

Summary
Primary liver cancer is a rising cause of cancer deaths in the US. Although immunotherapy with immune checkpoint inhibitors induces a potent response in a subset of patients, response rates vary among individuals. Predicting which patients will respond to immune checkpoint inhibitors is of great interest in the field. In a retrospective arm of the National Cancer Institute Cancers of the Liver: Accelerating Research of Immunotherapy by a Transdisciplinary Network (NCI-CLARITY) study, we use archived formalin-fixed, paraffin-embedded samples to profile the transcriptome and genomic alterations among 86 hepatocellular carcinoma and cholangiocarcinoma patients prior to and following immune checkpoint inhibitor treatment. Using supervised and unsupervised approaches, we identify stable molecular subtypes linked to overall survival and distinguished by two axes of aggressive tumor biology and microenvironmental features. Moreover, molecular responses to immune checkpoint inhibitor treatment differ between subtypes. Thus, patients with heterogeneous liver cancer may be stratified by molecular status indicative of treatment response to immune checkpoint inhibitors.
Keywords: cholangiocarcinoma, hepatocellular carcinoma, liver cancer, immunotherapy
Graphical abstract
Highlights
-
•
Molecular assessment of immune treatment response is feasible with FFPE specimens
-
•
Baseline tumors form stable gene clusters linked to survival and treatment response
-
•
Baseline clusters exhibit transcriptomic, mutational, and immune response differences
-
•
Few transcriptome changes occur in tumors after immunotherapy
Budhu et al. identify molecular indicators of the immunotherapy response of liver cancer patients, paramount for improved clinical management. Integrated transcriptome and genome assessment of FFPE specimens prior to and following immunotherapy among the NCI-CLARITY retrospective cohort shows stable molecular subtypes linked to survival, immune alterations, and treatment response.
Introduction
Primary liver cancers (PLCs), encompassing hepatocellular carcinoma (HCC), and biliary tract cancers (BTCs), including intra- and extrahepatic cholangiocarcinoma (iCCA and eCCA, respectively), are leading causes of cancer mortality worldwide with rising incidence in the US.1,2,3 While early-stage liver cancer may be curable by interventional options, including resection, ablation, or transplantation, most patients present with advanced disease, poor prognosis,4 and limited treatment options.
More recently, immunotherapy-based treatments, including immune checkpoint inhibitors (ICIs), have revolutionized PLC treatment. Positive results from large randomized phase III trials have provided three first-line ICI treatment options for advanced HCC patients (atezolizumab plus bevacizumab,5 tremelimumab plus durvalumab, and durvalumab monotherapy, found to be noninferior to sorafenib6) as well as gemcitabine/cisplatin plus durvalumab as the standard of care first-line therapy in advanced CCA.7 However, only 13%–30% of patients achieve an objective response rate to these immunotherapy regimens with suboptimal median survival.5,8,9 Therefore, identification of predicative biomarkers of treatment response and outcome is urgently needed to select patients who are likely to respond. However, because tumor biopsy collection during trials is lacking, and a limited number of patients undergo such regimens at individual clinical sites, our understanding of the molecular underpinnings of immunotherapy response in PLC patients remains poor. Additionally, most genomics studies have utilized frozen tumor tissue and, in some cases, adjacent non-tumor tissue as the starting material to interrogate tumor and microenvironmental molecular alterations;4,10,11,12,13 however, the majority of available specimens used for clinical diagnosis are formalin-fixed, paraffin-embedded (FFPE) tissue. Interrogating FFPE tissue may provide a more suitable route to efficiently identify relevant molecular alterations through establishment of routine diagnostic tests.
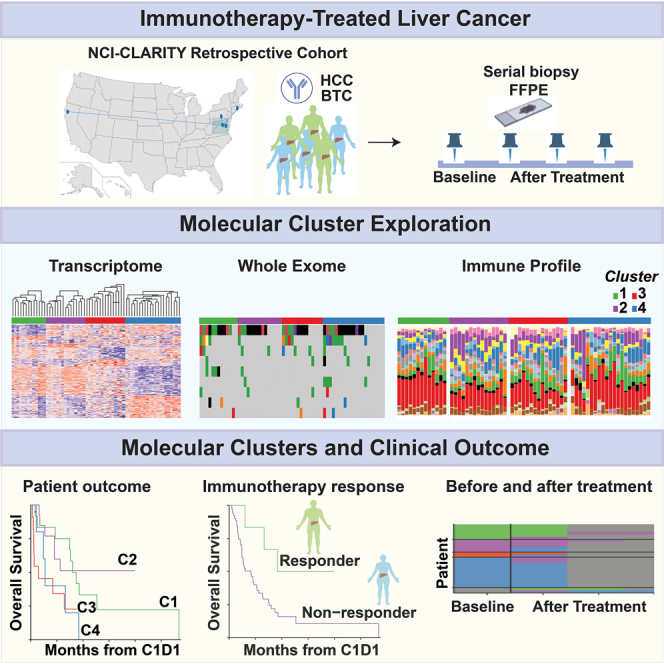
In this vein, we initiated the NCI-CLARITY study (National Cancer Institute Cancers of the Liver: Accelerating Research of Immunotherapy by a Transdisciplinary Network), a multisite prospective study of PLC clinical trial data and accompanying biospecimens, focused on exploring the underlying mechanisms that determine immunotherapy responses. Here, we report on a parallel retrospective arm of NCI-CLARITY entailing molecular classification of 230 primary HCC and BTC tumors (mainly iCCA) and adjacent non-tumor tissues, largely stored as FFPE attained before and following ICI treatment, using total RNA sequencing (RNA-seq) and whole-exome sequencing (WES) (Figure 1A). In this study, we established an FFPE-based workflow for comprehensive transcriptome and whole exome profiling of PLC to identify molecular features associated with ICI response and outcome. We identified four stable survival-related molecular subgroups defined by orthogonal axes of aggressive tumor biology and immune infiltration, the latter associated with ICI response. Furthermore, although the underlying molecular status of tumors prior to ICI treatment was largely maintained following treatment, some molecular responses tracked with patient outcome. Thus, outcome-related and intersecting molecular axes are apparent in FFPE liver tumors prior to ICI treatment, components of which could serve as indicators of treatment response.
Figure 1.

A baseline gene expression signature segregates primary liver tumors into high- and low-survival groups
(A) Overview of the patient cohort, available samples, and analyses. Subset numbers indicate those that pass RNA-seq quality control. PLC, primary liver cancer; HCC, hepatocellular carcinoma; BTC, biliary tract cancer.
(B) Kaplan-Meier curve of OS probability by survival signature-predicted risk group, all baseline tumors (low risk, n = 32 tumors; high risk n = 32 tumors).
(C) Kaplan-Meier curve of OS probability by survival signature-predicted risk group, HCC tumors (low risk, n = 30 tumors; high risk n = 11 tumors).
(B and C) The log rank p value is shown.
(D) Pathways enriched among signature genes, ordered by FDR-corrected p value. Genes were split by whether they were protective or not, and enriched gene sets are divided into MSigDB Hallmark (Hall) and c8 liver cell (LC) types.
(E) Kaplan-Meier curve of OS probability by risk group predicted for TCGA LIHC and CHOL primary tumors using the survival signature (low risk, n = 328 tumors; high risk, n = 79 tumors). The log rank p value is shown.
Results
Samples
We analyzed tumor biopsy specimens from 86 patients (56 HCC and 30 BTC) who received ICI treatment at five US clinical sites in a combined NCI-CLARITY retrospective cohort (Figure 1A). The broad diversity in treatment protocol, histological diagnosis, and prior treatment as well as etiology allowed us to comprehensively profile the variation present in this cohort (Table 1). Among these patients, 74 had a baseline tumor specimen prior to immunotherapy treatment (50 HCC and 24 BTC), 37 had a tumor specimen following immunotherapy treatment (17 HCC and 20 BTC), and 27 had an available specimen at both time points (13 HCC and 14 BTC). In addition, four HCC patients had serial tumors with up to five time points following immunotherapy treatment. RNA-seq and WES were performed on tumor and adjacent non-tumor tissue.
Table 1.
Patient characteristics
| HCC | BTC | Total | |
|---|---|---|---|
| Number of patients | 56 | 30 | 86 |
| Site | |||
| NIH Clinical Center | 22 (39%) | 23 (77%) | 45 (52%) |
| Georgetown University | 18 (32%) | 1 (3%) | 19 (22%) |
| Massachusetts General Hospital | 7 (13%) | 4 (13%) | 11 (13%) |
| University of California San Francisco | 7 (13%) | 2 (7%) | 9 (10%) |
| Thomas Jefferson University | 2 (4%) | 0 | 2 (2%) |
| Age | |||
| Age at diagnosis (median) | 35–84 (63) | 20–79 (63) | 20–84 (63) |
| Unknown | 8 (14%) | 4 (13%) | 12 (14%) |
| Sex | |||
| Male | 45 (80%) | 18 (60%) | 63 (73%) |
| Female | 11 (20%) | 12 (40%) | 23 (27%) |
| Race | |||
| White | 26 (46%) | 29 (97%) | 55 (64%) |
| Black or African American | 18 (32%) | 0 | 18 (21%) |
| Asian | 7 (13%) | 1 (3%) | 8 (9%) |
| Native Hawaiian or other Pacific Islander | 1 (2%) | 0 | 1 (1%) |
| Other | 2 (4%) | 0 | 2 (2%) |
| Unknown | 2 (4%) | 0 | 2 (2%) |
| Ethnicity | |||
| Hispanic or Latino | 3 (5%) | 1 (3%) | 4 (5%) |
| Not Hispanic or Latino | 53 (96%) | 28 (93%) | 81 (94%) |
| Unknown | 0 | 1 (3%) | 1 (1%) |
| Hepatitis B virus (HBV) status | |||
| No | 43 (77%) | 27 (90%) | 70 (81%) |
| Yes | 13 (23%) | 3 (10%) | 16 (19%) |
| Hepatitis C virus (HCV) status | |||
| No | 28 (50%) | 26 (87%) | 54 (63%) |
| Yes | 27 (48%) | 4 (13%) | 31 (36%) |
| Unknown | 1 (2%) | 0 | 1 (1%) |
| Steatohepatitis | |||
| No | 42 (75%) | 27 (90%) | 69 (80%) |
| Yes | 8 (14%) | 3 (10%) | 11 (13%) |
| Unknown | 6 (11%) | 0 | 6 (7%) |
| Alcohol abuse | |||
| No | 33 (59%) | 28 (93%) | 61 (71%) |
| Yes | 23 (41%) | 2 (7%) | 25 (29%) |
| Cirrhosis | |||
| No | 24 (43%) | 28 (93%) | 52 (60%) |
| Yes | 32 (57%) | 2 (7%) | 34 (40%) |
| Prior treatment | |||
| No | 16 (29%) | 1 (3%) | 17 (20%) |
| Yes | 32 (57%) | 27 (90%) | 59 (69%) |
| Unknown | 8 (14%) | 2 (7%) | 10 (12%) |
| Best RECIST v.1.1 response | |||
| CR | 1 (2%) | 0 | 1 (1%) |
| PR | 6 (11%) | 3 (10%) | 9 (10%) |
| SD | 25 (45%) | 12 (40%) | 37 (43%) |
| PD | 18 (32%) | 12 (40%) | 30 (35%) |
| Non-evaluable | 3 (5%) | 1 (3%) | 4 (6%) |
| Unknown | 3 (5%) | 2 (7%) | 5 (6%) |
| Immunotherapy category | |||
| Single-agent ICI | 15 (27%) | 9 (30%) | 24 (28%) |
| Single-agent ICI with VEGF inhibitor | 13 (23%) | 0 | 13 (15%) |
| Single-agent ICI with targeted therapy | 3 (5%) | 3 (10%) | 6 (7%) |
| Combination ICI | 24 (43%) | 18 (60%) | 42 (49%) |
| Combination ICI with targeted therapy | 1 (2%) | 0 | 1 (1%) |
BTC, biliary tract cancer; HCC, hepatocellular carcinoma; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; ICI, immune checkpoint inhibitor; TKI, tyrosine kinase inhibitor.
