

Abstract
BACKGROUND:
Macrophage activation plays a critical role in abdominal aortic aneurysm (AAA) development. However, molecular mechanisms controlling macrophage activation and vascular inflammation in AAA remain largely unknown. The objective of the study was to identify novel mechanisms underlying adenosine deaminase acting on RNA (ADAR1) function in macrophage activation and AAA formation.
METHODS:
Aortic transplantation was conducted to determine the importance of non-vascular ADAR1 in AAA development/dissection. Angiotensin II (Ang II) infusion of ApoE−/− mouse model combined with macrophage-specific knockout of ADAR1 was used to study ADAR1 macrophage-specific role in AAA formation/dissection. The relevance of macrophage ADAR1 to human AAA was examined using human aneurysm specimens. Moreover, a novel humanized AAA model was established to test the role of human macrophages in aneurysm formation in human arteries.
RESULTS:
Allograft transplantation of wild type abdominal aortas to ADAR1+/− recipient mice significantly attenuated AAA formation, suggesting that non-vascular ADAR1 is essential for AAA development. ADAR1 deficiency in hematopoietic cells decreased the prevalence and severity of AAA while inhibited macrophage infiltration and aorta wall inflammation. ADAR1 deletion blocked the classic macrophage activation, diminished NF-κB signaling, and enhanced the expression of a number of anti-inflammatory miRNAs. Mechanistically, ADAR1 interacted with Drosha to promote its degradation, which attenuated Drosha-DGCR8 interaction, and consequently inhibited pri- to pre-miRNA processing of microRNAs targeting IKKβ, resulting in an increased IKKβ expression and enhanced NF-κB signaling. Significantly, ADAR1 was induced in macrophages and interacted with Drosha in human AAA lesions. Reconstitution of ADAR1-deficient, but not the wild type, human monocytes to immunodeficient mice blocked the aneurysm formation in transplanted human arteries.
CONCLUSIONS:
Macrophage ADAR1 promotes aneurysm formation in both mouse and human arteries through a novel mechanism, i.e., Drosha protein degradation, which inhibits the processing of miRNAs targeting NF-kB signaling and thus elicits macrophage-mediated vascular inflammation in AAA.
Keywords: Adenosine deaminases acting on RNA1, Abdominal aortic aneurysm, macrophage, MicroRNA, humanized AAA model
Keywords: Vascular Biology, Aneurysm, Macrophage
Introduction
Aortic aneurysm is a bulging of the weakened area in the aortic wall, characterized by focal dilatation of the blood vessels. Aneurysms occurring in abdominal infrarenal aorta are known as abdominal aortic aneurysm (AAA) 1. AAA is closely associated with high morbidity and mortality in elderly population due to aortic dissection and rupture 2, representing a serious healthcare challenge worldwide. AAA is a chronic inflammatory disease involving inflammatory cell infiltration in adventitial and medial layers 3. The pathologic process of AAA is also associated with alteration and loss of vascular smooth muscle cells (SMC) as well as extracellular matrix remodeling. Current treatment is limited to surgical repair for eligible patients 4,5. No effective drug is currently available for AAA treatment, due to the limited understanding of molecular mechanisms governing the AAA development and progression.
Macrophages, as a critical player in immune system, give rise to diverse populations with phenotypic plasticity and functions under different pathophysiological conditions 6. The classically activated (M1) macrophages produce high levels of pro-inflammatory cytokines such as interleukin-1β (IL-1β), IL-6, TNFα, and inducible nitric oxide synthase (iNOS) 7. As key inflammatory mediators, M1 macrophages play critical roles in the pathogenesis of aortic aneurysm 8–10. Macrophages are present in aneurysmal lesions of human aortic walls and animal models of AAA 11–13. The macrophages accumulated in the adventitia of ApoE−/− mouse aorta with angiotensin II (Ang II) infusion are identified mainly as M1 population 14. Consistently, high levels of pro-inflammatory cytokines are detected in AAA tissues, which promote SMC phenotypic alteration and elastin degradation15,16. Inhibition of inflammatory cytokines, e.g., IL-1β and iNOS, has significantly reduced AAA formation 17,18. Despite a broad appreciation of aneurysm being an inflammation-related disease, the mechanisms regulating macrophage functions in AAA pathogenesis have not been sufficiently addressed.
Adenosine deaminase acting on RNA (ADAR) is a RNA editing enzyme catalyzing the conversion of adenosine to inosine 19. Among three members (ADAR1, 2 and 3) identified in mammalian cells, ADAR1 displays most versatile roles in physiological and pathological conditions 20–22. ADAR1-mediated A-to-I RNA editing alters RNA structure and gene coding sequence of proteins 23. Recent studies have also identified editing-independent functions of ADAR1 such as RNA binding 24,25.
Although ADAR1 in inflammation has been studied by several different groups, its functions in inflammation remain controversial. A few reports suggest an anti-inflammatory function of ADAR1 26–28, but the majority of the published studies show that ADAR1 promotes inflammatory response 21,29–33. However, the molecular mechanisms underlying ADAR1 functions in inflammation remain largely unknown. The current study is aimed to elucidate a new mechanism controlling ADAR1 function in activating macrophages in AAA development.
Our previous studies have shown that ADAR1 regulates AAA formation/dissection by modulating SMC phenotype 34. However, ADAR1+/− mice receiving WT donor aorta grafts with intact vascular ADAR1 are protected from AAA development, suggesting that hematopoietic ADAR1 is essential for AAA formation. Since macrophage activation and the related pro-inflammatory cytokine production occur at the initial phase of the AAA development and induce the transformation of inflammatory SMC phenotype 3,35, we used hematopoietic ADAR1-deficient mice to verify the role of macrophage ADAR1 in AAA formation. We found that hematopoietic ADAR1 deficiency significantly attenuates AAA development in mice. Mechanistically, ADAR1 inhibits the processing of primary to precursor microRNA (miRNAs) through a previously unknown editing-independent mechanism, which causes reduction of mature miRNAs that target nuclear factor-κB (NF-κB) signaling in inflammatory macrophages. More importantly, transfer of ADAR1-deficient human peripheral blood mononuclear cell (PBMCs) to immunodeficient mice significantly attenuates AAA formation in human arteries, indicating that ADAR1 is a novel protein factor essential for AAA development in human.
Methods
Animal procedures were performed in accordance with the Institutional Animal Care and Use Committee at the University of Missouri. All supporting data, analytical methods, and study materials developed from this group will be made available to other researchers for purposes of reproducing results or replicating the procedures. A detailed description of the methods and materials are provided in the Supplemental Methods in the Supplemental Materials.
Statistical analysis
All experiments were repeated at least for three times. All data represent independent data points but not technical replicates. Data are presented as the mean ± SD. Normality of data was assessed by the D’Agostino & Pearson normality test with alpha=0.05. For comparisons of two groups, student’s unpaired two-tailed t test was used for normally distributed data, and Mann-Whitney two tailed test was used for non-normally distributed data or for groups with n less than 7. For more than 2 groups, 1-way ANOVA with Tukey post-test analysis was used for normally distributed data, and Kruskal-Wallis test with Dunn’s multiple comparisons test was used for non-normally distributed data. Prism 9.0 (GraphPad Software, CA) or RStudio (Desktop 1.4.1717) was used for statistical analyses, and differences considered statistically significant when nominal P<0.05 or adjusted P<0.05 in case of multiple testing. However, the correcting for multiple testing across the entire body of the studies was not performed because both in vitro and in vivo experiments were performed, and various approaches were used in this study.
Results
Nonvascular ADAR1 is essential for AAA formation.
Although our previous study has shown that SMC ADAR1 is important for AAA formation, immune cell infiltration and inflammation are earlier events which triggers the later SMC phenotype modulation during AAA development 3,35. In order to test if ADAR1 in nonvascular cells plays a role in AAA formation, we performed a heterotopic transplantation by grafting abdominal aortas of ApoE−/− mice (wild type or WT) to WT or ADAR1+/− mice in ApoE−/− background (Figure 1A). The recipient aortas were ligated to detour blood flow to the donor aortas as previously described 34. One week later, Ang II was infused in these mice through an osmotic minipump for 28 days, and ultrasound was performed to observe the AAA formation and measure donor aorta diameters (Figure 1B). We observed a 24% mortality rate in WT recipient mice transplanted with WT aorta. However, ADAR1+/− recipient mice transplanted with WT aorta only showed 10% of mortality rate. Some of the mortality was likely due to post-anastomosis complications. The diameters of donor aortas in ADAR1+/− recipient mice were significantly smaller than that of WT recipient mice (Figure 1C). Verhoeff elastic staining of aorta sections showed significant dilation, media degeneration, and elastin fragmentation in donor aortas transplanted to the WT mice. However, these defects were attenuated in the aortas transplanted to the ADAR1+/− recipient mice (Figure 1D). As a result, the elastin degradation scores were significantly reduced in aortas transplanted to ADAR1+/− mice (Figure 1E). These results indicated that recipient nonvascular cell ADAR1 is essential for AAA formation in Ang II-infused WT mice.
Figure 1: Non-vascular ADAR1 contributed to AAA formation.
