

Abstract
Background
The majority of patients with childhood interstitial lung disease (chILD) caused by pathogenic variants in ATP binding cassette subfamily A member 3 (ABCA3) develop severe respiratory insufficiency within their first year of life and succumb to disease if not lung transplanted. This register-based cohort study reviews patients with ABCA3 lung disease who survived beyond the age of 1 year.
Method
Over a 21-year period, patients diagnosed as chILD due to ABCA3 deficiency were identified from the Kids Lung Register database. 44 patients survived beyond the first year of life and their long-term clinical course, oxygen supplementation and pulmonary function were reviewed. Chest CT and histopathology were scored blindly.
Results
At the end of the observation period, median age was 6.3 years (IQR: 2.8–11.7) and 36/44 (82%) were still alive without transplantation. Patients who had never received supplemental oxygen therapy survived longer than those persistently required oxygen supplementation (9.7 (95% CI 6.7 to 27.7) vs 3.0 years (95% CI 1.5 to 5.0), p=0.0126). Interstitial lung disease was clearly progressive over time based on lung function (forced vital capacity % predicted absolute loss −1.1% /year) and on chest CT (increasing cystic lesions in those with repetitive imaging). Lung histology pattern were variable (chronic pneumonitis of infancy, non-specific interstitial pneumonia, and desquamative interstitial pneumonia). In 37/44 subjects, the ABCA3 sequence variants were missense variants, small insertions or deletions with in-silico tools predicting some residual ABCA3 transporter function.
Conclusion
The natural history of ABCA3-related interstitial lung disease progresses during childhood and adolescence. Disease-modifying treatments are desirable to delay such disease course.
Keywords: ABCA3, rare lung diseases, paediatric interstitial lung disease
WHAT IS ALREADY KNOWN ON THIS TOPIC
ABCA3 deficiency is the most frequent genetic cause of childhood interstitial lung disease. Whereas two-thirds of the patients reported in the literature died early in life, very limited data are available on patients surviving beyond their first birthday.
WHAT THIS STUDY ADDS
During childhood and adolescence lung disease was progressive with an absolute loss of forced vital capacity % predicted of 1.1% per year and increasing cystic lesions on chest CT. At a median age of 6.3 years, 82% were still alive without lung transplantation, and disease course appeared dependent on the specific ABCA3 sequence variation.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Clinical trials with gene modifying and antifibrotic treatments are urgently needed.
Introduction
ABCA3 (ATP binding cassette subfamily A member 3) is a lipid transport protein anchored to the outer membrane of lamellar bodies (LBs) in alveolar type 2 lung cells.1 By transporting phosphatidylcholine and other lipids from the cytoplasm into LBs to assemble pulmonary surfactant, ABCA3 plays a critical part in normal surfactant metabolism. Mutated ABCA3 is a prominent genetic cause for diffuse interstitial or parenchymal lung disease in childhood (chILD).2 3
There is a strong genotype–phenotype correlation in chILD caused by ABCA3 sequence variants.2 3 Variants can be grouped according to their effect on ABCA3 function into ‘null’ and ‘hypomorphic’ variants. ‘Null’ variants do not produce functional protein due to frameshift variants or nonsense variants, or the deletion of an entire exon. ‘Hypomorphic’ mutations often result from missense variants, in-frame insertions or deletions and are predicted to result in an ABCA3 transporter protein with some residual functions.4
Reviewing the published literature, about two-thirds of the patients reported to date presented during the neonatal period, had a severe clinical course with global respiratory insufficiency and died in the first year of life.2 3 The literature search for children surviving beyond their first birthday retrieved very limited data from a total of 43 subjects from various case reports or miniseries.5–29 Patients surviving longer often carried a compound heterozygous genotype and had less oxygen supplementation during the neonatal period. Clinical characteristics of patients who survived longer have not been systematically reported yet.
Here, we detail the history, clinical phenotype, imaging and their relation to disease causing mutations of a cohort of 44 patients with ABCA3-related interstitial lung disease (ILD) who survived beyond infancy.
Methods
Patients
This cohort study investigates a group of patients with lung disease caused by ABCA3 variants. The patients were collected in the Kids Lung Register (KLR) database between January 2001 (establishment of KLR) and April 2021. We screened all patients with an ILD defined by the presence of tachydyspnoea, hypoxaemia and diffuse abnormalities on imaging2 30 and who were tested for ABCA3 variants. Included in this analysis were patients carrying homozygous or compound heterozygous ABCA3 variants. We focused on children who had survived without lung transplantation beyond the first year of life (online supplemental table S1). Patients who had died before the age of 1 year are listed in online supplemental table S2.
thorax-2022-219434supp001.pdf (948.2KB, pdf)
Genetic analysis
For genetic analyses, genomic DNA was isolated from whole blood (QIAmp DNA Mini Kit, Qiagen), and ABCA3 was analysed using Sanger sequencing.31 In some cases, ABCA3 variants were detected by whole exome analysis.32 Prediction of the pathogenicity of the variants was conducted with PROVEAN V.1.1.3,33 PolyPhen-234 and ClinVar.35 Since functional studies quantifying potential residual ABCA3 function in the previously unreported variants are not part of this work, their assignment as ‘hypomorphic’ variants is based on the in silicoprediction tools stated above and might not reflect true biological function.