Feasibility of molecular profiling using archival tissue
Genomic data studies on solid tumors have largely been generated using frozen tissue; however, most clinical tissue specimens are typically available as FFPE specimens. Although the fixation process introduces artifacts, including DNA fragmentation and base deamination,14 and FFPE samples often yield limited nucleic acid, reducing library complexity,15 use of archival FFPE tissue for comprehensive genomic analyses could greatly expand the pool of tumors available for analysis and be the premise for establishment of routine diagnostic tests.
To determine whether FFPE tissues could be used for transcriptomics analysis, we extensively analyzed biological and technical sources of variation among specimens. First, to determine the impact of technical variation on observed transcriptomic differences, we used variance partitioning to estimate the variation in expression explained by biological and technical variables for each gene across 201 primary liver tumors and adjacent non-tumor specimens (Figure S1A). We found that biological variation, such as patient of origin, histological diagnosis, or sample malignancy, outweighed technical variation among the most variable genes. We then quantified the impact of the amount of input RNA used in library creation on transcriptomic variation using four high-quality liver transplant samples from which multiple libraries were prepared using 5 ng or 100 ng of RNA from the same extraction. The number of expressed genes was higher for the 100-ng replicates than for the 5-ng replicates, as was expression correlation between replicates (Wilcoxon test, p = 0.03; Figure S1B); however, the samples clustered by patient regardless of input RNA amount (Figure S1C), confirming that true biological variation could be captured with low-input libraries.
We next investigated the impact of sample storage method, which may introduce systematic artifacts that could be conflated with true variation, using two sample sets where the same tumor was stored in multiple media. In the first set, six tumors were each stored in FFPE, RNALater (2 replicates), and OCT blocks (2 replicates) for a total of five replicates per tumor. Among these tumors, expression correlation was slightly higher between replicates with the same storage method (Wilcoxon test, p = 0.03; same storage comparisons n = 10, median correlation = 0.77; different storage comparisons n = 38, median correlation = 0.70). However, as before, samples still clustered by patient (Figure S1D), whereby expression variation because of patient of origin was far stronger than variation by storage method (Figure S1E). Thus, there was relatively little influence of storage method on expression variation. The second set consisted of 54 pairs of FFPE and frozen samples from the same tumor and time point. There was no significant difference in expression correlation between these storage pairs and FFPE sections from the same block (Wilcoxon test, p = 0.10) or tumors from a different time point from the same patient stored using the same method (Wilcoxon p = 0.87; Figure S1F). The correlation between the storage pairs decreased with de-duplicated read depth, confirming that concordance is greater with greater library complexity (Figure S1G). However, several pathways were enriched among the differentially expressed genes (DEGs) between FFPE and frozen pairs (Figure S1H), some of which have been observed previously.16 Together, these analyses confirmed the feasibility of using FFPE samples for transcriptomics analysis, provided that sufficient input RNA was available to generate complex libraries.
A gene signature predictive of survival on ICI therapy
The ability to predict response to ICIs prior to treatment would greatly benefit patients. Hence, we determined whether the molecular state of baseline liver tumors was predictive of patient response to treatment, considering overall survival (OS) following ICIs as a proxy. Baseline tumors of HCC and BTC were assessed together for the purpose of classifying tumors based on unified molecular features rather than histological subtypes.17 Survival risk prediction was employed to divide baseline tumors (n = 64) into high- and low-survival risk groups based on gene expression levels (Table S1) and resulted in predicted survival risk groups with significantly different OS (log rank test, p < 0.0001; Figure 1B). Although BTC had worse OS than HCC overall (by-diagnosis log rank test, p = 0.001), 11 of 41 HCC tumors were predicted to be high risk, and 2 of 23 BTC tumors were predicted to be low risk. To rule out confounding by histological diagnosis, we confirmed that risk group survival differences were still significant among HCC alone (log rank test, p < 0.001; BTC log rank test, p = 0.88 [data not shown]; Figure 1C). Furthermore, survival modeling on HCC tumors alone using the same input genes as the original model resulted in risk groups with significantly different OS (principal component [PC1-2], log rank test, p < 0.0001; 10-fold cross validation [10CV] permutation, p < 0.01) with similar gene weights as the original models (Pearson r = 0.85, p < 0.0001) (data not shown). Therefore, although the survival signature was trained on HCC and BTC, it reflected molecular states linked to survival in HCC alone. Gene set analysis on the signature genes showed that hepatocyte- and liver-function related gene sets were associated with good survival, while EpCAM+ bile duct gene sets were associated with poor survival, suggesting that the survival signature primarily distinguished less aggressive HCC from more aggressive HCC and BTC (Figure 1D). The survival signature was then used to predict OS in independent validation sets (namely, The Cancer Genome Atlas [TCGA] liver hepatocellular carcinoma [LIHC] and cholangiocarcinoma [CHOL] cohorts), which were not treated with ICIs (Figure 1E). The predicted risk groups had significantly different OS (log rank test, p = 0.02), although a far smaller proportion of HCC was assigned to the high-risk group. Taken together, a survival signature is apparent among patients treated with ICIs, potentially encoding underlying differences in prognosis in addition to response to ICIs.
Unsupervised molecular clusters
To provide greater resolution on baseline tumor molecular states, we next performed unsupervised consensus clustering on gene expression profiles. We identified four stable molecular clusters (Figures 2A and S2A–S2C), which largely corresponded to the survival risk groups (Figure S2D). Consensus clustering, removing extrahepatic CCA cases, yielded the exact same molecular clusters (Figure S2E). The molecular clusters also exhibited differences in OS, with cluster 1 (C1) and C2 having good survival and C3 and C4 having poor survival (Figure 2B). Survival differences by cluster were still evident when restricted to HCC (Figure 2C), but not in BTC alone, perhaps because of the small number of samples in C1 and C2 (Figure S2F). Interestingly, C4 exhibited progression-free survival (PFS) similar to C1 and C2, particularly when only HCC was considered (Figures 2D, S2G, and S2H), indicating that the diseases are potentially and temporarily controlled with ICIs. Thus, there were natural molecular divisions in baseline tumors related to patient outcome to ICI treatment.
Figure 2.

Baseline tumors form molecular clusters with differences in OS that correspond to survival risk groups
(A) Semi-supervised hierarchical clustering of baseline tumors on cluster-specific DEGs. Tumors were clustered using Euclidean distance with complete linkage within each cluster (C1, n = 13 tumors; C2, n = 15; C3, n = 15; C4, n = 21). Heatmap displays expression Z scores by row.
(B) Kaplan-Meier curve for OS probability since C1D1 of immunotherapy by molecular cluster, all baseline tumors (same numbers as in A).
(C) Kaplan-Meier curve for OS probability since C1D1 of immunotherapy by molecular cluster, HCC tumors only (C1, n = 12 tumors; C2, n = 13; C3, n = 11; C4, n = 5).
(D) Kaplan-Meier curve for PFS probability since C1D1 of immunotherapy by molecular cluster, HCC tumors (same numbers as in C).
(B–D) The overall log rank test p value is shown. The embedded table presents pairwise log rank test p values with Benjamini-Hochberg correction.
We then determined whether the molecular clusters were associated with clinical variables. The clusters exhibited some stratification by histological diagnosis, with C1–C3 primarily composed of HCC and C4 primarily of BTC (chi-square test, false discovery rate [FDR]-adjusted p = 0.008; Figure S3A), and the majority of the C4-specific DEGs were also differentially expressed between baseline HCC and BTC tumors (Figure S3B; Table S2). However, detailed analyses of cluster assignments and gene expression levels supported the joint clustering of HCC and BTC (Figures S3C–S3E). While a few clinical and technical variables distinguished the molecular clusters, such as enrichment of Child-Pugh score B in HCC samples in C4 (chi-square test, FDR-adjusted p = 0.045; Figures S3F-S3I; STAR Methods), there was no association with other demographic, etiological, or pathological features (Tables S3 and S4). Thus, molecular states could be shared by tumors with different histological diagnoses and were not confounded by other clinical variables or technical artifacts.
Next, we compared the identified molecular clusters with previously described subclasses18,19,20 and found a strong association (chi-square test with simulated p value, p < 0.05) between subclasses (Figure S4A). In particular, good-survival C1 and C2 were enriched in Hoshida subclass S3, representing differentiated tumors with high hepatocyte function, good survival, and enrichment of β-catenin mutations. They were also enriched for TCGA LIHC subclass iC2, representing low-grade tumors. In contrast, the poor-survival C3 and C4 were enriched for TCGA subclass iC1, characterized by high tumor grade, lower differentiation, proliferation, and poor survival, and C3 was enriched for the Hoshida S2 subclass, also characterized by proliferation. C4, which contained advanced HCC tumors and BTC, was also enriched for the Hoshida S1 subclass, characterized by enrichment of WNT signaling, vascular invasion, and epithelial-mesenchymal transition as well as the Yamashita EpCAM+ stemness signature. These results supported an axis of aggressive tumor biology or differentiation as the primary driver of the observed survival differences between molecular clusters.
RECIST response
We then explored Response Evaluation Criteria in Solid Tumors (RECIST) response in relation to the molecular clusters because RECIST is used extensively to provide a direct readout of response to immunotherapy treatment. We defined responders as patients with a complete response (CR) or partial response (PR) and non-responders as patients with stable disease (SD) or progressive disease (PD). Although the response rate was not significantly different across the molecular clusters overall and in HCC or BTC alone (chi-square test, p > 0.05; Figures 3A and S4B), all of the 6 responders were in good survival C2 and poor survival C4 (2 HCC and 2 HCC/2 BTC, respectively). There was no significant difference in response rate by diagnosis (chi-square test, p = 1). As expected, responders had better OS than non-responders (Figures 3B and S4C). Although no DEG was significant after multiple hypothesis correction, DEGs with an uncorrected p < 0.01 that were upregulated in responders vs. non-responders were enriched in immune-related gene sets (Table S5; Figure 3C). This was confirmed using CIBERSORTx, which showed an enrichment of CD8 T cells in responders (Wilcoxon test, uncorrected p < 0.05; Figures 3D and S4D). Therefore, responders appeared to have greater immune infiltration at baseline relative to non-responders, where immediate response to ICI treatment may be orthogonal to OS differences.
Figure 3.

Responders exhibit immune infiltration differences compared with non-responders
(A) Response (RECIST v.1.1) on immunotherapy by molecular cluster (C1, n = 13 tumors; C2, n = 15; C3, n = 15; C4, n = 21). Gray represents non-evaluable and unknown patients.
(B) Kaplan-Meier curve for OS probability since C1D1 of immunotherapy by response, all baseline tumors (responder, n = 6 tumors; non-responder, n = 51). The overall log rank test p value is shown.
(C) Pathways enriched among DEGs in responders vs. non-responders, ordered by FDR-corrected p value. Enriched gene sets are divided into MSigDB Hallmark and c8 LC types.
(D) CIBERSORTx cell type CD8 T cells, enriched in responders vs. non-responders (Wilcoxon test, uncorrected p < 0.05; numbers as in B). Boxplots: center line, median; box limits, first and third quartiles; whiskers, values ≤ 1.5 times the interquartile range from box limits; points, outliers.
