resistance to EGFR-targeted therapy in lung cancer. [DOI] [PubMed] [Google Scholar]
- 53.Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. Stages of embryonic development of the zebrafish. Dev. Dynam. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 54.Pettinato F., Mostile G., Battini R., Martinelli D., Madeo A., Biamino E., Frattini D., Garozzo D., Gasperini S., Parini R., et al. Clinical and radiological correlates of activities of daily living in cerebellar atrophy caused by PMM2 mutations (PMM2-CDG) Cerebellum. 2021;20:596–605. doi: 10.1007/s12311-021-01242-x/Published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Radenkovic S., Fitzpatrick-Schmidt T., Byeon S.K., Madugundu A.K., Saraswat M., Lichty A., Wong S.Y.W., McGee S., Kubiak K., Ligezka A., et al. Expanding the clinical and metabolic phenotype of DPM2 deficient congenital disorders of glycosylation. Mol. Genet. Metabol. 2021;132:27–37. doi: 10.1016/j.ymgme.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pfaffl M.W. 2001. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu S.H., Wang T., Wiggins K., Louie R.J., Merino E.F., Skinner C., Cassera M.B., Meagher K., Goldberg P., Rismanchi N., et al. Lysosomal cholesterol accumulation contributes to the movement phenotypes associated with NUS1 haploinsufficiency. Genet. Med. 2021;23:1305–1314. doi: 10.1038/s41436-021-01137-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hummel J., Strehmel N., Selbig J., Walther D., Kopka J. Decision tree supported substructure prediction of metabolites from GC-MS profiles. Metabolomics. 2010;6:322–333. doi: 10.1007/s11306-010-0198-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pang Z., Zhou G., Ewald J., Chang L., Hacariz O., Basu N., Xia J. Using MetaboAnalyst 5.0 for LC–HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat. Protoc. 2022;17:1735–1761. doi: 10.1038/s41596-022-00710-w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Deidentified metabolomics data have been deposited at National Metabolomics Data Repository (NMDR) Metabolomics workbench, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This study does not report a novel code.
-
•
Any additional information required to reanalyze the data reported in this paper such as values and complete statistical analysis used to create graphs in this paper as well as original western blot images are available from the lead contact upon request.