

Abstract
Aims/hypothesis
In diabetes, increased retinal oxidative stress is seen before the mitochondria are damaged. Phagocyte-like NADPH oxidase-2 (NOX2) is the predominant cytosolic source of reactive oxygen species (ROS). Activation of Ras-related C3 botulinum toxin substrate 1 (RAC1), a NOX2 holoenzyme member, is necessary for NOX2 activation and ROS generation. In this study we assessed the role of T cell lymphoma invasion and metastasis (TIAM1), a guanine nucleotide exchange factor for RAC1, in RAC1 and NOX2 activation and the onset of mitochondrial dysfunction in in vitro and in vivo models of glucotoxicity and diabetes.
Methods
RAC1 and NOX2 activation, ROS generation, mitochondrial damage and cell apoptosis were quantified in bovine retinal endothelial cells exposed to high glucose concentrations, in the retina from normal and streptozotocin-induced diabetic rats and mice, and the retina from human donors with diabetic retinopathy.
Results
High glucose activated RAC1 and NOX2 (expression and activity) and increased ROS in endothelial cells before increasing mitochondrial ROS and mitochondrial DNA (mtDNA) damage. N6-[2-[[4-(diethylamino)-1-methylbutyl]amino]-6-methyl-4-pyrimidinyl]-2-methyl-4,6-quinolinediamine, trihydrochloride (NSC23766), a known inhibitor of TIAM1–RAC1, markedly attenuated RAC1 activation, total and mitochondrial ROS, mtDNA damage and cell apoptosis. An increase in NOX2 expression and membrane association of RAC1 and p47phox were also seen in diabetic rat retina. Administration of NSC23766 to diabetic mice attenuated retinal RAC1 activation and ROS generation. RAC1 activation and p47phox expression were also increased in the retinal microvasculature from human donors with diabetic retinopathy.
Conclusions/interpretation
The TIAM1–RAC1–NOX2 signalling axis is activated in the initial stages of diabetes to increase intracellular ROS leading to mitochondrial damage and accelerated capillary cell apoptosis. Strategies targeting TIAM1–RAC1 signalling could have the potential to halt the progression of diabetic retinopathy in the early stages of the disease.
Keywords: Diabetic retinopathy, Mitochondrial dysfunction, NOX2, RAC1, TIAM1
Introduction
Diabetic retinopathy is a multi-factorial disease, and the pathogenesis of this slow-progressing disease remains unclear. Many metabolic abnormalities, including activation of the polyol pathway, protein kinase C, increase in oxidative stress and formation of advanced glycation end-products, are implicated in its development [1, 2]. We reported that an increase in oxidative stress in the retina and its capillary cells is observed before mitochondria become dysfunctional and apoptosis is accelerated [3, 4]. Mitochondrial superoxide radicals are considered to act as a common connection between the pathways triggered by hyperglycaemia, which induce vascular damage in diabetic complications including retinopathy [5]. However, the source of increased reactive oxygen species (ROS) in the retina, seen before mitochondrial damage, remains to be identified.
In addition to mitochondria as a potential source of ROS, superoxide radicals are also generated by NADPH oxidases (NOXs) and recent evidence implicates the phagocyte-like NOX2 in the generation of ROS, leading to cellular damage and apoptosis [6–9]. NOX2 is a multi-protein complex with gp91phox and p22phox representing the membranous core and p47phox, p40phox, p67phox and ras-related C3 botulinum toxin substrate 1 (RAC1) as the cytosolic core (Fig. 1). Cellular activation leads to translocation of the cytosolic core to the membrane for association with the membranous core for assembly of the holoenzyme and catalytic activation of NOX2. RAC1 activation also leads to its association with p67phox, triggering the translocation of RAC1–p67phox complex to the membrane [10]. However, putative cellular mechanisms underlying RAC1 activation in the diabetic retina remain to be defined.
Fig. 1.
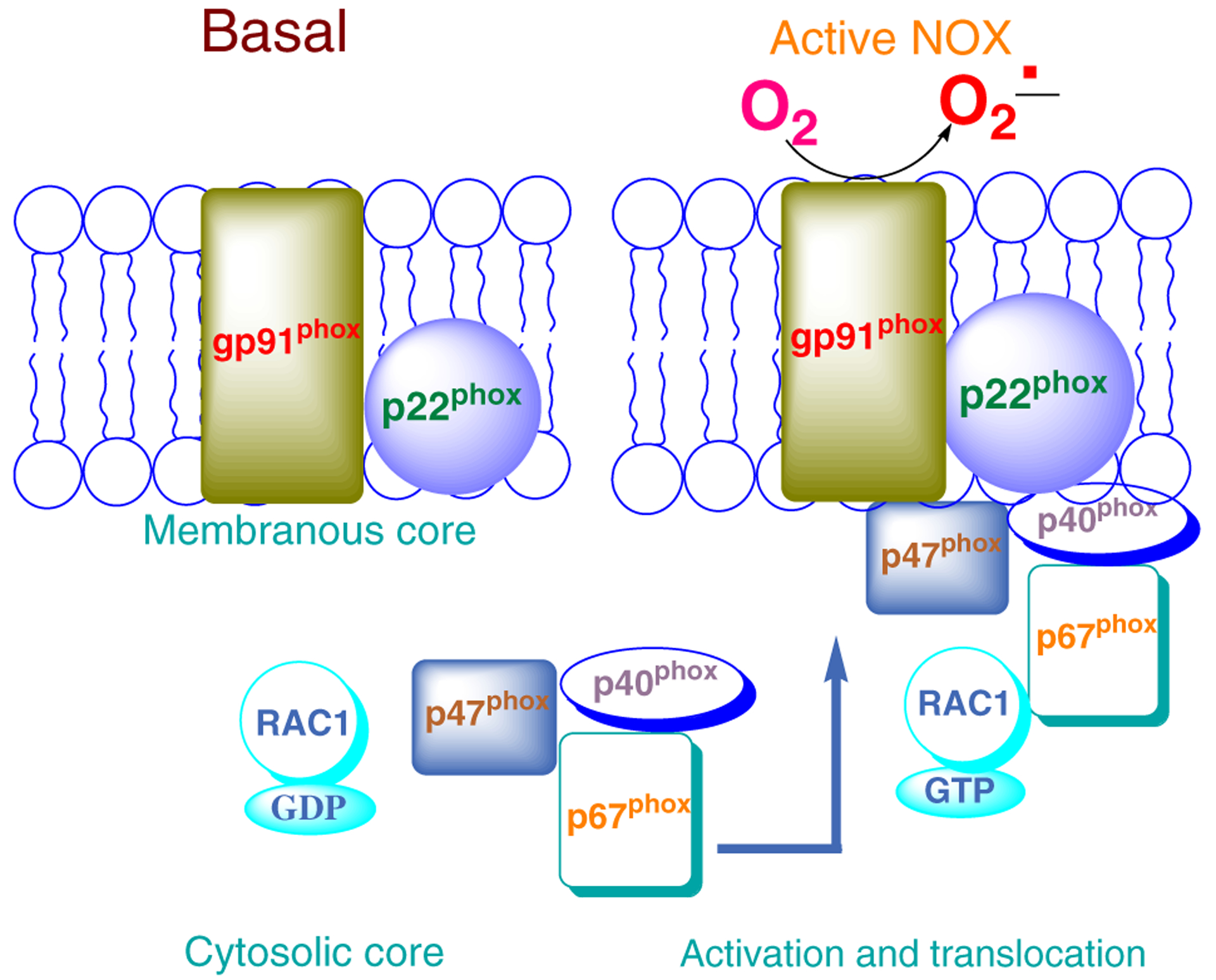
Schematic representation of NOX2 activation. The NOX2 holoenzyme consists of cytosolic and membranous core components. Upon activation, the cytosolic core translocates and associates with the membranous core to complete NOX2 holoenzyme assembly, resulting in its activation and generation of ROS
Most small G-proteins, including RAC1, undergo stimulus-specific activation–deactivation cycles during regulation of cell function. RAC1 is active in its GTP-bound conformation and returns to the GDP-bound inactive conformation following hydrolysis of GTP to GDP by its intrinsic GTPase activity. Furthermore, the activation–deactivation cycles of G-proteins are under the control of regulatory proteins/factors. For example, guanine nucleotide exchange factors (GEFs) mediate GDP/GTP exchange, while the GTPase functions are modulated by GTPase-activating proteins (GAPs). Several GEFs (T cell lymphoma invasion and metastasis [TIAM1], the ‘onc F’ proto-oncogene [VAV2] and triple functional domain protein [Trio]) are known to mediate RAC1 activation [11–13]. We recently identified TIAM1 as the candidate GEF for the activation of RAC1 in triggering the dysfunction and apoptosis of pancreatic beta cells under the duress of high glucose, lipids and proinflammatory cytokines [8, 14–16]. The role of the TIAM1–RAC1 axis in the regulation of retinal NOX2 in the pathogenesis of diabetic retinopathy remains to be elucidated.
