

SUMMARY
The nervous system requires metabolites and oxygen supplied by the neurovascular network, but this necessitates close apposition of neurons and endothelial cells. We find motor neurons attract vessels with long-range VEGF signaling, but endothelial cells in the axonal pathway are an obstacle for establishing connections with muscles. It is unclear how this paradoxical interference from heterotypic neurovascular contacts is averted. Through a mouse mutagenesis screen, we show that Plexin-D1 receptor is required in endothelial cells for development of neuromuscular connectivity. Motor neurons release Sema3C to elicit short-range repulsion via Plexin-D1, thus displacing endothelial cells that obstruct axon growth. When this signaling pathway is disrupted, epaxial motor neurons are blocked from reaching their muscle targets and concomitantly vascular patterning in the spinal cord is altered. Thus, an integrative system of opposing push-pull cues ensures detrimental axon-endothelial encounters are avoided while enabling vascularization within the nervous system and along peripheral nerves.
Graphical Abstract
In brief
Axons navigate complex environments where unwanted encounters with other cells might hinder their course. Martins et al. identify a selective obstacle-removal pathway based on short-range Sema3C/Plexin-D1 repulsive signaling that motor neurons employ to evade collisions with blood vessels while enabling assembly of interdependent neurovascular networks through attraction of endothelial cells.
INTRODUCTION
During embryonic development, neurons project through dense and heterogeneous environments guided by chemical and mechanical cues (Franze et al., 2013; Kolodkin and Tessier-Lavigne, 2011). Owing to concomitance of tissue morphogenesis and innervation, axons are confronted with a noisy and changing terrain, where true guideposts may be masked, and obstacles—in the forms of physical barriers or unwanted cell-cell contacts—interfere with pathfinding (Dodd and Jessell, 1988; Klose and Bentley, 1989; Tosney and Landmesser, 1985a). Despite these complications, neurons rarely make projection errors indicating that safeguard mechanisms are in place (Landmesser, 1978). This is in part achieved through cell-intrinsic switches that gate receptor signaling limiting detection to relevant cues (Bai et al., 2011; Bonanomi et al., 2019; Zang et al., 2021). In addition, neuronal growth cones form invasive structures and release matrix-degrading proteases to remove some of the physical constraints in tissues (Nichols and Smith, 2019; Santiago-Medina et al., 2015). However, while the instructive effect of tissue-derived signals on neuronal guidance have been studied in detail, much less is known on how axons affect the behavior of cells along their projection paths.
Coordinated interactions between axons and their cellular environment are manifested in the formation of apposed and interdependent neurovascular networks (Tam and Watts, 2010). This process begins in the embryo, when nerves and vessels attract each other through release of chemotropic factors or, alternatively, display independent but correlated responses to guidance cues (Carmeliet and Tessier-Lavigne, 2005; Eichmann and Brunet, 2014; James and Mukouyama, 2011). The significant overlap between periods of axon growth and angiogenesis suggests that the two processes must be harmonized to prevent interference (Erskine et al., 2017; James et al., 2009). Nonetheless, current models based on the principle of “one patterns the other” (Andreone et al., 2015), in which pre-established neural or vascular systems serve as template, do not resolve how axons negotiate their encounters with vessels when both are simultaneously engaged in pathfinding.
Navigating crowded tissues is particularly challenging for pioneer axons that set new routes in the absence of preexisting nerve tracts. This is the case for spinal motor neurons (MNs) that are the first to project through the mesenchymal environment, thereby laying down the basic pattern of peripheral nerve trajectories. MN subtypes that innervate functionally related muscle groups cluster together (Jessell, 2000). All motor axons travel initially in a common nerve until each subtype branches-off at defined choice-points to follow specific routes toward the appropriate target (Bonanomi, 2019; Tosney and Landmesser, 1985a). An evolutionarily conserved set of medial motor column (MMC) neurons with epaxial projections controls locomotion in limbless vertebrates and body posture in tetrapods (D’Elia and Dasen, 2018; Fetcho, 1987), while distinct MN subtypes that evolved from this ancestral population contact hypaxial or limb muscles.
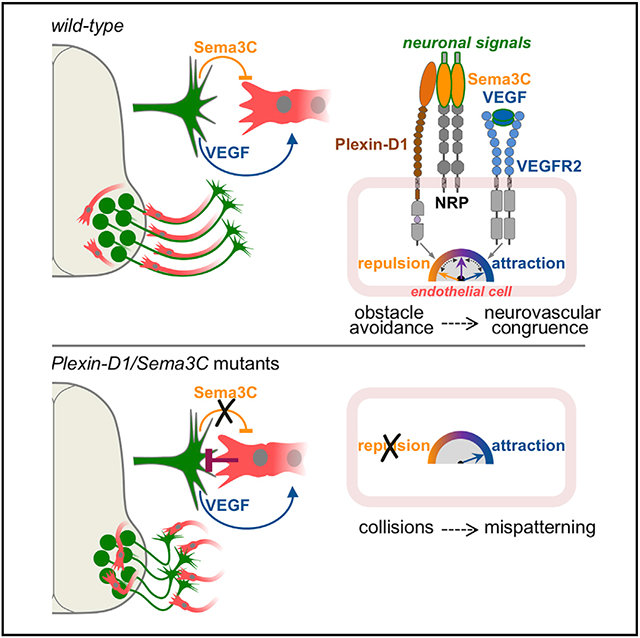
Taking advantage of the topographic organization of MN projections, we conducted a mouse forward genetic screen to isolate factors involved in neuromuscular connectivity. This analysis led to identification of the Drake mutant line harboring a missense mutation in Plexin-D1, a transmembrane receptor for Semaphorins. Plexin-D1 is best known as high-affinity receptor for Sema3E (Gu et al., 2005). The interaction is direct, unlike other class 3 secreted Semaphorins that bind to Neuropilins (NRPs) and rely on Plexins as signal-transducing co-receptors. By mediating Sema3E signaling, Plexin-D1 regulates vascular patterning, axon guidance, synapse formation, and metastasis (Oh and Gu, 2013). Here, through the study of Drake mutants with loss of function of Plexin-D1, we reveal an unexpected co-dependency between motor axons and vessels during pathfinding and show that neurons repurpose guidance signals to prevent detrimental interactions with endothelial cells (ECs). This crosstalk reconciles the opposing requirement for neurovascular congruency with the simultaneous need to overcome cellular barriers along axon projection paths.
RESULTS
An ENU-induced allele of Plexin-D1 causes cell-type-specific motor axon guidance defects
We designed an N-ethyl-N-nitrosourea (ENU)-mutagenesis mouse screen to identify recessive alleles that affect neuromuscular connectivity (Lewcock et al., 2007). On this basis, we isolated the mutant line Drake, which displayed MN projection defects characterized by large ectopic axon fascicles just outside the spinal cord (SC) (Figures 1A-1F and S1A-S1D). At all affected spinal roots, the presence of aberrant bundles was invariably associated with severe disruption of epaxial nerves, which were markedly thinner or absent (asterisks in Figures 1A-1D and S1A-S1D). In contrast, nerves supplying muscles in the limb or body wall were normal (Figure S1E; data not shown). Selective depletion of epaxial projections suggested that the ectopic proximal bundles were formed mostly—or completely—by MMC-subtype motor axons that had stalled shortly after exiting the SC (Figure 1G). Thus, the Drake mutation specifically affects targeting of a discrete set of axial MNs.
Figure 1. The ENU-induced Plexin-D1 allele Drake causes selective motor axon guidance defects.

(A–D) Whole mounts of E12.5 ISLMN::fGFP+ embryos (dorsal view of lumbar region). MNs in the spinal cord (SC) project through the ventral roots (VRs) to epaxial (Ax, asterisks) or limb (Lb) muscles. Drake homozygotes (Drk/Drk) display proximal motor axon bundles (arrowheads) and corresponding depletion of epaxial nerves. Control is Drk/+. Other nerve tracts are intact. A, anterior; P, posterior; M, medial; L, lateral.
(E and F) E12.5 transverse sections show the initial course of ISLMN::fGFP+ epaxial motor axons (arrowheads in E), which is halted in Drake mutants (arrowheads in F). βIII-tubulin, pan-neuronal marker. ISLMN::fGFP signal is higher in the MMC neurons relative to other subtypes. DRG, dorsal root ganglia.
(G) Motor axon stalling in Drake embryo.
(H) Plexin-D1 with C116S mutation in “Sema” ligand-binding domain. Other domains are labeled.
(I) Genomic DNA sequencing reveals homozygous T>A substitution in Plxnd1 exon 1 of Drake embryos, leading to C116S mutation.
(J) Evolutionary conservation of C116 (green, mouse sequence) and ENU-induced mutation (red).
(K) Lysates of E12.5 spinal cords and surrounding tissue. Plexin-D1 levels in Drake homozygous (C116S/C116S) or heterozygous (C116S/+) mutants are comparable to WT. Actin is a loading control.
(L) Alkaline phosphatase (AP)-tagged Sema3E reveals Plexin-D1 in E11.5 sagittal sections. Arrows point to intersomitic vessels. Binding is absent in C116S/C116S and Plxnd1 KO embryos.
(M) AP-Sema3E binding to COS-7 cells expressing WT Plexin-D1 but not C116S mutant. No signal is detected with control AP-Fc.
(N) Sema3E induces collapse of COS-7 cells expressing WT Plexin-D1 but not C116S mutant.
(O) Quantification of cell collapse. Mean ± SEM, Unpaired t test (***) p = 0.0002 WT untreated (untr.) versus Sema; (ns) p = 0.24 C116 untr. versus Sema. Sample size for this and following quantification is reported in STAR Methods.
Scale bars: 200 μm in (A) and (B); 100 μm in (C) and (D); 50 μm in (E) and (F); 50 μm in (M); and 50 μm in (N).
See also Figure S1.
RNA sequencing (RNA-seq) of Drake mutants identified an A>T transversion in the first exon of Plxnd1 (Figures 1H and 1I). Drake embryos were homozygous for the mutation that led to C116S substitution in the extracellular ligand-binding (Sema) domain of Plexin-D1. Cys-116 is part of a conserved set of cysteines that form disulfide bridges stabilizing the structure of the Sema domain (Antipenko et al., 2003; Love et al., 2003; Figures 1H, 1J, and S1G). Plexin-D1 transcript and protein levels were unchanged in Drake (Plxnd1C116S/C116S) embryos (Figures 1K and S1H) but surface targeting of Plexin-D1C116S was impaired (Figures S1I-S1L). The mutation abolished binding of Sema3E to Plexin-D1 (Figures 1L and 1M) and prevented induction of cell collapse (Figures 1N and 1O; Gu et al., 2005). Taken together, Plxnd1C116S is likely a severe loss-of-function allele of Plexin-D1 that abolishes receptor activity by interfering with ligand-binding, owing to altered folding and/or impaired membrane targeting. Plxnd1 knockout (KO) embryos exhibited motor axon defects similar to Drake mutants (Figure S1F), further indicating that the inactivating C116S mutation was responsible for the phenotypes.
Endothelial Plexin-D1 is required for motor neuron targeting and controls axon-vessel interactions
Plexin-D1 was enriched in ECs in both the SC and peripheral tissues but was undetectable in MNs in E10.5–E12.5 embryos (Figures 2A, 2B, S2A, S2B, and S6A). This distribution suggested that axon defects observed in Plxnd1 mutants might be non-cell-autonomous, caused by impaired Plexin-D1 activity in blood vessels rather than MNs. To test this possibility, we generated compound mutant embryos carrying one copy of Plxnd1C116S and one Plxnd1 “floxed” allele deleted from either ECs with TekEC::Cre, or from MNs with Olig2MN::Cre. While motor projections were normal in embryos lacking Plxnd1 in MNs, gene deletion in ECs resulted in axon guidance defects closely resembling those found in Drake and Plxnd1 KO embryos (Figures 2C-2F and S2C-S2E). The phenotype was fully penetrant, detected in all homozygous Drake or Plxnd1-null mutants and TekEC::Cre;Plxnd1C116/fl embryos but never in hemizygotes. A scoring system integrating the number of ectopic bundles and affected ventral roots (VRs), quantified in ISLMN::fGFP embryo whole mounts, confirmed that the severity of the phenotype was similar between Drake, Plxnd1−/− and TekEC::Cre;Plxnd1C116/fl embryos (Figure 2G). In addition, lack of complementation between Plxnd1C116S and KO alleles in compound hemizygotes demonstrated that Drake is a loss-of-function mutant of Plxnd1. These findings revealed a requirement for Plexin-D1 in ECs for targeting of MMC axons to epaxial muscles.
Figure 2. Endothelial Plexin-D1 is required for motor axon guidance and interaction with vessels.

(A) Immunohistochemistry for Plexin-D1 reveals restricted vascular expression at E11.5.
(B) In situ hybridization at E11.5 detects Plexin-D1 on blood vessels in the spinal cord (Sp), meninges (Men) and peripheral tissues (Per), but not in the MC (asterisk). Lower signal is visible in DRG.
(C–F) Aberrant motor axon bundles (arrowheads) and epaxial nerve thinning (asterisks) in E12.5 Plxnd1−/− whole mounts (dorsal view) (D) and following deletion of Plxnd1 from ECs (TekEC::Cre; Plxnd1C116S/fl) (F) but not MNs (Olig2MN::Cre; Plxnd1C116S/fl) (E). Control is Olig2MN::Cre; Plxnd1+/fl.
(G) Quantification of axon guidance phenotype. Mean ± SEM, ANOVA/Dunnett’s test (***) p < 0.00001 C116S, Plxnd1−/− and Plxnd1ECΔ versus Ctrl; (ns) Plxnd1MNΔ versus Ctrl; ANOVA/Tukey’s test (ns) p > 0.1 C116S versus Plxnd1−/− versus Plxnd1ECΔ.
(H and I) Whole mounts of embryos co-expressing motor (ISLMN::fGFP) and endothelial (KdkEC::Cherry) reporters (E12.5, dorsal view). Arrow marks the MMC choice point. Ectopically arranged ECs surround axon bundles in Plxnd1−/− (arrowheads in I). Epaxial nerves (Ax) are depleted (asterisk).
(J–K′) MMC axons (ISLMN::fGFPhigh) extend through vessels (CD31+) in E12.5 controls (J and J′; c-p, MMC choice point). Axons are blocked by ectopic EC clusters in mutants (K and K′, arrowheads) resulting in near-ablation of epaxial nerves (Ax, asterisk). Lb, limb nerve (ISLMN::fGFPlow).
(L and M) 3D two-photon imaging (E12.5). The rendered volume was clipped in M to visualize axon-EC apposition (arrowheads). Dashed lines mark the spinal cord margin.
(N and O) Intensity profile of ISLMN::fGFP (motor axons) and CD31 (vessels) along MMC trajectory reveals abnormal adjacent peaks in mutants (mean ± SEM).
(P–S′) Lumbar transverse sections of ISLMN::fGFP+ embryos stained for endothelial marker CD31 and motor/sensory neuron marker Isl1/2. MMC projections (Ax) intersect ECs migrating from the meninges (Men, arrowheads in Q′ and R′) along the common nerve path. Epaxial choice point (c-p, arrows in P–R′) In Plxnd1−/−, axons stall against ectopic EC clusters (arrowheads in S and S′). Asterisk marks residual epaxial axons.
(T) MMC axons are obstructed by ECs in Plxnd1 mutants. To simplify the diagram of axonal projections, MMC cell bodies are shown in a fictitious lateral position.
Scale bars: 200 μm in (A); 100 μm in (B); 200 μm in (C)–(F); 100 μm in (H) and (I); 50 μm in (J)–(K′); 50 μm in (L) and (M); and 50 μm in (P)–(S′).
See also Figure S2.
In control embryos, MMC axons grew through the VR populated with vessels (Figure 2H), whereas the abnormal axon bundles of Plxnd1 mutants were bordered by misplaced ECs (Figure 2I, arrowheads). Thus, while MMC axons normally extended through dispersed ECs avoiding direct contact (Figures 2J, 2J′, 2L, and 2N), their projection in mutant embryos was obstructed by an ectopic “endothelial barrier” positioned at the choice point for epaxial innervation (Figures 2K, 2K′, 2M, and 2O). In contrast, targeting of MNs supplying limb muscles (lateral MC [LMC]) or intercostal nerves (hypaxial MC [HMC]) was preserved despite close encounters with ECs (Figures S2F-S2M′). At lumbar level, the initial course of limb-innervating axons was devoid of vessels, whereas the first MMC axons veering from the common nerve intersected ECs abutting the meningeal vascular plexus, which had begun to enwrap the proximal nerve segment (Figures 2P-2Q′ and S2N-S2Q′). Nevertheless, MMC axons bypassed vessels in control embryos (Figures 2R, 2R′, S2R, and S2R′), while in Plxnd1 mutants they collided with EC clusters (Figure 2S, 2S′, and S2S-S2T′). These studies reveal a choreographed progression of neurovascular association within the VR and indicate that the interactions between MMC neurons and ECs require Plxnd1 signaling to ensure axial muscle innervation (Figure 2T).
Motor axons define vascular topography at the ventral root
Given the correlated development of axons and vessels at the VR, we asked whether motor nerves contributed to the spatial organization of the vascular network in this region. MNs were genetically ablated before they extended axons from the SC by combining Olig2MN::Cre, which is active in MN progenitors, with conditional diphtheria toxin A (DTA) allele (DTALSL). Lack of motor projections dramatically affected vascular patterning. In Olig2MN::Cre; DTALSL embryos, vessels failed to align precisely with spared sensory fibers and coalesced around motor exit points (Figures 3A-3D′ and S3A-S3C). The organization of the vascular network around the proximal motor nerve segment, characterized by vascular “rings” traversed by axons, collapsed in absence of motor nerves (Figures 3E-3G). Therefore, vascular topography of the VR zone is shaped by motor axons that co-develop with vessels establishing matching patterns.
