

Abstract
The transition from acute to chronic pain is critically important but not well understood. Here, we investigated the pathophysiological mechanisms underlying the transition from acute to chronic low back pain (LBP) and performed transcriptome-wide analysis in peripheral immune cells of 98 participants with acute LBP, followed for 3 months. Transcriptomic changes were compared between patients whose LBP was resolved at 3 months with those whose LBP persisted. We found thousands of dynamic transcriptional changes over 3 months in LBP participants with resolved pain but none in those with persistent pain. Transient neutrophil-driven up-regulation of inflammatory responses was protective against the transition to chronic pain. In mouse pain assays, early treatment with a steroid or nonsteroidal anti-inflammatory drug (NSAID) also led to prolonged pain despite being analgesic in the short term; such a prolongation was not observed with other analgesics. Depletion of neutrophils delayed resolution of pain in mice, whereas peripheral injection of neutrophils themselves, or S100A8/A9 proteins normally released by neutrophils, prevented the development of long-lasting pain induced by an anti-inflammatory drug. Analysis of pain trajectories of human subjects reporting acute back pain in the UK Biobank identified elevated risk of pain persistence for subjects taking NSAIDs. Thus, despite analgesic efficacy at early time points, the management of acute inflammation may be counterproductive for long-term outcomes of LBP sufferers.
INTRODUCTION
Chronic pain inflicts huge societal costs, in terms of management, loss of work productivity, and effects on quality of life (1). Chronic low back pain (LBP) is the most frequently reported chronic pain condition (2). LBP is a major problem worldwide: point, 1-month, and 1-year prevalence is 18, 31, and 38%, respectively (3). LBP ranks the highest of all chronic conditions in terms of years lived with disability, with its prevalence and burden increasing with age (4). Current treatments for LBP often target the immune system and include nonsteroidal anti-inflammatory drugs (NSAIDs), acetaminophen, and corticosteroids, although all of these drug classes are minimally effective at best (5). Despite advances in the understanding of social, psychological, and genetic factors, as well as brain processes associated with chronic LBP (6), we understand very little of the molecular mechanisms underlying the acute-to-chronic pain transition that might lead to more efficacious analgesic strategies.
Previous human genetic association studies and transcriptomic analysis of chronic LBP have been performed using candidate gene and genome-wide approaches, and they have provided evidence for the involvement of a variety of genes in many biological pathways (7–11). Increasing evidence suggests that the pathophysiology of chronic pain involves a complex interplay between the nervous and immune systems; that is, chronic pain is a neuroinflammatory disorder mediated by neuronal and non-neuronal cells alike (12). Circulating immune cells such as neutrophils, monocytes, and T cells are recruited to sites of tissue damage and/or inflammation and often also infiltrate the peripheral and central nervous systems (13, 14). Activation of these cells results in the expression of various inflammatory mediators, including cytokines/chemokines, lipids, and proteases, that act both directly on peripheral sensory or central second order neurons and indirectly on other immune or local cells to regulate pain. Microglia and astrocytes in the central nervous system act in a similar fashion, contributing to central sensitization and pain (15–18). The presence of these activated immune cells and glia, peripherally or centrally, is thought to contribute to the transition from acute to chronic pain (19–21).
Here, we used transcriptome-wide data to investigate the molecular pathophysiological mechanisms in peripheral blood immune cells at the transcriptome-wide level that underlie the transition of acute to chronic LBP, and we identified the protective effect of acute inflammatory responses against the development of chronic pain. We replicated our finding in an independent cohort of patients with another musculoskeletal pain condition, temporomandibular disorder (TMD). We then used rodent pain models to elucidate the mechanism mediating the transition from acute to chronic pain. Last, we analyzed a large human cohort (UK Biobank) to investigate the relationship between back pain and the use of anti-inflammatory drugs.
RESULTS
Differential gene expression in the LBP cohort
We assessed genome-wide transcriptomics in a cohort of 98 patients with LBP at the acute episode (t0) and a follow-up visit (t1) 3 months later. Study design, demographics, and patient characteristics are presented in fig. S1 (A and B) and table S1, and transcriptomics statistics are presented in table S2. Patients reported substantial, self-declared pain [0 to 10 numeric rating scale (NRS)] in their lower back at enrollment (t0: mean = 6.8, SD = 1.8, min = 4, and max = 10), but a much broader pain spectrum was observed at follow-up (t1: mean = 3.2, SD = 3.2, min = 0, and max = 10). We dichotomized study participants into two groups based on their pain scores at the second visit: those with resolved pain (“R”; n = 49) and those with persistent pain (“P”; n = 49) (fig. S1B). No differences were found on the pain outcome with drug classes either consumed between the clinical visits or used prophylactically (table S1).
We then tested differences between patient groups at the transcriptomics-wide level. At the first visit, there were no differentially expressed genes that reached genome-wide statistical significance between R and P patients (Fig. 1A, column “uncorrected,” and table S3A). By the time of the second visit, more than 1700 differentially expressed genes were detected at the genome-wide scale between R and P patients (Fig. 1B and table S3B). When time trajectories were considered, contrasting gene expression between the two visits in P patients identified no differentially expressed genes (Fig. 1C and table S3C), whereas in R patients, more than 5500 genes were differentially expressed (Fig. 1D and table S3D). This pattern remained after controlling for blood cell-type abundances, when we repeated analyses of differential expression of genes using estimated fractions of cell-type populations as additional covariables (22). A total of 1700 genes remained differentially expressed in those with resolved pain, whereas in those with persistent pain, there were still no changes (Fig. 1, column “corrected,” and table S4). Together, our genome-wide transcriptomics analyses suggest that the subjects who resolved pain over time have an abundance of active biological processes underlying recovery and these processes are partially driven by changes in blood cell composition.
Fig. 1. Differential expression of genes in study design contrasts.
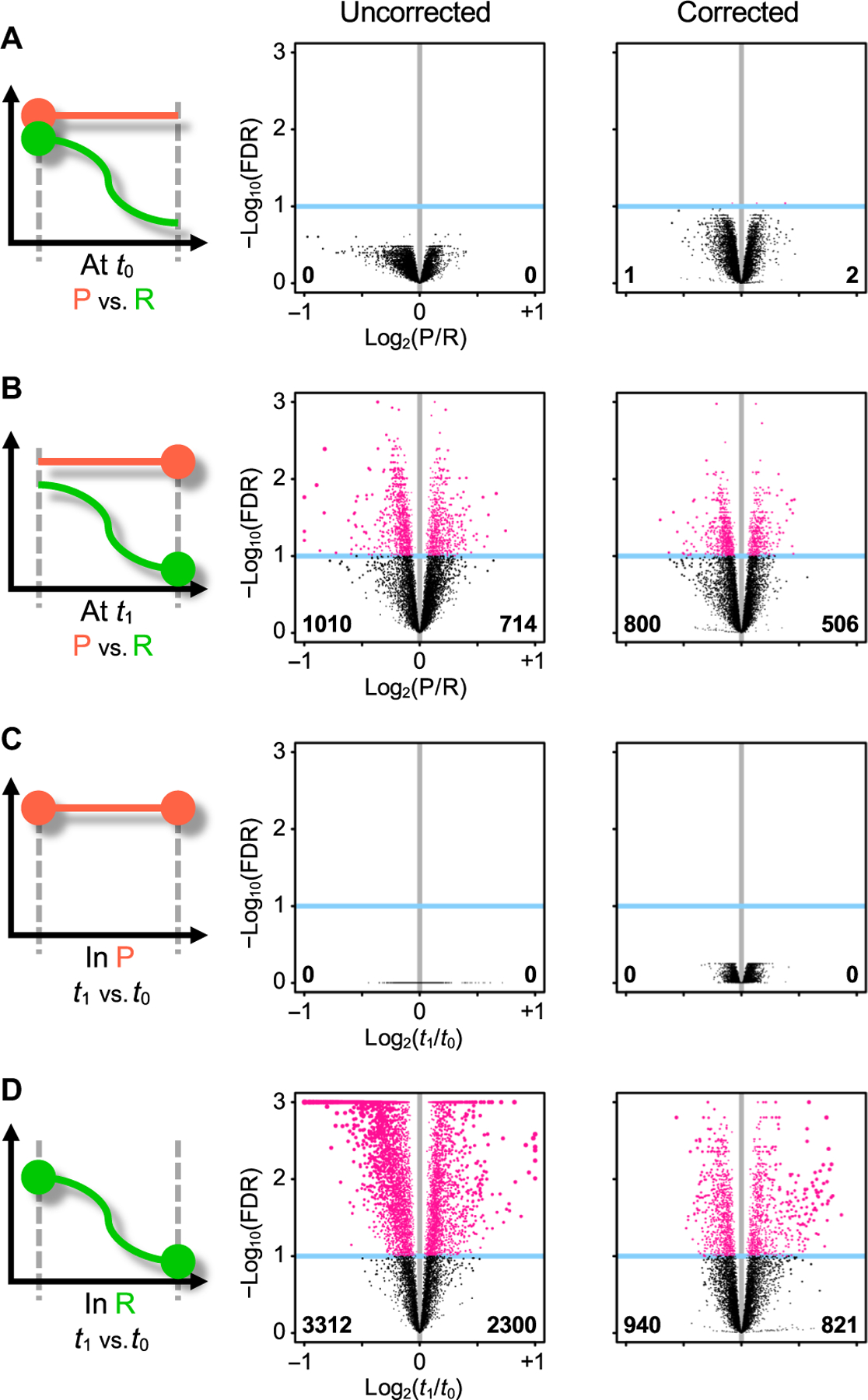
Study design pictograms (left) are juxtaposed to volcano plots (middle and right). The pictogram highlights the two contrasted conditions (large dots). The volcano plot shows statistical significance (y axis) as a function of fold change (x axis); each dot is a gene. Genes that would end up outside of the plot are squeezed inside. Vertical gray line indicates null fold change. Genes reaching statistical significance at the FDR 10% level (blue horizontal line) are highlighted in pink. Numbers in bold indicate counts of significantly differentially expressed genes that are down-regulated (lower left corner) or up-regulated (lower right corner). Volcano plots are shown uncorrected (middle column) and corrected (right column) for blood cell-type fractions. (A and B) Contrast between patients with persistent (P; orange) and resolved (R; green) pain outcomes (A) at t0 or (B) at t1. (C and D) Contrast between t1 and t0 in P patients (C) or in R patients (D).
