

Summary
Lung cancer in never-smokers (LCINS) presents clinicopathological and molecular features distinct from that in smokers. Tumor microenvironment (TME) plays important roles in cancer progression and therapeutic response. To decipher the difference in TME between never-smoker and smoker lung cancers, we conduct single-cell RNA sequencing on 165,753 cells from 22 treatment-naive lung adenocarcinoma (LUAD) patients. We find that the dysfunction of alveolar cells induced by cigarette smoking contributes more to the aggressiveness of smoker LUADs, while the immunosuppressive microenvironment exerts more effects on never-smoker LUADs’ aggressiveness. Moreover, the SPP1hi pro macrophage is identified to be another independent source of monocyte-derived macrophage. Importantly, higher expression of immune checkpoint CD47 and lower expression of major histocompatibility complex (MHC)-I in cancer cells of never-smoker LUADs imply that CD47 may be a better immunotherapy target for LCINS. Therefore, this study reveals the difference of tumorigenesis between never-smoker and smoker LUADs and provides a potential immunotherapy strategy for LCINS.
Keywords: lung cancer in never-smokers, adenocarcinoma, single-cell RNA sequencing, tumor microenvironment, CD47
Graphical abstract
Highlights
-
•
Cancer cells of never-smoker LUADs are less aggressive than smoker ones
-
•
Tumor microenvironment of never-smoker LUADs exerts more immunosuppressive properties
-
•
Cancer cells of never-smoker LUADs display higher expression of CD47
-
•
SPP1hi pro cells may be another independent source of monocyte-derived macrophages
Luo et al. employ single-cell RNA sequencing to identify more immunosuppressive microenvironment and higher expression of the immune checkpoint CD47 in never-smoker lung adenocarcinomas. These findings uncover the difference of tumorigenesis between never-smoker and smoker lung adenocarcinomas and provide a potential immunotherapy strategy for the former.
Introduction
Although tobacco smoking is one of the most important risk factors for lung cancer, there are still approximately 25% of patients with lung cancer who smoked fewer than 100 cigarettes in their lifetime, which is defined as lung cancer in never-smokers (LCINS).1 Compared with lung cancer in smokers (LCIS), LCINS exhibits distinct genomic architecture, including a lower tumor mutational burden (TMB), nucleotide base substitution dominated by C>T, and more frequent somatic alterations in EGFR, ERBB2, ALK, and ROS1.2,3 The prevalence of these genetic alterations confers more benefits from targeted therapy in LCINS, which may partially explain the better prognosis of LCINS patients.4,5 However, LCINS showed much less satisfactory response to anti-programmed death-ligand 1 (PD-L1) immunotherapy,6 suggesting that the tumor microenvironment (TME) of LCINS may be different from that of LCIS as TME plays important roles in immunotherapy. Therefore, understanding of the complex TME of LCINS should be helpful to improve its precise treatment strategies.
TME is a complex ecosystem composed of heterogeneous cancer cells, infiltrating immune cells, and stromal cells, whose interactions collectively determine tumor development as well as the response to therapy.7 For example, cytotoxic T lymphocytes in TME are often associated with favorable prognosis,8,9 as these T lymphocytes not only kill cancer cells but also inhibit tumor angiogenesis by secreting IFN-γ.10 Macrophage is a plastic and heterogeneous cell population in TME and plays a crucial role in tumor evolution and progression.11 Recently, single-cell RNA sequencing (scRNA-seq) has emerged as a powerful technology to investigate TME at a higher resolution and is widely employed to dissect the composition of TME in lung cancer.12,13,14,15,16,17,18,19 There are limited studies on the LCINS ecosystem,20 but a comprehensive depiction of the LCINS ecosystem especially for the Asian population at the single-cell resolution remains elusive.
Given that lung adenocarcinoma (LUAD) is the predominant histological type of LCINS,5 we conducted scRNA-seq to study the differences of TME between never-smoker and smoker LUADs. Our results provide a comprehensive insight into the diversity of cancer cells and immune cells, as well as the interaction among different cell types within the TME of never-smoker LUADs at the single-cell level. These findings improve our understanding of the oncogenesis of never-smoker LUADs and highlight potential therapeutic strategies for never-smoker LUADs.
Results
Increasing prevalence of never-smoker LUADs
To characterize never-smoker LUADs, we first performed a real-world analysis of 8,396 Chinese patients diagnosed with LUAD in West China Hospital, Sichuan University, from January 2009 to December 2016. More than half of the patients (59.4%) were never-smokers, with an increasing trend of proportion in all LUAD cases across 8 years (Figure S1A). Compared with smoker LUADs, never-smoker LUADs were more common among females (79.2% vs. 4.7%, p < 0.001; Figure S1B; Table S1) and were diagnosed at a younger age (mean age, 57.80 vs. 59.12 years, p < 0.001; Figure S1C; Table S1), which were similar to other studies.21,22,23 In terms of prognosis, never-smokers had significantly better overall survival (OS) than smokers (Figure S1D), which was also previously reported.4,24 By multivariate Cox regression analysis, presence of smoking history was an independent risk factor for poorer OS in LUAD patients (hazard ratio [HR], 1.31; 95% confidence interval [CI], 1.17–1.46; p < 0.001; Table S2). Besides, consistent with previous studies,2,3 analysis of genetic alterations in LUADs indicated that EGFR mutations were much more common in never-smoker LUADs (62.9% vs. 41.8%, p < 0.001; Figure S1E; Table S1), while KRAS was more frequently mutated in smoker ones (7.5% vs. 21.8%, p < 0.001; Figure S1E; Table S1). Moreover, PD-L1 expression was significantly lower in never-smoker LUADs than smoker ones (negative [<1%], 80.2% vs. 65.7%, p < 0.001; low expression [1%–49%], 15.0% vs. 20.7%, p < 0.001; high expression [≥50%], 4.8% vs. 13.6%, p < 0.001; Figure S1F; Table S1), supporting the worse response to anti-PD-L1 immunotherapy in never-smoker LUADs.6 Taken together, never-smoker LUADs exhibit distinct clinical and molecular features from smoker ones, highlighting the importance of exploring the TME of never-smoker LUADs at single-cell resolution.
Single-cell expression atlas and cell-type identification in LUAD
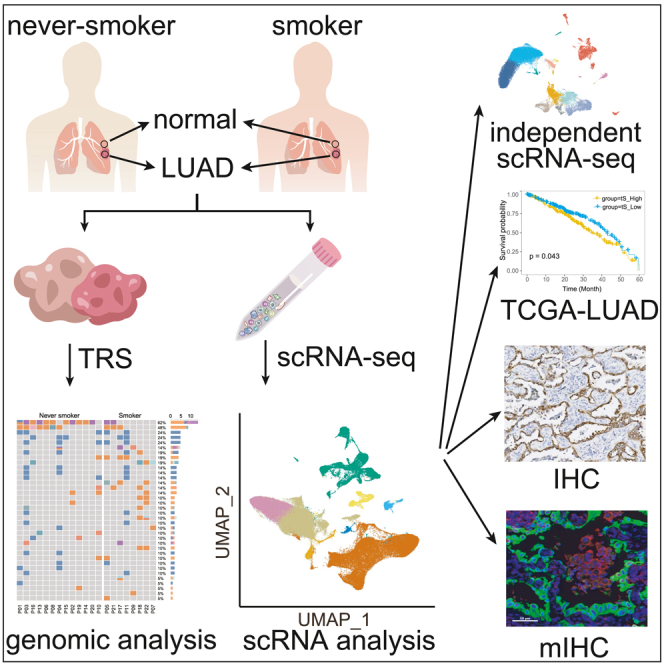
To elucidate the distinct TME of never-smoker LUADs, tumor specimens and their available paired distal normal lung tissues from 22 LUAD patients (13 never-smokers and nine smokers) were subjected to scRNA-seq (Figure 1A). Detailed clinical and pathological information, including age, gender, smoking status, and tumor-node-metastasis (TNM) stage, are listed in Table S3. First, we conducted 1,021-gene-panel targeted region sequencing (TRS) to confirm the smoking status of our enrolled patients at the genomic level. Consistent with previous studies,2,3 never-smoker LUADs harbored more EGFR mutations (84.6% vs. 25.0%, p = 0.018), fewer KRAS mutations (0.0% vs. 25.0%, p = 0.058), and lower TMB (median, 3.36 vs. 4.80 mutations/Mb, p = 0.08) than smoker ones in this study (Figures 1B and 7C; Table S4). Therefore, our enrolled samples represent LUADs with definite smoking status, and the scRNA-seq investigation on these samples will help to study the difference of TME between never-smoker and smoker LUADs.
Figure 1.

The mutational landscape and comprehensive single-cell atlas of never-smoker and smoker LUADs
(A) Illustration of the workflows in this study. nNS/nS, normal lung tissue from never-smoker/smoker; tNS/tS, tumor tissue from never-smoker/smoker; TRS, targeted region sequencing; scRNA-seq, single-cell RNA sequencing; TCGA, The Cancer Genome Atlas; IHC, immunohistochemistry; mIHC, multiplex immunohistochemistry.
(B) Mutational landscape of never-smoker and smoker LUADs included for scRNA-seq analysis. The frequencies of genetic alterations are shown on the right and presented in a decreasing order. The patient IDs are presented at the bottom. The colors denote different types of genetic alterations.
(C) UMAP view of 165,753 cells from 22 patients, colored by eight major cell types; epithelial cells are circled.
(D) UMAP view of cell types in different types of tissues.
(E) Dot plot of mean expression of canonical marker genes for eight major cell types from LUADs.
(F) Tissue preference of NK cells (left) and B lymphocytes (right) in tumor tissues and paired normal lung tissues from never-smokers (n = 10) and smokers (n = 7). The observed-to-expected (o/e) ratio is the relative score of observed cell numbers over expected cell numbers calculated by chi-squared test. Each box represents the interquartile range (IQR) between the 25th and 75th percentile with the mid-point of the data, and whiskers indicate the upper and lower value within 1.5 times the IQR. Dots represent different patients. Paired two-sided Wilcoxon signed-rank test, ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Asterisks represent the significance of differences in the following comparisons: all tumor tissues vs. all normal tissues (red); nNS vs. tNS and nS vs. tS (green).
Figure 7.

Cell-cell communication networks among cell types in LUAD
(A) Capacity for intercellular communications among cell types. The lines connect to the cell types that express the cognate receptors. The line thickness is proportional to the number of ligands when cognate receptors are present in the recipient cell types.
(B) Heatmap depicting the enrichment of the different cells in four tissue types (left), and the dot plot showing mean expression of genes associated with immunosuppression in each cell type (right).
(C) Violin plots with included boxplot of TMB in never-smoker and smoker LUADs; single-ended t test.
(D) Violin plots with included boxplot of relative PD-L1 expression in cancer cells from never-smoker and smoker LUADs; two-sided Wilcoxon signed-rank test. Outliers less than Q1 − 1.5 IQR or greater than Q3 + 1.5 IQR were removed.