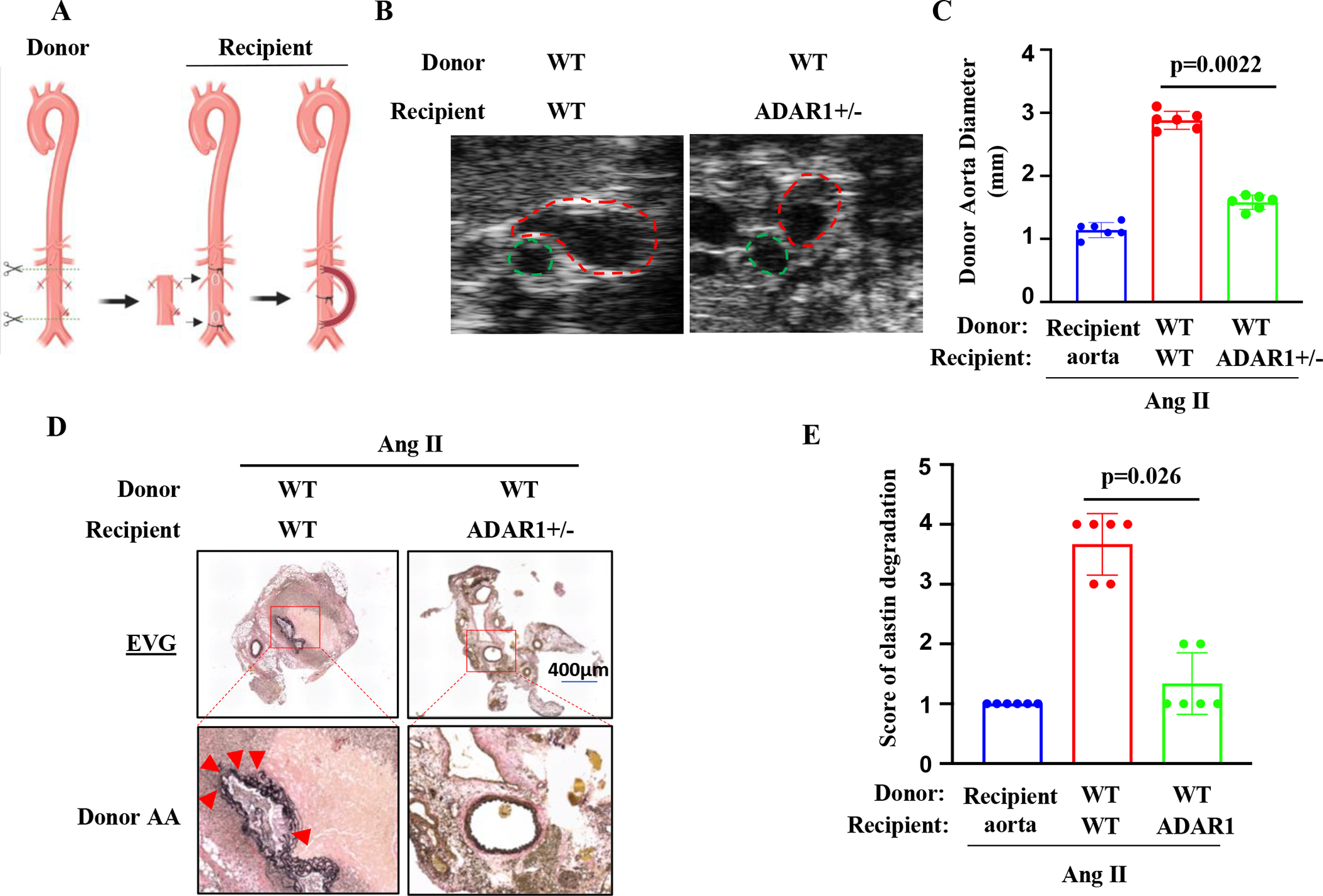
A, A graphic illustration of the mouse aortic transplant. Donor aortic segment was anastomosed to the recipient abdominal aorta in an end-to-side manner. The recipient aorta was ligated to diverge the blood flow through the donor aorta. Recipient mice were then infused with Ang II (1000 ng/kg/min) for 28 days. B, Representative transverse ultrasound images of abdominal aorta 28 days after the Ang II infusion. Red dash circles: donor aortas; Green dash circles: recipient’s own aortas. C, Maximal external diameters of donor aortas were measured by ultrasound imaging. P=0.0022, wild-type (WT) aortas to ADAR+/− vs. to WT recipients, n=6. D Representative images of Verhoeff’s elastic (EVG) staining of the transplanted aortas with aneurysm. The areas in the red boxes were shown with a higher magnification (4.2 folds) in the lower part of the panel. Arrows indicate elastin breaks. E: Quantification of elastin breaks in the donor aortas of Ang II-infused WT and ADAR1+/− recipient mice. P = 0.026, wild-type (WT) donor aortas to ADAR+/− vs. to WT recipients, n=6. Kruskal-Wallis test with Dunn multiple comparisons test was performed for C and E.
ADAR1 is predominantly expressed in macrophages in early-stage AAA lesions of the donor aortas.
To determine which ADAR1-expressing nonvascular cells are essential for the AAA formation, we collected donor aortas transplanted to the WT and ADAR1+/− mice that were infused with Ang II for 7 days. Cells in aortas were dissociated with enzymatic digestion and then immuno-stained with different antibodies followed by flow cytometry analyses to measure ADAR1 expression in different cell populations. Cells were gated by their expression of cell specific markers: CD45+CD11b+ for monocyte-derived macrophages, CD45-CD31+ for endothelial cells; CD45-CD31-FSP+ for fibroblasts; and CD45-α-SMA+ for SMCs. Interestingly, ADAR1 expression in endothelial cells, fibroblasts and SMCs did not show significant difference between the donor aortas transplanted to WT and ADAR1+/− recipient mice (Figure 2, A–B). However, ADAR1 levels were dramatically higher in monocyte-derived macrophages in donor aortas transplanted to WT as compared to the ADAR1+/− recipient mice (Figure 2, A–B, Figure S1). Immunostaining confirmed that ADAR1 was significantly induced in F4/80+ cells in the AAA lesion of donor aortas transplanted to the WT mice (Figure 2C). Consequently, ADAR1+F4/80+ macrophages were significantly more in donor aortas transplanted to WT mice than to ADAR+/− mice, suggesting that macrophage ADAR1 may play a critical role in the AAA development in donor aortas transplanted to the WT mice. Notably, ADAR1 was also expressed in SMCs in donor aortas, consistent with our previous report 34. The reason why SMC ADAR1 levels are similar in the two groups is because both donor aortas are WT, and thus their SMCs express ADAR1 similarly when the mice were infused with Ang II.
Figure 2: ADAR1 was highly expressed in monocyte-derived macrophages in wild type (WT), but not the ADAR+/− recipient mice.
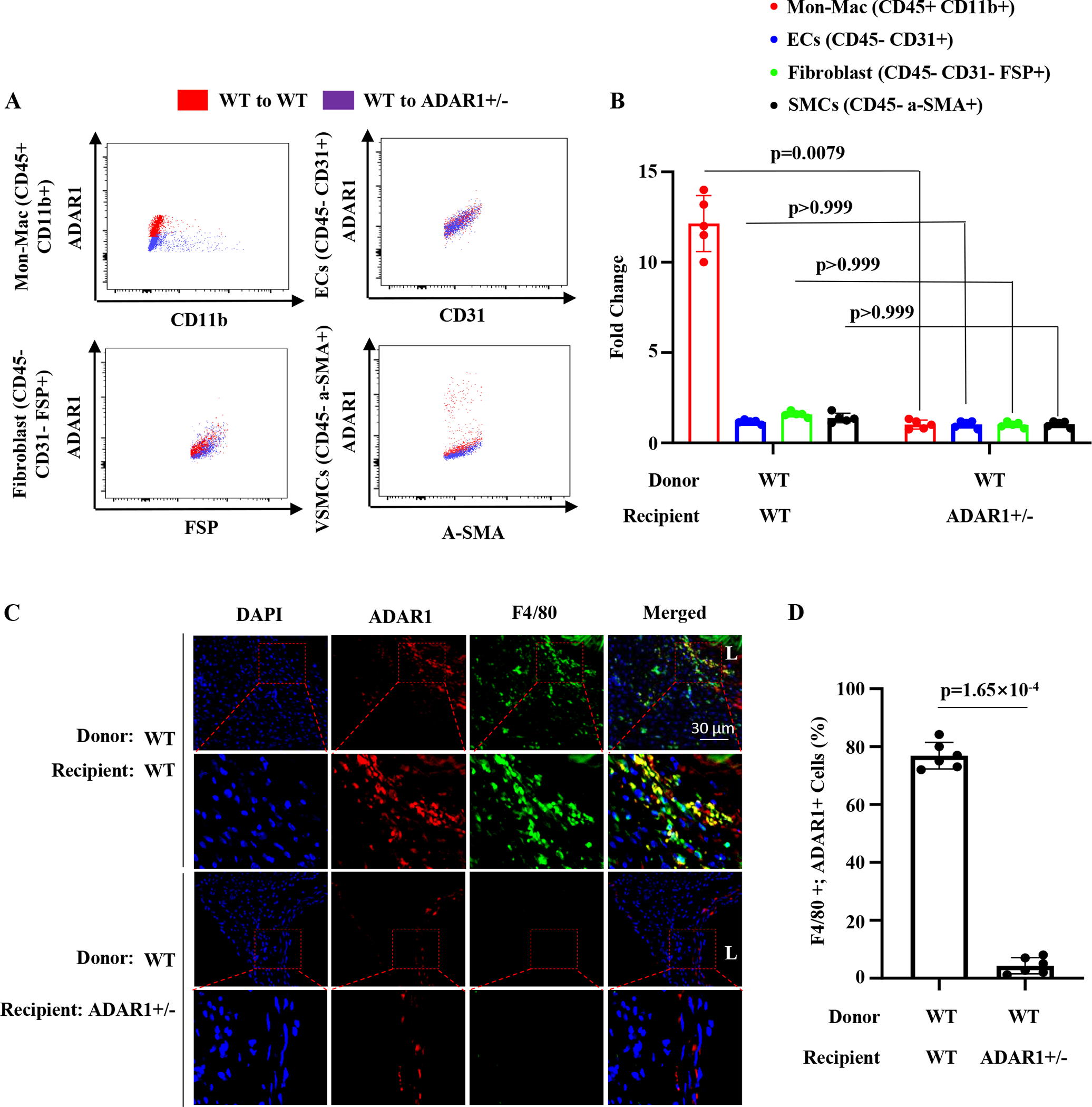
Recipient mice were infused with Ang II (1000 ng/kg/min) for 7 days. A, Recipient mouse abdominal aortic cells were isolated for Flow Cytometry analyses. ADAR1 levels were detected in monocyte-derived macrophages (CD45 +, CD11b+), endothelial cells (CD45-, CD31+), fibroblasts (CD45-, CD31-, FSP+) and smooth muscle cells (CD45-, α-SMA+) in aneurysm lesions. Red dots indicate ADAR1 levels in wild type aorta transplanted to wild type mice. Blue dots indicate ADAR1 levels in wild type aorta transplanted to ADAR1+/− mouse aorta. B, Quantification of ADAR1 levels on monocyte-derived macrophages and vascular cells. Macrophage ADAR1 levels, P=0.0079 (n=5), ADAR+/− vs. WT recipients; ADAR1 levels in endothelial, fibroblast, or smooth muscles, P > 0.999 (n=5), ADAR+/− vs. WT recipients. C, Abdominal aorta sections were co-immunostained with F4/80 and ADAR1 antibodies. The areas in the red boxes were shown with a higher magnification (2.8 folds) in the lower part of the panel. D, The percentage of ADAR1+;F4/80+ cells relative to total F4/80+ cells were averaged from 10 sections for each animal. P<0.001, ADAR+/− vs. WT recipients, n=6.
Since macrophage inflammation is an early event in aneurysm formation, we infused ApoE−/− mice with Ang II for 0, 3, 7, and 14 days in order to observe macrophage ADAR1 expression during the early stage of AAA formation. ADAR1 was barely detectable in control abdominal aorta. However, ADAR1 was induced in macrophages near the lumen side and adventitia 3 days after the Ang II infusion, suggesting that ADAR1 may be important for monocyte/macrophage infiltration into the AAA site (Figure S2). At day 7, ADAR1 was mainly expressed in adventitial macrophages (Figure S2). At day 14, ADAR1 appeared to be expressed in both the adventitia and media layers (Online Figure II), which is consistent with our previous findings34. These data further suggested that macrophage ADAR1 may play important roles in the initiation and progression of AAA formation.
ADAR1 deficiency in macrophages attenuates the development of AAA.
Because ADAR1 is prominently expressed in macrophages in AAA lesions (Figure 2), we generated ADAR1 hematopoietic cell-specific knockout mice (ADAR1mφ−/−) by crossing ADAR1fl/fl mice with LysM-Cre+/−;ApoE−/− (WT) mice. WT mice infused with Ang II for 28 days readily developed AAA/dissection with a 10% mortality rate, but ADAR1mφ−/− mice were resistant to aneurysm formation without lethality (Figure 3A). The AAA incidence was reduced from 66.67% in WT mice to 16.67% in ADAR1mφ−/− mice. These findings were corroborated by ultrasound data and ex vivo analyses, which confirmed the reduction in maximal aortic diameter in Ang II-infused ADAR1mφ−/− mice as compared to control WT mice (Figure 3, B–C and Figure S3–S4). Histological analyses showed that aortas from ADAR1mφ−/− mice had significantly less elastin fragmentation as compared to the WT mice (Figure 3, D–E). These results further demonstrated that macrophage ADAR1 plays an essential role in AAA formation/dissection.
