

Summary
Current antihypertensive options still incompletely control blood pressure, suggesting the existence of uncovered pathogenic mechanisms. Here, whether cytokine-like protein family with sequence similarity 3, member D (FAM3D) is involved in hypertension etiology is evaluated. A case-control study exhibits that FAM3D is elevated in patients with hypertension, with a positive association with odds of hypertension. FAM3D deficiency significantly ameliorates angiotensin II (AngII)-induced hypertension in mice. Mechanistically, FAM3D directly causes endothelial nitric oxide synthase (eNOS) uncoupling and impairs endothelium-dependent vasorelaxation, whereas 2,4-diamino-6-hydroxypyrimidine to induce eNOS uncoupling abolishes the protective effect of FAM3D deficiency against AngII-induced hypertension. Furthermore, antagonism of formyl peptide receptor 1 (FPR1) and FPR2 or the suppression of oxidative stress blunts FAM3D-induced eNOS uncoupling. Translationally, targeting endothelial FAM3D by adeno-associated virus or intraperitoneal injection of FAM3D-neutralizing antibodies markedly ameliorates AngII- or deoxycorticosterone acetate (DOCA)-salt-induced hypertension. Conclusively, FAM3D causes eNOS uncoupling through FPR1- and FPR2-mediated oxidative stress, thereby exacerbating the development of hypertension. FAM3D may be a potential therapeutic target for hypertension.
Keywords: FAM3D, hypertension, endothelial dysfunction, antihypertensive treatment
Graphical abstract
Highlights
-
•
Plasma FAM3D positively correlates with odds of hypertension in patients
-
•
Deficiency in FAM3D significantly ameliorates hypertension in mice
-
•
FAM3D causes eNOS uncoupling through endothelial FPR1/2-mediated oxidative stress
-
•
Targeting FAM3D reverses eNOS uncoupling and alleviates hypertension in mice
Shen et al. discover that the cytokine-like protein FAM3D is associated with hypertension etiology and that targeting endothelium-derived FAM3D or administration of FAM3D-neutralizing antibodies significantly restores endothelial function and lowers blood pressure in hypertensive mice. FAM3D may be a promising target for treating endothelial dysfunction as well as hypertension.
Introduction
Hypertension known as the elevation of blood pressure is a common cardiovascular disorder to potentially increase the risk of other serious comorbidities, such as myocardial infarction, stroke, and chronic renal failure.1,2 Despite sufficient effective antihypertensive options, an estimated 10%–15% of the general hypertensive population fails to exhibit reduced blood pressure and the alleviation of targeted organ damage, possibly due to a lack of response or intolerance to available antihypertensive agents.3 This unmet treatment need indicates the incompletely understood pathogenic mechanism of hypertension and prompts further investigation of additional targets and strategies.
Endothelial dysfunction, mainly characterized by reduced nitric oxide (NO) bioavailability, is a critical pathogenic hallmark of hypertension.4,5 NO generation in the endothelium is dependent on endothelial NO synthase (eNOS). The dysfunction or inhibition of eNOS causes NO unavailability and impairs endothelium-dependent vascular relaxation, further exacerbating the progression of hypertension.6,7,8,9,10,11 The major cause of eNOS dysfunction related to hypertension is uncoupling; uncoupled eNOS alternatively generates superoxide anions (O2−) rather than NO.6,12 In turn, NADPH oxidase-mediated oxidative stress is a major trigger for eNOS uncoupling and endothelial dysfunction.13 Moreover, oxidative stress-induced reactive oxygen species (ROS) directly react with NO to form reactive nitrogen species and reduce NO bioavailability.14 Consistently, modulating eNOS activity or targeting NADPH oxidases has been shown to rescue endothelial function and alleviate the progression of hypertension in animal models.15,16,17 Therefore, improving endothelial function is an attractive therapeutic strategy for hypertension, but current therapies specific to the endothelium are limited.
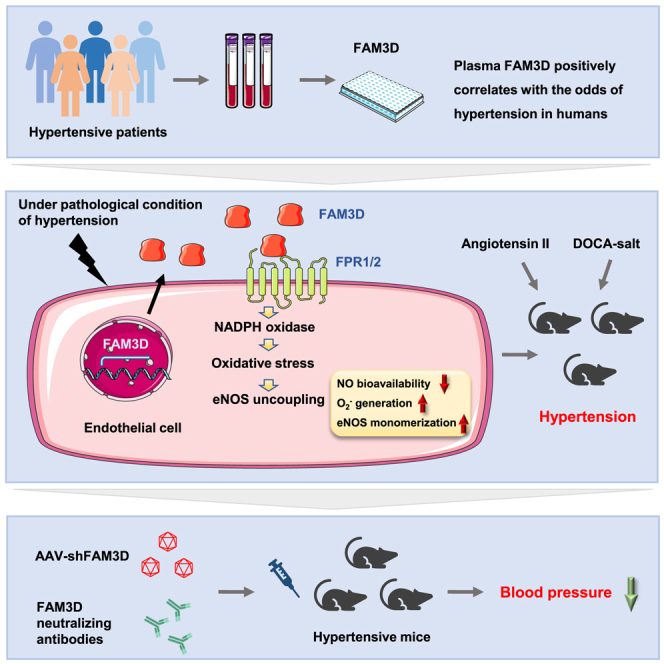
Family with sequence similarity 3, member D (FAM3D) is a cytokine-like protein with chemoattractant activity to recruit monocytes and neutrophils through its cell surface receptors FPR1 and FPR2.18,19 Our previous study identified FAM3D, which was significantly upregulated in endothelial cells (ECs) during the pathogenesis of abdominal aortic aneurysm (AAA), induced neutrophil recruitment and aggravated AAA formation through FPR1 and FPR2 signaling.20 Here, whether FAM3D was involved in the pathogenesis of hypertension was evaluated. A case-control study displayed that the FAM3D level was substantially elevated in plasma from patients with hypertension, with a positive association with odds of hypertension. Furthermore, we found that FAM3D induced eNOS uncoupling through FPR1- and FPR2-mediated oxidative stress, consequently causing endothelial dysfunction and exacerbating angiotensin II (AngII)-induced hypertension. Translationally, specific knockdown of FAM3D in ECs by adeno-associated virus or administration of FAM3D-neutralizing antibodies significantly reversed endothelial dysfunction and thereby ameliorated hypertension. Therefore, FAM3D may be a potential therapeutic target for improving endothelial function in response to antihypertensive treatment.
Results
The increase in FAM3D correlates with the pathogenesis of hypertension
Since FAM3D is a cytokine-like protein, we designed an age- and gender-matched case-control study between 80 cases and 80 responding controls (Table S1). For subject recruitment, participants with peripheral arterial disease, diabetes mellitus, chronic kidney disease, history of myocardial infarction, stroke, and tumor and those taking antihypertensive and lipid-lowering medications were excluded. Plasma FAM3D levels were measured to investigate whether FAM3D was involved in hypertension etiology. A significant increase in plasma FAM3D levels was observed in patients with hypertension (7.27 [5.33–10.70] ng/mL) compared with responding controls (5.54 [4.51–7.15] ng/mL) (Figure 1A). Moreover, the increase in plasma FAM3D positively correlated with systolic blood pressure in all patients with hypertension (n = 80, r = 0.3845, p = 0.0004) (Figure 1B). Furthermore, the associations between plasma FAM3D levels and hypertension status were estimated by univariable conditional logistic regressions and multivariable conditional logistic regressions adjusting potential confounding factors including fasting blood glucose, plasma total cholesterol, triglyceride, and heart rate. The odds ratios for the risk of hypertension were 1.06 (95% confidence interval [CI]: 1–1.13) and 1.09 (95% CI: 1.01–1.17) following an increase in the plasma FAM3D levels by one unit in the non-adjusted model and the multivariable-adjusted model, respectively (Table S2). Moreover, when FAM3D levels were regarded as categorical variables, odds ratios were calculated with the categorical tertiles. Compared with the lowest tertile of plasma FAM3D levels (<5.20 ng/mL), the multivariable-adjusted odds ratios of hypertension for the highest tertiles (≥7.89 ng/m:) were 7.34 (95% CI: 2.27–23.70) (Table S2). Collectively, these data indicated that plasma FAM3D levels positively correlated with odds of hypertension in humans.
Figure 1.

The elevation of FAM3D correlates with the pathogenesis of hypertension
(A) ELISA measurement of plasma FAM3D from individuals in the case-control study. N = 80 pairs. Data are represented as mean ± SEM. Mann-Whitney test.
(B) Spearman correlation analysis of plasma FAM3D levels and SBP in patients with hypertension. N = 80.
(C) ELISA measurement of plasma FAM3D levels in C57BL/6 mice treated with saline or AngII for 7 days. n = 5–6. Data are represented as mean ± SEM. Unpaired Student’s t test, ∗p < 0.05.
(D and E) Immunofluorescent staining of FAM3D in mesenteric arteries (E) and thoracic aortas (F) from C57BL/6 mice after saline or AngII infusion for 7 days. Scale bar, 40 μm.
(F) Immunofluorescent en face staining of FAM3D in the endothelial layer of thoracic aortas of C57BL/6 mice after saline or AngII infusion for 1 day or 7 days. Scale bar, 25 μm. n = 6. Data are represented as mean ± SEM. Unpaired Student’s t test, ∗p < 0.05.
(G) Immunofluorescent en face staining of FAM3D in the endothelial layer of thoracic aortas of sham-treated or DOCA-salt-induced mice. Scale bar, 25 μm. n = 6. Data are represented as mean ± SEM. Unpaired Student’s t test, ∗p < 0.05.
(H) Immunofluorescent en face staining of FAM3D in the endothelial layer of thoracic aortas of Wistar-Kyoto (WKY) or SHRs. Scale bar, 25 μm. n = 6. Data are represented as mean ± SEM. Unpaired Student’s t test, ∗p < 0.05.
Next, we evaluated the modulation of FAM3D expression in animal models, including AngII (490 ng/kg/min)-induced hypertensive mice (Table S3), deoxycorticosterone acetate (DOCA)-salt-induced hypertensive mice (Table S4) and spontaneously hypertensive rats (SHRs) (Table S5). As results, FAM3D was significantly elevated in plasma of AngII-infused mice (Figure 1C) and upregulated in aortic tissues in AngII- or DOCA-salt-induced hypertensive mice and SHRs (Figures S1A–S1D), suggesting that the upregulation of FAM3D might not be specific model dependent but a general alteration during the pathogenesis of hypertension. Since FAM3D is constitutively expressed in the gastrointestinal tract,21 we evaluated intestinal expression. FAM3D expressed in the colons did not exhibit obvious alteration in AngII- or DOCA-salt-induced hypertensive mice (Figures S1E and S1F). To further confirm the localization of upregulated FAM3D in arteries, we performed immunofluorescence staining on cross sections of thoracic aortas and mesenteric arteries from C57BL/6 mice after AngII infusion for 7 days (Figures 1D, 1E, S2A, and S2B). AngII infusion exclusively upregulated FAM3D expression in intimal ECs but not medial smooth muscle cells (vascular smooth muscle cells [VSMCs]). In addition, en face immunofluorescence staining of the arterial intima verified that endothelial FAM3D was upregulated in AngII-infused mice as early as day 1 of infusion (Figures 1F and S2C), indicating the potential pathogenic role of FAM3D. Similarly, endothelial FAM3D was also elevated in DOCA-salt-induced hypertensive mice (Figures 1G and S2D) and SHRs (Figures 1H and S2E). Thus, the upregulation of FAM3D expression in ECs may be associated with the pathogenesis of hypertension.
