

Summary
Tissue-residential natural killer (trNK) cells act as pioneering responders during infectious challenges. However, their discrimination with conventional NK (cNK) cells is still an issue. Through an integrative transcriptome comparison of the two NK subgroups from different tissues, we have defined two genesets capable of efficiently distinguishing them. Based on the two genesets, a fundamental difference between the activation of trNK and cNK is identified and further confirmed. Mechanistically, we have discovered a particular role of chromatin landscape in regulating the trNK activation. In addition, IL-21R and IL-18R are respectively highly expressed by trNK and cNK, indicating a role of cytokine milieu in determining their differential activation. Indeed, IL-21 is particularly critical in accessorily promoting trNK activation using a bunch of bifunctional transcription factors. Together, this study sheds light on the bona fide difference between trNK and cNK, which will further expand our knowledge about their distinct functionalities during immune responses.
Subject areas: Biological sciences, Molecular biology, Epigenetics, Immunology
Graphical abstract
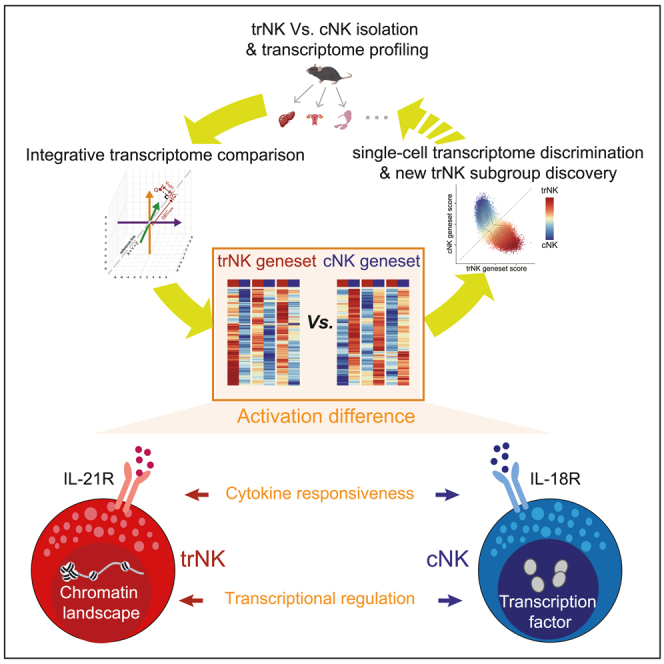
Highlights
-
•
Defining trNK and cNK genesets to efficiently distinguish the two NK populations
-
•
Revealing the essential role of chromatin landscape in determining trNK activation
-
•
Discovering the involvement of particular transcription factors in cNK activation
-
•
Identifying the different responsiveness of trNK and cNK to IL-21 and IL-18
Biological sciences; Molecular biology; Epigenetics; Immunology
Introduction
Natural killer (NK) cells are an important component of the innate immune system, playing critical roles against infectious challenges.1 They execute their roles mainly through producing inflammatory cytokines, cytotoxic granules, and proapoptotic molecules.2,3 In recent years, a particular NK subgroup, tissue-residential NK (trNK), has been discovered and attracted increasing attention. In contrast to the conventional NK (cNK) that patrol around the body, the trNK exclusively reside in peripheral tissues.4,5 This particular trait renders them the capacity to ignite an immediate response in local tissue after pathogen infections.6,7,8 In line with it, trNK deficiency usually leads to defective immune responses and delayed pathogen clearance.6,7
Though trNK are clearly defined based on the tissue residency, it is still challenging to distinguish them experimentally. Both trNK and cNK express the general NK cell markers NK1.1 and NKp46 and exhibit the cytotoxic activity, which distinguishes them from other innate lymphocytes such as type 1 innate lymphoid cells (ILC1).9,10,11 The liver trNK are also called liver ILC1s in many recent studies.7,12,13,14 Based on single-cell analysis, they should be the sample NK population with different nomenclature.15,16 And, given their cytotoxicity, we still prefer to take them as liver trNK. In addition, Clusters of Differentiation 49b (CD49b, or DX5) is usually considered as an exclusive marker of matured NK but is not expressed by ILC1.17 Usually, CD49a is utilized to distinguish trNK, especially in liver, uterus, and salivary glands (SGs). Nevertheless, it is not always a reliable marker of trNK in all tissues.18,19 Experimentally, circulatory immune cells, including cNK, can be distinguished through intravenous anti-CD45 antibody injection. After a short time, the anti-CD45 antibody is sufficient to label immune cells in the blood, but not in peripheral tissues.20,21 However, anti-CD45 antibody injection does not work well for the immune cells in lymphatic system.20 Thus, failure of the anti-CD45 labeling is not necessary to guarantee the tissue residency. Another more reliable method is parabiosis, a surgical model connecting the circulations of two congenic animals.22,23 After sufficient time of the parabiotic pairing, the circulatory immune cells will distribute evenly to the partnered animal, while the tissue-resident immune cells are still not exchanged.4,24 Nevertheless, this method requires a delicate surgical operation and is time-consuming. Thus, it is difficult to be prevalently used. Also, it is only suitable for animal studies but is not applicable for studying the tissue-resident immune cells in human. Therefore, a convenient and reliable method for the trNK and cNK discrimination is still required.
The functionalities of trNK and cNK exhibit many overlaps. After activation, trNK also produce effector cytokines and display a cytotoxic activity, though expression levels of the effector molecules are usually different from those of cNK.7,25,26 Given their tissue residency, the trNK activation should be tightly orchestrated, to avoid the overresponse-induced tissue damages. Nevertheless, the difference between trNK and cNK activation has yet to be comprehensively compared. Particularly, proinflammatory cytokines generated in local niches usually play important roles in promoting the activation of tissue-resident immune cells.27,28 Such proinflammatory cytokines, like interleukin (IL)-15, IL-18, and IL-21, are also involved in stimulating the NK activation.29,30 Thus, their roles in particularly promoting the trNK or cNK activation may need to be further examined.
Results
Identification of trNK and cNK genesets reflecting their fundamental difference
To unravel the fundamental difference between trNK and cNK, RNA sequencing (RNA-seq) was performed using the two NK subgroups sort-purified from liver, uterus, and SGs (Figures 1A and S1A). According to principal-component analysis (PCA), the trNK and cNK from these tissues could be roughly separated by principal component 2 (PC2) (16.4%), indicating a fundamental difference between them (Figures 1B and S1B). The transcriptome difference between these trNK and cNK subgroups in the three tissues was examined separately. The number of the differentially expressed genes between trNK and cNK (Transcripts Per Million (TPM) >10 in at least one group, fold change >2 and P. adj <0.05) was around 100–400 in each tissue (Figure S1C). However, we only discovered 27 trNK-specific genes and 23 cNK-specific genes across all three tissues, accounting for <20% of the total differentially expressed genes between trNK and cNK in each tissue (Figure S1D). It suggested that the major transcriptome difference between the two NK subgroups was associated with their tissue niche.
Figure 1.

Identification of trNK and cNK genesets reflecting their fundamental difference
(A) Workflow of transcriptome profiling of trNK and cNK from liver, uterus, and salivary glands (SGs).
(B) Principal component analysis (PCA) of trNK and cNK from liver, uterus, and SGs (n = 2 per group).
(C) Schematics of integrative transcriptome comparison of trNK and cNK from liver, uterus, and SGs.
(D) Definition of trNK and cNK genesets in a 3D coordinate system based on integrative transcriptome comparison.
(E) 2D visualization of trNK and cNK genesets in a coordinate system defined by x = GECave and y = VGEC.
(F) Heatmap displaying the expression of trNK and cNK genesets by the two NK subgroups from liver, uterus, and SGs.
The small trNK- and cNK-specific genesets were not satisfied for an intensive exploration of the two NK subgroups. In order to collect more trNK- and cNK-specific genes, we developed a method of integratively comparing their transcriptomes across the three tissues. Briefly, the gene expression comparison between trNK and cNK (log2 trNK/cNK) in liver, uterus, and SGs was assigned to three dimensions of a 3D coordinate system, respectively. Thus, the bona fide trNK- or cNK-specific genes should distribute in a zone close to the reference line x = y = z. We defined two parameters to identify the zone for the trNK- or cNK-specific genes: (1) average gene expression change (GECave) as defined by the distance of a gene’s projected coordinate on the reference line (x = y = z) to the origin (x = 0, y = 0, z = 0) and (2) variation of gene expression change (VGEC) as defined by the distance of a gene to the reference line (Figure 1C). Then, the trNK- and cNK-specific genes were defined as locating within the zone of |GECave| > 1, |GECave| > VGEC (Figure 1D). To simplify the visualization, GECave and VGEC were assigned to the x and y axes of a 2D system (Figure 1E). Based on this method, we identified 197 trNK-specific and 155 cNK-specific genes. Heatmap profiling further showed the consistency of their preferential expression in trNK or cNK across the three tissues, demonstrating the reliability of our method (Figure 1F). Collectively, through an integrative transcriptome comparison, we have identified two genesets representing the fundamental difference between trNK and cNK, which are termed trNK and cNK genesets hereafter.