Clinical data
Data on the clinical course and oxygen supplementation of the patients were retrieved from the register. Age at onset of chILD was defined as the time of first observed respiratory abnormalities or failure to thrive. Baseline referred to the relative clinical manifestation of the patients at their inclusion. The history and current oxygen supplementation were recorded at each clinic visit. Methods of oxygen supplementation included nasal cannula, face mask or various modes of ventilation support. Overall, we characterised four patterns of oxygen supplementation: (A) oxygen never required since onset of disease; (B) persistent oxygen required since onset of disease; (C) need for oxygen one or multiple times after disease onset, but not at the last visit and (D) need for oxygen one or multiple times including at the last visit.
Pulmonary function test information of patients in the register was collected from about age 4 years, onward at least baseline, 12 months and then annual data. Forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC) were expressed as per cent predicted using GLI-2012 reference values.10
High-resolution CT analysis
High-resolution CT (HR-CT) was centrally peer reviewed by agreement of three paediatric radiologists paediatric radiologists (JL-Z, BK and IK-S) and scored according to the criteria of Fleischner Society.36 The presence or absence of ground glass opacities (GGOs), diffuse or patchy, nodules or nodular opacities, focal consolidations (excluding dependent atelectasis), cystic parenchymal lesions, emphysema, bronchial wall thickening, (traction)-bronchiectasis, mosaic perfusion and mosaic attenuation, air trapping during expiration and fibrotic signs including linear or reticular opacities, honeycombing and architectural distortion, were differentiated.
Lung biopsy histology
All lung biopsies initially analysed by a local pathologist were centrally re-evaluated according to a standardised protocol by a pathologist trained and specialised in paediatric ILDs. Extent of histological changes was scored by severity (0=none, 1=discrete, 2=moderate and 3=strong) with respect to the localisation of the changes (see figure 5A) and the expression of individual histopathological features (see figure 5B).37
Statistics
Data sets retrieved from the data base were checked for missing or implausible values. All contributing investigators were asked in at least two rounds to complete the data as much as possible from local patient records. Statistical analysis was done with GraphPad Prism V.8. Grouped data were compared by unpaired t-tests, and Kaplan-Meier survival curves were assessed by log-rank tests. Imaging or histological data are available on request. As for the quantitative variables on lung function only baseline, 6 months and then annually tests were recorded in the register, all available tests were retrieved individually (from 17 of 26 patients able to perform test due to age) and recalculated into per cent predicted from litres according to the GLI reference values.10 For mixed model for repeated measures (MMRMs), generalised least squares with age was used for regression analysis in R, V.4.1.3. To visualise individual sources better, a linear regression line for each patient and for overall trend the regression line from MMRM was indicated in the figures.
Results
Patient cohort
Between January 2001 and April 2021, 398 of 1707 patients with chILD included in the KLR database were tested for ABCA3 sequence variations; the disease categories of those who were not tested are listed in online supplemental table S3. Genetic testing was discussed for all subjects and selection was advised by the chILD experts of the register, but the submitting physician made the final decision. Among these individuals 142 carried at least one ABCA3 variant and 79 carried 2 variants. Patients who received lung transplantation, died or were lost to follow-up in the first year of life were excluded (35/79), leaving 44/79 patients with biallelic variants included in the final cohort (figure 1A). 67.6% and 62.5% patients with biallelic variants classified as hypo/hypo or hypo/null genotype survived beyond 1 year, respectively, while only 15.4% null/null patients survived 1 year (figure 1B). In about half of the patients (n=21) lung disease started in the neonatal period (figure 2A). Fourteen patients carried homozygous ABCA3 variants, while 30 carried compound heterozygous variants. Grouping these patients according to the predicted functional consequences of the variants gave 37 patients carrying ‘hypo/hypo’, 5 ‘hypo/null’ and 2 ‘null/null’ variants (online supplemental table S1 and S4). The most common variant identified in our patients was p. E292V (allele frequency: 13/88), followed by p.R43H, p.R208W and p.G964D (allele frequency: 4/88). The frequency of the other variants was either 2/88 or 1/88 (online supplemental table S1). During the observation period, 8 of the 44 patients died or received lung transplantation (online supplemental table S1, figure 1B). Twenty-seven per cent (10/37) patients with ‘hypo/hypo’ genotype survived beyond 12 years of age.
Figure 1.