Based on evidence suggesting a causal role for hyperglycaemia-induced oxidative stress in the onset of mitochondrial dysfunction in the development of diabetic retinopathy [3, 4, 17, 18], we undertook the current study to assess the role of the TIAM1–RAC1–NOX2 signalling axis in ROS generation before the onset of mitochondrial damage. Using in vitro (bovine retinal endothelial cells) and in vivo (rodent) models of diabetic retinopathy, we established temporal relationships between diabetes-induced TIAM1–RAC1–NOX2 activation, mitochondrial dysfunction and cell apoptosis.
Methods
Bovine retinal endothelial cells
Endothelial cells were isolated from calf retina. Cells from the fifth to the sixth passage were incubated in DMEM containing 2% heat-inactivated fetal bovine serum, 10% Nu-serum, 50 μg/ml heparin, 1 μg/ml endothelial growth factor and antibiotic/antimycotic for 3–96 h in 5 or 20 mmol/l glucose in the absence or presence of 20 μmol/l N6-[2-[[4-(diethylamino)-1-methylbutyl]amino]-6-methyl-4-pyrimidinyl]-2-methyl-4,6-quinolinediamine, trihydrochloride (NSC23766; Calbiochem-EMD Millipore, Billerica, MA, USA). Cells incubated in mannitol instead of glucose served as osmotic controls [3, 19–21].
Rats and mice
Male Wistar rats (200–220 g, 6–8 weeks old) obtained from Harlan laboratories (Indianapolis, IN, USA) were rendered diabetic by streptozotocin (55 mg/kg body weight). To allow slow weight gain while maintaining hyperglycaemia (blood glucose levels of 20–25 mmol/l), a small dose of insulin (1–2 U; Humulin N; Lilly, IN, USA) was administered three to five times each week. Two months after induction of diabetes, the rats were killed by an overdose of pentobarbital (120 mg/kg) and the retina were isolated [3, 20].
Male C57BL/6J mice (~20 g, 6–7 weeks old) obtained from Jackson Laboratory (Bar Harbor, ME, USA), were rendered diabetic by streptozotocin injection (55 mg/kg) for 5 consecutive days [22, 23]. Those mice presenting with a blood glucose concentration of 14 mmol/l or higher, 1 day after the last injection of streptozotocin, were considered to be diabetic. A group of diabetic mice was administered NSC23766 (2.5 mg kg−1 day−1, i.p.) soon after establishment of diabetes. The mice were killed 2 weeks after initiation of NSC23766 treatment by carbon dioxide asphyxiation and the retina was isolated under a dissecting microscope and frozen immediately in liquid nitrogen. Age-matched normal mice served as controls.
The treatment of the animals conformed to the Association for Research in Vision and Ophthalmology Resolution on the Use of Animals in Research, and institutional guidelines.
Human retinal microvessels
Retinas were isolated from human post-mortem eyes (obtained from Midwest Eye Banks, IL, USA), and a small portion of each retina (~5 mm2) was incubated in 10 ml distilled water for 1 h at 37°C. This was followed by incubation with DNAse to remove neural and glial cells [24, 25]. The debris was cleaned from the vasculature under a microscope by repetitive inspiration and ejection through a Pasteur pipette and cleaned vasculature was used for analysis. Donors with established retinopathy (54–77 years of age) had diabetes of 10–30 years’ duration, and non-diabetic donors (55–77 years of age) served as controls (Table 1).
Table 1.
Characteristics of non-diabetic and diabetic human retina donors
| Donor type | Age (years) | Duration of diabetes (years) | Cause of death |
|---|---|---|---|
| Donors with diabetic retinopathy | 75 | 12 | Pulmonary oedema |
| 54 | 10 | Congestive heart failure | |
| 69 | 10 | Respiratory failure | |
| 75 | 30 | Myocardial infarction | |
| Non-diabetic donors | 57 | Myocardial infarction | |
| 70 | Stroke | ||
| 77 | Myocardial infarction | ||
| 55 | Subarachnoid haemorrhage |
These studies were conducted according the guidelines established by the US Department of Health and Human Services/NIH and approved by Wayne State University.
ROS
Total ROS levels were quantified fluorometrically using 2′,7′-dichlorofluorescein diacetate (DCHFDA; Sigma-Aldrich, St Louis, MO, USA). Briefly, 5–10 μg of protein (endothelial cells or retina) was incubated with 2 μmol/l of DCHFDA for 10 min and the resultant fluorescence was measured at 485 nm and 530 nm as excitation and emission wavelengths, respectively [20].
Mitochondrial ROS
Mitochondrial ROS were quantified in bovine retinal endothelial cells using MitoTracker Red (CM-H2XROS; Invitrogen, Pierce, Rockford, IL, USA), a mitochondria-selective dye that emits fluorescence when oxidised. At the end of the desired treatment, the cells were incubated with 400 nmol/l MitoTracker Red for 30 min and washed with PBS, followed by quantification of ROS at 579 nm (excitation) and 599 nm (emission) wavelengths [23, 26].
NOX2 activity
NOX2 activity was measured in cell homogenates (10 μg protein) by luminescence assay [27] using 20 μmol/l lucigenin as electron acceptor and 100 μmol/l NADPH. Specificity of NOX2 activation was determined by adding 0.2 mmol/l apocynin or 2.5 μmol/l gp91ds-tat peptide (or its inactive scrambled peptide) (AnaSpec, Fremont, CA, USA) in the assay medium [28].
RAC1 activation assay
RAC1 activation was determined by the pull-down assay kit (Cytoskeleton, Denver, CO, USA) and the relative amount of activated RAC1 (RAC1-GTP) was quantified by western blotting and densitometry [29, 30]. RAC1 activation in human retinal microvessels was quantified using a G-LISA (Cytoskeleton).
Translocation and membrane association of RAC1 and p47phox
Total membrane and soluble fractions were isolated from homogenates of bovine retinal endothelial cells or rat retinas by a single-step centrifugation method [28, 29] and the relative abundance of RAC1 and p47phox was quantified by western blotting and densitometry.
Isolation of mitochondria
Mitochondria were isolated using the Mitochondria Isolation kit from Invitrogen. These preparations are relatively free of other subcellular contaminants[23].
Mitochondrial dysfunction
Movement of BCL-2-associated X protein (BAX) into the mitochondria was estimated by western blot technique using COX IV as a loading standard [3, 19, 20].
Mitochondrial DNA damage
Mitochondrial DNA (mtDNA) damage was assessed by isolating total DNA using a DNeasy blood and tissue kit (Qiagen, Valencia, CA, USA), followed by performing extended-length PCR by amplifying long and short regions of mtDNA. The intensity of PCR gel photographs was measured using Un-Scan-It Gel digitising software, and the ratio of the long (13.4 kb) to short fragment (210 bp) of PCR amplicons was calculated. A decrease in the ratio of long to short amplicons indicated damage to the mtDNA [19, 20].
Mitochondrial DNA copy number
The mitochondrial DNA copy number was estimated by quantifying the genomic DNA levels of mtDNA-transcribed COX2, and normalising them with that of β-actin [31].
RNA isolation and gene expression
After extracting total RNA by Trizol reagent (Invitrogen, Grand Island, NY, USA), 1 μg RNA was used for cDNA preparation using the High Capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). The expression of genes encoding NOX2, RAC1 and p47phox was measured by SYBR green-based quantitative real-time PCR (qPCR) with melting curve analysis on ABI 7500. β-Actin was used as a housekeeping gene, and the transcript quantification was performed using the ΔΔCt method [23, 24, 26, 31]. Primers used are listed in electronic supplementary material (ESM) Table 1.