FAM3D deficiency ameliorates AngII-induced hypertension and related vascular pathologies
To examine the effect of FAM3D on hypertension, we compared the blood pressure of wild-type (WT) and FAM3D−/− mice after AngII infusion for 14 days through the non-invasive tail-cuff method and invasive radiotelemetry, respectively. Before AngII induction, these two groups of mice exhibited no differences in systolic/diastolic blood pressure (SBP/DBP), mean arterial pressure (MAP), and heart rate at baseline (Figure S3). Following AngII infusion, SBP, DBP, and MAP were gradually elevated in WT mice, whereas FAM3D deficiency significantly lowered the AngII-induced increase in blood pressure (Figures 2A–2D). Moreover, we performed telemetric monitoring of 24-h blood pressure in WT and FAM3D−/− mice after 14 days of AngII infusion. Consistently, FAM3D deficiency decreased the 24-h SBP and DBP in AngII-induced mice (Figures 2E and 2F). In addition, we observed hypertension-related vascular pathologies, including vascular stiffness and inflammation. AngII infusion significantly increased arterial pulse wave velocity (PWV) and adventitial collagen deposition in thoracic aortas, whereas FAM3D knockout inhibited these alterations, suggesting that FAM3D deficiency attenuated AngII-induced vascular stiffness (Figures 2G and 2H). Moreover, flow cytometric analysis showed that FAM3D depletion alleviated leukocyte infiltration in the aortic wall (Figure 2I), which was consistent with our previous study showing that FAM3D induced vascular leukocyte recruitment. These data suggested that FAM3D knockout mitigated AngII-induced hypertension and related vascular stiffness and inflammation.
Figure 2.

FAM3D deficiency ameliorates AngII-induced hypertension and related vascular pathologies
(A) SBP of WT and FAM3D−/− mice was measured by the tail-cuff method during AngII infusion for 14 days. n = 6–8. Data are represented as mean ± SEM. Repeated-measures analysis using a mixed-effects model, ∗p < 0.05.
(B–D) The SBP (B), DBP (C), and MAP (D) of WT and FAM3D−/− mice were measured by radiotelemetry during AngII infusion for 14 days. n = 5–6. Data are represented as mean ± SEM. Repeated-measures analysis using a mixed-effects model, ∗p < 0.05.
(E and F) 24-h recordings of SBP (E) and DBP (F) in WT and FAM3D−/− mice was measured by radiotelemetry following 7 days of AngII infusion. n = 3–5. Data are represented as mean ± SEM. Repeated-measures analysis using a mixed-effects model, ∗p < 0.05.
(G) Pulse wave velocity (PWV) in aortas from WT and FAM3D−/− mice treated with saline or AngII for 14 days. n = 5–6. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test, ∗p < 0.05.
(H) Representative Masson staining and quantification of adventitial collagen deposition in thoracic aortas from WT and FAM3D−/− mice treated with saline or AngII for 14 days. Scale bar, 100 μm. n = 5–6. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test, ∗p < 0.05.
(I) Flow cytometric analysis of CD45+ cells in aortas from WT and FAM3D−/− mice treated with saline or AngII for 14 days. n = 4–6. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test, ∗p < 0.05.
FAM3D impairs endothelium-dependent vasorelaxation to further enhance vasoconstriction
The dysregulation of arterial contraction and relaxation contributes to the pathogenesis of hypertension. Thus, we further examined whether FAM3D affected arterial contraction and relaxation. The vasoconstrictive response to phenylephrine was first evaluated in mesenteric resistance arteries from WT and FAM3D−/− mice. There was no difference in arterial contractility between the two groups of mice (Figures 3A and 3B). However, following 14 days of AngII infusion in vivo, FAM3D−/− arteries displayed markedly decreased vasoconstrictive responses to phenylephrine compared with WT vessels (Figure 3C). Furthermore, arteries were pretreated with the NO synthase inhibitor L-NG-nitroarginine methyl ester (L-NAME) to cause endothelial dysfunction. Obvious differences in the constrictive responses of arteries from AngII-infused WT and FAM3D−/− mice were abolished by L-NAME (Figure 3D), suggesting that FAM3D regulated vasocontraction by affecting endothelial function.
Figure 3.

FAM3D impairs endothelium-dependent vasorelaxation to further enhance vasocontraction
(A and B) Concentration-response curves of endothelium-dependent contraction (A) and endothelium-independent contraction (B) of mesenteric resistance arteries from WT and FAM3D−/− mice. n = 6. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test.
(C and D) Concentration-response curves of endothelium-dependent contraction (C) and endothelium-independent contraction (D) of mesenteric resistance arteries from WT and FAM3D−/− mice infused with AngII for 14 days. n = 6. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test. ∗p < 0.05.
(E and F) Concentration-response curves of endothelium-dependent relaxation (E) and endothelium-independent relaxation (F) of mesenteric resistance arteries from WT and FAM3D−/− mice. n = 5–6. Data are represented as mean ± SEM. Two-way ANOVA by Tukey’s multiple comparisons test.
(G and H) Concentration-response curves of endothelium-dependent relaxation (G) and endothelium-independent relaxation (H) of mesenteric arteries from WT and FAM3D−/− mice infused with AngII for 14 days. n = 6. Data are represented as mean ± SEM. Two-way ANOVA by Tukey’s multiple comparisons test. ∗p < 0.05.
We further compared vascular responses to acetylcholine and sodium nitroprusside to evaluate endothelium-dependent and -independent vasorelaxation, respectively. The WT and FAM3D−/− mesenteric arteries from mice without AngII infusion exhibited no differences in endothelium-dependent or -independent relaxation (Figures 3E and 3F). Moreover, FAM3D deficiency significantly enhanced vasorelaxation in arteries from AngII-infused mice (Figure 3G). In contrast, the endothelium-independent vasorelaxation of these arteries in response to sodium nitroprusside showed no obvious differences, suggesting that the distinct vasorelaxation between WT and FAM3D−/− mesenteric arteries may be related to endothelial function (Figure 3H). Meanwhile, we also evaluated endothelial hyperpolarizing factor (EDHF)-induced vasorelaxation on WT and FAM3D−/− mesenteric arteries. As results, no difference was observed in arteries from WT and FAM3D−/− mice with or without AngII infusion, excluding the potential effect of FAM3D on EDHF function (Figure S4).
Collectively, these results suggested that FAM3D specifically blunted endothelium-dependent arterial relaxation and further strengthened vasoconstriction.
FAM3D causes eNOS uncoupling in ECs in vitro
Coupled eNOS-dependent NO production is critical for endothelium-dependent vasorelaxation,22 and eNOS uncoupling reduces NO bioavailability and impairs vascular relaxation.6 Accordingly, we first measured intracellular NO production and extracellular NO release from ECs by DAF-FM DA staining and Griess assay, respectively. FAM3D dose-dependently decreased NO production and secretion in ECs (Figures S5A and S5B), whereas BH4, an inhibitor of eNOS uncoupling, significantly reversed the suppression of NO production by FAM3D in ECs (Figure S5C), indicating that eNOS uncoupling mediated the FAM3D-induced reduction in NO bioavailability. Despite the reduction in NO bioavailability, eNOS uncoupling caused alternative O2− generation and eNOS monomerization. Dihydroethidium (DHE) staining showed that FAM3D increased intracellular O2− generation in ECs, and L-NAME inhibited this effect of FAM3D, suggesting that FAM3D induced eNOS-dependent O2− production (Figure S5D). Moreover, western blot showed that FAM3D did not alter eNOS expression but induced eNOS monomerization in a time-dependent manner (Figure S5E). Thus, these data suggested that FAM3D caused eNOS uncoupling in vitro.
FAM3D deficiency reverses eNOS uncoupling to alleviate hypertension in vivo
We further validated FAM3D-induced eNOS uncoupling in vivo. FAM3D deficiency reversed the AngII infusion-induced reduction in NO, eNOS-dependent O2− production and eNOS monomerization (Figures 4A–4C), suggesting that FAM3D deficiency reversed eNOS uncoupling in AngII-infused hypertensive mice. Furthermore, 2,4-diamino-6-hydroxypyrimidine (DAHP) (10 mmol/L for 14 days) was administered in drinking water to reduce intracellular BH4 and cause eNOS uncoupling in vivo. DAHP administration abolished FAM3D deficiency-mediated restoration of NO bioavailability in response to AngII infusion and accordingly disrupted the protective effect of FAM3D deficiency against AngII-induced hypertension (Figures 4D and 4E). These results suggested that FAM3D deficiency alleviated hypertension through the restoration of eNOS coupling.
Figure 4.

FAM3D deficiency inhibits AngII-induced eNOS uncoupling in ECs of aortas in mice
(A) Representative DAF-FM DA en face staining and quantification of NO levels in ECs from the thoracic aortas of WT and FAM3D−/− mice after saline or AngII infusion for 14 days. n = 5–6. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test, ∗p < 0.05.
(B) Representative DHE en face staining and quantification of O2− in ECs from thoracic aortas in WT and FAM3D−/− mice infused with saline or AngII for 14 days. L-NAME (200 μmol/L) was ex vivo used to treat dissected aortas for 30 min before DHE staining. n = 3. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test, ∗p < 0.05.
(C) Representative western blot analysis and quantification of monomer/dimer eNOS ratios in the aortas of WT and FAM3D−/− mice treated with saline or AngII for 14 days. n = 5. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test, ∗p < 0.05.
(D) Representative DAF-FM DA en face staining and quantification of NO in ECs from the thoracic aortas of WT and FAM3D−/− mice infused with AngII and administered with or without 2,4- DAHP (10 mmol/L) in drinking water for 14 days. n = 5. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test, ∗p < 0.05.
(E) SBP was measured by the tail-cuff method in WT and FAM3D−/− mice infused with AngII and administered with or without DAHP (10 mmol/L) in drinking water for 14 days. n = 4–5. Data are represented as mean ± SEM. Repeated-measures analysis using a mixed-effects model followed by Tukey’s multiple comparisons test, ∗p < 0.05, WT + AngII vs. FAM3D−/− + AngII; ns, no significance, WT + AngII + DAHP vs. FAM3D−/− + AngII + DAHP.