The trNK and cNK genesets can efficiently discriminate the two NK subgroups
Next, we wondered whether the trNK and cNK genesets generated from the three tissues could also be utilized to discriminate NK subgroups in other tissues. Thus, RNA-seq of the typical cNK subgroup in spleen was performed to testify the possibility. Gene set enrichment analysis (GSEA) based on the trNK and cNK genesets showed that the splenic cNK preferentially expressed genes in the cNK geneset but not the trNK geneset, suggesting that the two genesets were also applicable for discriminating the splenic cNK (Figures 2A and S2A). The importance of the trNK and cNK genesets in discriminating the two NK subgroups was also assessed. Based on the Pearson correlation of whole transcriptome, the two NK subgroups in different tissues could hardly be accurately separated (Figure S2B). In contrast, using the trNK and cNK genesets, all the NK subgroups, including the splenic cNK, were separated according to their tissue residency (Figure 2B). Thus, the trNK and cNK genesets exerted an indispensable role in the transcriptomic discrimination of the two NK subgroups.
Figure 2.

The trNK and cNK genesets can efficiently discriminate the two NK subgroups
(A) Gene set enrichment analysis (GESA) showing expression of trNK and cNK genesets in splenic cNK.
(B) Pearson correlation analysis of trNK and cNK from liver, uterus, SGs, and spleen, using trNK and cNK genesets.
(C) Discrimination of trNK and cNK in liver, uterus, SGs, and spleen by single-sample GSEA (ssGSEA) scores, using trNK and cNK genesets.
(D) Assessment of trNK and cNK genesets in NK from liver, SGs, gut, and spleen (left) using single-cell transcriptome (scRNA-seq, GSE189807). ΔScore of each cell representing the difference between trNK and cNK geneset scores is calculated (right).
(E) Assessment of trNK and cNK genesets in NK cells of each tissue in (D).
(F) Scatterplot showing Itga1 (CD49a) and Itga2 (DX5) expression in NK cells (D).
Scoring based on the trNK and cNK genesets was also an alternative to discriminate an NK subgroup. The NK subgroups from liver, uterus, SGs, and spleen were reciprocally highly scored by their related genesets (Figure 2C). Heterogeneity of the NK population in different tissues had been intensively explored using single-cell RNA-seq (scRNA-seq) in recent years. We wondered whether the scoring could also be applied to the single-cell transcriptome, which would greatly expand the utilization of the trNK and cNK genesets. Thus, publicly available scRNA-seq data of NK cells from various tissues (Gene Expression Omnibus Series GSE189807) were revisited.31 Indeed, though the trNK and cNK genesets were generated based on bulk RNA-seq comparison, they were also applicable for scRNA-seq to discriminate trNK and cNK cells (Figures 2D and 2E). The result was further verified by an examination of Itga1 (encoding CD49a) and Itga2 (encoding DX5) expression (Figure 2F). In addition, it further demonstrated that the trNK and cNK genesets could be applied to discriminate the two NK subgroups from a broad range of tissues. On the other hand, same as we just observed, the trNK and cNK can hardly be clearly separated by whole transcriptomes, and the predominant difference between the single-cell clusters was not correlated with the discrimination between the two NK subgroups (Figures S2C and S2D). Consistent with a previous study, we also observed a subgroup of NK exhibiting an intermediate trNK and cNK feature, further arguing the usage of limited markers for the discrimination of the two NK subgroups (Figure 2D).31
We also testified the trNK and cNK genesets with publicly available scRNA-seq data of human NK cells (GSE70580, GSE150050).32,33 The cNK geneset did seem to be applicable for discriminating the cNK feature in human. However, the trNK geneset failed to provide a reliable assessment (Figure S2E). Thus, to discriminate the two NK subgroups in human, a particular integrative transcriptome comparison between human trNK and cNK was still required.
Overall, our data show that the trNK and cNK genesets we defined can be applied to discriminate the two NK subgroups from a broad range of tissues, based on either bulk or single-cell transcriptome.
The trNK and cNK genesets reveal a fundamental difference between the activation of the two NK subgroups
The high efficacy of the trNK and cNK genesets in the NK subgroup discrimination indicated that these genes reflected the principal differences between the two NK subgroups. Thus, a Gene Ontology (GO) analysis was performed to unravel the features enriched by them. Consistent with the traits of trNK in tissue residency and recruitment of other immune cells to the local tissue, the related features were significantly enriched by the trNK geneset (Figure 3A). We further profiled the genes related to the tissue residency (Figure S3A). Particularly, Zfp683, a known transcriptional regulator of tissue-resident lymphocytes, was upregulated in trNK.34,35 On the other hand, sphingosine-1-phosphate (S1P) receptors S1pr1, S1pr4, and S1pr5, crucial for immune cell trafficking, were preferentially expressed by cNK (Figure 3B).
Figure 3.

trNK and cNK genesets reveal a fundamental difference between the activation of the two NK subgroups
(A) Go analysis of trNK and cNK genesets.
(B–G) Relative expression of the indicated genes between trNK and cNK in liver, uterus, and SGs, grouped by lymphocyte migration-associated molecules (B), NK effector molecules (C), NK activation-related surface receptors (D), transcription factors (E), immune checkpoints (F), and cytokine receptors (G).
(H) Venn diagram showing differentially expressed genes between liver trNK and cNK (TPM >10 in at least one group, fold change >2 and P adj. < 0.05) 44 h after MCMV infection (GSE114827).
(I) Heatmap profiling differentially expressed genes in (H).
(J) Discrimination of MCMV infection-activated trNK and cNK (H) by GSEA, using trNK and cNK genesets.
(K) GO analysis of differentially expressed genes in liver trNK and cNK after MCMV infection (H).
Impressively, we also observed an intensive enrichment of “immune activation”-related features, especially by the trNK geneset, indicating that the activation of two NK subgroups might be primarily different (Figure 3A). In line with this, several immunosuppressive genes were found to be upregulated in trNK, while most immune activation-related genes were highly expressed by cNK (Figures S3B and S3C). To further verify this difference, signature genes associated with NK activation were examined. Among those effector molecules, Prf1 and Gzma were highly expressed by cNK in different tissues, while Gzmb was preferentially expressed by trNK (Figure 3C). NK activation was regulated by many surface and adaptor molecules. Among them, Klra1 (encoding Ly49a), Klrb1a (encoding CD161a), Klrc2 (encoding NKG2C), Klrg1 (encoding KLRG1), Klrk1 (encoding NKG2D), and Fyn were substantially downregulated in trNK (Figure 3D). Additionally, transcriptional regulators, including T-bet, Eomes, Nfil3, Runx3, and Id2, also participated in regulating the NK activation. Compared with cNK, trNK showed reduced expression of Tbx21 (encoding T-bet) and Eomes (Figure 3E), whereas immunosuppressive molecules Ctla4, Tigit, and Cd200r1 were significantly upregulated in trNK (Figure 3F).7 Collectively, the differential expression of these signature genes between the two NK subgroups indicated that trNK might prefer to maintain in an inactivated status. Stimulatory cytokines, such as IL-2, IL-15, IL-12, IL-18, and IL-21, were also involved in regulating NK activation.29,30 We found that Il18r1 and Il18rap (encoding the two subunits of IL-18R) were upregulated in cNK,36 while Il21r was particularly highly expressed by trNK,34 corroborating a potential difference between the trNK and cNK activation (Figure 3G).
Further, to demonstrate the difference between the two NK subgroups after activation, RNA-seq of Murine Cytomegalovirus (MCMV) infection-activated liver trNK and cNK (GSE114827) was revisited.37 A total of 7,128 genes were found to be truly expressed by either NK subgroup (TPM >10 in at least one group). Among them, 768 (10.8%) were upregulated in the MCMV infection-activated trNK, while 226 (3.2%) were increasingly expressed by the activated cNK (Figure 3H). Also, we found that many NK function-related genes were exclusively highly expressed by either of the two NK subgroups, corroborating their difference after activation (Figure 3I). In addition, the trNK and cNK genesets remained to be applicable for the discrimination of the activated NK subgroups, indicating that the principal difference between trNK and cNK including the activation difference was still maintained (Figure 3J). Further, their differentially expressed genes were obviously enriched in distinct pathways (Figure 3K).
Collectively, in addition to the well-known differences between trNK and cNK in cell adhesion and immune recruitment, our data also indicate a fundamental difference between them in activation.