Tumor-intrinsic and stromal differences between clusters
Despite similar survival profiles, differences in RECIST response within the good- and poor-survival clusters suggested underlying differences in tumor biology. We next sought to further elucidate which molecular features distinguished the stable molecular clusters, including tumor-intrinsic and microenvironmental features. To provide an overview of the cellular composition of each cluster, we first performed unsupervised principal-component analysis (PCA) on enrichment scores for liver cell-specific signature gene sets (MSigDB c8 module,21 gene sets). The four clusters clearly separated along PC1 and PC2 (Figure 4A). PC2 separated the good- and poor-survival clusters, with the strongest contribution from the hepatocyte gene set (good survival C1 and C2) and EpCAM+ bile duct gene sets (poor survival C3 and C4), recapitulating the original survival signature. There was strong concordance in the loadings of similar gene sets despite limited overlap in their constituent genes (binary score: bile duct cells, 0.48–0.81; hepatocytes, 0.41–0.93). In contrast, PC1 separated C1 and C3 from C2 and C4, with stronger contributions of stromal cell types. The involvement of several liver sinusoidal endothelial cells (LSECs), micro-vascular endothelial cells (MVECs), and stellate cell gene sets, all related to increased vascularization, was strongest in the direction of C4, indicating higher tumor aggressiveness. In contrast, C2 was associated with greater immune infiltration, with stronger contributions from B cell, natural killer (NK)/NK T cell, and Kupffer cell gene sets. This was further established using CIBERSORTx, which showed that, although there was high variability within clusters (Figure S4E), plasma cells, CD8+ T cells, monocytes (Wilcoxon test, uncorrected p < 0.01), naive B cells, and memory B cells were enriched within C2 relative to all other samples (Figures 4B and S4F). In contrast, M2 macrophages were enriched in C4 and eosinophils in C3. Together, these data indicated that, although tumor-intrinsic features (namely, tumor aggressiveness or stemness) appeared to distinguish the good- from poor-survival clusters, there existed orthogonal differences in the tumor microenvironment that may shape response to treatment.
Figure 4.

Tumor-intrinsic and stromal features distinguish molecular clusters
(A) PCA for baseline tumors (n = 64 tumors) on MSigDB c8 LC gene set enrichment scores (n = 31 gene sets), colored by molecular cluster. Arrows present scaled loadings for gene sets along PC1 and PC2.
(B) CIBERSORTx LM22 cell types enriched in a molecular cluster (Wilcoxon test vs. all other samples, uncorrected p < 0.01; C1, n = 13 tumors; C2, n = 15; C3, n = 15; C4, n = 21). M2 Mphage, M2 macrophage.
(C and D) LC gene sets (C) and MSigDB Hallmark gene sets (D) (n = 50 gene sets) enriched in each cluster (facet; FDR-adjusted p < 0.05, Wilcoxon test vs. all other samples). Rows display the Z score of the median single-sample Gene Set Enrichment Analysis (ssGSEA) enrichment score by cluster (column; same numbers as in B).
(E) Immune checkpoint expression by molecular cluster (same numbers as in B).
(B and E) Boxplots: center line, median; box limits, first and third quartiles; whiskers, values ≤ 1.5 times the interquartile range from box limits; points, outliers.
To further investigate high-level processes enriched in each tumor cluster, we profiled the enrichment of MSigDB liver cell gene sets and Hallmark gene sets (Figures 4C and 4D). As expected, C1 and C2 were enriched for hepatocytes and gene sets reflective of normal liver function. In contrast, C3 had lower levels of hepatocytes despite being predominately HCC. C2 also showed enrichment in B cell-related gene sets relative to other clusters (Figure S4G). C1 had lower enrichment for EpCAM+ bile duct cell gene sets, especially in comparison with C4, and the expression of EpCAM was also far lower in C1 (data not shown). C4 was most enriched for numerous pathways related to the inflammatory response, angiogenesis, and signaling pathways, and despite their differences in survival, C2 had the next-highest level. Interestingly, C2 was depleted in cell-cycle-related gene sets relative to all other samples. Gene set enrichment analysis therefore confirmed two axes of tumor-intrinsic and microenvironment features that distinguish the molecular clusters.
Expression of immune checkpoints has been extensively investigated as a predictive biomarker of immunotherapy response. Hence, we profiled the RNA expression of the three immune checkpoints that are targets of treatments used in this cohort (Figure 4E). PDCD1 and CD274 were differentially expressed across the clusters (Kruskal-Wallis test, p < 0.05) but CTLA4 was not. In particular, PDCD1 (i.e., PD-1, which may be associated with T cell exhaustion) had relatively lower expression in C1 than in other clusters, even when normalized by CD3 (Figure S4H). However, no significant differences in CD8 or PD-1 were observed by immunohistochemistry (Figure S4I; data not shown). Thus, although C1 had high OS, numerous lines of evidence suggested that it has less substantial or similar immune infiltration as C2, indicating additional underlying mechanisms corresponding to the high OS in C1.
Genomic alterations driving molecular clusters
To determine whether specific genomic alterations drove the observed transcriptomic states of the molecular clusters, we analyzed somatic variants identified by WES. As in transcriptome assessment, artifacts associated with the formalin fixation process also present a challenge for somatic variant calling. To determine whether FFPE samples could be used for exome analysis, we compared non-silent somatic variants called for the 54 FFPE/frozen pairs, treating the frozen sample as truth. The number of variants was much higher in the frozen samples than in FFPE samples (median 481.5 vs. 7.5 per sample), which was partially explained by low coverage in FFPE samples (median percent target region with >20 coverage, FFPE 91%, non-FFPE 99%; Figure S5A). In addition, the FFPE samples had poor precision (median 0.18, 9 missing) and extremely low recall (median 0.002; Figure S5B). For this reason, we used matched frozen samples where possible for the exome analysis, using the cluster and risk group assigned to the sample used in RNA-seq analysis (Figures S5C and S5D; STAR Methods).
Tumor mutation burden (TMB), particularly those contributing to neoantigens, has been shown to be predictive of immunotherapy response.22,23,24 In this cohort, the total number of somatic mutations per baseline tumor ranged from 0–3,027 (median 1,718), with a median proportion of non-silent mutations of 0.28. The transition/transversion (Ti/Tv) ratio and proportion of single-nucleotide polymorphisms (SNPs) with each base change was relatively consistent for non-FFPE samples, and FFPE samples exhibited a similar pattern with greater variation and a higher proportion of T>C variants (Figures S5E and S5F). The TMB (non-silent mutations per megabase of coding area, 35.7 Mb) was not significantly different across baseline clusters (Kruskal-Wallis test, p = 0.40), even when considering only non-FFPE samples (Kruskal-Wallis test, p = 0.21; Figure S5G). Therefore, TMB did not drive differences between molecular clusters in this cohort.
The landmark TCGA papers for PLC (LIHC and CHOL) identified several driver genes.19,25 To determine whether this cohort aligned with those results, we first profiled mutations in genes previously identified as drivers in HCC and BTC (Figure 5A). The rate of TP53 mutations in this cohort (36% for non-FFPE samples) was similar to TCGA LIHC19 (31%). However, TP53 mutation status was not significantly linked to OS in HCC (Figure S5H). Thus, differences in OS between the molecular clusters were not likely to be driven by TP53 mutation status. We next sought to determine whether any known driver genes were enriched in molecular clusters. Only CTNNB1 was differentially mutated, and it was enriched in C1 (Fisher’s exact test, uncorrected p < 0.01; Figure 5B). However, it appeared that the mutation rate of TP53, IDH1, and KRAS was qualitatively higher in C4. Among the known driver genes, all CTNNB1 and IDH1 mutations occurred within hotspots (FDR, p = 4.4e−8); KRAS, PIK3CA, and RP1L1 mutations were also enriched (FDR, p < 0.005; Figure S5I). The median variant allele frequency (VAF) of CTNNB1 and IDH1 mutations was also less than 50%, suggesting that they were true somatic mutations (Figure S5J). The majority of CTNNB1 and IDH1 mutations have been identified previously and are pathogenic or likely pathogenic (CLIN_SIG; CTNNB1: rs121913403, rs121913407, rs121913409; IDH1: rs121913499, rs121913500; Figures 5C and 5D). Therefore, driver mutations may have a moderate effect on cluster-specific molecular states, particularly for C1 and C4.
Figure 5.

Baseline tumor clusters exhibit genomic differences
(A) Oncoplot of known driver genes in baseline tumor samples (n = 64 tumors). Only genes with non-silent mutations in at least 3 samples are shown. Samples are ordered by the cluster assigned to the baseline tumor. The top bar plot presents the tumor mutation burden (TMB) per sample, defined as the number of non-synonymous mutations per megabase of coding region. The right bar plot presents the number and percentage of samples in which the gene is mutated.
(B) Mutated genes enriched in baseline tumor clusters (Fisher’s exact test, p < 0.01, cluster vs. all other samples, only genes with non-silent mutations in at least 3 samples). Bars are colored by the cluster in which the mutated gene is enriched (top; bottom/gray, all other samples), and fractions indicate the number of mutated samples over the total number of samples per cluster.
(C) Lollipop plot of non-silent CTNNB1 mutations in baseline tumors assigned to C1 (n = 4 patients, 5 mutations). Green circles represent missense mutations. Peach rectangles represent armadillo (ARM) domains. Transcript: NM_001098210.
(D) Lollipop plot of non-silent IDH1 mutations in baseline tumors (n = 3 patients/mutations). Green circles represent missense mutations. A peach rectangle represents PTZ00435 domains; a beige rectangle represents the PTS-1 signal domain. Transcript: NM_005896.
Because mutation signatures can reflect tumor etiology and lead to enrichment of particular driver mutations, we next determined their relationship with the identified molecular clusters. We identified de novo mutation signatures from all non-FFPE primary liver tumors and performed decomposition to known Catalogue of Somatic Mutations in Cancer (COSMIC) signatures (Figures S6A and S6B). The two main signatures in this cohort were SBS5, a clock-like signature increased in some cancers because of smoking and known to be relatively high in HCC, and SBS1, a clock-like signature that correlates with mitosis. The COSMIC signatures were relatively uniform across baseline tumors, except for LCP0074, which also included SBS15, representing defective DNA mismatch repair/microsatellite instability. This patient had the highest average TMB among non-FFPE samples. Therefore, mutation signatures were not significantly linked to molecular clusters.
Molecular changes following ICI treatment
Molecular changes after immunotherapy, including tumor-intrinsic transcriptional programs, increased immune infiltration, and changes to the surrounding stroma, may be important correlates of response to treatment. In addition to our baseline analyses, we utilized our extensive cohort to profile changes after ICI initiation. The cohort included 35 tumors obtained after treatment initiation, including 25 pairs of baseline and after-treatment tumors, with pairs collected at a relatively uniform interval (median 44 days; Figure S7A). Overall, there was little change in the tumor transcriptome following treatment, with most tumors retaining the same molecular cluster and risk group assignments as observed at baseline (Figures 6A and S7B). This was supported by the lack of variation because of sample time point across all primary liver samples (Figure S1A). However, in six patients, tumors that were assessed following treatment showed a change in molecular cluster, five of which also changed risk group, and these showed greater transcriptome changes (Wilcoxon test, p = 1.1e−5; Figure 6B). This change may represent variation in tumor sampling and tumor heterogeneity rather than a response to treatment because the samples that transitioned from poor- to good-survival clusters did not have improved OS relative to patients with the same histological diagnosis that transitioned in the opposite direction. Therefore, transcriptomic changes induced by ICI treatment generally did not overwhelm signatures because of patient of origin or baseline molecular state.
Figure 6.