Capillary cell apoptosis
Apoptosis was quantified by ELISA using the Cell Death Detection ELISAPLUS kit from Roche Diagnostics (Indianapolis, IN, USA). The mono- and oligonucleosomes generated from the apoptotic cells were quantified using monoclonal antibodies directed against DNA and histones, respectively, and the absorbance generated by the incubation with 2,2′-azino-di-(3-ethylbenzthiazoline sulfonate) diammonium salt was measured at 405 nm [23].
Statistical analysis
Statistical analysis of experimental data was carried out using Sigma Stat software (Jandel Scientific Corporation, San Rafael, CA, USA; Version 1.0). Data are expressed as means ± SD. The Shapiro–Wilk test was used to test for normal distribution of the data and for variables with normal distribution; Student’s t test was used for comparing two groups and analysis of variance followed by Bonferroni’s test was applied for multiple groups. For data that did not present normal distribution, the Mann–Whitney U test was used to compare two groups. A p value of <0.05 was considered statistically significant.
Results
Glucose-induced activation of NOX2 precedes mitochondrial damage, and its regulation by the TIAM1–RAC1 pathway
Incubation of bovine retinal endothelial cells with high glucose (20 mmol/l) increased total ROS levels by ~50% at 3 h and by over twofold at 24–96 h of glucose exposure compared with cells incubated in normal glucose (5 mmol/l). Incubation of cells with 20 mmol/l mannitol (96 h) as an osmotic control did not increase ROS levels (Fig. 2a). RAC1 undergoes activation–deactivation cycles for optimal regulation of cell function, and GTP loading onto RAC1 is facilitated by GEFs. Since TIAM1 is one of the known GEFs for RAC1, we assessed its role in glucose-induced mitochondrial dysregulation and cell apoptosis using NSC23766, a specific inhibitor of the TIAM1–RAC1 axis [13]. NSC23766 significantly inhibited total ROS levels in endothelial cells within 3 h of glucose exposure (Fig. 2a). However, NSC23766 did not affect basal ROS levels seen in retinal endothelial cells incubated with 5 mmol/l glucose (96 h; Fig. 2a).
Fig. 2.
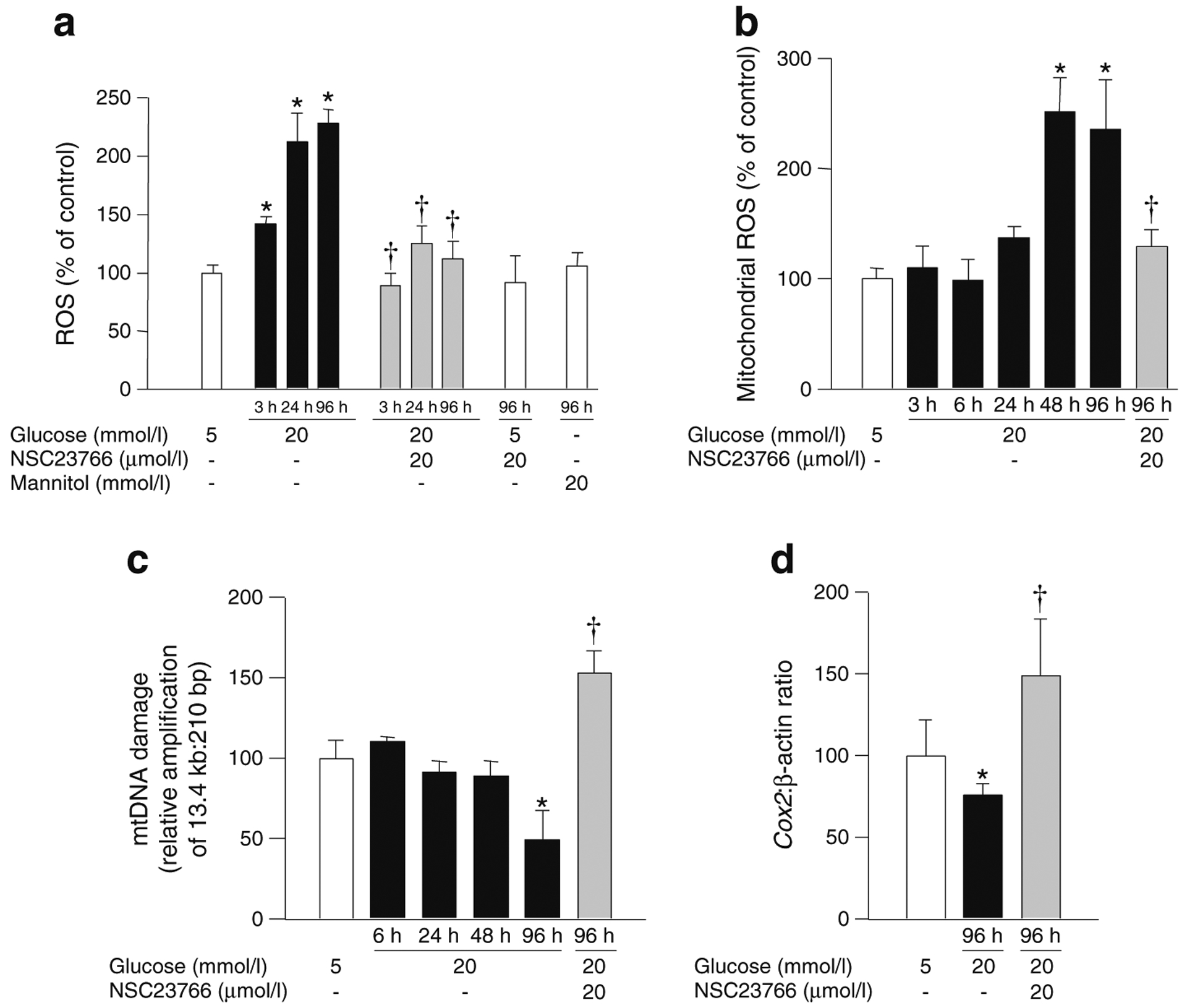
Glucose-induced mitochondrial damage in bovine retinal endothelial cells follows increase in cytosolic ROS. Cells were incubated with glucose for 3–96 h in the absence or presence of NSC23766 as indicated. (a) Total ROS levels were quantified fluorometrically. (b) Mitochondrial ROS levels were quantified fluorometrically using MitoTracker Red. (c) mtDNA damage was assessed by extended-length PCR by using primers for the long and short regions of mtDNA (see Methods). (d) mtDNA copy number was estimated by quantification of COX2 levels and normalised to β-actin. Each measurement was made in duplicate in three or more cell preparations. Values are expressed as mean ± SD of three to four different experiments; values obtained from cells in 5 mmol/l glucose are considered as 100%. *p<0.05 compared with 5 mmol/l glucose; †p<0.05 compared with 20 mmol/l glucose (no NSC23766)
In contrast to total ROS levels, glucose exposure for up to 24 h did not significantly affect mitochondrial ROS generation. However, at 48 h the mitochondrial ROS levels were elevated by ~150% and remained elevated after 96 h of high glucose insult (Fig. 2b). In cognate cellular preparations, mtDNA damage and copy number were not affected until 96 h of exposure to high glucose; the damage to the mtDNA was 50% higher (as indicated by decrease in the relative amplification ratio of 13.4 kb and 210 bp amplicons) and the copy number was ~30% lower compared with the cells incubated in normal glucose (Fig. 2c, d). NSC23766 also prevented increase in mitochondrial ROS levels at 96 h of high glucose insult (Fig. 2b). Similarly, the high-glucose-induced mtDNA damage and decrease in its copy number were prevented by NSC23766 (Fig. 2c, d). It is noteworthy that protection against high-glucose-induced mitochondrial defects by NSC23766 resulted in values higher than the control, suggesting a significant role for TIAM1–RAC1 in high-glucose-mediated damage to mitochondria. Cells incubated in normal glucose for 3–96 h had similar levels of total and mitochondrial ROS and mtDNA damage (data not shown).
