

Abstract
Abnormal aggregation of proteins into filamentous aggregates commonly associates with many diseases, such as Alzheimer’s disease, Parkinson’s disease and type-2 diabetes. These filamentous aggregates, also known as amyloids, can propagate their abnormal structures to either the same precursor molecules (seeding) or other protein monomers (cross-seeding). Cross-seeding has been implicated in the abnormal protein aggregation and has been found to facilitate the formation of physiological amyloids. It has risen to be an exciting area of research with a high volume of published reports. In this review article, we focus on the biophysical processes underlying the cross-seeding for some of the most commonly studied amyloid proteins. Here we will discuss the relevant literature related to cross-seeded polymerization of amyloid-beta, human islet amyloid polypeptide (hIAPP, or also known as amylin) and alpha-synuclein. SEVI (semen-derived enhancer of viral infection) amyloid formation by the cross-seeding between the bacterial curli protein and PAP248–286 is also briefly discussed.
1. Introduction
Many human diseases associate with the accumulation of abnormally folded proteins, known as amyloids [1]. Amyloids are stable and insoluble aggregates with β-sheet secondary structure [2]. More than fifty amyloid-forming proteins have been implicated in disease, which are collectively termed as proteinopathies [3]. The conversion of the normal functional proteins into amyloid fibrils typically follows nucleation-dependent polymerization which is often compared to crystallization [4,5]. Critical step in the nucleation-dependent polymerization is the assembly of misfolded proteins into nuclei (seeds). The nuclei are the smallest units that can promote (seed) the formation of amyloids by converting the normal cellular proteins into fibrils [6]. While in homogeneous polymerization, both the nuclei and precursor proteins are the same, in heterogeneous (cross-seeded) polymerization, the nuclei of one protein seed the aggregation of a different protein. Conversion of normal cellular proteins into amyloids by nucleation/seeding has been established as a central process explaining the “infectious” component of prion particles in prion diseases [7] and the spread of proteinaceous pathogenic inclusions in many neurodegenerative diseases [8,9].
Hetero- or cross-seeded polymerization between different amyloid proteins has been proposed as an explanation for many observations in proteinopathies. For example, heterogeneous (‘mixed’) pathology in which aggregates of multiple different proteins are found in the same tissue [10,11]. Diseases with ‘mixed’ pathologies are marked with increased severity and speedy progression, and manifest by complex phenotypes in which the affected individuals develop combined symptoms of different diseases [12,13]. Additional finding in support of cross-seeded polymerization, is that individuals diagnosed with one disorder are more susceptible to acquiring another [14,15]. Multiple studies summarized by Spires-Jones et al [16] now suggest that interactions between different amyloid proteins possibly occur in many neurodegenerative diseases. All these supports the notion that abnormal aggregation is possibly facilitated by the cross-seeded reactions between different amyloid proteins
Cross-seeded polymerization can also explain the formation of structurally distinct fibrils (polymorphs) in which one protein assembles in multiple different ways [17,18]. Furthermore, the unique fibril polymorphs can be linked to specific disease phenotypes [19–22]. In cross-seeded polymerization, the hetero- nuclei can stabilize distinct conformations and facilitate the formation of aggregates (strains) different than the ones formed by homogeneous polymerization. The cross-seeded aggregates have unique properties and can be generated naturally, inherited, or acquired by infection, carrying information to induce specific disease phenotype. Some examples of cross-seeded polymerization are summarized in Table 1.
Table 1.
Summary of cross-seeding between different amyloid proteins.
| Disease | Cross-seeded proteins | Reference |
|---|---|---|
| Prion Disease Parkinson’s Disease | PrP120-144 (human)-PrP120-144 (bank vole) | [23] |
| PrPC-α-synuclein | [24] | |
| α-synuclein (human)-α-synuclein (mouse) | [25] | |
| α-synuclein (mouse)-N- and C-terminus truncated | [26] | |
| α-synuclein (human) | ||
| α-synuclein (human)-Aβ | [27] | |
| α-synuclein (human)-Quinolinic acid | [28] | |
| Alzheimer’s Disease | Aβ42-PrPSC | [29] |
| Aβ40-Aβ42 | [30,31] | |
| Aβ40-hIAPP37 | [32] | |
| Aβ24-34-hIAPP19-29 S20G | [33] | |
| Aβ-ASC specks | [34] | |
| Aβ-exogenous amyloidogenic proteins: casein, fibroin, sericin, actin, IAPP | [35] | |
| Tau-Aβ42 | [36] | |
| Tau-α-synuclein | [37] | |
| Type 2 Diabetes | hIAPP37-Aβ42 | [38] |
| rIAPP37-hIAPP37 | [39] | |
| HIV infection | Curli - PAP248-286 | [40,41] |
In some cases amyloid formation can be beneficial for the organism [42,43]. Similar to the abnormal protein aggregation, cross-seeded polymerization can contribute to the formation of physiological amyloids in the lower organisms and may play a role in the adaptive evolution of higher organisms [44]. In this review we will focus on bacterial curli protein, which was found to promotes the conversion of PAP248–286 into the amyloid SEVI (semen-derived enhancer of viral infection) which markedly promotes HIV infection [40,41].
Although cross-seeded polymerization has been implicated in both abnormal functional amyloid formation, the general biochemical and biophysical principles of the cross-seeded reactions remain unclear. The protein-protein interactions underlying the cross-seeded reactions, and the complex interplay among different factors involved in the heteronucleation (e.g., amino sequence variation, structural compatibility, aggregation kinetics, and the protein microenvironments) are also poorly understood. For instance, it is puzzling that proteins with different degree of sequence similarity can cross-seed the amyloid formation of one other (Table 1). For example, amyloid-β (Aβ) and human islet amyloid polypeptide (hIAPP) share 50% sequence similarity and engage in cross-seeded reactions [32,33,38]. Surprisingly, proteins with much lower sequence similarity, such as Aβ and α-synuclein, bacterial curli protein and PAP248–286, or Aβ and the disease-related prion protein (PrPSC) can cross-seed the aggregation of one other [27,29,40]. Controversial findings have been also reported for the cross-seeded reactions of the same pair of proteins. For example, Aβ-hIAPP hetero-aggregation displays both cross-seeding-induced acceleration and inhibition of amyloid formation [38,45,46]. From a protein-protein viewpoint, intermolecular interactions between different amyloid proteins could be either competitive to promote cross-seeding or cooperative to delay cross-seeding. Furthermore, subtle changes in intermolecular interactions and sample environment can cause significant differences in structural conversions and mismatches between different amyloid aggregates, leading to a more complex or even contrasting scenario of amyloid cross-seeding. Similarly, controversial results regarding the toxicity of Aβ-hIAPP assemblies have been also observed. These inconsistencies can be explained with the complex conformational co-aggregation processes which involve a vast number of polymorphic structures of different sizes, conformations, and morphologies, and populations and are difficult to control.
Cross-seeded polymerization is a complex process which imposes significant challenges, it is still understudied topic. While amyloid cross-seeding systems is an area of intense studies, our understanding of the molecular mechanism of amyloid cross-seeding is limited. This provided the incentive for this review which summarizes the most recent and important findings in the area of amyloid cross-seeding and offers some personal perspectives for the mechanistic understanding of amyloid cross-seeding.
2. Unique β-sheet as general structural template for amyloid cross-seeding
Amyloid fibrils contain a common characteristic of cross-β-sheet structure, irrespective of their sequences [2,47,48]. The name cross-β-sheet originates from the characteristic cross-β patterns produced by the diffraction of oriented amyloid fibrils by x-rays. Distinguishing features of cross-β pattern of amyloid fibrils are the sharp meridional (4.7Å) and broad equatorial (10–12Å) reflections. The meridional 4.7 Å reflection (around y-axis) arises from the stacking of β-strands forming the β-sheets and the staggering between the paired β-sheets of the fibril core. The broad equatorial 10–12Å reflection (around x-axis) corresponds to the sheet-to-sheet distance of the paired β-sheets. Cross-β patterns of the amyloid fibrils signify that amyloids consist of paired β-sheets running along the fibril axis [2,47,48]. The common cross-β-sheet structures provide clues about the interaction between different amyloid proteins in the cross-seeded reactions and the formation of hybrid amyloid fibrils [49,50].
Similar to homogenous aggregation of the same amyloid proteins, the cross-seeded polymerization follows the same steps: structural conversion of precursor monomers, formation of semi-structured seeds, and growth of β-sheet-rich fibrils. However, in the case of the cross-seeded reactions, the polymorphic conformations and intermolecular forces are more complex than the homogenous aggregation due to the presence of different proteins. In contrast to the homogenous-seeding aggregation that always occurs, only some amyloid proteins can cross-seed the amyloid formation of one other. This suggests that cross-species barriers exist along the folding pathways of different amyloid proteins. Indeed, only some pairs of amyloid proteins promote amyloid aggregation and fibrillization, including tau and α-synuclein [37], tau and Aβ [36], Aβ and α-synuclein [27], Aβ and hIAPP [32,38], Aβ and PrPSC [29], and hIAPP and rat islet amyloid polypeptide (rIAPP) [39]. The cross-seeding-induced acceleration of amyloid aggregation likely bypasses the de novo formation of nuclei from monomers and proceeds in fast and exponential growth. Interestingly, it has been observed that pairs of amyloid proteins co-incubated together, may slow down or inhibit the aggregation of one other. Examples are hIAPP fragments (hIAPP30–37, hIAPP1–18, hIAPP8–18) and Aβ40 [51,52], hIAPP and insulin [53]. Inhibition or delay of amyloid cross-seeding usually involves a high energy barrier, for overcoming the mismatch between the different structures of amyloid aggregates.
Fundamentally, amyloid cross-seeding is presumably built on a primary principle of conformational compatibility between different amyloid aggregates. Such conformational compatibility depends on the aggregation differences in the folding kinetics and the populations of β-sheet structures formed by the different amyloid proteins. The unique β-sheet structure acts as general structural motifs and interaction sites for amyloid cross-seeding, where two cross-β sheets of different amyloid aggregates interact in a self-complementary fashion to form hybrid oligomeric and fibrillar aggregates. On one hand, highly populated β-sheet similarity allows to lower the cross-species barriers promoting mutual binding and recognition between different species, leading to amyloid hetero-assembly. On the other hand, intrinsic and complex protein-protein interactions could work in either competitive or corporative manner to enable different cross-seeding scenarios. In the latter case, different types of amyloid species can assemble via different folding pathways. There are the two aggregation models of template-assisted growth model and conformational-selection model for amyloid cross-seeding process (Fig. 1) [54]. For the template-assisted growth model, if one amyloid protein outperforms another one to form the more populated seeds in a faster way, these seeds will serve as structural templates to recruit and misfold the lower populated seeds of another amyloid proteins. The template-assisted cross-seeding is expected to show faster kinetics than homogenous seeding process by shortening the lag phase and accelerating the growth phase. In the case of the conformational-selection cross-seeding model, the amyloid aggregates of different sequences have similar folding kinetics and populated β-structure seeds, and amyloid cross-seeding will proceed through highly conformational similar heterogeneous seeds. Such conformational similar species possess the lower cross-species barriers allowing the interaction with one other and thus facilitating aggregation. Both models allow to (i) add homologous or heterogenous monomers or low-order aggregates to the tips of the existing fibrils and to elongate into new fibrils, and (ii) laterally associate heterogenous aggregates together via β-sheet packings. Regardless of amyloid sequence similarities, the selective co-assembly of conformational compatible amyloid species is critical for the onset and efficiency of amyloid cross-seeding.
Fig. 1.
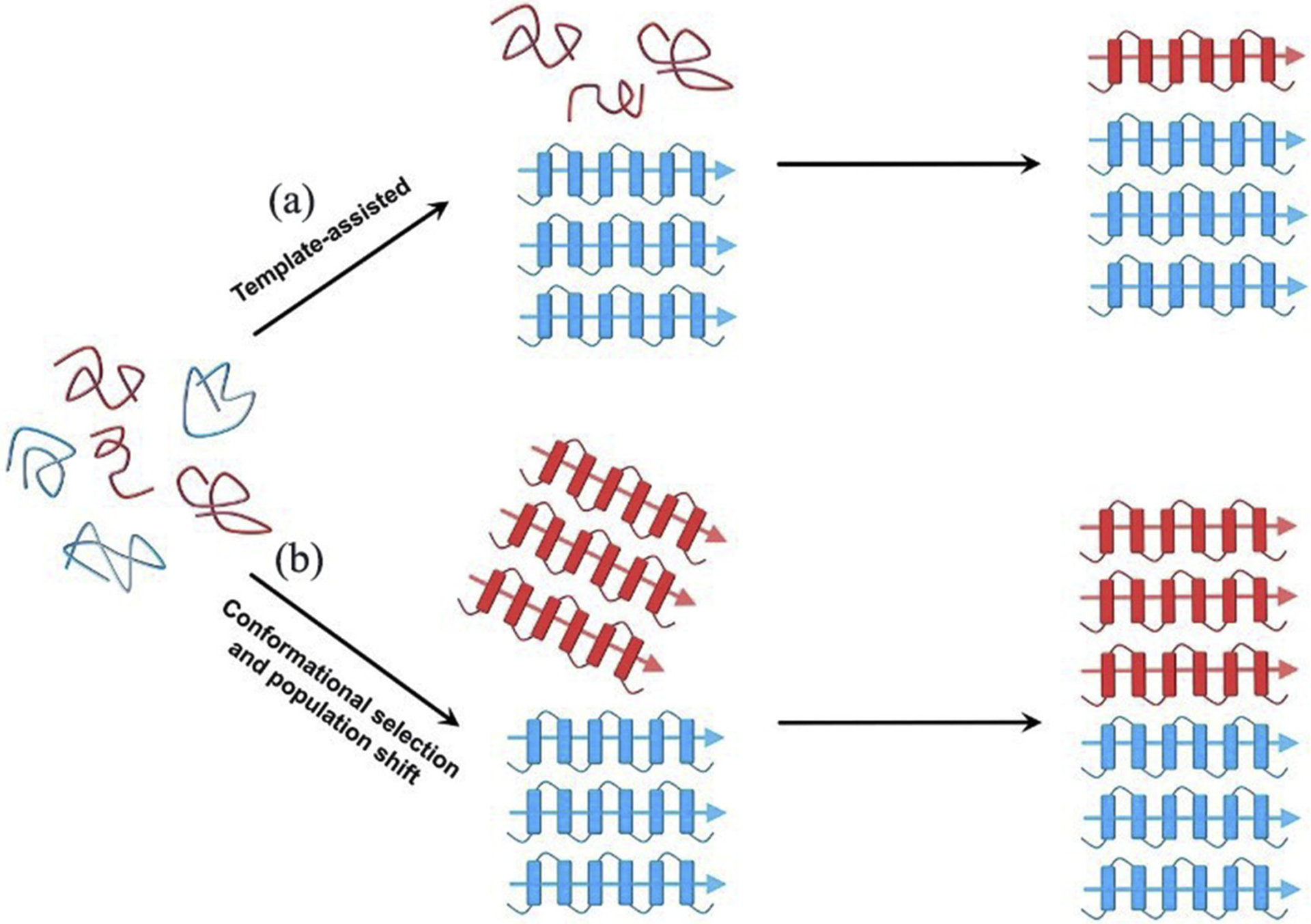
Hypothetical cross-seeding models via “template-assisted” and “conformational selection and population shift” mechanisms. The figure was adapted with permission from reference [54].
Amyloid proteins not only have intrinsic aggregation-prone regions to form β-sheet-rich structures, but also have conserved/functional regions that hinder their self-association. The β-sheet structure presents sites for diverse and favorable interfacial hetero-interactions which compete with the homo-interactions of amyloid self-aggregation. These interfacial interactions include electrostatic interactions, hydrogen bonds, hydrophobic contacts, π-π stacking, and residue ladders at cross-seeding interfaces. For instance, a hydrophobic segment of Aβ (KLVFFA) was found to interact strongly with the hydrophobic segments of tau (VQIINK, VQIVYK) [55] and hIAPP (TQRLANF, RLANFLVH) [56] via intermolecular β-sheet interactions (Fig. 2a). Moreover, β-sheets can pair with one other through the side chains of the amyloid segments which have geometric complementarity to form a dry interface within a bilayer. The resulting dry-interfaced β-sheets, also known as steric zippers, pack closely together (Fig. 2b). It was reported that steric zippers self-assembled by the same amyloidogenic segments cover diverse interfacial interactions, sequences, and β-sheet orientations [[57] 104]. At the atomic and structural level, polymorphic cross- seeding could occur through different modes of binding among these dry-interfaced β-sheets and each β-sheet in the pair can be contributed by different amyloid aggregates. Third, the β-strand-loop-β-strand motif is another structure that was observed in Aβ [[58] 845], hIAPP [[59] 844], β2-microglobulin [60], and human CA150 [61] fibrils. This motif consists of two pairs of β-sheets (Fig. 2c). Such turn-induced motif is advantageous, because it is additionally stabilized through the buried side-chain–side-chain interactions, and has two pairs of β-sheets for lateral cross-seeding. Polymorphic cross- seeding could occur through a variety of sheet-to-sheet interactions between same or different segments, and β-sheet packing and orientation. The wide conformational space that β-sheets can occupy and the abundance of multiple β-sheet-forming regions in the interacting proteins provide numerous possibilities for cross-seeded fibril assembly.
