

Abstract
Kidney fibrosis is a common step during chronic kidney disease (CKD), and its incidence has been increasing worldwide. Aberrant recovery after repeated acute kidney injury leads to fibrosis. The mechanism of fibrogenic changes in the kidney is not fully understood. Folic acid-induced kidney fibrosis in mice is an established in vivo model to study kidney fibrosis, but the mechanism is poorly understood. Moreover, the effect of higher concentrations of folic acid on kidney epithelial cells in vitro has not yet been studied. Oxidative stress is a common property of nephrotoxicants. Therefore, this study evaluated the role of folic acid-induced oxidative stress in fibrogenic changes by using the in vitro renal proximal tubular epithelial cell culture model. To obtain comprehensive and robust data, three different cell lines derived from human and mouse kidney epithelium were treated with higher concentrations of folic acid for both acute and long-term durations, and the effects were determined at the cellular and molecular levels. The result of cell viability by the MTT assay and the measurement of reactive oxygen species (ROS) levels by the DCF assay revealed that folic acid caused cytotoxicity and increased levels of ROS in acute exposure. The cotreatment with antioxidant N-acetyl cysteine (NAC) protected the cytotoxic effect, suggesting the role of folic acid-induced oxidative stress in cytotoxicity. In contrast, the long-term exposure to folic acid caused increased growth, DNA damage, and changes in the expression of marker genes for EMT, fibrosis, oxidative stress, and oxidative DNA damage. Some of these changes, particularly the acute effects, were abrogated by cotreatment with antioxidant NAC. In summary, the novel findings of this study suggest that higher concentrations of folic acid-induced oxidative stress act as the driver of cytotoxicity as an acute effect and of fibrotic changes as a long-term effect in kidney epithelial cells.
Graphical Abstract
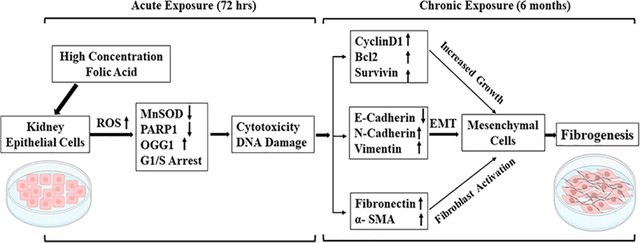
1. INTRODUCTION
Approximately 15% of the U.S. adult population is known to be affected by chronic kidney disease (CKD), and therefore this disease is still a major public health issue.1 Fibrosis is a common stage during the development of CKD. Repeated acute kidney injury is one of the leading causes of kidney fibrosis.2 Kidney fibrosis is a failed repair process after repeated acute kidney injury (AKI) that leads to continued deposition of the extracellular fibrotic matrix in the peritubular space in the kidney, thereby decreasing kidney function. Kidney fibrosis further progresses toward CKD and end-stage renal disease (ESRD).3 Currently, there is no effective therapy for kidney fibrosis.4 The mechanism of acute kidney injury transitioning to kidney fibrosis is not well understood.
There are many risk factors, including both endogenous and exogenous, for kidney disease. However, the generation of reactive oxygen species (ROS) causing oxidative stress is a common feature among most nephrotoxicants.5 Oxidative stress has also been implicated in the development of kidney fibrosis and CKD. However, the precise role of ROS and the molecular mechanism for oxidative stress-induced kidney fibrosis are not clear.
Folic acid (FA)-induced kidney fibrosis in mice is a widely used in vivo model to study acute kidney injury and kidney fibrosis. Administration of a high concentration of folic acid leads to the deposition of FA crystals in renal tubules, resulting in tubular necrosis, further leading to epithelial regeneration and scarring.6 Generation of oxidative stress has been reported during folic acid-induced AKI and fibrosis in animal mice models.7 However, the exact role of oxidative stress in the development of kidney fibrosis in this animal model is not well understood. Additionally, there is no in vitro study to evaluate whether FA induces oxidative stress in kidney epithelial cells, and if so then how elevated levels of oxidative stress contribute to fibrogenic changes is also not clear.
Folic acid does not occur naturally and is a synthetic form of naturally occurring folate. The active form folate is found in limited amounts in fruits and vegetables. Folic acid consumed in the form of fortified foods or supplements gets converted into the active form of tetrahydrofolate by the enzyme dihydrofolate reductase expressed in the intestine. Dihydrofolate subsequently gets converted into the methylated form 5-methyltetrahydrofolate (5MeTHF). However, because of the limited capacity of the enzyme, excessive intake of folic acid saturates the enzyme capacity for conversion and results in the appearance of unmetabolized folic acid (UMFA) in circulation.8 Folic acid at lower doses is known to have beneficial effects as an antioxidant by reducing the homocysteine levels.9 However, higher doses of FA can act as a pro-oxidant.10,11 Higher doses of folic acid are usually considered as the amount of FA received is much higher than the body usually can metabolize, and, because of this, it results in an unmetabolized form of folic acid (UMFA) in circulation.12 The recommended doses of folic acid as a prenatal multivitamin range from 400 to 500 μg in Australia to 800 μg/day in the U.S. and 1000 μg/day in Canada.12 Accumulating evidence from recent studies suggests that excessive intake of folic acid in the form of supplements and fortification in cereals may cause increased levels of unmetabolized folic acid (UMFA) in blood serum either because of saturation and/or inhibition of dihydrofolate reductase.13,14 The increased levels of UMFA can lead to disruption of the homocysteine to methionine conversion and therefore accumulation of unmetabolized homocysteine that can act as a pro-oxidant.10 Homocysteine during self-oxidization produces ROS directly11 or indirectly by activating NADPH oxidase.15 Homocysteine has also been shown to cause increased oxidative stress by mitochondrial dysfunction16 and oxidative stress-dependent collagen cross-linking modification.17 These reports suggest that FA at a lower dose is beneficial as an antioxidant, but the higher doses of FA can act as pro-oxidants.
Therefore, the objective of this study was to evaluate the role of FA-induced oxidative stress in cytotoxicity (as an indicator of AKI) and fibrogenic changes (as an indicator of fibrosis) in kidney epithelial cells using the in vitro cell culture model.
2. MATERIALS AND METHODS
2.1. Reagents.
Folic acid (FA), 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), 3,3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and N-acetyl cysteine (NAC) were purchased from Sigma-Aldrich (St. Louis, MO). Primary and secondary antibodies as well as DAPI (4′,6-diamidino-2-phenylindole) were purchased from Cell Signaling Technology.
2.2. Cell Culture and Treatments.
Human renal epithelial adenocarcinoma cells (Caki-1), renal cortex/proximal tubular epithelial cells (HK-2), and normal rat kidney epithelial (NRK) cells were purchased from ATCC. Caki-1 cells were maintained in Mccoy 5A medium with 10% fetal bovine serum (FBS) and 1% antibiotic. HK-2 cells were grown in serum-free keratinocyte medium (K-SFM) supplemented with human recombinant epidermal growth factor (EGF), bovine pituitary extract (BPE), and 1% antibiotic. NRK cells were propagated in DMEM/F12 growth medium with 5% FBS and 1% antibiotic.
For acute exposure, cells were treated with a range of concentrations (from 100 μM to 1 mM) of FA for 72 h. For long-term exposure, Caki-1 cells were treated for 6 months with 100, 200, and 400 μM of FA, which is hereafter termed as LT-Caki-1. HK-2 and NRK cells were exposed for the long-term (3 months) with 200 μM, 400 μM, and 1 mM FA. In the long-term treatment, semiconfluent cells were exposed to the respective concentrations of folic acid and were allowed to grow and reach the confluency of around 75% in a medium containing folic acid. These cells were then subcultured using a fresh medium and flask, and again treated with the same concentrations of folic acid that were used previously. Considering the mandatory fortification of cereal grain products with FA in the United States, reference concentrations of FA in human serum have been set at 5.8–32.8 ng/mL.18 Therefore, in this study, much higher concentrations of FA (the lowest concentration used was 100 μM, which is 44.14 μg/mL) were used to evaluate the adverse effects of higher concentrations of FA.
2.3. MTT Cell Viability Assay.
To assess the effect of acute and chronic exposure to FA on cell viability, we performed an MTT assay using Caki-1, HK-2, and NRK cells. The acute cytotoxic effect of FA was measured by treating parental control cells. Long-term FA-treated cells were also used in the MTT assay to determine the effect of long-term exposure to FA on these cells. Both untreated control and long-term FA-treated cells were seeded at a cell density of 4000 cells (for Caki-1 and NRK cells) or 5000 cells (for HK-2 cells) per well in 96-well plates. These different cell seeding numbers were selected after optimization as Caki-1 grows faster than HK-2 cells. After being seeded, cells were allowed to attach for 24 h, and they were treated with the respective concentrations of folic acid with and without NAC for 72 h. After 72 h of treatment, the medium was removed and washed with PBS. MTT solution at a concentration of 1 mg/mL in fresh media was added to each well and incubated for 3 h at 37 °C. To dissolve the formazan crystal formed in the viable cells, the MTT containing medium was removed, and 150 μL of DMSO per well was added. To dissolve the crystal, plates were incubated for approximately 5 min with gentle shaking. The intensity of the color formed was quantified by measuring the optical density using a plate reader at a wavelength of 570 and 630 nm. After the reference background was subtracted, the absorbance value was converted into a percentage as compared to the control. Each experiment was performed in triplicate and repeated twice.
2.4. DCF Fluorescence Assay.
The level of cellular reactive oxygen species (ROS) was quantified by using a 2′,7′-dichlorodihydro fluorescein diacetate (DCFH-DA) assay. Upon reaching into the cell, esterase deacetylates DCFH-DA further get oxidized by intracellular ROS into 2′,7′-dichlorofluorescein (DCF), which is highly fluorescent. The level of intracellular ROS determines the conversion of DCFH-DA to DCF. For the DCFH-DA assay, cells were seeded in 96-well plates, allowed to attach for 24 h, and then treated with various concentrations of FA both alone as well as in combination with antioxidant NAC. After 24 h of treatment, cell culture media from wells were removed, and cells were washed with the 1X PBS, and then DCFH-DA solutions (10 μM) in 1X PBS were added to each well and incubated for the next 30 min in a CO2 incubator. After incubation, cells were again washed with 1X PBS, and respective treatments were added to each group. H2O2 at 250 μM concentration was used as a positive control. The levels of ROS generated was measured by quantifying the DCF fluorescence at the emission wavelength of 535 nm using a plate reader. DCF fluorescence was measured at different time points starting from 5 min to an hour of exposure.