FAM3D leads to eNOS uncoupling through FPR1/2-mediated oxidative stress
Next, we examined how FAM3D induced eNOS uncoupling to cause endothelial dysfunction. Based on our previous finding that FPR1 and FPR2 mediate FAM3D-induced neutrophil recruitment,19,20 we used cyclosporin H (CsH) and WRW4 to antagonize FPR1 and FPR2, respectively. Both antagonists significantly inhibited FAM3D-induced eNOS monomerization and FAM3D-mediated impairment of NO bioavailability in ECs (Figures 5A and 5B), suggesting that FPR1 and FPR2 were involved in FAM3D-induced eNOS uncoupling and endothelial dysfunction. Oxidative stress is the major inducer of eNOS uncoupling. We observed that FAM3D increased intracellular ROS production from 15 min until 1 h after stimulation (Figure 5C), whereas both CsH and WRW4 efficiently inhibited ROS accumulation in ECs after FAM3D treatment for 1 h, suggesting that FAM3D-FPR1/2 induced obvious oxidative stress (Figure 5D). Of interest, L-NAME did not attenuate but instead further enhanced O2− generation induced by FAM3D at 1 h (Figure 5E), which was consistent with the finding that FAM3D did not induce eNOS uncoupling during this period (Figure S5E). These data collectively suggested that FAM3D directly induced oxidative stress prior to eNOS uncoupling. Moreover, the ROS inhibitor NAC and NADPH oxidase inhibitor apocynin could attenuate oxidative stress and markedly reversed FAM3D-induced NO reduction and eNOS monomerization (Figures 5F and 5G), indicating that FAM3D induced eNOS uncoupling through FPR1/2-mediated oxidative stress.
Figure 5.

FAM3D causes eNOS uncoupling through FPR1/2-mediated oxidative stress
(A) Representative western blot and quantification of monomer/dimer eNOS ratios in HUVECs treated with FAM3D (10 nmol/L) for 6 h after preincubation with CsH (1 μmol/L) and/or WRW4 (1 μmol/L) for 2 h. n = 4. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test, ∗p < 0.05.
(B) Quantification of intracellular NO by DAF-FM DA staining in human umbilical vein ECs (HUVECs) treated with FAM3D (10 nmol/L) for 6 h after preincubation with CsH (1 μmol/L) and/or WRW4 (1 μmol/L) for 2 h. n = 4. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test, ∗p < 0.05.
(C) Quantification of ROS by DCFH-DA staining in HUVECs treated with FAM3D (10 nmol/L) for various times. n = 3. Data are represented as mean ± SEM. One-way ANOVA followed by Dunnett’s multiple comparisons test, ∗p < 0.05.
(D) Quantification of ROS by DCFH-DA staining in HUVECs treated with FAM3D (10 nmol/L) for 60 min after preincubation with CsH (1 μmol/L) and/or WRW4 (1 μmol/L) for 2 h. n = 4. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test, ∗p < 0.05.
(E) Quantification of O2− generation by DHE staining in HUVECs treated with FAM3D (10 nmol/L) in the presence or absence of L-NAME (200 μmol/L) for 60 min. n = 3. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test, ∗p < 0.05.
(F) Quantification of intracellular NO by DAF-FM DA staining in HUVECs treated with FAM3D (10 nmol/L) for 6 h after preincubation with NAC (5 mmol/L) or apocynin (100 μmol/L) for 4 h. n = 4. Data are represented as mean ± SEM. Two-way ANOVA followed by Sidak’s multiple comparisons test, ∗p < 0.05.
(G) Representative western blot and quantification of monomer/dimer eNOS radios in HUVECs treated with FAM3D (10 nmol/L) for 6 h after preincubation with NAC (5 mmol/L) or apocynin (100 μmol/L) for 4 h. n = 3. Data are represented as mean ± SEM. Two-way ANOVA followed by Sidak’s multiple comparisons test, ∗p < 0.05.
Targeting FAM3D ameliorates hypertension
To further confirm the role of endothelial FAM3D and verify the potential translational significance of FAM3D in hypertension, we firstly infected C57BL/6 mice with an adeno-associated virus serotype 9 (AAV9) vector encoding endothelial-specific expressed FAM3D short hairpin RNA (shRNA) to knock down FAM3D in ECs in vivo, followed by 14-day AngII infusion at 2 weeks after AAV injection (Figure 6A). Consequently, endothelial FAM3D knockdown reversed eNOS uncoupling, as evidenced by the reduced eNOS monomerization and the increased NO bioavailability, and thereby lowered blood pressure in AngII-induced mice (Figures 6B–6D). Meanwhile, we applied DOCA-salt-induced models in mice with AAV-mediated specific knockdown of endothelial FAM3D (Figure 6E). As well, knockdown of endothelial FAM3D restored eNOS coupling and alleviated DOCA-salt-induced hypertension (Figures 6F–6H). These results indicated the pathogenic role of endothelium-derived FAM3D in hypertension etiology. To further target FAM3D in vivo, we used a FAM3D-neutralizing antibody (6D7) that had been shown to efficiently block FAM3D function in mice in our previous study.20 C57BL/6 mice were intraperitoneally injected with immunoglobulin G (IgG) or 6D7 during the induction of hypertension by AngII for 14 days or DOCA-salt for 21 days (Figures 7A and 7E). In both hypertension models, 6D7 administration greatly reversed eNOS uncoupling (Figures 7B, 7C, 7F, and 7G), while 6D7-mediated blockade of FAM3D subsequently lowered blood pressure elevated by either AngII or DOCA-salt induction (Figures 7D and 7H), indicating that FAM3D-neutralizing antibody may be a promising therapeutic strategy for hypertension intervention.
Figure 6.

Endothelial FAM3D knockdown alleviates AngII- and DOCA-salt-induced hypertension in mice
(A) Schematic diagram showing the injection of AAV9-Tie2-shRNA (shFAM3D or shControl, 5 × 1011 v.g./mice) via tail veins followed by 14 days of AngII induction in C57BL/6 mice.
(B) Representative western blot and quantification of monomer/dimer eNOS ratios and FAM3D in the aortas of C57BL/6 mice infected with AAV9-Tie2-shControl or AAV9-Tie2-shFAM3D followed by AngII infusion for 14 days. n = 5–6. Data are represented as mean ± SEM. Unpaired Student’s t test, ∗p < 0.05.
(C) Representative DAF-FM DA en face staining and quantification of NO in ECs from the thoracic aortas of C57BL/6 mice infected with AAV9-Tie2-shControl or AAV9-Tie2-shFAM3D followed by AngII infusion for 14 days. n = 5. Data are represented as mean ± SEM. Unpaired Student’s t test, ∗p < 0.05.
(D) SBP of C57BL/6 mice infected with AAV9-Tie2-shControl or AAV9-Tie2-shFAM3D followed by AngII infusion for 14 days. n = 5–6. Data are represented as mean ± SEM. Repeated-measures analysis using a mixed-effects model, ∗p < 0.05.
(E) Schematic diagram showing the injection of AAV9-Tie2-shRNA (shFAM3D or shControl, 5 × 1011 v.g./mice) via tail veins followed by 21 days of DOCA-salt treatment in C57BL/6 mice.
(F) Representative western blot and quantification of monomer/dimer eNOS ratios and FAM3D in the aortas of C57BL/6 mice infected with AAV9-Tie2-shControl or AAV9-Tie2-shFAM3D followed by DOCA-salt treatment for 21 days. n = 5–6. Data are represented as mean ± SEM. Unpaired Student’s t test, ∗p < 0.05.
(G) Representative DAF-FM DA en face staining and quantification of NO in ECs from the thoracic aortas of C57BL/6 mice infected with AAV9-Tie2-shControl or AAV9-Tie2-shFAM3D followed by DOCA-salt treatment for 21 days. n = 4. Data are represented as mean ± SEM. Unpaired Student’s t test, ∗p < 0.05.
(H) SBP of C57BL/6 mice infected with AAV9-Tie2-shControl or AAV9-Tie2-shFAM3D followed by DOCA-salt treatment for 21 days. n = 6. Data are represented as mean ± SEM. Repeated-measures analysis using a mixed-effects model, ∗p < 0.05.
Figure 7.

The FAM3D-neutralizing antibodies ameliorate AngII- and DOCA-salt-induced hypertension in mice
(A) Schematic diagram showing the administration of the FAM3D-neutralizing antibodies (6D7) or mouse IgG (100 μg/30 g) to C57BL/6 mice during 14 days of AngII infusion.
(B) Representative western blot and quantification of monomer/dimer eNOS ratios in the aortas of C57BL/6 mice that were intraperitoneally injected with mouse IgG or 6D7 during AngII infusion for 14 days. n = 5. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test, ∗p < 0.05.
(C) Representative DAF-FM DA en face staining and quantification of NO in ECs from the thoracic aortas of C57BL/6 mice that were intraperitoneally injected with mouse IgG or 6D7 during AngII infusion for 14 days. n = 3–4. Data are represented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test, ∗p < 0.05.
(D) SBP of C57BL/6 mice that were intraperitoneally injected with mouse IgG or 6D7 during AngII infusion for 14 days. n = 6–7. Data are represented as mean ± SEM. Repeated-measures analysis using a mixed-effects model, ∗p < 0.05.
(E) Schematic diagram showing the administration of the 6D7 or mouse IgG (100 μg/30 g) to C57BL/6 mice during 21 days of DOCA-salt treatment.
(F) Representative western blot and quantification of monomer/dimer eNOS ratios in the aortas of C57BL/6 mice that were intraperitoneally injected with mouse IgG or 6D7 during DOCA-salt treatment for 21 days. n = 5. Data are represented as mean ± SEM. Unpaired Student’s t test, ∗p < 0.05.
(G) Representative DAF-FM DA en face staining and quantification of NO in ECs from the thoracic aortas of C57BL/6 mice that were intraperitoneally injected with mouse IgG or 6D7 during DOCA-salt treatment for 21 days. n = 4. Data are represented as mean ± SEM. Unpaired Student’s t test, ∗p < 0.05.
(H) SBP of C57BL/6 mice that were intraperitoneally injected with mouse IgG or 6D7 during DOCA-salt treatment for 21 days. n = 6. Data are represented as mean ± SEM. Repeated-measures analysis using a mixed-effects model, ∗p < 0.05.
Discussion
To date, the mechanism of hypertension pathogenesis is still not fully understood. Approximately 10%–15% of patients with hypertension cannot be efficiently cured by current antihypertensive therapies,3 suggesting the presence of unknown critical modulatory factors or molecules associated with hypertension. In the current study, plasma FAM3D levels were significantly elevated in both patients with hypertension and hypertensive mice. Circulating FAM3D may be an independent risk factor for hypertension populations, as supported by the multivariable conditional logistic regression analysis of the case-control study. Furthermore, we identified the pathogenic role of cytokine-like protein FAM3D in hypertension.