Flow chart of patient inclusion and survival analysis of patients with two ABCA3 variants in KLR. (A) All patients were enrolled from Kids Lung Register. Numbers in dashed frame: excluded patients. Numbers in solid line frame: included patients. LTX: lung transplantation. (B) For patients who went through genetic tests and diagnosed with biallelic ABCA3 variants, their survival rate was analysed according to different genotypes: hypo/hypo, hypo/null and null/null. chILD,childhood interstitial lung disease; ILD, interstitial lung disease.
Figure 2.

Clinical follow-up of patients. (A) The age at the first manifestation of ABCA3-related chILD. Neo: first manifestation in neonatal period. 1m-1Y: from age 1-month-old to 1-year-old. (B) Absolute frequency (upper panel) and percentage of frequency (low panel) of patients’ requirement of oxygen at different visit age. Columns are cumulative frequency of patients during the indicated period. Baseline indicated patients in the neonatal period. 6w: 6 weeks; 12 m: 12 months; 2y: follow-up age from 1 to 2 years; 3-4y (and so forth): follow-up age from 3 to 4 years. (C) The distribution of patients according to the age of their last clinical follow-up (year). (D) O2 supplementation since onset of disease. According to oxygen supplementation changes from onset of disease to the last time of follow-up, patients were divided into four groups. From left to right, the columns represented the number of patients who never required oxygen since onset of disease; who always needed oxygen since onset of disease; who need oxygen once or multiple times after disease onset, but not at the last visit and who need oxygen once or multiple times after disease onset until the last follow-up time. *p<0.05 with Tukey’s test; **p<0.001. chILD, childhood interstitial lung disease.
Oxygen supplementation of patients at the last follow-up visit
We used patients’ oxygen requirement as a proxy for chronic respiratory insufficiency during survival. More than 50% of the patients needed additional oxygen in the first 6 months after baseline, while thereafter the frequency varied between 20% and 50% and dropped to about 10% in patients older than 16 years (figure 2B). At their last clinical evaluation, 9 patients were 1–3 years old, 10 were 3–6 years old, 7 were between 9 and 12 years, 4 were between 12 and 15 years, and 9 were older than 15 years (figure 2C). Overall, 15 of the 44 patients (34%) never required oxygen after disease onset and 10 patients (22.7%) were completely dependent on supplemental oxygen. Nineteen patients (43.2%) needed oxygen once or multiple times after disease onset, of these 11 did not use supplemental oxygen at their last clinical visit (figure 2D). Median survival for patients never requiring oxygen (9.7 years (95% CI 6.7 to 27.7)) was longer compared with those dependent on oxygen (3.0 years (95% CI 1.5 to 5.0)) (p=0.0126, figure 2D).
Pulmonary function test results reveal lung function decline over time
Serial PFT results were available in 17 of 26 patients able to perform lung function tests (figure 3). The individual level of lung function varied widely between patients and were larger than the intraindividual changes over time. Despite rising and falling levels at different tests, lung function in the majority of subjects (12/17) deteriorated, when comparing the first and last tests. In patient P1, expiration time was too short to obtain FEV1 values. This subject was omitted from regression analysis. MMRMs regression demonstrated an impact of age on FEV1 % predicted (p=0.0002) and on FVC % predicted (p=0.0171). Average loss for FVC % predicted was about 1.1% predicted per year and for FEV1 % predicted was about 1.7% predicted per year. Of interest was the observation that in addition to restrictive pattern of lung function impairment, we also noted obstructive pattern in several subjects (figure 3A,B).
Figure 3.

Pulmonary function results of patients with repeated tests 14 patients preformed repeated pulmonary function test. (A) Predicted forced vital capacity (FVC%). (B) Predicted forced expiratory volume (FEV1%). The individual patients’ measurements were indicated by different symbols consistent for each patient in (A) and (B) (P1 in blue; due to short expiration no FEV1 value was available; P1 thus not included in overall regression analysis. Other patients in grey). For illustration of individual courses, linear regression lines were given (in blue). For overall regression, a mixed model for repeated measures was calculated for FVC (% predicted) and FEV1 (% predicted) against time (red line in bold).
Chest HR-CT indicated GGO was a common and sustained pattern in all patients
Chest CTs were available in 23 patients. To analyse whether the CT patterns changed with age, CT scans were divided into those performed at less than 2 years of age and greater than 2 years. The three most common CT patterns in the younger group were GGOs, linear or reticular opacities and focal consolidations. In the older patients, the three most common CT patterns were GGO, linear or reticular opacities and cystic parenchymal lesions. The frequency of cystic parenchymal lesions in patients older than 2 years of age was higher compared with the younger group (p=0.0306), suggesting that the presence of cystic parenchymal lesions might serve as a potential progression marker of ABCA3 lung disease (figure 4A). Seven patients had repetitive CT scans, demonstrating the interindividual heterogeneity of findings and progression with time (figure 4B).