(E) Violin plots with included boxplot of relative CD47 expression in cancer cells from never-smoker and smoker LUADs; two-sided Wilcoxon signed-rank test.
(F) Multiplex IHC staining of PD-L1 and CD47 in LUAD tumor tissues from never-smokers and smokers. Nuclei with DAPI (blue), PanCK (green), PD-L1 (red), and CD47 (yellow). Scale bars, 50 μm. At least three independent samples per tissue types.
(G) Dot plot of MHC class I gene expression in the four types of tissue samples (nNS/nS/tNS/tS).
Tumor tissues and normal lung tissues from never-smokers/smokers are referred to as tNS/tS and nNS/nS, respectively. Based on the expression of canonical marker genes (Table S5), we cataloged 165,753 quality-control-passed cells into eight major cell types (Figures 1C–1E and S2A), including epithelial cells, immune cells (T lymphocytes, natural killer [NK] cells, B lymphocytes, myeloid cells, and mast cells), and stromal cells (fibroblasts and endothelial cells). We found that immune and stromal cells from different patients clustered together by cell types, while epithelial cells showed high heterogeneity among different patients (Figures 1C and S2B). The most abundant cells identified were T/NK lymphocytes and myeloid cells (Figures 1C and S2B–S2D; Table S6). Furthermore, we confirmed that tumor tissues had a lower proportion of NK cells and higher proportion of B lymphocytes than normal lung tissues (Figure 1F), indicating the activation of adaptive immune response in TME. Notably, NK cells in tNS declined more dramatically than those in tS, suggesting that the TMEs of never-smoker LUADs are more likely to impair the homing of NK cells.
Less aggressiveness of cancer cells within never-smoker LUADs
A total of 24,965 epithelial cells from tumors and normal lung tissues were obtained. Among them, epithelial cells from normal lung tissues were further clustered into five distinct subtypes, including alveolar type I (AT1: AGER and RTKN2), alveolar type II (AT2: SFTPC and SFTPD), basal (KRT15 and KRT17), ciliated (FOXJ1, CAPS, and TPPP3), and club cells (SCGB1A1) as well as one undefined population with high expression of genes related to club, ciliated, and lymphocyte infiltration (CCL4, CD52, CORO1A, and CXCR4) (Figures S3A and S3B; Table S5). Considering that there may be residual non-malignant cells in tumor tissues, we applied inferring copy number variations (inferCNVs) to distinguish cancer cells from non-malignant cells (Figure S3C). The Uniform Manifold Approximation and Projection (UMAP) plot demonstrated that the identified non-malignant cells from tumor tissues overlay with epithelial cells from normal lung tissues (Figures 2A and 2B). We observed that cancer cells exhibited strong heterogeneity among different patients, while epithelial cells from normal lung tissues showed no heterogeneity (Figures 2A, 2B, S3D, and S3E). To sharpen the difference between cancer cells and normal epithelial cells, we excluded these non-malignant cells from tumor tissues in subsequent analyses.
Figure 2.

Identification of transcriptional signatures for cancer cells from never-smoker and smoker LUADs
(A) UMAP view of all epithelial cells including epithelial cells from normal lung tissues, cancer cells, and non-malignant cells from tumor tissues identified by inferCNV analysis.
(B) UMAP view of subtypes of epithelial cells by tissue types.
(C) Boxplots showing the expression levels of AT2 signature across four tissue types. Two-sided Wilcoxon signed-rank test, ∗∗∗p < 0.001. Asterisks represent the significance of differences in the following comparisons: nNS vs. nS/tNS (black), nS vs. tS (red), and tNS vs. tS (green).
(D) mIHC staining of SFTPC in the four types of tissue samples (nNS/nS/tNS/tS). Nuclei with DAPI (blue) and SFTPC (green). Scale bars, 50 μm. The right side shows the SFTPC staining positivity in each tissue type. Each group contains three samples. Three random fields are derived from each sample. Two-sided Wilcoxon signed-rank test, ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Asterisks represent the significance of differences in the following comparisons: nNS vs. nS/tNS (black), nS vs. tS (red), and tNS vs. tS (green).
(E) Heatmap of signatures for tNS and tS identified by Seurat FindMarkers function (fold change >2).
(F) IHC staining of specific genes from tNS/tS signatures in the four types of tissue samples (nNS/nS/tNS/tS). Scale bars, 100 μm. The immunoreactive scores (IRSs) of IHC in each tissue type are shown at the bottom. Each group contains three samples. Three random fields are derived from each sample. Data are represented as mean ± SD. Two-sided Wilcoxon signed-rank test, ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Asterisks represent the significance of differences in the following comparisons: tNS vs. tS/nNS/nS (black), and tS vs. tNS/nNS/nS (red).
(G) Kaplan-Meier curves for the OS of TCGA-LUAD patients (n = 453), which were divided into two groups according to the mean expression of tS signature. Plus signs (+) indicate censored observations; p value was calculated using the two-sided log rank test.
(H–J) UMAP view of cancer cells, colored by clusters (H), patients (I), and smoking status (J).
(K) Boxplots showing the expression levels of the MHC-II signature across 16 clusters of cancer cells.
Alveolar cells comprise AT1 and AT2 subtypes, of which the latter is considered to be the origin of LUAD.25,26,27,28,29 Previous studies have shown that AT2 cells act as stem-like progenitor cells that contribute to alveolar renewal, repairment, and carcinogenesis.28 Using the established AT2 marker genes, including SFTPB, SFTPC, SFTPD, and SFTA3 (Table S7),28 we compared the expression of AT2 signature among four tissue types (tNS, tS, nNS, and nS). The results showed that tumor tissues (tNS/tS) expressed lower AT2 signatures than normal lung tissues (nNS/nS) (Figure 2C). Moreover, smoker tissues (tS/nS) demonstrated lower AT2 signatures than never-smoker tissues (tNS/nNS), regardless of tumor and normal lung tissues. These findings were validated by the multiplex immunohistochemistry (mIHC) staining of SFTPC, a marker of AT2 cells (Figure 2D) and by analyses of published LUAD single-cell data (GEO: GSE131907; Figure S4A).15 Given that cigarette smoke causes injury to the lung epithelium and is a definite exogenous carcinogen for lung cancer,30 our results suggested that smoking induces impairment of normal lung epithelial cells. In addition, we explored whether AT2 signature is a clinically relevant biomarker for patient survival and found a significant association between high expression of AT2 signature and improved OS in The Cancer Genome Atlas (TCGA) LUAD bulk RNA-seq dataset (p = 0.0045; Figure S4B), which could partially explain the better prognosis of never-smoker LUAD patients.
To deepen our understanding of the transcriptional signatures of cancer cells, we employed Seurat FindMarkers function and identified an upregulated 14 genes as tNS signature and 30 genes as tS signature, respectively (Figures 2E and S4C). Among them, four genes (CTSH, LPCAT1, GPR116, and PABPC1) in the tNS signature and one gene (MIF) in the tS signature were validated by immunohistochemistry (IHC) analysis (Figure 2F). Gene Ontology (GO) enrichment analysis revealed that different signal pathways were involved in never-smoker and smoker LUADs. For example, the tNS signature was associated with surfactant homeostasis and phospholipid biosynthesis and metabolism, while the tS signature was related to tumor biological properties including cell growth and migration (Figure S4D), implying more aggressiveness of tS cancer cells. Furthermore, higher expression of tS signature was associated with unfavorable survival (p = 0.043; Figure 2G).
To further zoom in cancer cells, we performed sub-clustering of all cancer cells and obtained 16 clusters. Most clusters from smokers were dominated by a single patient, while several clusters from never-smokers had cells from several patients. This indicates that cancer cells in smokers showed stronger heterogeneity than those in never-smokers (Figures 2H–2J and S4E). Intriguingly, we found the cluster C9 containing cancer cells from 17 patients, most of which were from never-smoker LUADs, suggesting that the expression pattern of C9 may partially represent common features of cancer cells in never-smoker LUADs. To decipher the transcriptional features of C9, we performed GO enrichment analysis and found that upregulated genes in C9 were involved in antigen processing and presentation especially via major histocompatibility complex (MHC) class II (Figure S4F). We further scored each cancer cell clusters with MHC-II marker genes (Table S7) and observed that C9 had a significant higher score of MHC-II signature (Figure 2K). Besides, we found another two tNS clusters, C3 and C15 (Figure 2K), also manifested with high MHC-II signature. Furthermore, we found that cancer cells from tNS had higher expression of MHC-II (Figure S4G), which was validated by mIHC staining in tNS and tS samples (Figure S4I) and by analysis of published scRNA-seq data from LUAD (GSE131907) (Figure S4H).15 Previous studies revealed that tumor-specific MHC-II molecules correlate with a better CD4+ T lymphocytes infiltration and activation in the tumor mass.31,32 In our study, we also found that higher proportions of both naive and active CD4+ T lymphocytes exist in tNS than in tS (Figure 7B left), indicating that cancer cells from never-smoker LUADs may prompt better CD4+ T cell infiltration and activation in the tumor mass to trigger anti-tumor immune response. Given that higher expression of MHC-II on cancer cells was associated with favorable prognosis in patients with cancer,31,32,33 higher expression of MHC-II in cancer cells of never-smoker LUADs may partially explain the better prognosis of never-smoker LUADs.
Identification of SPP1hi pro cells and their potential interconversion with MDMs
Myeloid cells (n = 47,580) were sub-clustered into 20 clusters that were subsequently assigned to known cell lineages based on the expression of classical marker genes: macrophages (tissue-resident macrophage [TRM; C0, C1, C5, C12, C15], monocyte-derived macrophage [MDM; C2, C4, C6, C8, C10, C13]), monocytes (CD14+ [C3] and CD16+ [C11] monocytes), dendritic cells (DCs; cDC1s [C17], cDC2s [C7, C14], activated DCs [C18], and pDCs [C16]), and neutrophils (C19) (Figures 3A, 3B, and S5A–S5C). Among these cell types, both CD14+ and CD16+ monocytes were depleted in tumor tissues (Figures 3F, S5D, and S5E). Macrophages have been reported to comprise two distinct lineages, TRM and MDM, with TRM self-renewed locally and short-lived MDM arising from adult hematopoietic stem cells,34,35 which were both found in our study (Figure 3A). Interestingly, we found another macrophage cluster (C9) with mixed characteristics, exhibiting high expression of cell-cycle genes (MKI67, STMN1, and TUBB), TRM marker genes (MARCO, FABP4, and C1QA), and MDM marker genes (APOE, SPP1, and CCL2) (Figures 3B and S5C), so we called this population of macrophage with potential proliferation property Mixed_mac.
Figure 3.

Identification of SPP1hi pro cells from myeloid cells
(A) UMAP view of myeloid cells, colored by cell subtypes.