Fig. 2.
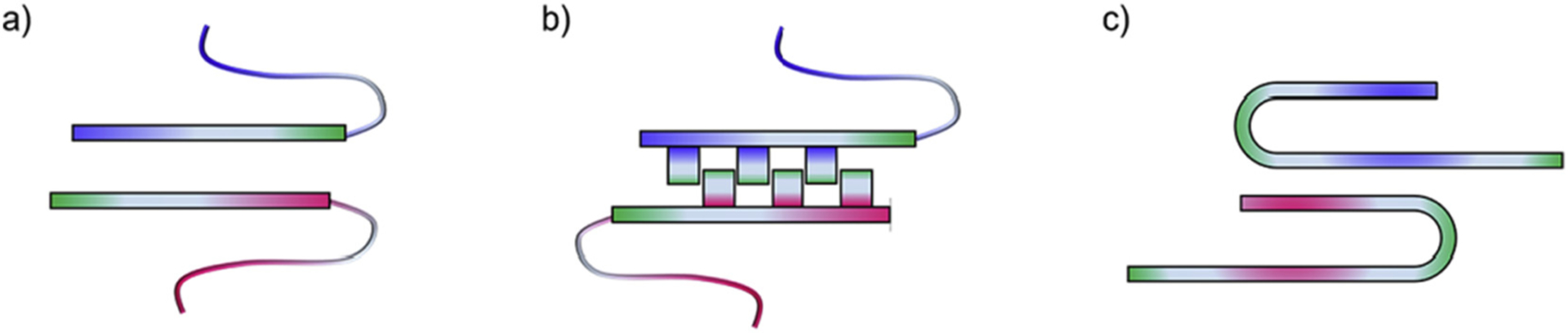
Hypothetical β-sheet-driven cross-seeding models via (a) β-structures, (b) dry-interfaced β-sheets, and (c) β-strand-loop-β-strands
3. Amyloid-beta and IAPP form toxic hetero-complexes
The complex in-vivo environment imposes multiple challenges in studying the cross-seeded reactions. In-vitro studies provide a systematic way to study the hetero-complexes, which form by cross-seeding between amyloid proteins. Such in-vitro studies are useful to understand the molecular processes underlying the pathological connections between different amyloid diseases. One such example is the common link between Alzheimer’s disease (AD) and type-2 diabetes mellitus (T2D) [62,63]. T2D and other pre-diabetic states of insulin resistance were found to associate with increased risk for sporadic AD [64,65]. Aβ and tau positive inclusions which typically form in the AD brains have been detected in pancreatic beta-cells. Reversely, amylin-positive inclusions have been identified in the brains of individuals with AD [65,66]. Immunohistological studies further suggest on possible cross-seeding interactions between the AD proteins (Aβ and tau) and T2D amylin [65]. These findings have been supported by many biochemical and biophysical studies [51,67]. Furthermore, cross-seeding and formation of Aβ-hIAPP and tau-hIAPP heterocomplexes have been proposed as a molecular explanation of T2M and AD [33,38,51,67–73]. A recent study demonstrated that hIAPP promotes the oligomerization of Aβ and results in hIAPP-Aβ42 heterocomplex, which images by transmission electron microscopy (TEM) are shown in Fig. 3 [74]. It is remarkable that these hIAPP-Aβ42 heterocomplexes have higher toxicity than amyloid-β or hIAPP alone [73]. To understand the effects of hIAPP on Aβ42 and the formation of the toxic heterocomplex, a non-fibril forming and non-toxic rIAPP was used as a control [74–76]. Time-lapsed NMR experiments revealed a small, stable and detectable hIAPP-Aβ42 heterocomplex, whereas samples of rIAPP-Aβ42 mixture contained well-resolved peaks indicative of absence of complex formation Fig. 4. These results were further confirmed by the size exclusion chromatography measurements. Interestingly, the co-incubation of Aβ with rIAPP results in the formation of oligomeric species with reduced toxicity. While these studies have provided insights into the role of cross-seeding effects and the potential link between AD and T2D, additional studies are needed to fully understand other factors involved in cross-seeding process, such as the lipid membrane, free lipids in the serum, metal ions (such as Zn2+ and Cu2+), other amyloid proteins and hormones. It is also worthwhile to investigate the molecular process that influences hIAPP transport from islet cells to the brain.
Fig. 3.
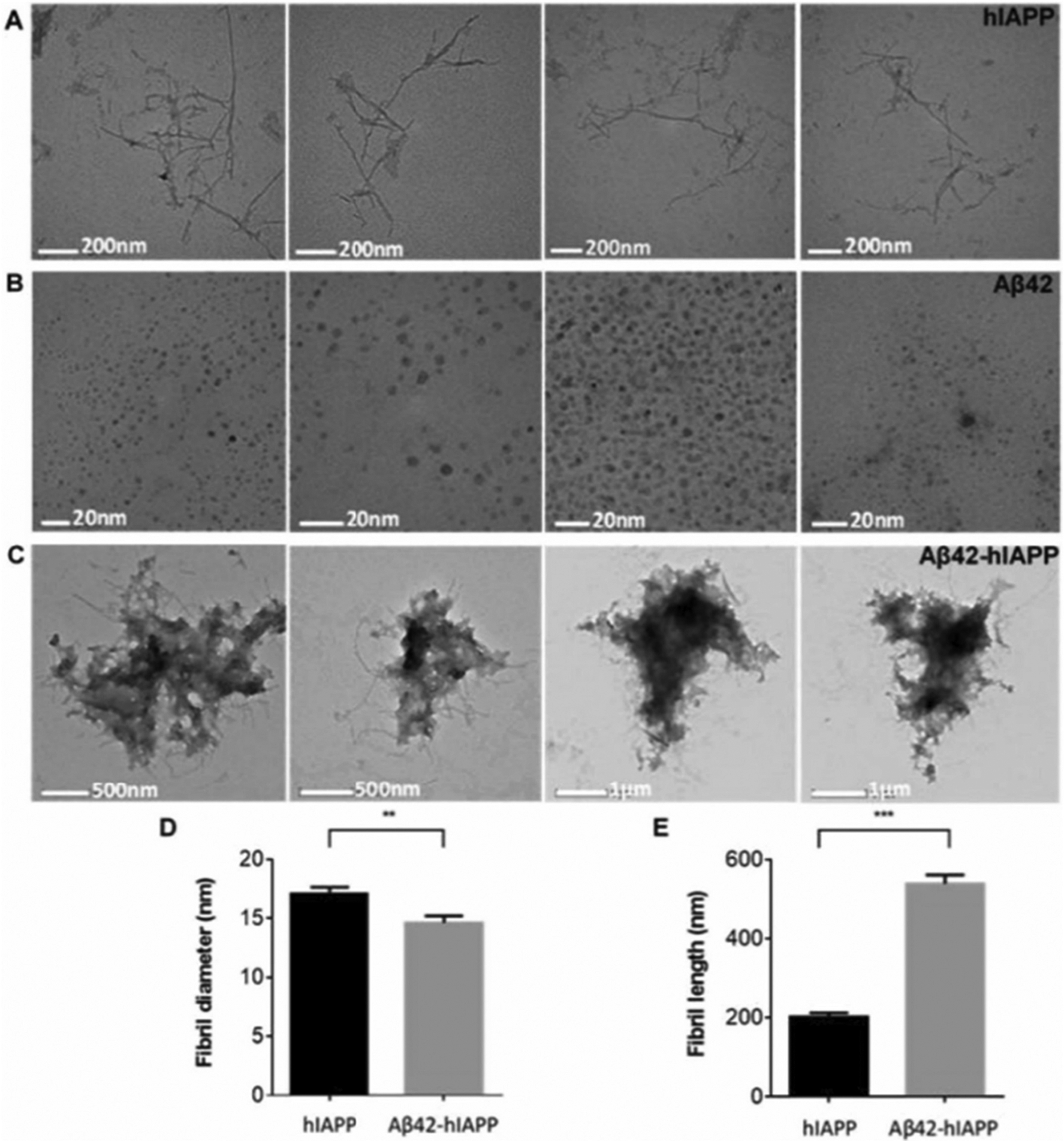
Electron micrographs of hIAPP, Aβ42 and Aβ42-hIAPP samples showing (A) fibril-like hIAPP (17 ± 0.5 nm in width, 202 ± 9.9 nm in length, n = 77) and spherical (B) Aβ42 oligomers (6 ± 0.2 nm in diameter, n = 207). (C) Aβ42-hIAPP formed large amorphous aggregates. Analysis of fibril (D) length and (E) diameter demonstrated that Aβ42-hIAPP (14 ± 0.6 nm in diameter, 539 ± 22.7 nm in length, n = 100) was significantly different from hIAPP (mean ± SEM, **p < 0.005, ***p < 0.001). Aβ42-hIAPP mixtures are large amorphous structures that are distinctly different from either spherical Aβ42 oligomers or fibril-like hIAPP. Scale bar (A) 200 nm, (B) 20 nm, (C) 500 nm/μm. The figure was adapted with permission from Reference [74].
Fig. 4.
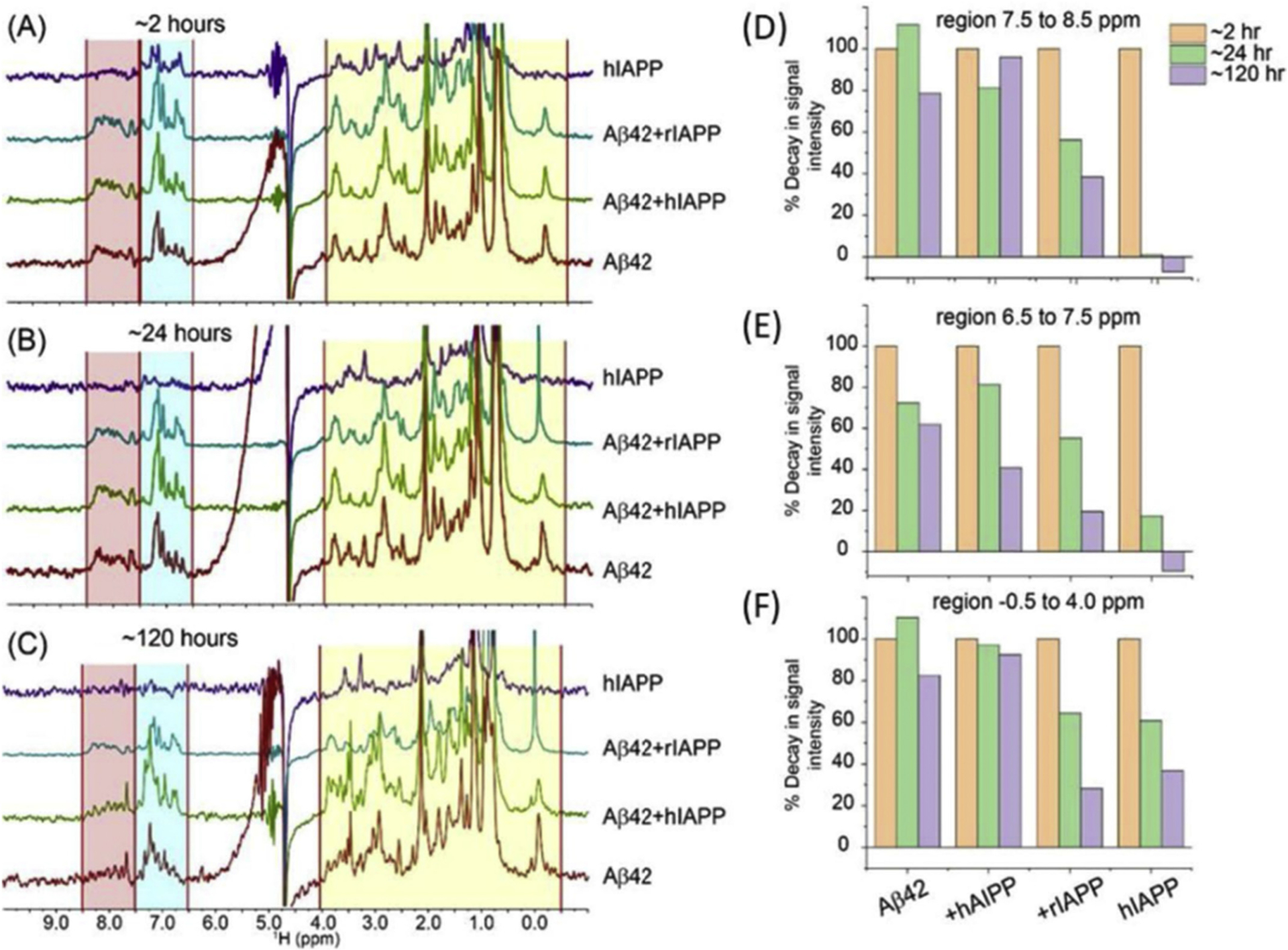
The aggregation kinetics of Aβ42-IAPP was followed by 1H NMR. (A–C) Time-lapse 1H NMR spectra of 25 μM Aβ42 co-incubated with 25 μM human-IAPP or rat-IAPP. NMR samples were prepared using 10 mM NaPi buffer, pH = 7.4 containing 10% D2O. Spectra were recorded on a 500 MHz NMR spectrometer at 25 ◦C. (D–F) Intensity decay of 1H NMR signal calculated from the spectra shown in (A–C) for 2, 24, and 120 hours incubations. The integrated intensities of the selected regions highlighted in (A–C) were analyzed using MestReNova. The figure was adapted with permission from reference [74].
4. Effect of environment factors on cross-seeding
Seeding and cross-seeding processes have multiple key roles in protein aggregation. These processes can initiate and stabilize multimeric intermediates, control the kinetics of aggregation and the structures of the amyloid fibrils and other species. While the in-vitro systems give many mechanistic insights on seeded aggregation of proteins, it has been a great challenge to study these processes in vivo. This is mainly because there are numerous different factors that need to be considered in the extremely complex and crowded system.
Amyloid formation is not always associated with a gain of toxic function. A number of studies have shown that the assembly of proteins into amyloid fibrils is part of their normal function in many microorganisms [77]. A variety of bacteria including bacillus, staphylococcus, salmonella and streptococcus, form amyloids in a controlled manner. While these functional amyloids exhibit a number of important properties, they can also cause abnormal aggregation of human proteins. Exposure to microorganisms such as bacteria, viruses, fungi, and yeasts may contribute to protein aggregation, phase separation and amyloid formation [77,78]. Bacterial proteins can cross-seed the aggregation of Aβ, IAPP, a peptide segment of prostatic acid phosphatase (PAP248–286), α-synuclein, and tau, whose aggregation is associated with fatal and incurable diseases [40]. In the following section, the cross-seeding effects of bacterial curli in the formation of semen-derived enhancer of viral infection (SEVI) amyloid fibrils is briefly described [79–85].
5. Bacterial curli cross-seeds the aggregation of PAP248–286 into SEVI (semen-derived enhancer of virus infection) amyloid formation making HIV more virulent
Studies have demonstrated that amyloid aggregates of PAP248–286 in human semen dramatically increase the infection efficiency of human immunodeficiency virus (HIV) [41 829]. Amyloid fibrils of PAP248–286 (called SEVI), but not the monomers, promote the interaction of the lipid membrane of the HIV virion with the host membrane and make the virus more infectious [86]. Biophysical studies revealed that PAP248–286 monomers are unstructured in solution and form α-helices in a membrane environment [87,88]. Because the nucleation step of SEVI formation is slow and PAP248–286 monomers can be degraded by proteolysis [89], additional cofactors present in the seminal plasma can catalyze the formation of SEVI amyloid fibrils. To investigate these factors, amyloid fibrils of bacterially-produced curli (curli or CsgA and CsgB) were used to cross-seed in vitro the aggregation of PAP248–286 monomers at low concentrations [40]. Biophysical and biochemical experiments showed that curli has a moderate effect on PAP248–286 lag-time of aggregation. However, the elongation rate of aggregation is greatly increased [40]. Imaging by transmission electron microscopy (TEM) revealed that fibrils cross-seeded with curli nucleation are longer compared to unseeded fibrils, see Fig. 5. Mechanistic aspects of this cross-seeding reaction are illustrated in Fig. 6. Thus, bacteria and other microorganisms along with PAP248–286 can synergistically facilitate HIV infection.