NSC23766 prevents glucose-induced activation of RAC1, NOX2 and mitochondrial dysfunction in retinal endothelial cells
Data shown in Fig. 3a demonstrate an approximately twofold stimulation in NOX2 activity in retinal endothelial cells within 3 h of high glucose exposure. At 24 h, the activation was increased threefold and the increase persisted up to 96 h compared with cells incubated in normal glucose. NOX2 activity in cells incubated with normal glucose for 3–96 h remained unaffected (not shown). High-glucose-induced NOX2 activity was attenuated by gp91ds-tat peptide, a specific inhibitor of NOX2 [28], but not by its inactive scrambled peptide, suggesting that high glucose specifically regulates NOX2 activity (Fig. 3b). NSC23766 elicited significant inhibitory effects on high-glucose-induced NOX2 activation, further confirming the role of TIAM1–RAC1 in this signalling step (Fig. 3b). High glucose exposure also increased the expression of NOX2 (Fig. 3c). Together, our findings suggest that NOX2 activation triggers mitochondrial dysfunction.
Fig. 3.
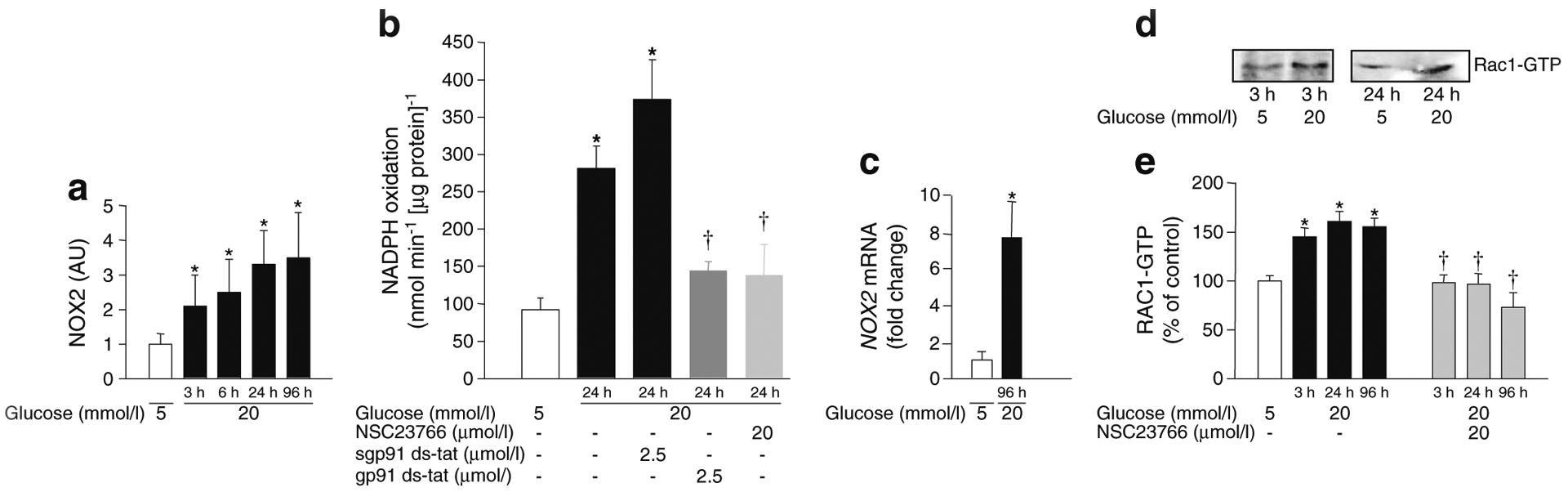
Time course of NOX2 and RAC1 activation in bovine retinal endothelial cells exposed to high glucose. Cells were incubated in 5 or 20 mmol/l glucose for 3–96 h. (a) Apocynin-sensitive NOX2 activity was measured by luminescence assay and is expressed as arbitrary units (AU). (b) Inhibition of glucose-induced NOX2 activity by gp91ds-tat peptide or NSC23766. (c) NOX2 mRNA levels assessed by SYBR green-based qPCR. (d) RAC1 activation (150 μg protein) was determined by pull-down assay. (e) Quantification of RAC1 activation [8, 11] in retinal endothelial cells incubated with low and high glucose in the absence or presence of NSC23766 as indicated. *p<0.05 vs 5 mmol/l glucose;†p<0.05 vs 20 mmol/l glucose (no NSC23766)
Since the activation of RAC1 is necessary for NOX2-mediated ROS generation, it was quantified in retinal endothelial cells incubated with high glucose for 3–96 h. A representative western blot (pull-down assay) demonstrating glucose-induced activation of RAC1 at 3 and 24 h is shown in Fig. 3d. Pooled data from multiple studies (Fig. 3e) suggested that RAC1 was activated by 50% within 3 h of high glucose exposure and that this increased activity persisted up to 96 h. Duration of incubation had no effect on RAC1 activation in cells incubated in normal glucose (not shown). NSC23766-mediated inhibition of increase in ROS in high glucose conditions (as shown in Fig. 2a) was accompanied by attenuation of glucose-induced activation of RAC1 (Fig. 3e).
NSC23766 prevents high glucose-induced apoptosis in bovine retinal endothelial cells
As we reported earlier [3], although an increase in apoptosis was not observed during the initial stages of glucose insult, apoptosis was approximately threefold higher following exposure of retinal endothelial cells to glucose for 96 h (Fig. 4). Based on these data, we conclude that high glucose promotes activation of NOX2 in retinal endothelial cells as early as 3 h, but mitochondrial dysfunction is not observed until the duration is extended to 96 h. Consistent with this, glucose-induced apoptosis of retinal capillary cells was inhibited by NSC23766 (Fig. 4), thus providing the first evidence for the involvement of the TIAM1–RAC1–NOX2 signalling cascade in glucose-induced mitochondrial dysregulation and cell death.
Fig. 4.