2.5. Cell Cycle Analysis.
To confirm the effect of folic acid on the growth pattern of cells, cell cycle analysis by flow cytometry was performed. After treatment as mentioned above, cells were harvested by trypsinization and collected by centrifugation, washed with 1X PBS, and then fixed into the 70% ethanol for 24 h at 4 °C. On the day of analysis, fixed cells were collected by centrifugation, washed with 1X PBS, and then stained with Guava cell cycle staining reagents for 30 min. Stained cells were then analyzed using a Guava Easy-Cyte HT flow cytometer by counting 5000 events.
2.6. RNA Isolation and Quantitative Real-Time Reverse Transcriptase-Polymerase Chain Reactions (qRT-PCR).
The effect of FA on the expression of genes at the transcript level was determined by quantitative real-time reverse transcriptase-polymerase chain reactions (qRT-PCR). The total RNA from cells was isolated using Trizol reagent (Invitrogen). Using the total RNA and one-step SYBR green RT-PCR kit (BioRad Inc.), qRT-PCR reaction mixtures were prepared, and PCR amplification was performed using the CFX96 real-time PCR detection system (BioRad Inc.). The following PCR amplification conditions were used: 50 °C for 15 min for reverse transcription, 95 °C for 5 min for the inactivation of reverse transcriptase, followed by 40 amplification cycles, each cycle consisting of step 1 denaturation at 95 °C for 10 s followed by primer annealing and extension at 60 °C for 30 s. The housekeeping gene GAPDH was also amplified as the internal control, and the Ct value of GAPDH from each sample was used for the normalization of each gene. Fold change in the expression of genes was calculated by the delta–delta Ct method.19 The primer sequences of each gene analyzed are given in Table 1.
Table 1.
List of Genes and Primer Sequence Used for Quantitative Real-Time Reverse Transcriptase-Polymerase Chain Reactions (qRT-PCR)
| gene | forward primer (5′—3′) | reverse primer (5′—3′) | size (base pair) |
|---|---|---|---|
| GAPDH | GGTGGTCTCCTCTGACTTCAACA | GTTGCTGTAGCCAAATTCGTTGT | 116 |
| cyclin D1 | AACTACCTGGACCGCTTCCT | CCACTTGAGCTTGTTCACCA | 204 |
| Bcl-2 | GGATGCCTTTGTGGAACTG | AGCCTGCAGCTTTGTTTCAT | 231 |
| survivin | AGCCAGATGACGACCCCATT | GCAACCGGCCGAATGCTTTT | 119 |
| MnSOD | GGGAGCACGCTTACTACCTT | GCTTACTGTATTCTGCAGTACTCT | 197 |
| GPx1 | TATCGAGAATGTGGCGTCCC | TCTTGGCGTTCTCCTGATGC | 143 |
| OGG1 | GGCTCAACTGTATCACCACTGG | GGCGATGTTGTTGTTGGAGGAAC | 228 |
| PARP1 | AAGGCGAATGCCAGCGTTAC | GCACTCTTGGAGACCATGTCA | 121 |
| E-cadherin | AACTACCTGGACCGCTTCCT | CCACTTGAGCTTGTTCACCA | 204 |
| alpha-SMA | CCCTTGAGAAGAGTTACGAGTTG | ATGATGCTGTTGTAGGTGGTTTC | 145 |
| vimentin | AGCCAGATGACGACCCCATT | GCAACCGGCCGAATGCTTTT | 119 |
| fibronectin | CTGGCCAGTCCTACAACCAG | CGGGAATCTTCTCTGTCAGC C | 118 |
| N-adherin | CCTTTCACTGCGGATACGTG | GATCCAGGGGCTTTGTCACC | 110 |
2.7. Western Blot Analysis.
Western blot analysis to confirm the gene expression at the protein level was performed with total protein lysates. Total cellular proteins were isolated from the respective groups by using RIPA lysis buffer, and the concentrations were determined by the Bradford assay. An equal amount of total protein from each sample was loaded on SDS-PAGE gel and separated by electrophoresis. Proteins were then transferred from the gel onto the PVDF membrane using a semidry transfer method. To prevent nonspecific binding, membranes were incubated with 5% nonfat dried milk in 1X Tris or 1X phosphate buffer saline for an hour. Membranes were then incubated with the diluted primary antibody in blocking buffer overnight at 4 °C. After primary antibody incubation, membranes were given three washes each for 5 min using washing buffer (1X PBS or TBS with 0.05% Tween 20). Membranes were then incubated with horseradish peroxidase conjugated-secondary antibody with appropriate dilutions for an hour at room temperature followed by three washes for 5 min each using washing buffer. To detect the chemiluminescence signal of protein bands, membranes were incubated with luminol reagent (Santa Cruz) developing solutions. Bands were visualized and documented by using the FluorChem gel documentation system. The intensity of the bands was quantified by using ImageJ software. The band intensity of the proteins of interest was normalized by the band intensity of GAPH as an internal control from each sample, and the graph was plotted using fold changes in arbitrary units.
2.8. Immunofluorescence Analysis.
Caki-1 cells and HK-2 cells were seeded on sterile coverslips in six-well plates and allowed to attach for about 48 and 72 h, respectively. Cells attached on coverslips were washed with 1X PBS and fixed by incubation in 100% methanol at −20 °C for an hour. Cells were again washed with 1X PBS and then incubated for 30 min in 0.2% Triton X-100 in PBS for permeabilization. After permeabilization, cells were washed with 1X PBS three times each for 5 min and incubated with the blocking solution containing 10% fetal bovine serum in 1X PBS for an hour at room temperature. After being blocked, cells were incubated with primary antibody (diluted in blocking solution) for an hour at room temperature. Subsequently, cells were washed with 1X PBS three times and incubated with Alexa Fluor 555 conjugated secondary antibody for 1 h at room temperature. Afterward, cells were washed with the 1X PBS three times and then incubated with DAPI at a 500 ng/mL final concentration for 2 min at room temperature for nucleus staining. Finally, cells were washed with 1X PBS three times and mounted on slides using mounting medium (90% glycerol in 1X PBS) and then sealed with tube sealant and stored at 4 °C for microscopic imaging.
2.9. Random Amplified Polymorphic DNA-Polymerase Chain Reaction (RAPD-PCR) Analysis.
To evaluate the genotoxicity at the DNA sequence level, RAPD-PCR analysis was performed using the total DNA. Total genomic DNA was isolated by the phenol–chloroform method as described earlier.20 DNA was quantified spectrophotometrically, and the quality was checked on the agarose gel. RAPD-PCR amplification was performed following our previously published method.21 The reaction mixture in a total volume of 25 μL included 1X enzyme assay buffer, 100 μM each of dNTPs (Applied Biosystems, Foster City, CA), 100 nM of random (10-bp) primer, 2.5 mM MgCl2, 0.5 U of AmpliTaq DNA polymerase (Applied Biosystems, Foster City, CA), and 75 ng of genomic DNA. PCR amplification was performed using the ABI GeneAmp PCR System 2700 with the following PCR amplification condition: initial denaturation at 92 °C for 3 min followed by 44 cycles with each cycle programmed for 1 min at 92 °C, 1 min at 34 °C, 2 min at 72 °C, and a final extension cycle of 15 min at 72 °C. After the completion of PCR, amplification products were separated by electrophoresis on 1.2% agarose gel. DNA bands on the gel were visualized by ethidium bromide staining and documented by using The FluorChem gel documentation system.
2.10. Statistical Analysis.
The statistical significance of the treatment group with the control was done using a two-tailed paired t test. The significance level was set at 0.05, and the difference having a p-value < 0.05 was considered the statistical significance.
3. RESULTS
3.1. Acute Exposure to High Concentrations of Folic Acid (FA) Causes Cytotoxicity.
To evaluate the effects of a high concentration of folic acid on cell viability, an MTT assay was performed. The result of the cell viability assay revealed that the lowest concentration (5 μM) of FA did not have any significant effect on the viability of the Caki-1 cells. However, the remaining higher concentrations (25–100 μM) of FA caused a concentration-dependent and statistically significant growth inhibition with the lowest growth inhibition of 23% at 25 μM of FA, whereas the highest of 37% was at 100 μM of FA (Figure 1A). For the HK-2 and NRK kidney epithelial cells, FA at concentrations up to 100 μM, however, did not affect the growth (Figure 1B and C). Higher concentrations of FA at 200 μM caused statistically significant growth inhibition by 10% of HK-2 cells as compared to the control (Figure 1B), but no significant changes in the growth of NRK cells. The maximum concentration used for FA tested was 1 mM, which resulted in significant growth inhibition of both HK-2 and NRK cells by 20% and 22%, respectively. The result suggests that Caki-1 kidney cancer epithelial cells with relatively faster growth rates are more sensitive to FA-induced cytotoxicity than are HK-2 and NRK normal kidney epithelial cells with slower growth rates.
Figure 1.
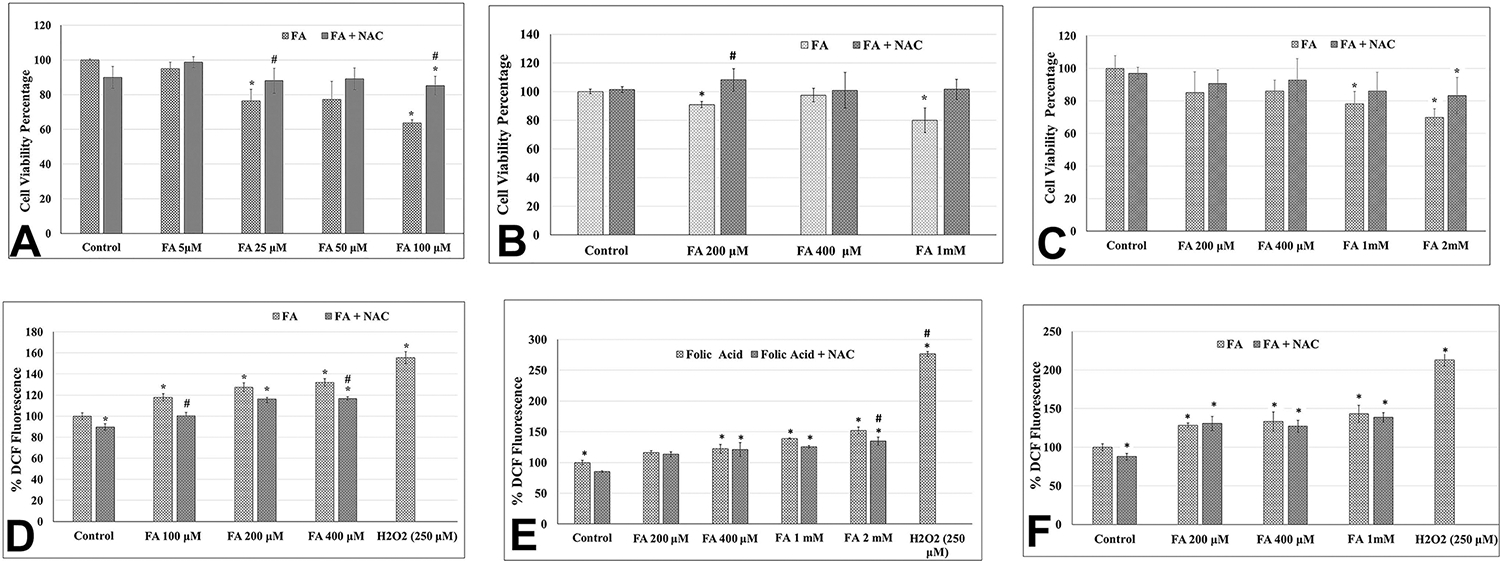
Effect of acute exposure to folic acid (FA) on the growth of kidney epithelial cells as measured by the MTT assay (upper panel: A–C) and on the level of intracellular ROS as measured by the DCF assay (lower panel: D–F). Upper panel: Bar graph representing the cell viability (in percentage) in FA-treated Caki-1 cells (A), HK-2 cells (B), and NRK cells (C) as compared to their respective untreated control cells (as 100%). Lower panel: Bar graph representing DCF fluorescence (in percentage) in FA-treated Caki-1 cells (D), HK-2 cells (E), and NRK cells (F) as compared to their respective untreated control cells (as 100%). Different concentrations (as given in the graph) of FA either alone or in combination with 25 μM NAC were used for cotreatment. Error bar showing the standard deviation (±SD) of the mean of triplicate values. Statistically significant (P < 0.05) changes as compared to control are shown by the symbol “*”. A statistically significant change in the FA and NAC cotreated group as compared to FA alone is shown by the symbol “#”.