Transcriptional regulation of trNK and cNK activation is fundamentally different
The difference between trNK and cNK after MCMV infection might be also caused by the difference of their niches.38 To further clarify the fundamental difference between trNK and cNK activation, a unified ex vivo stimulatory condition was considered. A short-time stimulation within hours was also preferred to avoid inducing alternative changes to trNK after leaving their tissue niche, which excluded the usage of stimulatory cytokines such as IL-15,12,13,15,39 whereas most other stimulatory manners, including co-culture with target cells, were inevitably affected by the differentially expressed receptors on trNK and cNK. Finally, phorbol 12-myristate 13-acetate (PMA) and ionomycin (PMA/ionomycin) that exerted a unified stimulatory effect on the trNK and cNK independent of their differentially expressed surface receptors were considered, although they were not supposed as a physiological condition. To compare the transcriptome changes between trNK and cNK during activation, resuspended liver lymphocytes were kept either unstimulated or stimulated by PMA/ionomycin for 5 h, and then the trNK and cNK were sort-purified for RNA-seq analysis (Figure S4A). As suggested by the transcriptome changes, the PMA/ionomycin stimulation substantially activated both trNK and cNK (Figures 4A and S4B). Nevertheless, the trNK and cNK genesets were still applicable for discriminating these ex vivo activated NK subgroups (Figures 3J and 4B). Since PMA/ionomycin were not supposed to activate NK in a physiological manner, transcriptomes of the NK subgroups activated ex vivo by PMA/ionomycin and in vivo after MCMV infection were compared. GSEA showed that both PMA and ionomycin activated trNK and cNK to a large extent and recapitulated their activation induced after MCMV infection, greatly alleviating our concern with the PMA/ionomycin stimulation (Figure 4C). After the PMA/ionomycin stimulation, genes upregulated and downregulated in trNK and cNK were obviously different (Figure S4C). To further reveal the activation difference between trNK and cNK, a GO analysis was performed using their activation-upregulated genes (Figures 4D and S4B). Between their enriched features, although there seemed to be some similarities, like “ribosome biogenesis”, “positive regulation of cytokine production”, and “cellular amino acid metabolic process”, the enrichment of “ribosome biogenesis” was substantially different. In addition, they also possessed differentially enriched features (Figure 4D). Collectively, using a unified ex vivo stimulation, we confirmed that the activation of trNK and activation of cNK were fundamentally different.
Figure 4.

Transcriptional regulation of trNK and cNK activation are fundamentally different
(A) Volcano plots of gene expression difference between unstimulated (Unstim) and PMA/ionomycin (PMA/Iono)-stimulated trNK (left) and cNK (right).
(B) Discrimination of trNK and cNK kept unstimulated (top) or stimulated by PMA/ionomycin (bottom) (A) by GSEA, using trNK and cNK genesets.
(C) GSEA comparison of ex vivo PMA/ionomycin stimulation-activated (A) and in vivo MCMV infection-activated (3I) trNK (left) or cNK (right).
(D) GO analysis of upregulated genes in PMA/ionomycin stimulation-activated trNK (left) and cNK (right).
(E) Venn diagram showing potential transcription factors (TFs) of the upregulated genes in PMA/ionomycin stimulation-activated trNK and cNK (A), according to a chromatin landscape analysis (S4C).
(F) Profiling of each potential transcription factor in (E), showing the percentage of their potentially regulated genes within the upregulated genes in PMA/ionomycin stimulation-activated trNK or cNK (left) and their expression in PMA/ionomycin-activated trNK or cNK (right).
(G) Overall expression changes of the potential transcription factors, grouped by trNK-specific, common, and cNK-specific (E), between trNK and cNK.
(H) Chromatin accessibility comparison between trNK and cNK, for regions related to the upregulated genes in PMA/ionomycin stimulation-activated trNK and containing binding motifs of trNK-specific (left) and common (right) potential transcription factors. Average chromatin accessibility is calculated on top and the peak heights and peak areas between trNK and cNK are compared in right.
(I) Chromatin accessibility comparison between trNK and cNK, for regions related to the upregulated genes in PMA/ionomycin stimulation-activated cNK and containing binding motifs of cNK-specific (left) and common (right) potential transcription factors. Average chromatin accessibility is calculated on top and the peak heights and peak areas between trNK and cNK are compared in right.
Transcription regulators played essential roles during NK activation. Thus, we next wondered whether the difference between trNK and cNK activation was caused by differential transcriptional regulation. To verify this assumption, chromatin landscapes of the two NK subgroups were examined, using a set of assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-Seq) data for liver trNK and cNK (GSE196716).25 Potential transcriptional regulators for the trNK and cNK activation-upregulated genes were predicted by scanning their binding motifs within the related chromatin-accessible regions (ChARs) (Figure S4D). In addition, expression levels of these potential transcriptional regulators were considered. Together, we identified 26 potential transcription regulators for the trNK activation and 15 potential transcription regulators for the cNK activation (Figure 4E). Among them, 11 were commonly utilized by both NK subgroups, while 15 were specific for the trNK activation and 4 were particular for the cNK activation (Figures 4F and S4E). As expected, the 4 transcription factors particular for cNK activation were highly expressed by cNK compared with trNK. However, the 11 trNK-specific and 15 common transcription factors displayed comparable expression between the two NK subgroups, indicating that their particular regulatory roles in trNK might be determined by their chromatin landscape (Figure 4G). Consistently, ChARs for those trNK activation-upregulated genes were more accessible (Figures S4F and S4G). Further, the ChARs containing binding motifs of the 11 trNK-specific and 15 common transcription factors displayed increased accessibility in trNK compared with cNK (Figure 4H). Interestingly, even for the cNK activation-upregulated genes, the related ChARs containing the binding motifs of the 4 cNK-specific and 15 common transcription factors also exhibited enhanced accessibility in trNK, suggesting a complicated correlation between these ChARs and the expression of their related genes in trNK (Figures 4I and S4G). Further, the complexity of trNK activation was also indicated by the presence of more regulatory relationships for those common features enriched by both the activation of trNK and cNK (Figure S4H).
Together, our results demonstrate a fundamental difference between trNK and cNK activation. The cNK activation is preferentially regulated by specific transcriptional regulators, while the trNK activation is complicated and is mostly relied on their chromatin landscape.
IL-21 and IL-18 play accessory roles in facilitating trNK and cNK activation, respectively
Cytokines also contributed to the NK activation.27,28 As mentioned earlier, the Il21r and Il18r1/Il18rap were preferentially highly expressed by trNK and cNK, respectively (Figure 3G). This differential Inteleukin-21 Receptor (IL-21R) and Inteleukin-18 Receptor (IL-18R) expression was also confirmed at protein level by flow cytometry, using trNK and cNK from liver, uterus, and SGs (Figures 5A–5D). Given the result, we wondered whether IL-21 and IL-18 signaling played distinct roles in regulating the trNK and cNK activation. The ex vivo liver trNK could hardly be activated by IL-21 or IL-18 stimulation alone in 5 h, as indicated by the subtle interferon (IFN)-γ and tumor necrosis factor alpha (TNF-α) production, while the ex vivo liver cNK were mildly activated by IL-18, but not IL-21 (Figures S5A and S5B). Considering that both cytokines were reported to promote the NK activation in previous studies,21 we speculated that these cytokines mainly exert accessory roles. Thus, their impacts on the PMA/ionomycin-stimulated trNK and cNK were further explored. As we suspected, together with the PMA/ionomycin stimulation, IL-21 preferentially further enhanced IFN-γ production in trNK, while IL-18 was more efficient in facilitating IFN-γ production in cNK (Figures 5E, 5F, and S5C). As reported, TNF-α was mainly produced by trNK, but the additional IL-21 or IL-18 stimulation did not increase its production too much (Figures 5E, 5F, and S5C). Collectively, these results suggest that IL-21 and IL-18 mainly play accessory roles in trNK and cNK activation, respectively.
Figure 5.

IL-21 and IL-18 play accessory roles in facilitating trNK and cNK activation, respectively
(A) Flow cytometric analysis of IL-21R expression on trNK and cNK from liver, uterus, and SGs. The same gating strategy as Figure S1A is used.
(B) Statistical calculation of IL-21 expression level on trNK and cNK from liver, uterus, and SGs, based on its mean fluorescence intensity (MFI) in (A) (n = 4 per group).
(C) Flow cytometric analysis of IL-18R1 expression on trNK and cNK from liver, uterus, and SGs. The same gating strategy as Figure S1A is used.
(D) Statistical calculation of IL-18R1 expression level on trNK and cNK from liver, uterus, and SGs, based on its mean fluorescence intensity (MFI) in (C) (n = 4 per group).
(E) Flow cytometric analysis of IFN-γ and TNF-α expression in liver trNK and cNK kept unstimulated or stimulated by the indicated conditions.
(F) Statistical calculation of IFN-γ and TNF-α expression changes in liver trNK and cNK under the indicated conditions, and relative to PMA/ionomycin stimulation alone induced IFN-γ and TNF-α expression changes in trNK and cNK, respectively (n = 4 per group). Data are shown as the mean ± SEM. P. values are calculated by unpaired t-test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Data are representative of at least three independent experiments.