Figure 3. Motor-endothelial interactions instruct vascular patterning in the spinal cord and ventral root.

(A–D′) Genetic ablation of MNs in E12 Olig2MN::Cre; DTALSL embryos disrupts vascular patterns at motor exit points (arrowheads) and along sensory fibers (βIII-tubulin, arrows). Controls are Cre negative. MNs (Hb9+) are efficiently eliminated (asterisks in B and D).
(E–G) 3D view of vessels surrounding ISLMN::fGFP+ motor nerves in E12 controls (arrows in E and F). This configuration is disrupted in absence of motor projections (G). Ax, epaxial nerve; Lb, limb nerve.
(H–K′) Vascular invasion of MCs (Isl1/2+, dashed lines) in Plxnd1 mutants (E12.5, lumbar level). Vessels encircle MCs in controls (Plxnd1+/−, H and H′) but intermingle with MNs in mutants (I–K′, arrows).
(L) Quantification ofvascular area (CD31 + px) within MCs at lumbar level. Mean, normalized to control ± SEM, ANOVA/Dunnett’s test (***) p < 0.00001 C116S and Plxnd1~’~ versus Ctrl.
(M) Quantification of vascular area (CD31+ px) in dorsal spinal cord. Mean, normalized to control ± SEM, ANOVA/Dunnett’s test (ns) p > 0.5.
(N) Vascular invasion of MCs in Plxnd1 mutants.
Scale bars: 100 μm in (A)–(B′); 50 μm in (C)–(D′); 100 μm in (E)–(G); and 100 μm in (H)μ(K′).
See also Figure S3.
Plexin-D1 controls vascularization of the motor columns
In addition to disrupted axon-endothelial interactions in the periphery, Plxnd1 mutant embryos displayed severe defects in vascular patterning within the SC. Specifically, the MCs, which at early embryonic stages are devoid of blood vessels (Himmels et al., 2017; James et al., 2009), became abnormally vascularized in mutants. Vessels normally formed half loops extending at the border of the MCs (Figures 3H and 3H′) and invaded this region only after E12.5 (see e.g., Figure 3A). Their intraspinal course depended on MC integrity, since vascularization was impaired following MN ablation (Figures S3D-S3E′). The stereotypical vascular pattern observed in control embryos was profoundly affected in Plxnd1C116S and Plxnd1-null homozygotes, as well as in embryos carrying EC-specific deletion of Plxnd1. Mutants displayed abnormal vessel ingression into the MC that resulted in significant increase in vascular density within this domain (Figures 3I-3L) accompanied by excessive filopodial sprouting indicative of deregulated angiogenesis (Figures S3F-S3G″). In contrast, vascularization of the dorsal SC was unaffected (Figures 3M, S3H, and S3I). Vascular invasion of the MC was observed at all segmental levels in Plxnd1 mutants (Figures S3L-S3R) and was concomitant with the first appearance of ectopic axon-endothelial bundles in the periphery at ~E11.5 (Figures S2S-S2T′). However, while axonal defects were restricted to MMC neurons, vessels spread irregularly throughout the MC, intermingling with the cell bodies of different MN subtypes (Figures S3I and S3L-S3Q′). Despite these changes, the number and columnar clustering of MNs, including MMC neurons, were not altered (Figures S3J-S3Q′). These results suggest that loss of Plexin-D1 in ECs impairs detection of ventral SC-derived signals, leading to premature and unrestrained vascular invasion of MNs (Figure 3N).
Motor axons repel endothelial cells through Plexin-D1
Our observations are consistent with direct MN-endothelial signaling mediated by Plexin-D1 but other cell types coexisting in the same territory might also contribute (Suter and Jaworski, 2019). To test unambiguously the interactions between MNs and ECs, we established a co-culture system in which human umbilical vein ECs (HUVECs) were seeded on top of mouse MN explants before axons had begun to extend. ECs initially adhered near the explant but progressively withdrew from the advancing axons, avoiding overlap, and eventually (~15 h later) formed a monolayer aligned with the axonal front (Figures 4A, 4B top, 4D, 4D′, and S4A-S4C′; Videos S1, S2, S3, and S4). EC death was not observed during this process. This opposing behavior indicated that ECs were repelled by motor axons, resulting in cell-free area that was measured to quantify the extent of repulsion (Figure 4C). The aversive signal was mostly associated with axons rather than cell bodies in the explant, since co-cultures grown on low-laminin substrate to prevent axon extension-while allowing EC adhesion/migration—displayed limited repulsion (Figures 4J, S4H, and S4H′). EC repulsion was strongly inhibited when Plxnd1 was knocked down (KD) in HUVECs prior to co-culture with motor explants (Figures 4B bottom, 4C, 4F, 4F′, 4J, S5L, and S5M; Videos S5 and S6), and this effect was recapitulated by treatment with a blocking antibody against Plexin-D1 (Figures 4J and S4I-S4J′). Cells lacking constitutive expression of Plxnd1, such as fibroblasts, were not repelled (Figures 4J and S4L). Therefore, Plexin-D1 is needed in ECs to detect repulsive signal(s) associated with motor axons. The repulsive activity was conserved in motor explants from chick embryos and also in this case depended on Plexin-D1 in ECs (Figures 4H, 4I, and 4K). Of note, Plxnd1 KD did not affect the rate of EC proliferation and migration per se (Figures 5M and S4Q).
Figure 4. Plexin-D1 is required for EC repulsion by motor axons.

(A) MN explant-EC repulsion assay.
(B) Live imaging of MN-EC co-cultures. (Top panels) HUVECs (EC) settle next to explants (0 h) but are repelled as axons extend. Dashed lines here and in following panels mark the EC front. (Bottom panels) Plxnd1 KD from ECs (siPlxnd1 EC) affects repulsion. “Control EC” transfected with non-targeting siRNA.
(C) EC repulsion quantified by measuring cell-free area after overnight (ON) growth. Mean, normalized to control ± SEM, Unpaired t test (***) p < 0.0001.
(D–G) ON co-cultures between Hb9MN::GFP motor explants and control (D and E) or siPlxnd1 EC (F and G) stained for EC marker VE-cadherin (VEcad) and neuronal βIII-tubulin.
(H and I) Co-cultures between motor explants from chick neural tube electroporated with GFP (green) and either control (H) or siPlxnd1 EC (I).
(J) EC repulsion after ON co-culture with mouse motor explants. “Control”: HUVEC WT or transfected with non-targeting siRNA; “siPlxnd1”: Plxnd1 KD; “anti-Plxnd1”: blocking antibody; “low-laminin” substrate; “MEF”/“3T3”: co-cultures with primary mouse embryonic fibroblasts or NIH-3T3. Mean, normalized to control ± SEM, ANOVA/Dunnett’s multiple comparison test (***) p < 0.0001 versus control.
(K) EC repulsion in co-cultures with chick motor explants. Mean, normalized to control ± SEM, ANOVA/Dunnett’s multiple comparison test (***) p < 0.0001 versus control.
(L) Axon extension from Hb9MN::GFP motor explants cultured alone or with either control or siPlxnd1 EC. Mean, normalized to control ± SEM, ANOVA/Dunnett’s test (***) p < 0.0001 versus control; unpaired t test (***) p < 0.0001 siPlxnd1 versus control.
(M) MN explants cultured with HUVEC-conditioned media versus control basal media. Mean, normalized to control ± SEM, Unpaired t test (ns) p = 0.61.
Scale bars: 200 μm in (B); 200 μm in (F) and (F’); 100 μm in (E) and (G); and 200 μm.
Figure 5. MN-derived Sema3C triggers EC repulsion through Plexin-D1/NRP receptors.

(A) Normalized RNA-seq counts for Semaphorins in Hb9MN::GFP+ MNs, GFP~ spinal cord cells (vSC) and DRG FACS-purified from E12.5 embryos. Sema3C is high in MNs while Sema3E levels are negligible.
(B) Sema3C detected in MCs by in situ hybridization at E11.5.
(C–D′) Vessels invade MCs (Hb9+, outlined) in Sema3C−/− (D and D′) but avoid this region in controls (Sema3C+/−, C and C′; E12.5 lumbar transverse sections).
(E) Quantification of vascular area (CD31+ px) within MCs. Mean, normalized to control ± SEM, ANOVA/Dunnett’s test (***) p < 0.00001 Sema3C−/− versus Ctrl; (ns) p > 0.1 Sema3E−/− and Sema4A−/− versus Ctrl.
(F–I) Proximal axon bundling (arrowheads) and epaxial nerve thinning (asterisks) in Sema3C−/− whole mounts (E12.5, dorsal view). Sema3E and Sema4A mutants are unaffected. Control is Sema3C heterozygous.
(J) Quantification of axon guidance phenotype. Mean ± SEM, ANOVA/Dunnett’s test (***) p = 0.0002 Sema3C−/− versus Ctrl; (ns) Sema3E−/− and Sema4A−/− versus Ctrl.
(K) Penetrance and severity of axon guidance defects in Sema3C mutants.
(L) Last frames (18 h) of scratch wound healing assay on control (siCtrl-transfected) or Plxnd1 KD HUVEC stimulated with Sema3C (lower panels) or left untreated (upper panels). Orange lines mark the initial wound margin; blue lines mark the migratory front. Both untreated control and siPlxnd1 EC migrate to close the gap. Sema3C inhibits migration of control but not siPlxnd1 EC.
(M) HUVEC migration rate in scratch assay. Mean ± SEM (see also Figure S5H).
(N and O) Hb9MN::GFP motor explants from either WT (N) or Sema3C−/− embryos (O) cultured ON with HUVEC.
(P) Quantification of EC repulsion in co-cultures with Sema3E or Sema3C KO motor explants. Repulsion from Sema3C heterozygous and KO explants (but not Sema3E−/−) is blunted. Mean, normalized to WT ± SEM, ANOVA/Dunnett’s test (***) p < 0.0001 Sema3C−/− versus Ctrl; (**) p = 0.0046 Sema3C+/~ versus Ctrl; (ns) p > 0.8 Sema3E+/~ and Sema3E−/− versus Ctrl.
(Q) Quantification of HUVEC repulsion from HEK cells overexpressing Sema3E (HEK:SemaE) or Sema3C (HEK:Sema3C). Representative images are shown in Figure S5P. Plxnd1 KD in HUVEC prevents repulsion from Sema3E and Sema3C. Nrp1 or Nrp2 KD impairs repulsion from Sema3C but not Sema3E. Mean, normalized to WT ± SEM, ANOVA/Dunnett’s test for HEK:Sema3C (***) p < 0.0001 siPlxnd1, siNrp1, siNrp2 versus Ctrl; for HEK:Sema3E (***) p < 0.0001 siPlxnd1 versus Ctrl; (ns) p > 0.5 siNrp1, siNrp2 versus Ctrl.
(R–T) Co-cultures between Hb9MN::GFP motor explants and control HUVEC (R) or HUVEC with KD of either Nrp1 (S) or Nrp2 (T).
(U) Silencing Nrp1 and Nrp2 impairs EC repulsion. Mean, normalized to WT ± SEM, ANOVA/Dunnett’s test (***) p = 0.0005 siNrp1 versus Ctrl; (***) p < 0.0001 siNrp2 versus Ctrl.
(V–Y) Co-cultures between motor explants and naive HEKs (V) or cells stably transfected with Plexin-D1 alone (W) or in combination with either NRP1 (Plxnd1/NRP1) (X) or NRP2 (Plxnd1/NRP2) (Y). HEKs are identified by F-actin (magenta) and nuclear (DAPI, cyan) staining. Motor axons grow over HEK-Ctrl and HEK-Plxnd1 but repel cells co-expressing Plexin-D1/NRPs. Dashed lines mark cell-free area.
(Z) Quantification of HEK cell repulsion. Mean, normalized to WT ± SEM, ANOVA/Dunnett’s test (***) p < 0.0001 Plxnd1/Nrp1 and Plxnd1/Nrp2 versus Ctrl; (ns) p = 0.44 Plxnd1 versus Ctrl.
Scale bars: 100 μm in (B); 100 μm in (C)x–(D’); 200 μm in (F)–(I); 400 μm in (L); 200 μm in (N)–(O); 200 μm in (R)–(T); and 200 μm in (V)–(Y).
See also Figure S5.
Motor axons rarely overshot the endothelial boundary, suggesting that ECs provided an unfavorable substrate (Figures 4D and 4E). Indeed, axon extension was reduced in co-cultures compared with explants grown alone (Figures 4L and S4G) and growth cones reaching the endothelial interface frequently exhibited a “collapsed” morphology indicative of avoidance behavior (Figure S4E). Axon growth was further impaired when explants were confronted with ECs silenced for Plxnd1, which they could not efficiently repel (Figure 4L). In this condition, axons were shorter, hyperfasciculated, and twisted at the interface with ECs; when they extended incoherently on the cell monolayer their growth cones were often abnormally enlarged (Figures 4G, S4D, and S4F). Thus, ECs appear to express non-permissive/repulsive factors that counter axon growth. These signals are likely to function at short range and/or in contact-dependent manner since motor axon elongation was not affected by HUVEC-conditioned media (Figure 4M).
We next asked how ECs responded when co-cultured with other populations of spinal neurons that, unlike MNs, project their axons exclusively within the central nervous system (CNS). Dorsal SC explants containing commissural interneurons were inefficient at repelling HUVECs, and their limited repulsive activity was independent of Plexin-D1 (Figures S4N-S4P). However, as for MNs, axon growth from dorsal explants was reduced in the presence of ECs (Figure S4N, compare with S4M). Likewise, SC interneurons inadvertently included within MN explants were unable to repel ECs but avoided crossing their boundary (Figure S4C′).
Taken together, these in vitro data indicate that signals from MNs repel ECs through Plexin-D1, and conversely that ECs express factors that hinder axon growth. The effect of disrupted balance between these opposing activities unmasked by Plxnd1 silencing in co-cultures is reminiscent of the phenotype observed in Plxdn1 mutant embryos, characterized by axons stalling against an “endothelial barrier.”
Motor neuron-derived Sema3C triggers endothelial cell repulsion via Plexin-D1/Neuropilin receptors
To identify relevant Plexin-D1 ligands that would mediate the repulsive effect of MNs on ECs, we first surveyed expression of Semaphorins in RNA-seq data of MNs (Bonanomi et al., 2019). MNs expressed members of the family but surprisingly lacked Sema3E—the canonical Plexin-D1 ligand—which was undetectable when phenotypes developed in Plxnd1 mutants (Figures 5A and S5B). Additional ligands for Plexin-D1 have been proposed (Feiner et al., 2001; Gitler et al., 2004; Hamm et al., 2016; Liu et al., 2016; Torres-Vázquez et al., 2004; Toyofuku et al., 2007). Among these, Sema3C was expressed at high levels in MNs (Figures 5A, 5B, S5A, and S5E) and was detected in all MN subtypes along the rostrocaudal axis, with relative enrichment in the MMC (Figure S6D; data not shown). Next, we tested whether KO embryos for candidate Plexin-D1 ligands displayed the phenotypes observed in Plxnd1 mutants. In addition to Sema3E and Sema3C mutants, we analyzed embryos KO for Sema4A—a Plexin-D1 ligand expressed in MNs (Figure 5A; Toyofuku et al., 2007). Loss of Sema3C resulted in abnormal vascular ingression into the MC, similar to Plxnd1 KO, whereas vessels remained excluded from this region in Sema3E−/− and Sema4A−/− embryos (Figures 5C-5E). However, vascular defects were less pronounced in Sema3C−/− compared with Plxnd1 mutants (Figure 5E compare with Figure 3L). Next, ISLMN::fGFP was crossed into Sema3E, Sema3C, and Sema4A KO mice to analyze motor projections. Axon targeting was normal in Sema3E−/− and Sema4A−/− embryos (Figures 5F-5H and 5J) despite disorganization of the intersomitic vasculature in Sema3E mutants (Figures S5C and S5D; Gu et al., 2005). In contrast, ~60% of Sema3C−/− embryos displayed motor axon defects resembling the phenotype of Plxnd1 mutants, albeit with reduced severity (Figures 5I-5K). Thus, loss of Sema3C causes vascular and axon guidance errors that phenocopy Plxnd1 mutation, suggesting that Sema3C signals via Plexin-D1 to regulate MN-EC interactions.
The incomplete penetrance of Sema3C KO might indicate compensation from other Semaphorins (Figures 5A, S5A, and S5F). We reasoned that examining directly the interaction between axons and ECs in vitro might reveal a requirement partially masked in vivo. A “scratch assay” showed that Sema3C inhibited EC migration through Plexin-D1 (Figures 5L and 5M). Sema3E−/− motor explants repelled ECs, whereas this response was hindered with explants from either Sema3C heterozygous (~20% decrease) or KO (~60% decrease) embryos (Figures 5N-5P, S5J, and S5K). Inefficient repulsion in Sema3C+/− co-cultures suggests that ECs are sensitive to intermediate decrease in Sema3C doses (Figures S5G and S5H). Motor axon extension was normal in Sema3C KO explants, indicating that impaired EC repulsion was not due to defective axon growth (Figure S5I). In conclusion, Sema3C accounts for a large portion of the repulsive activity of motor axons toward ECs detected in co-cultures.