Fig. 5.
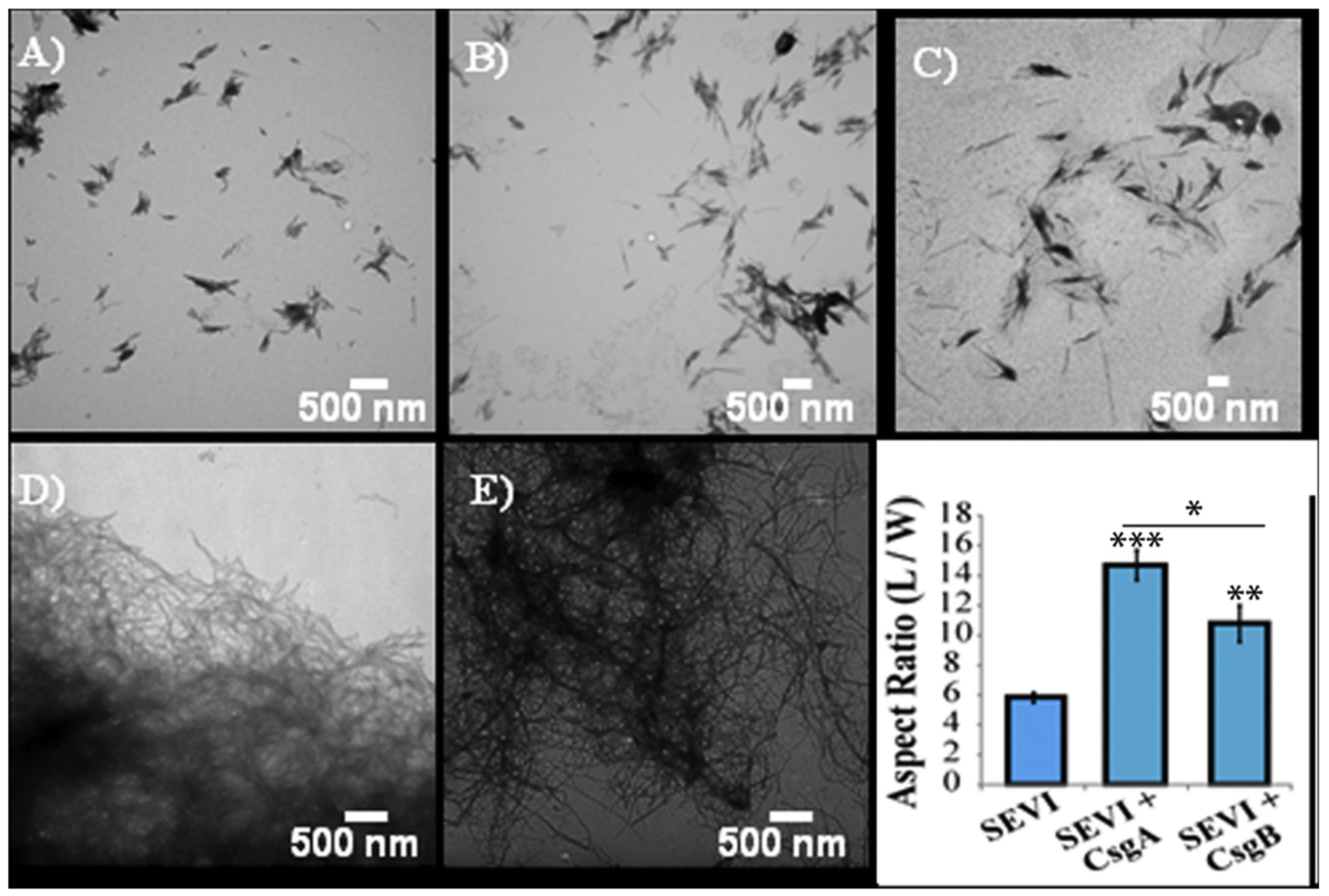
Cross-seeding of PAP248–286 with bacterial Curli results in longer SEVI fibrils. Transmission electron microscope (TEM) images of SEVI fibrils formed in the absence of curli (A) and in the presence of 5 mol% CsgA (B) and CsgB (C) fibrils. TEM images of CsgA (D) and CsgB (E) fibrils. (F) Aspect ratios of individual fibrils grown with and without curli nucleation/cross-seeding (n= 47, 23 and 37 for SEVI, SEVI+CsgA, and SEVI+CsgB respectively). Fibrils were grown at a concentration of 440 μM PAP248–286 at 37 ◦C with 1400 rpm orbital shaking for 7 days. P values were calculated using a two-tailed unpaired Student t-test against the control sample. The figure was adapted with permission from reference [40].
Fig. 6.
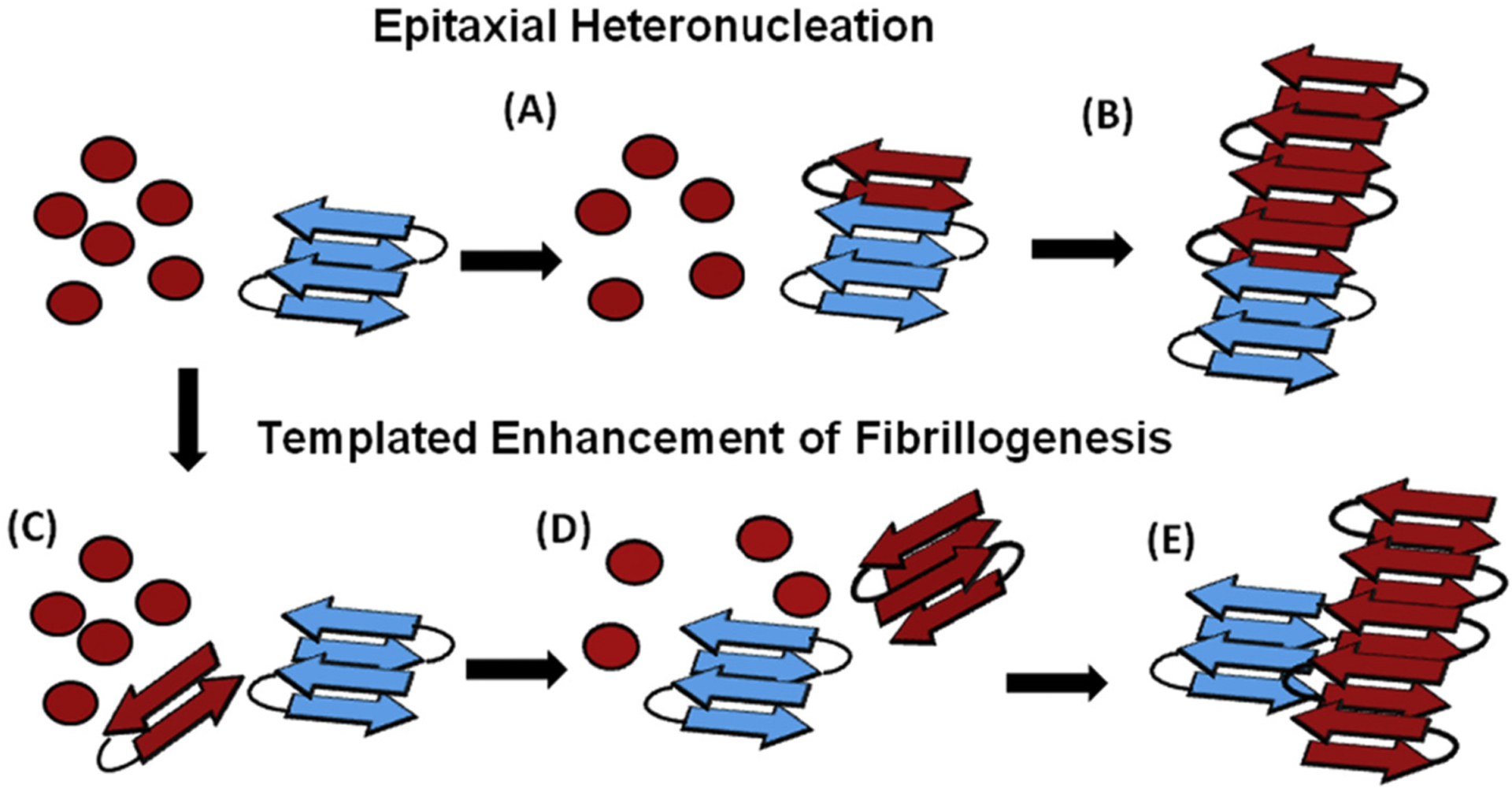
Proposed cross-seeded mechanisms of SEVI formation. Top row: Epitaxial Heteronucleation. (A) Binding of the PAP248–286 monomers (red) to the bacterial curli seed (blue) induces β-sheet formation of PAP248–286 (red). (B) Fibril growth proceeds epitaxially from the seeding nucleus to form fully matured amyloid fibrils. Bottom row: Possible mechanism for non-epitaxial heteronucleation. (C) A nucleus of the SEVI amyloid fibril forms independently of the curli fibril. (D) Growth of the SEVI fibril initially proceeds slowly due to unfavorable interactions between fibril subunits. (E) Lateral attachment of the nascent SEVI fibril to curli fibrils reduces repulsion between fibril subunits thereby enhancing the rate of fibrillogenesis. The curli seed may be incorporated into the final SEVI fibrils or may detach to catalyze further fibril extension events. The figure was adapted with permission from reference [40].
It has been also observed that bacterial curli proteins can cross-seed other amyloidogenic proteins. Studies have shown that the lag-times of Aβ40 and IAPP aggregation is reduced in the presence of bacterial curli [40]. These results suggest that cross-seeding by bacterial amyloid proteins may be ubiquitous. Although cross-seeding reactions of Aβ40 and IAPP with curli exhibit a complicated concentration dependent behavior, it would be valuable to investigate the involvement of other curli-like proteins produced by bacteria in the pathology of AD and T2D.
6. Aggregate co-occurrence and protein co-aggregation due to cross-seeding
Many studies have shown that different proteins can co-aggregate in the same inclusion or can co-occur as separate inclusions [11,12,90–92]. These observations can be explained by cross-seeding reactions, in which cross-seeds interact transiently with the umpolymerized protein (aggregate co-occurrence), or are permanently incorporated in the growing fibril (co-aggregation). Permanent incorporation of the cross-seeds in the fibril has been observed by high-speed AFM which showed the real-time growth of fibers in cross-seeded reactions between IAPP and Aβ [93]. High-resolution NMR studies also provided an evidence for the direct, transient interaction between the proteins, as described by the “dock-and-lock” mechanism [94]. Although the NMR approach can be applied to study many types of seeding and cross-seeding dynamic processes, studying transient cross-seeding, however, still remains elusive.
Hetero-interactions can be involved in different stages of fibril growth by either accelerating or inhibiting protein polymerization. For example, in hetero-reaction described by Chia et al., monomeric α-synuclein inhibits Aβ fibril formation by intervening with the fibril-catalyzed secondary nucleation, while cross-seeds of fibrillar α-synuclein accelerate the primary nucleation of Aβ fibrils [95]. The effects of cross-seeding by curli on SEVI, Aβ and IAPP have also been reported [40].
From a free-energy viewpoint, it is important to address the question of whether fibrils, formed by either transient or permanent cross-interactions, occupy a different global-energy minimum than the respective homo-nucleated fibrils. Because of the vast conformational flexibility and capacity for polymorphism, amyloid proteins typically undergo an exhaustive search of a huge sampling space, and need to overcome large energy barriers to realize amyloid cross-seeding. On the other hand, adding cross-seeds with conformations compatible to the seeded protein could lower the free-energy barriers through favored interactions, thus favoring cross-seeding even if the seeded protein has sequence which is different from the one of the cross-seeds.
7. Pathogenic nucleation of α-synuclein
7.1. Synucleinopathies
α-Synuclein precipitates in many terminal diseases collectively known as synucleinopathies. Abnormal α-synuclein is the main component of Lewy body (LB) and Lewy neurites (LN) [96,97]. These intraneuronal inclusions are the hallmark of dementia with Lewy bodies (DLB) and Parkinson’s disease (PD). Although both PD and DLB are manifested with LB pathology, these disorders have different phenotypes and progression. PD starts in the midbrain and is characterized by motor deficiency [98]. In contrast, DLB affects the cortex and manifests as dementia [99]. While in DLB and PD, α-synuclein inclusions are localized in different brain regions, and in the pure autonomic failure, LBs are mostly in the peripheral nervous system [100]. In the case of multiple system atrophy (MSA), α-synuclein forms predominantly glial cytoplasmic inclusions (GCI), or Papp-Lantos bodies, and less commonly nuclear inclusions in the oligodendrocytes [[101] 622, [102]]. Thus, α-synuclein inclusions are found in different brain regions, cell types, localize in different organelles, and associate with distinct diseases suggesting the underlying heterogeneity of the structure and function of these inclusions.
7.2. ‘Mixed’ pathologies
α-Synuclein inclusions often co-exist with other proteinaceous deposits [10,11,90,103]. A large proportion of inherited and sporadic AD have LB pathology [104–106]. α-Synuclein pathologies are also found in tau-proteinopathies with the highest occurrence in the progressive supranuclear palsy (PSP) [103]. Reversely, plaques of Aβ and tangles of tau, the hallmarks of AD, are common in synucleinopathies, such as PD and DLB [12,107,108]. Typically, ‘mixed’ pathologies associate with fast disease progression and rapid cognitive decline as compared to the single pathology cases [103,107,109]. Proteinopathic inclusions of other amyloid proteins, such as TDP43, have been found to co-exist with α-synuclein inclusions in PD, DLB [110–112], traumatic brain injury (TBI) [113], and chronic traumatic encephalopathy (CTE) [114]. α-Synuclein pathologies also co-localize with Huntingtin (Htt) protein inclusions in Huntington disease (HD) [115,116], and have been detected together with prion-protein plaques in sporadic and genetic cases of Creutzfeldt-Jakob disease (CJD) patients [117–120]. Currently, co-existence of different proteinaceous inclusions is recognized as a common feature of multiple neurodegenerative disorders [16,121]. However, the mechanism of ‘mixed’ pathology formation is still unclear.
7.3. Proteopathic cross-seeding
The occurrence of ‘mixed’ pathologies suggests an increased vulnerability to aggregation due to proteopathic cross-seeding, where aggregates of one pathogenic protein possibly nucleate, or cross-seed, the aggregation of other unrelated proteins [122]. Often α-synuclein acts as a seeding agent. In the case of tau and amyloid-β, α-synuclein promotes the formation amyloid species, which have heightened toxicity compared to unseeded species [27,37,49,95,122–128]. The evidence suggests that synthetic α-synuclein fibrils can induce the aggregation of TDP43, both in vitro and in vivo [10], and can cross-seed the aggregation of prion proteins and IAPP in vitro [24,129,130]. Conversely, Aβ, IAPP and the bacterial protein curli modulate the aggregation of α-synuclein [49,83,122,130,131]. It has also been observed that α-synuclein and tau act synergistically to facilitate the aggregation of one another [128]. Thus, in the cross-seeded reactions, α-synuclein can catalyze protein aggregation, be cross-seeded by other proteins, or act synergistically with other proteins to promote ‘mixed’ aggregation. However, we are just starting to understand the processes causing α-synuclein to act differently.
In support of proteopathic cross-seeding is the observation that the ultrastructure and the pattern of distribution of the ‘mixed’ pathologies changes compared to the single-pathology cases [122,125,132]. Additionally, the seeding capability of α-synuclein depends on its multi-merization state, as monomeric α-synuclein slows and prevents the aggregation of Aβ [95,133] and prion protein [134], while oligomeric α-synuclein nucleates the aggregation of Aβ and prion protein [27,37,49,95,122–127]. Although ‘mixed’ pathologies can be due to the failure of cellular proteostasis to simultaneously process the protein aggregates, the following evidence argues in favor of cross-seeding: i) one pathology (primary) impacts the prevalence of the other pathology (secondary) [10,11,90,103]; ii) the pathology of one protein accelerates the abnormal aggregation of another [135]; iii) the formation of the secondary pathology depends on the multimerization state of the nucleating protein [95,133]; and iv) the morphology of ‘mixed’ pathologies differs from the corresponding single-pathology cases [103,122,125,132]. This indicates that proteopathic cross-seeding can mechanistically explain the observed ‘mixed’ pathologies in multiple neurodegenerative disorders.