Figure 4.

Thorax HR-CT results. (A) HR-CT results of patients younger (n=9) and older (n=14) than 2 years old (total n=23). (B) Seven patients had repetitive HR-CT. Repeated results of each patient were presented as conjunct bars to observe CT patterns at different ages. Red blocks indicate the presence of a pattern in the CT scan at the age, grey blocks indicate the absence of the pattern. *p<0.05 with Tukey’s test indicates differences between age groups. HR-CT, high-resolution CT.
Various histology patterns in lung biopsies at the time of diagnosis
Due to advances in rapid genetic diagnosis of surfactant dysfunction disorders, nowadays lung biopsies are done very infrequently. Therefore, we included this treasure of data on histological analysis. Only seven patients had lung tissue obtained at initial diagnosis of chILD. Four patients had chronic pneumonitis of infancy (CPI, P1, P12, P17 and P43), one had chronic bronchiolitis (P29), one mixed non-specific interstitial pneumonia (mNSIP, P23) and one had desquamative interstitial pneumonia (DIP, P33). One patient had combined CPI and DIP (P1). Semiquantitative rating of the histopathological features showed a predominantly diffuse and heterogeneous distribution of the features, affecting mainly the alveoli (figure 5A). Four patients were observed to display airway abnormalities, including chronic lymphocytic inflammation, acute neutrophilic inflammation and follicular hyperplasia (figure 5B). The major histopathological deviations involved the alveolar/interstitial region. The most common and severe histological abnormalities were type II pneumocyte hyperplasia, alveolar enlargement and alveolar septal thickening (figure 5C).
Figure 5.
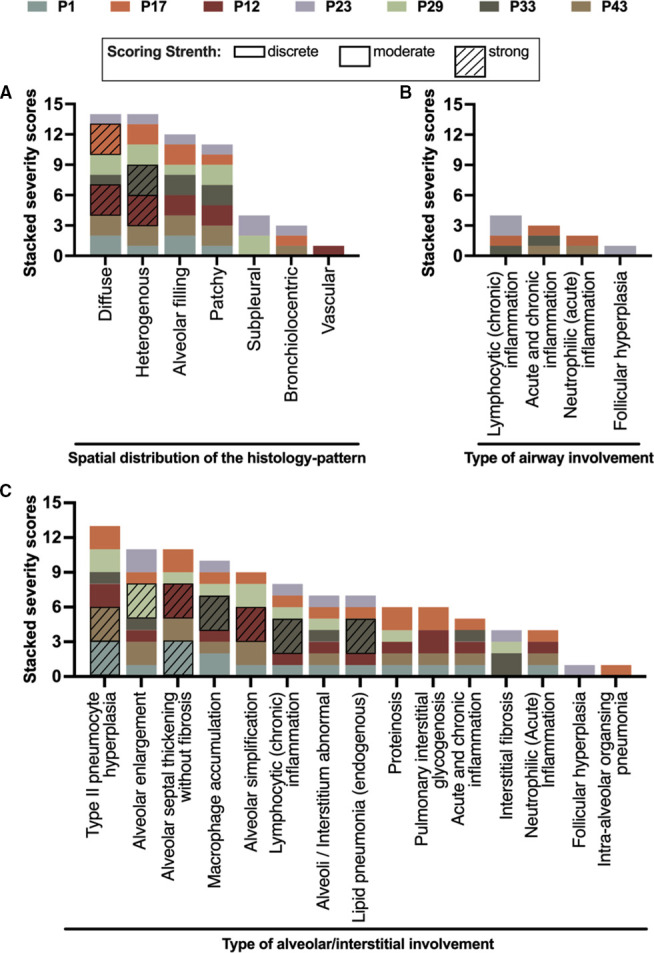
Histology of seven patients (each patient has a unique and different colour) had a lung biopsy. One pathologist trained in chILD reviewed all the histology, and analysed the pattern scores by severity (0=none, 1=discrete, 2=moderate and 3=strong). The severity is graphically expressed by the height of each column element on the y-axis. (A) Spatial distribution of the histopattern. (B). Type of airway involvement. (C). Type of alveolar/interstitial involvement. chILD, childhood interstitial lung disease.
Correlating survival and location of type of genetic variation in ABCA3
The variants identified scattered over the entire ABCA3 gene, among them was the known hotspot E292V (figure 6A). In the 44 patients described above who survived their first year of life, survival was independent of the domain location of their variants. To assess the relationship between domain location of variants and survival further, we included the 33 patients who died or received lung transplantation before the age of 1 year (figure 6A,B). We compared the locations of the allele frequencies between in these two survival groups. Patients with variants located in the intracellular helix 2 (IH2, p=0.012) and TMD2_P1 (p=0.016) had a longer survival, whereas patients with variants in helix 2 (p<0.0001) were more likely to die before the age of 1 year (figure 6C).