Immunotherapy treatment induces an overall increase in CD8 T cells and minor differences in immune infiltration between clusters
(A) PCA on paired baseline and after-treatment tumors (n = 25 pairs) with genes used in consensus clustering as features. Baseline clusters were assigned using consensus clustering; after-treatment clusters were assigned using a k-nearest neighbors model (STAR Methods). Lines link paired tumors.
(B) Pearson correlation between baseline and after-treatment tumor pairs (on median absolute deviation [MAD] > 2 genes), split by whether the cluster changes after treatment initiation and colored by baseline cluster (see A; no, n = 19; yes, n = 6). Boxplots: center line, median; box limits, first and third quartiles; whiskers, values ≤ 1.5 times the interquartile range from box limits; points, outliers.
(C) Enrichment score over time in paired tumors for the MSigDB Hallmark IL-6 JAK STAT signaling gene set.
(D) Absolute abundance over time in paired tumors for the CIBERSORTx cell type CD8 T cell.
(E) Normalized gene expression over time in paired tumors for CXCL1.
(F and G) Enrichment score over time in paired tumors for the MSigDB c8 C34 MHC class II positive (Pos) B cell gene set (F) or for the MSigDB Hallmark peroxisome gene set (G).
(C–G) Lines are colored by the predicted risk group or molecular cluster of the baseline tumor. Dashed lines represent BTC; solid lines represent HCC. n = 25 sample pairs.
(H) Tumor allele frequency (TAF) over time for non-silent CTNNB1 mutations in two patients assigned to C1 at baseline (LCP0091, rs121913407; LCP0105, rs121913403; both known pathogenic mutations).
(I and J) For four HCC patients with longitudinal after-treatment tumor sampling, (I) absolute abundance over time of the CIBERSORTx T cell CD8 cell type and (J) enrichment score over time of the Hallmark IL-6 JAK STAT3 signaling gene set. Samples are colored by the assigned molecular cluster.
Next, to identify any consistent time point-related transcriptomic changes across clusters with greater resolution, we performed differential expression analysis between baseline tumors (n = 64) and those following treatment (n = 35). The chemokine CXCL9 was the only significant DEG following treatment (FDR-adjusted p < 0.05; Figure S7C, left panel) and has been implicated previously in T cell attraction and immune checkpoint response.26 At a less stringent threshold (uncorrected p < 0.01; Table S6), genes upregulated following treatment were enriched in Hallmark and liver cell gene sets related to inflammation and immune signaling, including NK/NK T cells and endothelial cell types (Figure S7D). Interleukin-6 (IL-6) JAK STAT3 signaling also increased after treatment initiation (paired Wilcoxon test, uncorrected p < 0.01; Figure 6C), as did CD8 T cells and M1 macrophages (Figures 6D and S7C, right panel). Several of the most highly upregulated DEGs are expressed by immune cells (Human Protein Atlas; proteinatlas.org) and have known roles in T cell chemotaxis and activation (RefSeq summary; https://www.ncbi.nlm.nih.gov/refseq/), including CXCL10, CXCL11, and CXCR6. Among these, the chemokines were also members of the Hallmark IL-6 JAK STAT3 signaling, interferon gamma response, and inflammatory response gene sets. These results confirmed that ICI treatment induced an immune response, including increased CD8 T cell activity.
Because the baseline tumor states were linked to OS, we investigated whether they may influence molecular response immediately after treatment. To do so, we first explored DEGs whose change in expression following ICI treatment was different between baseline survival risk groups (Table S6). Although significant DEGs were not identified, low-risk tumors had an observed increase in follicular helper T cells following treatment with a concomitant decrease in high-risk tumors (uncorrected Wilcoxon p < 0.05 on delta, high vs. low risk; Figure S7E, left panel), with an opposite pattern observed for the adipogenesis pathway, related to immune imbalance and inflammation27 (Figure S7E, right panel). In high-risk tumors, there was a relative decrease in EpCAM+ bile duct cell gene set expression relative to low-risk tumors (DEGs with uncorrected p < 0.01, enriched pathways; Figure S7F). A top upregulated gene in the high-risk group was VWCE, with known expression in hepatocytes and a potential regulator of the -catenin signaling pathway,28 which is related to immune exclusion and resistance to ICI treatment.29,30 VWCE showed increased expression in high-risk samples following treatment with an opposite pattern observed in low-risk samples (Figure S7G). The top upregulated genes in the low-risk group included two genes from the survival signature, CXCL1 and CXCL8, which are members of several immune-related gene sets and whose expression increased strongly in low-risk samples following treatment (Figure 6E). Together, these results suggested that low- and high-risk tumors responded somewhat differently to ICI treatment, including a relatively higher immune response in low-risk tumors.
We also sought to identify transcriptomic changes because of ICI treatment that differ between molecular clusters by identifying DEGs with an interaction with the baseline cluster (Table S6). There were 39 genes differentially expressed following treatment initiation in C3 vs. C1 (adjusted p < 0.05), although there were relatively few C3 samples. Interestingly, the top 5 downregulated DEGs by log fold change (logFC) (uncorrected p < 0.01) for C2 vs. C1 were immunoglobulin genes (Figure S7H), and expression of the major histocompatibility complex (MHC) class II+ B cell gene set increased after treatment initiation in C1 relative to the other clusters (uncorrected Kruskal-Wallis test p < 0.05 on delta across clusters; Figure 6F). The reverse pattern was seen for the peroxisome gene set (Figure 6G), which has been linked to the immune response via T cell exhaustion and tumor metabolism.31,32 C2 tumors also showed increased expression of cell-cycle-related gene sets following treatment, from the previously noted relatively lower levels observed in this cluster compared with other clusters at baseline (DEGs with uncorrected p < 0.01, enriched pathways; Figure S7I). In two C1 patients with baseline CTNNB1 mutations, the same mutation was present in the tumor assessed following treatment but increased in allele frequency (Figure 6H). Thus, although C1 and C2 both exhibited high OS, they responded differently to ICI treatment.
Even in patients with strong immunotherapy responses, cancer relapse and recurrence can occur over time. We extended our profiling of response-related molecular features to longitudinal treatment time courses for four HCC patients. All patients were predicted to be low risk at baseline, and the majority maintained that classification over time; however, LCP0099 switched to C4 poor survival in the final time points (Figure S7B). As noted above, CD8 T cells increased following treatment; however, a gradual decrease was observed at time points more than 1 year after treatment initiation (Figure 6I). A similar pattern was observed for other immune cell types, including plasma cells and M2-polarized macrophages, and for inflammation-related Hallmark gene sets, including IL-6 JAK STAT3 signaling (Figure 6J). As expected, the switch from C2 to C4 in LCP0099 was accompanied by a decrease in hepatocyte-related gene sets relative to bile duct cell gene sets (data not shown). In another case, although both were low risk, the switch from C1 to C2 in LCP0042 was also accompanied by an increase in immune and endothelial cell gene sets as well as related Hallmark gene sets such as transforming growth factor (TGF-) signaling and angiogenesis (data not shown). Therefore, despite immediate responses to treatment, the effects gradually wore off over time, with some patients eventually changing molecular states, which might contribute to ICI resistance.
Discussion
In this study, we established an FFPE-based workflow to perform comprehensive transcriptome and whole-exome profiling of PLC to identify molecular features associated with ICI response and outcome. Among this sizable cohort of ICI-treated liver cancer patients, baseline tumors naturally segregated into four molecular clusters, all of which contained HCC and BTC, exhibited differences in outcome, and were distinguished by tumor-intrinsic and immune differences. Observed differences in the molecular status of each cluster to ICI treatment were intrinsic to the baseline tumor. There was no significant relationship of TMB with molecular clusters or treatment response.
While most genomic studies assess molecular alterations in frozen tissue specimens, most tissues collected in clinical studies are stored as FFPE. Here, we showed the feasibility of utilizing FFPE tissue to perform global genomic assessment at the transcriptomic and exomic levels. We investigated the impact of storage method on tumor transcriptome profiling, along with technical replicates at multiple levels of the pipeline. The results showed that FFPE can be used successfully for transcriptome analyses, provided that sufficient input RNA is available. In fact, the storage method had a relatively minor impact on the transcriptome relative to biological sources of variation. In contrast, concordance between frozen and FFPE pairs was poor for WES, indicating that archival tissue may necessitate a more targeted approach, such as gene panel profiling. Tissue specimen collection at various study sites had a minimal batch effect and did not significantly influence any of the results. Hence, we showed the feasibility of using FFPE tissue, rather than frozen tissue, to comprehensively profile the molecular status of liver cancer patients and relate these findings to clinical parameters, including outcome and treatment response. This established workflow could be implemented in future clinical studies/trials.
The evaluation of genomic data showed that tumor-intrinsic molecular pathways related to aggressive disease were differentially associated with the outcome-related molecular clusters identified in this study. While hepatocyte and liver function gene sets related to a more differentiated status were found in good-survival clusters, molecular signaling associated with aggressive disease, such as EpCAM signaling, was linked to the poor outcome cluster C4. EpCAM is associated with increased vascularization, cancer stemness, and poor survival.33,34 The WNT/-catenin pathway was also elevated in C4. WNT/-catenin pathway activity is deregulated in liver cancer and associated with enhanced proliferation and epithelial-to-mesenchymal transition, playing a critical role in cancer cell progression.35 However, WNT/-catenin-activating mutations, which have been linked to immune exclusion and ICI resistance in HCC,30 were more prevalent in good-survival C1. Furthermore, the impact of -catenin signaling on T cell exclusion and resistance to ICIs has been noted in other cancer types, such as melanoma.36 Therefore, the observed molecular pathways are consistent with those identified previously and highlight important underlying biological associations among these heterogeneous tumors.
Aside from tumor aberrations, alterations of immune cell populations were also observed among the molecular clusters identified in this study and varied among clusters with differences in ICI response. In line with previous findings, we observed an increase in immune cell populations, such as CD8+ T cells and M1 macrophages, after ICI treatment.37,38 Interestingly, although we identified two molecular clusters (C1 and C2) associated with good survival, they exhibited differences in immune infiltration at baseline and following ICI treatment. C2 had the greatest infiltration of CD8 T cells and plasma cells at baseline, while C1 showed increased enrichment of B cell gene sets after ICI initiation, representing a potential influx of B cells. Therefore, while various immune cells subsets and constituents are altered upon ICI treatment, it seems that multiple baseline states may be predictive of good survival. Further studies are warranted to explore these immune infiltrates to the tumor and to more specifically characterize the role of the surrounding microenvironment to determine the contribution of specific sets/subsets of immune cells on treatment response.
An important feature of this study is the availability of tissue specimens collected before and following ICI initiation. Our findings indicate that molecular features associated with outcome and response are largely intrinsic to the tumor status prior to ICI treatment. While immune infiltrates were apparent following ICI treatment, the changes were short lived, with reversion to the original baseline state after longitudinal assessment. Hence, the ability to interrogate tumors prior to ICI treatment may be helpful to stratify patients who are more likely benefit from ICI treatment and useful to define underlying molecular and immune conditions that are predictive of subsequent treatment response. These data support obtaining and incorporating baseline tumors in future clinical studies and clinical management of liver cancer, which is a common practice for many other cancer types. These findings also indicate the importance of exploring additional strategies to induce sustained immune changes in future studies.