Differential blood cell-type populations
We next estimated the changes in relative cell-type population fractions from our multiplexed RNA sequencing experiments (22), tracking cell-type population changes in different contrasts (Fig. 2A and table S5). In P participants, we did not detect any changes in blood cell-type populations over time (Fig. 2, column I). However, in R patients, we found statistically significant differences between the two visits in four cell types, with the largest difference observed in the numbers of neutrophils decreasing over time (P = 1.4 × 10−3; Fig. 2B). The other significant differences included increased number of CD8+ T cells (P = 1.5 × 10−3; Fig. 2C) and resting natural killer cells (P = 1.7 × 10−3; Fig. 2D) and decreased number of resting mast cells (P = 4.4 × 10−2; Fig. 2E).
Fig. 2. Blood cell-type fraction trajectories in time, in subjects with persistent or resolved pain.
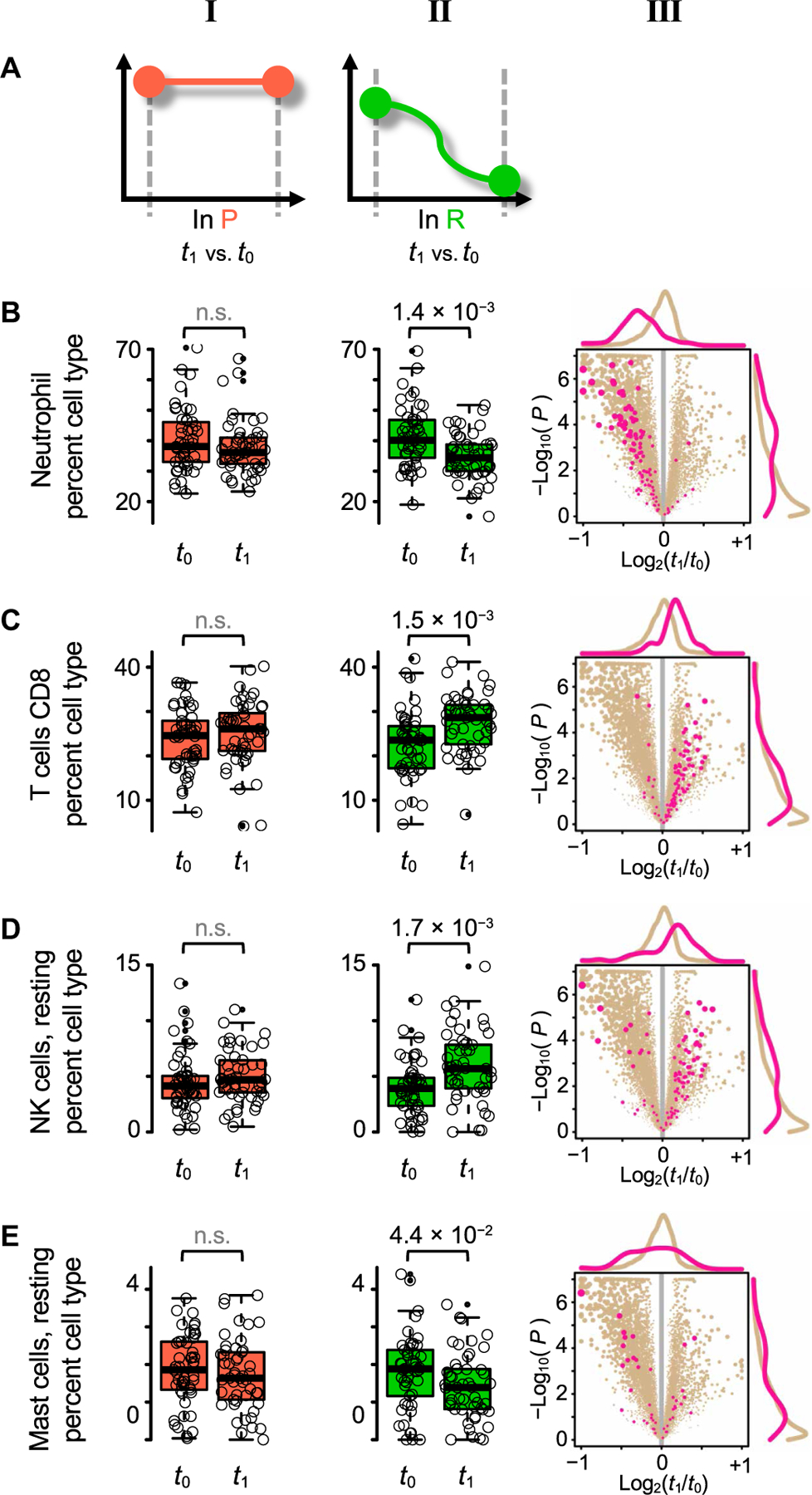
(A) Study design pictograms, showing contrasts in time in those with persistent pain (P; orange, column I), and in those with resolved pain (R; green, column II). (B to E) Box-and-whisker plots (columns I and II) showing the distributions of percent cell-type fraction at t0 and at t1. Cell-type fraction estimates inferred by CIBERSORT from transcriptomics data. P value obtained from logistic regression between the two time points shown on top; not significant (n.s.) when P > 0.05. Volcano plots (column III) for R patients, showing genes highly expressed in the corresponding row’s cell type (pink) versus all other genes (tan). (B) Neutrophils. (C) CD8+ T cells. (D) Natural killer (NK) cells, resting. (E) Mast cells, resting.
We also built a list of genes expressed by each cell type using CIBERSORT’s LM22 “pure” cell-type expression matrix. A gene was retained in the list if the expression level in that cell type was greater than the average across all other cell types (fig. S2). These genes are highlighted for differential expression in the volcano plots (Fig. 2, column III). Thus, changes in cell-type fractions can be tracked down to changes in gene expression, with matching change directions between fraction estimates and gene levels, with, again, the largest, most consistent, and most substantial contribution from neutrophil-specific genes down-regulated between visit t0 and t1.
Pathway analyses
We next analyzed biological pathways instead of individual differentially expressed genes (Fig. 3 and table S6). We found many biological pathways differentially expressed at a genome-wide level even in the comparisons where no individual genome-wide significant differentially expressed genes were identified, which can occur when a substantial amount of genes of the same pathway change expression in the same direction. Because we observed that active transcriptional processes underlay pain recovery over time, we focused on biological pathways at the acute stage that will yield the follow-up changes.
Fig. 3. Functional differences between the resolved and persistent pain groups.