Sema3C binds to Neuropilin-1 (NRP1) and Neuropilin-2 (NRP2) while Plexin-D1 functions as a co-receptor (Chen et al., 1998; Christie et al., 2021). Besides enabling signaling, Plexin-D1 enhanced binding of Sema3C to NRP (Gitler et al., 2004) and the C116S (Drake) mutation reduced this effect (Figures S5N and S5O). HUVECs with KD of either Plxnd1, Nrp1, or Nrp2 showed impaired repulsion from Sema3C released by HEK cells, confirming that while Plexin-D1 mediates Sema3E signaling directly, it relies on NRPs to transduce Sema3C signal (Figures 5Q and S5P). Consistent with this requirement, either Nrp1 or Nrp2 KD in HUVECs compromised repulsion from motor explants (Figures 5R-5U). The magnitude of the decrease was less pronounced than after Plxnd1 silencing, possibly due to redundancy and compensation between NRPs (Figure S5M) (Chen et al., 1998; Gitler et al., 2004; Gu et al., 2003; Plein et al., 2015). Therefore, Plexin-D1/NRP complexes are required in ECs to detect repulsive MN signals. To test sufficiency, we generated HEK cell lines expressing Plexin-D1 alone or together with either NRP1 or NRP2 and cultured them with motor explants. Naive HEKs, which lack Plxnd1, Nrp1, and Nrp2, did not respond to either Sema3E or Sema3C and were not repelled by motor axons (Figures 5V, S5Q, and S5R; data not shown). In contrast, HEKs stably transfected with these receptors responded predictably to Sema3E (HEK-Plxnd1) and Sema3C (HEK-Plxnd1/NRP1 or Plxnd1/NRP2) (Figures S5Q and S5R). When co-cultured with motor explants, HEKs expressing Plexin-D1 alone were not repelled, while those co-expressing Plexin-D1 and NRP gained repulsion (Figures 5W-5Z). Together, these studies show that Sema3C derived from MNs repels ECs through Plexin-D1/NRP complexes.
In silico screening of signaling interactions governing motor-endothelial crosstalk
Communication between MNs and ECs is impaired by loss of Plexin-D1. To gain insights into the underlying mechanisms, ECs from the SC and surrounding meninges were sorted from Plxnd1−/− embryos and control littermates based on KdrEC::Cherry reporter signal and analyzed by RNA-seq (Figure 6A). The meningeal sheath covering the SC and VRs was included in order to capture ECs associated with the proximal nerve region where abnormal axon-vessel interactions develop in mutants. From the same embryos, we sorted and sequenced ISLMN::fGFP+ MNs to identify secondary (non-cell autonomous) transcriptional changes resulting from loss of Plxnd1 in ECs (Figure 6A). EC profiling of Plxnd1 mutants identified moderate deregulation in genes involved in vascular development (Figures 6B and 6C; Table S1). Although MNs purified in parallel to ECs did not express Plxnd1 (Figure S6A), in mutant embryos, they displayed changes in a restricted set of genes related to axon guidance and cell-cell interactions (Figures 6D and 6E; Table S1). Sema3C and vascular endothelial growth factor (Vegfa) increased, suggesting that MNs react to perturbations resulting from loss of vascular Plxnd1 by modulating genes that signal directly to ECs. Expression-weighted cell-type enrichment (EWCE) showed that genes upregulated in Plxnd1−/− MNs were overrepresented in a MMC subset identified by single-cell RNA-seq (scRNA-seq) (Amin et al., 2021; Figures 6F, S6B, and S6C). Therefore, a significant degree of gene expression changes that developed in MNs as a result of Plxnd1 loss in the endothelium were confined to epaxial MNs, whose axons interacted abnormally with vessels in mutants.
Figure 6. Mutual signaling underlying MN-EC crosstalk.

(A) ECs (from spinal and meningeal vasculature) and MNs were FACS-purified from E12.5 embryos expressing ISLMN::fGFP+/KdrEC::Cherry reporters and analyzed by RNA-seq.
(B) Volcano plot of genes upregulated (160, red) or downregulated (65, blue) in Plxnd1−/− ECs (adjusted p < 0.01). Selected DEGs with highest significance are labeled.
(C) Gene ontology (GO) enrichment of DEGs in Plxnd1−/− ECs (Plxnd1 was removed). Dot plot shows the number of genes associated with GO term (x axis), gene count ratio (circle size) and enrichment significance (color scale). Selected upregulated (red) and downregulated (blue) genes are shown.
(D) Volcano plot of genes upregulated (52, red) or downregulated (23, blue) in MNs of Plxnd1−/− embryos (adjusted p < 0.01).
(E) GO enrichment of DEGs in MNs from Plxnd1−/− embryos.
(F) Stratification of Plxnd1−/− DEGs among MN subtypes by EWCE. Upregulated genes, red; downregulated genes, blue. (***) p = 0.0002 immature; p = 0.0005 MMCa.
(G) Counts of ligand-receptor interactions identified by CellPhoneDB between MN subtypes (E12.5) and ECs isolated from the mesenchyme of E11–E13 mouse embryos.
(H) GO of ligand-receptor pairs between MMC and EC identified by CellPhoneDB.
(I and J) Representation of the strongest ligand-receptor interactions between MMC and EC from a curated list of 675 pairs associated with cell guidance and angiogenesis. (I) Interactions between MMC ligands (31) and EC receptors (32). (J) Interactions between EC ligands (46) and MMC receptors (20). Color scale, “interaction scores.” Circle size, lowest percentage of cells of either type expressing a gene in the pair.
(K and L) Expression of receptors (K) and ligands (L) measured by RNA-seq of ECs isolated from either the meninges (Men_EC) or spinal cord (Sp_EC) of E12.5 embryos. Several ligands were higher in Men_ECs (20/46; adjusted p < 0.001) including ephrins and ECM factors. Most of these genes were expressed in HUVECs (Table S5).
Robust signaling was inferred between MMC neurons and ECs on the basis of expression of matched ligand-receptor genes, which were associated with neuronal guidance and vascular morphogenesis pathways (Figures 6G and 6H; Table S3). Among the strongest predicted interactions, we detected MMC-derived Sema3C coupled to endothelial Plexin-D1, NRP1, and NRP2 (Figures 6I and S6D; Table S4). MMC neurons expressed several additional factors that could either inhibit (Sema3A, Slit2, and EphrinB2) or promote (Vegfa and pleiotrophin) angiogenesis (Figures 6I and S6D; Table S4). Conversely, EC expressed signals that might influence axon growth, including adhesive ECM molecules (collagens, laminins, and perlecan/HSPG2) or repulsive factors such as Semaphorins (Sema6A and Sema6D) and ephrins (Efna1, Efna2, Efnb1, and Efnb2) (Figures 6J and S6E; Table S4). All EC receptors and ligands predicted to be involved in signaling with MMC neurons (Figures 6I and 6J) were detected in the intraspinal and meningeal endothelium, often displaying differential expression (Figures 6K, 6L, and S6F-S6J; Table S5), and were therefore present in vessels within the regions where neurovascular defects arise in Plxnd1 mutants (i.e., the SC and VR).
Plexin-D1 antagonizes VEGF to control motor neuron-endothelial interactions
Aberrant MC vascularization in Plxnd1 mutants suggests unmasking of attractive signals associated with MNs. The release of EC attractants from MNs was demonstrated by enhanced migration of HUVECs toward MN-conditioned media (Figures 7A, 7B, and 7D). Treatment with a neutralizing anti-VEGF164 antibody abolished this response (Figures 7C and 7D) but did not interfere with chemoattraction per se (Figure S7C). Thus, VEGF-A secreted from MNs (Figures S7A-S7B′) (Himmels et al., 2017; Mukouyama et al., 2005) attracts ECs at long range, overriding the repulsive signals from motor explants revealed in co-cultures. Since Plxnd1 KD did not perturb EC migration toward MN-conditioned media or recombinant VEGF, the two pathways appear to operate independently (Figures S7D-S7I). To address how antagonistic neuronal signals were integrated by ECs, we treated motor-endothelial co-cultures with anti-VEGF antibody. VEGF depletion partially restored repulsion of Plxnd1-KD HUVECs from motor explants (Figures 7E-7I′) and concomitantly improved axon growth (Figures 7I versus 7H). These results suggest that EC attraction toward MNs in the absence of efficient repulsion interferes with nerve projection. When attraction was attenuated by anti-VEGF treatment, Plexin-D1-independent repulsive signals associated with MNs could partially rescue axon-endothelial interactions.
Figure 7. Integration of opposing motor-derived stimuli by ECs controls axon targeting and SC vascularization.

(A–C) Crystal violet staining of HUVECs migrated to the bottom side of Transwell membrane. Migration is stimulated by MN-conditioned media in the lower compartment (B) relative to control basal media (A). The effect is prevented by neutralizing anti-VEGF164 antibody (VEGFNab) added to the lower chamber during migration (6 h) (C).
(D) Quantification of HUVEC migration. Mean, normalized to Ctrl ± SEM, ANOVA/Dunnett’s test (***) p < 0.0001 MN versus basal; (ns) p = 0.26 MN/VEGFNab versus basal.
(E) Quantification of cell repulsion in co-cultures between WT motor explants and control or siPxlnd1 HUVECs either treated with VEGFNAb or left untreated. Plxnd1 KD (siPlxnd1) prevents repulsion, and this effect is partially reversed by VEGF blockade (siPlxnd1/VEGFNab). VEGFNab does not affect repulsion of control cells (Ctrl/VEGFNab) compared with co-cultures in basal media (Ctrl). Mean, normalized to Ctrl ± SEM, ANOVA/Dunnett’s test (***) p < 0.0001 siPlxnd1 and siPlxnd1/VEGFNab versus Ctrl; (ns) p = 0.85 Ctrl/VEGFNab versus Ctrl; unpaired t test (***) p < 0.0001 siPlxnd1/VEGFNab versus siPlxnd1.
(F–I′) Motor explants co-cultured with siPlxnd1 or control HUVEC with or without VEGFNab. The fluorescent-secondary antibody used for VE-cadherin staining (red) also reveals VEGF in explants treated with VEGFNab due to matching host species of the primary antibodies (asterisks in G′ and I′).
(J–M′) Aberrant ingrowth of vessels into the MC (dashed lines) in Plxnd1 mutants (K and K′, arrow) is rescued by conditional deletion of Vegfa in MNs with Olig2MN::Cre (M and M′). Plxnd1−/− embryos heterozygous for Vegfa-floxed allele are not rescued (L and L′, arrow).
(N) Quantification of vascular area (CD31+ px) within MCs. Control and Plxnd1−/− measurements are also shown in Figure 3L. Mean, normalized to Ctrl ± SEM, ANOVA/Dunnett’s test (***) p < 0.00001 Plxnd1−/− and Plxnd1−/−;VEGFhet versus Ctrl; (ns) p > 0.1 0.1 Plxnd1−/−;VEGFflox versus Ctrl.
(O) Vascular invasion of Plxnd1 mutants is rescued by Vegfa inactivation in MNs.
(P–S) Axon bundling (arrowheads) and epaxial nerve thinning (asterisks) distinctive of Plxnd1 mutants are partially corrected by conditional deletion of Vegfa in MNs (E12.5 whole mounts, dorsal view).
(T) (Left) Quantification of axon guidance phenotype. Gradual rescue of Plxnd1−/− axon defects by deletion of one (Plxnd1−/−;VEGFhet) or two VEGF alleles (Plxnd1−/−;VEGFflox) in MNs. Mean ± SEM, ANOVA/Dunnett’s multiple comparison test (***) p < 0.0001 Plxnd1−/−;VEGFhet and Plxnd1−/−;VEGFflox versus Plxnd1−/−. (Right) Penetrance and severity of phenotype in Plxnd1/Vegfa compound mutants compared with Plxnd1−/−.
(U) Rescue of axon guidance errors of Plxnd1 mutants by Vegfa gene inactivation in MNs.
(V) (Left) Interactions between motor axons and vessels in WT and Plxnd1 mutant embryos (top). Dashed circles mark the choice point where epaxial MN axons use Sema3C/Plexin-D1 to displace meningeal ECs (ECMen) that cross their path (bottom panels). Push-pull signals (including Sema3C and VEGF) prevent disruptive interactions and instruct alignment. Loss of EC repulsion in Plxnd1 mutants results in unrestricted attraction to MNs and unmasks inhibitory vascular signals that obstruct axonal projection. In addition, vessels invade the MC. (Right) ECs detect Sema3C via Plexin-D1 in complex with NRPs. This repulsive pathway operates in parallel to VEGF/VEGFR2 attractive signaling. Conversely, ECs express both repellents and adhesive factors that signal to MNs.
Scale bars: 200 μm in (F)–(I′); 100 μm in (J)–(M′); and 100 μm in (P)–(S).
See also Figure S7.
This model was tested in vivo by generating Plxnd1 KO embryos in which Vegfa was conditionally deleted in MNs with Olig2MN::Cre and motor axons were traced with ISLMN::fGFP. First, we examined vascular patterns in compound mutants and found that while the peripheral vasculature was as disorganized as in Plxnd1−/− embryos, aberrant vascularization of the MC was corrected by homozygous deletion of Vegfa (Figures 7J-7O and S7J-S7M′). Therefore, VEGF released by MNs promotes ectopic vessel formation in the MCs when EC attraction is not counterbalanced by Plexin-D1-mediated repulsion. Likewise, the analysis of motor projections revealed that deletion of Vegfa resulted in dose-dependent correction of the distinctive axon guidance errors of Plxnd1−/− embryos, characterized by proximal stalling of epaxial nerves (Figures 7P-7U). These mouse genetic studies indicate that loss of Plxnd1 offsets the balance of attractive and repulsive signals between MNs and ECs, which can be partially recovered by lowering VEGF-driven attraction toward MNs (Figure 7V).
DISCUSSION
In his first descriptions of growth cones navigating in the embryonic nervous system, Ramón y Cajal captured two main problems of pathfinding: one is the “exquisite chemical sensitivity” that enables axons to select the correct trajectory; the other is their ability to “remove or overcome the obstacles” found along the path (Ramón y Cajal, 1909). Much of the molecular work in the field has focused on how axons respond to guidance cues, whereas the way they confront impediments is not well understood. We show that in order to avoid interference from heterotypic cellular encounters, neurons remodel the tissue microenvironment along their axonal routes by deploying classic guidance factors in a reversal of the typical ligand-receptor signaling orientation. We found MNs release Sema3C to displace ECs from their course. This aversive relationship was unexpected since the interactions between neurons and ECs are typically viewed through the principle of congruency achieved via mutual attraction (Andreone et al., 2015). Nevertheless, the synchronous development of nerves and vessels, together with their reciprocal affinity aimed at metabolic coupling, carry the risk of detrimental contacts that might perturb neural circuit wiring as well as vasculature patterning. The coexistence of angiogenesis and axonal outgrowth is also common in disease and injuries where vessels form close to nerve sprouts to promote repair but might obstruct regrowth due to their chaotic topography at the lesion site (Cattin et al., 2015; Dray et al., 2009; Muramatsu et al., 2012). It is possible that embryonic mechanisms exploited by axons to bypass vascular obstacles are reactivated during neovascularization of adult tissues.
Axon-endothelial push-pull prevents disruptive contacts and enables congruency
Motor axons avert inappropriate contacts with ECs by counter-balancing attraction mediated by VEGF with repulsion elicited via Sema3C. Neurovascular interactions appear to be mediated by a hierarchical, weighted signaling system that integrates opposing cues from both compartments in order to attain proximity yet preventing interference (Figure 7V). Sema3C/Plexin-D1 and VEGF (presumably via VEGFR2 receptor) constitute primary axes that act in concert with other neuronal/vascular pathways predicted by computational tools. Loss of Plexin-D1 does not affect EC attraction but unmasks vascular signals that obstruct axon projection. Conversely, when attraction is reduced (e.g., by VEGF inactivation), secondary, Plexin-D1-independent, repulsive factors from MNs restore to some extent axon-endothelial crosstalk. It is possible that these responses involve mechanical cues associated with nerves and vessels, which may become dominant when the balance of chemical signals is disrupted. Of note, the mechanosensing property of Plexin-D1, which is independent of Semaphorins, might provide an alternative means to control contacts with axons (Mehta et al., 2020).
EC repulsion prevails at the epaxial turning point while attraction outweighs past this region resulting in nerve-vessel alignment (Figure 7V). These choregraphed interactions might be influenced by the range of action of neurovascular cues. Our assays indicate that Sema3C-repulsion functions at short range, while VEGF-attraction operates at distance. Consistent with its limited diffusion, Sema3C elicits autocrine signaling in MNs (Sanyas et al., 2012). Despite all MNs express Sema3C, nerve-targeting defects in Plxnd1 and Sema3C mutants are restricted to epaxial MNs. The susceptibility of MMC axons might depend on the timing of pathfinding, which coincides with vascular patterning at the epaxial choice point. Other MN subtypes extend in advance of the developing vessels, whereas MMC axons undergo a “waiting period” and this delay may require that they have mechanisms to displace ECs occupying their late-forming trajectory (Figure 7V; Tosney and Landmesser, 1985b). Strong signaling crosstalk was predicted in silico between MMC and ECs. Hence, the molecular features of neurons and vessels, together with the spatiotemporal dynamics of their encounters, may dictate specificity and outcomes of neurovascular interactions. Axon-endothelial collisions in Plxnd1 mutants reverberate in secondary transcriptional changes within MNs. Interestingly, Sema3C and Vegfa were increased, suggesting that the signaling system controlling neurovascular communication might be under feedback regulation.