While molecular assessment of tissues provides important information regarding the status of tumors, additional features may also influence response to immunotherapy. Other patient characteristics and readouts from alternative biospecimens, such as tumor scans and blood- or urine-based metabolites,39,40 are more accessible than liver biopsies and could be considered predictors of response. For instance, the gut microbiome is known to affect drug metabolism and the immune system, modulating responses to ICIs.41 In addition, immune-related adverse events are associated with response and can be further explored.39,40,42 Future studies should include these additional features as well as extensive patient clinical data to predict response with greater accuracy. In fact, the prospective arm of NCI-CLARITY, currently underway, attempts to do just that, assessing and integrating multiple levels of “omics” from various biospecimen types along with clinical features to identify indicators of immunotherapy response among PLC patients.
Limitations of the study
There were limitations to the study. Because all patients were ICI treated, a comparison with a control non-ICI group could not be directly performed. Also, exploration of each ICI treatment category was not assessed because of limited sample size but can be evaluated in larger studies. Specimens were collected at various sites with various ICI-related treatment regimens, representing a highly variable cohort. Although major molecular differences were not observed among study sites, homogeneous clinical cohorts may produce more treatment-specific molecular targets. In addition, there should be a more comprehensive comparison between these data and others as they become available. While molecular features were related to ICI response, additional studies are needed to determine whether they serve as predictive biomarkers or signatures of response. Because specimens were analyzed retrospectively, clinical parameters could not be consistently assessed across all sites because some were not available or collected. Molecular assessment was performed on bulk tissue, whereas single-cell assessment could provide a more detailed portrait of tumor and immune status before and following treatment.43,44 Because of cancer heterogeneity, some molecular features could be masked by the lack of multi-region or longitudinal specimens, which may provide more in-depth readouts on tumor status and treatment response.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| NEBNext Ultra II Directional RNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7765 |
| SureSelect XT Reagent Kit (Human All Exon V7) | Agilent | Cat# 232859 |
| Deposited data | ||
| Raw total RNA-seq and whole exome sequencing data | This paper; dbGaP | dbGaP: phs003074.v1.p1 |
| Processed data | This paper; dbGap | dbGaP: phs003074.v1.p1 |
| Code for analysis of raw and processed data | This paper; Figshare | Figshare: https://doi.org/10.6084/m9.figshare.22595983 |
| TCGA HTSeq – Counts files | Genomic Data Commons Data Portal | RRID: SCR_014514 |
| TCGA survival data | cBioPortal | RRID: SCR_014555 |
| MSigDB Hallmark v7.4 and C8 v7.2 modules | Molecular Signatures Database | RRID: SCR_016863 |
| Molecular subclasses identified in previous studies | Candia et al., 202045 | Supplementary Data 7; https://doi.org/10.1038/s41467-020-18186-1 |
| Software and algorithms | ||
| CCBR Pipeliner | GitHub | RRID: SCR_002630 |
| DRAGEN | Illumina | RRID: SCR_010233 |
| survriskpred R package v0.2 | BRBArrayTools | RRID: SCR_010938 |
| ConsensusClusterPlus v1.56.0 | Bioconductor | RRID: SCR_016954 |
| ssGSEA2.0 | Broad Institute | RRID: SCR_016863 |
| CIBERSORTx | Stanford University | RRID: SCR_016955 |
| Limma v3.48.3 | Bioconductor | RRID: SCR_010943 |
| Maftools v2.8.5 | Bioconductor | RRID: SCR_006442 |
| Other | ||
| NCI-CLARITY study website | This paper | https://ccr.cancer.gov/liver-cancer-program/nci-clarity-study |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xin Wei Wang (xw3u@nih.gov).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human subjects
The NCI-CLARITY Retrospective cohort consisted of HCC and BTC patients undergoing clinical care at the NIH Clinical Research Center, Georgetown University, Thomas Jefferson University, UC San Francisco, and Massachusetts General Hospital. 93 patients contributed biospecimens included in this manuscript (NIH: 49, Georgetown: 22, Thomas Jefferson: 2, UCSF: 9, MGH: 11). A descriptive table of clinical features is provided in Table 1 for patients who received immunotherapy, including gender and age. The study was approved by institutional review boards at each site. All participants provided written informed consent.
Method details
Nucleic acid extraction
Tumor samples were stored as FFPE slides or blocks, frozen, in RNALater, or as OCT blocks. FFPE blocks were cut into slides. FFPE slides were created using RNase precautions with no heat treatment. Available H&E (hematoxylin and eosin) slides were annotated by a pathologist at the Molecular Histology Laboratory at NCI, and the % tumor content was estimated. FFPE slides were macro-dissected into tumor and non-tumor sections if both were present. Nucleic acid was extracted at the at the Molecular Histology Laboratory. DNA was extracted using the phenol method. RNA was extracted using the Qiagen RNeasy Mini Kit and DNase-treated.
A total of 282 samples had nucleic acid extracted. 274 had both RNA and DNA extracted, while four samples each had only RNA or DNA (non-tumor samples from the same patient, different timepoints).
Total RNA sequencing
Libraries were prepared using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina, using the 100ng or 5ng protocols based on RNA availability at the CCR Sequencing Facility. Samples with <5ng RNA (“low concentration”) were prepared using the 5ng protocol.
Sequencing data was generated by the CCR Sequencing Facility. 99 samples were sequenced on a NovaSeq S2 flowcell (2x151bp); 72 with good yield were then sequenced in greater depth on a NovaSeq S4 flowcell (2x151bp), and reads were combined. An additional 181 samples were sequenced on a NovaSeq S4 flowcell (2x101bp). Two RNA-seq libraries were sequenced in both rounds, for a total of 280 samples. Each sample produced 39 to 2,074 million pass-filter reads (median: 133 million).
Separately, 16 pilot liver transplant samples were sequenced on a NextSeq (2x76bp). Each sample produced 42–59 million pass-filter reads (median 53 million).
Pipeliner RNA-seq Quality Control Analysis (https://ccbr.github.io/pipeliner-docs/) was run on all RNA-seq fastq files (hg38). Reads were trimmed using Cutadapt46 (v1.18) to remove adapters and low-quality reads before mapping to hg38 using STAR47 (v. 2.5.2b) 2-pass alignment. Reads were assigned to a single read-group and duplicates flagged using Picard tools (v.2.17.11). The strandedness of the library was inferred using RSeQC48 (v. 2.6.4). Gene expression was quantified using RSEM49 (v. 1.3.0) with the inferred strandedness informing the forward probability.
RNA-seq samples passed QC if the percent of mapped bases in coding regions was at least 12.5% (240 of 280 non-pilot samples, all pilot samples). For non-pilot samples that passed QC, the number of de-duplicated reads per sample was 0.08–759 million (median: 39 million).
Whole exome sequencing
Libraries were prepared at the Genomics Core at NCI using the Agilent SureSelect XT (Human All Exon V7) kit.
A total of 278 samples were sequenced. 54 samples were sequenced on a NovaSeq S2 (2x151bp), 45 were sequenced on a NovaSeq S4 (2x151bp), and 179 samples were sequenced on a NovaSeq S4 (2x101bp). Each sample produced 0.09 to 475 million pass-filter reads per sample (median: 249 million).
Sequencing data was generated by the CCR Sequencing Facility. Reads were mapped and variants called using the DRAGEN pipeline in tumor-only mode with artifact flag enabled for all samples. The number of de-duplicated reads per sample ranged from 0.04 to 296 million reads (median: 151 million).
Variants were hard-filtered (FILTER: PASS only). Somatic variants were obtained by restricting to variants with a tumor read depth of >20, a variant read depth of >4, a tumor allele frequency of 5%, and a population frequency <0.1% in the gnomAD combined population, 1000 Genomes, and ExAC, and by removing variants within blacklist genes50 (perl/5.18.4).
Clinical and sample data
Clinical data was collected from each site using a standardized data dictionary and template divided into 13 forms. Data from the templates was stored in LabMatrix.
Clinical data was downloaded from LabMatrix on October 20, 2021 (v1.8). BMI was calculated using the formula 703∗(weight (lb)/height (in)ˆ2). Immunotherapy treatment names were standardized, and treatment categories were assigned based on immunotherapy agent(s) and any concurrent treatment(s). Prior treatment categories were assigned based on agent names. Treatment categories were not significantly different between HCC and BTC (Chi-squared test with simulated p value, p = 0.11), although only HCC patients received single agent ICI with VEGF inhibitors. Patients with entries in the Prior Treatment, Prior Surgery, or Radiation Treatment supplements were assigned “Yes” for the corresponding field in the Enrollment and Disease Status and Treatment forms. Overall survival censoring status was obtained from “Live/Dead Status”, and progression-free survival censoring status was obtained from “Disease Progression Status” obtained per investigator.
Image data, including % tumor estimates and pathologist comments, were exported from HALO on June 28, 2021. Tumor purity categories were assigned to each FFPE tumor and separate non-tumor sample based on estimated tumor purity and whether the slide was macrodissected (e.g., non-tumor sample was extracted from the same slide): Low (0–20% purity, “normal”), Mid (30–70% purity), High (80–100% purity or macrodissected), and Unevaluable (“na”,”?”).
The date of C1D1 and sample collection was available for NIH patients only. Time from C1D1 was calculated for all NIH samples.
Driver genes were identified from the TCGA-LIHC publication19 Figure 1 and the TCGA-CHOL publication25 Figure 1, for a total of 59 known driver genes.
Cancer Gene Census data was exported from COSMIC (https://cancer.sanger.ac.uk/census; August 17, 2021). Only genes known to be active in liver or biliary tract cancer were considered (somatic or germline; “hepat|cholang|biliary”).
Cohort sample structure
Primary liver tumors
Analysis was restricted to primary liver tumors and adjacent non-tumor from patients who received immunotherapy (230 samples from 86 patients, 199 of which pass RNA-seq QC). Excluded samples include 15 metastasis samples and 37 primary liver samples from patients who did not receive immunotherapy. However, 20 samples from the non-immunotherapy patients were included in the Georgetown storage test. BTC included CCA (19 intrahepatic, 2 extrahepatic, and 7 not otherwise specified patients), ampulla of vater cancer (2 patients), and gall bladder cancer (1 patient).
Replicates
Samples include replicates at multiple levels, including: 2 resequenced libraries (RNA-seq only); multiple sections from the same sample (9 tumor sections plus 6 associated non-tumor samples from 6 samples); and multiple nodules from the same timepoint (2 lesions sampled from the left lobe and right hepatic lobe, plus 2 associated non-tumor samples).
Tumors were also stored using multiple methods. 54 tumors from NIH were stored as both FFPE and frozen (45 pairs pass RNA-seq QC). An additional patient (LCP0074) had two samples stored in FFPE, RNALater, and OCT block, for a total of 9 samples (8 pass RNA-seq QC). This patient was included in both the main analysis and the Georgetown storage test.
Tumor selection
For most analyses, a single tumor per timepoint was selected for each patient. 74 patients had a baseline tumor, 37 had an after-treatment tumor, and 27 had both. In addition, four patients had after-treatment tumor time courses (8 additional tumors). One patient had a single tumor with an unknown timepoint, and one patient had only non-tumor samples sequenced.
For RNA-seq analysis, priority was given to FFPE samples when tumors were stored in multiple media and to replicates/nodules with a higher gene count (number of genes with any read). 64 patients had a baseline tumor that passed RNA-seq QC, 35 had a after-treatment tumor, and 25 had both. The 8 timecourse tumors passed QC. Baseline FFPE samples were evenly distributed between HCC and BTC (Chi-squared test with Yates’ continuity correction p value = 0.57).
For exome analyses, frozen samples were given priority over FFPE samples. 24 of the baseline FFPE tumors used in RNA-seq analyses were replaced with a matched frozen (n = 23) or RNALater (n = 1) sample. Additionally, 27 after-treatment FFPE samples were replaced with matched frozen samples.