Figure 3: ADAR1 deficiency in macrophages (ADAR1mφ−/−) ameliorated AAA formation.
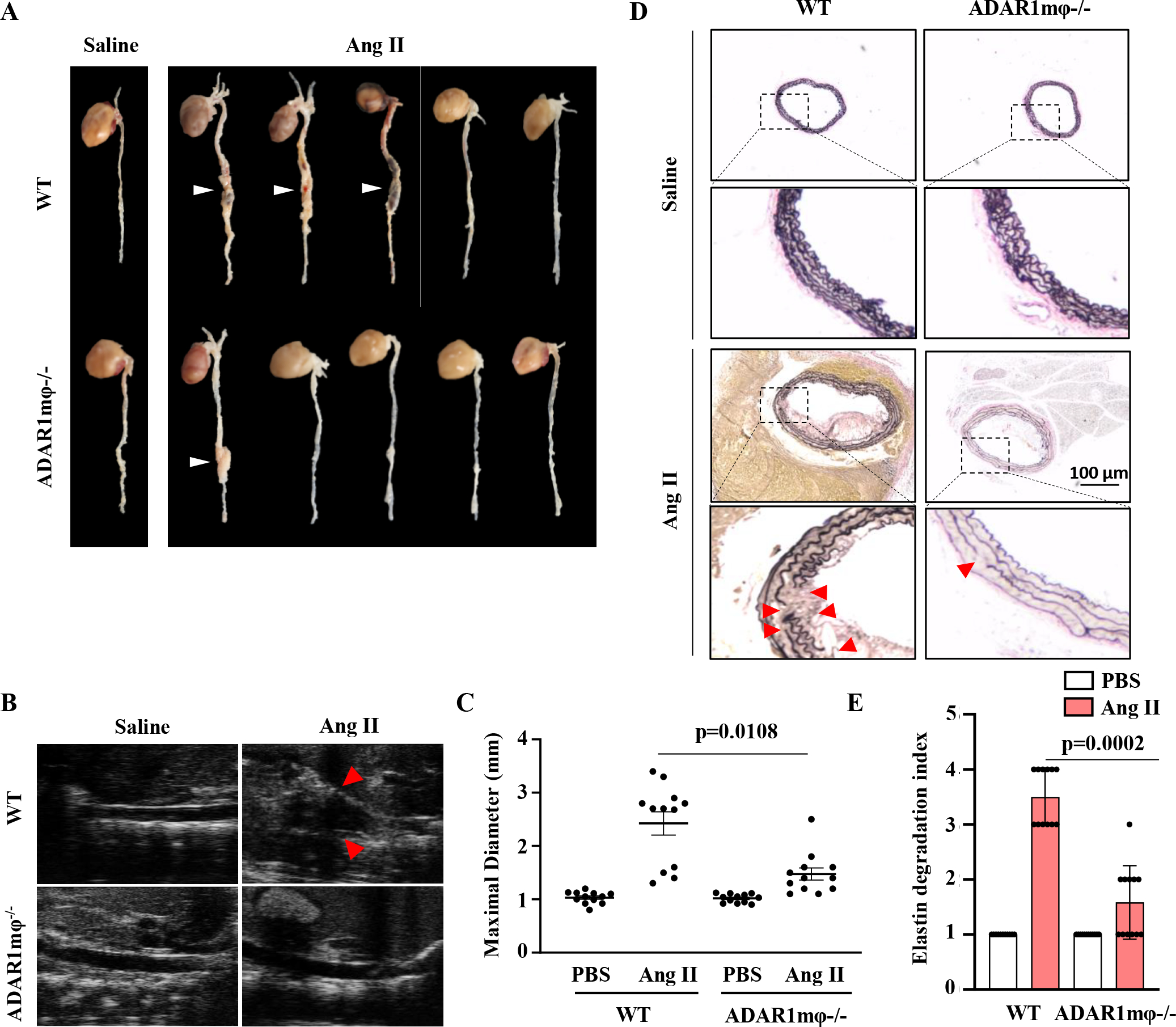
ApoE−/− (WT) and ADAR1mφ-/;ApoE−/− (ADAR1mφ−/− ) mice were infused with Ang II (1000 ng/kg/min) for 28 days. A, Representative aneurysm formation as marked by white arrowheads. B, Representative longitudinal ultrasound images of abdominal aorta. Arrowhead indicates the aneurysm. C, Quantitative analyses of maximal aorta diameters by ultrasound imaging. p= 0.0108, ADAR1mφ−/− vs. WT mice with Ang II infusion, n=12. D, Verhoeff’s elastic (EVG) staining of AAA tissues. The areas in the rectangle boxes are shown with a higher magnification (4.2 fold) in the lower part of the panel. Arrowheads indicate elastin breaks. E, Quantification of elastin degradation indexes in each group. P=0.0002, ADAR1mφ−/− vs. WT mice with Ang II infusion, n=12. Kruskal-Wallis test with Dunn multiple comparisons test was performed for C and E.
ADAR1mφ−/− modulates aorta inflammation and M1 macrophage activation in AAA.
Inflammation plays an important role in the pathogenesis of AAA. Proinflammatory cytokines such as IL-1β, IL-6, TNFα, and M1 macrophage marker iNOS are upregulated in AAA tissues and have been shown to induce synthetic/inflammatory SMC phenotype. Immunofluorescent staining showed that these cytokines were upregulated in AAA tissues in WT mice infused with Ang II for 7 days and largely colocalized with F4/80-positive cells (Figure 4A). However, their expression was diminished in ADAR1mφ−/− aortas (Figure 4A, Figure S5). Western blot analyses of proteins isolated from abdominal aortas confirmed that the increase of IL-1β, IL-6, TNFα and iNOS in AAA lesion of Ang II-infused WT mice was blocked in ADAR1mφ−/− aortas (Figure 4B, Figure S6, A–D). These results collectively indicated that macrophage ADAR1 deficiency attenuates AAA formation by inhibiting M1 macrophage activation-caused aortic wall inflammation.
Figure 4: ADAR1 promoted macrophage activation via regulating anti-inflammatory microRNA (miRNA) processing.
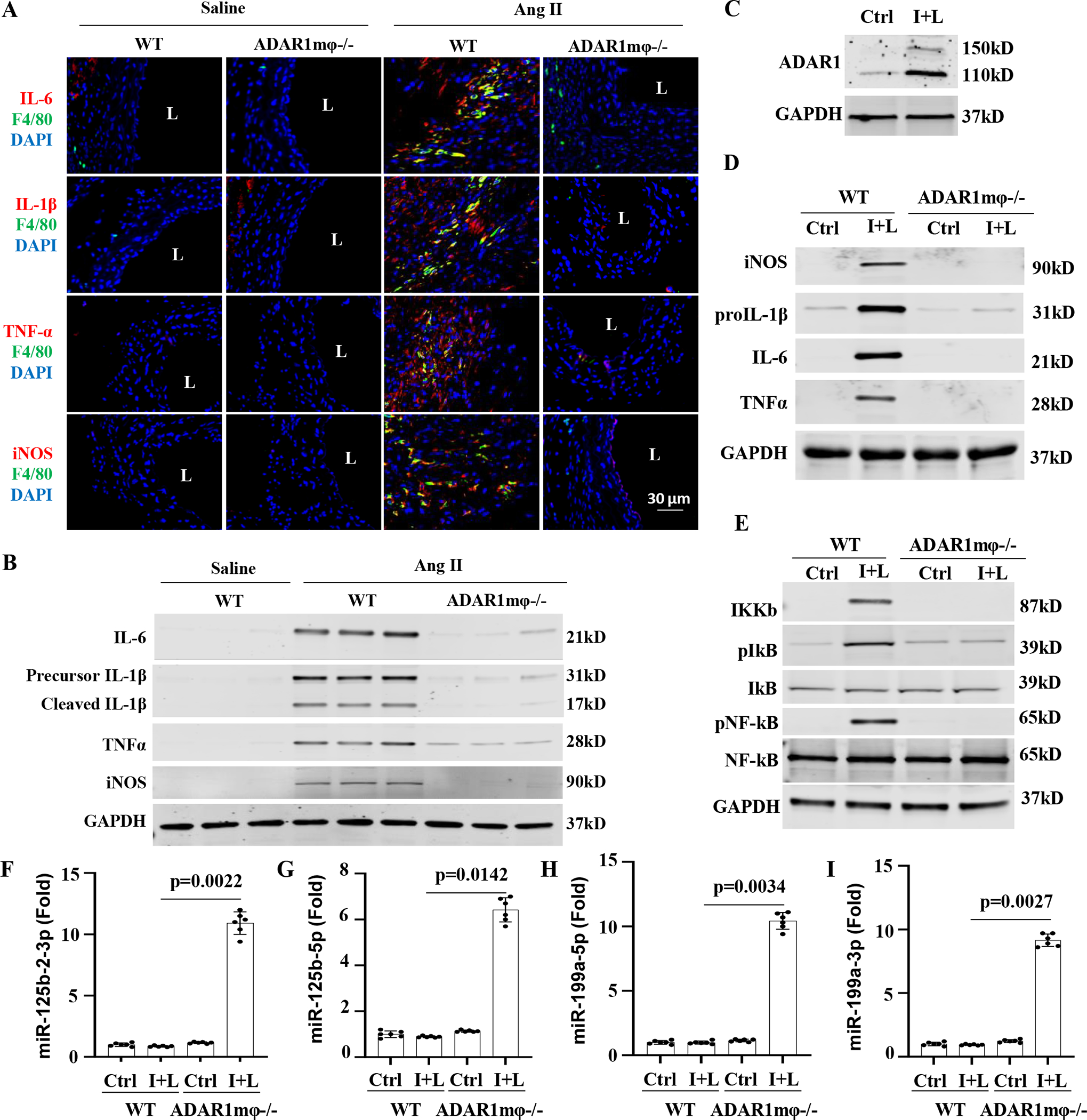
A-B, Mice were infused with angiotensin II (Ang II, 1000ng/kg/min) for 7 days. Abdominal aorta frozen sections were co-immunostained with F4/80 and IL-6, IL-1β, TNFα, or iNOS antibodies, respectively (A). The protein expression of IL-6, IL-1β, TNFα, and iNOS in mouse aorta were detected by Western blot, respectively (B). C, Bone marrow-derived macrophages (BMDMs) from WT mice were activated by interferon γ (IFNγ, 100 ng/mL) and lipopolysaccharides (LPS,100 ng/mL) (I+L) treatment for 6 h. ADAR1 protein expression was measured by Western blot. D, BMDMs isolated from WT or ADAR1mφ−/− mice were treated with vehicle (Ctrl) or I+L (100 ng/mL each) for 6 h to induce macrophage activation. The protein expression of iNOS and pro-inflammatory cytokines IL-1β, IL-6 and TNFα were determined by Western blot, respectively. E, ADAR1 deficiency (ADAR1mφ−/−) significantly inhibited I+L-induced NF-κB signaling. BMDMs isolated from WT or ADAR1mφ−/− mice were treated with vehicle (Ctrl) or I+L (100 ng/mL each) for 30 min. The protein expression and phosphorylation of NF-IKKβ, pIκB, NF-κB were measured by Western blot, respectively. Quantitative analyses for A-E are shown in online Figure V–VI (n=6). F-I, BMDMs isolated from WT or macrophage ADAR1 deficiency (ADAR1mφ−/−) mice were treated as in B. The mature miRNAs targeting NF-κB signaling, including miR-125b-2–3p (F), miR-125b-5p (G), miR-199a-5p (H), and miR-199a-3p (I) were assessed by RT-qPCR. p=0.0022 (F), 0.0142 (G), 0.0034 (H), and 0.0027 (I), ADAR1mφ−/− vs. WT cells treated with I+L, n=6. Kruskal-Wallis test with Dunn multiple comparisons test was performed for F to I.