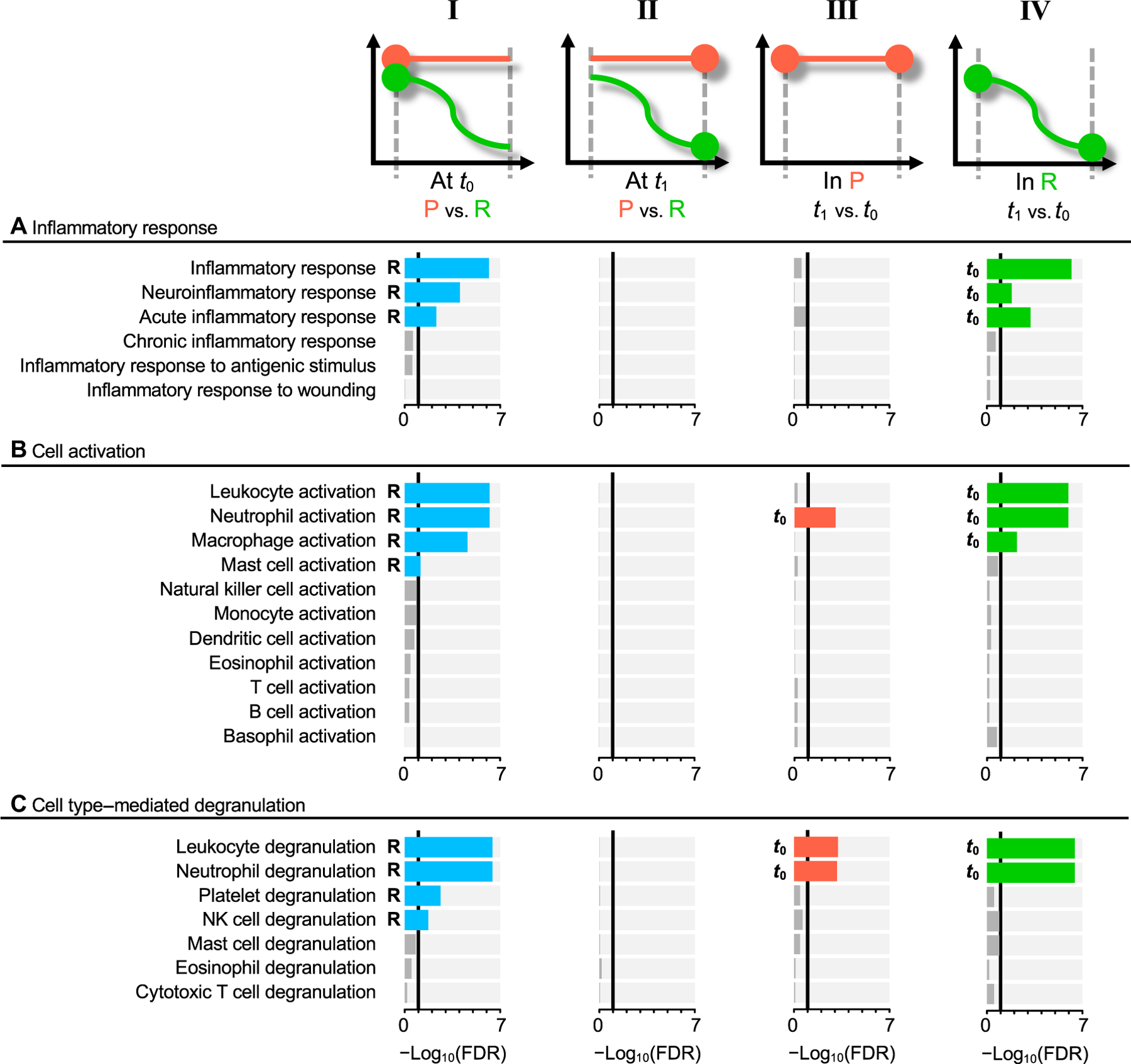
Functional differences assessed with selected pathways in Gene Ontology’s (GO) biological processes. Statistically significant pathways at the FDR 10% level are highlighted in nongray colors; blue at t0 (column I), orange for group P (column III), and green for group R (column IV). Assessment performed between the groups with resolved (R) and persistent (P) pain, at the first visit (column I), and at the second visit (column II). Functional trajectories between t0 and t1 are also shown for the P group (column III) and for the R group (column IV). (A) Pathways under inflammatory response (GO:0006954). (B) Pathways under leukocyte activation (GO:0045321). (C) Pathways under leukocyte degranulation (GO:0043299).
At the first visit (t0), the most differentially expressed pathways were related to cell activation and immune responses, and they were elevated in the R group. These processes seemed to be driven by neutrophil activation and degranulation, and by elevated inflammatory response. Although some of the leading-edge genes are shared between these two pathways, for the most part, they describe two different biological processes (fig. S3). This unexpected enhanced inflammatory response in R participants was consistently observed for other related inflammatory pathways (Fig. 3A, column I, and table S6, A and B). With time, there were barely any changes in the inflammatory response pathways in the P group (Fig. 3A, column III, and table S6, E and F). However, in the R group, the inflammatory response pathways were convincingly down-regulated over time, at t1 compared to t0 (Fig. 3A, column IV, and table S6, G and H).
The enhanced inflammatory pathways seemed to be driven by neutrophil activation through degranulation. We found leukocyte activation and degranulation pathways noticeably activated at t0 in R patients. Among leukocytes, neutrophils were the most activated, followed by macrophages and mast cells (Fig. 3B, column I, and table S6B). Degranulation of neutrophils showed the largest changes compared to platelet or natural killer cells (Fig. 3C, column I, and table S6B). Neutrophil activation and degranulation pathways were decreasing with time in both pain groups, although this decrease was more noticeable in R compared to P patients (Fig. 3 and table S6). The key molecules contributing to this dynamic regulation of cellular responses, with the largest expression at the acute stage and the fastest down-regulation by the second visit in the R group, are presented in table S7. Within the top hits, we found SLC11A1, a divalent metal transporter that is specific to myelomonocytic cells including neutrophils, monocytes, and macrophages (23, 24), and S100A8 and S100A9, neutrophil-specific genes coding for calcium-binding “alarmins” critical in the development and regulation of inflammation, which are known to comprise about 45% of the cytoplasmic proteins in neutrophils and function as homo- or heterodimers (25).
We found a positive correlation in transcriptional changes over time between R and P patients (fig. S4 and table S6), indicating that all individuals displayed similar biological responses and pathways, regardless of the pain outcome at the second visit (slope = +0.57, P < 2.2 × 10−16, r2 = 42%). The difference between groups was in the magnitude of the response: The R group response intensity was about 75% larger than that of the P group.
Replication of findings in an independent cohort
We replicated our findings using a prospective cohort of similar design (fig. S5A). The replication cohort comprised subjects with another musculoskeletal pain condition, TMD. Although the pathophysiology of TMD is likely not identical to LBP, we hypothesized that the active contribution of the immune system in the transition to chronic pain could be shared. At the first visit, all subjects displayed acute symptoms of TMD, whereas at a second visit, some subjects had their pain resolved (R), and in others, pain persisted (P). In this cohort as well, we observed a larger number of differentially expressed genes in subjects in the R group than in the P group (fig. S5, B and C) and elevated activity of inflammatory and neutrophil activation and degranulation pathways in the R group (fig. S5D and table S8). From CIBERSORT’s gene expression input matrix, we identified 100 genes whose expression in neutrophils is greater than the average across all other cell types. At t0, we found 80% of these genes to be more expressed in the R group in both LBP and TMD cohorts (table S8G) and about 75% of these genes to be more expressed at t0 than t1 in the resolved pain group, in both LBP and TMD cohorts (table S8H).
In addition, the TMD cohort allowed comparison with healthy controls and patients with chronic TMD (fig. S6 and table S8I). In comparison with healthy controls, the R group displayed a significantly higher inflammatory response at the acute stage [fgsea pathway enrichment score (ES) = +0.32 > 0, P = 1.1 × 10−5], whereas the P group displayed a significantly lower response (ES = −0.32 < 0, P = 1.3 × 10−7). By the time of the second visit, each pain group displayed a significantly reduced inflammatory response compared to the healthy group (P = 3.1 × 10−4 in R and P = 1.1 × 10−7 in P). The same pattern was observed for neutrophil activation and degranulation pathways.
We also observed up-regulated neutrophil activation and degranulation pathways in subjects with chronic TMD in comparison with healthy controls (ES = +0.19 > 0, P = 1.9 × 10−2 and 3.3 × 10−2, respectively; fig. S6), although to a lesser degree than for the R group at t0 (ES = +34 > 0, P = 4.6 × 10−7). These results indicate the importance of the up-regulation of inflammatory response at the acute stage of musculoskeletal pain as a protective mechanism against the development of chronic pain.
Impaired inflammatory response prolongs resolution of painful behavior in preclinical assays
Our human transcriptomics results suggested that active inflammatory responses, particularly those regulated by neutrophils, contribute to pain resolution. We hypothesized that inhibition of this active immune response will lead to the prolongation of pain and designed experiments to test this hypothesis in mice using pain assays featuring evidence of pain that is persistent but of finite duration.