Sema3C/Plexin-D1 controls axon-endothelial interactions and spinal cord vascularization
The effects of Sema3C on ECs have been studied in heart development, where Sema3C drives endothelial-to-mesenchymal transition (Plein et al., 2015). On the other hand, recombinant Sema3C countered pathological neovascularization in retinopathy (Yang et al., 2015) and inhibited tumor angiogenesis (Mumblat et al., 2015). Here, we demonstrate a requirement for Sema3C in physiological angiogenesis during CNS vascularization as well as in neurovascular interactions critical for nerve projection. Both motor axon targeting and SC vascularization rely on detection of opposing neuronal signals by ECs. Although mechanistically related, these appear to be separate processes as underscored by the fact that the axonal phenotypes of Sema3C and Plxnd1 mutants were restricted to epaxial nerves while vessels grew abnormally throughout all MN divisions. Since the onset of vessel ingression in the SC coincides with clustering of MNs and axon outgrowth, it has been proposed that MC vascularization is delayed in order to avoid interference with these processes (Himmels et al., 2017; James et al., 2009). One way, MNs prevent early vascular invasion is through expression of the decoy receptor sFlt1, which limits VEGF availability thereby countering EC attraction (Himmels et al., 2017). We found that sFlt1 levels were unchanged in the MC of Sema3C−/− embryos (data not shown) suggesting that VEGF-titration and Sema3C/Plexin-D1 function independently in a two-tiered system that defines timing and patterning of MC vascularization.
Targeted removal of cellular obstacles in directional migration
All forms of directional cell migration face the challenge of sensing and overcoming impediments. It has remained unclear whether specialized avoidance mechanisms are used to resolve encounters with obstacles during chemotaxis (Grima, 2007). Immune and cancer cells, similar to neuronal growth cones, exert protrusive forces and release proteases to facilitate locomotion in restrictive environments (Gaertner et al., 2022; Murphy and Courtneidge, 2011). However, these seemingly generic solutions might be inadequate alone to account for complex morphogenetic processes that rely on highly orchestrated multicellular interactions during organogenesis and tissue repair. It is therefore likely that obstacle-removal strategies based on selective targeting of signaling crosstalk with subsets of neighboring cells represent common mechanisms to ensure chemotactic movement in different biological contexts beyond nervous system wiring.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dario Bonanomi (bonanomi.dario@hsr.it).
Materials availability
Plasmids, cell lines and mice generated in this study are available from the lead contact upon request pending MTA approval.
Data and code availability
All original RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. The accession number is listed in the key resources table.
All original code is available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-Isl1/2 | Ericson et al., 1992 | N/A |
| rabbit anti-Hb9 | Thaler et al., 1999 | N/A |
| rabbit anti-GFP | Thermo Fisher Scientific | Cat# A-6455 RRID:AB_221570 |
| mouse anti-GFP | Thermo Fisher Scientific | Cat# A-11120 RRID: AB_221568 |
| chick anti-GFP | Abcam | Cat# ab13970 RRID:AB_300798 |
| mouse anti-β3 tubulin | Abcam | Cat# ab7751 RRID:AB_306045 |
| mouse anti-FLAG M2 | Sigma-Aldrich | Cat# F1804 RRID:AB_262044 |
| goat anti-Plexin-D1 | R&D Systems | Cat# AF4160 RRID:AB_2237261 |
| goat anti-VE-Cadherin | R&D Systems | Cat# AF938 RRID:AB_355726 |
| rabbit anti-VE-Cadherin | Abcam | Cat# ab33168 RRID: AB_870662 |
| rat anti-CD31 | BD Biosciences | Cat# 550274 RRID:AB_393571 |
| goat anti-CD31 | R&D Systems | Cat# AF3628 RRID:AB_2161028 |
| rabbit anti-Fsp1 | Millipore | Cat# 07-2274 RRID:AB_10807552 |
| rabbit anti-VAChT | Synaptic Systems | Cat# 139 103 RRID:AB_887864 |
| rabbit anti-GAPDH | Cell Signaling Technology | Cat# 2118 RRID:AB_561053 |
| rabbit anti-Pan-Actin | Cell Signaling Technology | Cat# 4968 RRID:AB_2313904 |
| rat anti-Semaphorin3C | R&D Systems | Cat# MAB1728 RRID:AB_2301533 |
| goat anti-NRP1 | R&D Systems | Cat# AF566 RRID:AB_355445 |
| goat anti-NRP2 | R&D Systems | Cat# AF2215 RRID:AB_2155371 |
| goat anti-VEGF164 | R&D Systems | Cat# AF564 RRID:AB_2212821 |
| donkey anti-goat IgG, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11055 RRID:AB_2534102 |
| donkey anti-goat IgG, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-21432 RRID:AB_2535853 |
| donkey anti-goat IgG, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21447 RRID:AB_2535864 |
| donkey anti-mouse IgG, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21202 RRID:AB_141607 |
| donkey anti-mouse IgG, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# 31570 RRID:AB_2536180 |
| donkey anti-mouse IgG, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31571 RRID:AB_162542 |
| donkey anti-rabbit IgG, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21206 RRID:AB_2535792 |
| donkey anti-rabbit IgG, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-31572 RRID:AB_162543 |
| donkey anti-rabbit IgG, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31573 RRID:AB_253618 |
| goat anti-rat IgG, Alexa Fluor 546 | Thermo Fisher Scientific | Cat# A-11081 RRID:AB_2534125 |
| ChromPure Goat IgG, whole molecule | Jackson ImmunoResearch Labs | Cat# 005-000-003 RRID:AB_2336985 |
| Chemicals, peptides, and recombinant proteins | ||
| ENU, N-Ethyl-N-nitrosourea | Sigma-Aldrich | Cat# N3385 |
| Laminin Mouse Protein | Gibco | Cat# #23017015 |
| Fibronectin, human | Corning | Cat# 354008 |
| Recombinant Human VEGF165 | Miltenyi Biotec | Cat# 130-109-395 |
| Recombinant Human GDNF | R&D Systems | Cat# 212-GD |
| Recombinant Mouse CXCL12/SDF-1 alpha | Prospec | Cat# CHM-324 |
| Recombinant Human/Murine/Rat BDNF | Peprotech | Cat# 450-02 |
| Recombinant Human Semaphorin 3C Fc | R&D Systems | Cat# 5570-S3 |
| complete, EDTA-free Protease Inhibitor Cocktail | Merck/Roche | Cat# 04693132001 |
| Halt Phosphatase Inhibitor Cocktail | Thermo Fisher Scientific | Cat# 78420 |
| DharmaFECT4 transfection reagent | Dharmacon | Cat# T-2004-01 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | Cat# 11668027 |
| Trizol LS Reagent | Thermo Fisher Scientific | Cat# 10296010 |
| Critical commercial assays | ||
| Papain Dissociation System | Worthington Biochemical | Cat# LK003153 |
| VECTASTAIN Elite ABC-HRP Kit, Peroxidase (Goat IgG) | Vector Laboratories | Cat# PK-6105 |
| DAB Substrate Kit, Peroxidase (HRP) | Vector Laboratories | Cat# SK-4100 |
| ClonExpress II One Step Cloning Kit | Vazyme | Cat# C112-01 |
| GENEART Site-Directed Mutagenesis System | Thermo Fisher Scientific | Cat# A13282 |
| M-MLV Reverse Transcriptase | Thermo Fisher Scientific | Cat# 28025013 |
| Random Hexamers | Thermo Fisher Scientific | Cat# N8080127 |
| SYBR Select Master Mix | Thermo Fisher Scientific | Cat# 4472913 |
| T7 Quick High Yield RNA Synthesis Kit | New England Biolabs | Cat# E2050S |
| mMESSAGE mMACHINE™ T7 Transcription Kit | Thermo Fisher Scientific | Cat# AM1344 |
| RNeasy Plus Mini kit | Qiagen | Cat# 74134 |
| RNase-Free DNase Set | Qiagen | Cat# 79254 |
| Illumina mRNA-Seq Sample Prep Kit | Illumina | Cat# RS-100-0801 |
| TruSeq RNA Library Preparation Kit v2 | Illumina | Cat# RS-122-2001 |
| Deposited data | ||
| RNA-seq data | This study | GEO: GSE207942 |
| Experimental models: Cell lines | ||
| HUVEC (Umbilical Vein Endothelial Cells) | Lonza | Cat# C2519A |
| AD-293 | Stratagene | Cat# 240085 |
| COS-7 | ATCC | Cat# CRL-1651 |
| NIH-3T3 | ATCC | Cat# CRL-1658 |
| Experimental models: Organisms/strains | ||
| Mouse: ISLMN::fGFP: Tg(Isl1-EGFP*)1Slp/J | Lewcock et al., 2007 | JAX:017952 |
| Mouse: Olig2::Cre | Dessaud et al., 2007 | N/A |
| Mouse: Hb9MN::GFP | Lee et al., 2004 | N/A |
| Mouse: MN218-2)::GFP | Amin et al., 2015 | N/A |
| Mouse: Tek::Cre (Tie2::Cre) | Kisanuki et al., 2001 | JAX:008863 |
| Mouse: Kdr::Cherry (Flk1-myr::mCherry) | Larina et al., 2009 | JAX:018542 |
| Mouse: Plexin-D1 flox | Zhang et al., 2009 | JAX:018319 |
| Mouse: Plexin-D1 knockout | This study | N/A |
| Mouse: R26-DTALSL (Rosa26-DTA176) | Wu et al., 2006 | JAX: 010527) |
| Mouse: Sema3C knockout | Feiner et al., 2001 | N/A |
| Mouse: Sema3E knockout | Gu et al., 2005 | N/A |
| Mouse: Sema4A knockout | This study | N/A |
| Mouse: VEGFflox | Gerber et al., 1999 | N/A |
| Oligonucleotides | ||
| gRNA Sema4A #1 5’-TGGGGTGAGTAGCGGGCATAAGG | This study Guide RNA | N/A |
| gRNA Sema4A #2 5’- GCAGCGTGTCAAAGTCTCGGAGG | This study Guide RNA | N/A |
| Mouse Plexin-D1 C116S mutagenesis (forward primer) 5’-GCAGGCCTCGAGCGAGCAC | This study | N/A |
| Mouse Plexin-D1 C116S mutagenesis (reverse primer) 5’-GTGCTCGCTCGAGGCCTGC | This study | N/A |
| control siRNA (Non-targeting Pool) | Dharmacon | Cat#D-001810-10-05 |
| Human PLXND1 (23129) siRNA SMARTpool | Dharmacon | Cat# L-014121-01-0005 |
| Human NRP1 (8829) siRNA SMARTpool | Dharmacon | Cat# L-019484-00-0005 |
| Human NRP2 (8828) siRNA SMARTpool | Dharmacon | Cat#L-017721-00-0005 |
| Primers for quantitative RT-PCR | This study | Table S6 |
| Recombinant DNA | ||
| Plasmid: CMV-PlexinD1-FLAG | this study | N/A |
| Plasmid: CMV-PlexinD1-C116S-FLAG | this study | N/A |
| Plasmid: CMV-Plexin-D1-FLAG-P2A-Puro | this study | N/A |
| Plasmid: PiggyBac-CAG-NRP1-HA | this study | N/A |
| Plasmid: PiggyBac-CAG-NRP2-HA | this study | N/A |
| Plasmid: pCMV-HAhyPBase | Yusa et al., 2011 | N/A |
| Plasmid: AP-Sema3E | Gu et al., 2005 | N/A |
| Plasmid: AP-Sema3C | Chen et al., 1998 | N/A |
| Plasmid: pN1-EGFP | Clontech | Addgene Cat# 6085-1 |
| Plasmid for in situ probe: mouse Plxnd1 (GenBank: NM_026376; 3544-6913bp) | Gu et al., 2005 | N/A |
| Plasmid for in situ probe: mouse Vegfa (GenBank: NM_001317041.1; 1431-1849bp) | Ruiz de Almodovar et al., 2011 | N/A |
| Plasmid for in situ probe: mouse Sema3C (GenBank: NM_013657.5; 546-1441bp) | this study | N/A |
| Plasmid for in situ probe: mouse Sema3E (GenBank: NM_011348; 1130-1920bp) | this study | N/A |
| Software and algorithms | ||
| STAR aligner | Dobin et al., 2013 | RRID:SCR_004463 |
| featureCounts | Liao et al., 2014 | RRID:SCR_012919 |
| ComBat | Zhang et al., 2020 | RRID:SCR_010974 |
| Bioconductor | Huber et al., 2015 | RRID:SCR_006442 |
| DESeq2 | Love et al., 2014 | RRID:SCR_015687 |
| Enrichr | Xie et al., 2021 | RRID:SCR_001575 |
| GeneCodis | Tabas-Madrid et al., 2012 | RRID:SCR_006943 |
| UMI-tools | Smith et al., 2017 | RRID:SCR_017048 |
| SEURAT | Hao et al., 2021 | RRID:SCR_007322 |
| Expression Weighted Celltype Enrichment (EWCE) | Skene and Grant, 2016 | https://doi.org/10.18129/B9.bioc.EWCE |
| CellPhoneDB | Efremova et al., 2020 | RRID:SCR_017054 |
| PROVEAN | Choi and Chan, 2015 | RRID:SCR_002182 |
| MOSAIK | Lee et al., 2014 | RRID:SCR_005486 |
| CRISPR Tool | Zhang Lab, MIT | http://crispr.mit.edu/ |
| Fiji | http://fiji.sc | RRID:SCR_002285 |
| ImageJ | https://imagej.net/ | RRID:SCR_003070 |
| Color Profiler (ImageJ plugin) | ImageJ | https://imagej.nih.gov/ij/plugins/color-profiler.html |
| FeatureJ (ImageJ plugin) | ImageJ | https://imagescience.org/meijering/software/featurej/ |
| Adobe Photoshop | Adobe Systems Inc. | RRID:SCR_014199 |
| Arivis Vision4D | Arivis-ZEISS | RRID:SCR_018000 |
| GraphPad Prism | GraphPad Software, Inc | RRID:SCR_002798 |
| Other | ||
| Endothelial Cell Growth Basal Medium-2 (EBM-2) | Lonza | Cat# CC-3156 |
| EGM-2 Endothelial factors | Lonza | Cat#CC-4176 |
| RNA scope probe for mouse Plxnd1 | ACD Bio-Techne | Cat# 405931 |
| Alexa Fluor 488 Phalloidin | Thermo Fisher Scientific | Cat# A12379 |
| Alexa Fluor 555 Phalloidin | Thermo Fisher Scientific | Cat# A34055 |
| Hydrogel Solution | Logos Biosystems | Cat# C1310X |
| Electrophoretic Tissue Clearing Solution | Logos Biosystems | Cat# C13001 |
| X-Clarity Mounting Solution | Logos Biosystems | Cat# C13101) |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse lines
Olig2::Cre (Dessaud et al., 2007); TekEC::Cre (Jax stock# 008863 (Kisanuki et al., 2001); ISLMN::fGFP (Jax stock# 017952 (Lewcock et al., 2007); Hb9MN::GFP (Lee et al., 2004); MN(218-2)::GFP (Amin et al., 2015); KdrEC::Cherry (Jax stock# 018542) (Larina et al., 2009); PlexinD1flox (Jax stock# 018319) (Zhang et al., 2009); Plxnd1-null allele was generated from crossing PlexinD1flox with ubiquitous EIIa-cre (Jax stock# 003724) (Lakso et al., 1996). R26-DTALSL (Jax stock# 010527) (Wu et al., 2006); Sema3C−/− (Feiner et al., 2001); Sema3E−/− (Gu et al., 2005); VEGFflox (Gerber et al., 1999). Sema4A−/− mice were generated with CRISPR/Cas9-mediated gene ablation using 2 guide RNAs (gRNA) targeting the Sema4A locus: gRNA#1 (5’-TGGGGTGAGTAGCGGGCATAAGG) targeted intron 1-2; gRNA#2 (5’- GCAGCGTGTCAAAGTCTCGGAGG) targeted exon 3. The two gRNAs were transcribed in vitro with T7 Quick High Yield RNA Synthesis Kit (New England Biolabs Cat# E2050S) and mixed at 15ng/μl each with 30ng/μl of in vitro transcribed, capped and polyadenylated Cas9 mRNA (mMESSAGE mMACHINE T7 Transcription Kit, Thermo Fisher Scientific Cat# AM1344) in microinjection buffer (10 mM Tris-HCl pH 7.4, 0.2 mM EDTA). The freshy prepared mixture was injected into the cytoplasm and pronucleus of CB6F1 zygotes (~2-3pl per embryo), which were reimplanted into CD1 recipient females. Founders were screened by PCR genotyping and confirmed by direct Sanger sequencing. The selected founder carried a ~2-kb genomic deletion encompassing exon 2, which contains the start codon, and part of exon 3. Hemizygous founders were bred for >4 generations with ISLMN::fGFP mice in CB6F1 back-ground. Plxnd1-null, Sema3-null, Sema3E-null, VEGFflox alleles were maintained in C57/BL6N strain. Sema3C mutants were also analyzed in CD1 strain. In some experiments Plxnd1 mutants were in CB6F1 strain. Phenotype expressivity was similar in different background strains. Embryos were generated by timed mating, and the day on of vaginal plug was designated as embryonic day 0.5 (E0.5). Data were collected from E10-E12.5 embryos of both sexes. There was no evidence of sex-related differences in the reported phenotypes. Mice were maintained in pathogen-free facilities under standard housing conditions with continuous access to food and water. All experimental procedures and handling of animals were approved by the Animal Research Committee of IRCCS San Raffaele Hospital and Salk Institute, and performed in compliance with IACUC guidelines.