Consistently, upon the stimulation of lipopolysaccharide (LPS) and interferon-Ƴ (IFNƳ), ADAR1 expression was remarkably elevated in bone marrow derived macrophages (BMDMs) (Figure 4C, Figure S6E). ADAR1 was also induced in macrophages that were activated by TNFα and IFNƳ (Figure S7A). We used LPS and IFNƳ because LPS is present in human or animal blood stream normally at a low dose due to the natural presence of gut microbiota 36, and interferon-Ƴ plays important roles in macrophage activation and AAA development 37. In line with the in vivo observations in AAA lesions, IFNγ+LPS induced the expression of IL-1β, IL-6, TNFα, and iNOS; but ADAR1 deficiency blunted the upregulation of these proinflammatory cytokines in BMDMs (Figure 4D, Figure S6, F–I). Since NF-κB signaling is very important for M1 macrophage activation38, we examined NF-kB signaling pathway in WT and ADAR1mφ−/− BMDMs. As shown in Figure 4E, IFNγ+LPS significantly increased IKKB expression along with the augmented IκB and NF-κB phosphorylation in WT BMDMs (Figure 4E, Figure S6, J–L). However, ADAR1mφ−/− dampened the IFNγ+LPS-activated NF-κB signaling, indicating that ADAR1 promotes the classical macrophage activation by targeting NF-κB signaling pathway. Similar results were observed in BMDMs that were activated by TNFα plus IFNƳ (Figure S7, B–C).
ADAR1 has been shown to regulate miRNA biogenesis through both editing-dependent and -independent mechanisms 39,40. Since ADAR1 promoted IKKβ expression without editing its pre-mRNA (Figure S8), and miRNAs have been shown to inhibit NF-κB signaling by negatively regulating IKKB-dependent IkB and NF-kB phosphorylation 41,42, we sought to determine whether ADAR1 regulates the expression of anti-inflammatory miRNAs in IFNγ+LPS-activated BMDMs. By screening a large number of miRNAs associated with AAA and inflammation 42–44, we found that IFNγ+LPS slightly altered, but did not significantly affect the levels of most anti-inflammatory miRNAs in WT BMDMs. However, ADAR1 deficiency caused a significant increase in the expression of a number of miRNAs (Figure S9). Of note, the levels of miR-125b and miR-199a, essential for suppressing NF-κB signaling42,45,46, were slightly decreased by IFNγ+LPS, but dramatically increased in ADAR1mφ−/− BMDMs following IFNγ+LPS stimulation (Figure 4, F–I).
ADAR1 deficiency promotes anti-inflammatory microRNA processing by increasing Drosha expression and its interaction with DGCR8.
Pri-miRNAs are firstly processed into precursor miRNAs (pre-miRNAs) which are further processed into mature miRNAs 47,48. We thus assessed the pri-miRNA and pre-miRNA levels of miR125b and miR-199a in WT and ADAR1mφ−/− BMDMs during classical macrophage activation. Interestingly, ADAR1mφ−/− did not significantly altered the pri-miRNA levels of miR-125b and miR-199a (Figure S10A), suggesting that ADAR1 did not affect the transcription of these miRNAs. However, the pre-miRNA levels of miR-125b and miR-199a were significantly increased in ADAR1mφ−/− BMDMs after IFNγ+LPS stimulation (Figure S10B), consistent with the mature miRNA levels. These data indicated that ADAR1 deficiency accelerated the pri-miRNA processing into pre-miRNAs while not affecting pri-miRNA transcription.
The pri- to pre-miRNA processing is mediated by RNase III enzyme Drosha in complex with pri-miRNA recognition factor DGCR8 39, we thus detected Drosha and DGCR8 expression in WT and ADAR1mφ−/− BMDMs activated by IFNγ and LPS. While DGCR8 expression was not affected, Drosha level was significantly increased in ADAR1mφ−/− BMDMs following IFNγ and LPS treatment (Figure S11A), suggesting that ADAR1 inhibits the pri- to pre-miRNA processing, at least partially, by decreasing Drosha protein level. Indeed, Drosha expression was reduced in IFNγ+LPS-treated WT BMDMs and PEMs in which ADAR1 was up-regulated (Figure 5A, Figure S11, A–C).
Figure 5: ADAR1 negatively regulates Drosha-DGCR8 interaction in macrophages and AAA lesions.
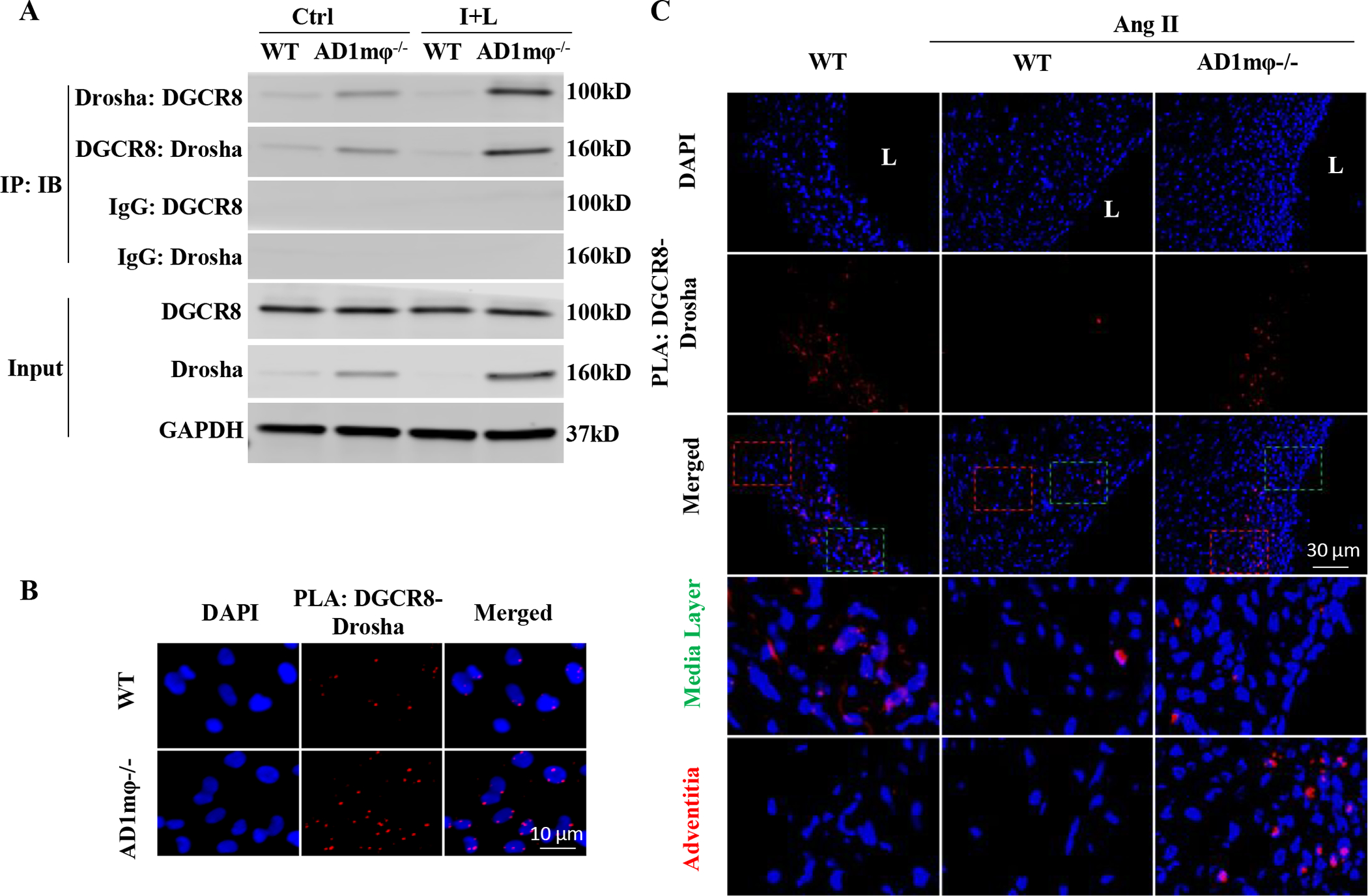
A-B, ADAR1mφ−/− (AD1mφ−/−) significantly enhanced Drosha-DGCR8 interaction in classically activated macrophages. BMDMs isolated from WT or AD6.31mφ−/− mice were treated with vehicle (Ctrl) or interferon γ (IFNγ, 100 ng/mL) and lipopolysaccharides (LPS,100 ng/mL) (I+L) for 6 h to induce macrophage classical activation. A, Coimmunoprecipitation assays were performed to detect the Drosha-DGCR8 interaction. Control (IgG), Drosha or DGCR8 antibodies were used for immunoprecipitation (IP), and immunoblotting (IB) was performed with DGCR8 and Drosha antibodies, respectively. B, In situ proximity ligation assays (PLA) were performed to confirm Drosha-DGCR8 interaction in AD1mφ−/− BMDMs activated by I+L (n=6). DAPI stains the nuclei. C, AD1mφ−/− enhanced the physical interaction between DGCR8 with Drosha in mouse AAA lesion. PLA was performed on mouse abdominal aorta or AAA sections by staining with both Drosha and DGCR8 antibodies (n=6). IgG staining was used as a negative control (online Figure XIV). DAPI stains nuclei. L: lumen. The media (green box) and adventitia areas (red box) in the merged images were enlarged in the lower panels (3.8 folds). Adventitia areas where macrophages accumulate show more Drosha-DGCR8 interaction. The quantifications of PLA signals in B and C are shown in online Figure XV.
To determine if the increased Drosha expression in ADAR1−/− macrophages mediates the pri-miRNA processing of miRNAs targeting Ikkβ-regulated NF-kB signaling, we knocked down Drosha expression in ADAR1−/− macrophages and examined its effect on the expression of IkkB and anti-inflammatory miRNAs. We found that knockdown of Drosha blocked the increase of the miRNAs while restored the IKKB expression downregulated by ADAR deficiency in IFNγ+LPS-treated macrophages (Figure S12). These data demonstrated that ADAR1 promotes macrophage activation via regulating anti-inflammatory miRNA processing through inhibiting Drosha expression.