(B) Heatmap of mean expression of known marker genes in each myeloid subtype.
(C) Trajectory of MDM, TRM, and Mixed_mac inferred by Monocle2.
(D) Volcano plot of differentially expressed genes between FABP4hi pro and SPP1hi pro cells.
(E) Dot plot of mean expression of selected marker genes in four populations of macrophages. FABP4, INHBA, and MARCO are the marker genes of TRM; SPP1 is the marker gene of MDM; and TYMS, STMN1, MKI67, BIRC5, and TUBB are proliferation-related marker genes.
(F) Tissue preference of monocytes (CD14+ and CD16+ monocytes), TRM, MDM, FABP4hi pro, and SPP1hi pro in tumor tissues and paired normal lung tissues from never-smokers (n = 10) and smokers (n = 7). The o/e ratio is the relative score of observed cell numbers over expected cell numbers calculated by chi-squared test. Paired two-sided Wilcoxon signed-rank test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Asterisks represent the significance of differences in the following comparisons: all tumor tissues vs. all normal tissues (red); nNS vs. tNS and nS vs. tS (green).
(G) mIHC staining of PanCK, CD68, and FABP4 in tumor tissues of never-smokers and smokers. Nuclei with DAPI (blue), PanCK (white), CD68 (green), and FABP4 (red). Each group contains three samples. Scale bar, 50 μm. The yellow arrow indicates the CD68+FABP4+ cells.
(H) Proportion of SPP1hi pro and MDM in the four types of tissue samples (nNS/nS/tNS/tS). Paired two-sided Wilcoxon signed-rank test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Asterisks represent the significance of differences in the following comparisons: all tumor tissues vs. all normal tissues (red); nNS vs. tNS and nS vs. tS (green).
(I) Trajectory of MDM, monocytes, and SPP1hi pro cells inferred by Monocle2.
To determine the transcriptional state of Mixed_mac, we performed trajectory analysis of three macrophage populations (TRM, MDM, and Mixed_mac). The results showed that the Mixed_mac cells can be divided into two parts: one (C9-1) located close to TRM and the other (C9-2) close to MDM (Figure 3C). Differential gene expression analysis between these two populations demonstrated that C9-1 expressed a high level of TRM marker genes (FABP4, MARCO, and INHBA), while C9-2 exhibited high expression of an MDM marker gene (SPP1) (Figures 3D and S5A); thus, we named these two populations of Mixed_mac FABP4hi pro and SPP1hi pro, respectively. Collectively, macrophages in our study were divided into four populations (TRM, MDM, FABP4hi pro, and SPP1hi pro) with distinct transcription patterns (Figures 3E and S6A).
Further distribution analysis of these four macrophage populations revealed that tumor tissues harbored less TRM and more MDM than normal lung tissues (Figures 3F, S5D, and S5E), consistent with the previous study.36 Moreover, TRM in tS remarkably decreased compared with that in nS, while there was no significant change between tNS and nNS (Figure 3F), which was validated by mIHC staining of CD68+FABP4+ cells in patient samples (Figure 3G). Importantly, we found that FABP4hi pro and TRM had similar distribution (Figure 3F), and SPP1hi pro showed the same tendency with MDM among four tissue types (Figures 3F and 3H).
Based on our trajectory result (Figure 3C), UMAP plot of myeloid cells (Figures 3A and S5A), and distribution tendency of these four macrophage populations (Figure 3F), we concluded that FABP4hi pro may refresh TRM, confirming the renewal ability of TRM.34,36 Likewise, we reasoned that SPP1hi pro could generate MDM. Given that MDM was reported to be derived only from monocytes,34,35 we performed trajectory analysis among MDMs, SPP1hi pro, and monocytes. The results showed that both SPP1hi pro cells and monocytes could be independent sources of MDMs (Figures 3I, S6B, and S6C). Moreover, SPP1hi pro cells were mainly found in tumor tissues, especially in tNS samples (Figure 3H), suggesting its contribution to more MDM production in TME to promote LUAD oncogenesis, especially for never-smokers. Importantly, single-cell analysis of another four independent never-smoker LUADs confirmed the existence of SPP1hi pro in TME (Figures S6D–S6G), implying the important role of SPP1hi pro in tumorigenesis.
Macrophages within never-smoker LUADs exert more immunosuppressive properties
Macrophages have high plasticity and are usually polarized into two phenotypes, the “classically activated” M1 and “alternatively activated” M2.37 M1 macrophages are considered as a pro-inflammatory and anti-tumor phenotype, while M2 macrophages act in an anti-inflammatory and pro-tumor role. Using M1/M2 marker genes (Table S7), we confirmed the co-existence of M1 and M2 macrophages in LUADs (Figure 4A). Both TRM and MDM in tumor tissues showed lower M1 scores and higher M2 scores than those in normal tissues (Figures 4B–4E), indicating the immunosuppressive property of TRM and MDM in LUADs. Moreover, we found that TRM in tNS had higher M2 score than that in tS (Figure 4E), which was contributed by higher expression of partial M2 marker genes, such as CTSA, CTSB, CTSC, CTSD, and CCL18 in tNS (Figure 4F). Besides, the anti-inflammatory score showed a similar tendency to the M2 score in TRM among four tissue types (Figures 4G and 4H). Collectively, these findings indicated that TRMs exert a stronger immunosuppressive and pro-tumorigenic role in never-smoker LUADs.
Figure 4.

Characterization of macrophage populations in LUADs
(A) Scatterplots of mean expression of M1 and M2 marker genes in four types of macrophages from tNS (up) and tS (bottom).
(B–E) Violin plots with included boxplot of the M1/M2 scores in MDM (B and D) and TRM (C and E). Two-sided Wilcoxon signed-rank test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Asterisks represent the significance of differences in the following comparisons: all tumor tissues vs. all normal lung tissues (brown), nS vs. nNS (black), tS vs. tNS (red), and tNS vs. nNS and tS vs. nS (green).
(F and H) Dot plot of mean expression of M2 signature (F) and anti-inflammatory signature (H) in TRM.
(G, I, and J) Violin plots with included boxplot of anti-inflammatory scores in TRM (G) and AP scores in TRM (I) and MDM (J). Two-sided Wilcoxon signed-rank test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Asterisks represent the significance of differences in the following comparisons: all tumor tissues vs. all normal lung tissues (brown), nS vs. nNS (black), tS vs. tNS (red), and tNS vs. nNS and tS vs. nS (green).
(K) Violin plots with included boxplot of M1/M2 scores, anti-inflammatory scores, and AP scores in SPP1hi MDM from tNS and tS. Two-sided Wilcoxon signed-rank test, ∗∗∗p < 0.001
(L) Violin plots with included boxplot of PD-L1 expression in SPP1hi MDM from tNS and tS. Two-sided Wilcoxon signed-rank test, ∗∗∗p < 0.001
Macrophages play an important role as antigen presentation (AP) cells in adaptive immune response. Based on AP marker genes (Table S7), we calculated AP score and found that TRM/MDMs within tumor tissues had a higher score than those in normal lung tissues (Figures 4I and 4J), indicating activation of adaptive tumor immunity. Moreover, we found that both TRMs and MDMs in tNS had significantly lower AP score than those in tS (Figures 4I and 4J), implying the immunosuppressive microenvironment represented by macrophages in never-smoker LUADs.
Based on the aforementioned SPP1 expression in macrophages, we observed that two MDM clusters (C2, C10) of cells expressed a dramatically high level of SPP1 (Figure S6A) and named them as SPP1hi MDM. Notably, this subset of MDM almost exclusively existed in tumor samples (Figure S5B), prompting us to explore its effect on TME of LUADs. We compared the scores of specific features (M1, M2, anti-inflammatory, and AP) between tNS and tS (Table S7). The results showed that tNS exerted lower M1 and AP scores and higher M2 and anti-inflammatory scores than tS (Figure 4K), indicating that SPP1hi MDM played more immunosuppressive and pro-tumorigenic roles in the TME of never-smoker LUADs. Moreover, the immunosuppressive role of SPP1hi MDM in tNS was further confirmed by its higher expression of PD-L1 (Figure 4L), a biomarker of response to anti-PD-L1 immunotherapy.38 Taken together, both TRM and MDM in never-smoker LUADs showed more immunosuppressive roles than those in smoker LUADs.
T/NK cells show more suppressive anti-tumor immunity in never-smoker LUADs
We performed sub-clustering on 80,194 T/NK cells to identify CD4+ T lymphocytes (naive CD4+, active CD4+, regulatory T [Treg]), CD8+ T lymphocytes (GZMB CD8+, GZMK CD8+, and exhausted T cells) and NK cells (CD16+ and CD16−) (Figures 5A, 5B, S7A, and S7B). Among these cells, Treg cells increased in tumor tissues (Figure 5C), similar to other reports.13,16,17,18 Moreover, Treg cells increased more dramatically in tNS than in tS (Figure 5D), which could be explained by more TRM in tNS (Figure 3F), as TRM is reported to recruit, differentiate, and expand Treg cells.36 As a predominant subgroup of cytotoxic NK cells, CD16+ NK cells decreased in tumor tissues, especially in tNS (Figure 5C). Combined with our previous observation that NK cells declined more dramatically in tNS (Figure 1E), we infer that impaired immunity mediated by NK cells is associated with both reduced numbers and impaired function. However, both GZMK+ and exhausted CD8+ T cells showed no significant difference between tumor and normal tissues (Figure 5C).
Figure 5.

Reprograming of T/NK lymphocytes toward pro-tumor phenotype in LUAD
(A) UMAP view of T/NK lymphocytes colored by cell subtypes.
(B) Heatmap of selected T/NK lymphocytes marker genes in each cell clusters. Top: tissue preference of each cluster. Bottom: relative expression map of known marker genes associated with each cell subset. Mean expression values were scaled by mean centering.
(C) Tissue preference of T/NK subtypes; the o/e ratio is the relative score of observed cell numbers over expected cell numbers calculated by chi-squared test. Paired two-sided Wilcoxon signed-rank test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Asterisks represent the significance of differences in the following comparisons: all tumor tissues vs. all normal tissues (red), and nNS vs. tNS and nS vs. tS (green).
(D) Boxplots showing the Treg fractions (in all CD4+ lymphocytes) across four tissue types. Paired two-sided Wilcoxon signed-rank test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Asterisks represent the significance of differences in the following comparisons: all tumor tissues vs. all normal lung tissues (brown), nS vs. nNS (black), tS vs. tNS (red), and tNS vs. nNS and tS vs. nS (green).
(E and F) Volcano plot of differentially expressed genes in GZMB+ CD8+ T lymphocytes between tumor tissues and normal lung tissues from never-smokers (E) or smokers (F). Two genes (GZMA and GZMH) elevated in tumor sample of never-smoker LUADs are outlined in red.
(G) Dot plot of mean expression level of cytotoxicity related genes in the four types of tissue samples (nNS/nS/tNS/tS).