Initial experiments used the classic steroidal anti-inflammatory drug, dexamethasone. Mechanical pain sensitivity was assessed before and at multiple time points after chronic constriction injury (CCI) of the sciatic nerve, injection of nerve growth factor (NGF) into the muscles of the low back, or inflammatory injury using the cell-mediated immunity stimulator, complete Freund’s adjuvant (CFA; inactivated Mycobacteria tuberculosis in oils). Dexamethasone or saline vehicle was administered daily for 6 days after CCI or CFA. All three injuries produced mechanical allodynia, that is, hypersensitivity to the evoking mechanical stimulus, lasting about 30 to 60 days (depending on the assay) in saline-treated mice, respectively (Fig. 4, A, D, and G). We observed that the steroid had no effect on hind paw allodynia at the end of the treatment period after CCI (t11 = 0.5, P = 0.63; Fig. 4B), as might be expected given that CCI does not produce hind paw inflammation. By contrast, dexamethasone produced robust inhibition of allodynia from NGF (t10 = 2.4, P = 0.04; Fig. 4D) and CFA (t13 = 3.0, P = 0.009; Fig. 4H) on day 6 after injection. However, dexamethasone delayed the recovery to baseline after CCI (t11 = 2.3, P = 0.04; Fig. 4, A and C), NGF (t10 = 2.5, P = 0.03; Fig. 4, D and F), and CFA (t13 = 2.7, P = 0.02; Fig. 4, G and I) such that the duration of the overall pain episode was increased by the steroid treatment, by twofold on average after CFA. To assess whether it was the anti-inflammatory or purely analgesic actions of dexamethasone that were responsible for this prolongation, we tested four other drugs: the NSAID diclofenac, and three analgesics with no known anti-inflammatory action, systemically administered gabapentin and morphine, and peripherally administered lidocaine. All four drugs reduced allodynia during their administration period (F4,49 = 6.1, P < 0.001; Fig. 4, J and K), but only diclofenac significantly prolonged the duration of the overall allodynia episode produced by CFA (F4,49 = 7.5, P < 0.001; Fig. 4, J and L). Diclofenac was also able to prolong the duration of CCI-induced allodynia (t13 = 3.0, P = 0.01; fig. S7).
Fig. 4. Prolongation of neuropathic and inflammatory pain by early anti-inflammatory treatment.
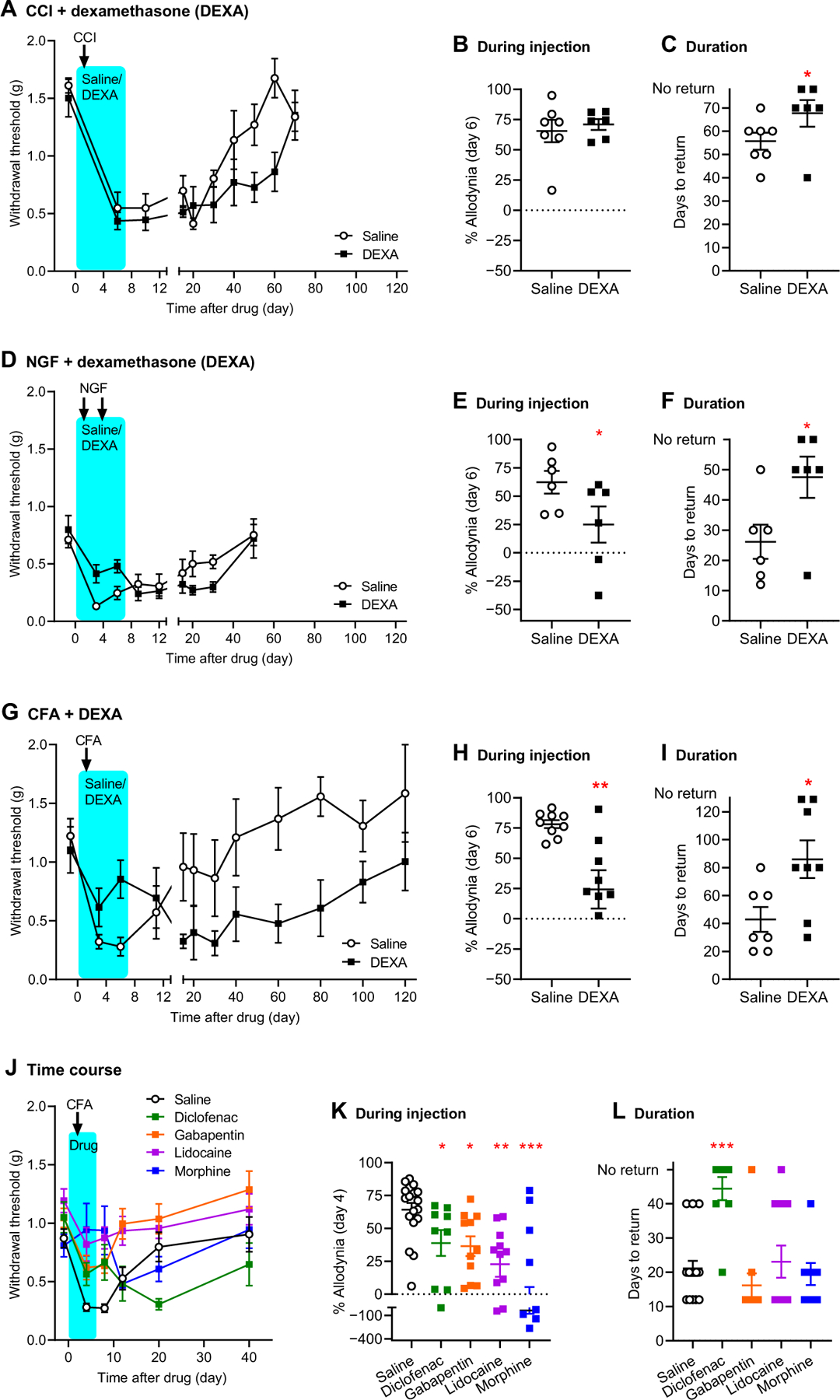
(A) Mechanical pain thresholds before and after chronic constriction injury (CCI) of the sciatic nerve in mice treated from days 0 to 6 with saline or dexamethasone (DEXA). Symbols represent means ± SEM hind paw withdrawal threshold (g). (B) Percentage of maximum possible allodynia (% allodynia) on day 6 after drug; see Materials and Methods for calculation details. Error bars represent SEM. (C) Days required to return to baseline thresholds; see Materials and Methods. Error bars represent SEM. Mice not returning to baseline by day 70 were assigned a value of 80. (D) Mechanical pain thresholds before and after hind paw injection of nerve growth factor (NGF) into the muscles of the low back in mice treated with saline or DEXA. Symbols as in (A). (E) Percent allodynia on day 6 after drug. (F) Days to return to baseline; mice not returning to baseline on day 50 were assigned a value of 60. (G) Mechanical pain thresholds before and after hind paw injection of complete Freund’s adjuvant (CFA) in mice treated with saline or DEXA. Symbols as in (A). (H) Percent allodynia on day 6 post-drug. (I) Days to return to baseline; mice not returning to baseline on day 120 were assigned a value of 140. (J) Mechanical pain thresholds before and after CFA in mice treated with saline, diclofenac, gabapentin, or lidocaine. Symbols as in (A). (K) Percent allodynia on day 4 after drug. (L) Days to return to baseline; mice not returning to baseline on day 40 were assigned a value of 50. *P < 0.05, **P < 0.01, and ***P < 0.001, compared to the corresponding saline group.
To directly assess the hypothesis that neutrophils are responsible for these effects, we performed two complementary experiments. First, we depleted neutrophils using an anti-Ly6G antibody, which causes specific but incomplete depletion (26). Whereas acute depletion of neutrophils using this antibody does not affect mechanical allodynia (27), prolonged administration of the antibody exacerbated allodynia (day 9: t10 = 2.4, P = 0.04; Fig. 5, A and B) and prolonged its duration (t10 = 8.7, P < 0.001; Fig. 5, A and C) in a fashion identical to that of dexamethasone. Next, we endeavored to determine whether the dexamethasone effect was neutrophil dependent by injecting neutrophils isolated from peripheral blood or the neutrophil-released proteins S100A8 or S100A9 into the hind paw (ipsilateral to CFA injection) of dexamethasone-treated mice. Neutrophil injection or injection of S100A8 or S100A9 prevented the development of allodynia entirely (comparison of dexamethasone-injected groups: F3,24 = 8.7, P < 0.001; Fig. 5, D and E) despite the administration of dexamethasone. Note that this experiment, performed in a different laboratory, also serves as a direct replication of the data shown in Fig. 4G. In the absence of dexamethasone, neither neutrophils nor S100A8/A9 significantly affected the duration of CFA allodynia (F15,130 = 1.1, P = 0.36; fig. S8). All reported mouse experiments were performed in both sexes, and no detectable interactions with sex were observed in any experiment (all P > 0.05).
Fig. 5. Involvement of neutrophils and neutrophil-released proteins S100A8 and S100A9 in pain resolution in the mouse.
