

Abstract
The possible link between hIAPP accumulation and β-cell death in diabetic patients has inspired numerous studies focusing on amyloid structures and aggregation pathways of this hormone. Recent studies have reported on the importance of early oligomeric intermediates, the many roles of their interactions with lipid membrane, pH, insulin and zinc on the mechanism of aggregation of hIAPP. The challenges posed by the transient nature of amyloid oligomers, their structural heterogeneity, and the complex nature of their interaction with lipid membranes have resulted in the development of a wide range of biophysical and chemical approaches to characterize the aggregation process. While the cellular processes and factors activating hIAPP-mediated cytotoxicity are still not clear, it has recently been suggested that its impaired turnover and cellular processing by proteasome and autophagy may contribute significantly towards toxic hIAPP accumulation and, eventually, β-cell death. Therefore, studies focusing on the restoration of hIAPP proteostasis may represent a promising arena for the design of effective therapies. In this review we discuss the current knowledge of the aggregation and pathology associated with hIAPP aggregation and point out the opportunities for therapy that a detailed biochemical, biophysical and cellular understanding of its aggregation may unveil.
Graphical Abstract
1. INTRODUCTION
One of the most striking features of type-2 diabetes (T2D) is the presence of highly abundant amyloid deposits of human islet amyloid polypeptide (hIAPP) around pancreatic β-cells in the islets of Langerhans. Because of the high tissue visibility of the amyloid deposits and their prevalence in diabetic patients, until recently it has been assumed that mature amyloid deposits of hIAPP play an important role in the pathogenesis of the disease. Although a firm relationship between the structure of different hIAPP aggregates in the pancreas and the severity of the disease has not yet been established, these studies have provided mechanistic insights on the link between hIAPP and T2D. Nevertheless, the 10% of T2D patients do not show hIAPP aggregates in pancreatic tissues.1–4 It is widely known that cells express an integrated array of proteolytic machinery that controls protein homeostasis (proteostasis) in a given range of adverse environmental conditions. As a matter of fact, the latest studies suggest that pathological conditions occur when the equilibrium between the production and the clearance of the involved proteins is unbalanced, or, in other words, when proteome surveillance by the proteolytic machinery of the cell is compromised.5 This notion, coupled with the failure of many clinical trials focusing on anti-aggregating drugs, suggests that the design of effective T2D therapies demands a deeper understanding of the structures of the various hIAPP assemblies as well as the mechanisms involved in proteome maintenance derangement.
This review covers the current knowledge about hIAPP and its role in T2D development with a special emphasis on: i) the structures of hIAPP aggregates and their role in damaging lipid membranes, ii) the mutual interactions of hIAPP with other amyloids, iii) the role of metal ions and small molecules in modifying hIAPP aggregation processes, and iv) hIAPP degradation routes. Finally, we also discuss about the emerging challenges and promising therapeutic opportunities in present and future research directions oriented to designing hIAPP proteostasis rescuers.