Considering the possible cross-seeding mechanisms, the interactions between the involved proteins can vary widely from transient to permanent. For example, multiple proteins have been shown to modulate α-synuclein aggregation by transient hetero- interactions [136,137]. Alternatively, the interacting proteins can permanently bind to form hetero- aggregates. For example, α-synuclein and Aβ directly interact with one other [49], form hetero-complexes, co-immunoprecipitate in the brains of individuals with AD/PD [126], and fragments of α-synuclein incorporate in the amyloid-β plaques [91,138]. Hetero-interactions are also involved in the propagation of the aberrant aggregates. For example, cellular prion protein has been found to act as a facilitator for the cellular intake and the spread of α-synuclein aggregates (fibrils) [139]. Thus, hetero-interactions between amyloid proteins can be involved in both cross-seeding and propagation. To further clarify the cross-seeding mechanism, we need to look for answers to questions such as: Where in the cell does the cross-seeding reaction take place? In the cross-seeded reactions, do the proteins interact transiently or permanently? What causes some proteins to co-aggregate or form separate inclusions? Why are ‘mixed’ pathologies manifested with higher toxicity? What other cellular factors facilitate/modulate the cross-seeded reactions? Do the cross-seeded aggregates propagate in the different way than homo-aggregates? What is the contribution of cross-seeding between α-synuclein and other unrelated proteopathic proteins to the etiology of neurodegenerative diseases? Addressing these very important and challenging questions is important in furthering our understanding of α-synuclein pathology and its relation to other commonly observed proteinopathies.
In addition to aggregated α-synuclein, many other cellular moieties, such as lipids, organelles, endomembrane structures, mixtures of vesicles, mitochondria, and lipids are sequestered in LBs [140,141]. Although the process of LB formation has been viewed as a major driver of neurodegeneration [142], little is known about the underlying mechanism of LB formation and the multimeric states of α-synuclein within the LBs. Additionally, of all components that co-precipitate with α-synuclein in LB, classifying the ones acting as initiators of LB formation and others that are recruited in the process of LB formation is still an open question.
7.4. Lipids as modulators of α-synuclein aggregation
Although interaction of α-synuclein with cellular membrane is necessary for its normal function [143–145], disturbance of the intricate interactions with lipids drives the abnormal aggregation and propagation of this protein [146–149]. Cross-seeded by the lipid membrane, α-synuclein can form multiple types of species. These species can be grouped into small-sized oligomers (on- or off- amyloid pathway) and amyloids (fibrils, intermediate-sized oligomers (protofibrils), and large fibrils) [147]. The off-pathway membrane-embedded oligomers have pore-like structure and have been shown to cause membrane leakage, which explains some aspects of the observed synuclein-related toxicity [150,151].
Impairment of the normal hetero-interactions between α-synuclein and the cellular membranes likely promotes the lipid-seeded aggregation of α-synuclein, and negatively impact cellular homeostasis. All α-synuclein mutants (P30A, E46K, H50Q, G51D, A53T, and A53E) found in familial PD [152–157] are located in the membrane-binding region at N-terminus and exhibit defective membrane binding. Typically, the impaired membrane interactions correlate with increased susceptibility to aggregation (P30A) [158], accelerated fibril formation, and toxicity (E46K, H50Q, A53T) of these mutants [159–162] α-Synuclein mutants which favor hetero-interactions with lipids are neurotoxic and their expression in mouse models associates with neurodegenerative and motor-deficient phenotypes [163].
The composition of the cell membrane largely influences the multimerization states and cross-seeded nucleation of α-synuclein. Negatively charged lipids modulate hetero-interactions with α-synuclein, and its aggregation [164,165]. Membranes enriched in lipids with short saturated acyl chains favor α-synuclein multimerization [147,166]. For example, galactosylceramidase deficiency and abnormal levels of phosphatidylcholine lead to α-synuclein accumulation [166–168]. Hybrid multimeric species formed between α-synuclein and lipids can cross-seed the abnormal conversion of α-synuclein and facilitate the spread of α-synuclein aggregation [169]. One the other hand, shifting lipid composition to the normal state by drugs decreases the pathogenic aggregation of α-synuclein, re-enforcing the notion that some lipids can cross-seed α-synuclein in synuclein-related proteinopathies [129].
Because α-synuclein affects multiple structural features of the membrane, such as curvature [170–172], thickness [173], and fluidity [174], variations of protein structure can lead to substantial membrane alternations [166]. Lipids and α-synuclein have interdependent relationships: while α-synuclein alters membrane structure and integrity, lipid composition modulates α-synuclein multimerization and aggregation. Studies suggest that α-synuclein easily forms multimeric states, which can grow into fibrils on lipid surfaces [175]. These multimeric states possibly result by the cross-seeded reactions between α-synuclein and certain types of lipids. Lipid-protein interactions add another layer of complexity when considering the cellular contributors to the abnormal aggregation of α-synuclein (see Fig. 7).
Fig. 7.
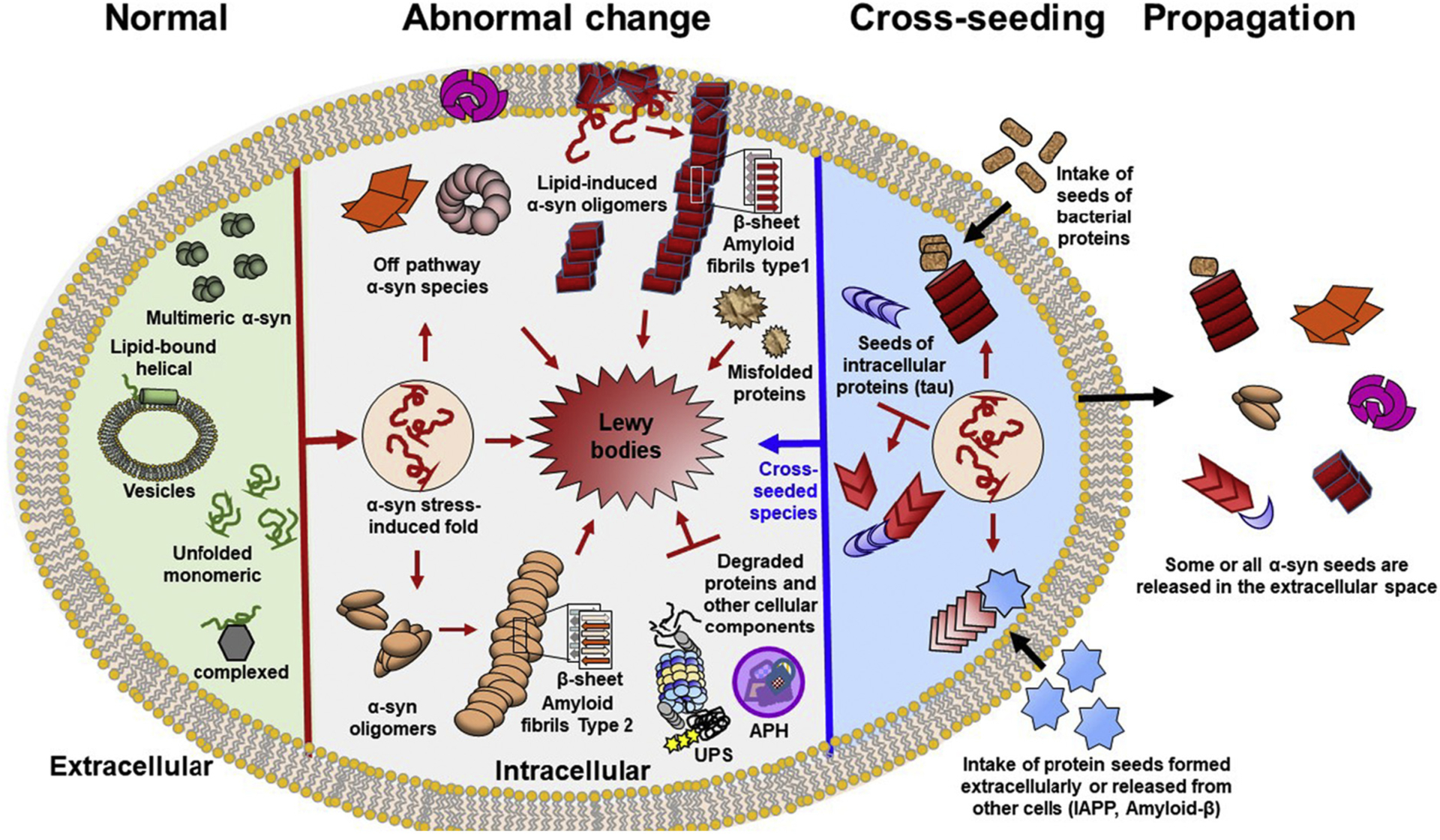
Schematic diagram of α-synuclein multimerization and Lewy body formation. For simplicity a single cell is shown. Homo- and hetero- multimers of α-synuclein are formed in both physiological (green) and pathological conditions (pink-red). Different shapes depict the structural heterogeneity of the pathogenic species. Cross-seeds are colored in blue and brown. Normally, α-synuclein is natively unfolded protein that adopts different conformations. This structural plasticity facilitates binding to many different proteins allowing α-synuclein to perform different functions. α-Synuclein also binds to vesicle membranes as a α-helical monomer. Multiple different stressors favor conformations that are prone to pathogenic multimerization. These factors include protein mutations, abnormal PTMs, metabolites, change of lipid composition, environmental pollutants (pesticides), and reactive oxygen species. Other triggers of multimerization are impaired ubiquitin-proteasome system (UPS) and chaperone function, as well as, dysregulation of autophagy (APH) and mitochondrial oxidative stress. The pathogenic multimerization results in off-pathway oligomers, or on- pathway oligomers. The on- pathway amyloid oligomers further grow into β-sheet-rich amyloid fibrils. Additionally some types of lipids can cross-seed the abnormal multimerization of α-synuclein (on- or off- pathway oligomers). Similar to lipids, other aggregation-prone proteins (tau, bacterial, Aβ, IAPP, prion etc) can cross-seed the multimerization of α-synuclein converting native α-synuclein and other cellular proteins into pathogenic entities. The end product of α-synuclein multimerization is Lewy bodies, which are large cellular inclusions with complex structures containing other multiple other components in addition to α-synuclein. The role of the non-synuclein components that can cross-seed α-synuclein multimerization and facilitate LB formation are poorly understood.
7.5. Cellular modifiers of α-synuclein aggregation
Protein modifications and cellular processes (metabolites and lipids) that favor α-synuclein fibril formation also associate with synucleinopathies. Cellular α-synuclein is modified by phosphorylation [176], acetylation [177,178], nitration [179], glycosylation [180], SUMO-ylation [181], ubiquitination, and truncation [182]. These post- translational modifications (PTMs) can either enhance or inhibit the formation of the aberrant structures. Various cellular macromolecules (RNA, heparan sulfates, Mn) have also been shown to modulate α-synuclein aggregation [183,184], and some of aspects of α-synuclein aggregation can be explained by cross-seeding processes.
Some biochemical and biophysical studies have shown that α-synuclein forms different types of multimeric species in response to specific set of conditions and cross-seeding systems, such as inorganic buffers, membranes, cytoplasm, extracellular space, various substances, modifiers and interactors PTMs, cellular metabolites, and other proteins. However, the relative contributions of each factor in α-synuclein multimerization or whether different factors act synergistically or independently in the complex cellular environments is generally not well understood. Further studies exploring these cross-seeding aspects and obtaining high-resolution insights on the amyloid aggregation of α-synuclein would be highly valuable for further studies to better understand the pathology of the associated amyloid diseases and possibly to develop treatment plans.
7.6. Oligomeric states of α-synuclein
α-Synuclein forms a wide range of abnormal species with varying sizes, structures, functions and toxicities [121]. These species can be oligomers (dimers, trimers, tetramers, etc.) [185], amyloid fibrils, or large aggregates with a complex composition (LBs or GCI) [186]. Although some oligomeric species have normal cellular function, many species (oligomeric or fibrillar) inflict negative effect on synaptic [186], axonal [187], mitochondrial [188], and protein homeostasis [189–191]. In the following sections, we focus on the amyloid fibrils.
7.7. Amyloid fibrils of α-synuclein are pathogenic
Early studies showed that intracellular inclusions of α-synuclein are filament structures reminiscent to amyloid fibrils [97,192]. It is now established that the amyloid fibrils of α-synuclein cause cellular toxicity. Intake of externally applied fibrils cause cellular toxicity and the formation of LB-like inclusions inside the cells [191]. Many familial mutants of α-synuclein which exhibit accelerated the fibril formation in vitro, associate with increased load of LB/LN pathology and neuro-degeneration [158–162]. Moreover, increased cellular levels of α-synuclein due to gene duplication and triplication cause familial synucleinopathies [152–155].
A direct link between the fibrillar state of α-synuclein and disease was established by injecting mice with α-synuclein fibrils. These fibril-injected mice develop LB-like pathology with PD-like symptoms [25,142,193,194]. This gain of toxic function was supported by the observation that α-synuclein knock-out mice show a mild phenotype and no PD-like symptoms [195–199]. As expected for a template seeding, fibril propagation is inhibited the removal of α-synuclein from the pool of proteins. α-Synuclein null mice injected with fibrils of recombinant α-synuclein did not develop neurodegeneration [200]. This evidence suggests that amyloid fibrils of α-synuclein can cause the formation of pathogenic inclusions and that cellular α-synuclein is a necessary component for initiation, growth and spread of these pathogenic inclusions.
7.8. Structural heterogeneity of fibrils and disease diversity
Multiple studies have shown that α-synuclein forms structurally distinct amyloid fibrils in which protein subunits assemble via β-sheet conformation in multiple different ways. Here, we hypothesize that the cross-seeded polymerization is a key contributing factor to the structural diversity of α-synuclein fibrils. α-Synuclein harbors multiple segments with high propensity for β-sheet [201,202] and contain many primitive residues (Ala, Thr, Val, Gly, Ile) with short aliphatic chains favor sheet-to-sheet interactions. The structural plasticity along with the presence of multiple β-sheet segments explains the ability of α-synuclein to assemble in many different types of fibrils. Simple changes in the fibril growth media, such as the ionic strength of the buffer, result in different fibril types [203–206]. Indeed, detailed structural studies showed that the full-length α-synuclein assemble into different types of fibrils distinguishable by their variable core structures [207–218]. The intrinsic structural plasticity also suggests that α-synuclein facilitate hetero-interactions with many different proteins, which in turn favor cross-seeded fibril formation.
Biochemical, biophysical and structural studies are now beginning to reveal the structural heterogeneity of in-vivo-formed species. Peng et al showed that α-synuclein species isolated from brain tissues of individuals with MSA have distinct structures, pattern of propagation and infectivity compared to species isolated from brain tissues with DLB [20]. Recently, cryoEM and NMR studies of α-synuclein fibrils purified from brain tissues of individuals with MDS, PD, and DLB provided valuable insights about the molecular organization of in-vivo fibrils [217,218]. These studies collectively suggest that α-synuclein forms a wide range of different structures in vivo in response to the complex cellular milieu and support the notion of structural heterogeneity as a key contributor to the distinct disease phenotypes. In summary cross-seeded polymerization may provide mechanistic insights on the origin of the structural polymorphism. Although challenging, it is central to identify and study the cross-seeding factors that contribute to the structural diversity of α-synuclein fibrils.
8. Effects of cross-seeding on amyloid fibrillation based on phase diagrams
Because numerous factors affect protein aggregation, it is important to develop simple approaches based on the major macroscopic properties of proteins to better understand the amyloid aggregation process. Two such macroscopic properties are the structure and solubility of amyloid-forming proteins (precursors). A specific structural feature of soluble precursors in conformational ensembles can easily form a nucleus that is required for initiation of amyloid fibril formation [219–221]. On the other hand, protein aggregation including amyloid fibril formation is driven by precursor solubility [222]. Both these properties share the macroscopic common processes of amyloid generation, the formation of amyloid seeds (i.e., templates) following nucleation and sequential growth of amyloid seeds. Thus, it is obvious that environment-sensitive changes in the structure and solubility influence amyloid formation.
Amyloid fibrillation is a thermodynamic and kinetic process. Precursors over their solubility limit (i.e., supersaturated state or super-saturation) form productive nuclei for amyloid seeds [221–223]. After ending of amyloid polymerization, the system thermodynamically reaches a two-state equilibrium (i.e., saturation), which has soluble precursors and insoluble amyloid fibrils. The concentration of the remaining soluble species is referred as solubility(thermodynamic) [221–224]. Precursors which concentrations are below solubility (i.e., unsaturation) do not aggregate. Interestingly, supersaturation has been shown to be a key player that controls the kinetics of amyloid fibrillation by modulating the nucleation [222–225]. Thus, even over solubility, supersaturation, the precursor amyloid proteins become kinetically soluble (i.e., kinetic solubility). Importantly, the change in solubility in response to ambient conditions such as temperature changes and the addition of other molecules is accompanied by a change in supersaturation (discussed below). It should also be noted that supersaturation is the strongest when it is the closest to the (thermodynamic) solubility curve as the metastability of supersaturation is the strongest (i.e., a kinetic barrier is the highest).