Cell lines
Human umbilical vascular ECs (HUVEC; Lonza Cat# C2519A) were cultured in EC Growth Basal Medium-2 (EBM-2; Lonza Cat# CC-3156) supplemented with EGM-2 Endothelial factors (Lonza Cat#CC-4176). Cells were discarded after passage 5. Immortalized cell lines were maintained in DMEM supplemented with 10% FBS, 1% L-Glutamine, 1% Penicillin/Streptomycin (Gibco). AD293 cells are a derivative of the HEK293 cell line (Stratagene Cat# 240085). COS-7 (Cat# CRL-1651) and NIH-3T3 (Cat# CRL-1658) were from the American Type Culture Collection (ATCC). Mouse embryonic fibroblasts (MEF) isolated from E13.5 Swiss Webster mice (Conner, 2001) were cultured in IMDM (Gibco) supplemented with 15% FBS (Euroclone Cat# ECS0180L), 1% L-Glutamine and 1% Penicillin/Streptomycin (Gibco). Cell cultures were maintained at 37°C with 5% CO2. HEK293 (AD293) lines stably expressing Plexin-D1 were generated by clonal selection with 1 μg/ml puromycin after Lipofectamine-based transfection of Plexin-D1-FLAG-P2A-Puro construct expressing C-terminal FLAG-tagged mouse Plexin-D1 and puromycin-resistance gene under the control of CMV promoter. To generate lines co-expressing Plexin-D1 and either NRP1 or NRP2, HEK-Plxnd1 stable clones were transfected using Lipofectamine 2000 with hyperactive piggyBac transposase [pCMV-HAhyPBase] (Yusa et al., 2011) and Piggy Bac donor plasmids expressing either C-terminal HA-tagged rat Nrp1 or Nrp2 under CAG promoter. Transfected cells were enriched through two rounds of FACS sorting after extracellular staining with 5μg of anti-NRP1 (R&D Systems Cat# AF566) and anti-NRP2 (R&D Systems Cat# AF2215) followed by Alexa647-conjugated secondary antibody.
Primary neuronal cultures
Mouse MN explants comprising LMC and MMC were prepared from the lumbar spinal cord of E12.5 embryos. Chick MN explants were prepared from the motor columns of embryos at Hamburger Hamilton (HH) stage 22-23. Explants were plated on 12mm Poly-D-Lysine (PDL)-treated coverslips (Corning Cat #354086) coated with 50μg/ml laminin (Gibco Cat# 23017015) and cultured in 4-well dishes (Nunc Cat#144444) with MN (MN) media: Neurobasal media containing B27 supplement (Gibco), 2mM L-Glutamine (Euroclone), 1% Penicillin/Streptomycin (Euroclone), 50μM Glutamic Acid (MilliporeSigma), 10ng/ml GDNF (R&D Systems Cat#212-GD). Mouse dorsal spinal cord explants were dissected from the dorsal-most region of the spinal cord of E12.5 embryos and cultured for ~20 hrs in MN media in which GDNF was replaced with 50ng/ml BDNF (Peprotech Cat#450-02).
METHOD DETAILS
ENU screen and mapping of Drake mutant
The design of the ENU-mutagenesis screen was previously described (Lewcock et al., 2007). Briefly, fifty DBA/2J mouse males (Jackson Labs) were given intraperitoneal injections of 100 mg/kg ENU (Sigma-Aldrich) once a week for 3 consecutive weeks starting at 8 weeks of age. Eight to ten weeks after injection, animals were mated to ISLMN::fGFP mice (maintained on CB6F1/J background, an F1 hybrid between BALB/cJ and C57BL/6J strains). Eighty-eight GFP+ F1 males were generated and used in the screen. Six to eight G3 litters derived from each F1 male were examined. Embryos at E12.5 were fixed for 2 hrs in 4% paraformaldehyde (PFA) in PBS, washed in PBS ON, then eviscerated and cleared by consecutive 2-4 hr-incubations (4°C) in 30%, 50%, 80% glycerol (in PBS) on an orbital shaker and flat-mounted between coverslips for GFP visualization under a fluorescence stereomicroscope. After >5 generations of backcrosses, the genomic region containing the Drake allele was restricted to a ~10Mb segment on chromosome 6 between dbSNP rs30015050 (108.8Mbp) and rs29870917 (118.5 Mbp) that contains ~200 genes. Total RNA was extracted with RNeasy Plus Mini kit (Qiagen Cat#74134) from the thoracic/brachial spinal cord region and surrounding mesenchymal tissue of three E12.5 mutant embryos, two control (i.e., unaffected) littermates, and one stage-matched DBA/2J embryo. Individual libraries for mRNA sequencing were prepared using Illumina mRNA-Seq Sample Prep Kit (Cat# RS-100-0801). Genome Analyzer IIx platform (Illumina) was used to perform 2x36 cycles of single-end sequencing. Reads were aligned to the reference genome with MOSAIK software to detect SNPs. RNA-seq did not reveal transcriptional and splicing changes in Drake mutants. To identify the ENU-asso-ciated base-change, SNPs detected on chromosome 6 in Drake mutants were filtered to remove (1) reference dbSNPs, (2) SNPs found in CB6F1/J control samples, (3) SNPs found in DBA/2J strain. The A>T transversion at 115,994,460 bp (GRCm38/mm10 assembly) of chromosome 6, in the first exon of Plxnd1 gene, was found in all reads aligned at that location in Drake embryos but never in controls or in the parental mouse strains. The resulting C116S mutation in Plexin-D1 is predicted as “deleterious” by PROVEAN software (score: −3.78; cutoff for neutral variants: −2.5).
DNA constructs
To generate CMV-PlexinD1-FLAG construct, a C-terminal FLAG-tag was fused to mouse Plxnd1 (GenBank: XR_868877.1) using ClonExpress II One Step Cloning Kit (Vazyme Cat# C112-01). After removal of the stop codon, a P2A-Puro cassette was inserted in frame downstream of FLAG-tag to obtain PlexinD-FLAG-P2A-Puro construct. For CMV-PlexinD1-C116S-FLAG, the C116S mutation was introduced with GENEART Site-Directed Mutagenesis System (Thermo Fisher Scientific Cat# A13282) with primers forward 5’-GCAGGCCTCGAGCGAGCAC and reverse 5’-GTGCTCGCTCGAGGCCTGC. Rat Nrp1 (GenBank: NM_145098.2) and rat Nrp2 (GenBank: NM_030869.4) fused to C-terminal HA-tag were cloned in a Piggy Bac donor plasmid under CAG promoter (Ballabio et al., 2020).
Neuronal explant-endothelial cell co-cultures
MN explants or dorsal spinal cord explants dissected from E12.5 mouse embryos were cultured as described above. After 5-6 hrs, 180,000 HUVECs detached with Accutase (Thermo Fisher Scientific Cat# A1110501) were added to the well containing explants. Co-cultures were fixed after ~15 hrs and processed for immunostaining. Co-cultures with other cell lines conformed to this experimental design except that cells were detached with Trypsin 0.25% (Gibco) and 100,000-150,000 MEF, NIH-3T3 or AD293 cells were added to the explants. For live imaging, motor explants were plated on coverslip-bottom 35mm dishes (Corning Cat# 354077) treated as described above and 50,000 HUVECs were seeded directly onto the coverslip after ~6 hrs. Co-cultures were monitored for 13-16 hrs with phase-contrast imaging on an inverted Zeiss Axiovert S100 TV2 microscope equipped with temperature/CO2/humidity-controlled environmental chamber (OkoLab) and autofocus module, and were fixed/stained at the end of the imaging session. For chick MN explants, pN1-EGFP construct (Clontech) was electroporated “in-ovo” in the neural tube of HH 17-18 embryos using a square wave electroporator (BTX) and eggs were incubated in a humidified and temperature-controlled chamber (37,5°C; humidity 55%). Explants were prepared at HH 22-23. When indicated, neutralizing anti-Plexin-D1 antibody (5μg/ml) (R&D Systems Cat# AF4160) was added to the co-cultures at the time of HUVEC seeding. Neutralizing anti-VEGF164 antibody (2.5μg/ml) (R&D Systems Cat# AF564) was added to motor explants 1 hr prior to HUVEC seeding. Both antibodies were maintained for the duration of the experiment. For “low-laminin” condition, PDL-treated coverslips were coated with 5 μg/ml laminin. For axon growth assays with HUVEC-conditioned media, HUVECs were cultured for 24 hrs in serum-free EBM-2 media with half concentration of factors. The media was collected, passed through 0.22μm filters and concentrated with 3kDa Amicon Ultra Centrifugal Filter Units (SigmaMillipore Cat# UFC900324). Motor explants were then cultured for 24 hrs in MN media supplemented with a volume of concentrated media estimated to be conditioned by the same number of HUVECs used in co-cultures (180,000 cells).
RNA interference in HUVEC
HUVECs at 60-70% confluency were transfected in 6-well plate with 5μM control siRNA (Non-targeting Pool; Dharmacon Cat#D-001810-10-05), 5μM Plxnd1 siRNA (Human PLXND1 (23129) siRNA– SMARTpool; Dharmacon Cat# L-014121-01-0005), 10μM Nrp1 siRNAs (Human NRP1 (8829) siRNA–SMARTpool; Dharmacon Cat# L-019484-00-0005) or 10μM Nrp2 siRNAs (Human NRP2 (8828) siRNA–SMARTpool; Dharmacon Cat#L-017721-00-0005) using 4μl of DharmaFECT4 transfection reagent (Dharmacon Cat# T-2004-01) in 400μl of Opti-MEM (Gibco Cat# 31985070) after 20 min incubation at RT. Cells were incubated ON with the transfection mixture and used 48 hrs later for co-cultures and other in vitro assays. Knockdown efficiency was tested routinely by quantitative RT-PCR and western blotting.
Collapse assay in COS-7 cells
COS-7 cells transfected in 6-well plate with 6μl of Lipofectamine 2000 and 2μg of plasmid expressing C-terminal FLAG-tagged Plexin-D1 (WT or C116 mutant), were transferred to PDL-coated coverslips and treated at 48 hrs after transfection with 7nM AP-Sema3E for 1 hr in Opti-MEM. After fixation with 4%PFA/4% sucrose for 30 min, cells were stained with anti-FLAG antibody and Alexa488-conjugated Phalloidin. Cells were classified as “collapsed” based on a ~70% average reduction in surface area and the presence of actin-rich protrusions.
Cell-cell repulsion assay
For HUVEC-HEK repulsion assays, HUVECs were transfected with the indicated siRNAs as described above and after 24 hrs 70,000 cells were transferred to 12mm PDL-coated coverslips. On the same day, AD293 were transfected in 24-well plate with pN1-EGFP (0.09μg) (Clontech) and either AP-Fc, or AP-Sema3E or AP-Sema3C (0,35μg) using with 1.3μl Lipofectamine 2000 [1:3 DNA:Lipofectamine ratio]. After 24 hrs, 2,000 transfected AD293 were seeded on top HUVEC monolayer. For HEK:Sema/HEK-Receptor assays, 2,000 AD293 cells co-transfected with GFP and AP-Sema constructs were seeded on a monolayer of naïve AD293 or cell lines stably expressing receptors genes. Co-cultures were fixed 16 hrs later and stained for GFP and F-actin (with Alexa555-conjugated Phalloidin). To quantify cell repulsion, the cell-free region around randomly selected GFP+ HEKs was manually traced using the freehand tool of ImageJ software, and the pixel area was normalized by the number of GFP+ HEKs at each site.
Scratch wound healing and cell proliferation assays
For scratch would healing assay, HUVECs transfected for 48 hrs with control or Plxnd1-targeting siRNAs were seeded in 96-well plate at 50,000 cells/well in complete EBM-2 media. After 24 hrs, the cell monolayer was scratched with a 96-pin Wound Maker (IncuCyte, Essen Bioscience). Cells were washed twice with PBS and incubated for 18 hrs in complete MN media with or without recombinant Sema3C-Fc (500 ng/ml; R&D Systems Cat# 5570-S3). The wound area was imaged every 15 min using the IncuCyte Zoom system and the rate of gap closure was quantified automatically with IncuCyte Analysis Software by measuring the relative wound confluence at each time point. For cell proliferation assay, 30,000-40,000 control or Plxnd1 knockdown HUVECs were seeded in 24-well plate containing either complete EBM-2 media or MN media, let recover for 1 hr and imaged with IncuCyte system for 36 hrs to automatically measure cell growth rates based on normalized confluence area.
Transwell migration assays
Approximately 30-40 motor explants were plated on coverslips in a 24-well plate as described above and cultured for 48 hrs to precondition MN media (600 μl). The lower side of Transwell inserts (8μm pore size; Corning Cat# CLS3422) was coated with 10 μg/ml fibronectin (Corning Cat# 354008) for 90 min and blocked with 0.2% BSA/PBS until the inserts were placed in the wells containing explants (or control wells with basal MN media). HUVECs (~75,000 cells) resuspended in plain EBM-2 containing 0.2% BSA were seeded in the upper chamber of the inserts and allowed to migrate for 6 hrs. For VEGF neutralization experiments, 2.5 μg/ml of anti-VEGF antibody (R&D Systems Cat# AF564) or control goat IgG (Jackson ImmunoResearch Cat# 005-000-003) were added to the explants 2 hrs before HUVEC plating and left in the lower chamber for the duration of the experiment. To study HUVEC migration in response to recombinant factors, the inserts were placed in wells containing plain EBM-2 (control) or EMB-2 supplemented with 100ng/ml human VEGF165 (Miltenyi Biotec Cat# 130-109-395) or 500ng/ml mouse SDF-1a/Cxcl12 (Prospec Cat# CHM-324). At the end of the experiment, cells that remained in the upper side of the insert membrane were mechanically removed with a cotton swab, while those migrated to the lower side were fixed with 4% PFA, stained with crystal violet and imaged with a Zeiss Axio Zoom.V16 stereo microscope.
AP-fusion protein binding assays
AP-fusion proteins were prepared from AD293 cells transfected with the indicated constructs (10μg in 10cm dish) using Lipofectamine 2000 (3μl Lipofectamine:1μg DNA). After transfection, cells were cultured in 5ml Opti-MEM and the media was collected and replaced for 3 consecutive days, passed through a 0.22μm cell strainer and concentrated on 30kDa Amicon Ultra Centrifugal Filter Units (SigmaMillipore Cat# UFC903024). Ligand concentration was determined as previously described (Flanagan and Leder, 1990). Cell binding assays were performed according to previous protocols (Gitler et al., 2004). COS-7 cells were transfected for 48hrs with the indicated constructs using Lipofectamine, washed with PBS, incubated at RT with AP-Sema3E (5nM for 75 min) or AP-Sema3C (1nM for 90 min), fixed for 15 min with 4% PFA (RT), washed and incubated in PBS at 65°C for 3 hrs to inactivate endogenous alkaline phosphatases. AP-Sema binding was detected by incubating cells at RT for 15-30 min in AP substrate buffer (100mM Tris pH9.5, 100mM NaCl, 50mNMgCl2, 0.1% Tween, 0.33mg/ml NBT and 0.05 mg/ml BCIP). AP signal intensity in individual cells was measured on inverted grayscale images using Fiji/ImageJ software. AP-Sema3E protein binding to tissue sections was performed as described (Feiner et al., 1997).
Western blotting
COS-7 cells transfected for 48 hrs in 6 well-plate with Plexin-D1 WT and C116S constructs (1.7μg) using Lipofectamine 2000 were lysed in 400μl/well SDS-lysis buffer (150mM NaCl, 50mM TrisHCl pH7.4, 10mM EDTA, 1% Tx-100, 0.1% SDS) supplemented with protease (Merck/Roche Cat# 4693132001) and phosphatase (Thermo Fisher Scientific Cat# 78420) inhibitor cocktails, cleared by centrifugation at 13,000 xg for 15 min (4°C) and resuspended in NuPAGE LDS Sample Buffer (Thermo Fisher Scientific Cat# NP0007). To detect Plexin-D1 in Drake mutants, the thoracic-brachial spinal cord segment and surrounding tissue from individual E12.5 embryos were homogenized in 200μl SDS-lysis buffer with protease/phosphatase inhibitors, cleared by centrifugation and resuspended in NuPAGE LDS Sample Buffer. The whole spinal cord, or isolated dorsal and ventral regions, from E12.5 embryos were lysed in 80-100 μl of modified RIPA buffer (150mM NaCl, 50mM TrisHCl pH7.4, 10mM EDTA, 1% NP40, 0.5% Sodium Deoxycholate) with protease/phosphatase inhibitors, cleared by centrifugation and resuspended in Laemmli Sample Buffer (Biorad Cat# #1610747). The same lysis buffer (80-100 μl/well) and procedures were used to prepare total protein extracts from siRNA-treated HUVECs and MN explants cultured for 48hrs on 12mm coverslips as described above. Protein samples were quantified with Bradford protein assay (Bio-Rad Cat #5000006), run on handcast 8-10% SDS-PAGE gels or NuPAGE 4-12% Bis-Tris gels (Thermo Fisher Scientific), transferred on nitrocellulose membranes (Amersham/Merck Cat# GE10600002) and probed with the indicated antibodies. Antibodies used for immunoblotting were: goat anti-PlexinD1 (1:500-1:800, R&D systems Cat# AF4160), rabbit anti-GAPDH (1:10,000, Cell Signaling Cat# 2118), rabbit anti pan-Actin (1:1000; Cell Signaling Cat# 4968), rat anti-Semaphorin3C (1:500; R&D Systems Cat# MAB1728).
Quantitative RT-PCR
RNA was extracted from the dorsal and ventral spinal cord of E12.5 embryos with RNeasy Mini Kit (Qiagen). cDNA was synthesized from 500ng RNA with M-MLV reverse transcriptase (Thermo Fisher Scientific Cat# 28025013) and random primers (Thermo Fisher Scientific Cat# N8080127), diluted to 40μl in molecular biology-grade water and used for real-time quantitative PCR with SYBR Select Master Mix (Thermo Fisher Scientific Cat#4472913). Real-time PCR reactions were performed in triplicate. Target mRNA levels were normalized to the GAPDH mRNA levels from the same RT reactions. Primer sequences are reported in Table S6.
Immunofluorescence, immunohistochemistry and in situ hybridization
Explants and cells were fixed with 4%PFA/4% sucrose in PBS for 30 min (RT), incubated for 2 hrs (RT) with primary antibodies diluted in 1% BSA/0.1% Tx-100 in PBS and, after 3 washes in PBS, with fluorophore-conjugated secondary antibodies in the same buffer for 1 hr (RT). Samples were washed in PBS, stained with DAPI and mounted with ProLong Diamond Antifade Mountant (Thermo Fisher Scientific Cat#P36965) or PermaFluor Mounting Medium (Thermo Fisher Scientific Cat# TA-030-FM).