CD8+ T lymphocytes exert a cytotoxic role mainly by secreting granzymes such as GZMB, GZMK, GZMH, and GZMM. Among them, GZMB was reported to exert the greatest cytotoxic capacity.39 We found that GZMK CD8+ T cells showed no significant difference between tumors and normal tissues, but GZMB CD8+ T cells were significantly depleted in tumor tissues, especially in tNS (Figure 5C), indicating that the adaptive immune response of cytotoxic CD8+ T cells was more suppressed in never-smoker LUADs than smoker ones. To further explore the dysregulated molecules in GZMB CD8+ T cells of never-smoker and smoker LUADs, we performed differential gene expression analysis and found that the expression of GZMA and GZMH was elevated in tNS (Figures 5E–5G). According to marker genes of granule-mediated cytotoxicity, we found that GZMB CD8+ T cells in tNS exhibited lower expression of GZMB but higher expression of GZMA, GZMH, and GZMM than those in tS (Figure 5G). These results suggested that the number of GZMB CD8+ T cells significantly decreased in tNS but also that their function may be impaired. Besides, GZMK CD8+ T cells expressed a lower level of cytotoxic markers (GNLY, NKG7, and FGFBP2) than GZMB CD8+ lymphocytes, but meanwhile expressed certain levels of exhaustion markers (LAG3 and PDCD1) (Figure 5B). It was reported that GZMK CD8+ subset was the hallmark of inflammatory aging in mice and humans,40 thus this GZMK CD8+ T population may represent the transition state from effector to exhausted T cells in LUADs.
Stromal cells exert different pro-tumor roles in never-smoker and smoker LUADs
Stromal cells within tissues include endothelial cells (ECs) and fibroblasts. First, we identified five distinct subtypes of ECs, including lymphatic, tip-like, stalk-like, and tumor ECs, as well as endothelial progenitor cells (Figures 6A and 6B). Consistent with previous studies,15,16 we also found that tumor ECs mainly existed in tumor tissues (Figure 6C). To gain more insights into ECs, we performed gene set variation analysis (GSVA) to compare expression profiles of ECs within tumors and normal lung tissues (Figure 6D). The top enriched pathways in tumor tissues included epithelial-mesenchymal transition, Wnt/β-catenin signaling, and angiogenesis, which may contribute to LUAD progression. We further compared expression profiles of ECs between tNS and tS samples and found that the myogenesis pathway was upregulated in the ECs of tS, while the apoptosis pathway was upregulated in the ECs of tNS (Figure 6E), suggesting that ECs play different roles in never-smoker and smoker LUADs.
Figure 6.

Subsets of stromal cells identified in this study
(A) UMAP view of ECs, colored by cell subtypes.
(B) Heatmap showing the mean expression of canonical marker genes in different endothelial cell subtypes.
(C) The number and proportion of major endothelial cell subsets in each tissue type.
(D and E) Differences in pathway activities scored per cell by GSVA between tumor and normal ECs (D) and between tumor ECs in smokers and never-smokers (E).
(F) UMAP view of fibroblast cells, colored by cell subtypes.
(G) Heatmap showing the mean expression of canonical marker genes in different fibroblast cell subtypes.
(H) The number and proportion of major fibroblast cell subsets in each tissue type.
(I and J) Differences in pathway activities scored per cell by GSVA between tumor and normal fibroblast cells (I) and between tumor fibroblast cells in smokers and never-smokers (J).
Nine distinct subtypes of fibroblasts were identified in our data (Figures 6F and 6G), including COL13A1+, inflammatory fibroblast, lipofibroblast, mesothelial, MMP-high, myofibroblast, pericyte, smooth muscle cell, and universal P16+ fibroblasts. Moreover, we found that tumor tissues have more myofibroblasts than normal lung tissues (Figure 6H), supporting the role of myofibroblasts as the cancer-associated fibroblast to promote tumor progression.41 The GSVA analyses showed that the angiogenesis pathway was enriched in tumor tissues compared with normal lung tissues (Figure 6I). Furthermore, myogenesis was not enriched in tNS samples but in tS samples (Figure 6J), suggesting the different roles of fibroblasts in never-smoker and smoker LUADs.
Cell-to-cell interactions reveal CD47 as an immunotherapy target for never-smoker LUADs
To delineate the cell-to-cell communications in the TME of LUADs, we initially constructed the cellular interaction network using CellPhoneDB to identify the expression of ligands/receptors among major cell types, and we found that extensive communications existed between macrophage cells (TRM, MDM, FABP4hi pro, and SPP1hi pro) and other cell types, especially cancer cells (Figure 7A). Moreover, the contents of four macrophage subtypes were all higher in tNS than in tS (Figure 7B, left). Thus, macrophages contribute more to tumor development in never-smoker LUADs.
Clinically, LCINS shows less sensitivity to anti-PD-L1 immunotherapy than LCIS.6 We first analyzed the most common predictive biomarkers, including TMB and PD-L1, in our never-smoker and smoker LUADs. The results revealed that, compared with tS, tNS harbored lower TMB (p = 0.08; Figure 7C) and had lower PD-L1 expression on cancer cells (p < 2.22e−16; Figure 7D), which were validated by mIHC microscopy (Figure 7F). These results can explain the unsatisfactory response to anti-PD-L1 immunotherapy in never-smoker LUADs.
To explore a better immunotherapy strategy for never-smoker LUADs, we examined the expression of immune checkpoints in different cell types. Compared with PD-L1, CD47 and CEACAM1 showed much higher expression on cancer cells. Interestingly, SIRPA, the ligand of CD47, exhibited much higher expression on four macrophage subtypes (MDM, SPP1hi pro, TRM, and FABP4hi pro) than other immune cells or stromal cells (Figure 7B, right). In addition, it is worth noting that cancer cells of never-smoker LUADs displayed higher CD47 expression than those of smoker ones (Figure 7E), which was validated by mIHC staining (Figure 7F). As our observation that four macrophage subtypes existed more in tNS than in tS (Figure 7B, left), we conclude that the immune checkpoint CD47-SIRPA axis may exert a greater immunosuppressive effect in never-smoker LUADs. Importantly, clinical trials of anti-CD47 antibody in solid tumors (including lung cancer) are in progress.42 Because tumors with lower expression of MHC-I molecules in cancer cells were reported to be more sensitive to anti-CD47 antibody,43 we compared the expression of MHC-I molecules between tNS and tS. The results showed that tNS had lower expression of MHC-I molecules (HLA-A, HLA-B, and HLA-C) (Figure 7G), implying that never-smoker LUADs could benefit more from anti-CD47 immunotherapy. Therefore, CD47 may be a potential immunotherapy target for LUADs, especially for never-smoker ones.
Discussion
Compared with LCIS, LCINS harbors a higher prevalence of targetable driver gene mutations and benefits more from targeted therapies.44 However, the effect of immunotherapy represented by anti-PD-L1 antibody is not satisfactory in LCINS.6 Previous studies have investigated the molecular characteristics of LCINS through genomic, transcriptomic, and proteomic sequencing on bulk tissues.2,44,45,46 As patients with different smoking status showed distinct response to immunotherapies, comparing TME of patients with different smoking status may provide some clues for this phenomenon. However, to the best of our knowledge, no study explores cancer cells and TME in LCINS at a single-cell level and investigates their effects on tumor response to immunotherapy. In this study, we provide a comprehensive single-cell transcriptomic atlas to investigate cancer cell heterogeneity, immune modulation, and cellular interactions in never-smoker and smoker LUADs, which includes the largest sample size of never-smoker LUADs from Asia. This study improves our understanding of the oncogenesis of never-smoker LUADs and provides potential immunotherapy strategies for this lung cancer subset.
Intriguingly, we identified a population of macrophages with proliferation potential within TME and named it SPP1hi pro because it shows high expression of cell-cycle genes (MKI67, STMN1, and TUBB) and SPP1, an MDM marker gene (Figures 3C–3E). MDM was reported to be derived only from monocytes,36,47 but the trajectory analysis in this study indicated that SPP1hi pro is another independent source of MDM besides monocytes (Figure 3I). Moreover, most SPP1hi pro cells existed in tumor tissues (Figure 3H), implying that TME may mediate the generation of SPP1hi pro cells. Thus, we conclude that some MDMs in TME may differentiate into SPP1hi pro cells, which, in turn, generate MDMs due to their proliferation potential. In addition, more SPP1hi pro cells in tNS can contribute to more MDMs, and more MDMs in TME can exhibit a stronger immunosuppressive role. For example, CCL20-treated IDO+ MDM could induce the generation of immunosuppressive Treg-like cells.48 Hence, we hypothesize that more SPP1hi pro in tNS may produce more MDM in tNS, thus tNS may have stronger immunosuppressive TME. However, the immunosuppressive role of SPP1hi in tumor, especially in tNS, needs further investigations.
In our study, we found that dysfunction of alveolar cells induced by cigarette smoking contributes more to the aggressiveness of smoker LUADs, while the immunosuppressive environment exerts more effects on never-smokers’ aggressiveness. First, the AT2 signature, which is involved in repair response to injury such as stimulation of cigarette smoke,26 was much more downregulated in smoker LUADs than in never-smoker ones (Figure 2C). Second, the upregulated genes in tS were mainly involved in cell growth and migration (Figure S4D), contributing to tumor aggressiveness. Third, never-smoker LUADs have a unique subpopulation of cancer cells expressing high levels of MHC-II molecules (Figures 2K and S4G–S4I), which participates in AP and activation of anti-tumor immunity.49 These findings suggested that cancer cells from smoker LUADs were more aggressive.
In never-smoker LUADs, a more immunosuppressive microenvironment was revealed by our following findings. (1) More significantly reduced and impaired GZMB CD8+ T and NK cells exist in tNS (Figures 1E, 5C, and 5E–5G). These two immune cells in TME mainly exert cytotoxic effects through direct killing of cancer cells as well as secreting effector cytokines (TNF-α and IFN-γ).39,50,51 (2) tNS had more TRM and MDM (Figures 3F and 7B left), and TRM in tNS displayed stronger M2 (Figures 4E and 4F) and anti-inflammatory phenotype (Figures 4G and 4H). In addition, SPP1hi MDMs, one subpopulation of MDMs, also showed stronger pro-tumor property in tNS (Figure 4K). M2-phenotype macrophages are widely acknowledged as one of the central suppressive populations and create tumor associated macrophage (TAM) within TME.52 For example, TAM exerts immunosuppressive roles by limiting the activity of tumor-infiltrating lymphocytes (TILs).53 Depletion of TAM in mice harboring mammary tumors resulted in more inflammatory chemokines, such as CCL2 and CXCL10, which are likely to enhance the entry of T cells into the tumor and their intratumoral migration.53 (3) tNS harbored more FABP4hi pro and SPP1hi pro cells (Figures 3F and 7B, left), which can generate more TRMs and MDMs, respectively (Figures 3I and S5A), leading to more immunosuppressive properties.