Recently, the phase diagrams have provided insights into the protein aggregation and liquid-liquid phase separation. The phase diagrams comprehensively explain phase behaviors of proteins under various conditions from the viewpoint of thermodynamics and kinetics [222–230]. Indeed, dynamic changes in phases of amyloid proteins depending on the conditions have been best characterized by phase diagrams. These phase diagrams include essential information about solubility, supersaturation and different phase regions (Fig. 8).
Fig. 8.
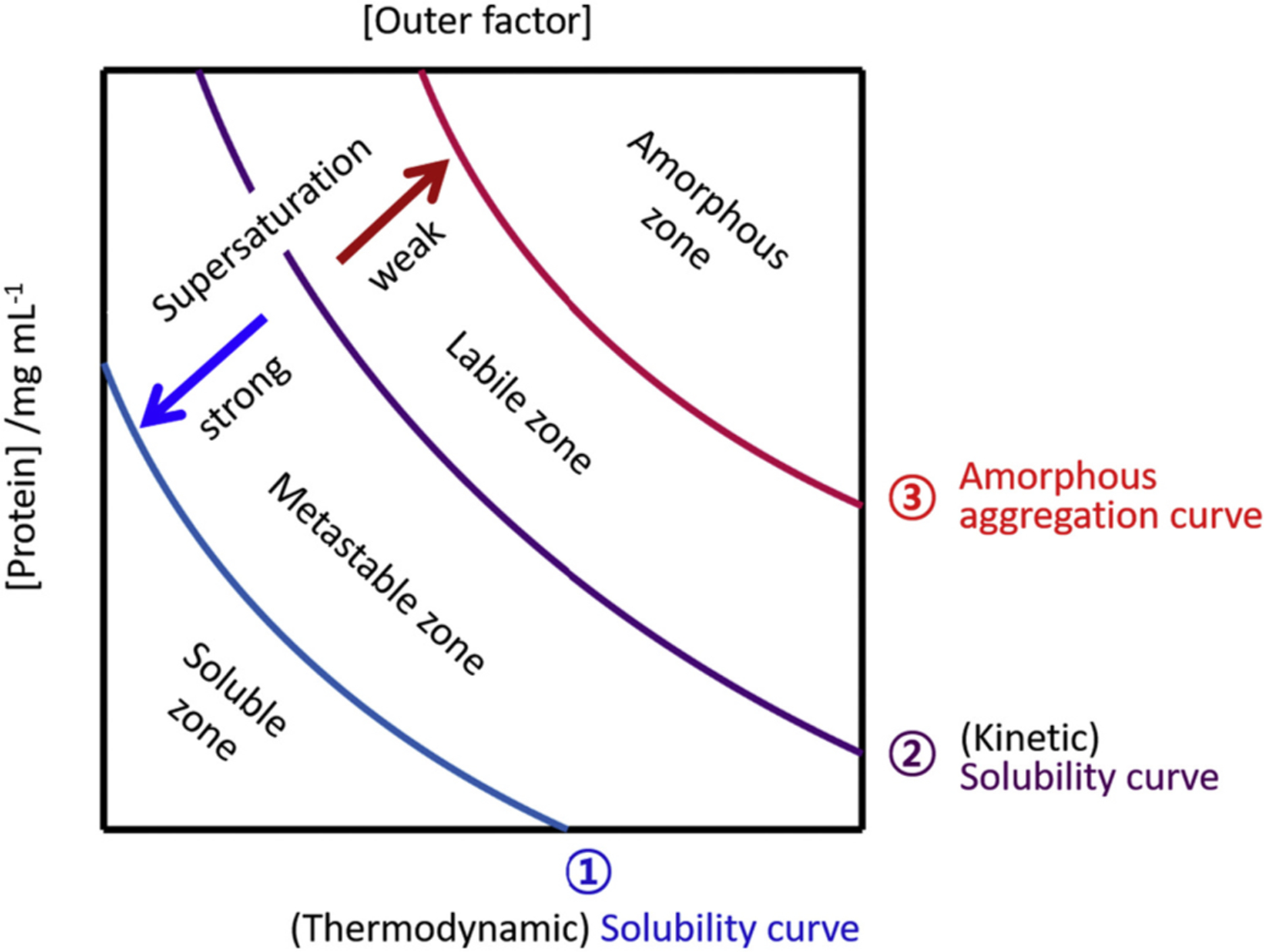
A phase diagram of nucleation-dependent amyloid formation. A typical phase diagram that explains the solubility and supersaturation-limited amyloid formation. Each region of the phase diagram designate the distinct phase properties of the amyloid proteins. These regions are labeled and separated by curves (labeled as 1, 2 and 3) which control the aggregation behaviors. The (thermodynamic) solubility curve in saturation is labeled as curve-1 (blue) and the (kinetic) solubility curve (or a metastability of supersaturation curve) is curve-2 (magenta). Curve-3 (red) is the amorphous aggregation curve. Strength of supersaturation is guided with two arrows. x and y axes indicate the concentration of proteins and external factors such as the concentration of salt, crowders, organic solvent, temperature, etc, respectively.
The soluble zone contains soluble molecular species only, and, thus, aggregation never takes place. In the metastable zone, only nucleation is allowable without amyloid elongation. On the one hand, both nucleation and elongation of amyloid fibrils are possible in the labile zone. Proteins in the amorphous aggregation zone rapidly precipitate in the form of amorphous aggregates in a nucleation-independent way. The four zones are defined by the three curves. The (thermodynamic) solubility curve largely divides the soluble and insoluble zones in equilibrium. The (kinetic) solubility curve is the border between the metastable and labile zones, and modulates not only nucleation and elongation but also supersaturation. Of note, strong supersaturation inhibits nucleation as it makes an energy barrier higher, and retardation of supersaturation results in the promotion of nucleation. The amorphous aggregation curve discriminates the labile and amorphous aggregation zones, and, also influences supersaturation.
Using phase diagrams, here we illustrate how cross-seeds, soluble molecular species or amyloid seeds of other amyloid proteins, impact amyloid generation (Fig. 9). Cross-seeds can promote or inhibit amyloidogenesis by shifting a curve and changing the area of a zone in the phase diagram. An example for boosting amyloid formation is illustrated in Fig. 9A. The addition of cross-seeds to the solution with a precursor shifts the kinetic solubility curve to the left, broadens the labile zone, and reduces the metastable zones. This new positioning of amyloid proteins to the labile zone from the metastable zone allows productive nucleation and elongation.
Fig. 9.
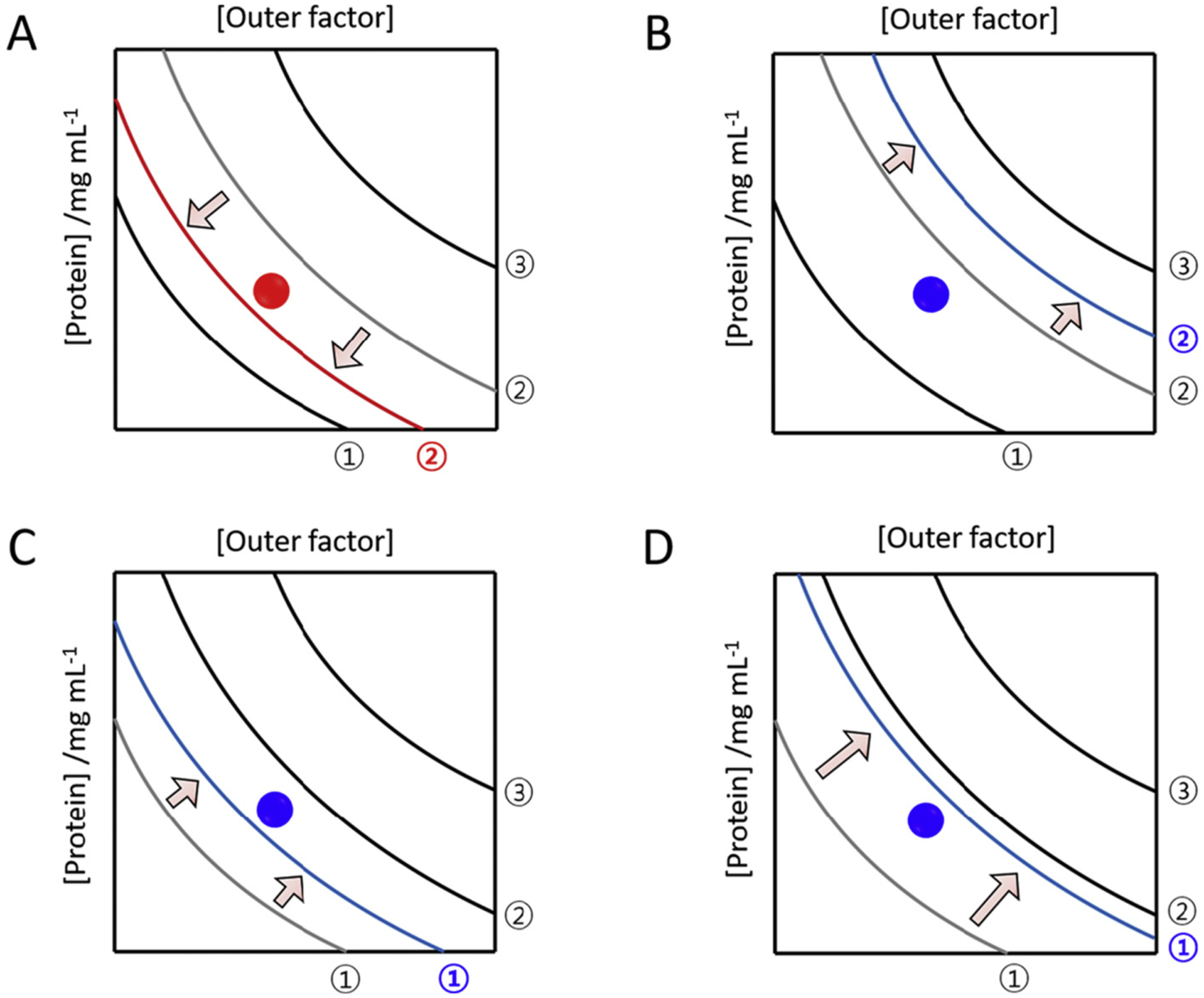
Phase diagrams showing the effects of cross-seeding on amyloid generation. Promotion (A) and inhibition (B–D) of amyloid generation due to cross-seeding are represented using phase diagrams. Amyloid proteins under given conditions are positioned with closed circles. It is assumed that the addition of cross seeds does not change the position of precursor amyloid proteins in the phase diagram. Curves in the presence of external amyloid seeds, or soluble amyloid proteins, are colored in red for promoting effects and blue for inhibiting effects. Curves prior to cross seeding are colored in gray and their corresponding shift is guided by arrows. Regions with distinct phase properties, curves (black and gray), and x and y axes are the same as explained in Fig. 8.
There are various ways to show an inhibitory action of cross-seeding as illustrated in Fig. 9B–D. For instance, interaction of cross-seeds with precursors shift the kinetic solubility curve to the right, which results in the extension of the metastable zone as shown in Fig. 9B. This, in turn, increases the metastability of supersaturation (i.e., supersaturation becomes stronger as shown in Fig. 8) impeding nucleation. Moreover, the shift of the thermodynamic solubility curve to the right can show the same supersaturation-related inhibitory mechanism by reducing the metastable zone (Fig. 9C). Increase of solubility fundamentally blocks amyloid formation by shifting the thermodynamic solubility curve to the right; i.e., the soluble zone are widened and the metastable zone is reduced. No significant effects of cross-seeding on amyloid formation may be explained without any marked changes in a phase diagram.
In this section, using a phase diagram, we have provided several possible examples that show the effects of cross-seeding on amyloid formation. A number of ways can be generated from the phase diagram to effectively affect amyloidogenesis and amyloidogenicity under various conditions. To further utilize the phase diagram for characterization of the cross-seeded aggregation, more case studies to determine the phase diagram of amyloid protein solubility and supersaturation is needed.
Acknowledgement
We acknowledge the funding support: NIH (9R01NS096785-06 to MII and AG048934 to AR) and UM Mcubed funding to MII and AR. Research YHL is supported by the National Research Foundation of Korea (NRF) grants funded by the Korean government [NRF-2018K1A3A1A39088040 and NRF-2019R1A2C1004954 (Y.-H.L.)], the National Research Council of Science & Technology (NST) grant funded by the Korea government (MSIP) (CAP-17-;05-KIGAM) (Y.-H.L.), and the Korea Basic Science Institute grant (C070410) (Y.-H.L.).
Abbreviations
- Aβ
Amyloid-β
- AD
Alzheimer’s disease
- α-synuclein
Alpha-synuclein
- CJD
Creutzfeldt-Jakob disease
- CTE
Chronic traumatic encephalopath
- DLB
Dementia with Lewy bodies
- GCI
lial cytoplasmic inclusions
- hIAPP
Human islet amyloid polypeptide
- HD
Huntington disease
- HIV
Human immunodeficiency virus
- Htt
Huntingtin
- LB
Lewy body
- LN
Lewy neurites
- MSN
Multiple system atrophy
- NAC
Non-amyloid-beta component
- PAP
Prostatic acid phosphatase
- PD
Parkinson’s disease
- PrPSC
Disease-related prion protein
- PSP
Progressive supranuclear pals
- PTMs
Post-translational modifications
- rIAPP
Rat islet amyloid polypeptide
- T2D
Type-2 diabetes mellitus
- TBI
Traumatic brain injury
- TEM
Transmission electron microscopy
- SEVI
Semen-derived enhancer of viral infection
- UPS
Ubiquitin-proteasome system
Footnotes
Declaration of Competing Interest
None.
CRediT author statement
Magdalena I. Ivanova: Conceptualization, Writing - original draft, Writing - review & editing. Yuxi Lin: Writing - original draft, Writing -review & editing. Young-Ho Lee: Conceptualization, Writing - original draft, Writing - review & editing. Jie Zheng: Conceptualization, Writing - original draft, Writing - review & editing. Ayyalusamy Ramamoorthy: Conceptualization, Writing - original draft, Writing - review & editing, Coordinating & Directing.
Data availability
No data was used for the research described in the article.