3.2. Folic Acid Causes Increased Levels of Reactive Oxygen Species (ROS).
To evaluate whether FA causes cytotoxicity and growth inhibition through the generation of ROS, a DCFH-DA assay was performed. A concentration-dependent increase in ROS levels was observed in FA-treated Caki-1, HK-2, and NRK cells as compared to the respective untreated control cells. FA at concentrations of 100, 200, and 400 μM increased the ROS levels by 18%, 25%, and 35%, respectively, in Caki-1 cells as compared to the untreated control cells (Figure 1D). FA at concentrations of 400 μM and 1 mM increased ROS production by 22% and 39%, respectively, in HK-2 cells (Figure 1E) and 30% and 45% in the NRK cells (Figure 1F). FA at 2 mM concentration also significantly increased ROS production by 52% in the HK-2 cells (Figure 1E).
3.3. Acute Exposure to Folic Acid Causes Cell Cycle Arrest and a Decrease in the S Phase.
To further confrm the growth inhibitory effects of FA, cell cycle analysis using flow cytometry was performed with Caki-1 cells. The result revealed a decrease in the population of cells in the S-phase from 15% in control to 10.85% and 7.74% in cells treated with 100 and 200 μM concentrations of FA, respectively (Figure 2).
Figure 2.
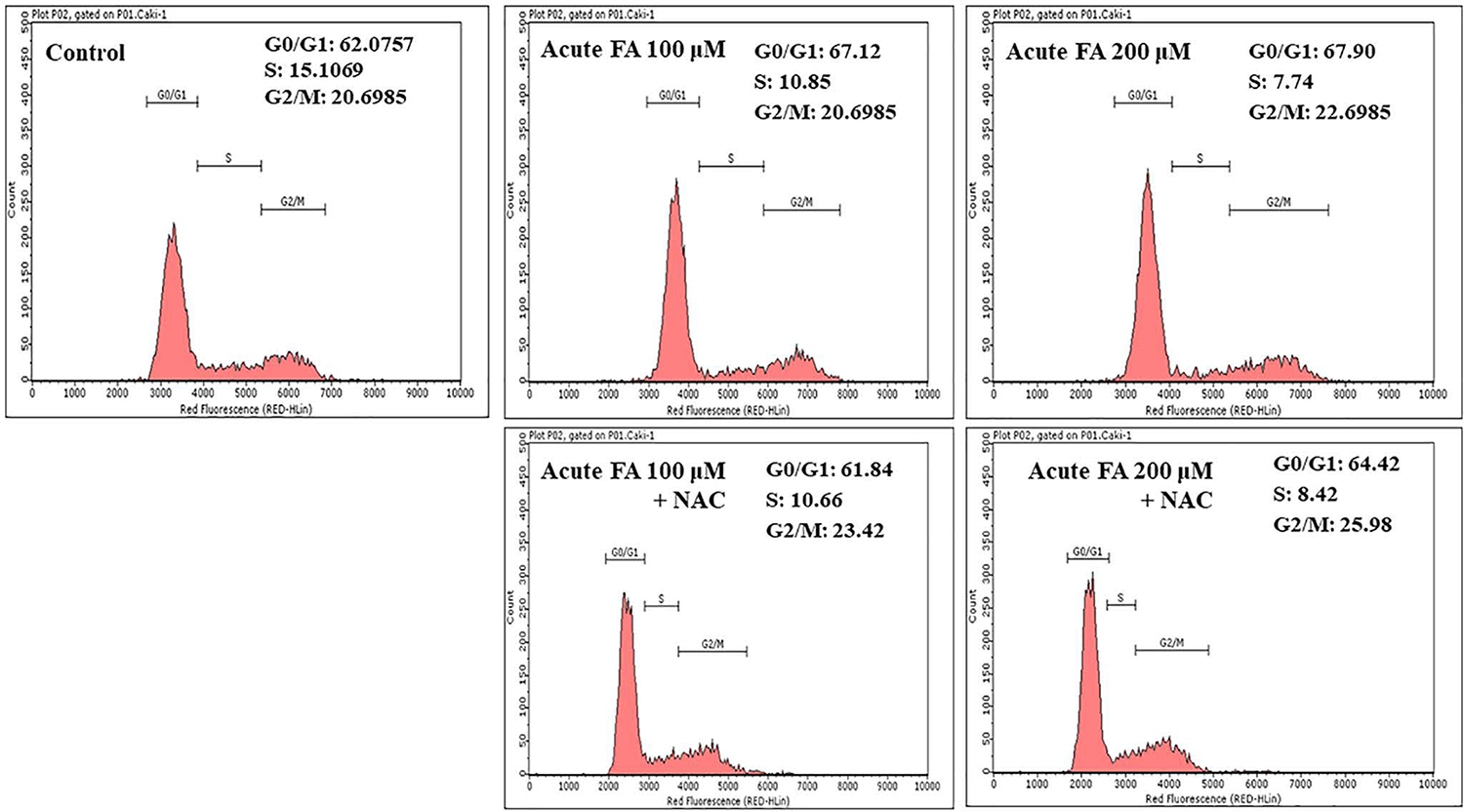
Histogram of flow cytometry data showing the effects of FA exposure on the cell population in G0/G1, S, and G2/M phases of the cell cycle from Caki-1 cells. Cells were given acute treatment with different concentrations of FA alone or cotreated with FA and NAC combination, and the population of the cells in various phases of the cell cycle was analyzed by flow cytometry as described in the Materials and Methods.
3.4. Scavenging Folic Acid-Induced ROS by Antioxidant NAC Abrogates the Cytotoxic Effect of Folic Acid.
To further confirm the pro-oxidant activity of FA in kidney epithelial cells, ROS levels were measured by the DCF assay using cells with coexposure to FA and antioxidant NAC. In Caki-1 cells, as compared to an 18% increase in ROS in 100 μM FA alone-treated cells, the cotreatment of NAC completely scavenged the ROS level back to the control level (Figure 1D). NAC partially scavenged the ROS levels from 25% and 35% in Caki-1 cells exposed to 200 and 400 μM concentrations of FA alone-treated cells to 16% and 17%, respectively, in cotreated Caki-1 cells (Figure 1D). Similarly, 22%, 39%, and 52% increases in ROS in HK-2 cells by 400 μM, 1 mM, and 2 mM, respectively, of FA were decreased by NAC cotreatment to 21%, 25%, and 35% (Figure 1E). In NRK cells, although the NAC cotreatment decreased the ROS levels, those decreases were statistically not significant (Figure 1F).
To evaluate whether FA-induced cytotoxicity is mediated by ROS, cell viability was further determined in cells coexposed to FA and NAC. FA at concentrations of 50 and 100 μM resulted in the decreased cell viability of Caki-1 cells by 23% and 37%, respectively, whereas cotreatment with NAC resulted in a 12% and a 14% decrease in cell viability, and thereby 11% and 23% protection in FA-induced cytotoxicity (Figure 1A). Similarly, treatment of HK-2 cells with 200 μM and 1 mM of FA alone resulted in decreased cell viability by 9% and 20%, respectively, whereas cotreatment with NAC resulted in 8% and 1% increases as compared to the untreated control, and thereby protection of 17% and 21%, respectively (Figure 1B). The protective effect of NAC in FA-induced cytotoxicity was also observed in NRK cells, but these protections were statistically not significant (Figure 1C).
3.5. Acute Exposure to Folic Acid Alters the Expression of Marker Genes for Cell Cycle, Cell Survival, Antioxidant, and Oxidative DNA Damage and Repair.
To further evaluate the cytotoxic and genotoxic effects of FA at the molecular level, the expression of representative marker genes for cell cycle, cell survival, antioxidant, and oxidative DNA damage and repair was determined by quantitative real-time PCR analysis only in Caki-1 cells (Figure 3A). Acute exposure to FA at 100 μM concentration resulted in a small but statistically significant increase by 1.5-fold in the expression of the survivin, whereas no significant change was observed in the expression of cyclin D1 and Bcl-2 (Figure 3A). A statistically significant decrease in the expression of the DNA damage marker gene PARP1 by 2-fold was observed, whereas an increase in the expression of oxidative DNA damage repair gene OGG1 by 2.8-fold was observed (Figure 3B). A decreased expression of antioxidant genes MnSOD by 1.8-fold and an increased expression of GPx1 by 1.4-fold were also statistically significant (Figure 3B).
Figure 3.