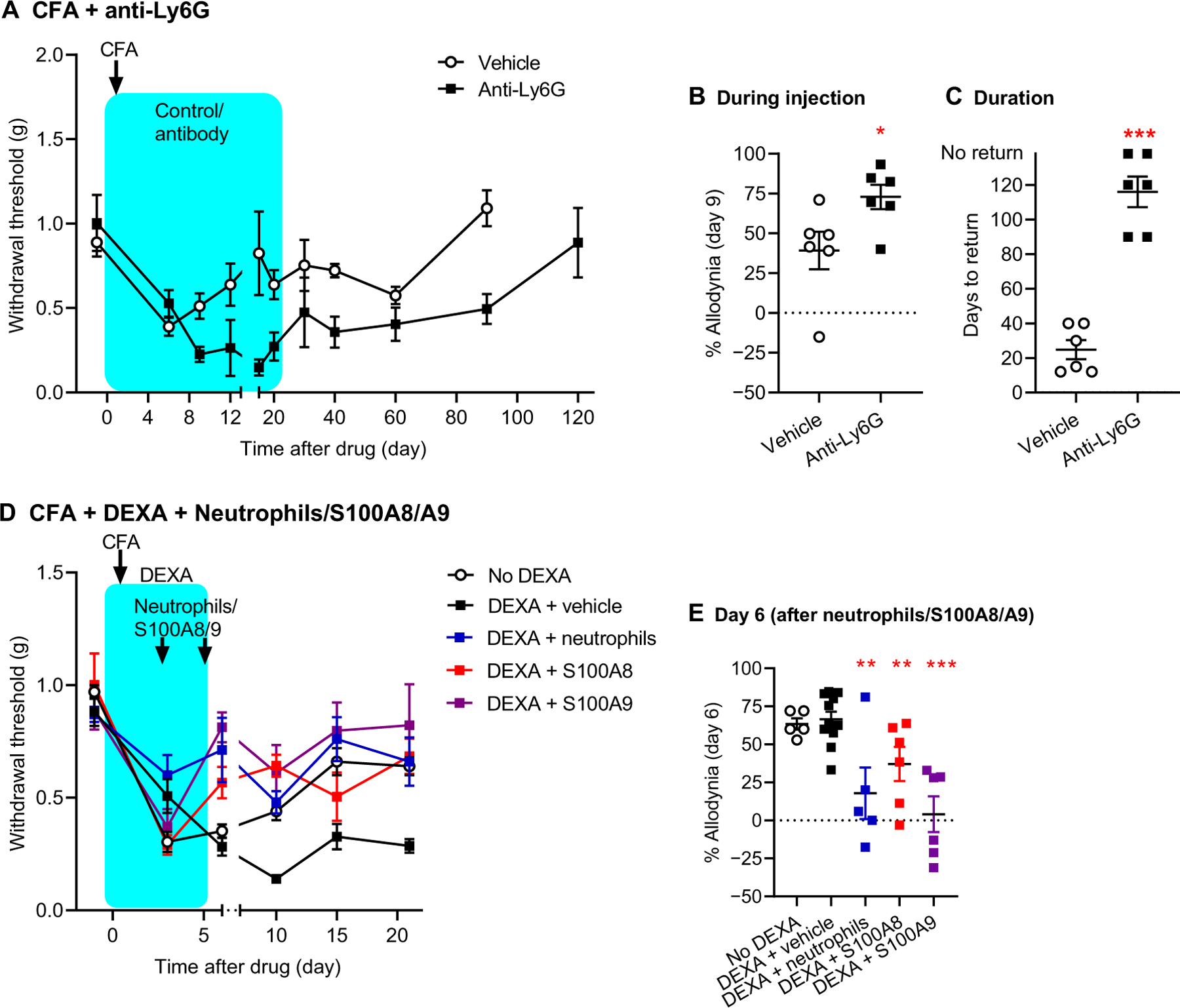
(A) Mechanical pain thresholds before and after CFA in mice treated with anti-Ly6G antibody or its isotype control (vehicle). Symbols represent means ± SEM hind paw withdrawal threshold (g). (B) Percent allodynia on day 6 after drug; see Materials and Methods for calculation details. (C) Days to return to baseline; see Materials and Methods. Mice not returning to baseline on day 120 were assigned a value of 140. (D) Mechanical pain thresholds before and after CFA in mice treated with DEXA (or saline), plus a hind paw injection of vehicle, isolated neutrophils, S100A8, or S100A9 on days 3 and 5 after DEXA. Symbols as in (A). (E) Percent allodynia on day 6 after DEXA. *P < 0.05, **P < 0.01, and ***P < 0.001, compared to the corresponding vehicle/no DEXA group.
Analgesic usage in human population studies
Finally, we examined the relationship between analgesic drug usage and back pain in a large human study from the UK Biobank project. We posited that drugs that inhibit inflammation might interfere with the natural recovery process, thus increasing the odds for chronic pain. To test this hypothesis, we compared several analgesic drug classes with available use information, including NSAIDs, paracetamol (acetaminophen), and antidepressants (Fig. 6 and table S9). We found that individuals with acute back pain were at 1.76-fold greater risk of developing chronic back pain if they reported NSAID usage (P = 2.0 × 10−5) than if they were not taking NSAIDs, adjusting for age, sex, ethnicity, and time interval between measurements (model 1). The increased risk for the development of chronic pain was maintained in the model that accounted for all drugs simultaneously [odds ratio (OR) = 1.78, P = 3.9 × 10−5; model 4]. No other analgesic medication category showed an association with the development of chronic back pain, either across models with the corresponding medication class variable adjusted for demographic covariates alone (models 2 and 3) or in the full model (model 4). We then considered further potential confounders for the development of chronic pain. Measures of pain intensity and higher psychological distress at the acute stage are two factors that have been shown repeatedly to be associated with the development of chronic pain (28, 29). Because pain intensity was not collected in the full UK Biobank cohort, we used the number of reported chronic pain body sites as a substitute for chronic pain intensity. Although pain intensity and anatomical extent of pain sites are different phenotypes, they are highly correlated and have been used previously in this capacity (30–32). When we adjusted our models using covariates that captured these potential confounders, all of the above observations held up, namely, a significantly elevated risk for chronic pain with NSAID usage (OR = 1.67, P = 3.0 × 10−4; model 5).
Fig. 6. Impact of drug class on the development of chronic pain in humans.
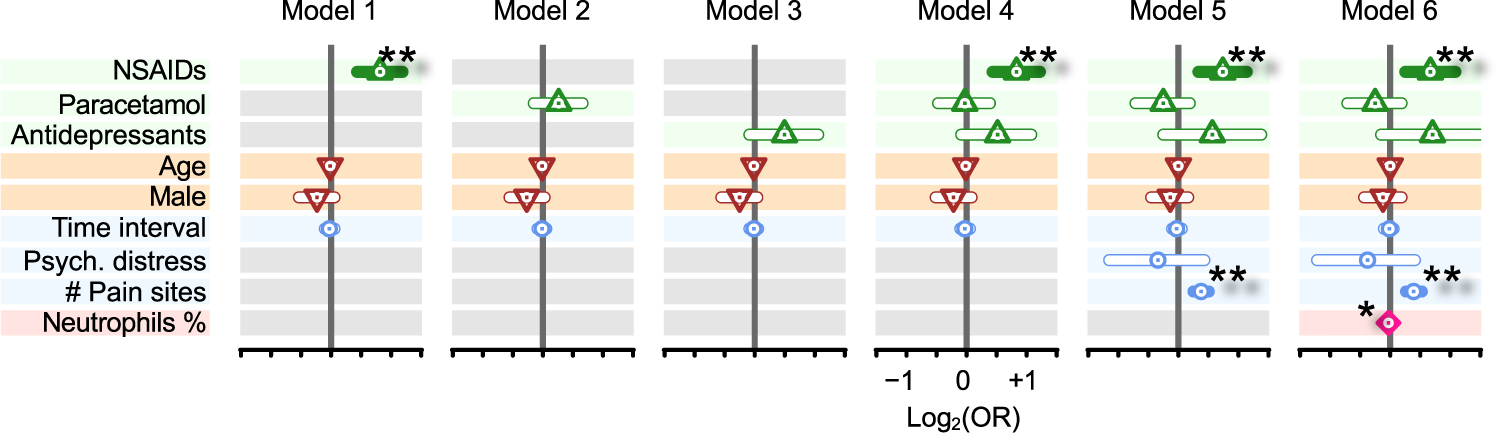
Forest plots track odds ratios (OR; log2 scaled) for the acute-to-chronic transition of back pain and several factors, including analgesic drug classes (up-pointing triangle; green), demographics (down-pointing triangle; brown), possible confounding factors (circle; blue), and percent of neutrophils (lozenge; pink). Models 1 to 3 test each analgesic drug class separately (gray background when factor is not included in the model), whereas model 4 tests all analgesic drug classes simultaneously. Model 5 takes into account possible confounding factors for analgesic drug intake. Model 6 includes neutrophil percentages. Vertical gray bars indicate OR = 1 [log2(OR) = 0]. ORs are indicated by dots inside shapes, whereas the 95% confidence intervals for OR are indicated by horizontal bars. Horizontal bars are filled when P < 0.05. *P < 5 × 10−2 and **P < 5 × 10−3.
Last, given the identification here of the crucial role of neutrophils in inflammatory mechanisms implicated in pain outcomes, we tested across leukocyte subset percentages at the acute pain state for association with the development of chronic back pain later in life by adding this explanatory variable to the full model. As expected, neutrophil percentage at the acute stage was inversely associated with chronic back pain (OR = 0.98; P = 0.02) after adjusting for usage of medications (model 6).