References
- [1].Chiti F, Dobson CM, Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade, Annu. Rev. Biochem 86 (2017) 27–68. [DOI] [PubMed] [Google Scholar]
- [2].Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC, Common core structure of amyloid fibrils by synchrotron X-ray diffraction, J. Mol. Biol 273 (1997) 729–739. [DOI] [PubMed] [Google Scholar]
- [3].Iadanza MG, Jackson MP, Hewitt EW, Ranson NA, Radford SE, A new era for understanding amyloid structures and disease, Nat. Rev. Mol. Cell Biol 19 (2018) 755–773. [DOI] [PubMed] [Google Scholar]
- [4].Jarrett JT, Lansbury PT Jr., Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 73 (1993) 1055–1058. [DOI] [PubMed] [Google Scholar]
- [5].Chatani E, Yamamoto N, Recent progress on understanding the mechanisms of amyloid nucleation, Biophys. Rev 10 (2018) 527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB, Amyloid β-protein fibrillogenesis Structure and biological activity of protofibrillar intermediates, J. Biol. Chem 274 (1999) 25945–25952. [DOI] [PubMed] [Google Scholar]
- [7].Cohen FE, Prusiner SB, Pathologic conformations of prion proteins, Annu. Rev. Biochem 67 (1998) 793–819, 4139 El Camino Way, PO Box 10139, Palo Alto, CA 94303–0139, USA. [DOI] [PubMed] [Google Scholar]
- [8].Prusiner SB, A unifying role for prions in neurodegenerative diseases, Science 336 (2012) 1511–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brettschneider J, Del Tredici K, Lee VM-Y, Trojanowski JQ, Spreading of pathology in neurodegenerative diseases: a focus on human studies, Nat. Rev. Neurosci 16 (2015) 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nonaka T, Masuda-Suzukake M, Hasegawa M, Molecular mechanisms of the co-deposition of multiple pathological proteins in neurodegenerative diseases, Neuropathology 38 (2018) 64–71. [DOI] [PubMed] [Google Scholar]
- [11].Rahimi J, Kovacs GG, Prevalence of mixed pathologies in the aging brain, Alzheimer’s Res. Therapy 6 (2014) 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Irwin DJ, Lee VM-Y, Trojanowski JQ, Parkinson’s disease dementia: convergence of [alpha]-synuclein, tau and amyloid-[beta] pathologies, Nat. Rev. Neurosci 14 (2013) 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Erkkinen MG, Kim M-O, Geschwind MD, Clinical neurology and epidemiology of the major neurodegenerative diseases, Cold Spring Harb. Perspect. Biol 10 (2018) a033118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P, Risk of dementia in diabetes mellitus: a systematic review, Lancet Neurol. 5 (2006) 64–74. [DOI] [PubMed] [Google Scholar]
- [15].Luchsinger JA, Reitz C, Patel B, Tang M-X, Manly JJ, Mayeux R, Relation of diabetes to mild cognitive impairment, Arch. Neurol 64 (2007) 570–575. [DOI] [PubMed] [Google Scholar]
- [16].Spires-Jones TL, Attems J, Thal DR, Interactions of pathological proteins in neurodegenerative diseases, Acta Neuropathol. 134 (2017) 187–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Close W, Neumann M, Schmidt A, Hora M, Annamalai K, Schmidt M, Reif B, Schmidt V, Grigorieff N, Fändrich M, Physical basis of amyloid fibril polymorphism, Nat. Commun 9 (2018) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tycko R, Physical and structural basis for polymorphism in amyloid fibrils, Protein Sci. 23 (2014) 1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Qiang W, Yau W-M, Lu J-X, Collinge J, Tycko R, Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes, Nature 541 (2017) 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Peng C, Gathagan RJ, Lee VM-Y, Distinct α-synuclein strains and implications for heterogeneity among α-synucleinopathies, Neurobiol. Dis 109 (2018) 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rasmussen J, Mahler J, Beschorner N, Kaeser SA, Häsler LM, Baumann F, Nystrom S, Portelius E, Blennow K, Lashley T, Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer’s disease, Proc. Natl. Acad. Sci 114 (2017) 13018–13023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Morales R, Abid K, Soto C, The prion strain phenomenon: molecular basis and unprecedented features, Biochim. Biophys. Acta (BBA)-Mol. Basis Dis 1772 (2007) 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang YM, Hall CK, Seeding and cross-seeding fibrillation of N-terminal prion protein peptides PrP(120–144), Protein Sci. 27 (2018) 1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Katorcha E, Makarava N, Lee YJ, Lindberg I, Monteiro MJ, Kovacs GG, Baskakov IV, Cross-seeding of prions by aggregated α-synuclein leads to transmissible spongiform encephalopathy, PLoS Pathog. 13 (2017) e1006563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Luk KC, Covell DJ, Kehm VM, Zhang B, Song IY, Byrne MD, Pitkin RM, Decker SC, Trojanowski JQ, Lee VM-Y, Molecular and biological compatibility with host alpha-synuclein influences fibril pathogenicity, Cell Rep. 16 (2016) 3373–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Terada M, Suzuki G, Nonaka T, Kametani F, Tamaoka A, Hasegawa M, The effect of truncation on prion-like properties of alpha-synuclein, J. Biol. Chem 293 (2018) 13910–13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ono K, Takahashi R, Ikeda T, Yamada M, Cross-seeding effects of amyloid β-protein and α-synuclein, J. Neurochem 122 (2012) 883–890. [DOI] [PubMed] [Google Scholar]
- [28].Tavassoly O, Sade D, Bera S, Shaham-Niv S, Vocadlo DJ, Gazit E, Quinolinic acid amyloid-like fibrillar assemblies seed alpha-synuclein aggregation, J. Mol. Biol 430 (2018) 3847–3862. [DOI] [PubMed] [Google Scholar]
- [29].Morales R, Estrada LD, Diaz-Espinoza R, Morales-Scheihing D, Jara MC, Castilla J, Soto C, Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases, J. Neurosci 30 (2010) 4528–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yoo BK, Xiao YL, McElheny D, Ishii Y, E22G pathogenic mutation of beta-amyloid (A beta) enhances misfolding of A beta 40 by unexpected prion-like cross talk between A beta 42 and A beta 40, J. Am. Chem. Soc 140 (2018) 2781–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jan A, Gokce O, Luthi-Carter R, Lashuel HA, The ratio of monomeric to aggregated forms of Aβ40 and Aβ42 is an important determinant of amyloid-β aggregation, fibrillogenesis, and toxicity, J. Biol. Chem 283 (2008) 28176–28189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Moreno-Gonzalez I, Edwards G III, Salvadores N, Shahnawaz M, Diaz-Espinoza R, Soto C, Molecular interaction between type 2 diabetes and Alzheimer’s disease through cross-seeding of protein misfolding, Mol. Psychiatry 22 (2017) 1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Krotee P, Griner SL, Sawaya MR, Cascio D, Rodriguez JA, Shi D, Philipp S, Murray K, Saelices L, Lee J, Common fibrillar spines of amyloid-β and human islet amyloid polypeptide revealed by microelectron diffraction and structure-based inhibitors, J. Biol. Chem 293 (2018) 2888–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, Griep A, Gelpi E, Beilharz M, Riedel D, Golenbock DT, Geyer M, Walter J, Latz E, Heneka MT, Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease, Nature 552 (2017) 355. [DOI] [PubMed] [Google Scholar]
- [35].Ono K, Takahashi R, Ikeda T, Mizuguchi M, Hamaguchi T, Yamada M, Exogenous amyloidogenic proteins function as seeds in amyloid β-protein aggregation, Biochim. Biophys. Acta (BBA) - Mol. Basis Dis 1842 (2014) 646–653. [DOI] [PubMed] [Google Scholar]
- [36].Vasconcelos B, Stancu I-C, Buist A, Bird M, Wang P, Vanoosthuyse A, Van Kolen K, Verheyen A, Kienlen-Campard P, Octave J-N, Heterotypic seeding of Tau fibrillization by pre-aggregated Abeta provides potent seeds for prion-like seeding and propagation of Tau-pathology in vivo, Acta Neuropathol. 131 (2016) 549–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Waxman EA, Giasson BI, Induction of intracellular tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of tau, J. Neurosci 31 (2011) 7604–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hu R, Zhang M, Chen H, Jiang B, Zheng J, Cross-seeding interaction between β-amyloid and human islet amyloid polypeptide, ACS Chem. Neurosci 6 (2015) 1759–1768. [DOI] [PubMed] [Google Scholar]
- [39].Hu R, Ren B, Zhang M, Chen H, Liu Y, Liu L, Gong X, Jiang B, Ma J, Zheng J, Seed-induced heterogeneous cross-seeding self-assembly of human and rat islet polypeptides, ACS Omega 2 (2017) 784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hartman K, Brender JR, Monde K, Ono A, Evans ML, Popovych N, Chapman MR, Ramamoorthy A, Bacterial curli protein promotes the conversion of PAP248–286 into the amyloid SEVI: cross-seeding of dissimilar amyloid sequences, PeerJ 1 (2013) e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Münch J, Rücker E, Ständker L, Adermann K, Goffinet C, Schindler M Wildum S, Chinnadurai R, Rajan D, Specht A, Semen-derived amyloid fibrils drastically enhance HIV infection, Cell 131 (2007) 1059–1071. [DOI] [PubMed] [Google Scholar]
- [42].Fowler DM, Koulov AV, Balch WE, Kelly JW, Functional amyloid–from bacteria to humans, Trends Biochem. Sci 32 (2007) 217–224. [DOI] [PubMed] [Google Scholar]
- [43].Van Gerven N, Van der Verren SE, Reiter DM, Remaut H, The role of functional amyloids in bacterial virulence, J. Mol. Biol 430 (2018) 3657–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hashimoto M, Ho G, Takamatsu Y, Wada R, Sugama S, Takenouchi T, Waragai M, Masliah E, Possible role of amyloid cross-seeding in evolvability and neurodegenerative disease, J. Parkinsons Dis 9 (2019) 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].O’Nuallain B, Williams AD, Westermark P, Wetzel R, Seeding specificity in amyloid growth induced by heterologous fibrils, J. Biol. Chem 279 (2004) 17490–17499. [DOI] [PubMed] [Google Scholar]
- [46].Yan LM, Velkova A, Tatarek-Nossol M, Andreetto E, Kapurniotu A, IAPP mimic blocks Aβ cytotoxic self-assembly: cross-suppression of amyloid toxicity of Aβ and IAPP suggests a molecular link between Alzheimer’s disease and type II diabetes, Angew. Chem. Int. Ed 46 (2007) 1246–1252. [DOI] [PubMed] [Google Scholar]
- [47].Malinchik SB, Inouye H, Szumowski KE, Kirschner DA, Structural analysis of Alzheimer’s β (1–40) amyloid: protofilament assembly of tubular fibrils, Biophys. J 74 (1998) 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Blake C, Serpell L, Synchrotron X-ray studies suggest that the core of the transthyretin amyloid fibril is a continuous β-sheet helix, Structure 4 (1996) 989–998. [DOI] [PubMed] [Google Scholar]
- [49].Mandal PK, Pettegrew JW, Masliah E, Hamilton RL, Mandal R, Interaction between Aβ peptide and α synuclein: molecular mechanisms in overlapping pathology of Alzheimer’s and Parkinson’s in dementia with Lewy body disease, Neurochem. Res 31 (2006) 1153–1162. [DOI] [PubMed] [Google Scholar]
- [50].Stancu I-C, Vasconcelos B, Terwel D, Dewachter I, Models of β-amyloid induced Tau-pathology: the long and “folded” road to understand the mechanism, Mol. Neurodegener 9 (2014) 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Andreetto E, Yan LM, Tatarek-Nossol M, Velkova A, Frank R, Kapurniotu A, Identification of hot regions of the Abeta-IAPP interaction interface as high-affinity binding sites in both cross- and self-association, Angew. Chem. Int. Ed. Eng 49 (2010) 3081–3085. [DOI] [PubMed] [Google Scholar]
- [52].Andreetto E, Yan LM, Caporale A, Kapurniotu A, Dissecting the role of single regions of an IAPP mimic and IAPP in inhibition of Abeta40 amyloid formation and cytotoxicity, Chembiochem: Eur. J. Chem. Biol 12 (2011) 1313–1322. [DOI] [PubMed] [Google Scholar]
- [53].Velkova A, Tatarek-Nossol M, Andreetto E, Kapurniotu A, Exploiting cross-amyloid interactions to inhibit protein aggregation but not function: nanomolar affinity inhibition of insulin aggregation by an IAPP mimic, Angew. Chem. Int. Ed. Eng 47 (2008) 7114–7118. [DOI] [PubMed] [Google Scholar]
- [54].Ren B, Zhang Y, Zhang M, Liu Y, Zhang D, Gong X, Feng Z, Tang J, Chang Y, Zheng J, Fundamentals of cross-seeding of amyloid proteins: an introduction, J. Mater. Chem B 7 (2019) 7267–7282. [DOI] [PubMed] [Google Scholar]
- [55].Miller Y, Ma B, Nussinov R, Synergistic interactions between repeats in tau protein and Aβ amyloids may be responsible for accelerated aggregation via polymorphic states, Biochemistry 50 (2011) 5172–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang M, Hu R, Chen H, Gong X, Zhou F, Zhang L, Zheng J, Polymorphic associations and structures of the cross-seeding of Aβ1–42 and hIAPP1–37 polypeptides, J. Chem. Inf. Model 55 (2015) 1628–1639. [DOI] [PubMed] [Google Scholar]
- [57].Riek R, Eisenberg DS, The activities of amyloids from a structural perspective, Nature 539 (2016) 227–235. [DOI] [PubMed] [Google Scholar]
- [58].Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R, 3D structure of Alzheimer’s amyloid-{beta}(1–42) fibrils, PNAS 102 (2005) 17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Luca S, Yau W-M, Leapman R, Tycko R, Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR, Biochemistry 46 (2007) 13505–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Iwata K, Fujiwara T, Matsuki Y, Akutsu H, Takahashi S, Naiki H, Goto Y, 3D structure of amyloid protofilaments of beta2-microglobulin fragment probed by solid-state NMR, PNAS 103 (2006) 18119–18124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ferguson N, Becker J, Tidow H, Tremmel S, Sharpe TD, Krause G, Flinders J, Petrovich M, Berriman J, Oschkinat H, Fersht AR, General structural motifs of amyloid protofilaments, PNAS 103 (2006) 16248–16253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jackson K, Barisone GA, Diaz E, Jin L.w., DeCarli C, Despa F, Amylin deposition in the brain: a second amyloid in Alzheimer disease? Ann. Neurol 74 (2013) 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Fawver JN, Ghiwot Y, Koola C, Carrera W, Rodriguez-Rivera J, Hernandez C, Dineley KT, Kong Y, Li J, Jhamandas J, Islet amyloid polypeptide (IAPP): a second amyloid in Alzheimer’s disease, Curr. Alzheimer Res 11 (2014) 928–940. [DOI] [PubMed] [Google Scholar]
- [64].Biessels GJ, Despa F, Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications, Nat. Rev. Endocrinol 14 (2018) 591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Despa F, Goldstein LB, Biessels GJ, Amylin as a potential link between type 2 diabetes and alzheimer disease, Ann. Neurol 87 (2020) 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Martinez-Valbuena I, Valenti-Azcarate R, Amat-Villegas I, Riverol M, Marcilla I, de Andrea CE, Sánchez-Arias JA, del Mar Carmona-Abellan M, Marti G, Erro ME, Amylin as a potential link between type 2 diabetes and alzheimer disease, Ann. Neurol 86 (2019) 539–551. [DOI] [PubMed] [Google Scholar]
- [67].Oskarsson ME, Paulsson JF, Schultz SW, Ingelsson M, Westermark P, Westermark GT, In vivo seeding and cross-seeding of localized amyloidosis: a molecular link between type 2 diabetes and Alzheimer disease, Am. J. Pathol 185 (2015) 834–846. [DOI] [PubMed] [Google Scholar]
- [68].Baram M, Atsmon-Raz Y, Ma B, Nussinov R, Miller Y, Amylin–Aβ oligomers at atomic resolution using molecular dynamics simulations: a link between type 2 diabetes and Alzheimer’s disease, Phys. Chem. Chem. Phys 18 (2016) 2330–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Berhanu WM, Yaşar F, Hansmann UH, In silico cross seeding of Aβ and amylin fibril-like oligomers, ACS Chem. Neurosci 4 (2013) 1488–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zhang M, Hu R, Chen H, Chang Y, Ma J, Liang G, Mi J, Wang Y, Zheng J, Polymorphic cross-seeding amyloid assemblies of amyloid-β and human islet amyloid polypeptide, Phys. Chem. Chem. Phys 17 (2015) 23245–23256. [DOI] [PubMed] [Google Scholar]
- [71].Ge X, Yang Y, Sun Y, Cao W, Ding F, Islet amyloid polypeptide promotes amyloid-beta aggregation by binding-induced helix-unfolding of the amyloidogenic core, ACS Chem. Neurosci 9 (2018) 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Gotz J, Lim Y-A, Eckert A, Lessons from two prevalent amyloidoses—what amylin and Aβ have in common, Front. Aging Neurosci 5 (2013) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhang M, Hu R, Ren B, Chen H, Jiang B, Ma J, Zheng J, Molecular understanding of Aβ-hIAPP cross-seeding assemblies on lipid membranes, ACS Chem. Neurosci 8 (2017) 524–537. [DOI] [PubMed] [Google Scholar]
- [74].Bharadwaj P, Solomon T, Sahoo BR, Ignasiak K, Gaskin S, Rowles J, Verdile G, Howard MJ, Bond CS, Ramamoorthy A, Amylin and beta amyloid proteins interact to form amorphous heterocomplexes with enhanced toxicity in neuronal cells, Sci. Rep 10 (2020) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Brender JR, Hartman K, Reid KR, Kennedy RT, Ramamoorthy A, A single mutation in the nonamyloidogenic region of islet amyloid polypeptide greatly reduces toxicity, Biochemistry 47 (2008) 12680–12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Nanga RPR, Brender JR, Xu J, Hartman K, Subramanian V, Ramamoorthy A, Three-dimensional structure and orientation of rat islet amyloid polypeptide protein in a membrane environment by solution NMR spectroscopy, J. Am. Chem. Soc 131 (2009) 8252–8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Bokranz W, Wang X, Tschäpe H, Römling U, Expression of cellulose and curli fimbriae by Escherichia coli isolated from the gastrointestinal tract, J. Med. Microbiol 54 (2005) 1171–1182. [DOI] [PubMed] [Google Scholar]