Figure 6.

Domain organisation of ABCA3 sequence variations observed in the cohort collected by the Kids Lung Register. (A) Frameshift, nonsense, missense, splice site, in-frame insertion/deletion variants were plotted according to the amino acid site, intronic variants not indicated. Variants plotted in blue: carrier survived beyond 1 year; plotted in orange: patients’ death before 1 year; edged yellow or blue dots: homozygous variants; dots with asterisk: compound heterozygous mutations. The position of TMDs and NBDs of ABCA3 were referred to the prediction algorithms of Onnée et al.40 (B). Age of survival of the mutations’ carrier (y-axis) in dependency of amino acid position (x-axis). For variants occurring in multiple patients, median survival age of carriers was indicated. Blue dots with yellow edge: homozygous variants; flat dots without edge: compound heterozygous variants. Number(s) inside dots: No of patients as listed in online supplemental table S1. Asterisk inside p.E292V dot indicated this variant was carried by multiple patients. (C) Allele frequency of variants for patients surviving >1 year or <1 year was counted in each domain and compared by Fisher’s exact test (*p<0.05, ***p<0.001). IHs, intracellular helices. TMD1_P1: part 1 of transmembrane domain 1, from p.K21 to p.R289. TMD1_P2: part 2 of transmembrane domain 1, from p.S301 to p.G450. TMD2_P1: part 1 of transmembrane domain 2, from p.M925 to p.Q1126. TMD2_P2: part 2 of transmembrane domain 2, from p.S1134 to p.E1325 (NP_001080.2). EHs, exocytoplasmic helices; IHs, intracellular helices; PHs, pinning helix; RDs, regulatory domains.
Generally, patients with homozygous mutation died earlier; an exception were patients P34 and P36 who carried homozygous p.G964D variants and survived into adulthood. For some variants the evidence of pathogenicity was weaker than for others; for these all supportive data from lung pathology, lavage measurements or in vitro data were detailed (online supplemental table S5).
Medication during the course
Medication information was available in 39 patients. Systemic glucocorticosteroids, hydroxychloroquine (HCQ) and macrolides were the major drugs used (online supplemental figure S1A). Steroids were often combined with HCQ and macrolides (online supplemental figure S1B). Due to lack of reliable time dependent data on treatment, no calculations on the impact of these treatments could be made.
Discussion
In this study, we present comprehensive clinical characteristics on 44 patients carrying biallelic ABCA3 variants and surviving beyond infancy. Repeated clinical observations of oxygen supplementation, lung function and clinically indicated chest HR-CTs were suitable outcome parameters describing the long-term course of molecularly defined ABCA3 lung disease during childhood and adolescence. Genotype was a prognostic indicator for the patients in our cohort. ‘Hypo/hypo’ variants were present in 37 patients and 5 patients carried ‘hypo/null’ variations. It must be noted that due to lack of comprehensive in vitro data the degree of residual function in variants characterised as ‘hypo’ likely varies and may possibly include functional ‘null’ variants. One ‘null/null’ patient with homozygous truncation variants died at age 1 year and 5 weeks, and another ‘null/null’ patient received lung transplantation at the age of 1.5 years. These findings were in accordance with published data.3 We attempted to link the position of the patients’ variants on ABCA3 protein domains to the maximal age of their carrier. Although variants located in helix2 and NBD2 were associated with shorter survival, domain distribution of the variants did not strongly indicate the prognosis of the patients.
ABCA3 deficiency is a severe condition even in survivors beyond the first year of life—4/44 of the patients in this cohort died before 5 years of age. We noticed later disease onset being associated with improved survival and less or even no need for oxygen supplementation. This observation is in agreement with recently published data on three cases of ABCA3 deficiency presenting in adulthood.27 Repeated lung function measurements were available in fourteen children beyond the age of 4 years. Overall, lung function impairment was restrictive or mixed obstructive/restrictive. Of interest, the level of FVC varied widely between different patients already at the start of measurement at age of 4–6 years. This very likely reflected the severity or extent of lung disease, which had developed during infancy and early childhood. We trusted and presented all clinically obtained measurements even at low age, as subjects were trained and values could be reproduced well. Over time and for the whole group of patients, the trajectories of lung function decreased from about 74.7% to 47.6% for FVC % predicted and to 34.3% for FEV1 % predicted during a 24-year period (from 4 to 28 years old), that is, yielding an average loss of FVC % predicted of 1.1% per year. These predictions are in accordance with the low level of lung function values reported for two other patients in the literature, a 41-year-old with a FEV1 % predicted of 49%,16 and a 61- year-old with a FVC % predicted of 49%.27
Up to date, there is very little data on CT pattern of patients with ABCA3 deficiency and its evolution beyond the first year of life. In children younger than 2 years typical features of fibrosis (honeycombing, architectural distortion, (traction)-bronchiectasis) were observed infrequently. All these features appeared to increase with age. In particular, the presence of cystic parenchymal lesions increased from approximately 10% to a prevalence of 60% in older patients. Previously, GGOs were reported to be the most common CT pattern,5 which matches with our data; we further observed a shift in the distribution of the GGOs from a diffuse in the younger patients (<1-year-old) to a patchy in older patients (>5-year-old). Taken together, CT imaging demonstrated a progression of the ILD over time.