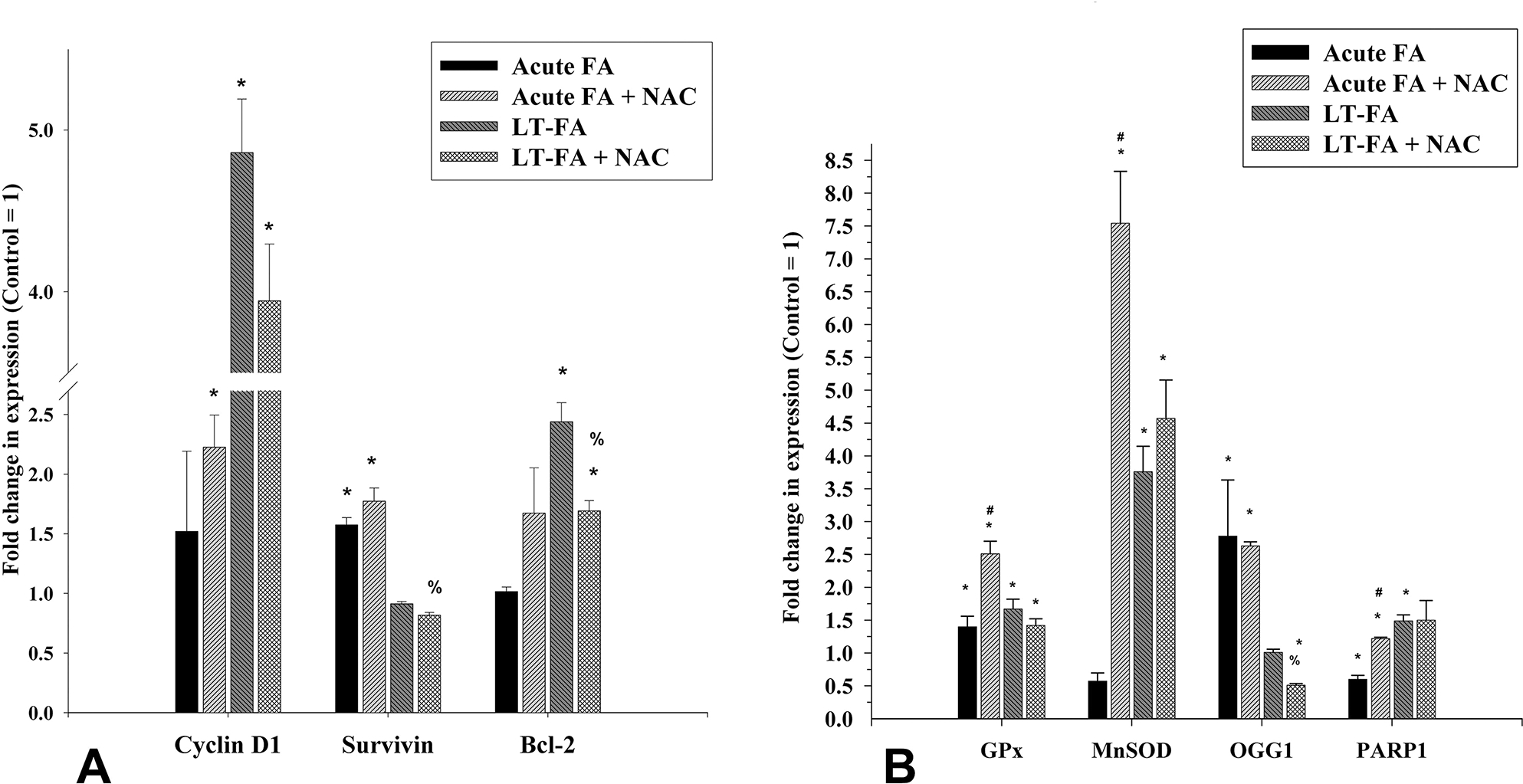
Effect of acute and long-term exposure to folic acid (FA) on the expression of marker genes for cell proliferation/survival (cyclin D1, survivin, and Bcl-2), antioxidant (GPx1 and MnSOD), and DNA damage (OGG1 and PARP1) in Caki-1 cells as measured by quantitative real-time PCR. Bar graphs represent the fold changes in the expression of the cell proliferation marker genes (A) and antioxidants as well as DNA repair genes (B) in acute (acute FA) and long-term (LT-FA) FA-treated Caki-1 cells as compared to the untreated control cells (control = 1-fold). Error bar showing the standard deviation (±SD) of the mean of triplicate values. Statistically significant (P < 0.05) changes in treated groups as compared to control are shown by the symbol “*”. The statistical significance of the cotreatment of NAC as compared to folic acid alone acute treatment is shown by the symbol “#”, whereas statistically significant changes in the cotreatment of NAC and FA as compared to FA alone in long-term treated cells are shown by the symbol “%”.
3.6. Acute FA Exposure-Induced Altered Expression of Genes Is Partially Restored by Antioxidant NAC.
Interestingly, the cotreatment of FA with NAC restored the expression of PARP1 to the control level, whereas it further increased the expression of cyclin D1, survivin, Bcl-2, GPx1, and MnSOD. For example, treatment with FA alone caused an increased expression of cyclin D1 and survivin by 1.5-fold each, and GPx1 by 1.4-fold, whereas cotreatment with NAC resulted in a further increase by 2.2-, 1.8-, and 2.5-folds, respectively (Figure 3A and B). As compared to the untreated control cells, the expressions of MnSOD and PARP1 in FA alone-treated cells were decreased by 1.8- and 2-fold, respectively, whereas the cotreatment with NAC resulted in increased expression of these two genes by 8- and 1.5-fold, respectively (Figure 3B). The expression of OGG1 was increased by 2.8-fold in FA-treated cells, whereas it was partially restored to 2.6-fold in NAC-cotreated cells (Figure 3B).
3.7. Long-Term Exposure to a High Concentration of Folic Acid Causes Increased Cell Growth.
In contrast to the cytotoxic effect of acute exposure to FA, chronic exposure to FA resulted in the increased growth and survival of Caki1 cells. Among the three concentrations of FA tested for chronic exposure, the 100 μM did not have any effect on cell viability, whereas the 200 and 400 μM of FA caused statistically insignificant but increased growth by 18% and 15%, respectively (Figure 4A).
Figure 4.
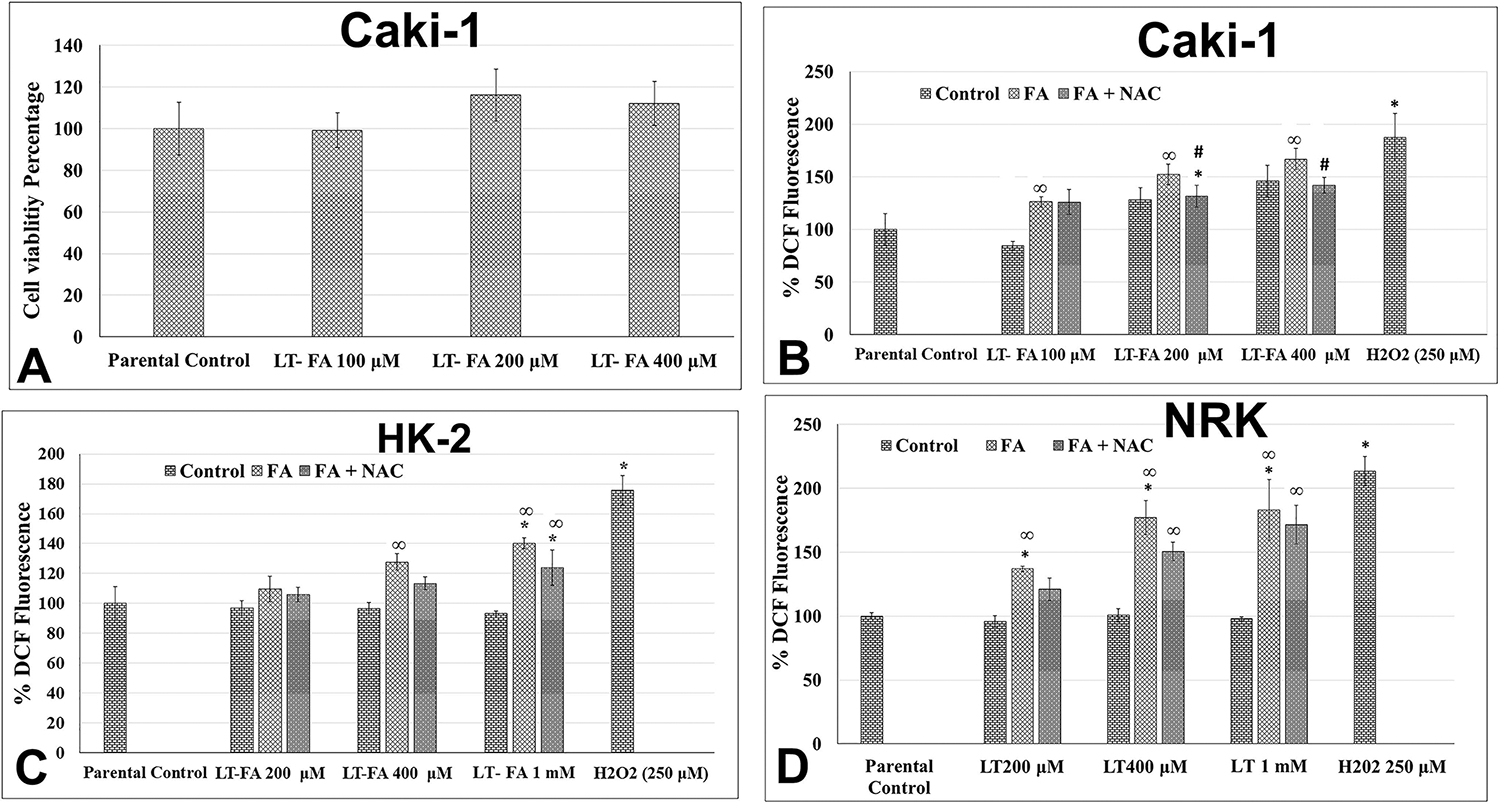
Effect of long-term exposure to folic acid (FA) on the growth and intracellular ROS production in kidney epithelial cells. Bar graph representing cell viability in percentage (control = 100%) in long-term FA-treated cells as measured by the MTT assay in Caki-1 cells (A), as well as intracellular levels of ROS in Caki-1 cells (B), HK-2 cells (C), and NRK cells (D). Different concentrations of FA as mentioned in the graph were used both alone and in combination with 25 μM of NAC. Error bars represent the standard deviation (±SD) of the mean of triplicate values. Statistically significant (P < 0.05) changes in treated groups as compared to control are shown by the symbol “*”. Statistically significant changes in the NAC and FA cotreatment groups as compared to FA alone are shown by the symbol “#”. Statistically significant changes in the NAC and FA cotreatment groups as compared to the untreated control are shown by the symbol “∞”.