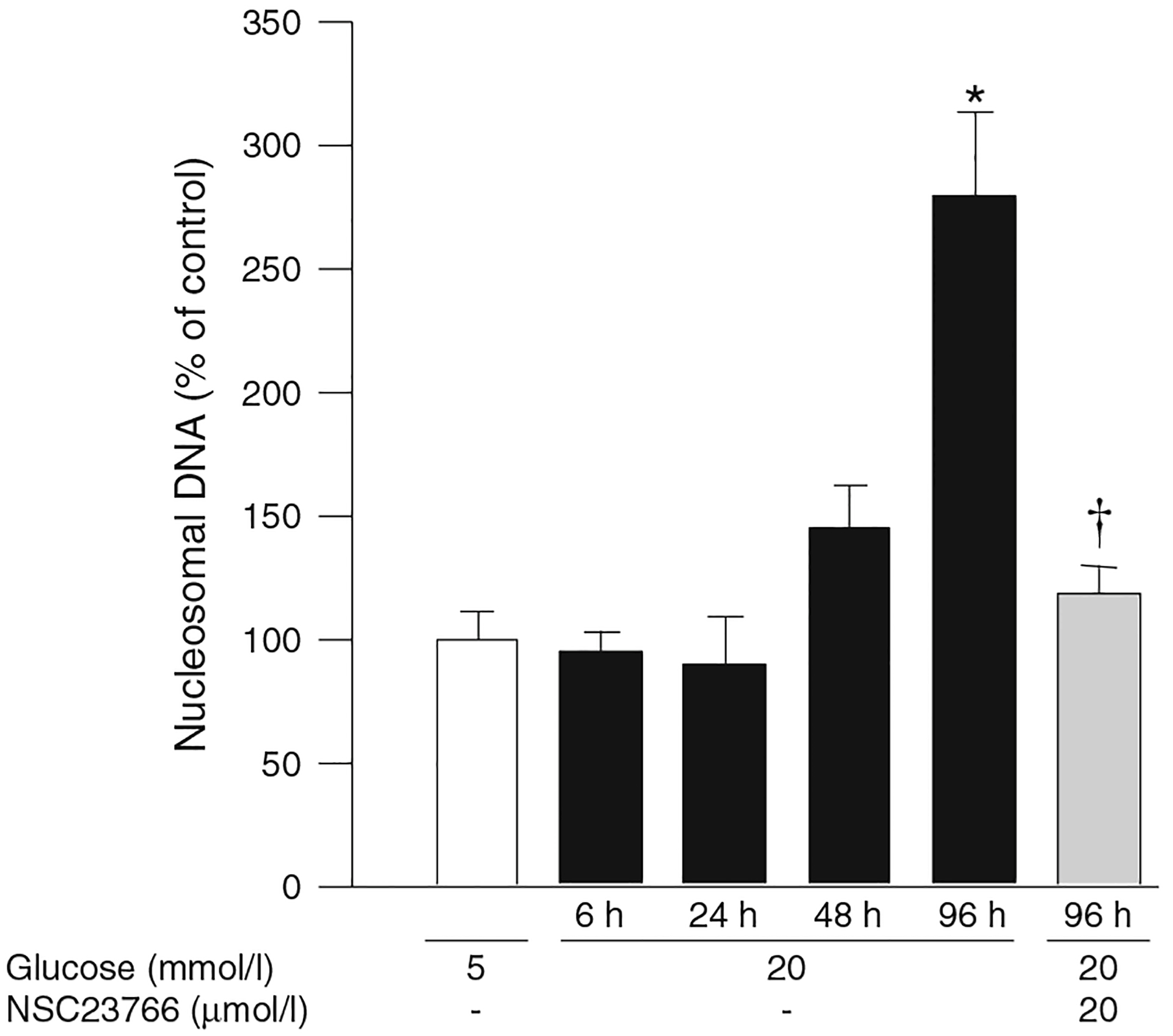
NSC23766 prevents glucose-induced accelerated apoptosis of retinal endothelial cells. Apoptosis was quantified by ELISA using the Cell Death Detection ELISAPLUS kit. Cells incubated in 5 mmol/l glucose (control), and the values obtained from those cells, are set at 100%. The results are represented as mean ± SD of three or more experiments, with each measurement made in duplicate. *p<0.05 vs 5 mmol/l and †p<0.05 vs 20 mmol/l glucose (no NSC23766)
Nox2 expression and membrane association of RAC1 and p47phox are increased in the retina from diabetic rats
Data shown in Fig. 5a demonstrate significant increase in the expression of retinal Nox2 in diabetic rats. Since translocation of the cytosolic core to the membrane fraction is necessary for NOX2 holoenzyme assembly and activation (Fig. 1), we determined association of RAC1 and p47phox with the membrane fraction in retina from control and streptozotocin-induced diabetic mice (2 months of duration of diabetes). Data shown in Fig. 5b demonstrate a significant increase in membrane-bound RAC1 and p47phox in the diabetic rats compared with the control rats. There was almost a threefold increase in membrane-bound RAC1 in bovine retinal endothelial cells incubated with high glucose (24 h; not shown). These findings suggest increased translocation of the cytosolic core of NOX2 to the membrane fraction under the duress of high glucose conditions in vitro and diabetic conditions in vivo.
Fig. 5.
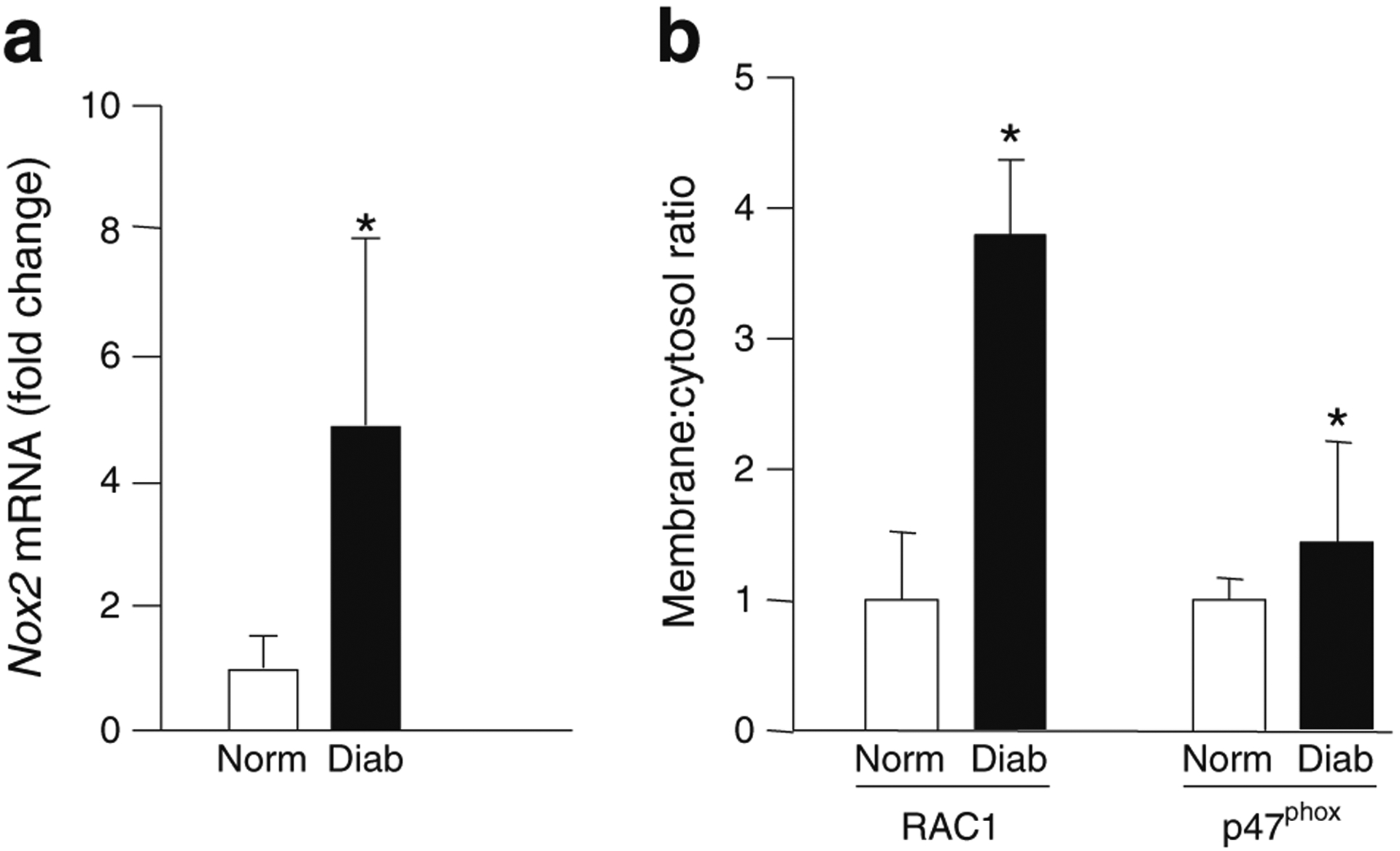
Increased Nox2 expression and translocation and membrane association of RAC1 and p47phox in retina from diabetic rats. (a) Nox2 mRNA level was quantified in retina by qPCR using β-actin as housekeeping protein. (b) Retinal lysates were subjected to a single-step centrifugation to separate the total particulate and soluble fractions. Relative abundance of RAC1 and p47phox was quantified by western blotting followed by densitometry, and expressed as membrane:cytosol ratio. The results are represented as mean ± SD from four or five rats in each group. Norm, non-diabetic (normal) rats; Diab, diabetic rats. *p<0.05 vs normal
NSC23766 prevents ROS generation and RAC1 activation in diabetic mice
To further validate our hypothesis, we quantified RAC1 activation, ROS levels and mitochondrial damage in the retina of diabetic mice following treatment with NSC23766. Consistent with our in vitro findings in bovine endothelial cells, retinal RAC1 was activated by 35% within 15 days of diabetes onset (Fig. 6a); this was accompanied by an increase in total ROS levels by over 90% (Fig. 6b). However, mitochondrial dysfunction was not observed at this duration as demonstrated by comparable levels of BAX in the retinal mitochondria from control and diabetic mice (Fig. 6c). Administration of NSC23766 ameliorated increase in RAC1 activation and ROS generation (Fig. 6a, b).
Fig. 6.