2. DIABETES: A MULTIFACTORIAL DISEASE
2.1. Diabetes
Diabetes mellitus is defined as a metabolic disease with increased blood glucose levels, which increases the risk of developing complications such as damage to the cardiovascular system, kidney, vision, and nerves. Therefore, diabetes mellitus is not a single disease but a group of disorders that have in common a deficiency of insulin. Diabetes can be divided into subgroups, which in turn are heterogeneous. The main forms of diabetes mellitus are type 1 diabetes (T1D), type 2 diabetes (T2D), and gestational diabetes. In addition to these three disorders, there are a large number of specific diabetes forms, some of which are hereditary, e.g., depending on mutation in the insulin gene or insulin receptor gene. T1D, constituting roughly 10% of diabetes cases, is an autoimmune disease leading to immune-mediated progressive destruction of pancreatic β-cells. Both humoral and cellular immune factors participate in this destruction. T1D most commonly starts at a young age but can debut at any age. Treatment of T1D is always with insulin, and the disease was initially called ‘insulin-dependent diabetes mellitus’ (IDDM). There is a genetic trait for T1D with the strongest association with the human leukocyte antigen (HLA) class II region6,7 that explains approximately 50% of the genetic background of T1D. The HLADR3/4-DQ8 is a high-risk genotype for the development of T1D8, but several susceptible and protective DR-DQ haplotypes have been identified.9 Maturity onset diabetes of the young (MODY) is a group of monogenic diabetes forms. These inherited disorders of non-autoimmune diabetes usually occur in adolescence or young adulthood10 and account for 2–3% of all diabetes cases. MODY characterized by the absence of autoantibodies against the β-cell antigens can occur without symptoms and is considered underdiagnosed. The most common MODY forms are MODY-1, which is caused by mutations in the transcriptional regulator HNF-4α gene11 and results in defective insulin release; MODY-2, caused by mutations in the glucokinase gene12 and results in a reduction in glucose sensing, leading to an increased blood glucose level but with an efficient remaining insulin secretory system; MODY-3, caused by mutations in the transcription factor HNF1-alpha gene13 which results in defective insulin release. By constrast with these types of diabetes, T2D constitutes more than 85% of diabetes cases and is clearly associated with aging and obesity. It is a globally occurring disease with rapidly increasing prevalence. The number of adult subjects with diabetes worldwide has been estimated to be 463 million while the number in 2045 is expected to rise to 700 million, 90% with T2D.14 Causes of this increase in prevalence include obesity associated with a sedentary (western) life style with over-consumption of food and too little exercise. There is also a strong, but complex genetic component involving a large number of genes. T2D is diagnosed by exclusion rather than by distinct criteria. If T1D or a monogenic form of diabetes is not present, the patient is assumed to have T2D. Most subjects with T2D have signs of metabolic syndrome, which includes obesity, particularly of abdominal type. The cause of obesity is multifactorial.15 T2D has a complex and insufficiently understood pathogenesis involving both β-cell failure and deficient peripheral sensitivity in adipose tissue and muscles to released insulin, often called insulin resistance.16 T2D is also associated with increased glucagon secretion and liver glucose production. The tendency to insulin resistance is at least partially hereditary, but obesity and sedentary lifestyle are important risk factors. Insulin resistance is an early event in the development of T2D and present already in a prediabetic stage, most often associated with abdominal obesity. Insulin resistance causes increasing insulin demand upon islet β-cells, which respond with increased insulin production and release. As long as this response is sufficient, blood glucose concentration is kept normal or near normal. It is still not completely understood how the impaired ability of β-cells to produce sufficient insulin gradually results in transition from prediabetes to overt T2D.17
T2D is heterogeneous, and at least five subgroups (or clusters) have been identified with different severities and tendencies to develop late complications.18,19 The clustering of these diseases has been performed from clinical features, and nothing is known regarding differences in islet lesions such as a reduction in β-cell mass or presence of islet amyloid. Such studies will be necessary for future islet researchers.
Although the impaired β-cell function associated with T2D is not fully understood, we know that several components may come into play.20 Even if the plasma insulin concentration can appear normal (or even high) when comparing obesity-matched individuals at similar plasma glucose levels, it is evident that insulin action is deficient in patients with T2D. While the loss of β-cells in T1D is accepted, it has been more challenging to demonstrate a reduced number of β-cells in T2D, although some analyses have shown a reduction in β-cell number in T2D21–23 and signs of malfunction in the remaining β-cells. Thus, the first-phase insulin response at an intravenous glucose challenge is already absent at the early stage of T2D24. There are also signs of aberrant proinsulin processing in T2D. In non-diabetic subjects, plasma proinsulin and processing intermediates constitute about 10% of circulating immunoreactive insulin but these levels are doubled in T2D.25 So far, there are no definite explanations for these defects.