For cryosectioning, embryos were fixed in 4% paraformaldehyde diluted in PBS for 2-3 hrs (4°C), washed extensively in PBS, cry-oprotected in 30% sucrose for 2-4 hrs at 4° C, and embedded in OCT compound (Sakura Cat# 4583). Cryosections (20-40μm) were incubated with primary (ON, 4°C) and secondary antibodies (2-3 hrs, RT) in PBS containing 1% BSA/0.2% Tx-100. Samples were washed in PBS, stained with DAPI and mounted with PermaFluor Mounting Medium or Fluorescent Mounting Medium (Dako Cat# Code S3023).
Immunohistochemistry for Plexin-D1 was carried out on 20μm sections. After washes in PBS, sections were dehydrated in an ascending series of methanol (MeOH 25% in PBS; MeOH 50% in PBS; MeOH 75% in H2O; MeOH 100%), incubated in 3% H2O2 in MeOH for 15 minutes at RT to block endogenous peroxidases, and rehydrated in a descending methanol series. Sections were incubated in blocking solution (1 mg/mL BSA; 10% FBS; 0.1% TritonX-100 in PBS) for 1h at RT, and then with primary goat anti-PlexinD1 diluted 1:200 in the same buffer ON at 4°C. After 3 washes in PBS, sections were incubated with secondary biotinylated rabbit anti-goat IgG (Vector lab Cat# PK-6105) diluted 1:200 in blocking solution for 90 min at RT. Immunoreactive signal was amplified with ABC working solution (Vector lab Cat# PK-6105) for 90min at RT. Chromogenic detection was performed with DAB peroxidase substrate (Vector lab Cat# SK-4100). Slides were mounted with Glycergel medium (Dako Cat# C0563).
Primary antibodies used for immunostaining were: chicken anti-GFP (1:500; Abcam Cat# ab13970), rabbit anti-GFP (1:1000; Thermo Fisher Scientific Cat# A6455), mouse anti-GFP (1:1000; Thermo Fisher Scientific Cat# A11120), mouse anti-FLAG (1:1000; Sigma-Aldrich Cat# F1804), goat anti-Plexin-D1 (1:200-1:500; R&D Systems Cat# AF4160), goat anti-VE-Cadherin (1:300; R&D Systems Cat# AF938), rabbit anti-VE-Cadherin (1:1000; Abcam Cat# ab33168), mouse anti-βIII Tubulin (1:1000; Abcam Cat#ab7751), rabbit anti-Fsp1 (1:250; Millipore Cat# 07-2274), rat anti-CD31 (1:1000; BD Biosciences Cat# 550274); goat anti-CD31 (1:1000; R&D systems Cat# AF3628); rabbit anti-VAChT (1:1000; Synaptic Systems Cat# 139103); rabbit anti-Isl1/2 1:2,500 (Ericson et al., 1992); rabbit anti-Hb9 #6055 1:8,000 (Thaler et al., 1999). Secondary antibodies were: goat or donkey anti-rabbit/mouse/goat/rat Alexa Fluor 488-, 555-, 647-conjugated (1:1000; Thermo Fisher Scientific). Alexa Fluor 488 Phalloidin (1:100; Thermo Fisher Scientific Cat# A12379) and Alexa Fluor 555 Phalloidin (1:100; Thermo Fisher Scientific Cat# A34055) were added with the secondary antibody mix.
In situ hybridization was carried out on 20 μm cryostat sections following standard procedures with digoxigenin-labeled RNA anti-sense probes: mouse Plxnd1 (GenBank: NM_026376; 3544-6913bp) (Gu et al., 2005); mouse Vegfa (GenBank: NM_001317041.1; 1431-1849bp) (Ruiz de Almodovar et al., 2011); mouse Sema3C (GenBank: NM_013657.5; 546-1441bp) cloned in pCRII-Topo (Thermo Fisher Scientific); mouse Sema3E (GenBank: NM_011348; 1130-1920bp) cloned in pCRII-Topo. RNA scope probe for mouse Plxnd1 (ACD Bio-Techne Cat# 405931) was used according to the manufacturer’s instructions,
Cell surface staining
COS-7 cells were transfected with C-terminal FLAG-tagged Plexin-D1 (WT or C116 mutant) and cultured for 48 hrs on PDL-treated coverslips. Surface-targeted Plexin-D1 was detected by staining live cells with goat anti-Plexin-D1 antibody to the extracellular domain (R&D Systems Cat #AF4160) diluted 1:200 in 3% BSA/DMEM at 11°C for 15 min. Cells were fixed with 4% PFA/4% Sucrose in PBS for 10min (RT), followed by incubation with Alexa555-conjugated anti-goat antibody (1:500; Thermo Fisher Scientific) in 1% BSA/PBS for 1 hr (RT). The total pool of Plexin-D1 was detected by intracellular staining with anti-FLAG antibody (1:1000; MilliporeSigma Cat# F1804) in 1% BSA/PBS containing 0.1% Tx-100 for 1 hr (RT), revealed with Alexa647-conjugated secondary antibody (1:000; Thermo Fisher Scientific) in the same buffer for 1hr (RT).
Embryo whole mount staining and tissue clearing
Whole mount embryo staining with rat anti-CD31 antibody (BD Biosciences) was performed as previously described (Zhang et al., 2009). For staining of thick tissue sections, embryos were fixed in 4% PFA ON at 4°C, dehydrated in an ascending methanol series (50% MeOH in PBS; 80% MeOH in H2O; 100% MeOH - 90min each at RT on a rocker), bleached with 6% H2O2 in MeOH ON at 4°C on a rocker, and rehydrated in a descending methanol series. Embryos were embedded in 5% low-melting agarose (GellyphorLM, Euroclone Cat# EMR911100) in PBS and sectioned with a vibratome at 1mm thickness. Agarose was removed under a dissection microscope and sections were incubated in PBSGT [0.2% gelatin, 0.5% TritonX-100, 0.01% Thimerosal (Sigma/Merck cat# T8784-5G) in PBS] for 24hrs at RT on a rocker. Primary antibodies were diluted in PBSGT with 10 mg/ml saponin and incubated for 7 days at RT on a rocker. Sections were washed 6 times over one day with PBSGT, then incubated with secondary antibodies in PBSGT with 10 mg/mL saponin ON at RT on a rocker. After 6 washes in PBSGT, sections were stored in PBS at 4°C until clearing.
Primary antibodies used for whole mount staining were: rat anti-CD31 (1:500; BD Biosciences); goat anti-CD31 (1:1000; R&D Systems); rabbit anti-GFP (1:5000; Thermo Fisher Scientific). Secondary antibodies (from Thermo Fisher Scientific) were: donkey anti-Rabbit AlexaFluor-488 (1:1000; Cat# A21206); goat anti-Rat AlexaFluor-546 (1:1000; Cat# A11081); donkey anti-goat AlexaFluor-555 (1:1000; Cat# A21432). Sections were cleared after staining with X-CLARITY Tissue Clearing System (Logos Biosystems). Samples were incubated in Hydrogel Solution (Logos Biosystems Cat# C1310X) for 24 hrs at 4°C, transferred in X-CLARITY Polymerization System for 3 hrs at 37°C, then washed several times in Electrophoretic Tissue Clearing Solution (Logos Biosystems Cat# C13001) and incubated ON at RT in the same solution for clarification. Samples were washed extensively in PBS and incubated in X-Clarity Mounting Solution (Logos Biosystems Cat# C13101) 1h at RT before imaging with a Nikon A1 MP multiphoton microscope with Coherent Chameleon Ultra II using Apo LWD 25 X (NA 1.1) Water Plan objective. 3D-image stacks were analyzed with Arivis 4D software.
Isolation and RNA-sequencing of motor neurons and endothelial cells
ECs and MNs were simultaneously isolated by FACS from E12.5 Plxnd1−/− embryos or control littermates (WT or Plxnd1+/−) harboring ISLMN::fGFP and KdrEC::Cherry reporters. The spinal cord and surrounding meninges –along with attached DRGs– were dissected, pooled according to genotype, and dissociated with Papain Dissociation System (Worthington Biochemical Cat# LK003153). After 15 min rotation at 37°C, tissues were mechanically triturated, centrifuged at 300 xg for 5 min and the reaction was blocked with DNase/albumin-inhibitor solution. Dissociated cells were pelleted, resuspended in Neurobasal media without phenol red (Gibco Cat# 12348017) supplemented with 2% Horse Serum/1% L-Glutamine, and passed through a 35mm cell strainer (BD Falcon Cat# 08-771-23). GFP+ MNs and Cherry+ ECs were purified on a BD Influx Cell Sorter and collected directly into RLT lysis buffer (Qiagen) containing β-mercaptoethanol. RNA was isolated with RNeasy Mini Kit and on-column DNase digestion (Qiagen RNase-Free DNase Set Cat# 79254). Each sample contained 100,000-300,000 GFP+ MNs or mCherry+ ECs collected from 2 control and 2 mutant embryos in each of 3 independent experiments. A total of 6 EC samples (3 control, 3 Plxnd−/−) and 6 MN samples (3 control, 3 Plxnd−/−) were quantified with Agilent 2100 Bioanalyzer prior to preparation of mRNA-sequencing libraries (50bp or 75bp single-end) with Illumina TruSeq RNA Library Preparation Kit (v2). Libraries were sequenced with Illumina NextSeq500 or HiSeq 2500 platform.
For RNA-seq of spinal and meningeal ECs, the ventral region of the spinal cord and meninges were dissected from E12.5 KdrEC::Cherry wild-type embryos, pooled separately and dissociated as described above. ECs (mCherry+) were isolated on BD Influx Cell Sorter and collected in Trizol LS Reagent (Thermo Fisher Scientific Cat# 10296010). Approximately 170,000 meningeal and ~40,000 spinal ECs were collected from an average of 7 embryos in each experiment. Stranded mRNA-sequencing libraries (75 bp single-end) were prepared with Illumina TruSeq RNA Library Preparation Kit (v2) from 5 RNA samples for each tissue. Libraries were sequenced with Illumina NextSeq 500 platform.
Microscopes
Fluorescence-assisted microdissection was performed with Zeiss Lumar V12 and Zeiss Axio ZoomV16 stereo microscopes. The same instruments were used for imaging anti-CD31 and AP-Sema3E staining in embryo whole-mounts. Confocal images were acquired with an Olympus Fluoview FV1000, Olympus Fluoview FV3000RS and Leica TCS SP8 SMD FLIM Laser Scanning Confocal microscopes. Thick cleared tissue sections were imaged with Nikon A1 MP multiphoton microscope with Coherent Chameleon Ultra II. For immunohistochemistry and in situ hybridization, color brightfield images were acquired with Zeiss AxioImager M2m microscope. Cell surface/intracellular staining images were acquired with Zeiss Axio Observer Z1 microscope equipped with Hamamatsu EM 9100 camera.
QUANTIFICATION AND STATISTICAL ANALYSIS
Computational analysis
RNA-sequencing data analysis
After trimming, reads were aligned using STAR aligner (Dobin et al., 2013) on the GRCm38 (mm10) genome and counted with featureCounts (Liao et al., 2014) on Gencode annotation release M22 (Harrow et al., 2012). Raw counts were batch-corrected with ComBat-seq (Zhang et al., 2020) and differential gene expression was assessed in R/BioConductor (Huber et al., 2015) using the DESeq2 package (Love et al., 2014). Gene ontology and pathway discovery were performed with Enrichr (Xie et al., 2021) and GeneCodis3 (Tabas-Madrid et al., 2012) gene enrichment tools. The accession number for the RNA-sequencing data reported in this paper is GEO: GSE207942.
Single-cell RNA-sequencing data of Hb9MN::GFP+ spinal MNs isolated from E12.5 embryos (Amin et al., 2021) (samples WT1 and WT2 at accession ArrayExpress: E- MTAB-10571) were aligned and counted as described above. UMI and barcodes were identified using UMI-tools (Smith et al., 2017). WT1 and WT2 datasets were analyzed independently and then integrated, following the Seurat V4 standard pipeline (Hao et al., 2021). Clusters were identified by a shared nearest neighbor (SNN) modularity optimization-based clustering algorithm at the resolution of 0.4. Single-cell RNA sequencing data from ECs isolated from limb of embryos at E11.0, E12.0 and E13.0 were analyzed by sub-setting the global count matrix downloaded from https://mouse-limb.cells.ucsc.edu/ (He et al., 2020). Gene counts were analyzed with the Seurat V4 standard pipeline. Gene expression in HUVEC was derived from two independent bulk RNA-seq datasets ([Madugundu et al., 2019] GEO: GSM3181634 and [Palikuqi et al., 2020] GEO: GSM3761214-217). Read counts were normalized by library size and gene length to obtain expression values, and averaged between replicates.
Expression Weighted Celltype Enrichment analysis
EWCE package with Bootstrap.enrichment.test (10,000 random gene sets) (https://doi.org/10.18129/B9.bioc.EWCE) was used to determine the relative distribution of DEGs (158 upregulated, 41 downregulated; DESeq2 adj. p < 0.1) identified by bulk RNA-seq of MNs isolated from Plxnd1−/− E12.5 embryos, within MN subtypes defined in the integrated Seurat object (WT1+WT2) from scRNA-seq MN data (Amin et al., 2021).
Cell-cell interaction analysis
After merging of Seurat objects derived from the reanalysis of scRNA-seq datasets of ECs (He et al., 2020) and MNs (Amin et al., 2021), gene counts were mapped to human gene homologs and normalized for the individual cell output. The resulting expression matrix was then analyzed using CellPhoneDB software (github.com/Teichlab/cellphonedb) (Efremova et al., 2020), using the statistical analysis method. A curated set of ligand-receptor pairs related to cell guidance and angiogenesis was obtained by filtering the mouse and human CellTalkDB database (http://tcm.zju.edu.cn/celltalkdb/index.php) against gene sets KEGG: hsa04360 (axon guidance), GO:0001525 (angiogenesis), GO:0042056 (chemoattractant activity), GO:0045499 (chemorepellent activity) and removing annotated interactions involving intracellular molecules (GNAI2, HRAS, ITGB1BP1, TLN1), genes not expressed in spinal or meningeal ECs (CD44, TLR4), and cis interactions (APP_DCC, ADAM10_EPHA3, CNTN2_NRP1), yielding a non-redundant list of 675 ligand-receptor pairs (Table S4). An “interaction score” = log10(ligand_expression) + log10(receptor_expression) was calculated between MMC neurons and ECs based on average expression of each gene of the pair in scRNA-seq data. Out of 1350 computed interactions (675 x 2), 134 displaying highest scores (top 10%) were retained for further analysis. Of these pairs, 66 had ligands expressed in MMC and receptors in ECs, while 68 had ligands expressed in ECs and receptors in MMC.
Image analysis and quantification
Images used for quantification were acquired using the same microscope settings and adjusted to the same background levels whenever possible. Images were processed with Fiji/ImageJ, Adobe Photoshop or Arivis 4D software. For quantification of motor axon guidance defects, a “phenotype score” was calculated as the product between the number of ectopic ISLMN::fGFP+ axon bundles and the number of affected motor roots manually counted in flat-mounted embryos under a fluorescence stereomicroscope. Axonal/vascular intensity profiles (Figures 2N and 2O) were measured with the Color Profiler plugin of Fiji/ImageJ software along a 200 px trace spanning the MMC choice point. Vascular density in Olig2MN::Cre; DTALSL vs. control embryos was measured across a 100 x 100 px ventral root area encompassing the motor exit point. CD31+ vessels present in this region were thresholded in Fiji/ImageJ software and the total vascular area (px) was measured. The same procedure was used to quantify spinal cord vascularization within the motor columns or dorsal regions manually defined based on marker expression and anatomical location. For MN quantification, the entire motor columns or MMC division were manually traced with “lasso” tool in Adobe Photoshop, based on marker expression (motor column: Hb9+/ISL1/2+; MMC: Hb9+/ISLMN::fGFPhigh/Isl1high) and mediolateral position. MN nuclei (Hb9+ or ISL+) were thresholded in Fiji/ImageJ and the total px area was measured as a proxy for cell count. A semi-automated quantification of motor explants outgrowth from was performed using the FeatureJ plugin of Fiji/ImageJ software to measure the total axonal area (#pixels) after removal of the signal associated with the cell bodies (Bonanomi et al., 2012). The extent of HUVEC repulsion in explant co-cultures was quantified by manually tracing and measuring the cell-free area (#px) around individual explants. For quantification of Plexin-D1 surface staining, individual transfected COS-7 cells were thresholded with Fiji/ImageJ and the ratio between mean fluorescence intensity of surface-associated and total signal (derived from intracellular staining) was calculated after subtraction of background fluorescence. For quantification of Transwell assays, tiled images spanning the bottom side of the insert membrane were thresholded with Fiji/ImageJ software to generate a binary mask of migrated cells, which were automatically counted with “analyze particles” function after defining a range size that was maintained for all conditions. Additional quantification approaches are described in the corresponding method section. Statistical analysis was performed with GraphPad Prism V8.4.0 software. Statistical details of the experiments are found in the figure legends.
Sample size for measurements reported in the Figures were:
Figure 1O: N transfected cells scored per condition: ~1000 from 2 independent experiments
Figure 2G: N embryos: Plxnd1C116S/+ [Ctrl] (18); Plxnd1C116S/C116S [C116S] (18); Plxnd1−/− (22); Olig2MN::Cre;Plxnd1C116S/fl [Plxnd1MNΔ] (9); TekEC::Cre; Plxnd1C116S/fl [Plxnd1ECΔ] (17).