Our major finding is that FAM3D directly causes eNOS uncoupling in hypertension. Endothelial dysfunction mediated by eNOS uncoupling is an important pathogenic process in the etiology of hypertension, as validated in various hypertensive animal models, including genetic hypertension (SHRs),23 AngII-induced hypertension,9 and DOCA-salt-induced hypertension,24 as well as patients with hypertension.25,26,27 Although the downregulation and uncoupling of eNOS reduce NO bioavailability and cause endothelial dysfunction, eNOS uncoupling is more harmful than a lack of eNOS expression due to alterative exacerbation of oxidative stress.6 Here, FAM3D caused eNOS uncoupling but did not alter its expression in ECs. A variety of mechanisms implicated in eNOS uncoupling involve oxidative stress-induced BH4 depletion,28 L-arginine deficiency,29 and eNOS S-glutathionylation.30 We demonstrated that ROS scavengers and BH4 supplementation abolished and reversed FAM3D-induced eNOS uncoupling, respectively, suggesting that oxidative stress-induced BH4 depletion might mediate the effect of FAM3D on eNOS uncoupling. Considering that the BH4 synthesis inhibitor DAHP mostly abolished the protective effect of FAM3D deficiency against hypertension, we further hypothesized that FAM3D modulated blood pressure mainly through inducing eNOS uncoupling. In addition, chemokine-mediated leukocyte recruitment and vascular inflammation also play critical roles in the etiology of hypertension.31,32 We previously reported that FAM3D was upregulated in ECs by proinflammatory stimuli and has chemoattractant activity to directly recruit neutrophils and monocytes.20 In the current study, FAM3D deficiency ameliorated leukocyte infiltration in the vascular wall, and the possibility that FAM3D may also modulate leukocyte-involved vascular inflammation during the pathogenesis of AngII-induced hypertension in mice could not be excluded. Of interest, BH4-deficiency-induced eNOS uncoupling contributes to other cardiovascular diseases, such as atherosclerosis and AAA.33 The current discovery of FAM3D-induced eNOS uncoupling might provide another explanation for our previous work associated with FAM3D-mediated AAA.20
FAM3D belongs to a cytokine-like family composed of 4 members, referred to as FAM3A, FAM3B, FAM3C, and FAM3D, which share a 4-helix-bundle structure.18 A previous study demonstrated that VSMC-specific deletion of FAM3A attenuated AngII-induced hypertension in mice. The related mechanism involves FAM3A, as a mitochondrial protein, intracellularly enhancing ATP production and release, further promoting VSMC phenotypic transition.34 In the current article, we found that FAM3D was not expressed in VSMCs and has no effect on VSMC contraction. In addition, direct effects of purified FAM3D protein and antihypertension function of FAM3D-neutralizing antibodies supported that FAM3D exerted extracellularly. Therefore, distinct from FAM3A, FAM3D may exert its pathogenic effects in an extracellular manner as a classic cytokine-like protein.
Another interesting finding of the current study is that FAM3D exerts its hypertensive effects probably in an endothelial autocrine manner. Endothelial autocrine growth factors or cytokines are essential for maintaining vascular homeostasis.35 VEGFA is physiologically expressed and released by ECs and, conversely, activates eNOS and induces endothelial NO production and vasodilation,36 whereas anti-VEGF therapies often trigger the development of systemic hypertension due to endothelial dysfunction.37,38 In addition, some vascular pathologies involve the regulation of endothelial autocrine signaling. Endothelial transforming growth factor β (TGF-β) induces eNOS uncoupling and endothelial-to-mesenchymal transition in an autocrine manner, further exacerbating atherosclerosis or AAA formation.39,40 Endothelial autocrine CXCL12 signaling promotes pulmonary vascular EC proliferation and exacerbates pulmonary arterial hypertension.41 FAM3D is highly expressed in the gastrointestinal tract and modulated gut microbiota.21 As gut microbiota facilitates AngII-induced vascular dysfunction and hypertension,42 we excluded alterations in intestinal FAM3D expression in hypertensive mice, suggesting that gastrointestinal FAM3D may not associate with hypertension etiology. In addition, we observed that FAM3D expression was upregulated in ECs but not VSMCs in arterial tissues, while endothelial FAM3D knockdown could alleviate AngII- or DOCA-salt-induced hypertension, indicating the pathogenic role of endothelium-derived FAM3D in hypertension etiology. In addition, FAM3D caused eNOS uncoupling extracellularly and depended on the cell surface receptors FPR1 and FPR2. Thus, these findings suggested that the potential pathological autocrine effect of endothelium-derived FAM3D caused endothelial dysfunction during the pathogenesis of hypertension.
Of note, EC-specific AT1a deficiency did not alter AngII-induced hypertension in mice, suggesting the endothelial AngII signaling plays a minor role in the modulation of blood pressure.43 Here, we uncovered that the alteration and effect of FAM3D in hypertension was not specific and direct in response to AngII. First, endothelial FAM3D was upregulated not only in AngII-induced mice and SHRs but also in DOCA-salt-induced hypertensive mice, which did not directly correlate with AngII induction.44 Second, targeting FAM3D significantly lowered blood pressure in both AngII- and DOCA-salt-induced hypertensive mice. This potentially explained why targeting endothelial FAM3D-induced eNOS uncoupling, rather than endothelial AngII signaling, significantly alleviated AngII-induced hypertension, although further investigation is required to explore the underlying general mechanism of modulation of endothelial FAM3D expression in distinct hypertensive models.
FPR1 and FPR2 are cell surface receptors of FAM3D. We previously reported that FPR1 and FPR2 on monocytes and neutrophils mediate the chemoattractant activity of FAM3D to recruit these cells and cause vascular inflammation.19,20 Notably, both FPR1 and FPR2 are expressed on ECs. Stimulation of FPR1 by N-fMLP induces ROS accumulation in ECs,45 while activation of FPR2 by the selective agonist WKYMVm stimulates ischemic neovasculogenesis by promoting homing of endothelial colony-forming cells.46 Furthermore, both FPR1 and FPR2 mediate mitochondrial N-formyl peptide-induced EC contraction and vascular leakage.47 Here, we confirmed that both FPR1 and FPR2 mediate FAM3D-induced oxidative stress and subsequent eNOS uncoupling. In addition, p47phox phosphorylation mediates FPR1 activation-induced oxidative stress in ECs,45 while endothelial p47phox-dependent NOX1 and NOX2 isoforms are involved in endothelial dysfunction and hypertension in mice.48 Our results also showed that the NADPH oxidase inhibitor apocynin inhibited FAM3D-induced endothelial dysfunction, suggesting the involvement of NADPH oxidases in endothelial dysfunction and hypertension regulated by FAM3D-FPR1/FPR2 signaling.
A recent study showed that FPR1 activation by N-formyl peptides originating from renal cell death induced spontaneous, premature hypertension, whereas administration of the FPR1 antagonist CsH markedly lowered blood pressure in Dahl salt-sensitive rats.49 Of interest, FPR1 deficiency or antagonism49,50 reduces vascular contractility but does not seem to affect endothelium-dependent relaxation. However, we found that FAM3D deficiency improved endothelium-dependent relaxation, further attenuating vasocontraction. This result suggested (1) that different endogenous agonists of FPR1 may exist in Dahl salt-sensitive rats vs. AngII-induced hypertensive mice and (2) that the pathogenic effects of FAM3D in hypertension also depend on FPR2 in addition to FPR1, although further confirmation is needed.
Targeting endothelial dysfunction is an effective potential approach to lower blood pressure, although classic antihypertension strategies mainly focus on altering cardiac output, effective circulating volume, and VSMC contractility. Clinically, direct administration of NO donors (e.g., nitroglycerin and sodium nitroprusside) efficiently compensated endothelial dysfunction and lowered blood pressure in patients with acute stroke but was unsuitable for long-term use due to the potential accumulation of peroxynitrites produced by excessive NO under hypertension-related oxidative stress conditions.51 In addition, oral administration of BH4 restored eNOS coupling and inhibited the increase in blood pressure in DOCA-salt-induced hypertension,52 while a pilot clinical trial (ClinicalTrials.gov: NCT00208780) supported the therapeutic effects of BH4 on hypertension.26 However, the chemical instability of BH4 to be easily oxidized to BH2 may limit its potential clinical use.6,53 Herein, we administered a FAM3D-neutralizing antibody or AAV9-shFAM3D to block FAM3D, which effectively alleviated endothelial dysfunction and lowered both AngII- and DOCA-salt-induced hypertension. Intervention on FAM3D, avoiding excessive supplement of NO or BH4, may be a prospective strategy to reverse endothelial dysfunction for antihypertensive therapy.
In conclusion, the cytokine-like protein FAM3D, which was upregulated in ECs under various pathogenic conditions of hypertension, directly caused eNOS uncoupling through FPR1- and FPR2-mediated oxidative stress, thereby exacerbating endothelial dysfunction and hypertension. Translationally, targeting endothelium-derived FAM3D or administration of a FAM3D-neutralizing antibody restored eNOS coupling and subsequently lowered blood pressure. Therefore, FAM3D might be a promising target for treating endothelial dysfunction in hypertension.