DISCUSSION
This study was designed and implemented to identify cellular and molecular mechanisms underlying acute-to-chronic pain transition in humans using data from a cohort of subjects with LBP. Our initial bioinformatics results indicated that there was a substantial difference in the time courses of transcriptomic changes in subjects with resolved pain compared to those with persistent pain. The trajectories show substantial differences: In the resolved pain group, several thousand genes were found to be differentially expressed over time, whereas there were no differences in the persistent pain group. Thus, our data suggest that active biological processes protect from transitioning to chronic pain after an acute pain episode.
To identify the initial processes that drive these differences in trajectories between the resolved and the persistent pain groups at the gene level, we compared functionally related sets of genes at the pathway level. We found neutrophil activation–dependent elevation of the inflammatory response at the acute stage of pain in subjects with resolved pain, which was decreased by the time of the second visit. Conversely, subjects with persistent pain did not show any changes in their inflammatory response. We replicated these findings in an independent TMD cohort. The shared pathophysiology between different chronic pain conditions has been argued through both high clinical comorbidity and shared genetic heritability (28, 33). The replication of our findings in the TMD cohort also suggests that our findings are likely to be applicable to other chronic pain conditions.
We did not identify any pathways with large negative correlations between the resolved and the persistent pain groups; the two pain groups showed strongly correlated processes. Instead, the difference between the two groups was in the magnitude of the regression slope, again suggesting that the resolved pain group’s response intensity was substantially larger than that of the persistent pain group. These results were in line with the observed differences in the number of differentially expressed genes over time between the groups, and reemphasized the perhaps counterintuitive concept that an active biological process underlies pain resolution rather than pain progression to chronic status. Our results suggest that this process is impaired in those who do not resolve acute pain over time and suggest time stratification of a cascade of processes resulting in a return to a normal, no-pain state (34)—in a fashion similar to timely processes involved in wound healing (35, 36)—and thus would require gene expression probing at many more time points to decipher the complete phenomenon of pain resolution. Nonetheless, our findings are in line with the observation that the beginning of the inflammatory process programs its resolution (34), and it is thus the failure to initiate an appropriate inflammatory response that may lead to chronic pain. This notion was further illustrated by our TMD cohort, which provided the advantage of the availability of a control no-pain group not available in the LBP cohort. We were able to confirm sharp up-regulation of neutrophil-related inflammatory response at the acute stage of the TMD pain-persisting group but not the TMD pain-resolving group and higher inflammatory states in patients with chronic pain.
Using three different assays of prolonged but resolving pain in the mouse, we confirmed that the acute treatment of inflammation with either the steroid, dexamethasone, or the NSAID, diclofenac—although both effectively reducing pain behavior during their administration—greatly prolonged the resolution of neuropathic, myofascial, and especially inflammatory pain states. Three analgesics without anti-inflammatory properties (gabapentin, morphine, and lidocaine) produced short-term analgesic effects without affecting the overall duration of the painful (allodynic) episode. We further showed the neutrophil dependence of these effects, with steroid-like pain prolongation being produced by neutrophil depletion and a complete blockade of allodynia produced by peripheral injection of neutrophils themselves. Furthermore, our mouse data confirmed the important roles of two neutrophil-specific proteins identified via human transcriptomics data, the alarmin proteins S100A8 and S100A9.
Last, we validated the negative consequences of anti-inflammatory drugs in a large human cohort from the UK Biobank project. In human subjects who reported acute back pain, we found that NSAIDs but not two other analgesic medications available for analyses increased risk to still report back pain 2 to 6 years later. Antidepressants were not associated with transition from acute to chronic pain despite the fact that patients who take antidepressants will generally have higher pain and higher psychological distress (37), two major risk factors for the development of chronic pain, in comparison with patients who take NSAIDs. Furthermore, even after these two variables were included as covariates in all logistic regression models considered, our findings that NSAID use (but no other analgesic class) increases risk of subsequent development of chronic back pain did not change. Last, consistent with our bioinformatics and animal model results, higher percentages of neutrophils at the acute pain state protected against chronic pain development.
Our study has several limitations. First, the LBP cohort did not have control subjects without any pain, preventing the comparison of transcriptomes of people who resolved pain to those never experiencing pain. Second, we did not evaluate pain in the LBP cohort before or after the 3-month time point, so it is possible that some subjects in the persisting pain group saw their pain resolve after 3 months but before 6 months. To better understand the inflammatory and pain trajectories, a new study with more frequent pain ratings and blood sampling would be required. Furthermore, to test the long-term consequences of NSAID use with regard to chronic pain development, a clinical trial on patients with acute pain specifically designed to address the question is needed. Although we controlled for potential confounders in our UK Biobank analysis, we did not have access to many important details such as pain intensities at various body sites and drug dosing.
Together, our results suggest that active immune processes confer adaptation at the acute pain stage, and impairment of such inflammatory responses in subjects with acute LBP (or TMD) increases the risk of developing chronic pain. These adaptive inflammatory responses are intrinsically transcriptionally driven, probably modified by both genetics and environmental factors, and can be inhibited by steroids and NSAIDs. These responses are transient, which is probably the main reason why they were previously overlooked. Our conclusions may have a substantial impact on medical treatment of the most common presenting complaints to health care professionals. Specifically, our data suggest that the long-term effects of anti-inflammatory drugs should be further investigated in the treatment of acute LBP and likely other pain conditions.
MATERIALS AND METHODS
Study design
The human LBP cohort is part of a larger study, PainOMICs, registered on clinicaltrials.gov (NCT02037763) and funded by the European Community in the Seventh Framework Programme (FP7)—THEME (HEALTH.2013.2.2.1–5—Understanding and controlling pain) to evaluate biomarkers related to pain. The protocol was approved by the Ethical Committee at the University Hospital of Parma (protocol number 43543; version 8). All patients signed a written informed consent before the enrolment and were followed up for 1 year.
The primary objective of this study was to investigate associations between genome-wide transcriptomics and the development of persistent chronic LBP in patients developing persistent chronic pain symptoms 3 months after an episode of acute LBP. Subjects enrolled in this study were a part of a larger protocol that follows the same design. For the current study, we retrospectively selected the first 50 patients with resolved pain and the first 50 patients with persistent pain. The patients were all Caucasian adults. Sample sizes were not estimated because of the hypothesis-free approach taken here, but our LBP cohort is similarly sized to other human transcriptomics studies in pain-identified group differences (10, 39–42).
The criterion for enrollment was the presence of acute LBP, that is, pain between the costal margins and the gluteal fold. All patients were evaluated using an NRS, a scale to assess pain from 0 to 10, where 0 is “no pain” and 10 is “worst pain imaginable,” and the painDETECT Questionnaire (43) to evaluate the neuropathic pain component. Inclusion criteria included a back pain level of ≥4 on the NRS, with a duration of no more than 6 weeks before the first visit, thus defining an acute phase. Exclusion criteria were history (in the past 6 months) of persistent chronic or acute LBP episodes, recent history (<1 year) of spinal fracture, pain in the back due to spinal tumor or infection, evidence of clinically unstable disease, severe psychiatric disorder (excluding mild depression) or mental impairment, and pregnancy.
Each patient was classified by clinical evaluation and diagnostic blocks to determine the pain generator using these six classes: facet joint pain, sacroiliac pain, discogenic pain, spinal stenosis, back pain with predominant radiculopathy, and nonspecific LBP (44). Patients were medically assessed and treated accordingly to physician’s decision, independently of the protocol. Drugs used and medical history were also recorded.
Clinicians followed a standardized protocol for treating acute LBP with drugs reducing acute inflammatory flare (NSAIDs and systemic steroids) plus opioids if the pain was severe and/or if it had a greater impairment on daily activity. The choice of drugs was driven only by clinical signs and not by any measures of inflammatory flare. Thus, all patients had the same treatment.
Patients were seen at two time points: at the time of enrollment (t0) and at a follow-up visit (t1) 3 months later. We defined the resolved pain group (R) patients as those who self-reported day-averaged pain of less than 4 on the NRS in the week before the follow-up visit; those reporting levels of 4 or higher were defined as the persistent pain group (P). The value of 4 was previously defined as an optimal cut point for “clinically significant” pain (45, 46), and in clinical practice, this cutoff is used to decide if pain may lead to functional and clinical disability and thus should be treated. The researcher who performed the laboratory analysis was masked to the group of patient analyzed.
Statistical analysis
Differential expression of genes in both LBP and TMD cohorts was assessed using moderated statistical tests implemented in the R statistical package DESeq2 (47). Each test was performed with the following covariables: sex, age, smoking status, and RNA Integrity number (RIN). Pathway enrichment scores were estimated using “fgsea,” with statistical significance assessed using a fast permutation scheme (48).
The meta-analysis was performed using a sample size–based analytical strategy, following the formula proposed by METAL (49). The sample sizes were n = 98 for LBP and n = 30 for TMD. For each pathway and study, the sign of the enrichment score combined with its associated P value was converted into a Z statistic. The overall Z statistic was obtained using a sample size–based weighted scheme. An overall P value was calculated from the overall Z.