Non-tumor samples
Samples include 43 adjacent non-tumor samples from FFPE slides, 34 of which passed RNA-seq QC. There were 35 tumor and adjacent non-tumor pairs (7 were WES- or RNA-only; including the replicates detailed above), including 24 that pass RNA-seq QC. For 2 tumors, separate non-tumor tissue was provided, and in one case, both separate and adjacent non-tumor was sequenced.
Input RNA pilot test
Libraries were prepared from four liver transplant samples using 5ng or 100ng of RNA, two replicates each, for a total of 16 libraries. All RNA-seq results pass QC. Expected gene counts were calculated using RSEM. Counts were loaded into a DGElist, filtered to remove lowly expressed genes (filterByExpr by input RNA amount and sample, library sizes recalculated), and voom transformed with quantile normalization. Genes were filtered to the top 4000 most variable genes by median absolute deviation (MAD).
Georgetown storage test
Six samples from four CCA patients were stored in three different storage media (FFPE, RNALater x 2, OCT x 2), for a total of 29 samples (27 pass RNA-seq QC). One patient (LCP0074) received immunotherapy and was also included in the main analysis.
Expected counts were loaded into a DGEList, filtered to remove lowly expressed genes (by patient), and voom transformed with quantile normalization. Genes were filtered to those with MAD >2 (n = 3,896).
Principal component analysis (PCA) was performed using prcomp() with centered and scaled matrices. Sample rotations, variable loadings, and the percent variance explained by each component were extracted from prcomp() output.
Primary liver transcriptome analysis
Expected counts per gene were calculated using RSEM. Counts were loaded into a DGElist, filtered to remove lowly expressed genes (n = 23,586 genes, filterByExpr by diagnosis and malignancy, library sizes recalculated), TMM normalized, and voom transformed with quantile normalization.
The MAD across all primary samples was calculated for each gene. Variance partitioning was performed on genes above MAD >2 (n = 4,002 genes) using the variancePartition package (R/4.0.3; edgeR, limma packages). 74% (n = 2,977) and 72% (n = 2,876) of the genes also have a MAD >2 within only HCC or BTC, respectively. Pathways enriched in genes with >20% of variance explained by study site (42 genes), amount of input RNA (12 genes), storage method (201 genes), or read depth (344 genes) were identified using the clusterProfiler enricher function on the MSigDB gene sets, with a minimum gene set size of 10. The only enriched pathway was HALLMARK_TNFA_SIGNALING_VIA_NFKB in the study site genes (q-value = 0.009).
FFPE vs. frozen storage test
Differentially expressed genes (DEGs) between pairs (n = 45) were identified using limma (lmFit/eBayes on normalized gene counts, blocked by biopsy). Pathways enriched in DEGs (each direction, adjusted p value <0.05) were identified using the clusterProfiler enricher function on the MSigDB gene sets, with a minimum gene set size of 10. Only pathways with an FDR-adjusted p value <0.05 were considered.
Survival analysis
Kaplan-Meier curves and log rank tests were generated using the survival and survminer packages (survfit(), survdiff(), pairwise_survdiff(), ggsurvplot(), and ggsurvplot_facet() functions). The proportional hazards assumption was tested using the coxph() and cox.zph() functions. Log rank p values were also calculated using pchisq on the results of survdiff() (lower tail = FALSE, df = n-1).
BRB-arrayTools survRiskPred
A model for overall survival was developed for unique baseline tumors (n = 64) using the survriskpred package (R implementation of BRB-ArrayTools function). Genes with a univariate Cox proportional hazards model p value <0.001 (n = 129) were used as variables in a principal component analysis. A multivariate Cox proportional hazards model was fit using the first several PCs as variables (1 to 1–3 PCs) to obtain a regression coefficient for each PC. A prognostic index was calculated for each sample using the average of each PC weighted by its regression coefficient. Samples were divided into two risk groups at the median prognostic index.
To evaluate the predictive value of the method, 10-fold cross-validation (CV) was performed from univariate gene selection to prognostic index assignment. A risk group was predicted for each sample using the iteration in which the sample was not selected (predictRiskTrainingModeE matrix). The CV process was repeated 100 times with randomly shuffled sample labels to generate a permutation p value for the log rank statistic between risk groups. The iteration with the lowest permutation p value was selected (PC1-2; permutation p value = 0.08, log rank p value = 0.016; coefficients PC1: 6.86, PC2: −0.725).
The risk group was predicted for all primary liver samples using the selected model, with intercept and prognostic index threshold obtained from the html output. The risk groups predicted using 10-fold CV and the final model were the same for 56/64 of the training samples. The proportion of tumors assigned to each risk group was not significantly different between the baseline tumors used to develop the signature and all tumor samples (Chi-squared test p = 0.76). Risk groups were not differently distributed by tumor storage method, sample timepoint, site, or de-duplicated read depth (Chi-squared test with simulated p value, p > 0.05). However, 91% of the non-tumor samples were assigned to the low-risk group.
Univariate hazard ratios and p values were calculated for each gene using Cox proportional hazards models (coxph() function). Gene loadings and weights were extracted from the loadingMatrixModE and genesInClassifierModE matrices. Genes with negative weight were considered protective because the coefficient for PC1, which had the largest weight, was positive. Pathways enriched among protective and non-protective genes in each signature were identified using enricher (minimum gene set size 10, FDR-adjusted p value <0.05).
SurvRiskPred was also performed on only HCC baseline tumors, clustering on genes with a univariate p value <0.001 across all baseline tumors, 1 to 1–3 PCs. The coefficients for the PC1-2 iteration were PC1: 6.195, PC2: 0.638.
TCGA survival analysis
A manifest was downloaded from GDC containing all HTSeq – Counts files with “liver and intrahepatic bile ducts”, “other and unspecified parts of biliary tract”, or “gallbladder” as the primary site (416 cases, 486 files; June 23, 2021). Files were downloaded using gdc-client. Metadata for TCGA-LIHC and TCGA-CHOL files was downloaded using gdc-client (407 cases, 469 files; June 23, 2021). Overall survival from diagnosis was obtained from the TCGA PanCancer Atlas datasets on cBioPortal (KM Plot: Overall Survival https://www.cbioportal.org/study/summary?id=lihc_tcga_pan_can_atlas_2018 and https://www.cbioportal.org/study/summary?id=chol_tcga_pan_can_atlas_2018, November 11, 2021; n = 408 patients) and merged on case ID (cases.0.submitter_id)/Patient ID.
Samples were filtered to primary tumors. Gene counts were loaded into a DGEList, filtered to remove lowly expressed genes (filterByExpr, by project, library sizes recalculated), TMM normalized, and voom transformed with quantile normalization.
A risk group was predicted for each sample using the SurvRiskPred model, using only genes that were present in both the signature and the TCGA gene expression matrix. Genes were matched using Ensembl IDs with the suffixes removed (includes 97% of genes from this study and 94% of TCGA genes). Six patients missing survival data were excluded.
Consensus clustering
Consensus clustering was performed on unique baseline tumors (n = 64) using Consensus Cluster Plus51 and median-centered gene expression restricted to genes with MAD >2 across all primary samples (n = 4,002 genes). Samples were clustered using k-means clustering (k = 2–10). 1000 resamplings were performed, and at each, 80% of samples and genes were sampled. The final consensus matrix was clustered using agglomerative hierarchical clustering on Euclidean distance with complete linkage. Cluster and item consensus results were calculated. A k = 4 solution was identified based on the delta area under the curve for the consensus score cumulative density function (elbow point) and cleanliness of clustering (proportion of non-ambiguous assignments).
Consensus clustering was also performed with the same parameters as above with: unique baseline tumors excluding extrahepatic CCA, genes with MAD >2 across remaining samples; HCC baseline tumors, genes with MAD >2 across all primary samples; BTC baseline tumors, genes with MAD >2 across all primary samples; and baseline tumors, genes with MAD >2 within baseline tumors. For the latter, 83% of the MAD >2 genes across all samples were shared with this gene set (n = 3,303). Consensus clustering using genes with MAD >2 within baseline samples recapitulated the clustering results using MAD >2 across all primary samples, identifying an optimal 4-cluster solution with only two samples assigned to different clusters.
K-nearest neighbors classifier
All primary liver samples were assigned to a molecular cluster using a K-nearest neighbors (KNN) model. The consensus clustering input genes (MAD >2) were pre-processed via centering and scaling across baseline tumors, which was then applied to the same genes across all primary tumor samples (pre-Process() and predict() functions). A KNN model was trained on the baseline input genes and cluster assignments and used to predict cluster assignments for all other samples. k = 7 neighbors were considered, with a seed of 9. The model was 91% accurate on input samples.
94% of non-tumor samples were assigned to cluster 2 (n = 32). Among tumor samples, the proportion of samples assigned to each cluster was not different than those generated using consensus clustering (Chi-squared test p = 0.10). The distribution of tumor samples by storage method, timepoint, and study site was not different across clusters (Chi-squared test with simulated p value, p > 0.05), but there was a difference by de-duplicated read depth (Kruskal-Wallis test p value = 1.2e-5), as for baseline tumors.
63% of FFPE/frozen storage pairs were assigned to the same molecular cluster using a k-nearest neighbors model, while 84% were assigned to the same risk group (Figures S5C and S5D; see STAR Methods). In contrast, sections from the same FFPE block were concordant 67% and 100% of the time, respectively. 85% of the samples used for baseline exome analysis had the same predicted molecular cluster as the corresponding sample used for RNA-seq analysis (51/60, 4 with no prediction due to poor quality RNA-seq sample), and 95% had the same predicted risk group (57/60). The proportion of FFPE samples was not different across molecular clusters (Chi-squared test p > 0.05).
Differentially expressed genes
Differentially expressed genes (DEGs) were identified between all pairs of molecular clusters using limma (lmFit/eBayes) on normalized expression values, with pairwise contrasts. Cluster-specific DEGs were those with an adjusted p value <0.05 and the same direction of change between each cluster and the other three clusters.
Diagnosis-specific DEGs were identified between HCC and BTC baseline tumors using limma (adjusted p value <0.05, 4,186 genes).
RECIST response
RECIST v1.1. was retrospectively assessed per investigator. Patients with unknown RECIST response or whose tumors were non-evaluable due to rapid decline were excluded from analyses.
DEGs were identified at baseline between responders and non-responders using limma (lmFit/eBayes). Pathways enriched in DEGs (each direction, uncorrected p value <0.01, n = 411 genes) were identified using the clusterProfiler enricher function on the MSigDB gene sets, with a minimum gene set size of 10. Only pathways with an FDR-adjusted p value <0.05 were considered.
Clinical/sample data enrichment in clusters
Select clinical data was tested for enrichment in baseline tumor clusters (all, just HCC, and just BTC; Table S3). Only baseline timepoints were considered for all tested data except Follow-up Treatment. Missing values were excluded, including “Unknown” entries and “Non-evaluable” entries for RECIST response. Text/list variables with multiple levels and data for multiple clusters were tested using a Chi-squared test (simulated p values). Numeric variables with data for at least one cluster were tested using a Kruskal-Wallis test. p-values were FDR-adjusted within each sample set.
Each prior treatment therapy category was also tested for association with baseline tumor clusters (Table S4). Patients whose prior treatment status was unknown were excluded (n = 6). Each therapy category with any variation was tested (i.e., patient received any prior treatment in the category vs. not; Chi-squared test with simulated p values). p-values were FDR-adjusted within each sample set. Association between clusters and the number of prior treatments was also tested (Kruskal-Wallis test p value = 0.40).