3.8. Long-Term Exposure to a High Concentration of Folic Acid Alters Intracellular ROS Levels.
Long-term treated Caki-1 cells without further acute treatment showed an increased level of ROS as compared to the parental control. For example, increases in the ROS levels by 28% and 45% were observed in 200 and 400 μM long-term FA-treated Caki-1 cells (Figure 4B). Additional acute treatment of those long-term FA-treated cells further increased the ROS levels in a concentration-dependent manner. For example, the levels of ROS in 100, 200, and 400 μM long-term FA-treated cells increased from 85%, 128%, and 145% to 126%, 152%, and 167%, respectively, after further one-time acute treatment of FA at the same concentrations (Figure 4B). This further acute FA treatment-mediated increase in ROS was scavenged by NAC cotreatment back to the level of long-term FA-treated (200 and 400 μM) Caki-1 cells (Figure 4B). In long-term FA-treated HK-2 and NRK cells, there was no change in ROS production as compared to their parental untreated control cells (Figure 4C and D). However, further acute treatment of these cells with FA increased the ROS levels. For example, the levels of ROS in 200 μM, 400 μM, and 1 mM long-term FA-treated HK-2 cells increased from the base level in untreated parental control to 9%, 27%, and 40%, respectively, after further one-time acute treatment of FA at the same concentrations (Figure 4C). Similarly, in long-term FA-treated NRK cells, there was no increase in the level of ROS as compared to the parental untreated control cells. However, one-time acute treatment with 200, 400, and 1 mM of FA resulted in statistically significant increased production of ROS by 36%, 77%, and 83%, respectively, from the base levels of ROS in their parental control cells (Figure 4D).
3.9. Long-Term Exposure to Folic Acid Causes Altered Expression of Marker Genes for Cell Cycle, Cell Survival, Antioxidant, and Oxidative DNA Damage and Repair.
To further confirm the MTT data showing increased growth of Caki-1 cells chronically exposed to FA, the expression of cyclin D1, survivin, and Bcl-2 was determined by qRT-PCR. The result revealed the 4.9-fold and 2.5-fold increased expression of cyclin D1 and Bcl-2, respectively, in FA-treated cells as compared to untreated control cells. Cotreatment of these cells with NAC resulted in a 3.94-fold and 1.7-fold increase of cyclin D1 and Bcl-2, respectively, as compared to the control, thereby suggesting partial protection from FA effects on the expression of these genes (Figure 3A). No significant change in the expression of survivin in FA-exposed cells was observed. A statistically significant increase by 3.8-fold in the expression of antioxidant genes MnSOD and by 1.5-fold in the expression of DNA damage marker PARP1 was observed in long-term FA-treated Caki-1 cells as compared to the control cells (Figure 3B). There was no significant change in the expression of DNA damage repair gene OGG1 in long-term FA-treated cells. NAC cotreatment did not have any significant effect on the expression of these genes.
3.10. Long-Term Exposure to Folic Acid Causes Altered Expression of the Epithelial–Mesenchymal Transition (EMT) and Fibrosis Marker Genes.
To evaluate the chronic exposure of FA to EMT and fibrogenic changes, the expression of marker EMT marker genes (E-cadherin, N-cadherin, and vimentin) and fibrosis marker genes (fibronectin and alpha-SMA) was analyzed by quantitative real-time PCR. Interestingly, the expression of fibronectin and alpha-SMA was increased by 11-fold and 5.7-fold in Caki-1 cells with chronic exposure to FA as compared to the untreated passage-matched control cells (Figure 5A). Treatment of NAC to cells with chronic exposure to FA did not have any significant effect on the expression of fibronection, whereas alpha-SMA expression was further increased to 7.1-fold (Figure 5A). Among the EMT marker genes, a 4-fold decreased expression of E-cadherin and a 2.2-fold increased expression of N-cadherin were observed in long-term FA-treated Caki-1 cells (Figure 5B). NAC treatment did not have any significant impact on the expression of these two genes. Expression of vimentin was increased by 6.3-fold, and the NAC treatment restored partially but statistically significant to the 3.2-fold level (Figure 5B).
Figure 5.
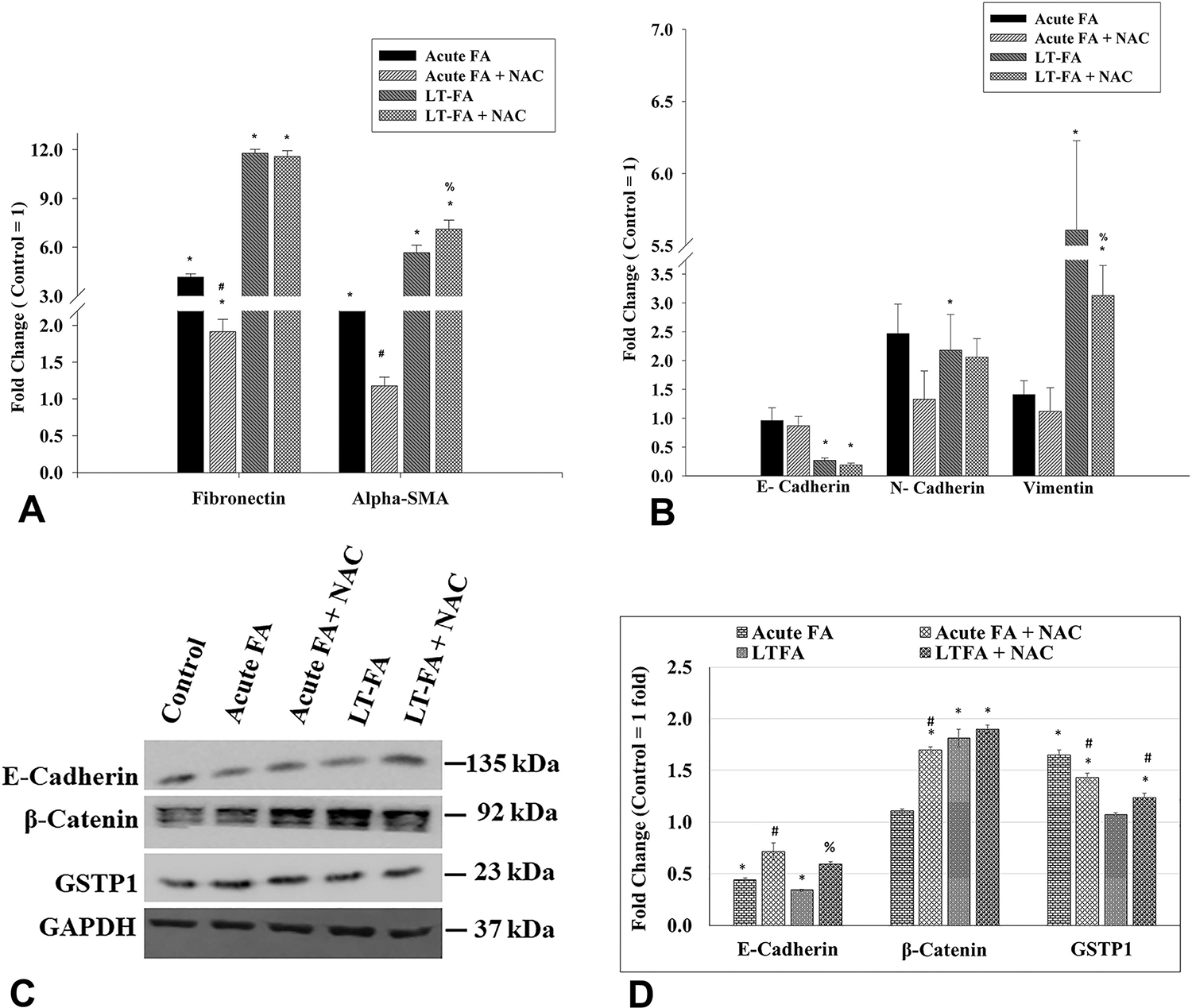
Effect of acute and long-term exposure to folic acid (FA) on the expression of marker genes transcripts for fibrosis and EMT (in Caki-1 cells as measured by quantitative real-time PCR (qRT-PCR)) (A,B) and on the expression of E-cadherin, β-catenin, and GSTP1 proteins as measured by Western blot analysis (C,D). Bar graphs represent the fold changes in the expression of gene transcripts of fibronectin and alpha-SMA (A), and E-cadherin, N-cadherin, and vimentin (B), in both acute (acute FA) and long-term (LT-FA) FA-treated Caki-1 cells as compared to untreated control cells (control = 1-fold). Images of Western blots of E-cadherin, β-catenin, GSTP1, and GAPDH (internal control) proteins in Caki-1 cells (C), and their band intensities (in arbitrary units) as measured by ImageJ software (D). The band intensity of each protein was normalized by the band intensity of the internal control GAPDH from each sample, and the bar graph was plotted using values in arbitrary units. Error bar showing the standard deviation (±SD) of the mean of triplicate values. Statistically significant (P < 0.05) changes in treated groups as compared to the control are shown by the symbol “*”. A statistical significance of the cotreatment of NAC as compared to folic acid alone acute treatment is shown by the symbol “#”, whereas statistically significant changes in the cotreatment of NAC and FA as compared to FA alone in long-term treated cells are shown by the symbol “%”.
3.11. Folic Acid Causes Altered Expression of GSTP1 and EMT Associated Proteins.
The expression of E-cadherin, β-catenin, and GSTP1 at the protein level was determined by Western blot analysis (Figure 5C). As compared to the control, the expression of E-cadherin was decreased by 2.3-fold and 2.9-fold in acute and long-term FA-treated cells, respectively (Figure 5C and D). As compared to 2.3-fold and 2.9-fold decreases in FA-alone treatment, the expression of E-cadherin in acute and long-term FA and NAC cotreated groups was 1.4-fold and 1.7-fold decreased, thereby suggesting a statistically significant restoration of expression by NAC (Figure 5C and D). There was no change in the expression of β-catenin in cells with acute treatment of FA. However, the long-term FA treatment resulted in a significant increase by 1.8-fold in the expression of β-catenin as compared to the untreated control group (Figure 5C and D). Acute cotreatment of Caki-1 cells with FA and NAC caused a further significant increase in the expression of β-catenin by 1.7-fold as compared to the untreated control (Figure 5C and D). In the long-term FA-treated group, the cotreatment with NAC did not cause any significant change as compared to FA alone (Figure 5C and D). Expression of GSTP1 was increased by acute exposure of FA, whereas there was no change in long-term FA-treated cells (Figure 5C and D). The cotreatment FA with NAC in acute exposure partially restored the FA-induced changes in GSTP1 protein levels.
3.12. Immunofluorescence Analysis Confirmed the Increased Expression of Fibrosis Marker Protein Fibronectin in Long-Term Folic Acid Exposed Caki-1 and HK-2 Cells.
The immunofluorescence analysis was performed to confirm the expression fibronectin, a most commonly used marker of fibrogenic changes, in long-term folic acid-treated Caki-1 and HK-2 cells, and the results are shown in a photomicrograph (Figure 6). Immunofluorescence staining of fibronectin revealed the increased intensity of staining of this protein and thereby suggested the increased expression of fibronectin in long-term folic acid exposed Caki-1 cells as compared to untreated control cells (Figure 6, upper two panels). The effect of FA on the expression of fibronectin protein was further confirmed by using another cell line HK-2 with long-term exposure to folic acid. The staining of fibronectin in long-term FA-treated HK-2 cells was much more intense as compared to the untreated passage-matched control, thereby confirming the increased expression of fibronectin in long-term FA-treated kidney epithelial cells (Figure 6, lower two panels).