Pain outcomes in the LBP cohort were regressed to blood cell-type fractions using the R statistical package function “glm,” using sex, age, and smoking status as covariables. Pain outcomes in the UK Biobank cohort were regressed to various drug classes and neutrophil fractions using the R statistical package function glm, individually and in combination, with age, sex, and ethnicity as covariables.
For transcriptomics, we relied on the false discovery rate (FDR) to correct for multiple testing because tests are not independent of one another. Significance levels are indicated in the text.
For animal experiments, a criterion α = 0.05 was used to determine statistical significance. Data were analyzed by Student’s t test or analysis of variance (ANOVA) followed by Tukey’s or Dunnett’s post hoc testing, as appropriate.
Supplementary Material
Acknowledgments:
We would like to thank all participants enrolled in this study through the Anesthesia, Critical Care and Pain Medicine Unit, Department of Medicine and Surgery at University of Parma. We also thank I. King for fruitful discussions. The current study was conducted under UK Biobank application 20802.
Funding:
This work was supported by Pfizer Canada Professorship in Pain Research (to L.D.); Canadian Excellence Research Chairs grant CERC09 (to L.D.); Canadian Institutes of Health Research grant 136975 (to P.A.T.); Canadian Institutes of Health Research Foundation grant 154281 (to J.S.M.); National Institute of Dental and Craniofacial Research grant R56DE025298 (to A.G.N. and G.D.S.); National Institute of Dental and Craniofacial Research grant U01DE017018 (to A.G.N., G.D.S., and L.D.); Canadian Institutes of Health Research grants PJT-173288, PJT-169671, and SCA-145102 (to N.G.); European Commission in the context of the Seventh Framework Program grant 602736 (to C.D. and M.A.); and Canadian Institutes of Health Research Strategy for Patient-Oriented Research in Chronic Pain grant SCA-145102 (to L.D.).
Footnotes
Competing interests: L.D. was/is a consultant for Duke University, ONO PHARMA USA Inc., Releviate Inc., and Orthogen AG. M.A. was/is a consultant for Health&RCB Srl and Clover Orthopedics Srl. P.A.T. is a shareholder of InflammatoRx Inc., a company developing an anti-S100A9 drug for the treatment of inflammatory bowel disease. All other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. Analysis code is available on github.com under project humanpaingeneticslab/code_2022y_A. The sequencing data were deposited in Gene Expression Omnibus under accession number GSE177034.
SUPPLEMENTARY MATERIALS
www.science.org/doi/10.1126/scitranslmed.abj9954
Materials and Methods
Figs. S1 to S8 Tables S1 to S9 References (48–67)
View/request a protocol for this paper from Bio-protocol.
REFERENCES AND NOTES
- 1.Gereau IV RW, Sluka KA, Maixner W, Savage SR, Price TJ, Murinson BB, Sullivan MD, Fillingim RB, A pain research agenda for the 21st century. J. Pain 15, 1203–1214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schopflocher D, Taenzer P, Jovey R, The prevalence of chronic pain in Canada. Pain Res. Manag 16, 445–450 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoy D, Bain C, Williams G, March L, Brooks P, Blyth F, Woolf A, Vos T, Buchbinder R, A systematic review of the global prevalence of low back pain. Arthritis Rheum 64, 2028–2037 (2012). [DOI] [PubMed] [Google Scholar]
- 4.GBD 2017 Disease and Injury Incidence and Prevalence Collaborators, Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1789–1858 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chou R, Deyo R, Friedly J, Skelly A, Weimer M, Fu R, Dana T, Kraegel P, Griffin J, Grusing S, Systemic pharmacologic therapies for low back pain: A systematic review for an American College of Physicians clinical practice guideline. Ann. Intern. Med 166, 480–492 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Vlaeyen JWS, Maher CG, Wiech K, Van Zundert J, Meloto CB, Diatchenko L, Battie MC, Goossens M, Koes B, Linton SJ, Low back pain. Nat. Rev. Dis. Primers 4, 52 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Freidin MB, Tsepilov YA, Palmer M, Karssen LC, Suri P, Aulchenko YS, Williams FMK; Group CMW, Insight into the genetic architecture of back pain and its risk factors from a study of 509,000 individuals. Pain 160, 1361–1373 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramesh D, D’Agata A, Starkweather AR, Young EE, Contribution of endocannabinoid gene expression and genotype on low back pain susceptibility and chronicity. Clin. J. Pain 34, 8–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Starkweather AR, Ramesh D, Lyon DE, Siangphoe U, Deng X, Sturgill J, Heineman A, Elswick RK Jr., S. G. Dorsey, J. Greenspan, Acute low back pain: Differential somatosensory function and gene expression compared with healthy no-pain controls. Clin. J. Pain 32, 933–939 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorsey SG, Renn CL, Griffioen M, Lassiter CB, Zhu S, Huot-Creasy H, McCracken C, Mahurkar A, Shetty AC, Jackson-Cook CK, Kim H, Henderson WA, Saligan L, Gill J, Colloca L, Lyon DE, Starkweather AR, Whole blood transcriptomic profiles can differentiate vulnerability to chronic low back pain. PLOS ONE 14, e0216539 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freidin MB, Tsepilov YA, Stanaway IB, Meng W, Hayward C, Smith BH, Khoury S, Parisien M, Bortsov A, Diatchenko L, Borte S, Winsvold BS, Brumpton BM, Zwart JA, Aulchenko YS, Suri P, Williams FMK, Pain HA-I, Sex- and age-specific genetic analysis of chronic back pain. Pain 162, 1176–1187 (2021). [DOI] [PubMed] [Google Scholar]
- 12.Ji R-R, Chamessian A, Zhang Y-Q, Pain regulation by non-neuronal cells and inflammation. Science 354, 572–577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kavelaars A, Heijnen CJ, Immune regulation of pain: Friend and foe. Sci. Transl. Med 13, eabj7152 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scholz J, Woolf CJ, The neuropathic pain triad: Neurons, immune cells and glia. Nat. Neurosci 10, 1361–1368 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Ji RR, Berta T, Nedergaard M, Glia and pain: Is chronic pain a gliopathy? Pain 154, S10–S28 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grace PM, Hutchinson MR, Maier SF, Watkins LR, Pathological pain and the neuroimmune interface. Nat. Rev. Immunol 14, 217–231 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grace PM, Tawfik VL, Svensson CI, Burton MD, Loggia ML, Hutchinson MR, The neuroimmunology of chronic pain: From rodents to humans. J. Neurosci 41, 855–865 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji RR, Nackley A, Huh Y, Terrando N, Maixner W, Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology 129, 343–366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chapman CR, Vierck CJ, The transition of acute postoperative pain to chronic pain: An integrative overview of research on mechanisms. J. Pain 18, 359.e1–359.e38 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Mifflin KA, Kerr BJ, The transition from acute to chronic pain: Understanding how different biological systems interact. Can. J. Anaesth 61, 112–122 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA, Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cellier M, Shustik C, Dalton W, Rich E, Hu J, Malo D, Schurr E, Gros P, Expression of the human NRAMP1 gene in professional primary phagocytes: Studies in blood cells and in HL-60 promyelocytic leukemia. J. Leukoc. Biol 61, 96–105 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Canonne-Hergaux F, Calafat J, Richer E, Cellier M, Grinstein S, Borregaard N, Gros P, Expression and subcellular localization of NRAMP1 in human neutrophil granules. Blood 100, 268–275 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J, S100A8/A9 in inflammation. Front. Immunol 9, 1298 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pollenus E, Malengier-Devlies B, Vandermosten L, Pham T-T, Mitera T, Possemiers H, Boon L, Opdenakker G, Matthys P, Van den Steen PE, Limitations of neutrophil depletion by anti-Ly6G antibodies in two heterogenic immunological models. Immunol. Lett 212, 30–36 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Ghasemlou N, Chiu IM, Julien JP, Woolf CJ, CD11b+Ly6G− myeloid cells mediate mechanical inflammatory pain hypersensitivity. Proc. Natl. Acad. Sci. U.S.A 112, E6808–E6817 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diatchenko L, Fillingim RB, Smith SB, Maixner W, The phenotypic and genetic signatures of common musculoskeletal pain conditions. Nat. Rev. Rheumatol 9, 340–350 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borsook D, Youssef AM, Simons L, Elman I, Eccleston C, When pain gets stuck: The evolution of pain chronification and treatment resistance. Pain 159, 2421–2436 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolfe F, Pain extent and diagnosis: Development and validation of the regional pain scale in 12,799 patients with rheumatic disease. J. Rheumatol 30, 369–378 (2003). [PubMed] [Google Scholar]