Additional IL-21 stimulation enhances the activation features of trNK
Next, we further examined the transcriptome changes of trNK and cNK in presence of the additional IL-21 or IL-18 stimulation (Figure 6A). First, with the additional IL-21 or IL-18, the two NK subgroups could still be discriminated by the trNK and cNK genesets, indicating that their fundamental difference was still maintained (Figure 6B). Next, whole transcriptome changes of trNK and cNK after the additional IL-21 or IL-18 stimulation were assessed. Compared with transcriptome changes caused by the PMA/ionomycin stimulation, the additional cytokine stimulation-induced transcriptome changes were much milder (Figure 6C). Nevertheless, in line with the differentially expressed cytokine receptors, PCA showed that the additional IL-21 stimulation substantially changed the transcriptome of PMA/ionomycin-activated trNK, while its impact on PMA/ionomycin-activated cNK was negligible (Figure 6D). On the other hand, the additional IL-18 stimulation altered the transcriptome of PMA/ionomycin-activated cNK more significantly (Figure 6D). Therefore, the transcriptome comparison corroborated the accessory roles of IL-21 and IL-18 in trNK and cNK activation, respectively.
Figure 6.

Additional IL-21 stimulation enhances the activation features of trNK
(A) Workflow of the RNA-seq analysis of ex vivo stimulated liver trNK and cNK. Sorting strategy is the same as Figure S4A.
(B) Pearson correlation between trNK and cNK under the indicated stimulatory conditions, using trNK and cNK genesets.
(C) Pearson correlation of trNK and cNK under the indicated stimulatory conditions according to their whole transcriptomes.
(D) PCA of trNK (left) and cNK (right) under the indicated stimulatory conditions (n = 2 per group). Dashed arrows in purple and blue respectively indicate additional IL-21 (purple) and IL-18 (blue) stimulation induced changes to PMA/ionomycin-activated trNK (right) and cNK (left).
(E) Heatmap profiling of upregulated (left) and downregulated (right) genes by additional IL-21 stimulation in trNK. The genes are screened based on, (1) reaching to the highest or lowest expression in trNK stimulated by additional IL-21, and (2) exhibiting at least 2-fold changes compared to either unstimulated or PMA/ionomycin-stimulated trNK.
(F) Go analysis of the upregulated (top) and downregulated (bottom) genes in trNK by additional IL-21 stimulation in (E).
Since the regulation of trNK activation was still elusive, the role of IL-21 in this process was particularly examined. Thus, genes affected by the additional IL-21 stimulation were screened according to two criteria: after the additional IL-21 stimulation (1) they reached to the highest or lowest expression level; (2) they showed at least 2-fold expression changes compared with either unstimulated or PMA/ionomycin-activated trNK. Finally, the additional IL-21 stimulation was found to correlate with 488 upregulated genes and 1,144 downregulated genes (Figure 6E). Further, a GO analysis showed that most PMA/ionomycin stimulation-induced changes in trNK were enhanced by the additional IL-21 stimulation, corroborating that IL-21 promoted the trNK activation (Figures 4D and 6F). In addition, genes involved in the major GO terms were profiled to assess the changes between unstimulated, PMA/ionomycin-stimulated, and additional IL-21-stimulated trNK. The result also demonstrated the role of IL-21 in facilitating trNK activation (Figure S6). Altogether, the additional IL-21 stimulation-induced transcriptome change suggests that IL-21 plays an important role in further promoting the trNK activation.
Bifunctional transcription factors are involved in mediating the transcriptome change in trNK induced by the additional IL-21 stimulation
Since chromatin landscape was found to play crucial roles during trNK activation, to unravel the underlying mechanism of additional IL-21-induced trNK further activation, we performed an ATAC-Seq with unstimulated, PMA/ionomycin-stimulated, and additional IL-21-stimulated trNK (Figure 7A). To interpret the additional IL-21 stimulation-induced gene expression changes in trNK, ChARs related to the additional IL-21 stimulation upregulated and downregulated genes were isolated. Among them, the concordant ChARs were further defined, based on two criteria: (1) ChARs related to the upregulated genes showed the highest accessibility or likewise ChARs related to the downregulated genes showed the lowest accessibility and (2) accessibility of these ChARs displayed at least 1.25-fold changes between the additional IL-21-stimulated trNK and the unstimulated or PMA/ionomycin-stimulated trNK (Figures 7B and S7A).40 Accordingly, we identified 294 concordant ChARs for the additional IL-21 stimulation-upregulated genes and 433 concordant ChARs for the additional IL-21 stimulation-downregulated genes (Figure 7C). Although their related genes did not account for a large proportion of the total upregulated or downregulated genes by the additional IL-21 stimulation, their enriched features recapitulated the additional IL-21 stimulation-induced changes in trNK, suggesting that the regulation of these genes might be still important (Figure 7D and 7E). Thus, potential transcription factors for these concordant ChAR-related genes were predicted, based on the appearance of their binding motifs and the expression levels (Figure 7F). Interestingly, using the concordant ChARs for either the additional IL-21 upregulated or downregulated genes, we had identified a similar bunch of transcription factors, including Zbtb17, Zfp281, Sp1, Sp2, Sp3, Egr1, Egr2, E2f4, and Ets1 (Figure 7F). Their expression remained almost unchanged at the different trNK activation conditions, indicating that their regulatory roles were mainly determined by the chromatin accessibility changes (Figure 7G). According to the literature, almost all these transcription factors were bifunctional, playing both transcriptional activation and suppression roles, consistent with the fact that the concordant ChAR-related genes, no matter upregulated or downregulated by the additional IL-21, were regulated by the same bunch of transcription factors (Figure S7B).41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57 Therefore, the additional IL-21 stimulation directs both gene upregulation and downregulation in trNK, using these bifunctional transcription factors.
Figure 7.

Bifunctional transcription factors are involved in mediating the transcriptome change in trNK induced by the additional IL-21 stimulation
(A) Workflow of the ATAC-Seq analysis of ex vivo stimulated liver trNK.
(B) Schematics showing screening of concordant ChARs for the upregulated or downregulated genes by additional IL-21 stimulation in trNK, and their related genes.
(C) Heatmap profiling of concordant ChARs for the upregulated (left) or downregulated (right) genes by additional IL-21 stimulation in trNK.
(D) Pie charts showing proportions of the concordant ChAR-related genes in upregulated (top) or downregulated (bottom) genes by additional IL-21 stimulation in trNK.
(E) Go analysis of the concordant ChAR-related genes upregulated (top) or downregulated (bottom) by additional IL-21 stimulation in trNK.
(F) Scatterplots of potential transcription factors for the concordant ChAR-related genes upregulated (left) or downregulated (right) by additional IL-21 stimulation in trNK, showing both sufficient expression and substantial regulatory roles.
(G) Profiling of each potential transcription factor in (F), showing the percentage of their potentially regulated genes within the upregulated or downregulated genes by additional IL-21 stimulation in trNK in trNK (left) and their expression in trNK under the indicated stimulatory conditions (right).
Discussion
The trNK subgroup has attracted increasing attention in recent years due to their particular roles in initiating immune responses in local tissues.6,7,26 In line with their tissue residency, they usually express certain adhesive molecules, like CD49a.38,58 Nevertheless, such markers cannot always accurately discriminate trNK in peripheral tissues. Here, through an integrative transcriptome comparison of the well-known trNK and cNK subgroups in liver, uterus, and SGs, we have defined two genesets for their discrimination. Though the genesets are generated by the bulk RNA-seq comparison of the two NK subgroups from the three familiar tissues, they are also applicable for discriminating NK subgroups in more tissues and are suitable for scRNA-seq analyses. Thus, in combination with the single-cell technology, discovery of new trNK subgroups in previously unstudied tissues will be easier. In addition, the integrative transcriptome comparison is also mathematically applicable for comparing trNK and cNK of more than three tissues (see STAR Methods). Therefore, with a growing knowledge of the transcriptomic difference between trNK and cNK in more tissues, the trNK and cNK genesets will be further upgraded, in a manner like that of an open-loop machine learning.
Activation of trNK is supposed to be orchestrated in a more precise manner, to avoid an overresponse-induced tissue damages. Consistently, NK cell activation-related features, such as “cell activation”, “cell proliferation”, “lymphocyte differentiation”, and “negative regulation of cell activation”, are significantly enriched in the trNK geneset, whereas such features are not obviously enriched in the cNK geneset. This result also indicates that activation of trNK is determined by a complicated mechanism, including both positive and negative regulations. In agreement, the trNK activation seems to be more difficult than the cNK activation under a unified PMA/ionomycin stimulation condition, as indicated by the milder IFN-γ production. Nevertheless, during infectious challenges, trNK activation can be dramatically enhanced to provide the host sufficient protection at an early stage, indicating that tissue environments also play critical roles in determining the activation difference between trNK and cNK.7,26
To comprehensively elucidate the activation of trNK and cNK, transcriptome profiling of the two NK subgroups under different activation status is performed. During MCMV infection, activated trNK and cNK in liver exhibit a significant difference. And, the trNK and cNK genesets are still applicable in discriminating them, suggesting that their fundamental difference is still maintained after activation. According to the literature, the tissue niches of trNK and cNK in liver are different, which may also lead to the difference in their activation.24 To exclude this interference, we performed another comparison of ex vivo activated trNK and cNK using a unified PMA/ionomycin stimulation condition. In consistent with the ex vivo activation, these ex vivo activated trNK and cNK also display a significant difference in transcriptome, and the trNK and cNK genesets remain applicable for their discrimination. Interestingly, the potential transcription factors involved in promoting gene upregulation in activated trNK are almost also comparably expressed by cNK, suggesting a particularly important role of the accessible chromatin in determining trNK activation. On the other hand, the four cNK-specific transcription factors are preferentially highly expressed in cNK. Thus, these distinct regulatory manners indicate that the activation of trNK and that of cNK are fundamentally different.