Clinically, a large proportion of lung cancer patients have no benefits from anti-PD-L1 immunotherapy.54 Among them, never-smoker LUAD patients are the representative ones with poor response, which could be explained by our results that tNS expressed lower PD-L1 and harbored lower TMB (Figures 7C and 7D). Thus, researchers are trying to discover other potential targets. In our LUAD cohort, we found much higher expression of the immune checkpoint CD47 than PD-L1 on cancer cells (Figure 7B right). Moreover, the CD47’s ligand, SIRPA, exhibited higher expression on four macrophage subtypes (MDM, SPP1hi pro, TRM, and FABP4hi pro) (Figure 7B right). Given that the interaction between CD47 on cancer cells and its ligand SIRPα on the surface of macrophages within the TME can make cancer cells release a "don’t-eat-me" signal, allowing cancer cells to escape immune surveillance.55 Thus, our results showed that targeting the CD47-SIRPα axis may trigger macrophage-mediated elimination of cancer cells. Interestingly, the clinical trials of anti-CD47 are ongoing in advanced solid tumors, including lung cancer.42 More importantly, we further found that cancer cells in never-smoker LUADs showed higher CD47 than smoker ones (Figure 7E), indicating that anti-CD47 immunotherapy may be a better strategy for never-smoker LUADs. Furthermore, we observed lower expression of MHC-I molecules (HLA-A/B/C) in cancer cells of never-smoker LUADs (Figure 7G), which can protect cancer cells from phagocytosis by expressing β2-microglobulin.43 Collectively, anti-CD47 immunotherapy targeting the interaction between cancer cells and macrophages might be a feasible treatment for never-smoker LUADs.
In conclusion, cancer cells showed less aggressive roles while the immune environment displayed more immunosuppressive roles in never-smoker LUADs. Moreover, a subtype of macrophage called SPP1hi pro was identified to be another independent source of MDM within TME, which plays an immunosuppressive role. Importantly, targeting the CD47-SIRPAα immune checkpoint axis may be a better immunotherapy strategy for never-smoker LUADs. Therefore, this study improves our understanding of the tumorigenesis of never-smoker LUADs and provides a potential immunotherapy strategy.
Limitations of the study
In this study, we identified a population of macrophage with proliferation property from our scRNA-seq data and named it SPP1hi pro, which was cross-validated by the single-cell transcriptome analysis of four independent never-smoker LUADs. Unfortunately, we failed to visualize these cells in patients’ samples using mIHC staining. This could be due to the small number of SPP1hi pro cells within the TME as well as the lower sensitivity of antibodies. Therefore, mIHC staining with more sensitive antibodies and a larger cohort of samples is needed to experimentally confirm this finding. In addition, we revealed higher expression of CD47 in never-smoker LUADs by our scRNA-seq analysis and mIHC staining, but it still needs further validation in an expanded cohort of clinical specimens. Moreover, the function and the immunotherapy potential of CD47 for never-smoker LUADs should be extensively investigated in animal models in future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-SFTPC | Millipore | Cat# AB3786; RRID: AB_91588 |
| Rabbit monoclonal anti-FABP4 | Abcam | Cat# ab92501; RRID: AB_10562486 |
| Mouse monoclonal anti-CD68 | BioLegend | Cat# 916104; RRID: AB_2616797 |
| Rat monoclonal anti-FOXP3 | Thermo Fisher Scientific | Cat# 14-4776-80; RRID: AB_467553 |
| Mouse monoclonal anti-PanCK | Abcam | Cat# ab7753; RRID: AB_306047 |
| Rabbit monoclonal anti-CD47 | Abcam | Cat# ab226837 |
| Rabbit recombinant anti-PD-L1 | Abcam | Cat# ab213524; RRID: AB_2857903 |
| Rabbit polyclonal anti-CTSH | Proteintech | Cat# 10315-1-AP; RRID: AB_2087534 |
| Rabbit polyclonal anti-GPR116 | ImmunoWay Biotechnology Company | Cat# YT1960 |
| Rabbit monoclonal anti-LPCAT1 | Abcam | Cat# ab214034 |
| Rabbit polyclonal anti-PABPC1 | Cell Signaling Technology | Cat# 4992; RRID: AB_10693595 |
| Rabbit polyclonal anti-MIF | Atlas Antibodies | Cat# HPA041219; RRID: AB_10794268 |
| Rabbit polyclonal anti-CD4 | Abcam | Cat# ab133616; RRID: AB_2750883 |
| Mouse monoclonal anti-MHC Class II | Abcam | Cat# ab55152; RRID: AB_944199 |
| Biological samples | ||
| Lung cancer tissue and normal lung tissue collection (n = 22) | West China Hospital, Sichuan University | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Bovine Serum Albumin (BSA) | Roche | Cat# 10711454001 |
| Collagenase I | Biosharp | BS163 |
| Collagenase IV | Biosharp | BS165 |
| DAPI (4′,6-diamidino-2-phenylindole) | Thermo Fisher Scientific | R37606 |
| Dimethyl Sulfoxide (DMSO) | MP Biomedicals | Cat# 196055 |
| Fetal Bovine Serum (FBS) | Gibco | Cat# 10099-141C |
| Formalin | Fisher Scientific | Cat# SF98-4 |
| Hank’s Balanced Salt Solution (HBSS) | Gibco | Cat# 14175095 |
| Phosphate Buffered Saline (PBS) | Bio-Channel | BC-BPBS-01 |
| Parafin | Sigma Aldrich | P3683-1KG |
| Red blood lysis buffer | Biosharp | BL503A |
| Critical commercial assays | ||
| Chromium™ Single Cell 3′ Library & Gel Bead Kit v2 | 10X Genomics | PN-120237 |
| Chromium™ Single Cell A Chip Kit | 10X Genomics | PN-120236 |
| Chromium™ Single Cell 3′ Library & Gel Bead Kit v3 | 10X Genomics | PN-1000075 |
| Chromium™ Single Cell B Chip Kit | 10X Genomics | PN-1000073 |
| Chromium™ i7 Multiplex Kit | 10X Genomics | PN-120262 |
| Dead Cell Removal Kit | Miltenyi Biotec | Cat# 130-090-101 |
| DAB substrate kit | Abcam | Cat# ab64238 |
| GeneReadTM DNA FFPE Kit | QIAGEN | Cat# 180134 |
| NEBNext Ultra DNA Library Prep kit | NEB | E7370S |
| KAPA library quantification universal kit | Roche | Cat# 07960140001 |
| OncoScreen™ Plus Cancer Mutation Profiling Tissue Kit | Guangzhou Burning Rock Biotech | LK103 (C) |
| Oncology Multi-Gene Variant Assay | Geneplus Medical Laboratory | BE201911001 |
| Qubit® dsDNA BR Assay Kit | Invitrogen | Q32850 |
| Qubit® dsDNA HS Assay Kit | Invitrogen | Q32854 |
| The Opal Polaris 7-Color Manual IHC Kit | Perkin Elmer | NEL861001KT |
| Deposited data | ||
| TCGA LUAD bulk RNA-seq | The Cancer Genome Atlas (TCGA) | https://portal.gdc.cancer.gov/projects/TCGA-LUAD |
| GSVA Hallmark gene sets | The Molecular Signatures Database (MsigDB) | https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp#H |
| scRNA-seq data from Kim N et al.15 | Gene Expression Omnibus (GEO) | GSE131907 |
| Software and algorithms | ||
| Burrows-Wheeler Aligner (BWA) | Li and Durbin, 200956 | http://bio-bwa.sourceforge.net/ |
| MuTect2 1.1.4 | Benjamin et al., 201957 | https://gatk.broadinstitute.org/hc/en-us/articles/4414594392347-Mutect2 |
| GATK 4.1.2.0 | Benjamin et al., 201957 | https://github.com/broadinstitute/gatk/releases |
| Cellranger 3.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| Seurat 4.0.0 | Hao et al., 202158 | https://satijalab.org/seurat/ |
| SingleR | Bioconductor | https://www.bioconductor.org/packages/release/bioc/html/SingleR.html |
| inferCNV | Bioconductor | http://www.bioconductor.org/packages/release/bioc/html/infercnv.html |
| Monocle 2.0 | Qiu et al., 201759 | https://github.com/cole-trapnell-lab/monocle-release |
| GSVA 1.38.2 | Bioconductor | https://www.bioconductor.org/packages/release/bioc/html/GSVA.html |
| survival package | R | https://cran.r-project.org/web/packages/survival/ |
| survminer package | R | https://cran.r-project.org/web/packages/survminer/ |
| CellphoneDB2 | Efremova et al., 202060 | https://www.cellphonedb.org/ |
| GraphPad Prism 8.0.2 | GraphPad Software lnc. | http://www.graphpad.com/scientific-software/prism/ |
| IBM SPSS statistics 25 | IBM | https://www-01.ibm.com/software/analytics/spss |
Resource availability
Lead contact
Further information and requests for reagents may be directed to and will be fulfilled by the lead contact Weimin Li (weimi003@scu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
LUAD patient cohorts and biological samples
For the real-world analysis of never-smoker LUADs, patients with pathologically diagnosed primary LUAD in West China Hospital, Sichuan University from January 2009 to December 2016 were enrolled. A total of 8,396 LUAD patients were divided into never-smokers and smokers (Table S1). Smoking status were categorized into never-smokers (individuals smoke <100 cigarettes in their lifetime) and smokers. We collected clinicopathological information of each patient, including age, gender, previous history of malignancy, family history of malignancy, tumor stage and treatment by retrospective review of the electronic medical record. We performed a long-term follow-up of the patients for up to ten years. OS was defined as the time from the date of LUAD diagnosis to death or the last follow-up. In addition, the primary tumor samples from pathology-confirmed LUAD patients between January 2016 and December 2018 were examined by the 56-gene-panel TRS (Burning Rock Biotech, Guangzhou, China) (n = 600, including 375 never smokers and 225 smokers) and by IHC staining with anti-PD-L1 antibody (SP142, Abcam, #ab228462) using the Ventana Benchmark system (n = 3155, including 2101 never smokers and 1054 smokers). The tumor proportion score (TPS) of PD-L1 was defined as the proportion of positive tumor cells of any intensity, which was categorized into three groups (<1%, 1–49% and 50%). The study was approved by the Institutional Review Board of West China Hospital of Sichuan University (2018.270 and 2019.638).
For scRNA-seq analysis, 22 patients, including 13 never-smokers and 9 smokers, were prospectively included in this study. These patients received surgical resection without neoadjuvant therapy at West China Hospital from May 2018 to November 2020, and were pathologically confirmed as LUAD. Tumor tissues with/without their matched distal normal lung tissues were collected during the surgery and all subjects have provided their written informed consent. Distal normal lung tissues were obtained at the periphery region of the overall specimen/lobe. The histology of tumors was diagnosed according to the 2021 WHO Classification of Lung Tumors by two experienced lung pathologists.61 The normal lung tissues were also assessed and only those with no cancer cell infiltration were included for scRNA-seq analysis. The detailed information of included patients and corresponding samples for scRNA-seq analysis are shown in Table S3.