In addition to affecting Drosha expression, ADAR1mφ−/− increased Drosha interaction with DGCR8, and their interaction was significantly enhanced in IFNγ+LPS-activated ADAR1mφ−/− BMDMs and PEMs (Figure 5A and Figure S11C). Similar results were observed in macrophages that was activated by TNFα and IFNƳ (Figure S13A). The increased Drosha-DGCR8 interaction in ADAR1mφ−/− BMDMs and PEMs was confirmed by in situ Proximity Ligation assay (PLA) (Figure 5B and Figure S11D, S14A & S15A). More importantly, the Drosha-DGCR8 interaction was barely detectable in AAA lesions of Ang II-infused WT mice but was significantly increased in ADAR1mφ−/− aortas where the AAA was attenuated (Figure 5C, Figure S14B & S15B). The increased Drosha-DGCR8 interaction was observed in adventitia layer of the ADAR1mφ−/− aortas (Figure 5C), consistent with the localization of activated macrophages in inflamed arteries. Interestingly, the Drosha-DGCR8 interaction was also observed in the media layer of WT control mice. However, the interaction was diminished in both the Ang II-infused WT and ADAR1mφ−/− mouse aortas (Figure 5C), suggesting ADAR1 non-editing function may also play a role in maintaining SMC homeostasis, which is an interesting subject for future study. Nevertheless, these results suggest that macrophage ADAR1 deficiency increases Drosha expression and further enhances its interaction with DGCR8, leading to the increased pri- to pre-miRNA processing of anti-inflammatory microRNAs, and consequently blocks macrophage activation in Ang II-infused mouse aortas.
ADAR1 interacts with Drosha and promotes its degradation.
In order to test how ADAR1 regulates Drosha expression, we first determined if ADAR1 interacts with Drosha. Coimmunoprecipitation assay showed that ADAR1 physically interacted with Drosha, and IFNγ+LPS stimulation significantly enhanced the ADAR1-Dorsha interaction in both BMDMs and PEMs (Figure 6A and Figure S16A). Similar results were observed in macrophages activated by TNFα and IFNƳ (Figure S13B). In Situ PLA confirmed the ADAR1-Drosha interaction in IFNγ+LPS-activated macrophages; but this interaction was diminished in ADAR1mφ−/− BMDMs and PEMs (Figure 6B, Figure S16B, S17A & S18A). To determine if ADAR1 physically interacts with Drosha in AAA lesions in vivo, we performed In Situ PLA in mouse abdominal aorta. Although ADAR1 barely interacts with Drosha in saline-infused WT mouse abdominal aorta, their interaction was significantly increased in Ang II-infused WT mouse aortas with AAA lesions while attenuated in ADAR1mφ−/− aortas (Figure 6C, Figure S17B & S18B).
Figure 6: ADAR1 binds and promotes Drosha degradation.
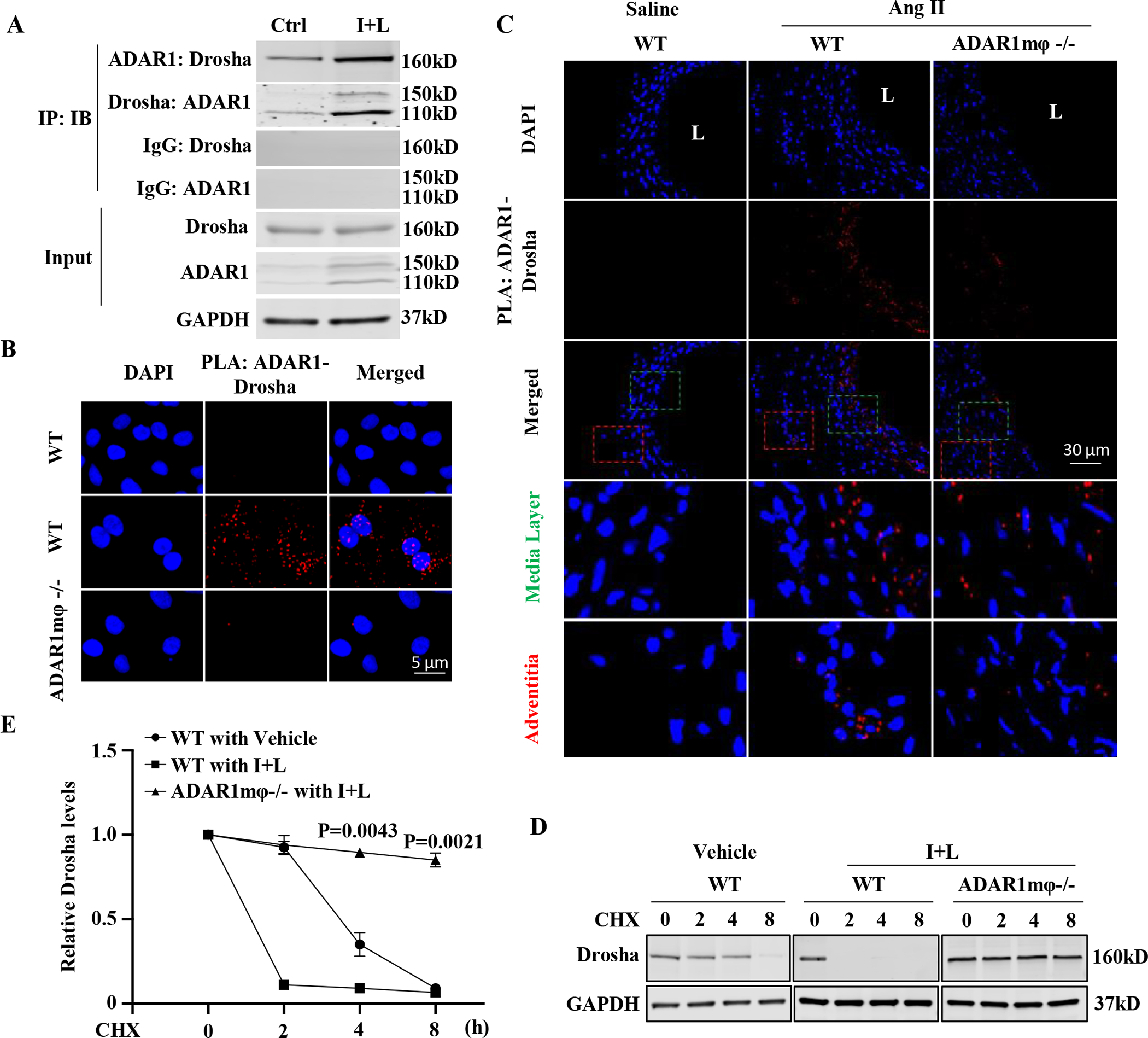
A-B, ADAR1 interacted with Drosha in activated macrophages. BMDMs were treated with vehicle (Ctrl) or interferon γ (IFNγ, 100 ng/mL) and lipopolysaccharides (LPS,100 ng/mL) (I+L) for 6 h. A, Coimmunoprecipitation assays were performed to detect the ADAR1-Drosha interaction. Control (IgG), ADAR1 or Drosha antibodies were used for immunoprecipitation (IP), and immunoblotting (IB) was performed with ADAR1 and Drosha antibodies, respectively. B, In situ Duolink proximity ligation assay (PLA) was performed to confirm the ADAR1-Drosha interaction. DAPI stains nuclei. C, ADAR1 interacted with Drosha in mouse AAA lesion. PLA was performed on mouse abdominal aorta or AAA sections by staining with both ADAR1 and Drosha antibodies. DAPI stains nuclei; L: lumen. IgG staining as a negative control is shown in online Figure XVII. The quantifications for PLA signals are shown in online Figure XVIII. The media (green box) and adventitia areas (red box) in the merged images were enlarged in the lower panels (3.8 folds). D-E, ADAR1mφ−/− inhibited I+L-induced Drosha degradation in BMDMs. BMDMs from WT or ADAR1mφ−/− mice were stimulated with I+L (100 ng/mL each) for 1 hour followed by cycloheximide (CHX,30 ug/mL) treatment for the times indicated. The Drosha protein levels were detected by Western blot (D) and quantified by normalizing to GAPDH (E). p=0.0043 (4 h) and 0.0021 (8 h), ADAR1mφ−/− vs. WT cells with I+L. Kruskal-Wallis test with Dunn multiple comparisons test was performed for E.
It appeared that ADAR1 binding to Drosha affects Drosha protein stability. When BMDMs were treated with cycloheximide that blocks protein synthesis, the Drosha protein was gradually degraded (Figure 6, D–E). IFNγ and LPS treatment accelerated the degradation process (Figure 6, D–E). However, ADAR1 deficiency blocked the IFNγ+LPS-accelerated Drosha protein degradation (Figure 6, D–E). These data indicated that ADAR1 deficiency increases Drosha level in activated macrophages by enhancing Drosha protein stability. To determine how ADAR1 mediates Drosha degradation, we tested if ADAR1 affects Drosha ubiquitination and found that ADAR1 deficiency dramatically inhibited Drosha protein ubiquitination in macrophages, especially in the activated macrophages (Figure S19). The effect of ADAR1 on the Drosha ubiquitination was further confirmed by detecting the direct interaction between Drosha and ubiquitin using PLA assay (Figure S20). These data indicated that ADAR1 promotes Drosha degradation by enhancing its protein ubiquitination.
ADAR1 is highly induced in macrophages, interacts with Drosha, coincident with the decreased Drosha-DGCR8 interaction in human AAA lesions.
To determine the relevance of macrophage ADAR1 expression and function in mouse model to human disease, we detected the ADAR1 expression in human AAA lesions. As shown in Figure 7A and Figure S21A, ADAR1 was highly upregulated in AAA lesions. Moreover, a major portion of ADAR1 was expressed in CD68+ macrophages. Other ADAR1 appeared to be in SMC layer as we reported previously 34. ADAR1 was not observed in endothelium of the healthy aorta or human AAA lesions (Figure S22). Importantly, although ADAR-Drosha interaction was scarcely present in healthy human aortas (Fig 7B), their interaction was significantly induced in human AAA lesions (Fig 7B, Figure S21B & S23). Conversely, Drosha-DGCR8 interaction was observed in the healthy aortas, but it was diminished in human AAA lesions (Fig 7B, Figure S21C & S23). These data suggest that ADAR1-mediated Drosha degradation and thus decreased Drosha-DGCR8 interaction in macrophages may play important roles in macrophage activation and further AAA formation in human patients.
Figure 7: Macrophage ADAR1 and its interaction with Drosha correlate with the development of human AAA.