In addition, we find that cytokine receptors IL-21R and IL-18R are respectively highly expressed on trNK and cNK, indicating that cytokine milieu also contributes to the difference in their activation. IL-21 or IL-18 mainly exerts accessory roles during the trNK and cNK activation. During PMA/ionomycin stimulation, their presence can further promote IFN-γ production. And, in line with the differential expression of their receptors between the two NK subgroups, additional IL-21 stimulation significantly promotes the trNK activation, while its impact on cNK activation is almost negligible. In contrast, additional IL-18 stimulation dramatically enhances cNK activation, but not the trNK activation. Transcriptome changes of PMA/ionomycin-activated trNK and cNK in presence of the additional IL-21 or IL-18 are also examined. Consistently, additional IL-21 stimulation seems to generate more transcriptome changes in trNK. Moreover, it further enhances the major features enriched in PMA/ionomycin-activated trNK. Together, these results suggest that IL-21 plays a particular role in facilitating trNK activation. An interesting question raised here is whether the IL-21 generated in vivo also has such a regulatory effect to determine the activation status of trNK.
Further, potential transcription factors involved in the additional IL-21-mediated trNK activation are predicted based on ATAC-Seq analysis. With both the additional IL-21 upregulated genes and downregulated genes, a similar bunch of transcription factors are identified, evidencing the reliability of our analysis. These potential transcription factors, Zbtb17, Zfp281, Sp1, Sp2, Sp3, Egr1, Egr2, E2f4, and Ets1, are all reported to exert bifunctional roles in both activating and repressing gene transcription.41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57 Nevertheless, the exact regulatory roles of these transcription factors during trNK activation still need to be further explored in the future. In addition, since these transcription factors only relate with a small proportion of the additional IL-21 stimulation upregulated or downregulated genes in trNK, other regulatory mechanisms during this process are also worth to be explored.
Limitations of the study
To compare the difference between trNK and cNK in activation, PMA/ionomycin stimulatin that can efficiently activate both populations within a short time is utilized in our study. The short-time stimulation also ensures that other features of the cells do not alter too much after leaving their tissue niche. Nevertheless, it also raises another inevitable issue that the PMA/ionomycin stimulation may be too strong compared with the physiological NK activation conditions. In future, the trNK and cNK activation under cytokine stimulation, such as IL-2, IL-7, IL-12, and IL-15, and the impacts of IL-21 and IL-18 during the process, may need to be further compared.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC anti-mouse CD45.2 | Thermo Fisher Scientific | Cat# 11-0454-82; RRID: AB_465061 |
| Brilliant Violet™ 421 anti-mouse CD19 | Thermo Fisher Scientific | Cat# 404-0193-82; RRID: AB_1659676 |
| Brilliant Violet™ 421 anti-mouse CD3ε | Thermo Fisher Scientific | Cat# 404-0031-82; RRID: AB_2925483 |
| eFluor™ 450 anti-mouse CD5 | Thermo Fisher Scientific | Cat# 48-0051-82; RRID: AB_1603250 |
| eFluor™ 450 anti-mouse Ly-6G/Ly6C | Thermo Fisher Scientific | Cat# 48-5931-82; RRID: AB_1548788 |
| NKp46 biotin | Thermo Fisher Scientific | Cat# 13-3351-82; RRID: AB_2572784 |
| APC-eFluor™ 780 anti-mouse NK1.1 | Thermo Fisher Scientific | Cat# 47-5941-82; RRID: AB_2735070 |
| PE anti-mouse CD49a | BioLegend | Cat# 142604; RRID: AB_10945158 |
| APC anti-mouse IL-21R | BioLegend | Cat# 131910; RRID: AB_2124133 |
| APC anti-mouse CD218a (IL-18Rα) | BioLegend | Cat# 157906; RRID: AB_2738885 |
| PE/Cyanine7 anti-mouse CD49b | BioLegend | Cat# 103518; RRID: AB_2566103 |
| FITC anti-mouse IFN-γ | BioLegend | Cat# 505806; RRID: AB_315400 |
| APC anti-mouse TNF-α | BioLegend | Cat# 506308; RRID: AB_315429 |
| Brilliant Violet 711™ anti-mouse CD186 (CXCR6) | BioLegend | Cat# 151111; RRID: AB_2721558 |
| TruStain FcX™ (anti-mouse CD16/32) | BioLegend | Cat# 101302; RRID: AB_312801 |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase type Ⅳ | Sigma-Aldrich | Cat# C4-22-1G |
| DNase I recombinant, RNase-free | Roche | Cat# 4716728001 |
| Brefeldin A | Thermo Fisher Scientific | Cat# 00-4506-51 |
| Fixable Viability Dye eFluor™ 506 | Thermo Fisher Scientific | Cat# 65-0866-14 |
| Brilliant Violet 785™ Streptavidin | BioLegend | Cat# 405249 |
| Recombinant Mouse IL-21 (carrier-free) | BioLegend | Cat# 574506 |
| Recombinant Mouse IL-18 | Sinobiological | Cat# 50073-MNCE |
| Critical commercial assays | ||
| TruePrep DNA Library Prep Kit V2 for Illumina | Vazyme | Cat# 502 |
| SMART-Seq® HT Kit | Takara | Cat# 634437 |
| NEBNext® Ultra™ DNA Library Prep Kit for Illumina® | New England Biolabs | Cat# E7370L |
| Deposited data | ||
| Data files for RNA and ATAC sequencing | This paper | GSE223677 |
| MCMV infection dataset | Quatrini et al.37 | SRP148791 |
| NK ATAC sequencing dataset | Nixon et al.25 | SRP359796 |
| Single cell NK dataset | Lopes et al., 202231 | GSE189807 |
| scRNA-seq of human tonsil NK | Björklund et al.33 | GSE70580 |
| scRNA-seq of human NK in different tissues | Mazzurana L et al.,32 | GSE150050 |
| Software and algorithms | ||
| Flowjo v10 | BD Biosciences | N/A |
| GraphPad Prism v9 | GraphPad Software | N/A |
| Illustrator | Adobe | N/A |
| DESeq2 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | |
| clusterProfiler | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html.org/packages/release/bioc/html/clusterProfiler.html | |
| ssGSEA | https://www.bioconductor.org/packages/release/bioc/vignettes/GSVA/inst/doc/GSVA.html | |
| Cytoscape | https://cytoscape.org | |
| Trimmomatic | https://www.usadellab.org/cms/?page=trimmomatic | |
| PICARD | https://broadinstitute.github.io/picard/ | |
| MACS | https://genomebiology.biomedcentral.com/articles/10.1186/gb-2008-9-9-r137 | |
| HOMER | http://homer.ucsd.edu/homer/ | |
| BEDtool | https://bedtools.readthedocs.io/en/latest/index.html | |
| Seurat | https://github.com/satijalab/seurat | |
| HISAT2 | http://daehwankimlab.github.io/hisat2/ | |
| bowtie2 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml | |
Resource availability
Lead contact
Further information and any related requests should be directed to and will be fulfilled by the lead contact, Chao Zhong (zhongc@pku.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice
Female C57BL/6 mice between 6 to 8 weeks of age were purchased from the Department of Laboratory Animal Science, Peking University Health Science Center (Beijing100191, China). All animals were maintained under specific pathogen-free (SPF) conditions. All experiments were approved by the Ethics Committee of Peking University Health Science Center.
Method details
Cell preparation
To prepare single-cell suspension from liver, the tissue was mechanically grinded and filtered through a 40 μm cell strainer. After centrifugation, the cell pellet was resuspended with 28% Percoll prepared in PBS, followed by another centrifugation at 2000 rpm for 20 min. Then, red blood cells in the pellet were lysed, and the liver cells were washed twice and resuspended in PBS containing 2% FBS. To prepare single-cell suspensions from uterus and SGs, the tissues were firstly cut into small pieces (∼5 mm2), and digested at 37°C in RPMI 1640 containing 1 mg/mL collagenase IV (Sigma-Aldrich), 0.1 mg/mL DNase I (Roche) and 5% fetal bovine serum (FBS) for 40 min. Then, the digested tissues were grinded and filtered through 40 μm cell strainers. After centrifugation, the cell pellets were washed twice and resuspended in PBS containing 2% FBS. To prepare single-cell suspension from spleen, the tissue was mechanically grinded and filtered through a 40 μm cell strainer. Then, splenocytes were washed twice and resuspended in PBS containing 2% FBS.