Method details
Tissue processing and preparation of single-cell suspensions
Immediately following collection, tumor tissues and distal normal lung tissues were immersed in Hank’s balanced salt solution (HBSS, Gibco), and rapidly transported to laboratory at a low temperature. These samples were quickly divided into two pieces; one-half was treated with enzymatic digestion and cell sorting for scRNA-seq, and the other was fixed in 10% neutral formalin solution and paraffin-embedded for TRS, IHC and mIHC staining.
For preparing single cells, tissues were minced with scissors on ice and digested well with 1 mg/mL collagenase I (BS163, Biosharp), 0.5 mg/mL collagenase IV (BS165, Biosharp) and HBSS. After 30 min of incubation at 37°C on a shaker, samples were filtered using the 40μm strainers (Corning), centrifuged at 500 x g for 5 min at 4°C and then removed the supernatant. The remaining cell pellet was treated with red blood lysis buffer (Biosharp) for 5 min to remove the red blood cells, followed by centrifugation once more for 5 min (500g, 4°C). After the removal of the supernatant, the samples were resuspended in the sorting buffer (0.04% BSA and PBS). The dead cells were removed using Dead Cell Removal Kit (Miltenyi Biotec, Germany) according to the manufacture’s protocol. The single-cell suspensions with a rate of cell viability greater than 80% were handled according to manufacturer’s instructions subsequently for scRNA-seq.
scRNA-seq library preparation and sequencing
The scRNA-seq libraries were prepared as per the manufacturer’s protocol, using the Chromium Single Cell 3′ Library, Gel Bead & Multiplex Kit and Chip Kit version 2 (10x Genomics) for patients P01-P12, and version 3 for patients P13-P22, respectively. The single-cell suspensions were loaded on the Chromium Single Cell Controller Instrument (10× Genomics) to generate single cell gel beads in emulsions (GEMs), aiming to capture a total of 8000–10000 cells per library. After the GEM generation, barcoded reverse transcription of RNA, cDNA clean-up, amplification by PCR for the appropriate number of cycles and purification with SPRIselect, the library was quantified using quantitative PCR (KAPA Illumina Quantification Kit) and quality controlled using LabChip GX Perkin-Elmer. Sequencing was carried out on a local Illumina NovaSeq6000 platform in West China Hospital.
Targeted region sequencing and genomic data analysis
Except for one insufficient tumor tissue, the genomic DNAs were extracted using GeneReadTM DNA FFPE Kit (QIAGEN) from these paraffin-embedded tumors and matched available normal lung tissues following the protocol of the manufacturer. The sequencing libraries were synthesized using the NEBNext Ultra DNA Library Prep Kit (NEB, Ipswich, MA) according to the manufactory’s protocol. TRS was performed using a customized 1021-gene-panel kit on the Gene+Seq-2000 Sequencing Platform. Sequencing reads were mapped to the human reference genome (hs37d5) using the Burrows-Wheeler Aligner (BWA).56 MuTect2 (version 1.1.4) and GATK(4.1.2.0) were applied to call single nucleotide variants (SNVs) and somatic indels, respectively. Variants with less than five high-quality supporting reads were removed. The called variants were annotated using ANNOVAR. The TMB was calculated based on the somatic non-synonymous variants.
Immunohistochemistry (IHC) staining and scoring
The 4-μm-thick formalin-fixed and parrffin-embedded (FFPE) tissue sections were dewaxed, hydrated, heat-induced antigen retrieved, and treated with 3% hydrogen peroxide for 15 min. Then, the sections were incubated overnight at 4°C with relevant primary antibody against CTSH (Proteintech, Cat#10315-1-AP), GPR116 (ImmunoWay Biotechnology Company, Cat#YT1961), LPCAT1 (Abcam, Cat#ab214034), PABPC1 (Cell Signaling Technology, Cat#4992) and MIF (Atlas Antibodies, Cat# HPA041219), respectively. After rinsed with distilled water, the sections were incubated with horseradish peroxidase-conjugated secondary antibody for 1 h and treated with diaminobenzidine (DAB) for color visualization. Finally, the sections were counterstained with hematoxylin, dehydrated, mounted and imaged using the AxioCamHR setup (Zeiss).
Semi-quantitative analysis was conducted using the immunoreactive score (IRS), which was calculated by multiplying the scores of percentage of positive cells (0–4) and scores of staining intensity (0–3). The scores of percentage of positive cells were defined as follows: 0, no positive cells; 1, <10% positive cells; 2, 10–50% positive cells; 3, 51–80% positive cells; and 4, >80% positive cells. The scores of staining intensity were defined as follows: 0, negative staining; 1, weak staining; 2, moderate staining; and 3, strong staining. All samples were scored by three persons in a blinded fashion, and the mean score was confirmed as the final IRS of each sample.
Multiplex immunohistochemistry (mIHC) staining
The 4 μm FFPE tissue sections were deparaffinized in xylene, rehydrated with graded ethanol and antigen retrieved. The stainings were performed using the Opal Polaris 7-Color Manual IHC Kit (Perkin Elmer) according to the manufacturer’s protocol. The Vectra Polaris multispectral Imaging System version 2 (PerkinElmer Vectra®) was used to scan the sections. Representative images were exported using QuPath software (version 0.3.2), and analyzed using inForm software (PerkinElmer). The antibodies used in these experiments were: SFTPC (Millipore, Cat# AB3786), FABP4 (Abcam, Cat# ab92501), CD68 (BioLegend, Cat# 916104), FOXP3 (Thermo Fisher Scientific, Cat# 14-4776-80) and PanCK (Abcam, Cat# ab7753), CD47 (Abcam, Cat# ab226837), PD-L1 (Abcam, Cat# ab213524), CD4 (Abcam, Cat# ab133616) and MHC-II (Abcam, Cat# ab55152).
Quantification and statistical analysis
Analysis of the real-world clinical data
Data were analyzed by IBM SPSS statistics 25, and plotted by GraphPad prism 8.0.2. Continuous variables were shown as mean ± SD, and were analyzed using two-sided Wilcoxon Signed-Ranked test. Categorical variables were presented as frequencies and percentages, and were analyzed using Chi-square test for intergroup comparisons. The Kaplan-Meier survival curves were generated for never-smoker and smoker LUAD patients. The Univariate and multivariate analyses were performed with the Cox proportional hazards model to identify independent prognostic factors affecting OS of the LUAD patients. The p value <0.05 was considered to be statistically significant.
Bioinformatics quality control and normalization of scRNA data
Cellranger version 3.0 (https://support.10xgenomics.com/single-cell-geneexpression/software/) was used to align FASTQ sequencing reads to the hg38 reference transcriptome, generating single-cell feature counts for each sample. We applied following quality measures on raw gene-cell-barcode matrix for each cell: gene count (ranging from 200 to 10,000), unique molecular identifiers (UMI, 300) and mitochondria gene ( 20%). In addition, we logarithmically quantified the complexity of cells according to the proportion of the number of gene detected per unit UMI, and discarded cells with a complexity less than 0.8. At the same time, we also filtered the genes expressed in less than 10 cells. For the remaining cells and genes, we defined relative expression by centering using the Seurat ScaleData function. The relative expression levels across the remaining subset of cells and genes were used for subsequent analysis.
Unsupervised dimensional reduction, clustering and cell annotation
Samples of patients P01-P12 were processed using V2 kits and samples of patients P13-P22 were processed using V3 kits. Hence, we used Seurat (v4.0.0) (https://satijalab.org/seurat/) canonical correlation analysis (CCA) to remove the batch effect of different kits. After data scale, we calculated principal components (PCs). We selected a subset of significant PCs using JackStraw and PCElBowPlot of Seurat and set different resolution values (0.4, 0.45, 0.5 … 0.8) for each PC. FindClusters and RunUMAP functions were used for cell clustering and UMAP visualization, respectively. We calculated the averaged Normalized Mutual Information (NMI) for each combination of PC and resolution, and the combination with the highest averaged NMI was chosen for the following analyses. To identify differentially expressed genes for cluster demarcation, the FindAllMarkers module was used, and genes expressed in more than 25% of the cells in each cluster were selected. Differentially expressed genes from each cluster were compared with sets of previously described cell-type markers to assign cell identities. The identities of cells in each cluster were determined by the combined results of SingleR automatic annotation, the differentially expressed genes of each cluster and the expression of known canonical marker genes listed in Table S5. The annotation results were checked manually and visualized by UMAP for the following analysis.
Tissue preference
We divided cells into four categories according to the origin of tissues: normal lung tissue from never-smoker (nNS), normal lung tissue from smoker (nS), tumor tissue from never-smoker (tNS), tumor tissue from smoker (tS). Firstly, we calculated proportion of each cell type in different tissues. We included 10 never smokers and 7 smokers with both tumor tissue and adjacent normal tissue, and calculated the o/e value of each cell type by Chi-square test. If the observed proportion of a cell type in a tissue type is much larger than the expression, it indicates that the cell type is enriched in the tissue type; otherwise, it indicates that the cell type is absent in the tissue type. We then performed a paired Wilcoxon test based on the o/e values of each cell type to see if a cell type differed significantly across tissue types.
Inference of CNV from scRNA-seq data
To exclude non-malignant tumor cells that may be contained in tumor tissues, we inferred the abnormality of copy number variations (CNVs) from the perturbation of chromosome gene expression. With the epithelial cells of adjacent normal tissues as the reference and the gene expression count matrix as input, the CNVs distribution was detected by inferCNV (https://github.com/broadinstitute/inferCNV). Genes with a mean count number of 0.1 or less across cells were excluded. We set the parameter cluster_by_groups False on account of obvious heterogeneity among the cancer cells from different samples, while the cells in cluster gathered with almost all samples were considered as non-malignant cells. In addition, the parameter denoise was set as TRUE in order to judge CNV more intuitively. The result of inferCNV was eventually visualized by heatmap.
Inference of the unsupervised trajectory
We first extracted the selected populations from the scRNA-seq data. Then we employed the Monocle (version 2)59 algorithm using high variable genes selected by the Seurat FindVariableFeature function as the input to determine the differential states between the selected populations. Using Monocle default parameters, a spanning tree was constructed by DDRTree algorithm for dimensionality reduction and cell ordering, so as to infer cell trajectories.
Marker gene selection specific to cancer cells
To identify specific genes for cancer cells of never-smoker or smoker LUADs, we used tNS/tS cancer cells to identify differentially expressed genes. We used the Seurat FindMarkers function to calculate the log2 fold change (log2FC) between the two groups (tNS cancer cells vs. Other Epithelial cells and tS cancer cells vs. Other Epithelial cells). The significance of the differences was determined using Wilcoxon test with Bonferroni correction. Genes were selected as signature genes according to the statistical thresholds: avg_log2FC > 1, p value <0.01, adjust p value (Bonferroni) < 0.01 and expressed more than 25% in either of the two cell groups. Specific genes were eventually visualized by heatmap.