Figure 6.
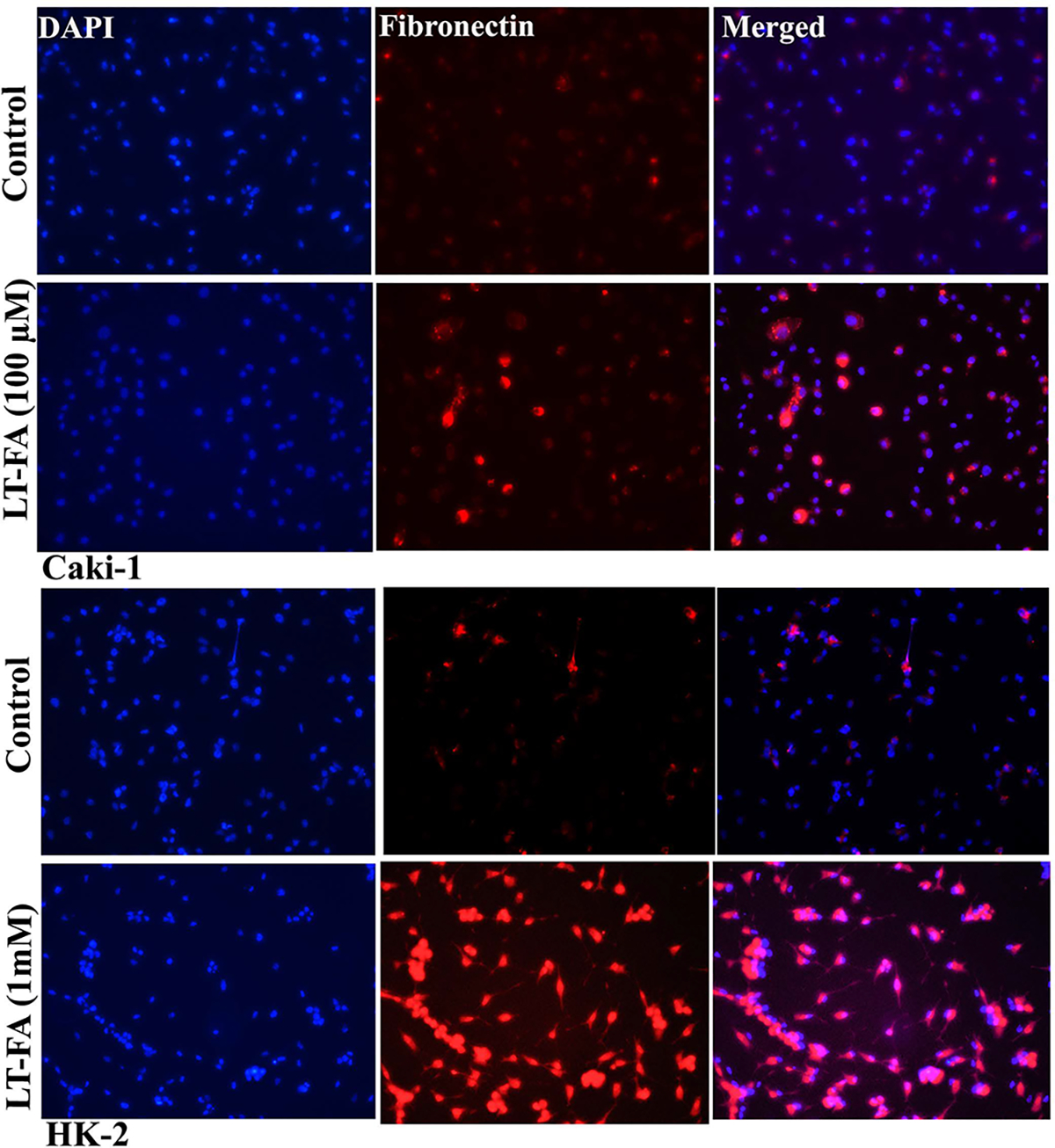
Microscopic images of the immunofluorescence detection of fibronectin in long-term FA-treated and untreated control Caki-1 and HK-2 cells. Representative immunofluorescence microscopic photographs showing the expression of fibronectin in untreated control and long-term FA-treated cells of Caki-1 (upper two panels) and HK-2 (lower two panels). Fibronectin staining is in red, whereas the nuclear staining by DAPI is in blue. The immunofluorescence staining of fibronectin was performed as described in the Materials and Methods. Scale bar = 100 μm.
3.13. RAPD Analysis Revealed Folic Acid-Induced DNA Damage.
The results of the RAPD-PCR fingerprint generated by using primer OPK 17 (5-CCCAGCTGTG-3′) revealed changes in the banding pattern in the 200 μM long-term FA-treated group of cells and therefore the target regions for FA-induced genotoxicity as compared to the untreated control cells. Among the four amplification products amplified in the RAPD fingerprint by OPK 17 primer, the three bands (2.5 kbp, 1.4 kbp, and 400 bp) were absent or undetectable in the RAPD fingerprint of 200 μM long-term FA-treated group of cells (lane number 6) as compared to the untreated control group of cells (lane number 2) (Figure 7). The band intensity of another amplification product of 1.3 kbp in the RAPD fingerprint from a 200 μM long-term FA-treated group of cells was significantly reduced as compared to its intensity in the untreated control group of cells (Figure 7). Interestingly, the long-term FA-treated cells when cotreated with NAC resulted in the restoration in the intensity of the 1.4 and 1.3 kbp bands (lane number 7), suggesting the protective effect of NAC in FA-induced genotoxicity in these cells (Figure 7). The RAPD fingerprint generated by another primer OPK19 (5′-CACAGGCGGA-3′) shows a similar fingerprint pattern, indicating unaffected genomic regions by FA treatment (Figure 7).
Figure 7.
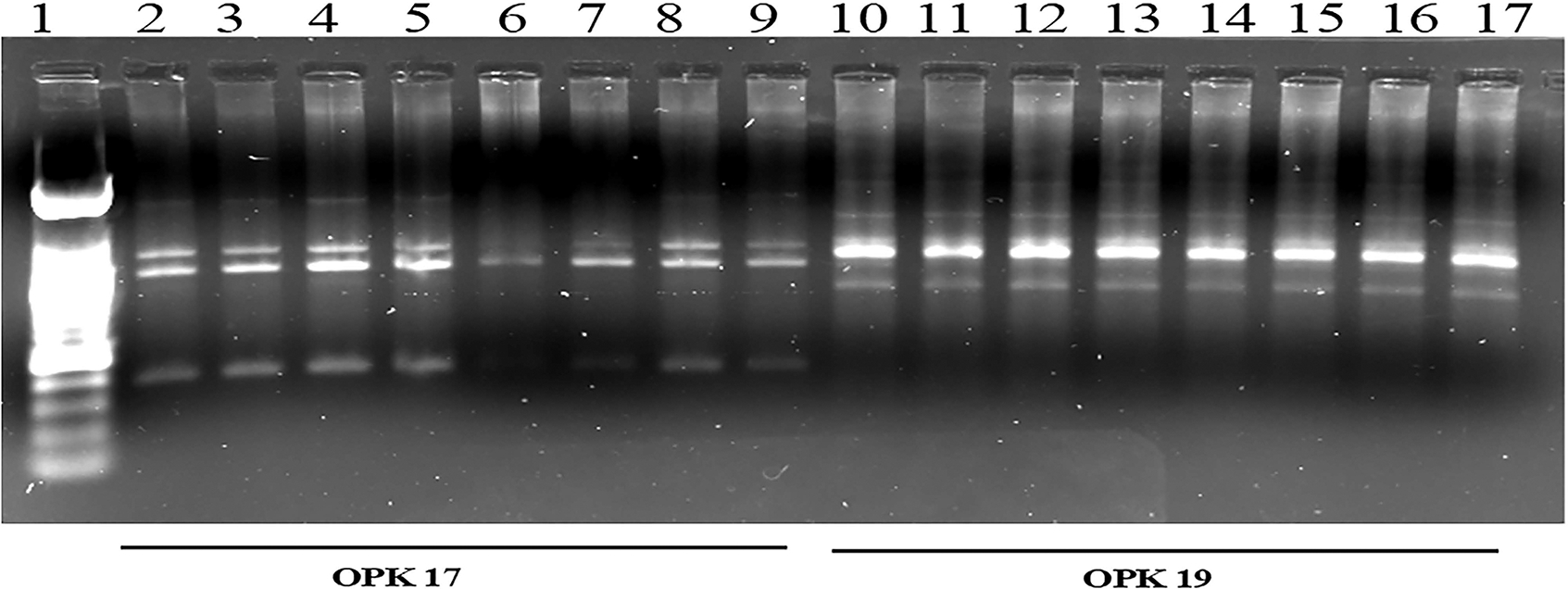
Representative RAPD fingerprints showing genotoxicity at the DNA sequence level in long-term FA-treated Caki-1 cells. RAPD-PCR fingerprints were generated by using primer OPK-17 (lanes 2–9) and OPK-19 (lanes 10–17). Samples in each lane are as follows: lane 1, DNA size marker; lanes 2 and 10, control; lanes 3 and 11, control + NAC; lanes 4 and 12, LT-FA 100 μM; lanes 5 and 13, LT-FA 100 μM + NAC; lanes 6 and 14, LT-FA 200 μM; lanes 7 and 15, LT-FA 200 μM + NAC; lanes 8 and 16, LT-FA 400 μM; and lanes 9 and 17, LT-FA 400 μM + NAC.
4. DISCUSSION
Although there are several reports on the analysis of AKI and fibrosis using a well-established folic acid-mice model, there is no report on the effect of FA in human or rodent kidney epithelial cells using the in vitro cell culture model. To our knowledge, this is the first report of an in vitro cell culture model-based analysis of the effects of higher concentrations of FA in both human and mouse kidney epithelial cells. The important findings of this study suggest that (a) FA at higher concentrations caused increased levels of oxidative stress in kidney epithelial cells, (b) FA altered the expression of antioxidant and oxidative DNA damage marker genes, and (c) acute exposure decreased growth, whereas long-term exposure caused increased survival, DNA damage, and fibrogenic changes. This study with an in vitro cell culture model not only confirmed the adverse effects of high concentrations of FA in the kidney of the animal model but also provides the underlying molecular mechanism. Most importantly, the data of this study showing protection by antioxidant NAC from FA-induced cytotoxicity, DNA damage, and changes in the expression of genes suggest that oxidative stress is a driver of the FA-induced acute cytotoxic effect and long-term fibrogenic changes in kidney epithelial cells.