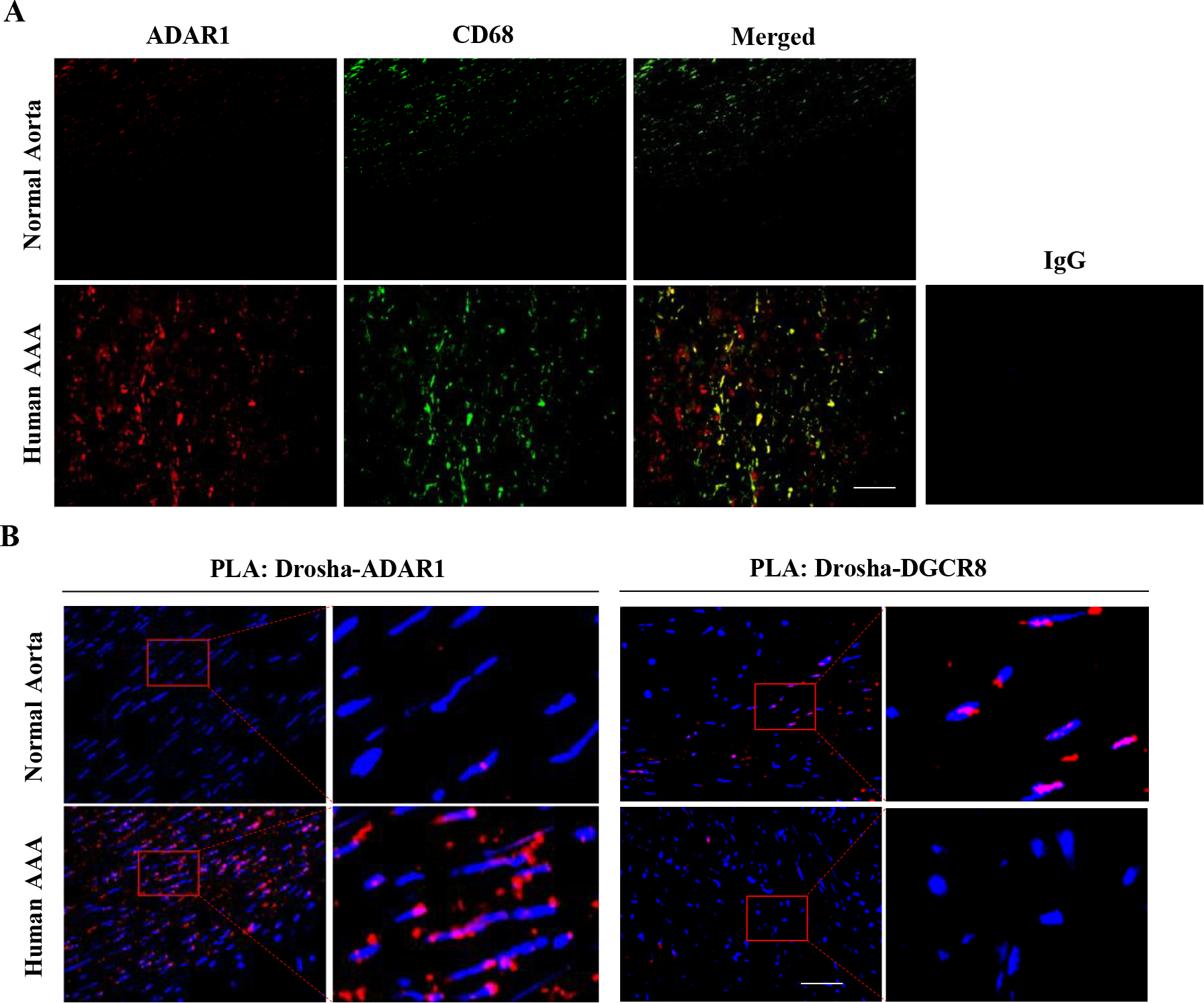
A, Normal healthy human abdominal aorta or AAA sections were co-immunostained with CD68 and ADAR1 antibodies. Red: ADAR1, Green: CD68. The areas in the red boxes were shown with a higher magnification (4.2 folds) in the left part of the panel. B, The increased ADAR1 interaction with Drosha correlated with decreased Drosha interaction with DGCR8 in human AAA lesions, as detected by In situ Duolink Proximity Ligation Assay. DAPI stains nuclei. Scale bar: 30 μm. The negative control of PLA assay is shown in online Figure XXIII. The quantitative analyses of immunostaining (A) and PLA signal (B) are shown in Online Figure XXI (n=6).
Transfer of ADAR1-deficient human monocytes attenuates AAA formation in human arteries transplanted to immunodeficient mice.
To further determine the role of macrophage ADAR1 in human AAA formation, we developed a humanized AAA model by transplanting the remnant human internal mammary arteries (IMA) to abdominal aorta of immunodeficient NOD SCID Gamma (NSG) mice through an end-to-end anastomosis. The mice were then reconstituted once every 7 days with control or ADAR1-deficient human peripheral blood mononuclear cells (PBMCs) labeled with green cell tracker. The PBMCs were isolated from the blood of the same individuals whose remnant IMAs were used for transplantation. The severe immunodeficiency of the NSG mice does not cause any rejection to the engraftment of human arteries and PBMCs. Control and ADAR1-deficient PBMCs were generated by transducing adenovirus expressing GFP (Ad-GFP) or ADAR1 shRNA (Ad-shADAR1). Ad-shADAR1 efficiently knocked down ADAR1 expression in PBMCs (Figure S24). Two weeks after the transplantation, Ang II was infused into the mice via osmotic minipumps for 28 days to observe the aneurysm formation (Figure 8A). Ultrasound imaging was performed to measure human artery diameters (Figure 8B). As shown in Figure 8C, the diameters of human arteries reconstituted with ADAR1-deficient PBMCs were significantly smaller than that with control PBMCs. HE and VEG staining showed significant adventitial inflammatory cell infiltration, media cell loss/degeneration, thrombus formation, and elastin degradation in human arteries with control PBMCs (Figure 8D). However, these defects were attenuated in arteries reconstituted with ADAR1-deficient PBMCs (Figure 8D). As a result, the elastin degradation scores were significantly reduced in arteries with ADAR1-deficient PBMCs (Figure 1E). These results indicated that ADAR1 in human PBMCs was essential for aneurysm formation/dissection in humanized AAA. Moreover, immunofluorescent staining showed that pro-inflammatory cytokines IL-1β, IL-6, TNFα, and iNOS were upregulated and largely colocalized with macrophages in human IMAs with control PBMC reconstitution and Ang II infusion for 7 days (Figure 8, F–J). However, the expression of these cytokines and iNOS was diminished in the arteries reconstituted with ADAR1-deificent PBMCs (Figure 8, F–J). These data further indicated that macrophage ADAR1 may play an important role in the development of AAA formation/dissection in human patients.
Figure 8. Human IMA was anastomosed to recipient abdominal aorta in an end-to-end manner in NSG mice.
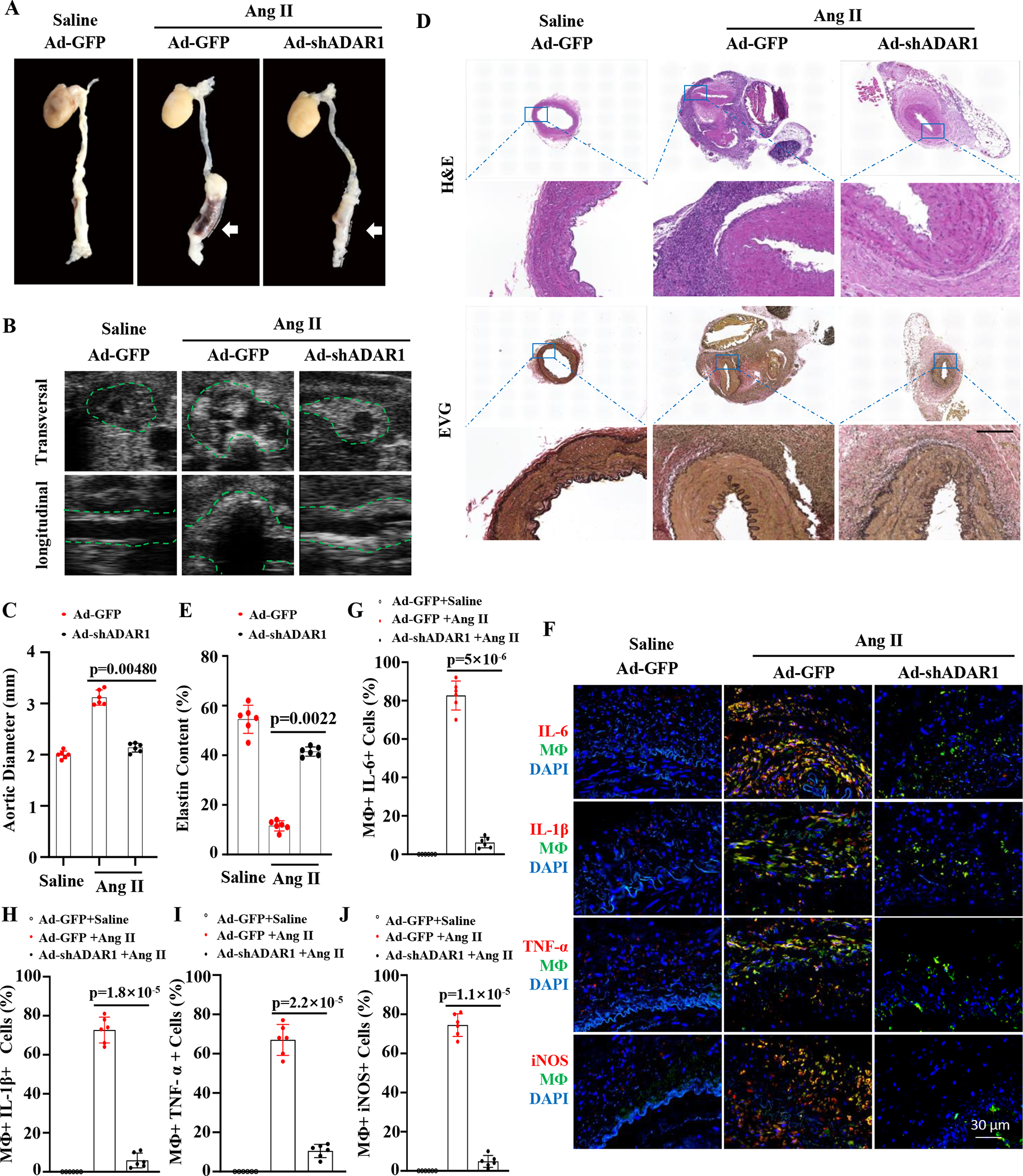
Monocyte stained with green dye were injected (I.V.) the first day after the transplantation and then once every 7 days thereafter. Two weeks after the transplantation, recipient mice were infused with Ang II (1000 ng/kg/min) for 28 days. A, Representative aneurysm formation as marked by white arrows. B, ADAR1 deficiency in monocytes diminished Ang II-infusion-caused aorta dilation in NSG mice as imaged by B mode ultrasound longitudinally and transversely. Green dash lines outline transplanted human IMA. C, Quantitative analyses of maximal external aortic diameters. D, Representative images of hematoxylin and eosin (H&E) or Verhoeff elastic (EVG) staining of AAA tissues. Scale bar=400 μm. E, Quantification of elastin contents shown in the EVG staining in aorta in D. F, Mice with human IMA transplants were infused with angiotensin II (Ang II, 1000ng/kg/min) for 7 days. Abdominal aorta frozen sections were immunostained (red) with IL-6, IL-1β, TNFα, or iNOS antibodies. Yellow indicates the colocalization of the cytokines with human monocytes/macrophages. G-J: The percentage of macrophages expressing IL6 (G), IL-1β (H), IL-6 (I) or iNOS (J) relative to the total macrophages were quantified for each grafted IMA. p=0.0048 (C), 0.0022 (E), 5×10−6 (G), 1.8×10−5 (H), 2.2×10−5 (I), and 1.1×10−5 (J), Recipient mice with ADAR1 shRNA adenoviral vector (Ad-shADAR1)-transduced monocytes vs. with Ad-GFP-transduced monocytes and Ang II infusion n=6. Mann-Whitney test (2-sided) was performed for E. Kruskal-Wallis test with Dunn multiple comparisons test was performed for C, G, H, I, and J.