- [78].Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ, Role of Escherichia coli curli operons in directing amyloid fiber formation, Science 295 (2002) 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Nicastro L, Tükel Ç, Bacterial amyloids: the link between bacterial infections and autoimmunity, Trends Microbiol. 27 (2019) 954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Friedland RP, Mechanisms of molecular mimicry involving the microbiota in neurodegeneration, J. Alzheimers Dis 45 (2015) 349–362. [DOI] [PubMed] [Google Scholar]
- [81].Ulusoy A, Phillips RJ, Helwig M, Klinkenberg M, Powley TL, Di Monte DA, Brain-to-stomach transfer of α-synuclein via vagal preganglionic projections, Acta Neuropathol. 133 (2017) 381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kim S, Kwon S-H, Kam T-I, Panicker N, Karuppagounder SS, Lee S, Lee JH, Kim WR, Kook M, Foss CA, Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease, Neuron 103 (2019) 627–641, e627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Chen SG, Stribinskis V, Rane MJ, Demuth DR, Gozal E, Roberts AM, Jagadapillai R, Liu R, Choe K, Shivakumar B, Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans, Sci. Rep 6 (2016) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Christensen LFB, Jensen KF, Nielsen J, Vad BS, Christiansen G, Otzen DE, Reducing the amyloidogenicity of functional amyloid protein FapC increases its ability to inhibit α-Synuclein fibrillation, Acs Omega 4 (2019) 4029–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Perov S, Lidor O, Salinas N, Golan N, Tayeb-Fligelman E, Deshmukh M, Willbold D, Landau M, Structural insights into curli CsgA cross-β fibril architecture inspire repurposing of anti-amyloid compounds as anti-biofilm agents, PLoS Pathog. 15 (2019) e1007978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Brender JR, Hartman K, Gottler LM, Cavitt ME, Youngstrom DW, Ramamoorthy A, Helical conformation of the SEVI precursor peptide PAP248–286, a dramatic enhancer of HIV infectivity, promotes lipid aggregation and fusion, Biophys. J 97 (2009) 2474–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Popovych N, Brender JR, Soong R, Vivekanandan S, Hartman K, Basrur V, Macdonald PM, Ramamoorthy A, Site specific interaction of the polyphenol EGCG with the SEVI amyloid precursor peptide PAP (248–286), J. Phys. Chem. B 116 (2012) 3650–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Nanga RP, Brender JR, Vivekanandan S, Popovych N, Ramamoorthy A, NMR structure in a membrane environment reveals putative amyloidogenic regions of the SEVI precursor peptide PAP248 286, J. Am. Chem. Soc 131 (2009) 17972–17979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Martellini JA, Cole AL, Svoboda P, Stuchlik O, Chen L-M, Chai KX, Gangrade BK, Sørensen OE, Pohl J, Cole AM, HIV-1 enhancing effect of prostatic acid phosphatase peptides is reduced in human seminal plasma, PLoS One 6 (2011) e16285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Irwin DJ, Hurtig HI, The contribution of tau, amyloid-beta and alpha-synuclein pathology to dementia in Lewy body disorders, J. Alzheimer’s Dis. Parkinsonism (2018) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Úeda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero D, Kondo J, Ihara Y, Saitoh T, Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease, Proc. Natl. Acad. Sci 90 (1993) 11282–11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Mori F, Tanji K, Odagiri S, Toyoshima Y, Yoshida M, Ikeda T, Sasaki H, Kakita A, Takahashi H, Wakabayashi K, Ubiquilin immunoreactivity in cytoplasmic and nuclear inclusions in synucleinopathies, polyglutamine diseases and intranuclear inclusion body disease, Acta Neuropathol. 124 (2012) 149–151. [DOI] [PubMed] [Google Scholar]
- [93].Prashant B, Solomon T, Sahoo BR, Katarzyna I, Gaskin S, Rowles J, Giuseppe V, Howard MJ, Bond CS, Ayyalusamy R, Amylin and beta amyloid proteins interact to form amorphous heterocomplexes with enhanced toxicity in neuronal cells, Sci. Rep. (Nat. Publ. Group) (2020) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Brender JR, Ghosh A, Kotler SA, Krishnamoorthy J, Bera S, Morris V, Sil TB, Garai K, Reif B, Bhunia A, Probing transient non-native states in amyloid beta fiber elongation by NMR, Chem. Commun 55 (2019) 4483–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Chia S, Flagmeier P, Habchi J, Lattanzi V, Linse S, Dobson CM, Knowles TP, Vendruscolo M, Monomeric and fibrillar α-synuclein exert opposite effects on the catalytic cycle that promotes the proliferation of Aβ42 aggregates, Proc. Natl. Acad. Sci 114 (2017) 8005–8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kim WS, Kågedal K, Halliday GM, Alpha-synuclein biology in Lewy body diseases, Alzheimer’s Res. Therapy 6 (2014) 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Goedert M, Jakes R, Spillantini MG, The synucleinopathies: twenty years on, J. Parkinsons Dis 7 (2017) S53–S71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Jankovic J, Parkinson’s disease: clinical features and diagnosis, J. Neurol. Neurosurg. Psychiatry 79 (2008) 368–376. [DOI] [PubMed] [Google Scholar]
- [99].Donaghy PC, McKeith IG, The clinical characteristics of dementia with Lewy bodies and a consideration of prodromal diagnosis, Alzheimer’s Res. Therapy 6 (2014) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Arai K, Kato N, Kashiwado K. i., Hattori T, Pure autonomic failure in association with human α-synucleinopathy, Neurosci. Lett 296 (2000) 171–173. [DOI] [PubMed] [Google Scholar]
- [101].Papp MI, Kahn JE, Lantos PL, Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome), J. Neurol. Sci 94 (1989) 79–100. [DOI] [PubMed] [Google Scholar]
- [102].Spillantini M, Goedert M, Synucleinopathies: past, present and future, Neuropathol. Appl. Neurobiol 42 (2016) 3–5. [DOI] [PubMed] [Google Scholar]
- [103].Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, Caswell C, Van Deerlin VM, Yan N, Yousef A, Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated, Brain 141 (2018) 2181–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Lippa CF, Fujiwara H, Mann DM, Giasson B, Baba M, Schmidt ML, Nee LE, O’Connell B, Pollen DA, George-Hyslop PS, Lewy bodies contain altered α-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes, Am. J. Pathol 153 (1998) 1365–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Hamilton RL, Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 Cases using α-synuclein immunohistochemistry, Brain Pathol. 10 (2000) 378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Trojanowski JQ, Emerging Alzheimer’s disease therapies: focusing on the future, Neurobiol. Aging 23 (2002) 985–990. [DOI] [PubMed] [Google Scholar]
- [107].McKeith I, Dickson DW, Lowe J, Emre M, O’brien J, Feldman H, Cummings J, Duda J, Lippa C, Perry E, Diagnosis and management of dementia with Lewy bodies third report of the DLB consortium, Neurology 65 (2005) 1863–1872. [DOI] [PubMed] [Google Scholar]
- [108].Armstrong RA, Cairns NJ, Lantos PL, β-Amyloid (Aβ) deposition in the medial temporal lobe of patients with dementia with Lewy bodies, Neurosci. Lett 227 (1997) 193–196. [DOI] [PubMed] [Google Scholar]
- [109].Marsh SE, Blurton-Jones M, Examining the mechanisms that link β-amyloid and α-synuclein pathologies, Alzheimer’s Res. Therapy 4 (2012) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Chanson J-B, Echaniz-Laguna A, Vogel T, Mohr M, Benoilid A, Kaltenbach G, Kiesmann M, TDP43-positive intraneuronal inclusions in a patient with motor neuron disease and Parkinson’s disease, Neurodegener. Dis 7 (2010) 260–264. [DOI] [PubMed] [Google Scholar]
- [111].Markopoulou K, Dickson DW, McComb R, Wszolek ZK, Katechalidou L, Avery L, Stansbury M, Chase B, Clinical, neuropathological and genotypic variability in SNCA A53T familial Parkinson’s disease, Acta Neuropathol. 116 (2008) 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, Arnold SE, Siderowf A, Grossman M, Leverenz JB, Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases, Acta Neuropathol. 114 (2007) 221–229. [DOI] [PubMed] [Google Scholar]
- [113].Delic V, Beck KD, Pang KC, Citron BA, Biological links between traumatic brain injury and Parkinson’s disease, Acta Neuropathol. Commun 8 (2020) 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Ling H, Morris HR, Neal JW, Lees AJ, Hardy J, Holton JL, Revesz T, Williams DD, Mixed pathologies including chronic traumatic encephalopathy account for dementia in retired association football (soccer) players, Acta Neuropathol. 133 (2017) 337–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].St-Amour I, Turgeon A, Goupil C, Planel E, Hébert SS, Co-occurrence of mixed proteinopathies in late-stage Huntington’s disease, Acta Neuropathol. 135 (2018) 249–265. [DOI] [PubMed] [Google Scholar]
- [116].Charles V, Mezey E, Reddy PH, Dehejia A, Young TA, Polymeropoulos MH, Brownstein MJ, Tagle DA, Alpha-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington’s disease patients and transgenic mouse models, Neurosci. Lett 289 (2000) 29–32. [DOI] [PubMed] [Google Scholar]
- [117].Haik S, Brandel J, Sazdovitch V, Delasnerie-Lauprêtre N, Peoc’h K, Laplanche JL, Privat N, Duyckaerts C, Kemeny J, Kopp N, Dementia with Lewy bodies in a neuropathologic series of suspected Creutzfeldt–Jakob disease, Neurology 55 (2000) 1401–1404. [DOI] [PubMed] [Google Scholar]
- [118].Kovacs GG, Seguin J, Quadrio I, Höftberger R, Kapás I, Streichenberger N, Biacabe AG, Meyronet D, Sciot R, Vandenberghe R, Genetic Creutzfeldt-Jakob disease associated with the E200K mutation: characterization of a complex proteinopathy, Acta Neuropathol. 121 (2011) 39–57. [DOI] [PubMed] [Google Scholar]
- [119].Vital A, Fernagut P-O, Canron M-H, Joux J, Bezard E, Martin-Negrier M-L, Vital C, Tison F, The nigrostriatal pathway in Creutzfeldt-Jakob disease, J. Neuropathol. Exp. Neurol 68 (2009) 809–815. [DOI] [PubMed] [Google Scholar]
- [120].Haik S, Privat N, Adjou K, Sazdovitch V, Dormont D, Duyckaerts C, Hauw J, Alpha-synuclein-immunoreactive deposits in human and animal prion diseases, Acta Neuropathol. 103 (2002) 516–520. [DOI] [PubMed] [Google Scholar]
- [121].Kayed R, Dettmer U, Lesńe SE, Soluble endogenous oligomeric α-synuclein species in neurodegenerative diseases: expression, spreading, and cross-talk, J. Parkinsons Dis (2020) 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM, Synergistic interactions between Aβ, tau, and α-synuclein: acceleration of neuropathology and cognitive decline, J. Neurosci 30 (2010) 7281–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Castillo-Carranza DL, Guerrero-Muñoz MJ, Sengupta U, Gerson JE, Kayed R, α-Synuclein oligomers induce a unique toxic tau strain, Biol. Psychiatry 84 (2018) 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Distinct α-synuclein strains differentially promote tau inclusions in neurons, Cell 154 (2013) 103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L, β-Amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease, Proc. Natl. Acad. Sci 98 (2001) 12245–12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Tsigelny IF, Crews L, Desplats P, Shaked GM, Sharikov Y, Mizuno H, Spencer B, Rockenstein E, Trejo M, Platoshyn O, Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases, PLoS One 3 (2008) e3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Khan SS, LaCroix M, Boyle G, Sherman MA, Brown JL, Amar F, Aldaco J, Lee MK, Bloom GS, Lesńe SE, Bidirectional modulation of Alzheimer phenotype by alpha-synuclein in mice and primary neurons, Acta Neuropathol. 136 (2018) 589–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM-Y, Initiation and synergistic fibrillization of tau and alpha-synuclein, Science 300 (2003) 636–640. [DOI] [PubMed] [Google Scholar]
- [129].Sorrentino ZA, Vijayaraghavan N, Gorion K-M, Riffe CJ, Strang KH, Caldwell J, Giasson BI, Physiological C-terminal truncation of α-synuclein potentiates the prion-like formation of pathological inclusions, J. Biol. Chem 293 (2018) 18914–18932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Horvath I, Wittung-Stafshede P, Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson’s disease, Proc. Natl. Acad. Sci 113 (2016) 12473–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Bassil F, Brown HJ, Pattabhiraman S, Iwasyk JE, Maghames CM, Meymand ES, Cox TO, Riddle DM, Zhang B, Trojanowski JQ, Amyloid-beta (Aβ) plaques promote seeding and spreading of alpha-synuclein and tau in a mouse model of Lewy body disorders with Aβ pathology, Neuron 105 (2020) 260–275, e266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Pletnikova O, West N, Lee MK, Rudow GL, Skolasky RL, Dawson TM, Marsh L, Troncoso JC, Aβ deposition is associated with enhanced cortical α-synuclein lesions in Lewy body diseases, Neurobiol. Aging 26 (2005) 1183–1192. [DOI] [PubMed] [Google Scholar]
- [133].Bachhuber T, Katzmarski N, McCarter JF, Loreth D, Tahirovic S, Kamp F, Abou-Ajram C, Nuscher B, Serrano-Pozo A, Müller A, Inhibition of amyloid-β plaque formation by α-synuclein, Nat. Med 21 (2015) 802. [DOI] [PubMed] [Google Scholar]
- [134].Shirasaka M, Kuwata K, Honda R, α-Synuclein chaperone suppresses nucleation and amyloidogenesis of prion protein, Biochem. Biophys. Res. Commun 521 (2020) 259–264. [DOI] [PubMed] [Google Scholar]
- [135].Maass A, Lockhart SN, Harrison TM, Bell RK, Mellinger T, Swinnerton K, Baker SL, Rabinovici GD, Jagust WJ, Entorhinal tau pathology, episodic memory decline, and neurodegeneration in aging, J. Neurosci 38 (2018) 530–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Chorell E, Andersson E, Evans ML, Jain N, Götheson A, Åden J, Chapman MR, Almqvist F, Wittung-Stafshede P, Bacterial chaperones CsgE and CsgC differentially modulate human α-synuclein amyloid formation via transient contacts, PLoS One 10 (2015) e0140194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Sanjeev A, Sahu RK, Mattaparthi VSK, Potential of mean force and molecular dynamics study on the transient interactions between α and β synuclein that drive inhibition of α-synuclein aggregation, J. Biomol. Struct. Dyn 35 (2017) 3342–3353. [DOI] [PubMed] [Google Scholar]
- [138].Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E, Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders, Am. J. Pathol 152 (1998) 367. [PMC free article] [PubMed] [Google Scholar]
- [139].Aulíc S, Masperone L, Narkiewicz J, Isopi E, Bistaffa E, Ambrosetti E, Pastore B, De Cecco E, Scaini D, Zago P, α-Synuclein amyloids hijack prion protein to gain cell entry, facilitate cell-to-cell spreading and block prion replication, Sci. Rep 7 (2017) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Lashuel HA, Do Lewy bodies contain alpha-synuclein fibrils? and Does it matter? A brief history and critical analysis of recent reports, in: Neurobiology of Disease, 2020, p. 104876. [DOI] [PubMed] [Google Scholar]
- [141].Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, Castaño-Díez D, Schweighauser G, Graff-Meyer A, Goldie KN, Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes, Nat. Neurosci 22 (2019) 1099–1109. [DOI] [PubMed] [Google Scholar]
- [142].Mahul-Mellier A-L, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, Leleu M, Knott GW, Lashuel HA, The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration, Proc. Natl. Acad. Sci 117 (2020) 4971–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Burŕe J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC, α-Synuclein promotes SNARE-complex assembly in vivo and in vitro, Science 329 (2010) 1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Logan T, Bendor J, Toupin C, Thorn K, Edwards RH, α-Synuclein promotes dilation of the exocytotic fusion pore, Nat. Neurosci 20 (2017) 681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Snead D, Eliezer D, Alpha-synuclein function and dysfunction on cellular membranes, Exp. Neurobiol 23 (2014) 292–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Uversky VN, Neuropathology, biochemistry, and biophysics of α-synuclein aggregation, J. Neurochem 103 (2007) 17–37. [DOI] [PubMed] [Google Scholar]
- [147].Mori A, Imai Y, Hattori N, Lipids: key players that modulate α-synuclein toxicity and neurodegeneration in Parkinson’s disease, Int. J. Mol. Sci 21 (2020) 3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Inoshita T, Cui C, Hattori N, Imai Y, Regulation of membrane dynamics by Parkinson’s disease-associated genes, J. Genet 97 (2018) 715–727. [PubMed] [Google Scholar]
- [149].Varkey J, Langen R, Membrane remodeling by amyloidogenic and nonamyloidogenic proteins studied by EPR, J. Magn. Reson 280 (2017) 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Volles MJ, Lansbury PT, Vesicle permeabilization by protofibrillar α-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism, Biochemistry 41 (2002) 4595–4602. [DOI] [PubMed] [Google Scholar]