Cell stimulation
Single-cell suspension of liver was centrifuged and resuspended in RPMI 1640 containing 10% FBS and 10 ng/ml recombinant murine IL-15. Then, the cells were kept unstimulated or stimulated by phorbol 12-myristate 13-acetate (PMA, 50 ng/mL) and ionomycin (1 μg/mL), in presence or absence of IL-21 (20 ng/mL) or IL-18 (10 ng/mL), for 5 hours at 37°C in 5% CO2 atmosphere. A protein transport inhibitor Brefeldin A (Thermo Fisher Scientific) was added after the first hour.
Cell staining
Resuspended cells in PBS containing 2% FBS were incubated with anti-CD16/32 antibody first for Fc receptor blockade. Then, the cells were stained by antibodies to their surface molecules for 30 min at 4°C. For intracellular cytokine staining, the cells were fixed with 4% paraformaldehyde in PBS and permeabilized using 0.1% Triton X-100 in PBS for 10 minutes at room temperature. After washing, cytokine antibodies were added into the cell suspension. Antibodies used for cell staining are, antibodies specific to mouse CD45.2 (104), CD19 (eBio1D3), CD3 (145-2C11), CD5 (53-7.3), Gr-1 (RB6-8C5), NKp46 (29A1.4), NK1.1 (PK136) purchased from Thermo Fisher Scientific, and antibodies specific to mouse CD49a (HMα1), CXCR6 (SA051D1), IL-21R (4A9), IL-18Rα (A17071D), DX5 (DX5), IFN-γ (XMG1.2), TNF-α (MP6-XT22) and CD16/32 (93) purchased from Biolegend.
Flow cytometry
The stained cells were washed twice and resuspended in PBS containing 2% FBS before the analysis. Flow cytometry was performed on LSRFortessa (BD Biosciences). Data were analyzed with FlowJo software (BD Biosciences).
Cell sorting
The stained cells were washed twice and resuspended in RPMI 1640 containing 10% FBS. Cell sorting was performed on FACSAria III (BD Biosciences) using 4-way-purity sort mode.
In liver, trNK were gated based on live Lineage-NK1.1+NKp46+CD49a+DX5-, while cNK were gated based on live Lineage-NK1.1+NKp46+CD49a-DX5+. The liver trNK were also termed as ILC1 by other people, but we prefer to define them as trNK according to their cytotoxicity and localized development.12,14,39
In uterus, trNK were gated based on live Lineage-NK1.1+NKp46+CD49a+CXCR6-, while cNK were gated based on live Lineage-NK1.1+NKp46+CD49a-CXCR6-, according to a previous study.59
In SGs, trNK were gated based on live Lineage-NK1.1+NKp46+CD49a+DX5+, while cNK were gated based on live Lineage-NK1.1+NKp46+CD49a-DX5+.36
RNA-sequencing
NK cells were directly sorted into lysis buffer, and then mRNA reverse transcription and cDNA amplification were performed using a SMART-Seq HT Kit (Takara) according to the manufacturer’s instruction. Libraries were prepared with NEB Next Ultra DNA Prep Kit (New England Biolabs). Quantity and quality of the libraries were assessed on Agilent 2100 Bioanalyzer system. Library sequencing was performed at paired-end 150 bp on Illumina NovaSeq platform.
ATAC-sequencing
10,000 sort purified cells were collected and pelleted by centrifugation at 600 g for 10 min. The pellets were resuspended in 50 μL lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40) and kept on ice for 3 minutes, followed by a centrifugation at 600 g for 10 min to collect nuclei. Then, nuclei were resuspended in transposition reaction buffer. DNA fragmentation and library preparation were performed using TruePrep DNA Library Prep Kit V2 for Illumina (Vazyme) according to the manufacturer’s instruction. Quantity and quality of libraries were assessed on Agilent 2100 Bioanalyzer system. Library sequencing was performed at paired-end 150 bp on Illumina NovaSeq platform.
Quantification and statistical analysis
Public data acquisition
Raw data for gene expression profiles (RNA-Seq) of MCMV infected liver trNK and cNK were retrieved from Sequence Read Archive (SRA) under accession number SRP148791. Chromatin accessibility profiles (ATAC-Seq) of mouse liver trNK and cNK were retrieved from SRA under accession number SRP359796. Single-cell gene expression profiles (scRNA-Seq) of NK cells collected from different mouse tissues were retrieved from Gene Expression Omnibus (GEO) under accession numbers GSE189807.
RNA-Seq data processing
RNA-Seq reads in fastq format were aligned to mm10 assembly of mouse genome using HISAT2, quantified by RSEM. Transcripts-per-million (TPM) values for gene-level counts were calculated using R-package scuttle. Genes with confirmed expression were selected based on TPM> 10 in all repeats of a group, and were used for further analyses. Similarities between different NK subgroups were analyzed by principal component analysis (PCA) and Pearson correlation. Differential gene expression analysis was performed by DESeq2 pipeline. Gene Set Enrichment Analysis (GSEA) was performed using R-package fgsea, and normalized enrichment scores (NES) and P values were calculated. Gene Ontology (GO) analysis was performed by over-representation test with clusterProfiler package. To assess pathway activity in different NK subgroups, a non-parametric and unsupervised software algorithm single-sample GSEA (ssGSEA) in GSVA package was used. Transcriptional regulatory network between transcription factors and enriched pathways was constructed with Cytoscape.
Identification of trNK and cNK genesets by integrative transcriptome comparison
To integrate transcriptome comparisons of trNK and cNK in different tissues, gene expression change (GEC) between the two NK subgroups (log2 trNK/cNK) in each tissue was assigned to each axis of a coordinate system. Particularly, here GECs of trNK and cNK in liver, uterus and SGs were respectively assigned to x, y and z axis of a three-dimensional (3D) coordinate system (see Figures 1C and 1D). To calculate average GEC of trNK and cNK (GECave) and variation of GECs among all tissues (VGEC), a diagonal reference line (x = y = z) was introduced. GECave was defined by the distance from the origin to the projection of a gene on the reference line, while VGEC was defined by the projection distance of a gene to the reference line. After calculation, GECave equaled the mean GEC in these tissues, (GECliver + GECuterus + GECSGs)/3, and VGEC representing the Euclidean distance from a gene’s coordinate (GECliver, GECuterus, GECSGs) to its projected point on the reference line (GECave, GECave, GECave) equaled [(GECliver - GECave)2 + (GECuterus - GECave)2 +(GECSGs - GECave)2]1/2. Mathematically, this method could also be applied to integrative transcriptome comparison of trNK and cNK in more tissues, based on the following function.
Finally, trNK geneset was defined by GECave > 1 and VGEC < GECave, while cNK geneset was defined by GECave < -1 and VGEC < -GECave. The comparison result could be better visualized in a 2D coordinate system, with GECave assigned to x axis and VGEC assigned to y axis.
Single-cell RNA-Seq data processing
Raw count matrix of single-cell RNA-Seq (scRNA-Seq) was processed using R-package Seurat to obtain qualified cells with normalized gene expression. Single-cell clustering was performed with shared nearest neighbors (SNN) algorithm, and dimension reduction was operated with Uniform Manifold Approximation and Projection (UMAP) algorithm. Module scores for gene expression in single cell was calculated with AddModuleScore function in Seurat. Exact classification of NK cells in each tissue was performed using FindClusters function in Seurat.
ATAC-Seq data processing
ATAC-Seq quality trimming and primer removal were performed with Trimmomatic, using the following parameters: LEADING:15, TRAILING:15, SLIDINGWINDOW:4:15, MINLEN:36. Trimmed reads were aligned to mm10 assembly of mouse genome using Bowtie2. Then, the aligned reads were sorted using samtools, and duplicates were removed using PICARD. Peak-calling for ATAC-Seq was performed with MACS on bam files, based on a q-value threshold of 0.01. Consensus peaks from all NK subgroups were merged to create a raw peak universe. Genomic regions were annotated to their neighboring genes with HOMER. ATAC-Seq reads in each peak were quantified using BEDtool. TPM values for peak-level counts were calculated with R-package scuttle.
Concordant chromatin accessible regions (ChARs) were defined as ChARs exhibiting concordant accessibility changes (fold change > 1.25) with the expression changes (fold change > 2) of their annotated genes. GO analysis for concordant ChARs was conducted using their annotated genes.
Motif analysis
Motif analysis to obtain gene-transcription factor (TF) pairs was performed by MEME Suit. Proportion of genes in a geneset (> 15%) possessing binding motifs of a specific TF and expression of the TF (TPM > 10) were both considered to identify potential TFs for the geneset.
Statistics
Data were analyzed by GraphPad Prism Software. Mean ± SEM are presented in figures. P. values were calculated by unpaired t-test. P. values above 0.05 were considered not significant, ∗P< 0.05, ∗∗P< 0.01, ∗∗∗P< 0.001, ∗∗∗∗P< 0.0001.