Discussion
AAA is a life-threatening disease associated with substantial morbidity and mortality due to aortic rupture. Our previous study has shown that SMC ADAR1 is important for AAA formation. However, accumulation of pro-inflammatory macrophages is an earlier event preceding to the SMC phenotype alteration in AAA development 14. Moreover, macrophage-produced cytokines such as TNF-α and IL-1β trigger the inflammatory/synthetic SMC phenotype formation 34. Our aorta transplantation studies show that nonvascular ADAR1 deficiency significantly attenuates the AAA formation. Since ADAR1 is expressed predominantly in the macrophage populations in the grafted aorta at the early stage of AAA development, the AAA attenuation is most likely attributed to the ADAR1 deficiency in macrophages. Indeed, ADAR1 is significantly upregulated in activated macrophages and promotes classical macrophage activation with pro-inflammatory responses in BMDMs and PEMs in vitro. In vivo, ADAR1 hematopoietic cell-specific deficiency remarkably inhibits the aneurysm formation along with reduced aortic dilation and elastin degradation. ADAR1 deficiency in hematopoietic cells also reduces the expression of pro-inflammatory cytokines including iNOS, IL-1β, IL-6 and TNFα, suggesting that ADAR1 promotes vascular inflammation. Although ADAR1 functions in macrophages and inflammatory response remain controversial 29,49–51, our results clearly show that ADAR1 is indispensable for classical macrophage activation and inflammatory response during AAA development. The discrepancy among other studies is likely due to the use of different sources of macrophages or different approaches to block ADAR1 expression.
It appears that AAA development is orchestrated by the paracrine effect of the adventitia macrophages on vascular smooth muscle phenotypic switch. This notion is supported by our previous and current studies showing that 1) IL-1β downregulates SMC marker gene expression 34; 2) ADAR1 deficiency inhibits IL-1β and TNFα production in adventitia macrophages in both the transplanted human IMA and mouse AAA lesions; and 3) adventitia IL-1β and TNFα levels are inversely correlated to SMC contractile protein expression in the healthy aorta and AAA tissues (Figure S25–S29). Therefore, targeting the paracrine effect of proinflammatory cytokines produced due to the increased ADAR1 in macrophages could effectively halt the AAA progression.
Our studies have also discovered a novel RNA editing-independent mechanism controlling ADAR1 function in macrophage activation. ADAR1 has been shown to regulate innate immune response by both editing-dependent and -independent mechanisms 52. Our data show that ADAR1 promotes macrophage activation by activating IKKβ-IκB-NF-κB signaling pathway independent of its RNA editing activity. Specifically, ADAR1 deficiency in macrophages increases miR-125b and miR-199a levels. Although the direct effects of miR-125b and miR-199a on the overall aortic wall inflammation during AAA development are not tested in this study, these microRNAs are known to negatively regulate NF-κB signaling 41,42,53. Interestingly, only pre- and mature, but not pri- miR-125b and pri-miR-199a levels are augmented in ADAR1-deficient macrophages, indicating that ADAR1 blocks the pri- to pre- miRNA processing of these miRNAs. Importantly, miR-125b and miR-199a levels are elevated in Ang II-infused ADAR1mφ−/− mouse aorta along with reduced inflammation and AAA formation, demonstrating that blocking pri- to pre-miRNA processing is an important mechanism for ADAR1 to regulate the innate immune response in AAA development.
ADAR1 appears to interact with Drosha to promote its degradation and thus inhibits the pri- to pre-miRNA processing of miRNAs targeting NF-κB signaling. Drosha and DGCR8 are critical factors for pri- to pre- miRNA processing 39. Drosha protein expression is attenuated in Ang II-infused mouse aortas (Figure S30). However, ADAR1 deficiency remarkably promotes Drosha, but not DGCR8, expression during macrophage activation. Mechanistically, ADAR1 physically interacts with Drosha, which promotes Drosha protein degradation through ubiquitination, reduces Drosha levels, and thus Drosha-DGCR8 interaction. Therefore, ADAR1 blocks pri- to pre-miRNA processing of miR-125b and miR-199a through two mechanisms: causing Drosha degradation and blocking Drosha-DGCR8 interaction. Together, our study reveals a previously unknown function for ADAR1, i.e., regulating protein stability. How ADAR1 mediates Drosha ubiquitination is a subject for future investigation.
Our results from the in vitro and in vivo animal studies are relevant to human AAA development. ADAR1 is highly upregulated in macrophages in human AAA lesions. ADAR1 also interacts with Drosha, and this interaction is conversely correlated to the Drosha-DGCR8 interaction in human AAA lesions, suggesting that ADAR1 may regulate macrophage activation and AAA development in humans through a similar mechanism as in mice. More significantly, ADAR1-deficient human monocytes attenuate the aneurysm formation of human artery when transplanted to immunosuppressed mice, suggesting that our results have a high potential to be translated into the prevention or treatment of AAA in humans.
Supplementary Material
Novelty and Significance.
What Is Known?
Abdominal aortic aneurysm (AAA) has high morbidity and mortality associated with aorta rupture and dissection, especially in the elderly population. Pharmacological approaches to limit AAA progression and rupture have thus far proven ineffective.
Macrophage activation, especially the classically activated macrophages, play critical roles in the initiation of AAA development.
ADAR1 (adenosine deaminases acting on RNA 1) has both RNA editing and non-editing functions. ADAR1 is upregulated in pathological artery wall following injury or exposed to aneurysm insults. ADAR1 promotes neointima formation by its RNA-editing function while endorses the induction of inflammatory SMC phenotype through its non-editing function.
What New Information Does This Article Contribute?
Nonvascular ADAR1, especially macrophage ADAR1, is essential for AAA development. ADAR1 deficiency in macrophages diminished angiotensin II-induced AAA formation/dissection in ApoE−/− mice.
ADAR1 promotes classic macrophage activation by interacting with Drosha to facilitate its degradation, which attenuates Drosha-DGCR8 interaction and consequently inhibits pri- to pre-miRNA processing of microRNAs targeting IKKβ, resulting in increased IKKβ expression and enhanced NF-κB signaling in macrophages, leading to vascular inflammation.
ADAR1 is induced in macrophages and interacts with Drosha in human AAA lesions, which contributes to the human AAA development.
AAA is a progressive vascular disease with >150,000 new cases and 10,000 deaths annually in the United States. Both open surgical and endovascular repair are associated with significant short- and long-term morbidity and mortality. Currently, there is no effective pharmacological treatment for AAA. Our previous study discovered that ADAR1 promotes inflammatory SMC phenotype and AAA formation. However, the goal of these studies is focused on macrophage activation because immune cell infiltration and inflammation are earlier events triggering the later SMC phenotype modulation. In this study, by using aortic transplantation, macrophage-specific deficient mice, and angiotensin II-induced AAA model, we show that macrophage ADAR1 plays an essential role in AAA development. We further show that ADAR1 promotes macrophage activation by interacting with Drosha to facilitate its degradation, which attenuates Drosha-DGCR8 interaction and consequently inhibits pri- to pre-miRNA processing of microRNAs targeting IKKβ, resulting in increased IKKβ expression and enhanced NF-κB signaling, causing vascular inflammation and aneurysm development. Moreover, we have generated a humanized AAA model and demonstrated that ADAR1 plays a critical role in human AAA formation. Our findings provide key insights into the mechanism of AAA development and identify macrophage ADAR1 as a potential new therapeutic target to hinder AAA growth and rupture.
Acknowledgements
We thank the cardiac surgery team in the Department of Surgery, University of Missouri for providing human mammary arteries for the generation of humanized AAA model. Graph Abstract was created with BioRender.com.
Sources of Funding:
This work was supported by grants from National Institutes of Health (HL117247, HL119053, HL135854, and HL147313) and the University of Missouri School of Medicine TRIUMPH Initiative Funding. Dunpeng Cai is recipient of American Heart Association Postdoctoral Fellowship (#657293).
Non-standard Abbreviations and Acronyms
- AAA
abdominal aortic aneurysm
- SMC
smooth muscle cells
- ADAR
adenosine deaminase acting on RNA
- NO
nitric oxide
- iNOS
inducible nitric oxide synthase
- WT
Wild type
- LPS
lipopolysaccharide
- BMDM
bone marrow derived macrophage
- PEM
peritoneal macrophage
- IFNƳ
interferon-Ƴ
- PLA
proximity ligation assay
- TNFα
tumor necrosis factor alpha
- NF-kB
nuclear factor kappa B
- DGCR8
DiGeorge syndrome critical region 8
- IKKβ
inhibitor of nuclear factor kappa-B
- IkB
IkappaB
- miRNA
microRNA
Footnotes
Disclosures: None
References
- 1.Golledge J, Muller J, Daugherty A, Norman P. Abdominal aortic aneurysm: pathogenesis and implications for management. Arterioscler Thromb Vasc Biol. 2006;26:2605–2613. doi: 10.1161/01.ATV.0000245819.32762.cb [DOI] [PubMed] [Google Scholar]
- 2.Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011;8:92–102. doi: 10.1038/nrcardio.2010.180 [DOI] [PubMed] [Google Scholar]
- 3.Ma D, Zheng B, Suzuki T, Zhang R, Jiang C, Bai D, Yin W, Yang Z, Zhang X, Hou L, et al. Inhibition of KLF5-Myo9b-RhoA Pathway-Mediated Podosome Formation in Macrophages Ameliorates Abdominal Aortic Aneurysm. Circ Res. 2017;120:799–815. doi: 10.1161/CIRCRESAHA.116.310367 [DOI] [PubMed] [Google Scholar]
- 4.Dias NV, Ivancev K, Malina M, Resch T, Lindblad B, Sonesson B. Does the wide application of endovascular AAA repair affect the results of open surgery? Eur J Vasc Endovasc Surg. 2003;26:188–194. doi: 10.1053/ejvs.2002.1866 [DOI] [PubMed] [Google Scholar]
- 5.Warner CJ, Roddy SP, Chang BB, Kreienberg PB, Sternbach Y, Taggert JB, Ozsvath KJ, Stain SC, Darling RC, 3rd. Regionalization of Emergent Vascular Surgery for Patients With Ruptured AAA Improves Outcomes. Ann Surg. 2016;264:538–543. doi: 10.1097/SLA.0000000000001864 [DOI] [PubMed] [Google Scholar]
- 6.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dale MA, Ruhlman MK, Baxter BT. Inflammatory cell phenotypes in AAAs: their role and potential as targets for therapy. Arterioscler Thromb Vasc Biol. 2015;35:1746–1755. doi: 10.1161/ATVBAHA.115.305269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah PK. Inflammation, metalloproteinases, and increased proteolysis: an emerging pathophysiological paradigm in aortic aneurysm. Circulation. 1997;96:2115–2117. [DOI] [PubMed] [Google Scholar]