Clusters were also tested for association with sample variables using Chi-squared tests with simulated p values and Kruskal-Wallis tests. The clusters were not associated with storage method, sequencing batch, or RIN or with estimated tumor purity (FFPE only, n = 44). The associations with read depth (Figure S3I) and amount of input RNA (Chi-squared test p value = 0.04, 4 missing entries) were significant.
ssGSEA gene set analysis
The ssGSEA2.0 repo was cloned (https://github.com/broadinstitute/ssGSEA2.0). GMT files for the Hallmark (v7.4) and C8 (v7.2) modules were downloaded from MSigDB52 (as Gene Symbols; http://www.gsea-msigdb.org/gsea/downloads.jsp). The C8 gene sets were restricted to single cell-derived liver cell type signatures from Aizarani et al.21(n = 31). The TPM gene expression matrix output by RSEM was converted to GCT format. ssGSEA was performed using ssgsea-cli.R to generate gene set enrichment scores for each sample. The FDR-corrected p value was <0.05 for all gene sets and samples. The median score for each cluster and a Z score across medians was calculated for each cluster.
Cluster-specific gene sets were identified using one-vs-all Wilcox tests for each cluster on ssGSEA enrichment scores. p-values were FDR-adjusted. The logFC was calculated as the log2 ratio of the mean score for that cluster vs. the mean score for all other samples.
CIBERSORTx
CIBERSORTx was performed to estimate the proportion of immune cells in a bulk sample. The TPM gene expression matrix output by RSEM was reformatted to include only gene symbol. Immune infiltration was profiled using CIBERSORTx53 with the LM22 signature matrix in absolute mode with batch correction. The relative abundance of each cell type was calculated as a proportion of the absolute score. All but one primary liver sample had a p value <0.05 (n = 200).
Cluster-specific cell types were identified using one-vs-all Wilcox tests for each cluster on absolute abundances. p-values were FDR-adjusted. The logFC was calculated as the log2 ratio of the mean abundance for that cluster vs. the mean abundance for all other samples.
Nearest template prediction
HCC samples were assigned to molecular subclasses identified in earlier studies18,19,20,54 using Nearest Template Prediction. Characteristic genes for each subclass were downloaded from Candia et al.,45 Supplementary Data 7. The GenePattern module NearestTemplatePrediction (v4) was used to predict subclasses (default parameters, not weighted; September 8, 2021). The RSEM TPM expression matrix in GCT format (uniqued gene names) was used as input. All predictions had significant p values (p < 0.05, Bonferroni or BH FDR-corrected).
After-treatment analysis
The time interval between the baseline and first after-treatment tumors was calculated for all NIH Clinical Center samples (23 pairs).
Cluster assignments for paired baseline tumors were not significantly different than for all baseline tumors (Chi-squared test with simulated p value, p value = 0.32). The correlation between paired baseline and after-treatment tumors was not significantly different by baseline molecular cluster (Kruskal-Wallis test p value = 0.18) or risk group (Wilcox test p value = 0.47). Tumor pairs with the same storage method were not more similar than those with different methods (Wilcox test p value = 0.27); nor was the sample correlation correlated with after-treatment read depth (Pearson correlation p value = 0.08) or the interval between biopsies (p value = 0.53); nor was it different between HCC and BTC (Wilcox test p value = 0.77) or by best RECIST response (Kruskal-Wallis p value = 0.24). The sample correlation was also not related to overall survival (log rank test p value = 0.23).
DEGs were identified between baseline (n = 64) and after-treatment tumors (n = 35) using limma lmFit/eBayes with blocking by patient. Additionally, DEGs with interactions with baseline risk group or molecular cluster were identified, using only after-treatment tumors with a matched baseline tumor (n = 25).
Pathways enriched in DEGs (each direction, uncorrected p value <0.01) were identified using the clusterProfiler enricher function on the MSigDB gene sets, with a minimum gene set size of 10. Only pathways with an FDR-adjusted p value <0.05 were considered.
Gene sets and CIBERSORT cell types with significant changes after treatment were identified using paired Wilcox tests on matched baseline and after-treatment pairs (n = 25). Interactions with baseline risk group or molecular cluster were identified using Wilcox or Kruskal-Wallis tests on the delta between baseline and after-treatment samples.
Gene expression locations were obtained from the Human Protein Atlas (https://www.proteinatlas.org/).
Exome analysis
Maftools was used to plot and analyze somatic variants. Non-silent variants followed the default classification by maftools (Variant_Classification).
The titv() maftools function was used to calculate the proportion of SNPs belonging to each base substitution, including synonymous mutations.
Differentially mutated genes between clusters were identified using the clinicalEnrichment() function, including the 63 baseline samples with any variant and genes with at least 3 mutated samples (n = 1,484). Mutated genes were considered cluster-specific with an uncorrected p value of <0.01 between the cluster and all other samples. The function plotEnrichmentResults() was used to plot cluster-specific mutated genes.
The oncodrive() maftools function was used to identify drivers and cluster-specific genes with an enrichment of non-silent mutations in hotspots. Pre-defined values were used for the background model. The lollipopPlot() maftools function was used to produce lollipop plots for mutations of interest. The function plotVaf() was used to plot the variant allele frequency of genes of interest.
SigProfiler
SigProfilerMatrixGeneratorFunc was used to generate mutation matrices from somatic variants for all non-FFPE primary liver samples, including technical replicates (n = 92; GRCh38, exome = TRUE, filtered to exome target bed file). Signatures were extracted from the SBS96 matrix using SigProfilerExtractor (GRCh38, exome = TRUE, 1–10 signatures, 100 iterations). The suggested solution (2 signatures) was decomposed into COSMIC signatures. Etiologies for each COSMIC signature were obtained from the COSMIC website (https://cancer.sanger.ac.uk/signatures/sbs/).
Immunohistochemistry
30 cases with available FFPE 5um tissue sections were stained for CD8 or PD-1. Immunohistochemistry for CD8 was performed on a Ventana Benchmark instrument using an anti-CD8 AB (SP57, which is a rabbit monoclonal antibody (dilution RTU (ready to use). The PD-1 immunohistochemistry was performed on a Dako Autostainer, with antigen retrieval with a pH6 Citrate buffer with an autoclave for 20 min, antibody hybridized for 30 min at room temperature with a dilution of 1:100, and detected with Dako Envision + (Mouse) with DAB applied for 5 min. The PD-1 antibody is Cell Signaling PD1 (EH33), a mouse monoclonal. Appropriate negative and positive controls were performed for both markers. Cases were scored blindly using a scoring matrix of the following categories: none (0), rare (1), scattered/individual (2), groups (3) or aggregates (4).
Quantification and statistical analysis
Statistics
Details for each statistical analysis are provided under method details, in the results, and in the figure legends. Rationales for samples included in each analysis are described under “cohort sample structure”. Significance was defined as p value <0.05 except where explicitly noted. Kaplan-Meier curves were tested for the proportional hazards assumption (see “survival analysis”).
All Wilcox test p values are two-sided.
Software versions
Python libraries used included numpy, pandas, and bcftools. R packages used include: ggplot2 (v3.3.5), gridExtra (v2.3), openxlsx (v4.2.4), plyr (v1.8.6), RColorBrewer (v1.1.2), reshape2 (v1.4.4), limma (v3.48.3), edgeR (v3.34.1), ggrepel (v0.9.1), circlize55 (v0.4.13), variancePartition (v1.22.0), ComplexHeatmap56 (v2.8.0), ConsensusClusterPlus (v1.56.0), survival (v3.2.13), VennDiagram (v1.7.0), tidyr (v1.1.4), clusterProfiler57 (v4.0.5), preprocessCore (v1.54.0), survminer (v0.4.9), NMF (v0.23.0), caret (v6.0.90), class (v7.3.19), tidytext (v0.3.2), maftools (v2.8.5), scales (v1.1.1), and survriskpred (v0.2). Analysis and visualization were performed using custom scripts in R (v. 4.1.1).
Additional resources
NCI-CLARITY study website: https://ccr.cancer.gov/liver-cancer-program/nci-clarity-study.
Acknowledgments
T.F.G. and X.W.W. are supported by the Intramural Research Program of the NIH, NCI (ZIA BC 011343, ZIA BC 010877, and ZIA BC 011870). The study was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. C.M. received a training fellowship from the Cholangiocarcinoma Foundation. Collection of clinical data was supported in part by the Advanced Biomedical Computational Science (ABCS) group, NCI Frederick. Biospecimen and clinical data management was supported in part by Labmatrix (BioFortis Inc.). Images and associated metadata management was supported in part by the NCI HALO Image Analysis Resource. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). Palantir Foundry was used in the integration, harmonization, and analysis of specimen and clinical data inside the secure NIH Integrated Data Analysis Platform (NIDAP). This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, Department of Health and Human Services, under Contracts 75N91019D00024 and HHSN261201800001I. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Author contributions
A.B., T.F.G., and X.W.W. conceptualized the study and acquired funding. A.B. and E.C.P. administered the project and wrote the original draft. T.F.G., A.H., L.G., R.K.K., H.D., R.L., K.P., C.M., and C.X., provided patient samples. E.C.P., L.K., K.Z., I.B., L.M., and C.X. performed data curation. D.E.K., S.M.H., B.T., J.S., X.W., Y.Z., T-W.S., S.C., Y.K., K.Y., A.C.W., E.F.E., and M.F. performed pathology, sequencing, or specimen preparation. E.C.P and M.R. performed formal analysis. E.C.P performed software development and visualization. M.T. provided computational resources and supervision of analyses. All authors reviewed and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: May 23, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101052.
Contributor Information
Tim F. Greten, Email: tim.greten@nih.gov.
Xin Wei Wang, Email: xw3u@nih.gov.
Supplemental information
Data and code availability
-
•
De-identified total RNA-seq and whole exome sequencing data is available at dbGaP: phs003074.v1.p1. Authorized access is required for individual level data as determined by the informed consent under which the data and samples were collected. These data can be requested through dbGaP.