Figures 2N and 2O: N ventral roots: Ctrl (9; 6 embryos); Plxnd1−/− (14; 9 embryos).
Figure 3L: N motor columns: Control [Ctrl] (60; 19 embryos, heterozygous or WT littermates); Plxnd1C116S/C116S [C116S] (22; 6 embryos); Plxnd1−/− (58; 12 embryos).
Figure 3M: N spinal cords: Ctrl (34; 23 embryos, heterozygous or WT littermates); Plxnd1C116S/C116S [C116S] (10; 6 embryos); Plxnd1−/− (25; 16 embryos).
Figure 4C: N explants: Control (26); siPlxnd1 (23).
Figure 4J: N explants: Control (73); siPlxnd1 (50); anti-Plxnd1 (30); low laminin (35); MEF (47); 3T3 (28).
Figure 4K: N explants: Control (43); siPlxnd1 (23); anti-Plxnd1 (18).
Figure 4L: N explants: no-EC (23); control EC (66); siPlxnd1 EC (69).
Figure 4M: N explants: control media (25); conditioned media (23).
Figure 5E: N motor columns: Control [Ctrl] (36; 9 embryos, Sema3C+/− or WT littermates); Sema3C−/− (66; 7 embryos); Sema3E−/− (54; 5 embryos); Sema4A−/− (52; 5 embryos).
Figure 5J: N embryos: Sema3C, Sema3E, Sema4A heterozygous [Ctrl] (>30); Sema3E−/− (17); Sema4A−/− (9); Sema3C−/− (41).
Figure 5K: 41 Sema3C−/− embryos.
Figure 5M: N independent wells from 3 experiments: Ctrl/untr. (26); Ctrl/Sema3C (21); siPlxnd1/untr. (28); siPlxnd1/Sema3C (20)
Figure 5P: N explants: WT (74); Sema3E+/− (57); Sema3E−/− (60); Sema3C+/− (42); Sema3C+/− (113).
Figure 5Q: N fields of view: >95 from 3 independent experiments.
Figure 5U: N explants: control (37); siNrp1 (37); siNrp2 (65).
Figure 5Z: N explants: control HEK (29); HEK-Plxnd1 (34); HEK-Plxnd1/Nrp1 (55); HEK-Plxnd1/Nrp2 (40)
Figure 7D: N=Transwells from >3 experiments: basal (13); motor-conditioned media [MN] (11), MN/VEGFNAb (6)
Figure 7E: N=explants: Ctrl/untr. (25); Ctrl/VEGFNab (39); siPlxnd1/untr. (54); siPlxnd1/VEGFNab (70).
Figure 7N: N motor columns: Control [Ctrl] (60; 19 embryos, Plxnd1 heterozygous or WT littermates); Plxnd1−/− (58; 12 embryos); Plxnd1−/−;VEGFhet (82; 4 embryos); Plxnd1−/−;VEGFflox (61; 6 embryos).
Figure 7T: N embryos: Plxnd1−/− (22) [also shown in Figure 2G]; Plxnd1−/−;VEGFhet (14); Plxndr−/−;VEGflox (7)
Figure S1K: N cells: WT (89), C116 (78).
Figure S1L: N=~150 cells from 3 experiments.
Figure S3C: N ventral roots: Ctrl (29; 8 embryos); DTA (28; 9 embryos).
Figures S3J and S3K: N MCs: Ctrl (41; 11 embryos, heterozygous or WT littermates); Plxnd1C116S/C116S [C116S] (14; 4 embryos); Plxnd1−/− (29; 7 embryos).
Figure S3R: N MCs: Control [Ctrl] (34; 8 embryos, heterozygous or WT littermates); Plxnd1−/− (49; 10 embryos).
Figure S4O: N explants: motor (61); dorsal (39).
Figure S4P: N explants: control EC (40); siPlxnd1 EC (27).
Figure S4Q: N, 12 wells per condition.
Figure S5H: N embryos: WT (3), Sema3C+/− (10).
Figure S5I: N explants: Ctrl (48), Sema3C−/− (42).
Figure S5M: N experiments: siPlxnd1 (4) siNrp1 (6) siNrp2 (6).
Figure S5O: N, cells from 3 experiments: Plexin-D1WT (478), NRP1 (836), Plexin-D1WT/NRP1 (959), Plexin-D1C116S/NRP1 (1026).
Figure S5R: N=20-35 fields of view.
Figure S7C: N=Transwells from 3 experiments: basal media (4); Cxcl12 (9) Cxcl12/VEGFNAb (7).
Figure S7H: N=Transwells from >2 experiments: Ctrl/basal (5); Ctrl/MN (4); siPlxnd1/basal (5); siPlxnd1/MN (4).
Figure S7I: N= 4 Transwells per group from 2 experiments.
Supplementary Material
Highlights.
Plexin-D1 is required in ECs to establish axial MN connectivity
MNs release attractive and repulsive signals that pattern blood vessels
Motor axons displace endothelial obstacles with Sema3C/Plexin-D1 signaling
Neurovascular crosstalk links axon targeting of neuronal subtypes to vascularization
ACKNOWLEDGMENTS
We thank Joe Lewcock for the design of the mouse screen; Karen Lettieri and Miriam Gullo for technical assistance; Shawn Driscoll for bioinformatic analysis; Jonathan Raper and Sophie Chauvet for Sema3C KO mice; Christiana Ruhrberg and Genentech, Inc. for Vegfa-flox mice; Chenghua Gu for AP-Sema3C and Plxnd1 riboprobe plasmids; Carmen Ruiz de Almodóvar and Peter Carmeliet for Vegfa riboprobe; Roman Giger for Nrp1-HA and Nrp2-HA plasmids; Akiyoshi Uemura for mouse Plexin-D1 clone; Luca Tiberi for PiggyBac system plasmids; Andrea Ditadi for MEFs; Nasun Hah and the Next-Generation Sequencing Core of Salk Institute; Yelena Dayn and the Transgenic Core Facility of Salk Institute; and the advanced microscopy laboratory (ALEMBIC) of San Raffaele Hospital for instrumentation and expertise. L.F.M. was supported by fellowship PD/BD/114170/2016 from the Foundation for Science and Technology Portugal. S.d.P. was supported by AIRC/CRUK/ AECC Accelerator Award (ECRIN-M3) A29370. Funding sources: European Research Council Starting Grant 335590 and Giovanni Armenise-Harvard Foundation Career Development Award to D.B.; Howard Hughes Medical Institute Investigator Grant, National Institute of Neurological Disorders and Stroke (NINDS) RO1 NS123160-01, the Sol Goldman Charitable Trust, and Benjamin H. Lewis Chair in Neuroscience to S.L.P.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.neuron.2022.09.021.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Amin ND, Bai G, Klug JR, Bonanomi D, Pankratz MT, Gifford WD, Hinckley CA, Sternfeld MJ, Driscoll SP, Dominguez B, et al. (2015). Loss of motoneuron-specific microRNA-218 causes systemic neuromuscular failure. Science 350, 1525–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin ND, Senturk G, Costaguta G, Driscoll S, O’Leary B, Bonanomi D, and Pfaff SL (2021). A hidden threshold in motor neuron gene networks revealed by modulation of miR-218 dose. Neuron 109, 3252–3267.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreone BJ, Lacoste B, and Gu C (2015). Neuronal and vascular interactions. Annu. Rev. Neurosci 38, 25–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antipenko A, Himanen JP, van Leyen K, Nardi-Dei V, Lesniak J, Barton WA, Rajashankar KR, Lu M, Hoemme C, Püschel AW, et al. (2003). Structure of the semaphorin-3A receptor binding module. Neuron 39, 589–598. [DOI] [PubMed] [Google Scholar]
- Bai G, Chivatakarn O, Bonanomi D, Lettieri K, Franco L, Xia C, Stein E, Ma L, Lewcock JW, and Pfaff SL (2011). Presenilin-dependent receptor processing is required for axon guidance. Cell 144, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabio C, Anderle M, Gianesello M, Lago C, Miele E, Cardano M, Aiello G, Piazza S, Caron D, Gianno F, et al. (2020). Modeling medullo-blastoma in vivo and with human cerebellarorganoids. Nat. Commun 11, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanomi D. (2019). Axon pathfinding for locomotion. Semin. Cell Dev. Biol 85, 26–35. [DOI] [PubMed] [Google Scholar]
- Bonanomi D, Chivatakarn O, Bai G, Abdesselem H, Lettieri K, Marquardt T, Pierchala BA, and Pfaff SL (2012). Ret is a multifunctional coreceptor that integrates diffusible- and contact-axon guidance signals. Cell 148, 568–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanomi D, Valenza F, Chivatakarn O, Sternfeld MJ, Driscoll SP, Aslanian A, Lettieri K, Gullo M, Badaloni A, Lewcock JW, et al. (2019). p190RhoGAP filters competing signals to resolve axon guidance conflicts. Neuron 102, 602–620.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, and Tessier-Lavigne M (2005). Common mechanisms of nerve and blood vessel wiring. Nature 436, 193–200. [DOI] [PubMed] [Google Scholar]
- Cattin AL, Burden JJ, Van Emmenis L, Mackenzie FE, Hoving JJ, Garcia Calavia N, Guo Y, McLaughlin M, Rosenberg LH, Quereda V, et al. (2015). Macrophage-induced blood vessels guide Schwann cell-mediated regeneration of peripheral nerves. Cell 162, 1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, He Z, Bagri A, and Tessier-Lavigne M (1998). Semaphorin-neuropilin interactions underlying sympathetic axon responses to class III semaphorins. Neuron 21, 1283–1290. [DOI] [PubMed] [Google Scholar]
- Choi Y, and Chan AP (2015). PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie SM, Hao J, Tracy E, Buck M, Yu JS, and Smith AW (2021). Interactions between semaphorins and plexin-neuropilin receptor complexes in the membranes of live cells. J. Biol. Chem 297, 100965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner DA (2001). Mouse embryo fibroblast (MEF) feeder cell preparation. Curr. Protoc. Mol. Biol Chapter 23. Unit 23 22. [DOI] [PubMed] [Google Scholar]
- D’Elia KP, and Dasen JS (2018). Development, functional organization, and evolution of vertebrate axial motor circuits. Neural Dev. 13, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessaud E, Yang LL, Hill K, Cox B, Ulloa F, Ribeiro A, Mynett A, Novitch BG, and Briscoe J (2007). Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature 450, 717–720. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd J, and Jessell TM (1988). Axon guidance and the patterning of neuronal projections in vertebrates. Science 242, 692–699. [DOI] [PubMed] [Google Scholar]
- Dray C, Rougon G, and Debarbieux F (2009). Quantitative analysis by in vivo imaging of the dynamics of vascular and axonal networks in injured mouse spinal cord. Proc. Natl. Acad. Sci. USA 106, 9459–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc 15, 1484–1506. [DOI] [PubMed] [Google Scholar]
- Eichmann A, and Brunet I (2014). Arterial innervation in development and disease. Sci. Transl. Med 6, 252ps259. [DOI] [PubMed] [Google Scholar]
- Ericson J, Thor S, Edlund T, Jessell TM, and Yamada T (1992). Early stages of motor neuron differentiation revealed by expression of homeobox gene Islet-1. Science 256, 1555–1560. [DOI] [PubMed] [Google Scholar]
- Erskine L, François U, Denti L, Joyce A, Tillo M, Bruce F, Vargesson N, and Ruhrberg C (2017). VEGF-A and neuropilin 1 (NRP1)shape axon projections in the developing CNS via dual roles in neurons and blood vessels. Development 144, 2504–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiner L, Koppel AM, Kobayashi H, and Raper JA (1997). Secreted chick semaphorins bind recombinant neuropilin with similar affinities but bind different subsets of neurons in situ. Neuron 19, 539–545. [DOI] [PubMed] [Google Scholar]
- Feiner L, Webber AL, Brown CB, Lu MM, Jia L, Feinstein P, Mombaerts P, Epstein JA, and Raper JA (2001). Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development 128, 3061–3070. [DOI] [PubMed] [Google Scholar]
- Fetcho JR (1987). A review of the organization and evolution of motoneurons innervating the axial musculature of vertebrates. Brain Res. 434, 243–280. [DOI] [PubMed] [Google Scholar]
- Flanagan JG, and Leder P (1990). The kit ligand: a cell surface molecule altered in steel mutant fibroblasts. Cell 63, 185–194. [DOI] [PubMed] [Google Scholar]
- Franze K, Janmey PA, and Guck J (2013). Mechanics in neuronal development and repair. Annu. Rev. Biomed. Eng 15, 227–251. [DOI] [PubMed] [Google Scholar]
- Gaertner F, Reis-Rodrigues P, de Vries I, Hons M, Aguilera J, Riedl M, Leithner A, Tasciyan S, Kopf A, Merrin J, et al. (2022). WASp triggers mechanosensitive actin patches to facilitate immune cell migration in dense tissues. Dev. Cell 57, 47–62.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD, Radtke F, Aguet M, and Ferrara N (1999). VEGF is required for growth and survival in neonatal mice. Development 126, 1149–1159. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Lu MM, and Epstein JA (2004). PlexinD1 and semaphorin signaling are required in endothelial cells for cardiovascular development. Dev. Cell 7, 107–116. [DOI] [PubMed] [Google Scholar]
- Grima R (2007). Directed cell migration in the presence of obstacles. Theor. Biol. Med. Model 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Rodriguez ER, Reimert DV, Shu T, Fritzsch B, Richards LJ, Kolodkin AL, and Ginty DD (2003). Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev. Cell 5, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Yoshida Y, Livet J, Reimert DV, Mann F, Merte J, Henderson CE, Jessell TM, Kolodkin AL, and Ginty DD (2005). Semaphorin 3E and plexin-D1 control vascular pattern independently of neuropilins. Science 307, 265–268. [DOI] [PubMed] [Google Scholar]
- Hamm MJ, Kirchmaier BC, and Herzog W (2016). Sema3d controls collective endothelial cell migration by distinct mechanisms via Nrp1 and PlxnD1. J. Cell Biol 215, 415–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. (2012). GENCODE: the reference human genome annotation for the ENCODE Project. Genome Res. 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Williams BA, Trout D, Marinov GK, Amrhein H, Berghella L, Goh ST, Plajzer-Frick I, Afzal V, Pennacchio LA, et al. (2020). The changing mouse embryo transcriptome at whole tissue and single-cell resolution. Nature 583, 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmels P, Paredes I, Adler H, Karakatsani A, Luck R, Marti HH, Ermakova O, Rempel E, Stoeckli ET, and Ruiz de Almodóvar C (2017). Motor neurons control blood vessel patterning in the developing spinal cord. Nat. Commun 8, 14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramón y Cajal S (1909). Histologie du systeme nerveux de l’homme et des vertébrés1 (Maloine; ). [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James JM, Gewolb C, and Bautch VL (2009). Neurovascular development uses VEGF-A signaling to regulate blood vessel ingression into the neural tube. Development 136, 833–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James JM, and Mukouyama YS (2011). Neuronal action on the developing blood vessel pattern. Semin. Cell Dev. Biol 22, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessell TM (2000). Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat. Rev. Genet 1, 20–29. [DOI] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, and Yanagisawa M (2001). Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol 230, 230–242. [DOI] [PubMed] [Google Scholar]
- Klose M, and Bentley D (1989). Transient pioneer neurons are essential for formation of an embryonic peripheral nerve. Science 245, 982–984. [DOI] [PubMed] [Google Scholar]
- Kolodkin AL, and Tessier-Lavigne M (2011). Mechanisms and molecules of neuronal wiring: a primer. Cold Spring Harb. Perspect. Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, and Westphal H (1996). Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. USA 93, 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmesser L (1978). The development of motor projection patterns in the chick hind limb. J. Physiol 284, 391–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larina IV, Shen W, Kelly OG, Hadjantonakis AK, Baron MH, and Dickinson ME (2009). A membrane associated mCherry fluorescent reporter line for studying vascular remodeling and cardiac function during murine embryonic development. Anat. Rec, 292 (Hoboken), pp. 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Jurata LW, Funahashi J, Ruiz EC, and Pfaff SL (2004). Analysis of embryonic motoneuron gene regulation: derepression of general activators function in concert with enhancer factors. Development 131, 3295–3306. [DOI] [PubMed] [Google Scholar]
- Lee WP, Stromberg MP, Ward A, Stewart C, Garrison EP, and Marth GT (2014). MOSAIK: a hash-based algorithm for accurate next-generation sequencing short-read mapping. PLoS One 9, e90581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewcock JW, Genoud N, Lettieri K, and Pfaff SL (2007). The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron 56, 604–620. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu X, Uemura A, Fukushima Y, Yoshida Y, and Hirashima M (2016). Semaphorin 3G provides a repulsive guidance cue to lymphatic endothelial cells via Neuropilin-2/PlexinD1. Cell Rep. 17, 2299–2311. [DOI] [PubMed] [Google Scholar]