Limitations of the study
Our study has limitations that should be considered. First, we demonstrated the upregulation of FAM3D in ECs in distinct hypertensive mouse models, but the underlying mechanism regulating FAM3D expression during the pathogenesis of hypertension is lacking direct evidence. Further exploration is needed. Additionally, we discovered that FAM3D directly caused endothelial dysfunction in human ECs in vitro and hypertensive mice in vivo, whereas whether FAM3D correlates with endothelial function in patients with hypertension has not been explored, but it is feasible to perform. Third, we started to administer AAV or neutralizing antibodies to target FAM3D before hypertension induction in mice, thus the protective effects we observed preferably suggested the preventative potential of targeting FAM3D. More translationally, targeting FAM3D by AAV or neutralizing antibodies could be performed following the establishment of hypertension in mice, which might be more relevant to the clarification of potential therapeutic efficacy. Moreover, humanization of mouse FAM3D-neutralizing antibodies is required to be accomplished before attempting to translate into them human hypertension.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-FAM3D | R&D Systems | Cat# MAB2869; RRID: AB_2262535 |
| Alexa Fluor 647 anti-mouse CD144 (VE-cadherin) | BioLegend | Cat# 138005; RRID: AB_10568319 |
| Mouse anti-CD31 | Santa Cruz Biotechnology | Cat# sc-376764; RRID: AB_2801330 |
| Alexa Fluor 488 donkey anti-goat IgG | Life Technologies | Cat# A32814; RRID: AB_2762838 |
| Alexa Fluor 555 goat anti-mouse IgG | Life Technologies | Cat# A21422; RRID: AB_141822 |
| Fluorescein isothiocyanate (FITC) anti-mouse CD45 | BioLegend | Cat# 103107; RRID: AB_312972 |
| Mouse anti-eNOS | Santa Cruz | Cat# sc-376751; RRID: AB_2832203 |
| Chemicals, peptides, and recombinant proteins | ||
| Angiotensin II | Sigma–Aldrich | A9525 |
| Deoxycorticasterone acetate (DOCA) | Innovative Research | M-121 |
| DAF-FM DA fluorescent dyes | Beyotime Institute of Biotechnology | S0019 |
| DHE fluorescent dyes | Beyotime Institute of Biotechnology | S0063 |
| DCFH-DA fluorescent dyes | Beyotime Institute of Biotechnology | S0033S |
| Osmotic minipumps | Alzet Corporation | Model 2001 or Model 2002 |
| Acetylcholine | Sigma-Aldrich Chemical | 60-31-1 |
| L-NAME | Sigma-Aldrich Chemical | 51298-62-5 |
| Sodium nitroprusside | Sigma-Aldrich Chemical | 13755-38-9 |
| Phenylephrine | Sigma-Aldrich Chemical | P1240000 |
| Apamin | MedChemexpress | HY-P0256 |
| TRAM-34 | MedChemexpress | HY-13519 |
| Indomethacin | MedChemexpress | Hy-14397 |
| Critical commercial assays | ||
| CircuLex Human FAM3D ELISA Kit | Marine Biological Laboratory | CY-8201 |
| Mouse FAM3D ELISA Kit | CUSABIO | CSB-EL008229MO |
| Trichrome stain Kit | Solarbio Life Sciences | G1340 |
| Nitrate/nitrite colorimetric assay kit | Beyotime Institute of Biotechnology | S0021 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 mice, FAM3D−/− | He et al., 201820 | https://doi.org/10.1161/ATVBAHA.118.311289 |
| Rat: Wistar-Kyoto rats | Vital River | 21005A |
| Rat: spontaneously hypertensive rats | Vital River | 21006A |
| Software and algorithms | ||
| GraphPad Prism 9.0 software | Graphpa by dotmatics | https://www.graphpad.com/ |
| Odyssey infrared imaging system software | LI-COR Biosciences | https://www.licor.com/ |
| QuantStudio Design & Analysis Software | Thermo Fisher Scientific | https://www.thermofisher.cn/cn/zh/home/life-science/pcr/real-time-pcr/real-time-pcr-instruments/quantstudio-3-5-real-time-pcr-system/quantstudio-3.html |
| LAS X software | Leica | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| LabChart 8 Reader software | ADINSTRUMENTS | https://www.adinstruments.com/products/labchart-reader |
| FlowJo 7 software | FLOWJO | https://www.flowjo.com/ |
| ImageJ software | ImageJ | https://github.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Yi Fu (yi.fu@bjmu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human study
All participants were from an atherosclerosis cohort survey conducted in Beijing, China, and definitions of diseases were described previously.54 In brief, community residents aged ≥40 years were recruited between December 2011 and April 2012. A case-control study based on baseline survey was designed to investigate the association of plasma FAM3D level and hypertension status. Initially, participants with peripheral arterial disease, diabetes mellitus, chronic kidney disease, history of myocardial infarction, stroke, tumor, taking antihypertensive and lipid-lowering medications were excluded. Then cases were selected from hypertensives with following criteria: systolic blood pressure (SBP) ≥ 140 mmHg and/or DBP ≥90 mmHg. Controls were selected from normotensives (both SBP <120 mmHg and DBP <80 mmHg), and then matched cases with 1:1 ratio for the same gender, similar age (±1 year) and body mass index (±1 kg/m2). Finally, a total of 160 subjects (80 pairs) were used in this analysis. This study was approved by the ethics committee of Peking University First Hospital, and performed in accordance with the principles of the Declaration of Helsinki. All participants provided written informed consent. Considering high-fat diet feeding causes instant elevation of FAM3D in humans,34 a venous blood sample was obtained from the forearm of each participant after an overnight fasting and plasma were separated and stored at −80°C FAM3D level was measured using frozen plasma with an ELISA kit. The study project was approved by Peking University First Hospital (Ethics No. 2021[302]) and informed consent was obtained from subjects. The detailed information of all the above patients are listed in Table S1.
Animals
All animal studies and experimental procedures followed the guidelines of the Institutional Animal Care and Use Committee of Peking University Health Science Center, an ethical organization authorized by the Beijing Municipal Science & Technology Commission. C57BL/6 mice were purchased from Vital River (Beijing, China). Eight-week-old male Wistar-Kyoto (WKY) rats and spontaneously hypertensive rats (SHR) were purchased from Huafukang Co. (Beijing, China). FAM3D−/− mice were generated on a C57BL/6 background by TALEN-mediated gene targeting.20 Twelve-week-old FAM3D−/− and littermate male mice were infused with AngII (490 ng/kg per minute) or saline for 1, 7 or 14 days via subcutaneously implanted osmotic minipumps.55 For DOCA-salt-induced hypertensive model, as previous,56 the left kidney was removed from 6-week old male mice, and after 2 days, DOCA pellets were implanted followed by feeding water containing 0.9% NaCl. Sham mice were also nephrectomized, but not received DOCA pellets implantation and given tap water. For knockdown of endothelium-derived FAM3D, mice were injected with adeno-associated viral vector serotype 9 (AAV9) vectors encoding Tie2 promoter-driven FAM3D shRNA in an amount of 5×1011 v.g./mice via tail veins two weeks before induction of hypertension models. Equal amount of AAV9 vectors encoding Tie2 promoter-driven control shRNA served as negative controls.
Cell culture
The procedures for the isolation of human-derived primary cells were approved by the Ethics Committee of the Peking University Health Science Center. As described previously,20 human umbilical vein endothelial cells (HUVECs) were isolated from umbilical cords. The umbilical cord was washed with PBS 3 times, clamped at both ends, filled with 0.25% trypsin, and then placed at 37°C. After 15 min, the umbilical cord was gently kneaded to digest endothelial cells, and then rinsed with 5% endothelial cell medium (ECM) to collect endothelial cells. HUVECs were maintained in 20% ECM at passages 1 to 3 and then in 5% ECM after passages 3. The cells were digested and passaged with 0.05% trypsin, and cells at passages 4 to 6 were used for experiments. Cell cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Method details
Western blots
Mouse tissues and cells were lysed in RIPA buffer. The protein concentrations were quantified using a BCA Protein Assay Kit. Equal amounts of total protein per sample were separated on an 8% or 12% SDS–PAGE gel and transferred onto nitrocellulose filter membranes (Pall, Port Washington, NY, USA). The membranes were blocked with 5% BSA. The membranes were incubated with primary antibodies and appropriate IRDye-conjugated secondary antibodies. The immunofluorescent signal was detected by an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA).
To detect eNOS monomers/dimers, nondenatured tissue and cell lysates were dissolved in ice-cold buffer (Tris-HCl, 50 mmol/L, pH 8; NaCl, 180 mmol/L; EDTA, 0.5 mmol/L; NP40, 0.2%; phenylmethylsulfonyl fluoride, 100 mmol/L; DTT, 1 mmol/L) without adding β-mercaptoethanol and heat denaturation, and then separated on an 8% SDS–PAGE gel and transferred onto nitrocellulose filter membranes at a low temperature, as previously described.57
Real-time PCR
Total RNA was extracted from tissues using TRIzol reagent. Equal amounts (2 μg) of total RNA were reverse transcribed to cDNA using NovoScript II Reverse Transcriptase. Real-time polymerase chain reaction (PCR) amplification was performed using SYBR Green 2× PCR mix and recorded with a QuantStudio 3 Real-Time PCR Instrument (Thermo Fisher Scientific, Waltham, MA, USA). The amplification reactions were programmed at 94°C for 7 min followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The data were analyzed using the ΔΔCT method with QuantStudio Design & Analysis Software (Thermo Fisher Scientific, Waltham, MA, USA) and normalized to β-actin as an internal control. Primer sequence information is listed in Table S6.
Histological analysis of mouse arteries
Mice were euthanized by CO2 inhalation, followed by perfusion with PBS and fixation with 4% paraformaldehyde (PFA). Thoracic aortas and mesenteric arteries were dissected and embedded in OCT compound and then cut into frozen serial sections (10 μm thick, approximately 400 μm apart). For immunofluorescent en face staining, thoracic aortas without PVAT were dissected and opened by a longitudinal cut and then inserted into a silicone disc with thin needles, followed by further fixation with 4% PFA for 15 minutes at room temperature and permeabilization with 0.1% Triton X-100 in PBS for 15 minutes.
Masson’s trichrome staining was performed on aortic cross-sections using a Trichrome Stain Kit according to the manufacturer’s protocol. The sections were incubated in mordant solution in 60°C for 1 hour and then washed with running water for 10 minutes. Celestite blue solution was dropped onto the sections for 2 minutes and washed with distilled water twice for 30 seconds. May hematoxylin solution was dropped onto the section for 1 minute and washed with distilled water twice for 30 seconds. The sections were differentiated with acid differentiation solution for several seconds and were rinsed in running water for 10 minutes. Then ponceau-acid fuchsin solution was dropped onto the sections and stained for 10 minutes. Phosphmolybic acid solution was treated with for 10 minutes and discarded. Then the sections were stained directly with aniline blue solution for 2 minutes and rinsed in acetic acid solution for 2 minutes. Finally, the sections were dehydrated and mounted.
Immunofluorescent staining was performed on arterial cross-sections or longitudinally opened thoracic aortas. The samples were blocked with 3% bovine serum albumin for 1 hour at room temperature and then incubated with goat anti-FAM3D antibodies (1:200 dilution, 2 μg/mL) and/or Alexa Fluor 647 anti-mouse VE-cadherin (1:200 dilution) for 24 hours at 4°C and then washed with Krebs solution for 3 times. Alexa Fluor 488 donkey anti-goat IgG (1:500 dilution, 2 μg/mL) secondary antibodies were incubated for 2 hours at room temperature and then washed with Krebs solution for 3 times, followed by nuclear staining with DAPI (1:1000 dilution, 5 μg/mL) for 5 minutes at room temperature. The longitudinally opened thoracic aortas were mounted on slides using antifade mounting solution, with the intima facing up. Immunofluorescent staining of arterial cross-sections or the endothelial layers of thoracic aortas was observed and acquired using a confocal microscope (Leica, Wetzlar, Germany).
Mouse blood pressure measurement and monitoring
Mouse blood pressure was measured according to the recommendation of the American Heart Association council on high blood pressure research.58 A noninvasive computerized CODA tail-cuff blood pressure system (Kent Scientific, Torrington, CT, USA) was used to measure mouse SBP. The equipment was kept clean and free from foreign scents and blood odors. The investigator was blinded to the experimental groups when performing the measurements, and the mice were tested in a randomized order. All mice underwent training sessions from 1 to 4 PM on 7 consecutive days to become accustomed to the tail-cuff procedure. During the 14 days of Ang II induction, SBP was recorded every 1-2 days for AngII-induced hypertensive model and every 4 days for DOCA-salt-induced hypertensive model, and 5 measurements were performed daily from 1-4 PM on each mouse.