Identification of signature genes
For comparisons between two selected specific groups (two mixed populations of macrophages estimated by the trajectory analysis, tumor cells vs. normal cells of GZMB CD8+ T lymphocytes in never-smokers or smokers) we calculated the log2 fold change (log2FC) between the selected two groups using the Seurat FindMarkers function. The significance of the differences was determined using Wilcoxon test with Bonferroni correction. Genes were selected as signature genes according to the statistical thresholds: average log2 fold change (avg_log2FC) > 1, p value <0.01, adjust p value (Bonferroni) < 0.01 and expressed more than 25% in either of the two cell groups. Differential genes were eventually visualized by volcano maps.
Signature scoring
Marker genes associated with specific signatures, including AT2, M1/M2, anti-inflammatory, and antigen presentation (Table S7) were curated form literature. The normalized weighted mean expression of those genes was calculated by Seurat’s AddModuleScore.
Gene set variation analysis (GSVA)
Pathway analyses were predominantly performed on the 50 hallmark pathways described in the molecular signature database (MSigDB).62 Pathway activity estimates were obtained using the GSVA package (version 1.38.2). The GSVA algorithm obtains the pathway enrichment score matrix by the given gene expression matrix and the marker gene set downloaded from MSigDB (H.all.v7.2). Then we used the lmFit analysis of the limma package to obtain the different pathways. p value was calculated by the limma package and FDR was also provided.
Survival analysis
RNA-seq and clinical data of LUAD samples were obtained from TCGA to evaluate the prognostic effects of gene sets derived from specific cell states. The RNA-seq data of 494 LUAD tumors were divided into two groups according to the mean expression of the target genes. Survival curves were fitted using the Kaplan–Meier formula in the R package ‘survival’, and visualized using the ggsurvplot function of the R package ‘survminer’.
Cell-cell interaction analysis
We mapped the receptor-ligand pairs using CellphoneDB2 (www.cellhonedb.org)60 onto our cell subsets to identify cell-cell interactions. This method infers the potential interaction strength between two cell subsets based on gene expression level, and provides the significance through permutation test (1000 times). For the following analysis, we selected the interaction relationship of p < 0.05 returned by CellPhoneDB, and removed the interaction pairs with collagens.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (to W.M.L., 92159302; to Y.P., 81821002), Fundamental Research Funds for the Central Universities (to W.M.L., SCU2022D025), Science and Technology Foundation of Sichuan Province, China (to Y.P., 2022YFS0046 and 2022ZYD0128; to W.X.L., 2022NSFSC0842; to Y.L.L., 2020YFS0572), and the 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (to Y.P., ZYGD20008; to W.M.L., ZYGD20009).
Author contributions
Conceptualization, Y.P. and W.M.L.; methodology, W.X.L., Z.Z., Y.J., and L.Y.; validation, W.X.L., Z.Z., Y.J., L.Y., Y.Y., M.L.Y., and Y.Q.L.; formal analysis, W.X.L., Z.Z., Y.J., L.Y., T.F., and Y.T.P.; investigation, W.X.L., Z.Z., Y.J., and L.Y.; resources, W.X.L., X.X., S.C., G.W.C., L.Y., and W.M.L.; data curation, W.X.L., Z.Z., Y.J., L.Y., T.F., Y.Y., M.L.Y., Y.Q.L., and G.W.; writing – original draft, W.X.L., Z.Z., Y.J., L.Y., and Y.Y.; writing – review & editing, W.X.L., Z.Z., Y.J., L.Y., Z.F.W., T.F., Y.Y., C.D.W., L.Z., Y.L.L., Y.P., and W.M.L.; visualization, Z.Z., Y.J., L.Y., and Y.Y.; supervision, Y.P. and W.M.L.; funding acquisition, Y.L.L., Y.P., and W.M.L.
Declaration of interests
The authors declare no competing interests.
Published: June 9, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101078.
Contributor Information
Yong Peng, Email: yongpeng@scu.edu.cn.
Weimin Li, Email: weimi003@scu.edu.cn.
Supplemental information
Data and code availability
-
•
The data of this study has been submitted to the Genome Sequence Archive for human (GSA for human) database. The accession number for the single cell sequencing data and 1021-gene-panel targeted region sequencing data reported in this paper is HRA002953, and the accession number for the single cell sequencing data used for verification in this study is HRA002992. They are publicly accessible in https://ngdc.cncb.ac.cn/gsa-human/, and the download of these data requires Data Access Committee (DAC) approval.
-
•
This paper does not generate the original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Sun S., Schiller J.H., Gazdar A.F. Lung cancer in never smokers--a different disease. Nat. Rev. Cancer. 2007;7:778–790. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- 2.Govindan R., Ding L., Griffith M., Subramanian J., Dees N.D., Kanchi K.L., Maher C.A., Fulton R., Fulton L., Wallis J., et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gou L.Y., Niu F.Y., Wu Y.L., Zhong W.Z. Differences in driver genes between smoking-related and non-smoking-related lung cancer in the Chinese population. Cancer. 2015;121(Suppl 17):3069–3079. doi: 10.1002/cncr.29531. [DOI] [PubMed] [Google Scholar]
- 4.Kerrigan K., Wang X., Haaland B., Adamson B., Patel S., Puri S., Akerley W. Real world characterization of advanced non-small cell lung cancer in never smokers by actionable mutation status. Clin. Lung Cancer. 2021;22:260–267.e2. doi: 10.1016/j.cllc.2021.01.013. [DOI] [PubMed] [Google Scholar]
- 5.Casal-Mouriño A., Valdés L., Barros-Dios J.M., Ruano-Ravina A. Lung cancer survival among never smokers. Cancer Lett. 2019;451:142–149. doi: 10.1016/j.canlet.2019.02.047. [DOI] [PubMed] [Google Scholar]
- 6.El-Osta H., Jafri S. Predictors for clinical benefit of immune checkpoint inhibitors in advanced non-small-cell lung cancer: a meta-analysis. Immunotherapy. 2019;11:189–199. doi: 10.2217/imt-2018-0086. [DOI] [PubMed] [Google Scholar]
- 7.Binnewies M., Roberts E.W., Kersten K., Chan V., Fearon D.F., Merad M., Coussens L.M., Gabrilovich D.I., Ostrand-Rosenberg S., Hedrick C.C., et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018;24:541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katsuta E., Yan L., Opyrchal M., Kalinski P., Takabe K. Cytotoxic T-lymphocyte infiltration and chemokine predict long-term patient survival independently of tumor mutational burden in triple-negative breast cancer. Ther. Adv. Med. Oncol. 2021;13 doi: 10.1177/17588359211006680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reissfelder C., Stamova S., Gossmann C., Braun M., Bonertz A., Walliczek U., Grimm M., Rahbari N.N., Koch M., Saadati M., et al. Tumor-specific cytotoxic T lymphocyte activity determines colorectal cancer patient prognosis. J. Clin. Invest. 2015;125:739–751. doi: 10.1172/jci74894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Almeida P.E., Mak J., Hernandez G., Jesudason R., Herault A., Javinal V., Borneo J., Kim J.M., Walsh K.B. Anti-VEGF treatment enhances CD8(+) T-cell antitumor activity by amplifying hypoxia. Cancer Immunol. Res. 2020;8:806–818. doi: 10.1158/2326-6066.Cir-19-0360. [DOI] [PubMed] [Google Scholar]
- 11.Vitale I., Manic G., Coussens L.M., Kroemer G., Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30:36–50. doi: 10.1016/j.cmet.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Baslan T., Hicks J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat. Rev. Cancer. 2017;17:557–569. doi: 10.1038/nrc.2017.58. [DOI] [PubMed] [Google Scholar]
- 13.Guo X., Zhang Y., Zheng L., Zheng C., Song J., Zhang Q., Kang B., Liu Z., Jin L., Xing R., et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018;24:978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 14.He D., Wang D., Lu P., Yang N., Xue Z., Zhu X., Zhang P., Fan G. Single-cell RNA sequencing reveals heterogeneous tumor and immune cell populations in early-stage lung adenocarcinomas harboring EGFR mutations. Oncogene. 2021;40:355–368. doi: 10.1038/s41388-020-01528-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim N., Kim H.K., Lee K., Hong Y., Cho J.H., Choi J.W., Lee J.I., Suh Y.L., Ku B.M., Eum H.H., et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020;11:2285. doi: 10.1038/s41467-020-16164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lambrechts D., Wauters E., Boeckx B., Aibar S., Nittner D., Burton O., Bassez A., Decaluwé H., Pircher A., Van den Eynde K., et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018;24:1277–1289. doi: 10.1038/s41591-018-0096-5. [DOI] [PubMed] [Google Scholar]
- 17.Lavin Y., Kobayashi S., Leader A., Amir E.A.D., Elefant N., Bigenwald C., Remark R., Sweeney R., Becker C.D., Levine J.H., et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. 2017;169:750–765.e17. doi: 10.1016/j.cell.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinjab A., Han G., Treekitkarnmongkol W., Hara K., Brennan P.M., Dang M., Hao D., Wang R., Dai E., Dejima H., et al. Resolving the spatial and cellular architecture of lung adenocarcinoma by multiregion single-cell sequencing. Cancer Discov. 2021;11:2506–2523. doi: 10.1158/2159-8290.Cd-20-1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu F., Fan J., He Y., Xiong A., Yu J., Li Y., Zhang Y., Zhao W., Zhou F., Li W., et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021;12:2540. doi: 10.1038/s41467-021-22801-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trefzer T.B., Schneider M.A., Jechow K., Chua R.L., Muley T., Winter H., Kriegsmann M., Meister M., Eils R., Conrad C. Intratumoral heterogeneity and immune modulation in lung adenocarcinoma in female smokers and never smokers. Cancer Res. 2022;82:3116–3129. doi: 10.1158/0008-5472.Can-21-3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelosof L., Ahn C., Gao A., Horn L., Madrigales A., Cox J., McGavic D., Minna J.D., Gazdar A.F., Schiller J. Proportion of never-smoker non-small cell lung cancer patients at three diverse institutions. J. Natl. Cancer Inst. 2017;109 doi: 10.1093/jnci/djw295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toh C.K., Ong W.S., Lim W.T., Tan D.S.W., Ng Q.S., Kanesvaran R., Seow W.J., Ang M.K., Tan E.H. A decade of never-smokers among lung cancer patients-increasing trend and improved survival. Clin. Lung Cancer. 2018;19:e539–e550. doi: 10.1016/j.cllc.2018.03.013. [DOI] [PubMed] [Google Scholar]