Currently, there are no biomarkers for disease progression in ABCA3 deficiency. To aid clinical management of patients, we provide longitudinal data on lung function and chest HR-CT scans in a suitable number of patients.
Our data suggest that pathogenic variants in some domains of ABCA3 like IH2, TMD1 and TMD2 may be linked to longer survival. Further data are necessary to confirm this. Genetic analysis of a blood sample allows little invasive molecular diagnosis of ABCA3 deficiency. Thus, today lung biopsies are rarely performed, therefore the biopsies were mainly done during the first decade of our observation period and were now reread in a standardise manner by a single specialized pathologist. Except for a patient with some evidence for fibrosis due to the pattern of mNSIP, none of the subjects had fibrosing lung disease on histology. The major hallmark was the predominant involvement of the alveolar and interstitial region, with a type II pneumocyte hyperplasia, alveolar septal thickening, macrophage accumulation, often minor alveolar proteinosis or lipid pneumonia pattern. Immune cell infiltration was mainly lymphocytic, which might give a clue for anti-inflammatory treatment. Of importance, alveolar simplification indicated lung developmental abnormalities. Overall, these findings matched the characteristic histology patterns of surfactant dysfunction syndromes.5 38
This study has some limitations. First, the composition of the study cohort was dependent on the nature of patients submitted to the register. Subjects with a different or very mild phenotype might not have been tested for ABCA3 deficiency and thus not included into the study; this concerns in addition some of those patients we could not assess due to lack of data, genetic material or consent (online supplemental table S3). On the other hand, we may have included patients with a fitting clinical or pathological phenotype, however, with weak or lacking data on variant pathogenicity for ABCA3 deficiency (online supplemental table S5). An unbiased population-based approach would be necessary to solve these issues. However, this may be costly and has other, in particular ethical problems due to the detection of asymptomatic carriers or variants of unknown significance. Second, age and follow-up time were broadly variable between subjects, yielding incomplete longitudinal data and asymptomatic children might no longer be seen at clinical visits. Third, caution is necessary when correlating genotype and associated outcome. In patients carrying compound heterozygous pathogenic variants the effect of the two mutations may be asymmetrical or additional yet unknown interactions may play a role. Due to the limited observation times for some variants and the absence of detailed treatment recordings, associated prognosis can be erroneous. The strength of this investigation includes the generation of a good size cohort of subjects over a period of 20 years with standardised assessment of HR-CT imaging and histopathology by experts specialised in paediatric radiology and pathology and blinded to the molecular diagnosis.
This study provides details on the longitudinal history of ABCA3-related ILD of patients surviving beyond infancy into early adulthood. We provide evidence for progressive fibrosing lung disease based on lung function course and HR-CT changes. Such knowledge in rare and molecularly defined entities is very important to define the characteristics of cohorts for interventional trials of novel treatment approaches.39
Footnotes
Contributors: MG managed the KLR database and designed the project. MG and YL analysed the data and wrote the manuscript. JL-Z, BK, JD and IK-S analysed the imaging data. SR-H analysed histology data. ES, KK, FG, MEF, KM, IP, FG, FS, XY and CK contributed to data retrieval and register organisation. JC, MW, AM-G, AT-V, JL, KK, NR, SM, TS, AA, NR, MP, FS, LN, KA, SB, CK-R, EP, EDM, SAP, IC, MK and NS contributed cases, discussed the study results and contributed to the final manuscript. MG acts as the guarantor of the study.
Funding: This work was supported by a grant from Ludwig-Maximilians-Universität München-China Scholarship Council (LMU-CSC) Scholarship Programme (grant to YL, 201608500066), the German Research Fund (DFG Gr 970/9-1) and chILD UK (http://www.childlungfoundation.org/).
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
This study involves human participants and was approved by the ethics committee of the Ludwig-Maximilians-University Munich (EK 111-13, 20-0329). Participants gave informed consent to participate in the study before taking part.