- 30.Barbero M, Fernandez-de-Las-Penas C, Palacios-Cena M, Cescon C, Falla D, Pain extent is associated with pain intensity but not with widespread pressure or thermal pain sensitivity in women with fibromyalgia syndrome. Clin. Rheumatol 36, 1427–1432 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Martin LJ, Smith SB, Khoutorsky A, Magnussen CA, Samoshkin A, Sorge RE, Cho C, Yosefpour N, Sivaselvachandran S, Tohyama S, Cole T, Khuong TM, Mir E, Gibson DG, Wieskopf JS, Sotocinal SG, Austin JS, Meloto CB, Gitt JH, Gkogkas C, Sonenberg N, Greenspan JD, Fillingim RB, Ohrbach R, Slade GD, Knott C, Dubner R, Nackley AG, Ribeiro-da-Silva A, Neely GG, Maixner W, Zaykin DV, Mogil JS, Diatchenko L, Epiregulin and EGFR interactions are involved in pain processing. J. Clin. Invest 127, 3353–3366 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meloto CB, Benavides R, Lichtenwalter RN, Wen X, Tugarinov N, Zorina-Lichtenwalter K, Chabot-Dore AJ, Piltonen MH, Cattaneo S, Verma V, Klares R 3rd, Khoury S, Parisien M, Diatchenko L, Human pain genetics database: A resource dedicated to human pain genetics research. Pain 159, 749–763 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Serhan CN, Savill J, Resolution of inflammation: The beginning programs the end. Nat. Immunol 6, 1191–1197 (2005). [DOI] [PubMed] [Google Scholar]
- 34.St Laurent G 3rd, Seilheimer B, Tackett M, Zhou J, Shtokalo D, Vyatkin Y, Ri M, Toma I, Jones D, McCaffrey TA, Deep sequencing transcriptome analysis of murine wound healing: Effects of a multicomponent, multitarget natural product therapy-Tr14. Front. Mol. Biosci 4, 57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seifert AW, Monaghan JR, Voss SR, Maden M, Skin regeneration in adult axolotls: A blueprint for scar-free healing in vertebrates. PLOS ONE 7, e32875 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roughan WH, Campos AI, Garcia-Marin LM, Cuellar-Partida G, Lupton MK, Hickie IB, Medland SE, Wray NR, Byrne EM, Ngo TT, Martin NG, Renteria ME, Comorbid chronic pain and depression: Shared risk factors and differential antidepressant effectiveness. Front. Psych 12, 643609 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bratus-Neuenschwander A, Castro-Giner F, Frank-Bertoncelj M, Aluri S, Fucentese SF, Schlapbach R, Sprott H, Pain-associated transcriptome changes in synovium of knee osteoarthritis patients. Genes (Basel) 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo Y, Walsh AM, Fearon U, Smith MD, Wechalekar MD, Yin X, Cole S, Orr C, McGarry T, Canavan M, Kelly S, Lin TA, Liu X, Proudman SM, Veale DJ, Pitzalis C, Nagpal S, CD40L-dependent pathway is active at various stages of rheumatoid arthritis disease progression. J. Immunol 198, 4490–4501 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Held M, Karl F, Vlckova E, Rajdova A, Escolano-Lozano F, Stetter C, Bharti R, Forstner KU, Leinders M, Dusek L, Birklein F, Bednarik J, Sommer C, Uceyler N, Sensory profiles and immune-related expression patterns of patients with and without neuropathic pain after peripheral nerve lesion. Pain 160, 2316–2327 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Theken KN, Hersh EV, Lahens NF, Lee HM, Li X, Granquist EJ, Giannakopoulos HE, Levin LM, Secreto SA, Grant GR, Detre JA, FitzGerald GA, Grosser T, Farrar JT, Variability in the analgesic response to ibuprofen is associated with cyclooxygenase activation in inflammatory pain. Clin. Pharmacol. Ther 106, 632–641 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Freynhagen R, Baron R, Gockel U, Tolle TR, painDETECT: A new screening questionnaire to identify neuropathic components in patients with back pain. Curr. Med. Res. Opin 22, 1911–1920 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Allegri M, Montella S, Salici F, Valente A, Marchesini M, Compagnone C, Baciarello M, Manferdini ME, Fanelli G, Mechanisms of low back pain: A guide for diagnosis and therapy. F1000Res 5, F1000 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirschfeld G, Zernikow B, Variability of “optimal” cut points for mild, moderate, and severe pain: Neglected problems when comparing groups. Pain 154, 154–159 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Shafshak TS, Elnemr R, The visual analogue scale versus numerical rating scale in measuring pain severity and predicting disability in low back pain. J. Clin. Rheumatol 27, 282–285 (2021). [DOI] [PubMed] [Google Scholar]
- 45.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sergushichev A, An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 10.1101/060012 [Preprint]. 2016. [DOI]
- 47.Willer CJ, Li Y, Abecasis GR, METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dagostino C, De Gregori M, Gieger C, Manz J, Gudelj I, Lauc G, Divizia L, Wang W, Sim M, Pemberton IK, MacDougall J, Williams F, Van Zundert J, Primorac D, Aulchenko Y, Kapural L, Allegri M, PainOmics G, Validation of standard operating procedures in a multicenter retrospective study to identify -omics biomarkers for chronic low back pain. PLOS ONE 12, e0176372 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liao Y, Smyth GK, Shi W, featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 51.O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, Astashyn A, Badretdin A, Bao Y, Blinkova O, Brover V, Chetvernin V, Choi J, Cox E, Ermolaeva O, Farrell CM, Goldfarb T, Gupta T, Haft D, Hatcher E, Hlavina W, Joardar VS, Kodali VK, Li W, Maglott D, Masterson P, McGarvey KM, Murphy MR, O’Neill K, Pujar S, Rangwala SH, Rausch D, Riddick LD, Schoch C, Shkeda A, Storz SS, Sun H, Thibaud-Nissen F, Tolstoy I, Tully RE, Vatsan AR, Wallin C, Webb D, Wu W, Landrum MJ, Kimchi A, Tatusova T, DiCuccio M, Kitts P, Murphy TD, Pruitt KD, Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44, D733–D745 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.George NI, Bowyer JF, Crabtree NM, Chang CW, An iterative leave-one-out approach to outlier detection in RNA-seq data. PLOS ONE 10, e0125224 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.The Gene Ontology Consortium, The gene ontology resource: 20 years and still Going strong. Nucleic Acids Res 47, D330–D338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G, Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R, InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 16, 169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Slade GD, Ohrbach R, Greenspan JD, Fillingim RB, Bair E, Sanders AE, Dubner R, Diatchenko L, Meloto CB, Smith S, Maixner W, Painful temporomandibular disorder: Decade of discovery from OPPERA Studies. J. Dent. Res 95, 1084–1092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schiffman E, Ohrbach R, Truelove E, Look J, Anderson G, Goulet JP, List T, Svensson P, Gonzalez Y, Lobbezoo F, Michelotti A, Brooks SL, Ceusters W, Drangsholt M, Ettlin D, Gaul C, Goldberg LJ, Haythornthwaite JA, Hollender L, Jensen R, John MT, De Laat A, de Leeuw R, Maixner W, van der Meulen M, Murray GM, Nixdorf DR, Palla S, Petersson A, Pionchon P, Smith B, Visscher CM, Zakrzewska J, Dworkin SF; International RDC/TMD Consortium Network, International Association for Dental Research; Orofacial Pain Special Interest Group, International Association for the Study of Pain, Diagnostic criteria for temporomandibular disorders (DC/TMD) for clinical and research applications: Recommendations of the International RDC/TMD Consortium Network* and orofacial pain special interest groupdagger. J. Oral Facial Pain Headache 28, 6–27 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Slade GD, Greenspan JD, Fillingim RB, Maixner W, Sharma S, Ohrbach R, Overlap of five chronic pain conditions: Temporomandibular disorders, headache, back pain, irritable bowel syndrome, and fibromyalgia. J. Oral Facial Pain Headache 34, s15–s28 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bennett GJ, Xie YK, A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33, 87–107 (1988). [DOI] [PubMed] [Google Scholar]
- 61.La Porta C, Tappe-Theodor A, Differential impact of psychological and psychophysical stress on low back pain in mice. Pain 161, 1442–1458 (2020). [DOI] [PubMed] [Google Scholar]
- 62.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL, Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Meth 53, 55–63 (1994). [DOI] [PubMed] [Google Scholar]
- 63.Mogil JS, Ritchie J, Sotocinal SG, Smith SB, Croteau S, Levitin DJ, Naumova AK, Screening for pain phenotypes: Analysis of three congenic mouse strains on a battery of nine nociceptive assays. Pain 126, 24–34 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Allen NE, Sudlow C, Peakman T, Collins R, Biobank UK, UK Biobank data: Come and get it. Sci. Transl. Med 6, 224ed224 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, Liu B, Matthews P, Ong G, Pell J, Silman A, Young A, Sprosen T, Peakman T, Collins R, UK Biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLOS Med 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brown LF, Kroenke K, Theobald DE, Wu J, Tu W, The association of depression and anxiety with health-related quality of life in cancer patients with depression and/or pain. Psychooncology 19, 734–741 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Verma V, Khoury S, Parisien M, Cho C, Maixner W, Martin LJ, Diatchenko L, The dichotomous role of epiregulin in pain. Pain 161, 1052–1064 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.