- 23.Cufari M.E., Proli C., De Sousa P., Raubenheimer H., Al Sahaf M., Chavan H., Shedden L., Niwaz Z., Leung M., Nicholson A.G., et al. Increasing frequency of non-smoking lung cancer: presentation of patients with early disease to a tertiary institution in the UK. Eur. J. Cancer. 2017;84:55–59. doi: 10.1016/j.ejca.2017.06.031. [DOI] [PubMed] [Google Scholar]
- 24.Cho J., Choi S.M., Lee J., Lee C.H., Lee S.M., Kim D.W., Yim J.J., Kim Y.T., Yoo C.G., Kim Y.W., et al. Proportion and clinical features of never-smokers with non-small cell lung cancer. Chin. J. Cancer. 2017;36:20. doi: 10.1186/s40880-017-0187-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z., Li Z., Zhou K., Wang C., Jiang L., Zhang L., Yang Y., Luo W., Qiao W., Wang G., et al. Deciphering cell lineage specification of human lung adenocarcinoma with single-cell RNA sequencing. Nat. Commun. 2021;12:6500. doi: 10.1038/s41467-021-26770-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nabhan A.N., Brownfield D.G., Harbury P.B., Krasnow M.A., Desai T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science. 2018;359:1118–1123. doi: 10.1126/science.aam6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X., Rock J.R., Lu Y., Futtner C., Schwab B., Guinney J., Hogan B.L.M., Onaitis M.W. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc. Natl. Acad. Sci. USA. 2012;109:4910–4915. doi: 10.1073/pnas.1112499109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desai T.J., Brownfield D.G., Krasnow M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature. 2014;507:190–194. doi: 10.1038/nature12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mainardi S., Mijimolle N., Francoz S., Vicente-Dueñas C., Sánchez-García I., Barbacid M. Identification of cancer initiating cells in K-Ras driven lung adenocarcinoma. Proc. Natl. Acad. Sci. USA. 2014;111:255–260. doi: 10.1073/pnas.1320383110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duma N., Santana-Davila R., Molina J.R. Non-small cell lung cancer: epidemiology, screening, diagnosis, and treatment. Mayo Clin. Proc. 2019;94:1623–1640. doi: 10.1016/j.mayocp.2019.01.013. [DOI] [PubMed] [Google Scholar]
- 31.Park I.A., Hwang S.H., Song I.H., Heo S.H., Kim Y.A., Bang W.S., Park H.S., Lee M., Gong G., Lee H.J. Expression of the MHC class II in triple-negative breast cancer is associated with tumor-infiltrating lymphocytes and interferon signaling. PLoS One. 2017;12 doi: 10.1371/journal.pone.0182786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forero A., Li Y., Chen D., Grizzle W.E., Updike K.L., Merz N.D., Downs-Kelly E., Burwell T.C., Vaklavas C., Buchsbaum D.J., et al. Expression of the MHC class II pathway in triple-negative breast cancer tumor cells is associated with a good prognosis and infiltrating lymphocytes. Cancer Immunol. Res. 2016;4:390–399. doi: 10.1158/2326-6066.Cir-15-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Axelrod M.L., Cook R.S., Johnson D.B., Balko J.M. Biological consequences of MHC-II expression by tumor cells in cancer. Clin. Cancer Res. 2019;25:2392–2402. doi: 10.1158/1078-0432.Ccr-18-3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hashimoto D., Chow A., Noizat C., Teo P., Beasley M.B., Leboeuf M., Becker C.D., See P., Price J., Lucas D., et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schulz C., Gomez Perdiguero E., Chorro L., Szabo-Rogers H., Cagnard N., Kierdorf K., Prinz M., Wu B., Jacobsen S.E.W., Pollard J.W., et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 36.Casanova-Acebes M., Dalla E., Leader A.M., LeBerichel J., Nikolic J., Morales B.M., Brown M., Chang C., Troncoso L., Chen S.T., et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. 2021;595:578–584. doi: 10.1038/s41586-021-03651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vogel D.Y.S., Glim J.E., Stavenuiter A.W.D., Breur M., Heijnen P., Amor S., Dijkstra C.D., Beelen R.H.J. Human macrophage polarization in vitro: maturation and activation methods compared. Immunobiology. 2014;219:695–703. doi: 10.1016/j.imbio.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 38.Zhao Y., Harrison D.L., Song Y., Ji J., Huang J., Hui E. Antigen-presenting cell-intrinsic PD-1 neutralizes PD-L1 in cis to attenuate PD-1 signaling in T cells. Cell Rep. 2018;24:379–390.e6. doi: 10.1016/j.celrep.2018.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garzón-Tituaña M., Arias M.A., Sierra-Monzón J.L., Morte-Romea E., Santiago L., Ramirez-Labrada A., Martinez-Lostao L., Paño-Pardo J.R., Galvez E.M., Pardo J. The multifaceted function of granzymes in sepsis: some facts and a lot to discover. Front. Immunol. 2020;11:1054. doi: 10.3389/fimmu.2020.01054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mogilenko D.A., Shpynov O., Andhey P.S., Arthur L., Swain A., Esaulova E., Brioschi S., Shchukina I., Kerndl M., Bambouskova M., et al. Comprehensive profiling of an aging immune system reveals clonal GZMK(+) CD8(+) T cells as conserved hallmark of inflammaging. Immunity. 2021;54:99–115.e12. doi: 10.1016/j.immuni.2020.11.005. [DOI] [PubMed] [Google Scholar]
- 41.Otranto M., Sarrazy V., Bonté F., Hinz B., Gabbiani G., Desmoulière A. The role of the myofibroblast in tumor stroma remodeling. Cell Adh. Migr. 2012;6:203–219. doi: 10.4161/cam.20377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sikic B.I., Lakhani N., Patnaik A., Shah S.A., Chandana S.R., Rasco D., Colevas A.D., O'Rourke T., Narayanan S., Papadopoulos K., et al. First-in-Human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 2019;37:946–953. doi: 10.1200/jco.18.02018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barkal A.A., Weiskopf K., Kao K.S., Gordon S.R., Rosental B., Yiu Y.Y., George B.M., Markovic M., Ring N.G., Tsai J.M., et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 2018;19:76–84. doi: 10.1038/s41590-017-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Devarakonda S., Li Y., Martins Rodrigues F., Sankararaman S., Kadara H., Goparaju C., Lanc I., Pepin K., Waqar S.N., Morgensztern D., et al. Genomic profiling of lung adenocarcinoma in never-smokers. J. Clin. Oncol. 2021;39:3747–3758. doi: 10.1200/jco.21.01691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang T., Joubert P., Ansari-Pour N., Zhao W., Hoang P.H., Lokanga R., Moye A.L., Rosenbaum J., Gonzalez-Perez A., Martínez-Jiménez F., et al. Genomic and evolutionary classification of lung cancer in never smokers. Nat. Genet. 2021;53:1348–1359. doi: 10.1038/s41588-021-00920-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y.J., Roumeliotis T.I., Chang Y.H., Chen C.T., Han C.L., Lin M.H., Chen H.W., Chang G.C., Chang Y.L., Wu C.T., et al. Proteogenomics of non-smoking lung cancer in east asia delineates molecular signatures of pathogenesis and progression. Cell. 2020;182:226–244.e17. doi: 10.1016/j.cell.2020.06.012. [DOI] [PubMed] [Google Scholar]
- 47.Lahmar Q., Keirsse J., Laoui D., Movahedi K., Van Overmeire E., Van Ginderachter J.A. Tissue-resident versus monocyte-derived macrophages in the tumor microenvironment. Biochim. Biophys. Acta. 2016;1865:23–34. doi: 10.1016/j.bbcan.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 48.Ye L.Y., Chen W., Bai X.L., Xu X.Y., Zhang Q., Xia X.F., Sun X., Li G.G., Hu Q.D., Fu Q.H., Liang T.B. Hypoxia-induced epithelial-to-mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res. 2016;76:818–830. doi: 10.1158/0008-5472.Can-15-0977. [DOI] [PubMed] [Google Scholar]
- 49.Neefjes J., Jongsma M.L.M., Paul P., Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011;11:823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- 50.Philip M., Schietinger A. CD8(+) T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 2022;22:209–223. doi: 10.1038/s41577-021-00574-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Myers J.A., Miller J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021;18:85–100. doi: 10.1038/s41571-020-0426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderson N.R., Minutolo N.G., Gill S., Klichinsky M. Macrophage-based approaches for cancer immunotherapy. Cancer Res. 2021;81:1201–1208. doi: 10.1158/0008-5472.Can-20-2990. [DOI] [PubMed] [Google Scholar]
- 53.Becht E., de Reyniès A., Giraldo N.A., Pilati C., Buttard B., Lacroix L., Selves J., Sautès-Fridman C., Laurent-Puig P., Fridman W.H. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin. Cancer Res. 2016;22:4057–4066. doi: 10.1158/1078-0432.Ccr-15-2879. [DOI] [PubMed] [Google Scholar]
- 54.Luo W., Wang Z., Zhang T., Yang L., Xian J., Li Y., Li W. Immunotherapy in non-small cell lung cancer: rationale, recent advances and future perspectives. Precis. Clin. Med. 2021;4:258–270. doi: 10.1093/pcmedi/pbab027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Catalán R., Orozco-Morales M., Hernández-Pedro N.Y., Guijosa A., Colín-González A.L., Ávila-Moreno F., Arrieta O. CD47-SIRPα Axis as a biomarker and therapeutic target in cancer: current perspectives and future challenges in nonsmall cell lung cancer. J. Immunol. Res. 2020;2020 doi: 10.1155/2020/9435030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benjamin D., Sato T., Cibulskis K., Getz G., Stewart C., Lichtenstein L. Calling Somatic SNVs and Indels with Mutect2. bioRxiv. 2019:861054. doi: 10.1101/861054. [DOI] [Google Scholar]
- 58.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qiu X., Mao Q., Tang Y., Wang L., Chawla R., Pliner H.A., Trapnell C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods. 2017;14:979–982. doi: 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 61.Nicholson A.G., Tsao M.S., Beasley M.B., Borczuk A.C., Brambilla E., Cooper W.A., Dacic S., Jain D., Kerr K.M., Lantuejoul S., et al. The 2021 WHO classification of lung tumors: impact of advances since 2015. J. Thorac. Oncol. 2022;17:362–387. doi: 10.1016/j.jtho.2021.11.003. [DOI] [PubMed] [Google Scholar]
- 62.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The data of this study has been submitted to the Genome Sequence Archive for human (GSA for human) database. The accession number for the single cell sequencing data and 1021-gene-panel targeted region sequencing data reported in this paper is HRA002953, and the accession number for the single cell sequencing data used for verification in this study is HRA002992. They are publicly accessible in https://ngdc.cncb.ac.cn/gsa-human/, and the download of these data requires Data Access Committee (DAC) approval.
-
•
This paper does not generate the original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.