Our data revealed increased levels of oxidative stress in kidney epithelial cells exposed to FA. Several reports suggest that folic acid, at low concentration, produces beneficial effects and it acts as an antioxidant.9 However, it is well-established that high-dose FA causes oxidative stress as evidenced by lipid peroxidation in animal models.22 The basis for the high-dose FA-induced oxidative stress is not fully understood. In this context, a recent study using the C. elegans model suggests that high-dose FA results in the accumulation of unmetabolized FA, causing disruption in the methionine cycle that results in the accumulation of homocysteine and ultimately oxidative stress.23 Increased levels of homocysteine can increase the ROS levels by self-oxidation as well as through activation of NADPH oxidase.11,24 It is also known that the kidney has a higher affinity for folate than other organs because kidney epithelial cells contain much higher levels of folate receptors than other cells, and therefore there is accumulation of FA in the kidneys.25 Additionally, among the different cellular compartments, the mitochondria, a major source of ROS, accumulate approximately 40% of total folate inside the cell.26,27 Antioxidant NAC has been shown to abrogate mitochondrial dysfunction and oxidative stress and kidney damage in the FA-induced AKI model in mice.27 These previous reports and our findings suggest that FA at higher concentrations can act as a pro-oxidant in kidney epithelial cells potentially through its accumulation in mitochondria, a major source of intracellular ROS. The observation of decreased expression of antioxidant genes MnSOD in cells with acute exposure to FA and its restoration by NAC further suggests that FA can increase mitochondrial ROS indirectly by decreasing the antioxidant system in the kidney. In contrast, an increased expression of glutathione transferase P1 (GSTP1) was observed in cells after acute exposure to FA. GSTP1 is a cytoplasmic enzyme that plays an important role in the detoxification process and in protection from oxidative damage. Similar to the finding of this study, increased expression of GSTP1 was also reported from a mice model of acute renal failure induced by Ischemia-reperfusion injury.28 The increased expression of GSTP1 could be in response to increased oxidative stress and oxidative DNA damage.
The folic acid-induced kidney fibrosis model is a well-established model for studying AKI and kidney fibrosis because it recapitulates the pathological processes of kidney disease and is highly reproducible. However, the mechanism of FA-induced AKI and fibrosis is not clear. The cell culture model is an ideal and commonly used in vitro model for mechanistic study. Although numerous studies used in vivo mice models of FA-induced AKI and kidney fibrosis, surprisingly there is no in vitro cell culture-based study analyzing the effect of FA in kidney cells. In this context, this is the first study using the in vitro cell culture of human and mouse kidney epithelial cells to analyze the effect of FA. The result of this study revealed that acute exposure to folic acid causes cytotoxicity and genotoxicity, whereas long-term exposure resulted in fibrogenic changes. This observation is similar to the acute effect of AKI and the long-term effect of fibrosis in a well-established folic acid-induced kidney disease animal model of mice or rats.25,29 Although acute exposure to higher concentrations of FA was cytotoxic to all three kidney epithelial cell lines used in this study, the concentrations at which FA was cytotoxic were different for these three cell lines. The concentration required for cytotoxicity is higher in NRK cells and HK-2 cells derived from normal kidneys than in Caki-1 cells derived from kidney tumors, which could be due to the normal cell’s slow growth rate. FA at the same concentration caused greater growth inhibition and cytotoxic effects in Caki-1 cells that have a relatively faster growth rate as compared to the HK-2 and NRK cells (both derived from the normal kidney). To our knowledge, this is the first report on the acute and long-term effects of higher concentrations of folic acid using the in vitro model of kidney epithelial cells as well as the underlying molecular mechanism.
The role of FA-induced ROS in cytotoxicity, genotoxicity, and fibrogenic changes was the next logical question that was addressed in this study. The mechanistic basis for FA-induced AKI and kidney fibrosis in animal mice models is still unclear. Previous studies suggest that a high dose of folic acid is nephrotoxic because it accumulates in the kidney via a folate receptor, which is highly abundant in kidneys.25 The higher levels of FA result in the deposition of FA crystals in lumens obstructing the kidney tubules and therefore direct toxicity to the proximal tubular epithelial cells.30 The previous study has shown increased levels of oxidative stress in the in vivo folic acid-induced kidney fibrosis models and other in vivo models of kidney diseases.31 However, the role of oxidative stress in the induction of AKI and fibrosis is unclear. In this context, this study for the first time using an in vitro cell culture model provided evidence for the role of a high concentration of FA-induced oxidative stress as a driver of cytotoxicity, which is also observed in AKI in vivo, and fibrogenic changes in kidney epithelial cells. The result of FA-induced cytotoxicity, production of ROS, and changes in the expression of marker genes confirm these adverse effects of FA, and partial abrogation of most of these effects by antioxidant NAC provides evidence for the role of FA-induced ROS in cytotoxicity and fibrogenic changes. Interestingly, in long-term FA-treated cells, the increased ROS level persisted until 2 weeks even without further FA treatment. This could potentially be due to the accumulation of unmetabolized FA in the cells; however, this needs to be confirmed with further experimentation. This finding is similar to the previous report of unmetabolized FA causing increased ROS in the C. elegans model.23 The observations of DNA damage as detected by RAPD analysis and the altered expression of oxidative DNA damage repair gene OGG1 and DNA strand break sensor PARP1 further suggest FA-induced oxidative DNA damage as a basis for the FA-induced cytotoxicity.
To further understand the molecular basis of FA-induced fibrogenic changes, marker genes for EMT and fibrosis were also analyzed in this study. The role of kidney tubular epithelial cells and EMT has been a topic of debate in kidney fibrosis.32,33 Renal proximal tubular cell injury is the common pathological process reported in folic acid-induced AKI. The transition of AKI to the CKD and kidney fibrosis depends upon the nature and extent of kidney injury.34 If the injury is repairable, tubular epithelial cells try to repair and recover from the injury and get back to normal function. However, persistent and repetitive injuries cause an inappropriate repair process due to cellular reprogramming that in the long term leads to chronic kidney disease and fibrosis.35 The role of kidney tubular epithelial cells in kidney fibrosis has been a topic of debate. Earlier reports suggest the reprogramming of injured tubular epithelial cells into mesenchymal cells through the EMT process that leads to the onset and progression of kidney fibrosis.36 However, relatively recent reports suggest that epithelial cells themselves do not get converted into the extracellular matrix producing mesenchymal cells but produce inflammatory cytokines that activate the resident fibroblast into myofibroblast causing kidney fibrosis.37 In either case, tubular epithelial cells are important, either directly converting them into mesenchymal phenotype or initiating other inflammatory chemokines, or activating and differentiating the resident fibroblast into ECM-producing myofibroblasts.38
Our data of Western blot analysis revealed the downregulation of epithelial marker protein E-cadherin in both acute and long-term treated cells, and the expression was restored by antioxidant NAC, suggesting the role of oxidative stress in the regulation of this EMT marker protein. Similarly, the mesenchymal marker gene N-cadherin and vimentin, as well as the fibrosis marker genes such as fibronectin and alpha-SMA, were also upregulated at the transcript level in both acute and long-term FA-treated cells. The immunofluorescence staining of fibronectin in long-term FA-treated cells further confirmed the fibrogenic changes by folic acid in kidney epithelial cells. The expressions of these genes at transcript levels were partially restored by antioxidant NAC mostly in acutely treated but not in long-term FA-treated cells, suggesting that scavenging FA-induced oxidative stress can protect the initiation of EMT and fibrogenic process in the initial stages, but the long-term repeated exposure to oxidative stress permanently reprograms this gene expression that cannot be restored by antioxidant. Therefore, our data support the role of kidney tubular epithelial cells through EMT in FA-induced fibrogenic changes. Additionally, for the first time, this study provides evidence for the regulation of EMT and fibrosis marker genes by oxidative stress during FA-induced fibrogenic changes in the vitro cell culture model.
Among the various signaling pathways, β-catenin has been shown to play an important role in the regulation of EMT and fibrosis-related genes.39,40 The β-catenin pathway regulates multiple target genes including the upregulation of fibronectin via the snail-mediated pathway41,42 and the downregulation of E-cadherin.43 The finding of this study of a decreased expression of E-cadherin in both acute and long-term FA-treated cells but an increased expression of the mesenchymal markers N-cadherin and vimentin in long-term FA-treated cells and their partial restoration by antioxidant NAC further suggest FA-induced oxidative stress as a driver of EMT and fibrogenic changes in kidney epithelial cells. Additionally, the increased expression of β-catenin in long-term FA-treated cells and its partial restoration by NAC further suggest that FA-induced oxidative stress, through the β-catenin pathway, regulates the fibrogenic changes in these cells. Sustained activation of WNT-β-catenin signaling has been shown to promote kidney fibrosis.44 A link between oxidative stress and β-catenin during CKD has also been reported.45 Together, these previous reports and the result of increased level of β-catenin in FA-treated cells and its restoration by NAC suggest that FA potentially through oxidative stress causes β-catenin pathway-mediated fibrogenic changes in kidney epithelial cells.
In summary, this is the first study with the in vitro cell culture model to evaluate the effects of higher concentrations of FA in kidney epithelial cells. The novel findings of this study suggest that FA-induced ROS acts as a driver for the acute genotoxicity and cytotoxicity as well as long-term exposure associated with EMT and fibrogenic changes in kidney epithelial cells. These effects of FA observed in the in vitro cell culture model are similar to the acute effect of FA-induced AKI and the long-term effects of fibrosis in animal models. The results of this study suggest the potential of antioxidants in abrogating the pro-oxidant nephrotoxicants-induced AKI and fibrosis. The findings of this study also have significance in understanding the higher FA exposure that has recently been reported to cause an increased unmetabolized form of FA in serum and a high homocysteine level.
ACKNOWLEDGMENTS
This work was partially supported by a grant (R15DK121362-;01A1 to K.P.S.) from NIDDK.
Footnotes
The authors declare no competing financial interest.
Contributor Information
Ramji Kandel, Department of Environmental Toxicology, The Institute of Environmental and Human Health (TIEHH), Texas Tech University, Lubbock, Texas 79409, United States.