Blood pressure was also monitored using implanted radiotelemetry probes (PA-C10, Data Science International, Tilburg, The Netherlands).59 Model PA-C10 transmitters were inserted into the left carotid artery of each mouse under anesthesia. Then, the mice were allowed to recover for 10 days before the implantation of osmotic minipumps containing saline or AngII. BP was monitored before and after minipump implantation on a Dataquest ART Silver acquisition system (Data Sciences International). Continuous measurements of SBP, DBP and MBP were performed daily every 10 seconds from 1 to 4 PM, and the averages of these 3-hour measurements were recorded. In addition, 24-hour monitoring of SBP and DBP was performed using measurements taken at 10-second intervals, and the averages of the measurements every 5 minutes were recorded.
Pulse wave velocity measurement
Pulse wave velocity (PWV) was measured 2 weeks after saline or Ang II infusion by using a high-resolution ultrasound instrument (Vevo 2100, Visual Sonics, Toronto, Canada), as described previously.60 Ascending and descending aortic peak velocities were measured in the pulse wave (PW) Doppler-mode aortic arch view. PWV was obtained from the B-mode and Doppler-mode aortic arch views and was calculated as (D: aortic arch distance; T: transit time).
Flow cytometry
Leukocytes in aortic vessels were analyzed by flow cytometry as previously described.20,61 Aortic vessels were cleaned off fatty tissue, minced, and digested into single cells with aortic dissociation enzyme solution (Collagenase I, 450 U/ml; Hyaluronidase type I-S, 60 U/ml; DNase I, 60 U/ml; Collagenase XI, 125 U/ml). After washing off the digest with Krebs solution (NaCl, 119 mmol/L; NaHCO3, 25 mmol/L; KCl, 4.7 mmol/L; KH2PO4, 1.2 mmol/L; CaCl2, 2.5 mmol/L; MgCl2, 1 mmol/L; D-glucose, 11 mmol/L) for 3 times, single-cell suspensions were stained with FITC-anti-CD45 (1:200) in Krebs solution for 30 minutes on ice. Then the cells were resuspended in 300 μl Kreb’s solution after washing out the antibody and analyzed using a BD FACSCalibur system (BD Biosciences, San Diego, CA).
Isometric force measurement in wire myography
Fresh mouse mesenteric arteries were dissected and stored in ice-cold Krebs solution (119 mmol/L NaCl, 25 mmol/L NaHCO3, 4.7 mmol/L KCl, 1.2 mmol/L KH2PO4, 2.5 mmol/L CaCl2, 1 mmol/L MgCl2, and 11 mmol/L D-glucose). Mesenteric arteries (about 1 mm in length) were mounted on wire myographs (Danish Myo Technology, Aarhus, Denmark) by 25-μm diameter wire to record changes in isometric tension, as described by previous studies.62,63 To test VSMC integrity, vasocontraction in response to phenylephrine (0.003 nmol/L to 30 μmol/L) was evaluated in mesenteric arterial rings with or without pretreatment with L-NAME (100 μmol/L) for 30 minutes. Endothelium-dependent relaxation in response to acetylcholine (ACh, 0.003 to 10 μmol/L) was examined in rings that had been precontracted with phenylephrine (Phe, 1 μmol/L). After being washed 4 times, the rings were incubated with NG-nitro-L-arginine methyl ester (L-NAME, 100 μmol/L) for 30 minutes. Then, endothelium-independent relaxation in response to sodium nitroprusside (SNP, 0.001 to 10 μmol/L) were examined after the rings were precontracted with Phe (1 μmol/L). For endothelial hyperpolarizing factor (EDHF)-induced relaxation in response to Ach was examined in rings that were also precontracted with Phe (1 μmol/L) after combined incubation of L-NAME (100 μmol/L) and PGI2 inhibitor indomethacin (INDO, 10 μmol/L) for 30 minutes, and then were precontracted with Phe (1 μmol/L) after combined incubation of L-NAME, INDO and EDHF inhibitor (Apamin, 1 μmol/L; Tram-34, 1 μmol/L) for 30 minutes. Following the previous study,64 the active wall tension (△T) was applied to indicate the extent of vasocontraction and vasorelaxation. Calculation formula includes and (Tactive: wall tensions in activating solution; Trelax: wall tensions in relaxing solutions; T: wall tension; F: measurement of force, g: mean vessel segment length).
Purification of FAM3D protein
Human FAM3D protein was purified following our previous study.20 The plasmids pcDNA3.1-human FAM3D-Myc-His were transfected into HEK293T cells using jetPEI (PT-101, Polyplus, Berkeley, CA, USA) according to the manufacturer’s instructions. Sixteen hours after transfection, the culture medium was replaced with OptiMEM (Invitrogen, Waltham, MA, USA), and the cells were cultured for another 3 days. The conditioned medium was harvested, concentrated and replaced with binding buffer. then FAM3D-Myc-His proteins were purified by the Ni Sepharose High Performance system according to the manufacturer’s instructions (GE Healthcare, Danderyd, Sweden). Briefly, using a gravity column, binding buffer and nickel beads were combined three times, followed by elution of nontarget protein with 5 mM, 15 mM and 30 mM imidazole eluate, and then target proteins were eluted with 100 mM imidazole eluent. The target protein eluate was replaced with PBS and finally concentrated.
Measurement of endothelial NO production
Intracellular NO production was measured in endothelial cells or the arterial intima using DAF-FM DA (excitation at 488 nm and emission at 520 nm).65,66 HUVECs were treated and incubated with 10 μmol/L DAF-FM DA in serum-free ECM at 37°C for 30 minutes. After being washed 3 times with PBS, the cells were fixed with 4% PFA for 15 minutes at room temperature. Images were acquired using a Cytation 5 system (Bio Tek, VT, USA). In addition, fresh thoracic aortas were incubated with 50 μmol/L DAF-FM DA in Krebs-HEPES buffer at room temperature for 45 minutes with shaking in the dark. After being washed 3 times with Krebs buffer, the aortas were fixed with 4% PFA for 15 minutes at room temperature. Then, the aortas were opened by a longitudinal cut for subsequent staining of the endothelial marker VE-cadherin as described above. Images were taken under a confocal microscope (Leica, Wetzlar, Germany).
NO secretion from endothelial cells into the culture media was measured using a Griess assay.67 Briefly, 5 × 104 HUVECs per well were seeded in a 96-well plate and allowed to attach for 24 hours. Following the various treatments, a nitrate/nitrite colorimetric assay kit was used to measure extracellular NO levels in the cell culture media. Total nitrite levels were measured at 540 nm after adding Griess reagents using a Varioskan microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
Detection of O2- generation in endothelial cells
O2− generation was measured by DHE staining in HUVECs and the endothelial layer of aortic tissues with or without preincubation with L-NAME (200 μmol/L) for 30 minutes. For cultured cell staining, DHE (1 μmol/L) in serum-free ECM was added and incubated for 30 minutes at 37°C. After being washed 3 times with PBS, the cells were digested with 0.05% trypsin, resuspended with 300 μL serum-free ECM and measured by flow cytometry using a BD FACSCalibur system (BD Biosciences, San Diego, CA). In addition, fresh thoracic aortas were incubated with 10 μmol/L DHE in Krebs-HEPES buffer at room temperature for 45 minutes with shaking in the dark. After being washed 3 times with Krebs-HEPES buffer, the aortas were fixed with 4% PFA for 15 minutes at room temperature. The aortas were opened by a longitudinal cut for subsequent staining of the endothelial marker VE-cadherin as described above. Images were taken under a confocal microscope (Leica, Wetzlar, Germany) with excitation at 535 nm and emission at 610 nm.
Detection of ROS production in HUVECs
ROS production in HUVECs was assessed using the fluorescent dye DCFH-DA and flow cytometry.68 HUVECs were stimulated and loaded with 1 μmol/L of DCFH-DA in serum-free ECM for 30 min at 37°C. After being washed 3 times with PBS, the cells were digested with 0.05% trypsin, resuspended with 300 μL serum-free ECM and measured by flow cytometry using a BD FACSCalibur system (BD Biosciences, San Diego, CA).
Quantification and statistical analysis
Values are presented as the mean ± standard error (SEM) for normally distributed data and median (1/4 interquartile range (IQR)-3/4 IQR) for skew distributed data. Statistical analyses were performed using GraphPad Prism 9.0 software (GraphPad Software, San Diego, CA, USA). For statistical comparisons, we first evaluated whether the data were normally distributed using the Shapiro–Wilk normality test. Then, we used Student’s t-tests for two-group comparisons of normally distributed data. If the data was is skewed distribution, we used Mann-Whitney test. In addition, we used ANOVA followed by post hoc analysis for comparisons among normally distributed data from more than two groups. Specifically, Dunnett's multiple comparisons test was used to compare each group with a control group, while Tukey's multiple comparisons test was used to compare each group with every other group. In addition, Sidak’s multiple comparisons test was performed to compare each test group with its own control after two-way ANOVA. In all cases, a statistically significant difference was present when the two-tailed probability was less than 0.05. The details of the statistical analysis used in each experiment are presented in the corresponding figure legends.
Acknowledgments
This research was supported by funding from the National Natural Science Foundation of China of P.R. China (NSFC, 81922009, 81921001, 31930056, and 81900412), the Key R&D Program of Sichuan Province (2021YFSY0038), the National Key R&D Program of China (2019YFA0801600), and the Scientific Research Starting Foundation of Peking University (BMU2022RCZX011).
Author contributions
Conceptualization, Y.S., W.K., and Y.F.; validation, Y.S., Z.D., S.Z., R.D., J.H., N.X., Z.G., X.Y., J.T., and L.H.; formal analysis, Y.S., Z.D., F.F., and K.L.; investigation, Y.S. and Z.D.; visualization, Y.S. and Z.D.; writing – original draft, Y.S. and Y.F.; writing – review & editing, W.K. and Y.F.; resources, F.F., L.L., F.Y., Y.T., J.X., Y.W., and Y.Z.; funding acquisition, L.H., Z.Y., W.K., Y.Z., and Y.F.; supervision, W.K., Y.Z., and Y.F.; project administration, Y.F. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: June 9, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101072.
Contributor Information
Wei Kong, Email: kongw@bjmu.edu.cn.
Yan Zhang, Email: drzhy1108@163.com.