Acknowledgments
We thank the animal and flow cytometry facilities at Peking University Institute of Systems Biomedicine for technical support. We thank members of the Zhong group for inspiring discussion and all contributions. This study is supported by funding from the National Key Research and Development Program of China (2022YFA1103602, 2022YFA0806400), the National Natural Science Foundation of China (No. 31770957, No. 91842102, No. 32170896), the Natural Science Foundation of Beijing (No. 18G10645), and the Shenzhen Innovation Committee of Science and Technology (JCYJ20220818100401003).
Author contributions
C.Z. conceived the project. L.H. performed bioinformatic analyses. M.H. performed sequencing library preparation and flow cytometry analysis. Y.D. helped with cell sorting. J.G., Z.H., Y.Z., Y.Z., and J.H. helped with some experiments. L.H., M.H., and C.Z. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: June 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107187.
Supplemental information
Data and code availability
-
•
The sequencing data analyzed in this study are deposited in GEO or SRA and are publicly available. Accession numbers are listed in the key resources table.
-
•
The paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Mujal A.M., Delconte R.B., Sun J.C. Natural Killer Cells: From Innate to Adaptive Features. Annu. Rev. Immunol. 2021;39:417–447. doi: 10.1146/annurev-immunol-101819-074948. [DOI] [PubMed] [Google Scholar]
- 2.Horowitz A., Stegmann K.A., Riley E.M. Activation of natural killer cells during microbial infections. Front. Immunol. 2011;2:88. doi: 10.3389/fimmu.2011.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Long E.O., Kim H.S., Liu D., Peterson M.E., Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013;31:227–258. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sojka D.K., Plougastel-Douglas B., Yang L., Pak-Wittel M.A., Artyomov M.N., Ivanova Y., Zhong C., Chase J.M., Rothman P.B., Yu J., et al. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. Elife. 2014;3:e01659. doi: 10.7554/eLife.01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou J., Tian Z., Peng H. Tissue-resident NK cells and other innate lymphoid cells. Adv. Immunol. 2020;145:37–53. doi: 10.1016/bs.ai.2019.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Shannon J.P., Vrba S.M., Reynoso G.V., Wynne-Jones E., Kamenyeva O., Malo C.S., Cherry C.R., McManus D.T., Hickman H.D. Group 1 innate lymphoid-cell-derived interferon-gamma maintains anti-viral vigilance in the mucosal epithelium. Immunity. 2021;54:276–290.e5. doi: 10.1016/j.immuni.2020.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weizman O.E., Adams N.M., Schuster I.S., Krishna C., Pritykin Y., Lau C., Degli-Esposti M.A., Leslie C.S., Sun J.C., O'Sullivan T.E. ILC1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell. 2017;171:795–808.e12. doi: 10.1016/j.cell.2017.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams N.M., Sun J.C. Spatial and temporal coordination of antiviral responses by group 1 ILCs. Immunol. Rev. 2018;286:23–36. doi: 10.1111/imr.12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun J.C., Lanier L.L. NK cell development, homeostasis and function: parallels with CD8(+) T cells. Nat. Rev. Immunol. 2011;11:645–657. doi: 10.1038/nri3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diefenbach A., Colonna M., Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity. 2014;41:354–365. doi: 10.1016/j.immuni.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eberl G., Colonna M., Di Santo J.P., McKenzie A.N.J. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science. 2015;348:aaa6566. doi: 10.1126/science.aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai L., Vienne M., Tang L., Kerdiles Y., Etiennot M., Escalière B., Galluso J., Wei H., Sun R., Vivier E., et al. Liver type 1 innate lymphoid cells develop locally via an interferon-gamma-dependent loop. Science. 2021;371:eaba4177. doi: 10.1126/science.aba4177. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y., Wang X., Hao X., Li B., Tao W., Zhu S., Qu K., Wei H., Sun R., Peng H., Tian Z. Ly49E separates liver ILC1s into embryo-derived and postnatal subsets with different functions. J. Exp. Med. 2022;219:e20211805. doi: 10.1084/jem.20211805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Censo C., Marotel M., Mattiola I., Müller L., Scarno G., Pietropaolo G., Peruzzi G., Laffranchi M., Mazej J., Hasim M.S., et al. Granzyme A and CD160 expression delineates ILC1 with graded functions in the mouse liver. Eur. J. Immunol. 2021;51:2568–2575. doi: 10.1002/eji.202149209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ducimetière L., Lucchiari G., Litscher G., Nater M., Heeb L., Nuñez N.G., Wyss L., Burri D., Vermeer M., Gschwend J., et al. Conventional NK cells and tissue-resident ILC1s join forces to control liver metastasis. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2026271118. e2026271118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang L., Peng H., Zhou J., Chen Y., Wei H., Sun R., Yokoyama W.M., Tian Z. Differential phenotypic and functional properties of liver-resident NK cells and mucosal ILC1s. J. Autoimmun. 2016;67:29–35. doi: 10.1016/j.jaut.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Geiger T.L., Sun J.C. Development and maturation of natural killer cells. Curr. Opin. Immunol. 2016;39:82–89. doi: 10.1016/j.coi.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao Y., Souza-Fonseca-Guimaraes F., Bald T., Ng S.S., Young A., Ngiow S.F., Rautela J., Straube J., Waddell N., Blake S.J., et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017;18:1004–1015. doi: 10.1038/ni.3800. [DOI] [PubMed] [Google Scholar]
- 19.Park E., Patel S., Wang Q., Andhey P., Zaitsev K., Porter S., Hershey M., Bern M., Plougastel-Douglas B., Collins P., et al. Toxoplasma gondii infection drives conversion of NK cells into ILC1-like cells. Elife. 2019;8:e47605. doi: 10.7554/eLife.47605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson K.G., Mayer-Barber K., Sung H., Beura L., James B.R., Taylor J.J., Qunaj L., Griffith T.S., Vezys V., Barber D.L., Masopust D. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 2014;9:209–222. doi: 10.1038/nprot.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson K.G., Sung H., Skon C.N., Lefrancois L., Deisinger A., Vezys V., Masopust D. Cutting edge: intravascular staining redefines lung CD8 T cell responses. J. Immunol. 2012;189:2702–2706. doi: 10.4049/jimmunol.1201682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamran P., Sereti K.I., Zhao P., Ali S.R., Weissman I.L., Ardehali R. Parabiosis in mice: a detailed protocol. J. Vis. Exp. 2013 doi: 10.3791/50556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright D.E., Wagers A.J., Gulati A.P., Johnson F.L., Weissman I.L. Physiological migration of hematopoietic stem and progenitor cells. Science. 2001;294:1933–1936. doi: 10.1126/science.1064081. [DOI] [PubMed] [Google Scholar]
- 24.Gasteiger G., Fan X., Dikiy S., Lee S.Y., Rudensky A.Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. 2015;350:981–985. doi: 10.1126/science.aac9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nixon B.G., Chou C., Krishna C., Dadi S., Michel A.O., Cornish A.E., Kansler E.R., Do M.H., Wang X., Capistrano K.J., et al. Cytotoxic granzyme C-expressing ILC1s contribute to antitumor immunity and neonatal autoimmunity. Sci. Immunol. 2022;7:eabi8642. doi: 10.1126/sciimmunol.abi8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klose C.S.N., Flach M., Möhle L., Rogell L., Hoyler T., Ebert K., Fabiunke C., Pfeifer D., Sexl V., Fonseca-Pereira D., et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. 2014;157:340–356. doi: 10.1016/j.cell.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 27.Sun H., Sun C., Xiao W., Sun R. Tissue-resident lymphocytes: from adaptive to innate immunity. Cell. Mol. Immunol. 2019;16:205–215. doi: 10.1038/s41423-018-0192-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan X., Rudensky A.Y. Hallmarks of Tissue-Resident Lymphocytes. Cell. 2016;164:1198–1211. doi: 10.1016/j.cell.2016.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huntington N.D., Cursons J., Rautela J. The cancer-natural killer cell immunity cycle. Nat. Rev. Cancer. 2020;20:437–454. doi: 10.1038/s41568-020-0272-z. [DOI] [PubMed] [Google Scholar]