-
•
All original code is available at Figshare: https://doi.org/10.6084/m9.figshare.22595983.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Altekruse S.F., McGlynn K.A., Reichman M.E. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J. Clin. Oncol. 2009;27:1485–1491. doi: 10.1200/JCO.2008.20.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 3.Van Dyke A.L., Shiels M.S., Jones G.S., Pfeiffer R.M., Petrick J.L., Beebe-Dimmer J.L., Koshiol J. Biliary tract cancer incidence and trends in the United States by demographic group. Cancer. 2019;125:1489–1498. doi: 10.1002/cncr.31942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Llovet J.M., Kelley R.K., Villanueva A., Singal A.G., Pikarsky E., Roayaie S., Lencioni R., Koike K., Zucman-Rossi J., Finn R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers. 2021;7:6. doi: 10.1038/s41572-020-00240-3. [DOI] [PubMed] [Google Scholar]
- 5.Finn R.S., Ryoo B.Y., Merle P., Kudo M., Bouattour M., Lim H.Y., Breder V., Edeline J., Chao Y., Ogasawara S., et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase III trial. J. Clin. Oncol. 2020;38:193–202. doi: 10.1200/JCO.19.01307. [DOI] [PubMed] [Google Scholar]
- 6.Abou-Alfa G.K., Lau G., Kudo M., Chan S.L., Kelley R.K., Furuse J., Sukeepaisarnjaroen W., Kang Y.-K., Van Dao T., De Toni E.N., et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evidence. 2022;1 doi: 10.1056/EVIDoa2100070. [DOI] [PubMed] [Google Scholar]
- 7.Oh D.-Y., Ruth He A., Qin S., Chen L.-T., Okusaka T., Vogel A., Kim J.W., Suksombooncharoen T., Ah Lee M., Kitano M., et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evidence. 2022;1 doi: 10.1056/EVIDoa2200015. [DOI] [PubMed] [Google Scholar]
- 8.Yau T., Hsu C., Kim T.Y., Choo S.P., Kang Y.K., Hou M.M., Numata K., Yeo W., Chopra A., Ikeda M., et al. Nivolumab in advanced hepatocellular carcinoma: sorafenib-experienced Asian cohort analysis. J. Hepatol. 2019;71:543–552. doi: 10.1016/j.jhep.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 9.Yau T., Kang Y.K., Kim T.Y., El-Khoueiry A.B., Santoro A., Sangro B., Melero I., Kudo M., Hou M.M., Matilla A., et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the CheckMate 040 randomized clinical trial. JAMA Oncol. 2020;6 doi: 10.1001/jamaoncol.2020.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma L., Hernandez M.O., Zhao Y., Mehta M., Tran B., Kelly M., Rae Z., Hernandez J.M., Davis J.L., Martin S.P., et al. Tumor cell biodiversity drives microenvironmental reprogramming in liver cancer. Cancer Cell. 2019;36:418–430.e6. doi: 10.1016/j.ccell.2019.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Budhu A., Forgues M., Ye Q.H., Jia H.L., He P., Zanetti K.A., Kammula U.S., Chen Y., Qin L.X., Tang Z.Y., Wang X.W. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10:99–111. doi: 10.1016/j.ccr.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 12.Budhu A., Roessler S., Zhao X., Yu Z., Forgues M., Ji J., Karoly E., Qin L.X., Ye Q.H., Jia H.L., et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology. 2013;144:1066–1075.e1. doi: 10.1053/j.gastro.2013.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoshida Y., Nijman S.M.B., Kobayashi M., Chan J.A., Brunet J.P., Chiang D.Y., Villanueva A., Newell P., Ikeda K., Hashimoto M., et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385–7392. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Do H., Dobrovic A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clin. Chem. 2015;61:64–71. doi: 10.1373/clinchem.2014.223040. [DOI] [PubMed] [Google Scholar]
- 15.Levin Y., Talsania K., Tran B., Shetty J., Zhao Y., Mehta M. Optimization for sequencing and analysis of degraded FFPE-RNA samples. J. Vis. Exp. 2020 doi: 10.3791/61060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wehmas L.C., Hester S.D., Wood C.E. Direct formalin fixation induces widespread transcriptomic effects in archival tissue samples. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-71521-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dotto G.P., Rustgi A.K. Squamous cell cancers: a unified perspective on biology and genetics. Cancer Cell. 2016;29:622–637. doi: 10.1016/j.ccell.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoshida Y., Nijman S.M.B., Kobayashi M., Chan J.A., Brunet J.-P., Chiang D.Y., Villanueva A., Newell P., Ikeda K., Hashimoto M., et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385–7392. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas Research Network Electronic address wheeler@bcmedu. Cancer Genome Atlas Research Network Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–1341.e23. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamashita T., Forgues M., Wang W., Kim J.W., Ye Q., Jia H., Budhu A., Zanetti K.A., Chen Y., Qin L.-X., et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68:1451–1461. doi: 10.1158/0008-5472.CAN-07-6013. [DOI] [PubMed] [Google Scholar]
- 21.Aizarani N., Saviano A., Sagar M.L., Mailly L., Durand S., Herman J.S., Pessaux P., Grün D., Grün D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature. 2019;572:199–204. doi: 10.1038/s41586-019-1373-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samstein R.M., Lee C.H., Shoushtari A.N., Hellmann M.D., Shen R., Janjigian Y.Y., Barron D.A., Zehir A., Jordan E.J., Omuro A., et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019;51:202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jardim D.L., Millis S.Z., Ross J.S., Woo M.S.A., Ali S.M., Kurzrock R. Landscape of cyclin pathway genomic alterations across 5,356 prostate cancers: implications for targeted therapeutics. Oncol. 2021;26:e715–e718. doi: 10.1002/onco.13694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duffy M.J., Crown J. Biomarkers for predicting response to immunotherapy with immune checkpoint inhibitors in cancer patients. Clin. Chem. 2019;65:1228–1238. doi: 10.1373/clinchem.2019.303644. [DOI] [PubMed] [Google Scholar]
- 25.Farshidfar F., Zheng S., Gingras M.C., Newton Y., Shih J., Robertson A.G., Hinoue T., Hoadley K.A., Gibb E.A., Roszik J., et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 2017;18:2780–2794. doi: 10.1016/j.celrep.2017.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.House I.G., Savas P., Lai J., Chen A.X.Y., Oliver A.J., Teo Z.L., Todd K.L., Henderson M.A., Giuffrida L., Petley E.V., et al. Macrophage-derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin. Cancer Res. 2020;26:487–504. doi: 10.1158/1078-0432.CCR-19-1868. [DOI] [PubMed] [Google Scholar]
- 27.Ramadori P., Kam S., Heikenwalder M. T cells: friends and foes in NASH pathogenesis and hepatocarcinogenesis. Hepatology. 2022;75:1038–1049. doi: 10.1002/hep.32336. [DOI] [PubMed] [Google Scholar]
- 28.Lian Z., Liu J., Li L., Li X., Clayton M., Wu M.C., Wang H.Y., Arbuthnot P., Kew M., Fan D., Feitelson M.A. Enhanced cell survival of Hep3B cells by the hepatitis B x antigen effector, URG11, is associated with upregulation of beta-catenin. Hepatology. 2006;43:415–424. doi: 10.1002/hep.21053. [DOI] [PubMed] [Google Scholar]
- 29.Berraondo P., Ochoa M.C., Olivera I., Melero I. Immune desertic landscapes in hepatocellular carcinoma shaped by β-catenin activation. Cancer Discov. 2019;9:1003–1005. doi: 10.1158/2159-8290.CD-19-0696. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz de Galarreta M., Bresnahan E., Molina-Sánchez P., Lindblad K.E., Maier B., Sia D., Puigvehi M., Miguela V., Casanova-Acebes M., Dhainaut M., et al. β-Catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019;9:1124–1141. doi: 10.1158/2159-8290.CD-19-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scharping N.E., Rivadeneira D.B., Menk A.V., Vignali P.D.A., Ford B.R., Rittenhouse N.L., Peralta R., Wang Y., Wang Y., DePeaux K., et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 2021;22:205–215. doi: 10.1038/s41590-020-00834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raggi C., Taddei M.L., Sacco E., Navari N., Correnti M., Piombanti B., Pastore M., Campani C., Pranzini E., Iorio J., et al. Mitochondrial oxidative metabolism contributes to a cancer stem cell phenotype in cholangiocarcinoma. J. Hepatol. 2021;74:1373–1385. doi: 10.1016/j.jhep.2020.12.031. [DOI] [PubMed] [Google Scholar]
- 33.Yamashita T., Honda M., Nakamoto Y., Baba M., Nio K., Hara Y., Zeng S.S., Hayashi T., Kondo M., Takatori H., et al. Discrete nature of EpCAM+ and CD90+ cancer stem cells in human hepatocellular carcinoma. Hepatology. 2013;57:1484–1497. doi: 10.1002/hep.26168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamashita T., Ji J., Budhu A., Forgues M., Yang W., Wang H.Y., Jia H., Ye Q., Qin L.X., Wauthier E., et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–1024. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deldar Abad Paskeh M., Mirzaei S., Ashrafizadeh M., Zarrabi A., Sethi G. Wnt/β-Catenin signaling as a driver of hepatocellular carcinoma progression: an emphasis on molecular pathways. J. Hepatocell. Carcinoma. 2021;8:1415–1444. doi: 10.2147/JHC.S336858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luke J.J., Bao R., Sweis R.F., Spranger S., Gajewski T.F. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin. Cancer Res. 2019;25:3074–3083. doi: 10.1158/1078-0432.CCR-18-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim C.G., Hong M.H., Kim K.H., Seo I.-H., Ahn B.-C., Pyo K.-H., Synn C.-B., Yoon H.I., Shim H.S., Lee Y.I., et al. Dynamic changes in circulating PD-1+ CD8+ T lymphocytes for predicting treatment response to PD-1 blockade in patients with non-small-cell lung cancer. Eur. J. Cancer. 2021;143:113–126. doi: 10.1016/j.ejca.2020.10.028. [DOI] [PubMed] [Google Scholar]
- 38.Huang J., Zhang L., Chen J., Wan D., Zhou L., Zheng S., Qiao Y. The landscape of immune cells indicates prognosis and applicability of checkpoint therapy in hepatocellular carcinoma. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.744951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nam D., Chapiro J., Paradis V., Seraphin T.P., Kather J.N. Artificial intelligence in liver diseases: improving diagnostics, prognostics and response prediction. JHEP Rep. 2022;4 doi: 10.1016/j.jhepr.2022.100443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alsaleh M., Barbera T.A., Reeves H.L., Cramp M.E., Ryder S., Gabra H., Nash K., Shen Y.L., Holmes E., Williams R., Taylor-Robinson S.D. Characterization of the urinary metabolic profile of cholangiocarcinoma in a United Kingdom population. Hepat. Med. 2019;11:47–67. doi: 10.2147/HMER.S193996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwabe R.F., Greten T.F. Gut microbiome in HCC - mechanisms, diagnosis and therapy. J. Hepatol. 2020;72:230–238. doi: 10.1016/j.jhep.2019.08.016. [DOI] [PubMed] [Google Scholar]
- 42.Monge C., Xie C., Steinberg S.M., Greten T.F. Clinical indicators for long-term survival with immune checkpoint therapy in advanced hepatocellular carcinoma. J. Hepatocell. Carcinoma. 2021;8:507–512. doi: 10.2147/JHC.S311496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma L., Wang L., Khatib S.A., Chang C.W., Heinrich S., Dominguez D.A., Forgues M., Candia J., Hernandez M.O., Kelly M., et al. Single-cell atlas of tumor cell evolution in response to therapy in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Hepatol. 2021;75:1397–1408. doi: 10.1016/j.jhep.2021.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma L., Heinrich S., Wang L., Keggenhoff F.L., Khatib S., Forgues M., Kelly M., Hewitt S.M., Saif A., Hernandez J.M., et al. Multiregional single-cell dissection of tumor and immune cells reveals stable lock-and-key features in liver cancer. Nat. Commun. 2022;13:7533. doi: 10.1038/s41467-022-35291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Candia J., Bayarsaikhan E., Tandon M., Budhu A., Forgues M., Tovuu L.O., Tudev U., Lack J., Chao A., Chinburen J., Wang X.W. The genomic landscape of Mongolian hepatocellular carcinoma. Nat. Commun. 2020;11:4383. doi: 10.1038/s41467-020-18186-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. j. 2011;17:10. [Google Scholar]
- 47.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L., Wang S., Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- 49.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shyr C., Tarailo-Graovac M., Gottlieb M., Lee J.J.Y., van Karnebeek C., Wasserman W.W. FLAGS, frequently mutated genes in public exomes. BMC Med. Genomics. 2014;7:64. doi: 10.1186/s12920-014-0064-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilkerson M.D., Hayes D.N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D., et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roessler S., Jia H.-L., Budhu A., Forgues M., Ye Q.-H., Lee J.-S., Thorgeirsson S.S., Sun Z., Tang Z.-Y., Qin L.-X., Wang X.W. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010;70:10202–10212. doi: 10.1158/0008-5472.CAN-10-2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gu Z., Gu L., Eils R., Schlesner M., Brors B. Circlize Implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 56.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 57.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
De-identified total RNA-seq and whole exome sequencing data is available at dbGaP: phs003074.v1.p1. Authorized access is required for individual level data as determined by the informed consent under which the data and samples were collected. These data can be requested through dbGaP.
-
•
All original code is available at Figshare: https://doi.org/10.6084/m9.figshare.22595983.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.