Yi Fu, Email: yi.fu@bjmu.edu.cn.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Oparil S., Acelajado M.C., Bakris G.L., Berlowitz D.R., Cífková R., Dominiczak A.F., Grassi G., Jordan J., Poulter N.R., Rodgers A., Whelton P.K. Hypertension. Nat. Rev. Dis. Prim. 2018;4 doi: 10.1038/nrdp.2018.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrison D.G., Coffman T.M., Wilcox C.S. Pathophysiology of hypertension: the mosaic theory and beyond. Circ. Res. 2021;128:847–863. doi: 10.1161/CIRCRESAHA.121.318082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oparil S., Schmieder R.E. New approaches in the treatment of hypertension. Circ. Res. 2015;116:1074–1095. doi: 10.1161/CIRCRESAHA.116.303603. [DOI] [PubMed] [Google Scholar]
- 4.Gallo G., Volpe M., Savoia C. Endothelial dysfunction in hypertension: current concepts and clinical implications. Front. Med. 2021;8 doi: 10.3389/fmed.2021.798958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ambrosino P., Bachetti T., D'Anna S.E., Galloway B., Bianco A., D'Agnano V., Papa A., Motta A., Perrotta F., Maniscalco M. Mechanisms and clinical implications of endothelial dysfunction in arterial hypertension. J. Cardiovasc. Dev. Dis. 2022;9 doi: 10.3390/jcdd9050136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Q., Youn J.Y., Cai H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 2015;33:1128–1136. doi: 10.1097/HJH.0000000000000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leo F., Suvorava T., Heuser S.K., Li J., LoBue A., Barbarino F., Piragine E., Schneckmann R., Hutzler B., Good M.E., et al. Red blood cell and endothelial eNOS independently regulate circulating nitric oxide metabolites and blood pressure. Circulation. 2021;144:870–889. doi: 10.1161/CIRCULATIONAHA.120.049606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrizzo A., Conte G.M., Sommella E., Damato A., Ambrosio M., Sala M., Scala M.C., Aquino R.P., De Lucia M., Madonna M., et al. Novel potent decameric peptide of spirulina platensis reduces blood pressure levels through a PI3K/AKT/eNOS-Dependent mechanism. Hypertension. 2019;73:449–457. doi: 10.1161/HYPERTENSIONAHA.118.11801. [DOI] [PubMed] [Google Scholar]
- 9.Li H., Li Q., Zhang Y., Liu W., Gu B., Narumi T., Siu K.L., Youn J.Y., Liu P., Yang X., Cai H. Novel treatment of hypertension by specifically targeting E2F for restoration of endothelial dihydrofolate reductase and eNOS function under oxidative stress. Hypertension. 2019;73:179–189. doi: 10.1161/HYPERTENSIONAHA.118.11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galougahi K.K., Liu C.C., Gentile C., Kok C., Nunez A., Garcia A., Fry N.A.S., Davies M.J., Hawkins C.L., Rasmussen H.H., Figtree G.A. Glutathionylation mediates angiotensin II-induced eNOS uncoupling, amplifying NADPH oxidase-dependent endothelial dysfunction. J. Am. Heart Assoc. 2014;3 doi: 10.1161/JAHA.113.000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H., Yuan Z., Wang B., Li B., Lv H., He J., Huang Y., Cui Z., Ma Q., Li T., et al. COMP (cartilage oligomeric matrix protein), a novel PIEZO1 regulator that controls blood pressure. Hypertension. 2022;79:549–561. doi: 10.1161/HYPERTENSIONAHA.121.17972. [DOI] [PubMed] [Google Scholar]
- 12.Wu Y., Ding Y., Ramprasath T., Zou M.H. Oxidative stress, GTPCH1, and endothelial nitric oxide synthase uncoupling in hypertension. Antioxidants Redox Signal. 2021;34:750–764. doi: 10.1089/ars.2020.8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griendling K.K., Camargo L.L., Rios F.J., Alves-Lopes R., Montezano A.C., Touyz R.M. Oxidative stress and hypertension. Circ. Res. 2021;128:993–1020. doi: 10.1161/CIRCRESAHA.121.318063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tejero J., Shiva S., Gladwin M.T. Sources of vascular nitric oxide and reactive oxygen species and their regulation. Physiol. Rev. 2019;99:311–379. doi: 10.1152/physrev.00036.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szczepaniak P., Siedlinski M., Hodorowicz-Zaniewska D., Nosalski R., Mikolajczyk T.P., Dobosz A.M., Dikalova A., Dikalov S., Streb J., Gara K., et al. Breast cancer chemotherapy induces vascular dysfunction and hypertension through NOX4 dependent mechanism. J. Clin. Invest. 2022;132 doi: 10.1172/JCI149117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie H.H., Zhou S., Chen D.D., Channon K.M., Su D.F., Chen A.F. GTP cyclohydrolase I/BH4 pathway protects EPCs via suppressing oxidative stress and thrombospondin-1 in salt-sensitive hypertension. Hypertension. 2010;56:1137–1144. doi: 10.1161/HYPERTENSIONAHA.110.160622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Virdis A., Colucci R., Fornai M., Duranti E., Giannarelli C., Bernardini N., Segnani C., Ippolito C., Antonioli L., Blandizzi C., et al. Cyclooxygenase-1 is involved in endothelial dysfunction of mesenteric small arteries from angiotensin II-infused mice. Hypertension. 2007;49:679–686. doi: 10.1161/01.HYP.0000253085.56217.11. [DOI] [PubMed] [Google Scholar]
- 18.Zhu Y., Xu G., Patel A., McLaughlin M.M., Silverman C., Knecht K., Sweitzer S., Li X., McDonnell P., Mirabile R., et al. Cloning, expression, and initial characterization of a novel cytokine-like gene family. Genomics. 2002;80:144–150. doi: 10.1006/geno.2002.6816. [DOI] [PubMed] [Google Scholar]
- 19.Peng X., Xu E., Liang W., Pei X., Chen D., Zheng D., Zhang Y., Zheng C., Wang P., She S., et al. Identification of FAM3D as a new endogenous chemotaxis agonist for the formyl peptide receptors. J. Cell Sci. 2016;129:1831–1842. doi: 10.1242/jcs.183053. [DOI] [PubMed] [Google Scholar]
- 20.He L., Fu Y., Deng J., Shen Y., Wang Y., Yu F., Xie N., Chen Z., Hong T., Peng X., et al. Deficiency of FAM3D (family with sequence similarity 3, member D), A novel chemokine, attenuates neutrophil recruitment and ameliorates abdominal aortic aneurysm development. Arterioscler. Thromb. Vasc. Biol. 2018;38:1616–1631. doi: 10.1161/ATVBAHA.118.311289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang W., Peng X., Li Q., Wang P., Lv P., Song Q., She S., Huang S., Chen K., Gong W., et al. FAM3D is essential for colon homeostasis and host defense against inflammation associated carcinogenesis. Nat. Commun. 2020;11:5912. doi: 10.1038/s41467-020-19691-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Félétou M., Köhler R., Vanhoutte P.M. Nitric oxide: orchestrator of endothelium-dependent responses. Ann. Med. 2012;44:694–716. doi: 10.3109/07853890.2011.585658. [DOI] [PubMed] [Google Scholar]
- 23.Li H., Witte K., August M., Brausch I., Gödtel-Armbrust U., Habermeier A., Closs E.I., Oelze M., Münzel T., Förstermann U. Reversal of endothelial nitric oxide synthase uncoupling and up-regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J. Am. Coll. Cardiol. 2006;47:2536–2544. doi: 10.1016/j.jacc.2006.01.071. [DOI] [PubMed] [Google Scholar]
- 24.Du Y.H., Guan Y.Y., Alp N.J., Channon K.M., Chen A.F. Endothelium-specific GTP cyclohydrolase I overexpression attenuates blood pressure progression in salt-sensitive low-renin hypertension. Circulation. 2008;117:1045–1054. doi: 10.1161/CIRCULATIONAHA.107.748236. [DOI] [PubMed] [Google Scholar]
- 25.Rossi R., Chiurlia E., Nuzzo A., Cioni E., Origliani G., Modena M.G. Flow-mediated vasodilation and the risk of developing hypertension in healthy postmenopausal women. J. Am. Coll. Cardiol. 2004;44:1636–1640. doi: 10.1016/j.jacc.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 26.Porkert M., Sher S., Reddy U., Cheema F., Niessner C., Kolm P., Jones D.P., Hooper C., Taylor W.R., Harrison D., Quyyumi A.A. Tetrahydrobiopterin: a novel antihypertensive therapy. J. Hum. Hypertens. 2008;22:401–407. doi: 10.1038/sj.jhh.1002329. [DOI] [PubMed] [Google Scholar]
- 27.Jablonski K.L., Racine M.L., Geolfos C.J., Gates P.E., Chonchol M., McQueen M.B., Seals D.R. Dietary sodium restriction reverses vascular endothelial dysfunction in middle-aged/older adults with moderately elevated systolic blood pressure. J. Am. Coll. Cardiol. 2013;61:335–343. doi: 10.1016/j.jacc.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chuaiphichai S., Crabtree M.J., McNeill E., Hale A.B., Trelfa L., Channon K.M., Douglas G. A key role for tetrahydrobiopterin-dependent endothelial NOS regulation in resistance arteries: studies in endothelial cell tetrahydrobiopterin-deficient mice. Br. J. Pharmacol. 2017;174:657–671. doi: 10.1111/bph.13728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu Y., Rajapakse A.G., Montani J.P., Yang Z., Ming X.F. p38 mitogen-activated protein kinase is involved in arginase-II-mediated eNOS-uncoupling in obesity. Cardiovasc. Diabetol. 2014;13:113. doi: 10.1186/s12933-014-0113-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen C.A., Wang T.Y., Varadharaj S., Reyes L.A., Hemann C., Talukder M.A.H., Chen Y.R., Druhan L.J., Zweier J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468:1115–1118. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mikolajczyk T.P., Szczepaniak P., Vidler F., Maffia P., Graham G.J., Guzik T.J. Role of inflammatory chemokines in hypertension. Pharmacol. Ther. 2021;223 doi: 10.1016/j.pharmthera.2020.107799. [DOI] [PubMed] [Google Scholar]