- 30.Gotthardt D., Trifinopoulos J., Sexl V., Putz E.M. JAK/STAT Cytokine Signaling at the Crossroad of NK Cell Development and Maturation. Front. Immunol. 2019;10:2590. doi: 10.3389/fimmu.2019.02590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lopes N., Galluso J., Escalière B., Carpentier S., Kerdiles Y.M., Vivier E. Tissue-specific transcriptional profiles and heterogeneity of natural killer cells and group 1 innate lymphoid cells. Cell Rep. Med. 2022;3:100812. doi: 10.1016/j.xcrm.2022.100812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mazzurana L., Czarnewski P., Jonsson V., Wigge L., Ringnér M., Williams T.C., Ravindran A., Björklund Å.K., Säfholm J., Nilsson G., et al. Tissue-specific transcriptional imprinting and heterogeneity in human innate lymphoid cells revealed by full-length single-cell RNA-sequencing. Cell Res. 2021;31:554–568. doi: 10.1038/s41422-020-00445-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bjorklund A.K., Forkel M., Picelli S., Konya V., Theorell J., Friberg D., Sandberg R., Mjosberg J. The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing (vol 17, pg 451, 2016) Nat. Immunol. 2016;17:740. doi: 10.1038/ni0616-740a. [DOI] [PubMed] [Google Scholar]
- 34.Yomogida K., Bigley T.M., Trsan T., Gilfillan S., Cella M., Yokoyama W.M., Egawa T., Colonna M. Hobit confers tissue-dependent programs to type 1 innate lymphoid cells. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2117965118. e2117965118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mackay L.K., Minnich M., Kragten N.A.M., Liao Y., Nota B., Seillet C., Zaid A., Man K., Preston S., Freestone D., et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. 2016;352:459–463. doi: 10.1126/science.aad2035. [DOI] [PubMed] [Google Scholar]
- 36.Filipovic I., Chiossone L., Vacca P., Hamilton R.S., Ingegnere T., Doisne J.M., Hawkes D.A., Mingari M.C., Sharkey A.M., Moretta L., Colucci F. Molecular definition of group 1 innate lymphoid cells in the mouse uterus. Nat. Commun. 2018;9:4492. doi: 10.1038/s41467-018-06918-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quatrini L., Wieduwild E., Escaliere B., Filtjens J., Chasson L., Laprie C., Vivier E., Ugolini S. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat. Immunol. 2018;19:954–962. doi: 10.1038/s41590-018-0185-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng H., Jiang X., Chen Y., Sojka D.K., Wei H., Gao X., Sun R., Yokoyama W.M., Tian Z. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J. Clin. Invest. 2013;123:1444–1456. doi: 10.1172/Jci66381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friedrich C., Taggenbrock R.L.R.E., Doucet-Ladevèze R., Golda G., Moenius R., Arampatzi P., Kragten N.A.M., Kreymborg K., Gomez de Agüero M., Kastenmüller W., et al. Effector differentiation downstream of lineage commitment in ILC1s is driven by Hobit across tissues. Nat. Immunol. 2021;22:1256–1267. doi: 10.1038/s41590-021-01013-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu L., Zhao X., Li P., Zeng Y., Zhang Y., Shen Y., Wang Y., Sun X., Lai B., Zhong C. Proximal and Distal Regions of Pathogenic Th17 Related Chromatin Loci Are Sequentially Accessible During Pathogenicity of Th17. Front. Immunol. 2022;13:864314. doi: 10.3389/fimmu.2022.864314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bakovic M., Waite K.A., Vance D.E. Functional significance of Sp1, Sp2, and Sp3 transcription factors in regulation of the murine CTP:phosphocholine cytidylyltransferase alpha promoter. J. Lipid Res. 2000;41:583–594. [PubMed] [Google Scholar]
- 42.Collins S., Lutz M.A., Zarek P.E., Anders R.A., Kersh G.J., Powell J.D. Opposing regulation of T cell function by Egr-1/NAB2 and Egr-2/Egr-3. Eur. J. Immunol. 2008;38:528–536. doi: 10.1002/eji.200737157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng Y., Desjardins C.A., Cooper O., Kontor A., Nocco S.E., Naya F.J. EGR1 Functions as a Potent Repressor of MEF2 Transcriptional Activity. PLoS One. 2015;10:e0127641. doi: 10.1371/journal.pone.0127641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fidalgo M., Shekar P.C., Ang Y.S., Fujiwara Y., Orkin S.H., Wang J. Zfp281 functions as a transcriptional repressor for pluripotency of mouse embryonic stem cells. Stem Cell. 2011;29:1705–1716. doi: 10.1002/stem.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo J., Xue Z., Ma R., Yi W., Hui Z., Guo Y., Yao Y., Cao W., Wang J., Ju Z., et al. The transcription factor Zfp281 sustains CD4(+) T lymphocyte activation through directly repressing Ctla-4 transcription. Cell. Mol. Immunol. 2020;17:1222–1232. doi: 10.1038/s41423-019-0289-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsu J., Arand J., Chaikovsky A., Mooney N.A., Demeter J., Brison C.M., Oliverio R., Vogel H., Rubin S.M., Jackson P.K., Sage J. E2F4 regulates transcriptional activation in mouse embryonic stem cells independently of the RB family. Nat. Commun. 2019;10:2939. doi: 10.1038/s41467-019-10901-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsu J., Sage J. Novel functions for the transcription factor E2F4 in development and disease. Cell Cycle. 2016;15:3183–3190. doi: 10.1080/15384101.2016.1234551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee C.G., Kwon H.K., Kang H., Kim Y., Nam J.H., Won Y.H., Park S., Kim T., Kang K., Rudra D., et al. Ets1 suppresses atopic dermatitis by suppressing pathogenic T cell responses. JCI Insight. 2019;4:e124202. doi: 10.1172/jci.insight.124202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mavrothalassitis G., Ghysdael J. Proteins of the ETS family with transcriptional repressor activity. Oncogene. 2000;19:6524–6532. doi: 10.1038/sj.onc.1204045. [DOI] [PubMed] [Google Scholar]
- 50.Möröy T., Saba I., Kosan C. The role of the transcription factor Miz-1 in lymphocyte development and lymphomagenesis-Binding Myc makes the difference. Semin. Immunol. 2011;23:379–387. doi: 10.1016/j.smim.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 51.Rashkovan M., Vadnais C., Ross J., Gigoux M., Suh W.K., Gu W., Kosan C., Möröy T. Miz-1 regulates translation of Trp53 via ribosomal protein L22 in cells undergoing V(D)J recombination. Proc. Natl. Acad. Sci. USA. 2014;111:E5411–E5419. doi: 10.1073/pnas.1412107111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Staller P., Peukert K., Kiermaier A., Seoane J., Lukas J., Karsunky H., Möröy T., Bartek J., Massagué J., Hänel F., Eilers M. Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 2001;3:392–399. doi: 10.1038/35070076. [DOI] [PubMed] [Google Scholar]
- 53.Shen N., Jiang S., Lu J.M., Yu X., Lai S.S., Zhang J.Z., Zhang J.L., Tao W.W., Wang X.X., Xu N., et al. The Constitutive Activation of Egr-1/C/EBPa Mediates the Development of Type 2 Diabetes Mellitus by Enhancing Hepatic Gluconeogenesis. Am. J. Pathol. 2015;185:513–523. doi: 10.1016/j.ajpath.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 54.Phan D., Cheng C.J., Galfione M., Vakar-Lopez F., Tunstead J., Thompson N.E., Burgess R.R., Najjar S.M., Yu-Lee L.Y., Lin S.H. Identification of Sp2 as a transcriptional repressor of carcinoembryonic antigen-related cell adhesion molecule 1 in tumorigenesis. Cancer Res. 2004;64:3072–3078. doi: 10.1158/0008-5472.Can-03-3730. [DOI] [PubMed] [Google Scholar]
- 55.DeLuca P., Majello B., Lania L. Sp3 represses transcription when tethered to promoter DNA or targeted to promoter proximal RNA. J. Biol. Chem. 1996;271:8533–8536. doi: 10.1074/jbc.271.15.8533. [DOI] [PubMed] [Google Scholar]
- 56.Kumbrink J., Kirsch K.H., Johnson J.P. EGR1, EGR2, and EGR3 Activate the Expression of Their Coregulator NAB2 Establishing a Negative Feedback Loop in Cells of Neuroectodermal and Epithelial Origin. J. Cell. Biochem. 2010;111:207–217. doi: 10.1002/jcb.22690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Law A.Y.S., Yeung B.H.Y., Ching L.Y., Wong C.K.C. Sp1 Is a Transcription Repressor to Stanniocalcin-1 Expression in TSA-Treated Human Colon Cancer Cells, HT29. J. Cell. Biochem. 2011;112:2089–2096. doi: 10.1002/jcb.23127. [DOI] [PubMed] [Google Scholar]
- 58.Tang L., Peng H., Zhou J., Chen Y., Wei H., Sun R., Yokoyama W.M., Tian Z. Differential phenotypic and functional properties of liver-resident NK cells and mucosal ILC1s. J. Autoimmun. 2016;67:29–35. doi: 10.1016/j.jaut.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 59.Erick T.K., Anderson C.K., Reilly E.C., Wands J.R., Brossay L. NFIL3 Expression Distinguishes Tissue-Resident NK Cells and Conventional NK-like Cells in the Mouse Submandibular Glands. J. Immunol. 2016;197:2485–2491. doi: 10.4049/jimmunol.1601099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The sequencing data analyzed in this study are deposited in GEO or SRA and are publicly available. Accession numbers are listed in the key resources table.
-
•
The paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.