References
- 1. Shulenin S, Nogee LM, Annilo T, et al. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med 2004;350:1296–303. 10.1056/NEJMoa032178. [DOI] [PubMed] [Google Scholar]
- 2. Kröner C, Wittmann T, Reu S, et al. Lung disease caused by ABCA3 mutations. Thorax 2017;72:213–20. 10.1136/thoraxjnl-2016-208649 [DOI] [PubMed] [Google Scholar]
- 3. Wambach JA, Casey AM, Fishman MP, et al. Genotype-Phenotype correlations for infants and children with ABCA3 deficiency. Am J Respir Crit Care Med 2014;189:1538–43. 10.1164/rccm.201402-0342OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beers MF, Knudsen L, Tomer Y, et al. Aberrant lung remodeling in a mouse model of surfactant dysregulation induced by modulation of the Abca3 gene. Ann Anat 2017;210:135–46. 10.1016/j.aanat.2016.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doan ML, Guillerman RP, Dishop MK, et al. Clinical, radiological and pathological features of ABCA3 mutations in children. Thorax 2008;63:366–73. 10.1136/thx.2007.083766 [DOI] [PubMed] [Google Scholar]
- 6. Yokota T, Matsumura Y, Ban N, et al. Heterozygous ABCA3 mutation associated with non-fatal evolution of respiratory distress. Eur J Pediatr 2008;167:691–3. 10.1007/s00431-007-0542-8 [DOI] [PubMed] [Google Scholar]
- 7. Young LR, Nogee LM, Barnett B, et al. Usual interstitial pneumonia in an adolescent with ABCA3 mutations. Chest 2008;134:192–5. 10.1378/chest.07-2652 [DOI] [PubMed] [Google Scholar]
- 8. Copertino M, Barbi E, Poli F, et al. A child with severe pneumomediastinum and ABCA3 gene mutation: a puzzling connection. Arch Bronconeumol 2012;48:139–40. 10.1016/j.arbres.2011.11.008 [DOI] [PubMed] [Google Scholar]
- 9. Flamein F, Riffault L, Muselet-Charlier C, et al. Molecular and cellular characteristics of ABCA3 mutations associated with diffuse parenchymal lung diseases in children. Hum Mol Genet 2012;21:765–75. 10.1093/hmg/ddr508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Quanjer PH, Stanojevic S, Cole TJ, et al. Multi-Ethnic reference values for spirometry for the 3-95-yr age range: the global lung function 2012 equations. Eur Respir J 2012;40:1324–43. 10.1183/09031936.00080312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kitazawa H, Moriya K, Niizuma H, et al. Interstitial lung disease in two brothers with novel compound heterozygous ABCA3 mutations. Eur J Pediatr 2013;172:953–7. 10.1007/s00431-013-1977-8 [DOI] [PubMed] [Google Scholar]
- 12. Thavagnanam S, Cutz E, Manson D, et al. Variable clinical outcome of ABCA3 deficiency in two siblings. Pediatr Pulmonol 2013;48:1035–8. 10.1002/ppul.22698 [DOI] [PubMed] [Google Scholar]
- 13. Thouvenin G, Nathan N, Epaud R, et al. Diffuse parenchymal lung disease caused by surfactant deficiency: dramatic improvement by azithromycin. BMJ Case Rep 2013;2013. doi: 10.1136/bcr-2013-009988. [Epub ahead of print: 24 Jun 2013]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Turcu S, Ashton E, Jenkins L, et al. Genetic testing in children with surfactant dysfunction. Arch Dis Child 2013;98:490–5. 10.1136/archdischild-2012-303166 [DOI] [PubMed] [Google Scholar]
- 15. Akimoto T, Cho K, Hayasaka I, et al. Hereditary interstitial lung diseases manifesting in early childhood in Japan. Pediatr Res 2014;76:453–8. 10.1038/pr.2014.114 [DOI] [PubMed] [Google Scholar]
- 16. Epaud R, Delestrain C, Louha M, et al. Combined pulmonary fibrosis and emphysema syndrome associated with ABCA3 mutations. Eur Respir J 2014;43:638–41. 10.1183/09031936.00145213 [DOI] [PubMed] [Google Scholar]
- 17. Hallik M, Annilo T, Ilmoja M-L. Different course of lung disease in two siblings with novel ABCA3 mutations. Eur J Pediatr 2014;173:1553–6. 10.1007/s00431-013-2087-3 [DOI] [PubMed] [Google Scholar]
- 18. Williamson M, Wallis C. Ten-Year follow up of hydroxychloroquine treatment for ABCA3 deficiency. Pediatr Pulmonol 2014;49:299–301. 10.1002/ppul.22811 [DOI] [PubMed] [Google Scholar]
- 19. Jackson T, Wegner DJ, White FV, et al. Respiratory failure in a term infant with cis and trans mutations in ABCA3. J Perinatol 2015;35:231–2. 10.1038/jp.2014.236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mukhtar GMA, Al Otaibi WH, Al-Mobaireek KFA, et al. Adenosine triphosphate-binding cassette member A3 gene mutation in children from one family from Saudi Arabia. Ann Thorac Med 2016;11:227–9. 10.4103/1817-1737.182900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ota C, Kimura M, Kure S. Abca3 mutations led to pulmonary fibrosis and emphysema with pulmonary hypertension in an 8-year-old girl. Pediatr Pulmonol 2016;51:E21–3. 10.1002/ppul.23379 [DOI] [PubMed] [Google Scholar]