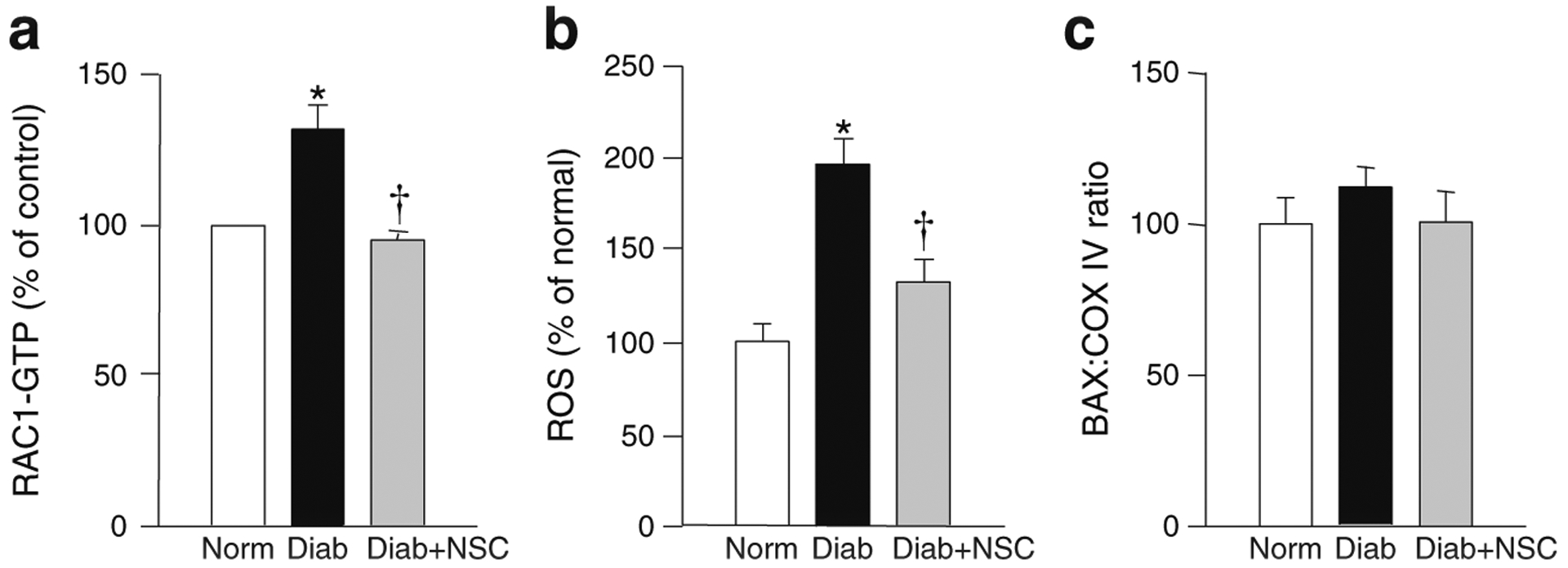
NSC23766 administration protects mouse retina from diabetes-induced ROS generation and RAC1 activation. Retina from diabetic mice, administered with or without NSC23766 (as indicated in Methods) was analysed. RAC1 activation (a), total ROS levels (b) and BAX translocation into the mitochondrial fraction (c) were quantified as described in the Methods. The values are presented as mean±SD and each measurement was made in duplicate in five or six mice per group, and the values from normal mice (Norm) are considered as 100%. Diab, diabetic mice; NSC, NSC23766. *p<0.05 vs normal; †p<0.05 vs diabetes
Rac1 is activated in the retinal microvasculature of human donors with diabetic retinopathy
To transition from the in vitro and animal models to the human disease, retinal microvessels from non-diabetic donors and those with diabetic retinopathy were analysed. As shown in Fig. 7, retinal microvessels from donors with diabetic retinopathy had significantly high levels of NOX2 expression, over 50% more active (GTP-bound) RAC1 and a more than 2.5-fold increase in RAC1 mRNA compared with the non-diabetic controls. Similarly, p47phox expression (protein and mRNA levels), was also elevated by 40–70% in the retinal microvasculature of diabetic individuals (Fig. 7), suggesting upregulation of the members of NOX2 holoenzyme in diabetic retinopathy.
Fig. 7.
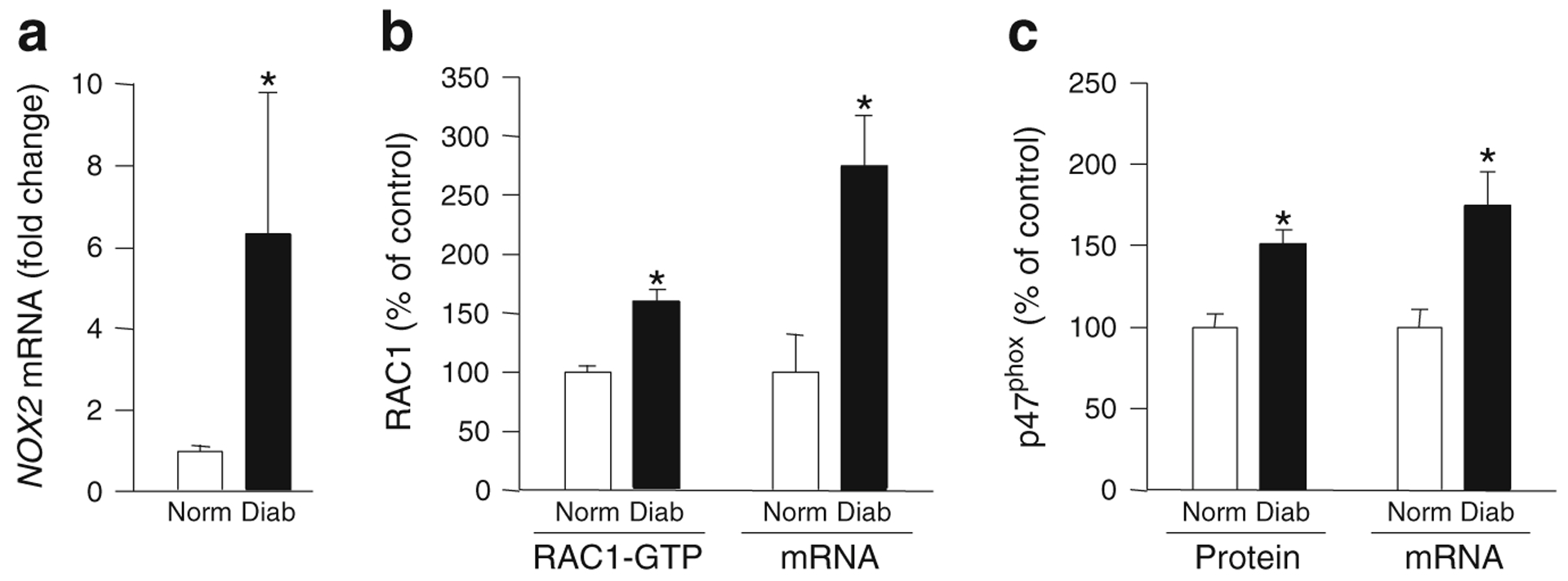
Expression of NOX2 and p47phox and RAC1 activation are increased in the retinal microvasculature from human donors with diabetic retinopathy. Microvessels from the retina of diabetic individuals with documented retinopathy (Diab) and age-matched non-diabetic individuals (Norm) were analysed for expression of NOX2 (a), RAC1 activation (RAC1-GTP) and mRNA levels (b) and p47phox expression (protein and mRNA levels) (c). Each measurement was performed in duplicate in the retinal microvessels from non-diabetic and diabetic donors (n=4 each). *p<0.05 vs non-diabetic donors
Discussion
The main objective of the current study was to investigate the regulatory role of NOX2 in glucose-induced metabolic dysfunction of the retinal capillary cells. We used in vitro as well as in vivo models to accomplish this goal. We made the following observations: (1) high glucose activates the NOX2 signalling cascade including the expression, activation and membrane association of RAC1 and p47phox; (2) NOX2 activation is seen before mitochondria are damaged; (3) pharmacological inhibition of TIAM1 significantly attenuates high-glucose-mediated RAC1 activation, mitochondrial ROS generation and cell apoptosis; (4) diabetes-induced increase in total ROS and RAC1 activation in the retina can be prevented by administration of NSC23766 in mice and (5) the retinal vasculature from human donors with diabetic retinopathy exhibit increased expression and activation of RAC1 and p47phox. Thus, our findings provide the first evidence to indicate that TIAM1–RAC1-mediated NOX2 activation is an early signalling event in increasing cytosolic ROS, which in turn triggers mitochondrial damage leading to cell apoptosis and the onset of diabetic retinopathy.