- Love CA, Harlos K, Mavaddat N, Davis SJ, Stuart DI, Jones EY, and Esnouf RM (2003). The ligand-binding face of the semaphorins revealed by the high-resolution crystal structure of SEMA4D. Nat. Struct. Biol 10, 843–848. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq datawith DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madugundu AK, Na CH, Nirujogi RS, Renuse S, Kim KP, Burns KH, Wilks C, Langmead B, Ellis SE, Collado-Torres L, et al. (2019). Integrated transcriptomic and proteomic analysis of primary human umbilical vein endothelial cells. Proteomics 19, e1800315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta V, Pang KL, Rozbesky D, Nather K, Keen A, Lachowski D, Kong Y, Karia D, Ameismeier M, Huang J, et al. (2020). The guidance receptor plexin D1 is a mechanosensor in endothelial cells. Nature 578, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukouyama YS, Gerber HP, Ferrara N, Gu C, and Anderson DJ (2005). Peripheral nerve-derived VEGF promotes arterial differentiation via neuropilin 1-mediated positive feedback. Development 132, 941–952. [DOI] [PubMed] [Google Scholar]
- Mumblat Y, Kessler O, Ilan N, and Neufeld G (2015). Full-length Semaphorin-3C is an inhibitor of tumor lymphangiogenesis and metastasis. Cancer Res. 75, 2177–2186. [DOI] [PubMed] [Google Scholar]
- Muramatsu R, Takahashi C, Miyake S, Fujimura H, Mochizuki H, and Yamashita T (2012). Angiogenesis induced by CNS inflammation promotes neuronal remodeling through vessel-derived prostacyclin. Nat. Med 18, 1658–1664. [DOI] [PubMed] [Google Scholar]
- Murphy DA, and Courtneidge SA (2011). The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat. Rev. Mol. Cell Biol 12,413–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols EL, and Smith CJ (2019). Pioneer axons employ Cajal’s battering ram to enter the spinal cord. Nat. Commun 10, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh WJ, and Gu C (2013). The role and mechanism-of-action of Sema3E and Plexin-D1 in vascular and neural development. Semin. Cell Dev. Biol 24, 156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikuqi B, Nguyen DT, Li G, Schreiner R, Pellegata AF, Liu Y, Redmond D, Geng F, Lin Y, Gómez-Salinero JM, et al. (2020). Adaptable haemodynamic endothelial cells for organogenesis and tumorigenesis. Nature 585, 426–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plein A, Calmont A, Fantin A, Denti L, Anderson NA, Scambler PJ, and Ruhrberg C (2015). Neural crest-derived SEMA3C activates endothelial NRP1 for cardiac outflow tract septation. J. Clin. Invest 125, 2661–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz de Almodovar C, Fabre PJ, Knevels E, Coulon C, Segura I, Haddick PC, Aerts L, Delattin N, Strasser G, Oh WJ, et al. (2011). VEGF mediates commissural axon chemoattraction through its receptor Flk1. Neuron 70, 966–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Medina M, Gregus KA, Nichol RH, O’Toole SM, and Gomez TM (2015). Regulation of ECM degradation and axon guidance by growth cone invadosomes. Development 142, 486–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyas I, Bozon M, Moret F, and Castellani V (2012). Motoneuronal Sema3C is essential for setting stereotyped motor tract positioning in limb-derived chemotropic semaphorins. Development 139, 3633–3643. [DOI] [PubMed] [Google Scholar]
- Skene NG, and Grant SGN (2016). Identification of Vulnerable Cell Types in Major Brain Disorders Using Single Cell Transcriptomes and Expression Weighted Cell Type Enrichment. Frontiers in Neuroscience 10, 16. 10.3389/fnins.2016.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter TACS, and Jaworski A (2019). Cell migration and axon guidance at the border between central and peripheral nervous system. Science 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas-Madrid D, Nogales-Cadenas R, and Pascual-Montano A (2012). GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 40, W478–W483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam SJ, and Watts RJ (2010). Connecting vascular and nervous system development: angiogenesis and the blood-brain barrier. Annu. Rev. Neurosci 33, 379–408. [DOI] [PubMed] [Google Scholar]
- Thaler J, Harrison K, Sharma K, Lettieri K, Kehrl J, and Pfaff SL (1999). Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron 23, 675–687. [DOI] [PubMed] [Google Scholar]
- Torres-Vázquez J, Gitler AD, Fraser SD, Berk JD, Van NP, Fishman MC, Childs S, Epstein JA, and Weinstein BM (2004). Semaphorin-plexin signaling guides patterning of the developing vasculature. Dev. Cell 7, 117–123. [DOI] [PubMed] [Google Scholar]
- Tosney KW, and Landmesser LT (1985a). Development of the major pathways for neurite outgrowth in the chick hindlimb. Dev. Biol 109, 193–214. [DOI] [PubMed] [Google Scholar]
- Tosney KW, and Landmesser LT (1985b). Growth cone morphology and trajectory in the lumbosacral region of the chick embryo. J. Neurosci 5, 2345–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku T, Yabuki M, Kamei J, Kamei M, Makino N, Kumanogoh A, and Hori M (2007). Semaphorin-4A, an activator for T-cell-mediated immunity, suppresses angiogenesis via Plexin-D1. EMBO J. 26, 1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Wu Y, and Capecchi MR (2006). Motoneurons and oligodendrocytes are sequentially generated from neural stem cells but do not appear to share common lineage-restricted progenitors in vivo. Development 133, 581–590. [DOI] [PubMed] [Google Scholar]
- Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. (2021). Gene set knowledge discovery with Enrichr. Curr. Protoc 1, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WJ, Hu J, Uemura A, Tetzlaff F, Augustin HG, and Fischer A (2015). Semaphorin-3C signals through Neuropilin-1 and PlexinD1 receptors to inhibit pathological angiogenesis. EMBO Mol. Med 7, 1267–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K, Zhou L, Li MA, Bradley A, and Craig NL (2011). A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. USA 108, 1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang Y, Chaudhari K, and Bashaw GJ (2021). New insights into the molecular mechanisms of axon guidance receptor regulation and signaling. Curr. Top. Dev. Biol 142, 147–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Parmigiani G, and Johnson WE (2020). ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform 2, lqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Singh MK, Degenhardt KR, Lu MM, Bennett J, Yoshida Y, and Epstein JA (2009). Tie2Cre-mediated inactivation of plexinD1 results in congenital heart, vascular and skeletal defects. Dev. Biol 325, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. The accession number is listed in the key resources table.
All original code is available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-Isl1/2 | Ericson et al., 1992 | N/A |
| rabbit anti-Hb9 | Thaler et al., 1999 | N/A |
| rabbit anti-GFP | Thermo Fisher Scientific | Cat# A-6455 RRID:AB_221570 |
| mouse anti-GFP | Thermo Fisher Scientific | Cat# A-11120 RRID: AB_221568 |
| chick anti-GFP | Abcam | Cat# ab13970 RRID:AB_300798 |
| mouse anti-β3 tubulin | Abcam | Cat# ab7751 RRID:AB_306045 |
| mouse anti-FLAG M2 | Sigma-Aldrich | Cat# F1804 RRID:AB_262044 |
| goat anti-Plexin-D1 | R&D Systems | Cat# AF4160 RRID:AB_2237261 |
| goat anti-VE-Cadherin | R&D Systems | Cat# AF938 RRID:AB_355726 |
| rabbit anti-VE-Cadherin | Abcam | Cat# ab33168 RRID: AB_870662 |
| rat anti-CD31 | BD Biosciences | Cat# 550274 RRID:AB_393571 |
| goat anti-CD31 | R&D Systems | Cat# AF3628 RRID:AB_2161028 |
| rabbit anti-Fsp1 | Millipore | Cat# 07-2274 RRID:AB_10807552 |
| rabbit anti-VAChT | Synaptic Systems | Cat# 139 103 RRID:AB_887864 |
| rabbit anti-GAPDH | Cell Signaling Technology | Cat# 2118 RRID:AB_561053 |
| rabbit anti-Pan-Actin | Cell Signaling Technology | Cat# 4968 RRID:AB_2313904 |
| rat anti-Semaphorin3C | R&D Systems | Cat# MAB1728 RRID:AB_2301533 |
| goat anti-NRP1 | R&D Systems | Cat# AF566 RRID:AB_355445 |
| goat anti-NRP2 | R&D Systems | Cat# AF2215 RRID:AB_2155371 |
| goat anti-VEGF164 | R&D Systems | Cat# AF564 RRID:AB_2212821 |
| donkey anti-goat IgG, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11055 RRID:AB_2534102 |
| donkey anti-goat IgG, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-21432 RRID:AB_2535853 |
| donkey anti-goat IgG, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21447 RRID:AB_2535864 |
| donkey anti-mouse IgG, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21202 RRID:AB_141607 |
| donkey anti-mouse IgG, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# 31570 RRID:AB_2536180 |
| donkey anti-mouse IgG, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31571 RRID:AB_162542 |
| donkey anti-rabbit IgG, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21206 RRID:AB_2535792 |
| donkey anti-rabbit IgG, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-31572 RRID:AB_162543 |
| donkey anti-rabbit IgG, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31573 RRID:AB_253618 |
| goat anti-rat IgG, Alexa Fluor 546 | Thermo Fisher Scientific | Cat# A-11081 RRID:AB_2534125 |
| ChromPure Goat IgG, whole molecule | Jackson ImmunoResearch Labs | Cat# 005-000-003 RRID:AB_2336985 |
| Chemicals, peptides, and recombinant proteins | ||
| ENU, N-Ethyl-N-nitrosourea | Sigma-Aldrich | Cat# N3385 |
| Laminin Mouse Protein | Gibco | Cat# #23017015 |
| Fibronectin, human | Corning | Cat# 354008 |
| Recombinant Human VEGF165 | Miltenyi Biotec | Cat# 130-109-395 |
| Recombinant Human GDNF | R&D Systems | Cat# 212-GD |
| Recombinant Mouse CXCL12/SDF-1 alpha | Prospec | Cat# CHM-324 |
| Recombinant Human/Murine/Rat BDNF | Peprotech | Cat# 450-02 |
| Recombinant Human Semaphorin 3C Fc | R&D Systems | Cat# 5570-S3 |
| complete, EDTA-free Protease Inhibitor Cocktail | Merck/Roche | Cat# 04693132001 |
| Halt Phosphatase Inhibitor Cocktail | Thermo Fisher Scientific | Cat# 78420 |
| DharmaFECT4 transfection reagent | Dharmacon | Cat# T-2004-01 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | Cat# 11668027 |
| Trizol LS Reagent | Thermo Fisher Scientific | Cat# 10296010 |
| Critical commercial assays | ||
| Papain Dissociation System | Worthington Biochemical | Cat# LK003153 |
| VECTASTAIN Elite ABC-HRP Kit, Peroxidase (Goat IgG) | Vector Laboratories | Cat# PK-6105 |
| DAB Substrate Kit, Peroxidase (HRP) | Vector Laboratories | Cat# SK-4100 |
| ClonExpress II One Step Cloning Kit | Vazyme | Cat# C112-01 |
| GENEART Site-Directed Mutagenesis System | Thermo Fisher Scientific | Cat# A13282 |
| M-MLV Reverse Transcriptase | Thermo Fisher Scientific | Cat# 28025013 |
| Random Hexamers | Thermo Fisher Scientific | Cat# N8080127 |
| SYBR Select Master Mix | Thermo Fisher Scientific | Cat# 4472913 |
| T7 Quick High Yield RNA Synthesis Kit | New England Biolabs | Cat# E2050S |
| mMESSAGE mMACHINE™ T7 Transcription Kit | Thermo Fisher Scientific | Cat# AM1344 |
| RNeasy Plus Mini kit | Qiagen | Cat# 74134 |
| RNase-Free DNase Set | Qiagen | Cat# 79254 |
| Illumina mRNA-Seq Sample Prep Kit | Illumina | Cat# RS-100-0801 |
| TruSeq RNA Library Preparation Kit v2 | Illumina | Cat# RS-122-2001 |
| Deposited data | ||
| RNA-seq data | This study | GEO: GSE207942 |
| Experimental models: Cell lines | ||
| HUVEC (Umbilical Vein Endothelial Cells) | Lonza | Cat# C2519A |
| AD-293 | Stratagene | Cat# 240085 |
| COS-7 | ATCC | Cat# CRL-1651 |
| NIH-3T3 | ATCC | Cat# CRL-1658 |
| Experimental models: Organisms/strains | ||
| Mouse: ISLMN::fGFP: Tg(Isl1-EGFP*)1Slp/J | Lewcock et al., 2007 | JAX:017952 |
| Mouse: Olig2::Cre | Dessaud et al., 2007 | N/A |
| Mouse: Hb9MN::GFP | Lee et al., 2004 | N/A |
| Mouse: MN218-2)::GFP | Amin et al., 2015 | N/A |
| Mouse: Tek::Cre (Tie2::Cre) | Kisanuki et al., 2001 | JAX:008863 |
| Mouse: Kdr::Cherry (Flk1-myr::mCherry) | Larina et al., 2009 | JAX:018542 |
| Mouse: Plexin-D1 flox | Zhang et al., 2009 | JAX:018319 |
| Mouse: Plexin-D1 knockout | This study | N/A |
| Mouse: R26-DTALSL (Rosa26-DTA176) | Wu et al., 2006 | JAX: 010527) |
| Mouse: Sema3C knockout | Feiner et al., 2001 | N/A |
| Mouse: Sema3E knockout | Gu et al., 2005 | N/A |
| Mouse: Sema4A knockout | This study | N/A |
| Mouse: VEGFflox | Gerber et al., 1999 | N/A |
| Oligonucleotides | ||
| gRNA Sema4A #1 5’-TGGGGTGAGTAGCGGGCATAAGG | This study Guide RNA | N/A |
| gRNA Sema4A #2 5’- GCAGCGTGTCAAAGTCTCGGAGG | This study Guide RNA | N/A |
| Mouse Plexin-D1 C116S mutagenesis (forward primer) 5’-GCAGGCCTCGAGCGAGCAC | This study | N/A |
| Mouse Plexin-D1 C116S mutagenesis (reverse primer) 5’-GTGCTCGCTCGAGGCCTGC | This study | N/A |
| control siRNA (Non-targeting Pool) | Dharmacon | Cat#D-001810-10-05 |
| Human PLXND1 (23129) siRNA SMARTpool | Dharmacon | Cat# L-014121-01-0005 |
| Human NRP1 (8829) siRNA SMARTpool | Dharmacon | Cat# L-019484-00-0005 |
| Human NRP2 (8828) siRNA SMARTpool | Dharmacon | Cat#L-017721-00-0005 |
| Primers for quantitative RT-PCR | This study | Table S6 |
| Recombinant DNA | ||
| Plasmid: CMV-PlexinD1-FLAG | this study | N/A |
| Plasmid: CMV-PlexinD1-C116S-FLAG | this study | N/A |
| Plasmid: CMV-Plexin-D1-FLAG-P2A-Puro | this study | N/A |
| Plasmid: PiggyBac-CAG-NRP1-HA | this study | N/A |
| Plasmid: PiggyBac-CAG-NRP2-HA | this study | N/A |
| Plasmid: pCMV-HAhyPBase | Yusa et al., 2011 | N/A |
| Plasmid: AP-Sema3E | Gu et al., 2005 | N/A |
| Plasmid: AP-Sema3C | Chen et al., 1998 | N/A |
| Plasmid: pN1-EGFP | Clontech | Addgene Cat# 6085-1 |
| Plasmid for in situ probe: mouse Plxnd1 (GenBank: NM_026376; 3544-6913bp) | Gu et al., 2005 | N/A |
| Plasmid for in situ probe: mouse Vegfa (GenBank: NM_001317041.1; 1431-1849bp) | Ruiz de Almodovar et al., 2011 | N/A |
| Plasmid for in situ probe: mouse Sema3C (GenBank: NM_013657.5; 546-1441bp) | this study | N/A |
| Plasmid for in situ probe: mouse Sema3E (GenBank: NM_011348; 1130-1920bp) | this study | N/A |
| Software and algorithms | ||
| STAR aligner | Dobin et al., 2013 | RRID:SCR_004463 |
| featureCounts | Liao et al., 2014 | RRID:SCR_012919 |
| ComBat | Zhang et al., 2020 | RRID:SCR_010974 |
| Bioconductor | Huber et al., 2015 | RRID:SCR_006442 |
| DESeq2 | Love et al., 2014 | RRID:SCR_015687 |
| Enrichr | Xie et al., 2021 | RRID:SCR_001575 |
| GeneCodis | Tabas-Madrid et al., 2012 | RRID:SCR_006943 |
| UMI-tools | Smith et al., 2017 | RRID:SCR_017048 |
| SEURAT | Hao et al., 2021 | RRID:SCR_007322 |
| Expression Weighted Celltype Enrichment (EWCE) | Skene and Grant, 2016 | https://doi.org/10.18129/B9.bioc.EWCE |
| CellPhoneDB | Efremova et al., 2020 | RRID:SCR_017054 |
| PROVEAN | Choi and Chan, 2015 | RRID:SCR_002182 |
| MOSAIK | Lee et al., 2014 | RRID:SCR_005486 |
| CRISPR Tool | Zhang Lab, MIT | http://crispr.mit.edu/ |
| Fiji | http://fiji.sc | RRID:SCR_002285 |
| ImageJ | https://imagej.net/ | RRID:SCR_003070 |
| Color Profiler (ImageJ plugin) | ImageJ | https://imagej.nih.gov/ij/plugins/color-profiler.html |
| FeatureJ (ImageJ plugin) | ImageJ | https://imagescience.org/meijering/software/featurej/ |
| Adobe Photoshop | Adobe Systems Inc. | RRID:SCR_014199 |
| Arivis Vision4D | Arivis-ZEISS | RRID:SCR_018000 |
| GraphPad Prism | GraphPad Software, Inc | RRID:SCR_002798 |
| Other | ||
| Endothelial Cell Growth Basal Medium-2 (EBM-2) | Lonza | Cat# CC-3156 |
| EGM-2 Endothelial factors | Lonza | Cat#CC-4176 |
| RNA scope probe for mouse Plxnd1 | ACD Bio-Techne | Cat# 405931 |
| Alexa Fluor 488 Phalloidin | Thermo Fisher Scientific | Cat# A12379 |
| Alexa Fluor 555 Phalloidin | Thermo Fisher Scientific | Cat# A34055 |
| Hydrogel Solution | Logos Biosystems | Cat# C1310X |
| Electrophoretic Tissue Clearing Solution | Logos Biosystems | Cat# C13001 |
| X-Clarity Mounting Solution | Logos Biosystems | Cat# C13101) |