- 22. Tan JK, Murray C, Schultz A. Abca3 lung disease in an ex 27 week preterm infant responsive to systemic glucocorticosteroids. Pediatr Pulmonol 2016;51:E1–3. 10.1002/ppul.23260 [DOI] [PubMed] [Google Scholar]
- 23. Akil N, Fischer AJ. Surfactant deficiency syndrome in an infant with a C-terminal frame shift in ABCA3: a case report. Pediatr Pulmonol 2018;53:E12–14. 10.1002/ppul.23994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El Boustany P, Epaud R, Grosse C, et al. Unusual long survival despite severe lung disease of a child with biallelic loss of function mutations in ABCA-3. Respir Med Case Rep 2018;23:173–5. 10.1016/j.rmcr.2018.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manali ED, Legendre M, Nathan N, et al. Bi-allelic missense ABCA3 mutations in a patient with childhood ILD who reached adulthood. ERJ Open Res 2019;5. doi: 10.1183/23120541.00066-2019. [Epub ahead of print: 22 07 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cho J-G, Thakkar D, Buchanan P, et al. ABCA3 deficiency from birth to adulthood presenting as paediatric interstitial lung disease. Respirol Case Rep 2020;8:e00633. 10.1002/rcr2.633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klay D, Platenburg MGJP, van Rijswijk RHNAJ, et al. ABCA3 mutations in adult pulmonary fibrosis patients: a case series and review of literature. Curr Opin Pulm Med 2020;26:293–301. 10.1097/MCP.0000000000000680 [DOI] [PubMed] [Google Scholar]
- 28. Nishida D, Kawabe S, Iwata N, et al. ABCA3 deficiency dramatically improved by azithromycin administration. Pediatr Int 2021;63:602–4. 10.1111/ped.14487 [DOI] [PubMed] [Google Scholar]
- 29. Zhang W, Zhiyong L, Yiming L, et al. A novel synonymous ABCA3 variantidentified in a chinese family with lethal neonatal respiratory failure. BMC Med 2021;14:256. 10.1016/j.envpol.2021.116891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kurland G, et al. , An official American Thoracic Society clinical practice guideline . Classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med 2013;188:376–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schindlbeck U, Wittmann T, Höppner S, et al. ABCA3 missense mutations causing surfactant dysfunction disorders have distinct cellular phenotypes. Hum Mutat 2018;39:841–50. 10.1002/humu.23416 [DOI] [PubMed] [Google Scholar]
- 32. Schuch LA, Forstner M, Rapp CK, et al. FARS1-related disorders caused by bi-allelic mutations in cytosolic phenylalanyl-tRNA synthetase genes: look beyond the lungs! Clin Genet 2021;99:789–801. 10.1111/cge.13943 [DOI] [PubMed] [Google Scholar]
- 33. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015;31:2745–7. 10.1093/bioinformatics/btv195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–9. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Landrum MJ, Chitipiralla S, Brown GR, et al. ClinVar: improvements to accessing data. Nucleic Acids Res 2020;48:D835–44. 10.1093/nar/gkz972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hansell DM, Bankier AA, MacMahon H, et al. Fleischner Society: glossary of terms for thoracic imaging. Radiology 2008;246:697–722. 10.1148/radiol.2462070712 [DOI] [PubMed] [Google Scholar]
- 37. Verma H, Nicholson AG, Kerr KM, et al. Alveolar proteinosis with hypersensitivity pneumonitis: a new clinical phenotype. Respirology 2010;15:1197–202. 10.1111/j.1440-1843.2010.01848.x [DOI] [PubMed] [Google Scholar]
- 38. Brasch F, Schimanski S, Mühlfeld C, et al. Alteration of the pulmonary surfactant system in full-term infants with hereditary ABCA3 deficiency. Am J Respir Crit Care Med 2006;174:571–80. 10.1164/rccm.200509-1535OC [DOI] [PubMed] [Google Scholar]
- 39. Deterding R, Young LR, DeBoer EM, et al. Nintedanib in children and adolescents with fibrosing interstitial lung diseases. Eur Respir J 2022. doi: 10.1183/13993003.01512-2022. [Epub ahead of print: 22 Sep 2022]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Onnée M, Fanen P, Callebaut I, et al. Structure-based understanding of ABCA3 variants. Int J Mol Sci 2021;22:10282–96. 10.3390/ijms221910282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
thorax-2022-219434supp001.pdf (948.2KB, pdf)
Data Availability Statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information.