- 10.Raffort J, Lareyre F, Clement M, Hassen-Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. 2017;14:457–471. doi: 10.1038/nrcardio.2017.52 [DOI] [PubMed] [Google Scholar]
- 11.Tieu BC, Lee C, Sun H, Lejeune W, Recinos A 3rd, Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009;119:3637–3651. doi: 10.1172/JCI38308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Ait-Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S, Huang J, Offenstadt G, Combadiere C, Renia L, et al. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest. 2010;120:422–432. doi: 10.1172/JCI38136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qin Z, Bagley J, Sukhova G, Baur WE, Park HJ, Beasley D, Libby P, Zhang Y, Galper JB. Angiotensin II-induced TLR4 mediated abdominal aortic aneurysm in apolipoprotein E knockout mice is dependent on STAT3. J Mol Cell Cardiol. 2015;87:160–170. doi: 10.1016/j.yjmcc.2015.08.014 [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Schmidt J, Ryschich E, Mueller-Schilling M, Schumacher H, Allenberg JR. Inducible nitric oxide synthase is present in human abdominal aortic aneurysm and promotes oxidative vascular injury. J Vasc Surg. 2003;38:360–367. [DOI] [PubMed] [Google Scholar]
- 16.Johnston WF, Salmon M, Su G, Lu G, Stone ML, Zhao Y, Owens GK, Upchurch GR Jr., Ailawadi G. Genetic and pharmacologic disruption of interleukin-1beta signaling inhibits experimental aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2013;33:294–304. doi: 10.1161/ATVBAHA.112.300432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oller J, Mendez-Barbero N, Ruiz EJ, Villahoz S, Renard M, Canelas LI, Briones AM, Alberca R, Lozano-Vidal N, Hurle MA, et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat Med. 2017;23:200–212. doi: 10.1038/nm.4266 [DOI] [PubMed] [Google Scholar]
- 18.Da Ros F, Carnevale R, Cifelli G, Bizzotto D, Casaburo M, Perrotta M, Carnevale L, Vinciguerra I, Fardella S, Iacobucci R, et al. Targeting Interleukin-1beta Protects from Aortic Aneurysms Induced by Disrupted Transforming Growth Factor beta Signaling. Immunity. 2017;47:959–973 e959. doi: 10.1016/j.immuni.2017.10.016 [DOI] [PubMed] [Google Scholar]
- 19.Licht K, Jantsch MF. Rapid and dynamic transcriptome regulation by RNA editing and RNA modifications. J Cell Biol. 2016;213:15–22. doi: 10.1083/jcb.201511041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song C, Sakurai M, Shiromoto Y, Nishikura K. Functions of the RNA Editing Enzyme ADAR1 and Their Relevance to Human Diseases. Genes (Basel). 2016;7. doi: 10.3390/genes7120129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature. 2019;565:43–48. doi: 10.1038/s41586-018-0768-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Functions Nishikura K. and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nemlich Y, Baruch EN, Besser MJ, Shoshan E, Bar-Eli M, Anafi L, Barshack I, Schachter J, Ortenberg R, Markel G. ADAR1-mediated regulation of melanoma invasion. Nat Commun. 2018;9:2154. doi: 10.1038/s41467-018-04600-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bahn JH, Ahn J, Lin X, Zhang Q, Lee JH, Civelek M, Xiao X. Genomic analysis of ADAR1 binding and its involvement in multiple RNA processing pathways. Nat Commun. 2015;6:6355. doi: 10.1038/ncomms7355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu S, Xie J, Zhao B, Hu X, Li X, Zhang B, Wang X, Wang Y, Jiang J, Yin W, et al. ADAR1 prevents small intestinal injury from inflammation in a murine model of sepsis. Cytokine. 2018;104:30–37. doi: 10.1016/j.cyto.2018.01.020 [DOI] [PubMed] [Google Scholar]
- 27.Wang G, Wang H, Singh S, Zhou P, Yang S, Wang Y, Zhu Z, Zhang J, Chen A, Billiar T, et al. ADAR1 Prevents Liver Injury from Inflammation and Suppresses Interferon Production in Hepatocytes. Am J Pathol. 2015;185:3224–3237. doi: 10.1016/j.ajpath.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ben-Shoshan SO, Kagan P, Sultan M, Barabash Z, Dor C, Jacob-Hirsch J, Harmelin A, Pappo O, Marcu-Malina V, Ben-Ari Z, et al. ADAR1 deletion induces NFkappaB and interferon signaling dependent liver inflammation and fibrosis. RNA Biol. 2017;14:587–602. doi: 10.1080/15476286.2016.1203501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu Y, Wang H, Zhang J, Ma X, Meng J, Li Y, Hou Z, Luo X. Adenosine deaminase that acts on RNA 1 p150 in alveolar macrophage is involved in LPS-induced lung injury. Shock. 2009;31:410–415. doi: 10.1097/SHK.0b013e31817c1068 [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Gao X, Hu J, Xie Y, Zuo Y, Xu H, Zhu S. ADAR1p150 Forms a Complex with Dicer to Promote miRNA-222 Activity and Regulate PTEN Expression in CVB3-Induced Viral Myocarditis. Int J Mol Sci. 2019;20. doi: 10.3390/ijms20020407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, Dao Thi VL, Shilvock AR, Hoffmann HH, Rosenberg BR, et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell. 2018;172:811–824 e814. doi: 10.1016/j.cell.2017.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lazzari E, Mondala PK, Santos ND, Miller AC, Pineda G, Jiang Q, Leu H, Ali SA, Ganesan AP, Wu CN, et al. Alu-dependent RNA editing of GLI1 promotes malignant regeneration in multiple myeloma. Nat Commun. 2017;8:1922. doi: 10.1038/s41467-017-01890-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dong N, Dong C, Xiong S. Janus effects of ADAR1 on CVB3-induced viral myocarditis at different infection stages. Int J Cardiol. 2016;223:898–905. doi: 10.1016/j.ijcard.2016.08.315 [DOI] [PubMed] [Google Scholar]
- 34.Cai D, Sun C, Zhang G, Que X, Fujise K, Weintraub NL, Chen SY. A Novel Mechanism Underlying Inflammatory Smooth Muscle Phenotype in Abdominal Aortic Aneurysm. Circ Res. 2021;129:e202–e214. doi: 10.1161/circresaha.121.319374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tazume H, Miyata K, Tian Z, Endo M, Horiguchi H, Takahashi O, Horio E, Tsukano H, Kadomatsu T, Nakashima Y, et al. Macrophage-derived angiopoietin-like protein 2 accelerates development of abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2012;32:1400–1409. doi: 10.1161/ATVBAHA.112.247866 [DOI] [PubMed] [Google Scholar]
- 36.Wassenaar TM, Zimmermann K. Lipopolysaccharides in Food, Food Supplements, and Probiotics: Should We be Worried? Eur J Microbiol Immunol (Bp). 2018;8:63–69. doi: 10.1556/1886.2018.00017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiong W, Zhao Y, Prall A, Greiner TC, Baxter BT. Key roles of CD4+ T cells and IFN-gamma in the development of abdominal aortic aneurysms in a murine model. J Immunol. 2004;172:2607–2612. doi: 10.4049/jimmunol.172.4.2607 [DOI] [PubMed] [Google Scholar]
- 38.Hou Y, Lin H, Zhu L, Liu Z, Hu F, Shi J, Yang T, Shi X, Zhu M, Godley BF, et al. Lipopolysaccharide increases the incidence of collagen-induced arthritis in mice through induction of protease HTRA-1 expression. Arthritis Rheum. 2013;65:2835–2846. doi: 10.1002/art.38124 [DOI] [PubMed] [Google Scholar]
- 39.Nishikura K A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol. 2016;17:83–96. doi: 10.1038/nrm.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fei J, Cui XB, Wang JN, Dong K, Chen SY. ADAR1-Mediated RNA Editing, A Novel Mechanism Controlling Phenotypic Modulation of Vascular Smooth Muscle Cells. Circ Res. 2016;119:463–469. doi: 10.1161/CIRCRESAHA.116.309003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai L, Gu L, Di W. MiR-199a attenuates endometrial stromal cell invasiveness through suppression of the IKKbeta/NF-kappaB pathway and reduced interleukin-8 expression. Mol Hum Reprod. 2012;18:136–145. doi: 10.1093/molehr/gar066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy AJ, Guyre PM, Pioli PA. Estradiol suppresses NF-kappa B activation through coordinated regulation of let-7a and miR-125b in primary human macrophages. J Immunol. 2010;184:5029–5037. doi: 10.4049/jimmunol.0903463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adam M, Raaz U, Spin JM, Tsao PS. MicroRNAs in Abdominal Aortic Aneurysm. Curr Vasc Pharmacol. 2015;13:280–290. doi: 10.2174/15701611113119990015 [DOI] [PubMed] [Google Scholar]
- 44.Borek A, Drzymala F, Botor M, Augusciak-Duma AM, Sieron AL. Roles of microRNAs in abdominal aortic aneurysm pathogenesis and the possibility of their use as biomarkers. Kardiochir Torakochirurgia Pol. 2019;16:124–127. doi: 10.5114/kitp.2019.88601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dai L, Gu L, Di W. MiR-199a attenuates endometrial stromal cell invasiveness through suppression of the IKKβ/NF-κB pathway and reduced interleukin-8 expression. Mol Hum Reprod. 2012;18:136–145. doi: 10.1093/molehr/gar066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou W, Pal AS, Hsu AY, Gurol T, Zhu X, Wirbisky-Hershberger SE, Freeman JL, Kasinski AL, Deng Q. MicroRNA-223 Suppresses the Canonical NF-κB Pathway in Basal Keratinocytes to Dampen Neutrophilic Inflammation. Cell Rep. 2018;22:1810–1823. doi: 10.1016/j.celrep.2018.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishikura K. Editor meets silencer: crosstalk between RNA editing and RNA interference. Nat Rev Mol Cell Biol. 2006;7:919–931. doi: 10.1038/nrm2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hundley HA, Bass BL. ADAR editing in double-stranded UTRs and other noncoding RNA sequences. Trends Biochem Sci. 2010;35:377–383. doi: 10.1016/j.tibs.2010.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J, Xie J, Liu S, Li X, Zhang D, Wang X, Jiang J, Hu W, Zhang Y, Jin B, et al. ADAR1 attenuates allogeneic graft rejection by suppressing miR-21 biogenesis in macrophages and promoting M2 polarization. FASEB J. 2018;32:5162–5173. doi: 10.1096/fj.201701449R [DOI] [PubMed] [Google Scholar]
- 50.Rossetti C, Picardi E, Ye M, Camilli G, D’Erchia AM, Cucina L, Locatelli F, Fianchi L, Teofili L, Pesole G, et al. RNA editing signature during myeloid leukemia cell differentiation. Leukemia. 2017;31:2824–2832. doi: 10.1038/leu.2017.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heale BS, Eulalio A, Schulte L, Vogel J, O’Connell MA. Analysis of A to I editing of miRNA in macrophages exposed to Salmonella. RNA Biol. 2010;7:621–627. doi: 10.4161/rna.7.5.13269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heraud-Farlow JE, Walkley CR. The role of RNA editing by ADAR1 in prevention of innate immune sensing of self-RNA. J Mol Med (Berl). 2016;94:1095–1102. doi: 10.1007/s00109-016-1416-1 [DOI] [PubMed] [Google Scholar]
- 53.Zhou W, Pal AS, Hsu AY, Gurol T, Zhu X, Wirbisky-Hershberger SE, Freeman JL, Kasinski AL, Deng Q. MicroRNA-223 Suppresses the Canonical NF-kappaB Pathway in Basal Keratinocytes to Dampen Neutrophilic Inflammation. Cell Rep. 2018;22:1810–1823. doi: 10.1016/j.celrep.2018.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.