- 32.Murray E.C., Nosalski R., MacRitchie N., Tomaszewski M., Maffia P., Harrison D.G., Guzik T.J. Therapeutic targeting of inflammation in hypertension: from novel mechanisms to translational perspective. Cardiovasc. Res. 2021;117:2589–2609. doi: 10.1093/cvr/cvab330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chuaiphichai S., Rashbrook V.S., Hale A.B., Trelfa L., Patel J., McNeill E., Lygate C.A., Channon K.M., Douglas G. Endothelial cell tetrahydrobiopterin modulates sensitivity to Ang (angiotensin) II-induced vascular remodeling, blood pressure, and abdominal aortic aneurysm. Hypertension. 2018;72:128–138. doi: 10.1161/HYPERTENSIONAHA.118.11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Wit N.J.W., IJssennagger N., Oosterink E., Keshtkar S., Hooiveld G.J.E.J., Mensink R.P., Hammer S., Smit J.W.A., Müller M., van der Meer R. Oit1/Fam3D, a gut-secreted protein displaying nutritional status-dependent regulation. J. Nutr. Biochem. 2012;23:1425–1433. doi: 10.1016/j.jnutbio.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Ricard N., Bailly S., Guignabert C., Simons M. The quiescent endothelium: signalling pathways regulating organ-specific endothelial normalcy. Nat. Rev. Cardiol. 2021;18:565–580. doi: 10.1038/s41569-021-00517-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee S., Chen T.T., Barber C.L., Jordan M.C., Murdock J., Desai S., Ferrara N., Nagy A., Roos K.P., Iruela-Arispe M.L. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Touyz R.M., Herrmann J. Cardiotoxicity with vascular endothelial growth factor inhibitor therapy. NPJ Precis. Oncol. 2018;2:13. doi: 10.1038/s41698-018-0056-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Touyz R.M., Herrmann S.M.S., Herrmann J. Vascular toxicities with VEGF inhibitor therapies-focus on hypertension and arterial thrombotic events. J. Am. Soc. Hypertens. 2018;12:409–425. doi: 10.1016/j.jash.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen P.Y., Qin L., Li G., Wang Z., Dahlman J.E., Malagon-Lopez J., Gujja S., Cilfone N.A., Kauffman K.J., Sun L., et al. Endothelial TGF-beta signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 2019;1:912–926. doi: 10.1038/s42255-019-0102-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang K., Wang Y., Siu K.L., Zhang Y., Cai H. Targeting feed-forward signaling of TGFbeta/NOX4/DHFR/eNOS uncoupling/TGFbeta axis with anti-TGFbeta and folic acid attenuates formation of aortic aneurysms: novel mechanisms and therapeutics. Redox Biol. 2021;38 doi: 10.1016/j.redox.2020.101757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yi D., Liu B., Wang T., Liao Q., Zhu M.M., Zhao Y.Y., Dai Z. Endothelial autocrine signaling through CXCL12/CXCR4/FoxM1 Axis contributes to severe pulmonary arterial hypertension. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22063182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karbach S.H., Schönfelder T., Brandão I., Wilms E., Hörmann N., Jäckel S., Schüler R., Finger S., Knorr M., Lagrange J., et al. Gut microbiota promote angiotensin II-induced arterial hypertension and vascular dysfunction. J. Am. Heart Assoc. 2016;5 doi: 10.1161/JAHA.116.003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rateri D.L., Moorleghen J.J., Knight V., Balakrishnan A., Howatt D.A., Cassis L.A., Daugherty A. Depletion of endothelial or smooth muscle cell-specific angiotensin II type 1a receptors does not influence aortic aneurysms or atherosclerosis in LDL receptor deficient mice. PLoS One. 2012;7 doi: 10.1371/journal.pone.0051483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gavras H., Brunner H.R., Laragh J.H., Vaughan E.D., Jr., Koss M., Cote L.J., Gavras I. Malignant hypertension resulting from deoxycorticosterone acetate and salt excess: role of renin and sodium in vascular changes. Circ. Res. 1975;36:300–309. doi: 10.1161/01.res.36.2.300. [DOI] [PubMed] [Google Scholar]
- 45.Cattaneo F., Castaldo M., Parisi M., Faraonio R., Esposito G., Ammendola R. Formyl peptide receptor 1 modulates endothelial cell functions by NADPH oxidase-dependent VEGFR2 transactivation. Oxid. Med. Cell. Longev. 2018;2018 doi: 10.1155/2018/2609847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heo S.C., Kwon Y.W., Jang I.H., Jeong G.O., Yoon J.W., Kim C.D., Kwon S.M., Bae Y.S., Kim J.H. WKYMVm-induced activation of formyl peptide receptor 2 stimulates ischemic neovasculogenesis by promoting homing of endothelial colony-forming cells. Stem Cell. 2014;32:779–790. doi: 10.1002/stem.1578. [DOI] [PubMed] [Google Scholar]
- 47.Wenceslau C.F., McCarthy C.G., Webb R.C. Formyl peptide receptor activation elicits endothelial cell contraction and vascular leakage. Front. Immunol. 2016;7:297. doi: 10.3389/fimmu.2016.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lassègue B., Clempus R.E. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003;285:R277–R297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 49.Edwards J.M., Roy S., Galla S.L., Tomcho J.C., Bearss N.R., Waigi E.W., Mell B., Cheng X., Saha P., Vijay-Kumar M., et al. FPR-1 (formyl peptide receptor-1) activation promotes spontaneous, premature hypertension in Dahl salt-sensitive rats. Hypertension. 2021;77:1191–1202. doi: 10.1161/HYPERTENSIONAHA.120.16237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wenceslau C.F., McCarthy C.G., Szasz T., Calmasini F.B., Mamenko M., Webb R.C. Formyl peptide receptor-1 activation exerts a critical role for the dynamic plasticity of arteries via actin polymerization. Pharmacol. Res. 2019;141:276–290. doi: 10.1016/j.phrs.2019.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Varon J. Treatment of acute severe hypertension: current and newer agents. Drugs. 2008;68:283–297. doi: 10.2165/00003495-200868030-00003. [DOI] [PubMed] [Google Scholar]
- 52.Landmesser U., Dikalov S., Price S.R., McCann L., Fukai T., Holland S.M., Mitch W.E., Harrison D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H., Forstermann U. Pharmacological prevention of eNOS uncoupling. Curr. Pharmaceut. Des. 2014;20:3595–3606. doi: 10.2174/13816128113196660749. [DOI] [PubMed] [Google Scholar]
- 54.Fan F., Qi L., Jia J., Xu X., Liu Y., Yang Y., Qin X., Li J., Li H., Zhang Y., Huo Y. Noninvasive central systolic blood pressure is more strongly related to kidney function decline than peripheral systolic blood pressure in a Chinese community-based population. Hypertension. 2016;67:1166–1172. doi: 10.1161/HYPERTENSIONAHA.115.07019. [DOI] [PubMed] [Google Scholar]
- 55.Nosalski R., Siedlinski M., Denby L., McGinnigle E., Nowak M., Cat A.N.D., Medina-Ruiz L., Cantini M., Skiba D., Wilk G., et al. T-Cell-Derived miRNA-214 mediates perivascular fibrosis in hypertension. Circ. Res. 2020;126:988–1003. doi: 10.1161/CIRCRESAHA.119.315428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pérez-Rivera A.A., Fink G.D., Galligan J.J. Vascular reactivity of mesenteric arteries and veins to endothelin-1 in a murine model of high blood pressure. Vasc. Pharmacol. 2005;43:1–10. doi: 10.1016/j.vph.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 57.Lobysheva I., Rath G., Sekkali B., Bouzin C., Feron O., Gallez B., Dessy C., Balligand J.L. Moderate caveolin-1 downregulation prevents NADPH oxidase-dependent endothelial nitric oxide synthase uncoupling by angiotensin II in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2011;31:2098–2105. doi: 10.1161/ATVBAHA.111.230623. [DOI] [PubMed] [Google Scholar]
- 58.Kurtz T.W., Griffin K.A., Bidani A.K., Davisson R.L., Hall J.E., Subcommittee of, P. Public Education of the American Heart Association Council on High Blood Pressure, R. Recommendations for blood pressure measurement in humans and experimental animals: part 2: blood pressure measurement in experimental animals: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Arterioscler. Thromb. Vasc. Biol. 2005;25:e22–e33. doi: 10.1161/01.ATV.0000158419.98675.d7. [DOI] [PubMed] [Google Scholar]
- 59.Kawarazaki W., Mizuno R., Nishimoto M., Ayuzawa N., Hirohama D., Ueda K., Kawakami-Mori F., Oba S., Marumo T., Fujita T. Salt causes aging-associated hypertension via vascular Wnt5a under Klotho deficiency. J. Clin. Invest. 2020;130:4152–4166. doi: 10.1172/JCI134431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bendall J.K., Alp N.J., Warrick N., Cai S., Adlam D., Rockett K., Yokoyama M., Kawashima S., Channon K.M. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ. Res. 2005;97:864–871. doi: 10.1161/01.RES.0000187447.03525.72. [DOI] [PubMed] [Google Scholar]
- 61.Wenzel P., Knorr M., Kossmann S., Stratmann J., Hausding M., Schuhmacher S., Karbach S.H., Schwenk M., Yogev N., Schulz E., et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–1381. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y., Tan N., Zong Y., Li L., Zhang Y., Liu J., Wang X., Han W., Liu L. LncRNA ENSMUST00000155383 is involved in the improvement of DPP-4 inhibitor MK-626 on vascular endothelial function by modulating cacna1c-mediated Ca(2+) influx in hypertensive mice. Front. Mol. Biosci. 2021;8 doi: 10.3389/fmolb.2021.724225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu J., Wang L., Tian X.Y., Liu L., Wong W.T., Zhang Y., Han Q.B., Ho H.M., Wang N., Wong S.L., et al. Unconjugated bilirubin mediates heme oxygenase-1-induced vascular benefits in diabetic mice. Diabetes. 2015;64:1564–1575. doi: 10.2337/db14-1391. [DOI] [PubMed] [Google Scholar]
- 64.Mulvany M.J., Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- 65.Park S.H., Belcastro E., Hasan H., Matsushita K., Marchandot B., Abbas M., Toti F., Auger C., Jesel L., Ohlmann P., et al. Angiotensin II-induced upregulation of SGLT1 and 2 contributes to human microparticle-stimulated endothelial senescence and dysfunction: protective effect of gliflozins. Cardiovasc. Diabetol. 2021;20:65. doi: 10.1186/s12933-021-01252-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang J., Cai C., Zheng T., Wu X., Wang D., Zhang K., Xu B., Yan R., Gong H., Zhang J., et al. Endothelial scaffolding protein ENH (enigma homolog protein) promotes PHLPP2 (pleckstrin homology domain and leucine-rich repeat protein phosphatase 2)-mediated dephosphorylation of AKT1 and eNOS (endothelial NO synthase) promoting vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2020;40:1705–1721. doi: 10.1161/ATVBAHA.120.314172. [DOI] [PubMed] [Google Scholar]
- 67.Sundar U.M., Ugusman A., Chua H.K., Latip J., Aminuddin A. Piper sarmentosum promotes endothelial nitric oxide production by reducing asymmetric dimethylarginine in tumor necrosis factor-alpha-induced human umbilical vein endothelial cells. Front. Pharmacol. 2019;10:1033. doi: 10.3389/fphar.2019.01033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eligini S., Barbieri S.S., Cavalca V., Camera M., Brambilla M., De Franceschi M., Tremoli E., Colli S. Diversity and similarity in signaling events leading to rapid Cox-2 induction by tumor necrosis factor-alpha and phorbol ester in human endothelial cells. Cardiovasc. Res. 2005;65:683–693. doi: 10.1016/j.cardiores.2004.10.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.