NOX2, a phagocytic enzyme, is also found in non-phagocytic cells, including vascular endothelial cells, smooth muscle cells and pancreatic islet β cells [8, 32, 33]. Our recent findings in islet β cells demonstrated that the NOX2 pathway promotes islet dysfunction under glucolipotoxic conditions by increasing oxidative stress and mitochondrial dysregulation [14, 34]. In endothelial cells, NOX2 has been identified as one of the major sources of ROS production and, at a molecular level, endothelial NOX is analogous to NOX2 complex [35, 36]. In the retina, NOX2 is present in endothelial cells and in pericytes, the two cell types that are the main target of pathology associated with diabetic retinopathy [27, 36–38], and in retinal endothelial cells ischaemia increases the expression of the catalytic subunit, gp91phox [27]. A key role for mitochondria in increased retinal ROS levels in diabetes has also been demonstrated [3–5]. NOX2 is also implicated in increased ROS production, which can be prevented by the deletion of NOX2 or treatment of mice with apocynin [27, 38, 39]. Furthermore, in the pathogenesis of diabetic retinopathy, NOX2 is primarily related to retinal vascular inflammation as NOX2-mediated activation of signal transducer and activator of transcription 3 is shown to increase levels of vascular endothelial growth factor and break down the blood–retina barrier [38, 40–42]. Our current findings demonstrate that NOX2-mediated increase in ROS in diabetes is an early event, which precedes mitochondrial ROS production, and that inhibition of NOX2 attenuates mitochondrial damage and capillary cell apoptosis.
Activation–deactivation of RAC1 is under the control of GEFs and GAPs. TIAM1 represents the GEF for RAC1 activation and downstream signalling events, including NOX2 activation, mitochondrial dysfunction and cell apoptosis, in pancreatic β cells incubated with high glucose, lipids and proinflammatory cytokines [14, 15, 43]. Our current findings imply that TIAM1 has a regulatory role in glucose-induced RAC1 and NOX2 activation, mitochondrial ROS generation and cell apoptosis. Our in vivo model, showing that NSC23766 treatment of diabetic mice largely restores RAC1 activation and ROS generation to normal levels, further implicates TIAM1 in the RAC1 activation mechanism.
The role of RAC1-mediated NOX2 activation and cellular dysfunction has been reported in the diabetic heart and kidney. Increased RAC1 expression and NOX2 activity are seen in the heart within 7 days of induction of streptozotocin-induced diabetes in rodents, and gene silencing of Rac1 significantly inhibits these abnormalities, suggesting a regulatory role for RAC1 in NOX2 activation. Treatment of db/db mice with NSC23766 inhibits NOX2 activity and cell apoptosis, leading to partial restoration of myocardial function. Pharmacological inhibition by NSC23766 or overexpression of inactive mutants of gp91phox and p47phox prevents glucose-induced damage to cardiomyocytes [44, 45]. Thus, these findings in cardiac myocytes [44, 45] and in pancreatic β cells [8, 14–16] are consistent with our current findings in the retina demonstrating the role of the TIAM1–RAC1 axis in glucose-induced NOX2 activation, ROS generation and cell apoptosis. To further support the role of RAC1 in diabetic retinopathy, our results clearly demonstrate that RAC1 is activated in the retinal microvasculature, the site of histopathology, obtained from human donors with diabetic retinopathy. Data presented here, and published work from other laboratories [16, 36, 46, 47], demonstrate that RAC1 activation leads to mitochondrial damage.
We conclude that hyperglycaemic conditions promote the activation of the TIAM1–RAC1 signalling module during early stages of diabetic retinopathy, which leads to the activation of NOX2 and an associated increase in intracellular ROS. This, in turn, damages mitochondria resulting in cell apoptosis and the development of diabetic retinopathy. Once the mtDNA is damaged, it initiates the vicious cycle of ROS generation [20], and breaking of this vicious cycle could be a daunting task. Thus, strategies to inhibit TIAM1–RAC1 signalling in particular and NOX2 activation in general to achieve the desired levels of inhibition of this signalling pathway could have potential to inhibit the development and progression of retinopathy in the early stages of the disease, and eventually spare diabetic patients from losing their sight as a result of this devastating disease.
Supplementary Material
Funding
This work was supported by grants from the National Institutes of Health (EY014370, EY017313 and EY022230 to RK; DK74921 and EY022230 to AK), JDRF (5-2012-313 to RK and 5-2012-257 to AK) and the Department of Veterans Affairs (1BX000469 to AK) and by unrestricted funds to the Department of Ophthalmology from Research to Prevent Blindness (to RAK). AK is the recipient of a Senior Research Career Scientist Award from the Department of Veterans Affairs.
Abbreviations
- BAX
BCL-2-associated X protein
- DCHFDA
2′,7′-Dichlorodihydrofluorescein diacetate
- GAPs
GTPase-activating proteins
- GEFs
Guanine nucleotide exchange factors
- mtDNA
Mitochondrial DNA
- NOX
NADPH oxidase
- qPCR
Quantitative real-time PCR
- RAC1
Ras-related C3 botulinum toxin substrate 1
- ROS
Reactive oxygen species
- TIAM1
T cell lymphoma invasion and metastasis
Footnotes
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
Electronic supplementary material The online version of this article (doi:10.1007/s00125014-3194-z) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Contributor Information
Renu A. Kowluru, Department of Ophthalmology, Wayne State University, Detroit, MI, USA
Anjaneyulu Kowluru, Department of Pharmaceutical Sciences, Eugene Applebaum College of Pharmacy and Health Sciences, Wayne State University, 259 Mack Avenue, Detroit, MI 48201, USA; John D. Dingell VA Medical Center, Detroit, MI, USA.
Rajakrishnan Veluthakal, Department of Ophthalmology, Wayne State University, Detroit, MI, USA.
Ghulam Mohammad, Department of Ophthalmology, Wayne State University, Detroit, MI, USA.
Ismail Syed, Department of Pharmaceutical Sciences, Eugene Applebaum College of Pharmacy and Health Sciences, Wayne State University, 259 Mack Avenue, Detroit, MI 48201, USA.
Julia M. Santos, Department of Ophthalmology, Wayne State University, Detroit, MI, USA
Manish Mishra, Department of Ophthalmology, Wayne State University, Detroit, MI, USA.