Kamaleshwar P. Singh, Department of Environmental Toxicology, The Institute of Environmental and Human Health (TIEHH), Texas Tech University, Lubbock, Texas 79409, United States
REFERENCES
- (1).Prevention, C. f. D. C. a. Chronic Kidney Disease in the United States; U.S. Department of Health and Human Services: Atlanta, GA, 2021. [Google Scholar]
- (2).Parr SK; Siew ED Delayed Consequences of Acute Kidney Injury. Adv. Chronic Kidney Dis 2016, 23 (3), 186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hsu CY; McCulloch CE; Fan D; Ordoñez JD.; Chertow GM.; Go AS. Community-based incidence of acute renal failure. Kidney Int. 2007, 72 (2), 208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Breyer MD; Susztak K The next generation of therapeutics for chronic kidney disease. Nat. Rev. Drug Discov 2016, 15 (8), 568–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ozbek E Induction of Oxidative Stress in Kidney. International Journal of Nephrology 2012, 2012, 465897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bosch RJ; Woolf AS; Fine LG Gene transfer into the mammalian kidney: direct retrovirus-transduction of regenerating tubular epithelial cells. Exp. Nephrol. 1993, 1, 49–54. [PubMed] [Google Scholar]
- (7).Ortiz A; Sanchez-Niño MD.; Izquierdo MC; Martin-Cleary C; Garcia-Bermejo L; Moreno JA; Ruiz-Ortega M; Draibe J; Cruzado JM; Garcia-Gonzalez MA; et al. Translational value of animal models of kidney failure. Eur. J. Pharmacol. 2015, 759, 205–220. [DOI] [PubMed] [Google Scholar]
- (8).Kalmbach RD; Choumenkovitch SF; Troen AM; D’Agostino R; Jacques PF; Selhub J Circulating folic acid in plasma: relation to folic acid fortification. Am. J. Clin. Nutr. 2008, 88 (3), 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Joshi R; Adhikari S; Patro BS; Chattopadhyay S; Mukherjee T Free radical scavenging behavior of folic acid: evidence for possible antioxidant activity. Free Radic Biol. Med. 2001, 30 (12), 1390–1399. [DOI] [PubMed] [Google Scholar]
- (10).Koseki K; Maekawa Y; Bito T; Yabuta Y; Watanabe F High-dose folic acid supplementation results in significant accumulation of unmetabolized homocysteine, leading to severe oxidative stress in Caenorhabditis elegans. Redox Biol. 2020, 37, 101724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Olszewski AJ; McCully KS Homocysteine metabolism and the oxidative modification of proteins and lipids. Free Radic Biol. Med. 1993, 14 (6), 683–693. [DOI] [PubMed] [Google Scholar]
- (12).Ledowsky C; Mahimbo A; Scarf V; Steel A Women Taking a Folic Acid Supplement in Countries with Mandatory Food Fortification Programs May Be Exceeding the Upper Tolerable Limit of Folic Acid: A Systematic Review. Nutrients 2022, 14, 2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hu J; Wang B; Sahyoun NR Application of the Key Events Dose-response Framework to Folate Metabolism. Crit Rev. Food Sci. Nutr 2016, 56 (8), 1325–1333. [DOI] [PubMed] [Google Scholar]
- (14).Choi JH; Yates Z; Veysey M; Heo YR; Lucock M Contemporary issues surrounding folic Acid fortification initiatives. Prev Nutr Food Sci. 2014, 19 (4), 247–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Siow YL; Au-Yeung KK; Woo CW; O, K. Homocysteine stimulates phosphorylation of NADPH oxidase p47phox and p67phox subunits in monocytes via protein kinase Cbeta activation. Biochem. J. 2006, 398 (1), 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhang X; Huang Z; Xie Z; Chen Y; Zheng Z; Wei X; Huang B; Shan Z; Liu J; Fan S; et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic Biol. Med. 2020, 160, 552–565. [DOI] [PubMed] [Google Scholar]
- (17).Thaler R; Agsten M; Spitzer S; Paschalis EP; Karlic H; Klaushofer K; Varga F Homocysteine suppresses the expression of the collagen cross-linker lysyl oxidase involving IL-6, Fli1, and epigenetic DNA methylation. J. Biol. Chem. 2011, 286 (7), 5578–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pfeiffer CM; Caudill SP; Gunter EW; Osterloh J; Sampson EJ Biochemical indicators of B vitamin status in the US population after folic acid fortification: results from the National Health and Nutrition Examination Survey 1999–2000. Am. J. Clin. Nutr. 2005, 82, 442–450. [DOI] [PubMed] [Google Scholar]
- (19).Schmittgen TD; Livak KJ Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3 (6), 1101–1108. [DOI] [PubMed] [Google Scholar]
- (20).Gupta RC Nonrandom binding of the carcinogen N-hydroxy-2-acetylaminofluorene to repetitive sequences of rat liver DNA in vivo. Proc. Natl. Acad. Sci. U. S. A. 1984, 81 (22), 6943–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Singh KP Analysis of Toxicants-Induced Alterations in DNA Methylation by Methylation-Sensitive-Random Amplified Polymorphic DNA-Polymerase Chain Reaction (MS-RAPD-PCR). Methods Mol. Biol. 2020, 2102, 213–224. [DOI] [PubMed] [Google Scholar]
- (22).Doi K; Okamoto K; Negishi K; Suzuki Y; Nakao A; Fujita T; Toda A; Yokomizo T; Kita Y; Kihara Y; et al. Attenuation of folic acid-induced renal inflammatory injury in platelet-activating factor receptor-deficient mice. American journal of pathology 2006, 168 (5), 1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Koseki K; Maekawa Y; Bito T; Yabuta Y; Watanabe F High-dose folic acid supplementation results in significant accumulation of unmetabolized homocysteine, leading to severe oxidative stress in Caenorhabditis elegans. Redox Biology 2020, 37, 101724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tyagi N; Sedoris KC; Steed M; Ovechkin AV; Moshal KS; Tyagi SC Mechanisms of homocysteine-induced oxidative stress. Am. J. Physiol Heart Circ Physiol 2005, 289 (6), H2649–2656. [DOI] [PubMed] [Google Scholar]
- (25).Yan L-J Folic acid-induced animal model of kidney disease. Animal Models and Experimental Medicine 2021, 4 (4), 329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Che R; Yuan Y; Huang S; Zhang A Mitochondrial dysfunction in the pathophysiology of renal diseases. American Journal of Physiology-Renal Physiology 2014, 306 (4), F367–F378. [DOI] [PubMed] [Google Scholar]
- (27).Aparicio-Trejo OE; Reyes-Fermín LM; Briones-Herrera A; Tapia E; León-Contreras JC; Hernández-Pando R; Sánchez-Lozada LG; Pedraza-Chaverri J Protective effects of N-acetylcysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free Radic Biol. Med. 2019, 130, 379–396. [DOI] [PubMed] [Google Scholar]
- (28).Leonard MO; Kieran NE; Howell K; Burne MJ; Varadarajan R; Dhakshinamoorthy S; Porter AG; O’Farrelly C; Rabb H; Taylor CT Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia-reperfusion injury. FASEB J. 2006, 20 (14), 2624–2626. [DOI] [PubMed] [Google Scholar]
- (29).Stallons LJ; Whitaker RM; Schnellmann RG Suppressed mitochondrial biogenesis in folic acid-induced acute kidney injury and early fibrosis. Toxicol. Lett. 2014, 224 (3), 326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Fink M; Henry M; Tange JD Experimental folic acid nephropathy. Pathology 1987, 19 (2), 143–149. [DOI] [PubMed] [Google Scholar]
- (31).Yang HC; Zuo Y; Fogo AB Models of chronic kidney disease. Drug Discov Today Dis Models 2010, 7 (1–2), 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sheng L; Zhuang S New Insights Into the Role and Mechanism of Partial Epithelial-Mesenchymal Transition in Kidney Fibrosis. Front Physiol 2020, 11, 569322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zeisberg M; Kalluri R The role of epithelial-to-mesenchymal transition in renal fibrosis. J. Mol. Med. (Berl) 2004, 82 (3), 175–181. Sheng L.; [DOI] [PubMed] [Google Scholar]; Zhuang S. New Insights Into the Role and Mechanism of Partial Epithelial-Mesenchymal Transition in Kidney Fibrosis. Frontiers in Physiology 2020, 11, Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Jiang K; Ponzo TA; Tang H; Mishra PK; Macura SI; Lerman LO Multiparametric MRI detects longitudinal evolution of folic acid-induced nephropathy in mice. Am. J. Physiol Renal Physiol 2018, 315 (5), F1252–f1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Yang L; Besschetnova TY; Brooks CR; Shah JV; Bonventre JV Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nature Medicine 2010, 16 (5), 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Rout-Pitt N; Farrow N; Parsons D; Donnelley M Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respiratory Research 2018, 19 (1), 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Grande MT; Sanchez-Laorden B; Lopez-Blau C; De Frutos CA; Boutet A; Arevalo M; Rowe RG; Weiss SJ; Lopez-Novoa JM; Nieto MA Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21 (9), 989–997. [DOI] [PubMed] [Google Scholar]
- (38).Borthwick LA; Wynn TA; Fisher AJ Cytokine mediated tissue fibrosis. Biochim. Biophys. Acta 2013, 1832 (7), 1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).He W; Dai C; Li Y; Zeng G; Monga SP; Liu Y Wnt/beta-catenin signaling promotes renal interstitial fibrosis. Journal of the American Society of Nephrology: JASN 2009, 20 (4), 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhou D.; Tan RJ.; Fu H.; Liu Y. Wnt/β-catenin signaling in kidney injury and repair: a double-edged sword. Laboratory Investigation 2016, 96 (2), 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zhou D; Tan RJ; Fu H; Liu Y Wnt/beta-catenin signaling in kidney injury and repair: a double-edged sword. Lab Invest 2016, 96 (2), 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hao S; He W; Li Y; Ding H; Hou Y; Nie J; Hou FF; Kahn M; Liu Y Targeted inhibition of beta-catenin/CBP signaling ameliorates renal interstitial fibrosis. J. Am. Soc. Nephrol 2011, 22 (9), 1642–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Tan RJ; Zhou D; Zhou L; Liu Y Wnt/β-catenin signaling and kidney fibrosis. Kidney international supplements 2014, 4 (1), 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hao S.; He W.; Li Y.; Ding H.; Hou Y.; i J.; Hou FF.; Kahn M.; Liu Y. Targeted inhibition of β-catenin/CBP signaling ameliorates renal interstitial fibrosis. Journal of the American Society of Nephrology: JASN 2011, 22 (9), 1642–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Liang S; Yadav M; Vogel KS; Habib SL A novel role of snail in regulating tuberin/AMPK pathways to promote renal fibrosis in the new mouse model of type II diabetes. FASEB BioAdvances 2021, 3 (9), 730–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Schunk SJ; Floege J; Fliser D; Speer T WNT-beta-catenin signalling - a versatile player in kidney injury and repair. Nat. Rev. Nephrol 2021, 17 (3), 172–184. [DOI] [PubMed] [Google Scholar]
- (45).Zhou L; Chen X; Lu M; Wu Q; Yuan Q; Hu C; Miao J; Zhang Y; Li H; Hou FF; et al. Wnt/beta-catenin links oxidative stress to podocyte injury and proteinuria. Kidney Int. 2019, 95 (4), 830–845. [DOI] [PMC free article] [PubMed] [Google Scholar]