- [151].Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT, α-Synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils, J. Mol. Biol 322 (2002) 1089–1102. [DOI] [PubMed] [Google Scholar]
- [152].Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG, The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia, Ann. Neurol 55 (2004) 164–173. [DOI] [PubMed] [Google Scholar]
- [153].Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, Alpha-synuclein p. H50Q, a novel pathogenic mutation for Parkinson’s disease, Mov. Disord 28 (2013) 811–813. [DOI] [PubMed] [Google Scholar]
- [154].Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Mutation in the α-synuclein gene identified in families with Parkinson’s disease, Science 276 (1997) 2045–2047. [DOI] [PubMed] [Google Scholar]
- [155].Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schols L, Riess O, AlaSOPro mutation in the gene encoding α-synuclein in Parkinson’s disease, Nat. Genet 18 (1998) 106–108. [DOI] [PubMed] [Google Scholar]
- [156].Lesage S, Anheim M, Letournel F, Bousset L, Honoŕe A, Rozas N, Pieri L, Madiona K, Dürr A, Melki R, G51D α-synuclein mutation causes a novel Parkinsonian–pyramidal syndrome, Ann. Neurol 73 (2013) 459–471. [DOI] [PubMed] [Google Scholar]
- [157].Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Pöyhönen M, Paetau A, A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology, Neurobiol. Aging 35 (2014) 2180, e2181–2180. e2185. [DOI] [PubMed] [Google Scholar]
- [158].Li J, Uversky VN, Fink AL, Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein, Biochemistry 40 (2001) 11604–11613. [DOI] [PubMed] [Google Scholar]
- [159].Khalaf O, Fauvet B, Oueslati A, Dikiy I, Mahul-Mellier A-L, Ruggeri FS, Mbefo MK, Vercruysse F, Dietler G, Lee S-J, The H50Q mutation enhances α-synuclein aggregation, secretion, and toxicity, J. Biol. Chem 289 (2014) 21856–21876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Crowther RA, Goedert M, Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein, FEBS Lett. 576 (2004) 363–368. [DOI] [PubMed] [Google Scholar]
- [161].Tsigelny IF, Sharikov Y, Kouznetsova VL, Greenberg JP, Wrasidlo W, Overk C, Gonzalez T, Trejo M, Spencer B, Kosberg K, Molecular determinants of α-synuclein mutants’ oligomerization and membrane interactions, ACS Chem. Neurosci 6 (2015) 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH, A novel α-synuclein missense mutation in Parkinson disease, Neurology 80 (2013) 1062–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Nuber S, Rajsombath M, Minakaki G, Winkler J, Müller CP, Ericsson M, Caldarone B, Dettmer U, Selkoe DJ, Abrogating native α-Synuclein tetramers in mice causes a L-DOPA-responsive motor syndrome closely resembling Parkinson’s disease, Neuron 100 (2018) 75–90, e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Perrin RJ, Woods WS, Clayton DF, George JM, Interaction of human α-synuclein and Parkinson’s disease variants with phospholipids structural analysis using site-directed mutagenesis, J. Biol. Chem 275 (2000) 34393–34398. [DOI] [PubMed] [Google Scholar]
- [165].Bussell R, Eliezer D, Effects of Parkinson’s disease-linked mutations on the structure of lipid-associated α-synuclein, Biochemistry 43 (2004) 4810–4818. [DOI] [PubMed] [Google Scholar]
- [166].Galvagnion C, Brown JW, Ouberai MM, Flagmeier P, Vendruscolo M, Buell AK, Sparr E, Dobson CM, Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein, Proc. Natl. Acad. Sci 113 (2016) 7065–7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Marshall MS, Bongarzone ER, Beyond Krabbe’s disease: The potential contribution of galactosylceramidase deficiency to neuronal vulnerability in late-onset synucleinopathies, J. Neurosci. Res 94 (2016) 1328–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Seyfried T, Choi H, Chevalier A, Hogan D, Akgoc Z, Schneider J, Sex-related abnormalities in substantia nigra lipids in Parkinson’s disease, ASN Neuro 10 (2018), 1759091418781889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Cholak E, Bucciarelli S, Bugge K, Johansen NT, Vestergaard B, Arleth L, Kragelund BB, Langkilde AE, Distinct α-synuclein: lipid co-structure complexes affect amyloid nucleation through fibril mimetic behavior, Biochemistry 58 (2019) 5052–5065. [DOI] [PubMed] [Google Scholar]
- [170].Trexler AJ, Rhoades E, α-Synuclein binds large unilamellar vesicles as an extended helix, Biochemistry 48 (2009) 2304–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC, Petrlova J, Voss JC, Stamou DG, Steven AC, Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins, J. Biol. Chem 285 (2010) 32486–32493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Westphal CH, Chandra SS, Monomeric synucleins generate membrane curvature, J. Biol. Chem 288 (2013) 1829–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Leftin A, Job C, Beyer K, Brown MF, Solid-state 13C NMR reveals annealing of raft-like membranes containing cholesterol by the intrinsically disordered protein α-synuclein, J. Mol. Biol 425 (2013) 2973–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Ouberai MM, Wang J, Swann MJ, Galvagnion C, Guilliams T, Dobson CM, Welland ME, α-Synuclein senses lipid packing defects and induces lateral expansion of lipids leading to membrane remodeling, J. Biol. Chem 288 (2013) 20883–20895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Rabe M, Soragni A, Reynolds NP, Verdes D, Liverani E, Riek R, Seeger S, On-surface aggregation of α-synuclein at nanomolar concentrations results in two distinct growth mechanisms, ACS Chem. Neurosci 4 (2013) 408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, Meijer L, Kahle PJ, Haass C, Constitutive phosphorylation of the Parkinson’s disease associated α-synuclein, J. Biol. Chem 275 (2000) 390–397. [DOI] [PubMed] [Google Scholar]
- [177].Watson MD, Lee JC, N-terminal acetylation affects α-synuclein fibril polymorphism, Biochemistry 58 (2019) 3630–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Trexler AJ, Rhoades E, N-terminal acetylation is critical for forming α-helical oligomer of α-synuclein, Protein Sci. 21 (2012) 601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Ischiropoulos H, Oxidative modifications of a-synuclein, Ann. N. Y. Acad. Sci 991 (2003) 93–100. [DOI] [PubMed] [Google Scholar]
- [180].Guerrero E, Vasudevaraju P, Hegde ML, Britton G, Rao K, Recent advances in α-synuclein functions, advanced glycation, and toxicity: implications for Parkinson’s disease, Mol. Neurobiol 47 (2013) 525–536. [DOI] [PubMed] [Google Scholar]
- [181].Savyon M, Engelender S, SUMOylation in α-synuclein homeostasis and pathology, Front. Aging Neurosci (2020) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Sorrentino ZA, Giasson BI, The emerging role of α-synuclein truncation in aggregation and disease, J. Biol. Chem (2020) 011743, jbc. REV120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Maïza A, Chantepie S, Vera C, Fifre A, Huynh MB, Stettler O, Ouidja MO, Papy-Garcia D, The role of heparan sulfates in protein aggregation and their potential impact on neurodegeneration, FEBS Lett. 592 (2018) 3806–3818. [DOI] [PubMed] [Google Scholar]
- [184].Yan D-Y, Liu C, Tan X, Ma Z, Wang C, Deng Y, Liu W, Xu Z-F, Xu B, Mn- Induced neurocytes injury and autophagy dysfunction in alpha-synuclein wild-type and knock-out mice: highlighting the role of alpha-synuclein, Neurotox. Res 36 (2019) 66–80. [DOI] [PubMed] [Google Scholar]
- [185].Kalia LV, Kalia SK, McLean PJ, Lozano AM, Lang AE, α-Synuclein oligomers and clinical implications for Parkinson disease, Ann. Neurol 73 (2013) 155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Lashuel HA, Overk CR, Oueslati A, Masliah E, The many faces of α-synuclein: from structure and toxicity to therapeutic target, Nat. Rev. Neurosci 14 (2013) 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Volpicelli-Daley LA, Effects of α-synuclein on axonal transport, Neurobiol. Dis 105 (2017) 321–327. [DOI] [PubMed] [Google Scholar]
- [188].Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, Hu X, McCoy J, Chu CT, Burton EA, α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease, Sci. Transl. Med 8 (2016) 342ra378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T, Schneider BL, Lee MK, Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo, J. Neurosci 32 (2012) 3306–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Xilouri M, Brekk OR, Stefanis L, Autophagy and alpha-synuclein: relevance to P arkinson’s disease and related synucleopathies, Mov. Disord 31 (2016) 178–192. [DOI] [PubMed] [Google Scholar]
- [191].Hinault M-P, Cuendet AFH, Mattoo RU, Mensi M, Dietler G, Lashuel HA, Goloubinoff P, Stable α-synuclein oligomers strongly inhibit chaperone activity of the Hsp70 system by weak interactions with J-domain co-chaperones, J. Biol. Chem 285 (2010) 38173–38182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M, α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies, Proc. Natl. Acad. Sci 95 (1998) 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Volpicelli-Daley LA, Luk KC, Lee VM, Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite–like aggregates, Nat. Protoc 9 (2014) 2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van Den Haute C, Melki R, Baekelandt V, α-Synuclein strains cause distinct synucleinopathies after local and systemic administration, Nature 522 (2015) 340. [DOI] [PubMed] [Google Scholar]
- [195].Al-Wandi A, Ninkina N, Millership S, Williamson SJ, Jones PA, Buchman VL, Absence of α-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice, Neurobiol. Aging 31 (2010) 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein, J. Neurosci 22 (2002) 8797–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Gureviciene I, Gurevicius K, Tanila H, Role of α-synuclein in synaptic glutamate release, Neurobiol. Dis 28 (2007) 83–89. [DOI] [PubMed] [Google Scholar]
- [198].Liu S, Ninan I, Antonova I, Battaglia F, Trinchese F, Narasanna A, Kolodilov N, Dauer W, Hawkins RD, Arancio O, α-synuclein produces a long-lasting increase in neurotransmitter release, EMBO J. 23 (2004) 4506–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho W-H, Castillo PE, Shinsky N, Verdugo JMG, Armanini M, Ryan A, Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system, Neuron 25 (2000) 239–252. [DOI] [PubMed] [Google Scholar]
- [200].Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM-Y, Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice, Science 338 (2012) 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Thompson MJ, Sievers SA, Karanicolas J, Ivanova MI, Baker D, Eisenberg D, The 3D profile method for identifying fibril-forming segments of proteins, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 4074–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Goldschmidt L, Teng PK, Riek R, Eisenberg D, Identifying the amylome, proteins capable of forming amyloid-like fibrils, Proc. Natl. Acad. Sci 107 (2010) 3487–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Verasdonck J, Bousset L, Gath J, Melki R, Böckmann A, Meier BH, Further exploration of the conformational space of α-synuclein fibrils: solid-state NMR assignment of a high-pH polymorph, Biomol. NMR Assign 10 (2016) 5–12. [DOI] [PubMed] [Google Scholar]
- [204].Pinotsi D, Buell AK, Galvagnion C, Dobson CM, Kaminski Schierle GS, Kaminski CF, Direct observation of heterogeneous amyloid fibril growth kinetics via two-color super-resolution microscopy, Nano Lett. 14 (2013) 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M, Molecular-level secondary structure, polymorphism, and dynamics of full-length α-synuclein fibrils studied by solid-state NMR, Proc. Natl. Acad. Sci. U. S. A 102 (2005) 15871–15876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Bockmann A, Meier BH, Melki R, Structural and functional characterization of two alpha-synuclein strains, Nat. Commun 4 (2013) 2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Chen M, Margittai M, Chen J, Langen R, Investigation of alpha-synuclein fibril structure by site-directed spin labeling, J. Biol. Chem 282 (2007) 24970–24979. [DOI] [PubMed] [Google Scholar]
- [208].Vilar M, Chou HT, Luhrs T, Maji SK, Riek-Loher D, Verel R, Manning G, Stahlberg H, Riek R, The fold of alpha-synuclein fibrils, Proc. Natl. Acad. Sci. U. S. A 105 (2008) 8637–8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Solid-state NMR structure of a pathogenic fibril of full-length human [alpha]-synuclein, Nat. Struct. Mol. Biol 23 (2016) 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Guerrero-Ferreira R, Taylor NM, Mona D, Ringler P, Lauer ME, Riek R, Britschgi M, Stahlberg H, Cryo-EM structure of alpha-synuclein fibrils, Elife 7 (2018) e36402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, Sangwan S, Guenther EL, Johnson LM, Zhang M, Structure of the toxic core of [agr]-synuclein from invisible crystals, Nature 525 (2015) 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Zhao K, Li Y, Liu Z, Long H, Zhao C, Luo F, Sun Y, Tao Y, Su X. d., Li D, Parkinson’s disease associated mutation E46K of α-synuclein triggers the formation of a distinct fibril structure, Nat. Commun 11 (2020) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Boyer DR, Li B, Sun C, Fan W, Sawaya MR, Jiang L, Eisenberg DS, Structures of fibrils formed by α-synuclein hereditary disease mutant H50Q reveal new polymorphs, Nat. Struct. Mol. Biol 26 (2019) 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Ni X, McGlinchey RP, Jiang J, Lee JC, Structural insights into α-synuclein fibril polymorphism: Effects of Parkinson’s disease-related C-terminal truncations, J. Mol. Biol 431 (2019) 3913–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Li B, Ge P, Murray KA, Sheth P, Zhang M, Nair G, Sawaya MR, Shin WS, Boyer DR, Ye S, Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel, Nat. Commun 9 (2018) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Li Y, Zhao C, Luo F, Liu Z, Gui X, Luo Z, Zhang X, Li D, Liu C, Li X, Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy, Cell Res. 28 (2018) 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, Matsubara T, Tomita T, Ando T, Hasegawa K, Structures of α-synuclein filaments from multiple system atrophy, Nature (2020) 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Strohäker T, Jung BC, Liou S-H, Fernandez CO, Riedel D, Becker S, Halliday GM, Bennati M, Kim WS, Lee S-J, Structural heterogeneity of α-synuclein fibrils amplified from patient brain extracts, Nat. Commun 10 (2019) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Neudecker P, Robustelli P, Cavalli A, Walsh P, Lundström P, Zarrine-Afsar A, Sharpe S, Vendruscolo M, Kay LE, Structure of an intermediate state in protein folding and aggregation, Science 336 (2012) 362–366. [DOI] [PubMed] [Google Scholar]
- [220].Sakurai K, Maeno A, Lee Y-H, Akasaka K, Conformational properties relevant to the amyloidogenicity of β2-microglobulin analyzed using pressure-and salt-dependent chemical shift data, J. Phys. Chem. B 123 (2019) 836–844. [DOI] [PubMed] [Google Scholar]
- [221].Lin Y, Sahoo BR, Ozawa D, Kinoshita M, Kang J, Lim MH, Okumura M, Huh YH, Moon E, Jang JH, Diverse structural conversion and aggregation pathways of Alzheimerʼs amyloid-β (1–40), ACS Nano 13 (2019) 8766–8783. [DOI] [PubMed] [Google Scholar]
- [222].Lin Y, Kardos JZ, Imai M, Ikenoue T, Kinoshita M, Sugiki T, Ishimori K, Goto Y, Lee Y-H, Amorphous aggregation of cytochrome c with inherently low amyloidogenicity is characterized by the metastability of supersaturation and the phase diagram, Langmuir 32 (2016) 2010–2022. [DOI] [PubMed] [Google Scholar]
- [223].Lin Y, Lee Y-H, Yoshimura Y, Yagi H, Goto Y, Solubility and supersaturation-dependent protein misfolding revealed by ultrasonication, Langmuir 30 (2014) 1845–1854. [DOI] [PubMed] [Google Scholar]
- [224].Ikenoue T, Lee Y-H, Kardos J, Yagi H, Ikegami T, Naiki H, Goto Y, Heat of supersaturation-limited amyloid burst directly monitored by isothermal titration calorimetry, Proc. Natl. Acad. Sci 111 (2014) 6654–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Terakawa MS, Lin Y, Kinoshita M, Kanemura S, Itoh D, Sugiki T, Okumura M, Ramamoorthy A, Lee Y-H, Impact of membrane curvature on amyloid aggregation, Biochim. Biophys. Acta (BBA)-Biomembr 1860 (2018) 1741–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Nam G, Lin Y, Lim MH, Lee Y-H, Key Physicochemical and Biological Factors of the Phase Behavior of Tau, Chem, 2020. [Google Scholar]
- [227].Saio T, Okumura M, Lee Y-H, Solution NMR spectroscopy for investigation of liquid-liquid phase separation, J. Korean Magn. Reson. Soc 24 (2020) 47–52. [Google Scholar]
- [228].Mioduszewski Ł, Cieplak M, Protein droplets in systems of disordered homopeptides and the amyloid glass phase, Phys. Chem. Chem. Phys 22 (2020) 15592–15599. [DOI] [PubMed] [Google Scholar]
- [229].Shakya A, Park S, Rana N, King JT, Liquid-liquid phase separation of histone proteins in cells: role in chromatin organization, Biophys. J 118 (2020) 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Shin Y, Brangwynne CP, Liquid phase condensation in cell physiology and disease, Science (2017) 357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.