References
- 1.Frank RN (2004) Diabetic retinopathy. N Engl J Med 350:48–58 [DOI] [PubMed] [Google Scholar]
- 2.Kowluru RA (2005) Diabetic retinopathy: mitochondrial dysfunction and retinal capillary cell death. Antiox Redox Signal 7:1581–1587 [DOI] [PubMed] [Google Scholar]
- 3.Kowluru RA, Abbas SN (2003) Diabetes-induced mitochondrial dysfunction in the retina. Investig Ophthalmol Vis Sci 44:5327–5334 [DOI] [PubMed] [Google Scholar]
- 4.Kowluru RA (2013) Mitochondria damage in the pathogenesis of diabetic retinopathy and in the metabolic memory associated with its continued progression. Curr Med Chem 20:3226–3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brownlee M (2005) The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54:1615–1625 [DOI] [PubMed] [Google Scholar]
- 6.Babior BM, Lambeth JD, Nauseef W (2002) The neutrophil NADPH oxidase. Arch Biochem Biophys 397:342–344 [DOI] [PubMed] [Google Scholar]
- 7.Bokoch GM, Knaus UG (2003) NADPH oxidases: not just for leukocytes anymore. Trends Biochem Sci 28:502–508 [DOI] [PubMed] [Google Scholar]
- 8.Syed I, Kyathanahalli CN, Jayaram B et al. (2011) Increased phagocyte-like NADPH oxidase and reactive oxygen species generation in type 2 diabetic ZDF rat and human islets: role of Rac1-Jun NH2-terminal kinase 1/2 signaling pathway in mitochondrial dysregulation in the diabetic Islet. Diabetes 60:2843–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bokoch GM, Zhao T (2006) Regulation of the phagocyte NADPH oxidase by Rac GTPase. Antioxid Redox Signal 8:1533–1548 [DOI] [PubMed] [Google Scholar]
- 10.Sarfstein R, Gorzalczany Y, Mizrahi A et al. (2004) Dual role of Rac in the assembly of NADPH oxidase, tethering to the membrane and activation of p67phox: a study based on mutagenesis of p67phox-Rac1 chimeras. J Biol Chem 279:16007–16016 [DOI] [PubMed] [Google Scholar]
- 11.Veluthakal R, Madathilparambil SV, McDonald P, Olson LK, Kowluru A (2009) Regulatory roles for Tiam1, a guanine nucleotide exchange factor for Rac1, in glucose-stimulated insulin secretion in pancreatic beta-cells. Biochem Pharmcol 77:101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowluru A (2010) Small G-proteins in islet -cell function. Endocr Rev 31:52–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Rijssel J, van Buul JD (2012) The many faces of the guanine-nucleotide exchange factor trio. Cell Adhes Migr 6:482–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Syed I, Kyathanahalli CN, Kowluru A (2011) Phagocyte-like NADPH oxidase generates ROS in INS 832/13 cells and rat islets: role of protein prenylation. Am J Physiol 300:756–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subasinghe W, Syed I, Kowluru A (2011) Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic beta-cells: evidence for regulation by Rac1. Am J Physiol 300:12–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohammed AM, Syeda K, Hadden T, Kowluru A (2013) Upregulation of phagocyte-like NADPH oxidase by cytokines in pancreatic beta-cells: attenuation of oxidative and nitrosative stress by 2-bromopalmitate. Biochem Pharmacol 85:109–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jarrett SG, Lin H, Godley BF, Boulton ME (2008) Mitochondrial DNA damage and its potential role in retinal degeneration. Prog Retin Eye Res 27:596–607 [DOI] [PubMed] [Google Scholar]
- 18.Trudeau K, Molina AJ, Guo W, Roy S (2010) High glucose disrupts mitochondrial morphology in retinal endothelial cells: implications for diabetic retinopathy. Am J Pathol 177:447–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santos JM, Tewari S, Goldberg AFX, Kowluru RA (2011) Mitochondria biogenesis and the development of diabetic retinopathy. Free Radic Biol Med 51:1849–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santos JM, Tewari S, Kowluru RA (2012) A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Radic Biol Med 53:1729–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong Q, Kowluru RA (2011) Epigenetic changes in mitochondrial superoxide dismutase in the retina and the development of diabetic retinopathy. Diabetes 60:1304–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanwar M, Chan PS, Kern TS, Kowluru RA (2007) Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci 48:3805–3811 [DOI] [PubMed] [Google Scholar]
- 23.Kowluru RA, Mohammad G, Dos Santos JM, Zhong Q (2011) Abrogation of MMP9 gene protects against the development of retinopathy in diabetic mice by preventing mitochondrial damage. Diabetes 60:3023–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohammad G, Kowluru RA (2012) Diabetic retinopathy and signaling mechanism for activation of matrix metalloproteinase-9. J Cell Physiol 227:1052–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tewari S, Zhong Q, Santos JM, Kowluru RA (2012) Mitochondria DNA replication and DNA methylation in the metabolic memory associated with continued progression of diabetic retinopathy. Investig Ophthalmol Vis Sci 53:4881–4888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tewari S, Santos JM, Kowluru RA (2012) Damaged mitochondrial DNA replication system and the development of diabetic retinopathy. Antioxid Redox Signal 17:492–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Shabrawey M, Bartoli M, El-Remessy AB et al. (2005) Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am J Pathol 167:599–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Touyz RM, Chen X, Tabet F, Yao G et al. (2002) Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res 90:1205–1213 [DOI] [PubMed] [Google Scholar]
- 29.Kowluru A, Veluthakal R (2005) Rho guanosine diphosphate-dissociation inhibitor plays a negative modulatory role in glucose-stimulated insulin secretion. Diabetes 54:3523–3529 [DOI] [PubMed] [Google Scholar]
- 30.Veluthakal R, Kaur H, Goalstone M, Kowluru A (2007) Dominant negative alpha subunit of farnesyl- and geranyl-transferase inhibits glucose-stimulated insulin secretion from insulin-secreting INS-cells. Diabetes 56:204–210 [DOI] [PubMed] [Google Scholar]
- 31.Santos JM, Kowluru RA (2011) Role of mitochondria biogenesis in the metabolic memory associated with the continued progression of diabetic retinopathy. Invest Ophthalmol Vis Sci 52:8791–8798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lassegue B, Griendling KK (2010) NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 30: 653–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takac I, Schroder K, Brandes RP (2012) The Nox family of NADPH oxidases: friend or foe of the vascular system? Curr Hypertens Rep 14:70–78 [DOI] [PubMed] [Google Scholar]
- 34.Kowluru A (2011) Friendly, and not so friendly, roles of Rac1 in islet ß-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochem Pharmacol 81:965–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li JM, Shah AM (2002) Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem 277:19952–19960 [DOI] [PubMed] [Google Scholar]
- 36.Saito Y, Geisen P, Uppal A, Hartnett ME (2007) Inhibition of NAD(P)H oxidase reduces apoptosis and avascular retina in an animal model of retinopathy of prematurity. Mol Vis 13:840–853 [PMC free article] [PubMed] [Google Scholar]
- 37.Cacicedo JM, Benjachareowong S, Chou E, Ruderman NB et al. (2005) Palmitate-induced apoptosis in cultured bovine retinal pericytes: roles of NAD(P)H oxidase, oxidant stress, and ceramide. Diabetes 54:1838–1845 [DOI] [PubMed] [Google Scholar]
- 38.Al-Shabrawey M, Rojas M, Sanders T et al. (2008) Role of NADPH oxidase in retinal vascular inflammation. Invest Ophthalmol Vis Sci 49:3239–3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du Y, Veenstra A, Palczewski K, Kern TS (2013) Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc Natl Acad Sci U S A 110:16586–16591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Al-Shabrawey M, Bartoli M, El-Remessy AB et al. (2008) Role of NADPH oxidase and Stat3 in statin-mediated protection against diabetic retinopathy. Invest Ophthalmol Vis Sci 49:3231–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tawfik A, Sanders T, Kahook K, Akeel S, Elmarakby A, Al-Shabrawey M (2009) Suppression of retinal peroxisome proliferator-activated receptor gamma in experimental diabetes and oxygen-induced retinopathy: role of NADPH oxidase. Invest Ophthalmol Vis Sci 50:878–884 [DOI] [PubMed] [Google Scholar]
- 42.Wilkinson-Berka JL, Rana I, Armani R, Agrotis A (2013) Reactive oxygen species, Nox and angiotensin II in angiogenesis: implications for retinopathy. Clin Sci (Lond) 124:597–615 [DOI] [PubMed] [Google Scholar]
- 43.Syed I, Jayaram B, Subasinghe W, Kowluru A (2010) Tiam1/Rac1 signaling pathway mediates palmitate-induced, ceramide-sensitive generation of superoxides and lipid peroxides and the loss of mitochondrial membrane potential in pancreatic beta-cells. Biochem Pharmacol 80:874–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen E, Li Y, Li Y et al. (2009) Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes 58:2386–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Zhu H, Shen E, Wan L, Arnold JM, Peng T (2010) Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 59:2033–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas PS, Kim J, Nunez S, Glogauer M, Kaartinen V (2010) Neural crest cell-specific deletion of Rac1 results in defective cell-matrix interactions and severe craniofacial and cardiovascular malformations. Dev Biol 340:613–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Valente AJ, Yoshida T, Clark RA, Delafontaine P, Siebenlist U, Chandrasekar B (2013) Advanced oxidation protein products induce cardiomyocyte death via Nox2/Rac1/superoxi-dedependent TRAF3IP2/JNK signaling. Free Radic Biol Med 60:125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.