2.1.1. Normal glucose regulation
Glucose homeostasis is maintained by a complex hormonal system that controls short and long-term blood glucose regulation, thereby preventing hyperglycemia and hypoglycemia. Islets of Langerhans contain β-cells and α-cells which secrete the polypeptide hormones insulin and glucagon, respectively. Insulin released in response to glucose binds to its receptor, and this results in the translocation of the glucose transporter GLUT-4 from the cytoplasm to the cell membrane that facilitates glucose uptake in muscles and adipose tissue.26,27 In humans, glucose enters the hepatocytes via the glucose transporter GLUT2 using an insulin independent process.28 During the postprandial state, glucose is used for glycogen synthesis in the liver and muscle. In the presence of insulin, glucose is used for free fatty acid (FFA) and triglyceride synthesis in the hepatocytes. FFA released from the liver is redistributed to the adipocytes. In the liver, the GLUT-2 transporter allows bidirectional transport of glucose in and out of the cells. Glucagon, released in response to hypoglycemia binds to its receptor present on hepatocytes and stimulates glycogenolysis and gluconeogenesis, thereby increasing the blood glucose level. Sustained high blood glucose levels are dangerous and increase the risk for complications, including cardiovascular disease, kidney and nerve damage, and vision problems (see also https://www.idf.org/aboutdiabetes/complications.html).29
2.1.2. Medical treatment for T2D
There are many options for the treatment of T2D.20 Early in the disease, it is often possible to mitigate symptoms by changing lifestyle, leading to a reduction in weight and increasing physical activity, which reduces insulin resistance. It is also possible to increase the production or release of insulin from the pancreas. Sulfonylurea derivatives are examples of this mode of action, and there are several new drugs acting this way. For example, metformin works by reducing insulin resistance and liver glucose production and is one of the most commonly used treatments in T2D. In some of the patients, insulin production ceases gradually making insulin treatment necessary.30
Thiazolidinediones, also known as glitazones31, act through the nuclear receptor peroxisome proliferator-activity receptor gamma (PPAR) and promote insulin sensitivity32. PPAR is ubiquitously expressed with enrichment in adipose tissues where it regulates uptake and storage of lipids in peripheral tissue and reduces the lipotoxicity. PPAR controls the differentiation of stem cells into adipocytes. Glitazones are used in combination with other diabetes drugs such as metformin or sulfonylureas. Treatment is in some individuals associated with severe side effects such as edema, weight gain, congestive heart failure.33
The polypeptide hormone glucagon-like peptide-1 (GLP-1) is an incretin released from the proglucagon molecule after posttranslational processing with the prohormone convertase PC1/3 and secreted from the intestinal L-cells in response to nutrients.34 In the islet, GLP-1 stimulates glucose-dependent insulin secretion and insulin gene transcription and inhibits glucagon secretion. Outside the pancreas, GLP-1 decreases gastric emptying and reduces food intake. In T2D especially, the insulinotropic action of GLP-1 is lost. The half-life of GLP-1 is short since the peptide is proteolytically degraded by the enzyme dipeptidyl peptidase-4 (DPP-4).35 Administration of DPP-4 inhibitors results in a prolonged activity of GLP-1. A group of GLP-1 analogs has been produced, retaining the biological activity of GLP-1, but with an extended half time.36 GLP-1 receptor agonists are useful incretin mimics since their administration will not cause hypoglycemia, a critical side effect of insulin administration. Also, metformin increases postprandial GLP-1 secretion, and therefore GLP-1 may be responsible for glucose-lowering observed during metformin treatment.37
Glucose is filtered out of the blood through glomeruli under normal conditions and reabsorbed by sodium-glucose linked transporter 2 (SGLT2), located in proximal convoluting tubules. Blocking SGLT-2 increases glucose excretion and lowers blood glucose concentration. Treatment with SGLT-2 inhibitor reduces HbA1c comparable to other tablet drugs, and some weight reduction has been observed.38 Several reports suggest that treatment with SGLT-2 reduces the risk of death from cardiovascular disease35 and the development of kidney failure.39 This means that treatment also reduces the risk of developing complications. A combination of GLP-1 agonist and an SGLT2-inhibitor resulted in additive effects on lowering HbA1c and body weight and reducing cardiovascular events.40
An upcoming drug is AM83341 (see also https://ml-eu.globenewswire.com/Resource/Download/265c351d-c721-43e0-ab7d-ab841a44d72a), and this IAPP-analog showed exciting results in a randomized phase-2 clinical trial that lasted for 26 weeks. The drug was shown to be well-tolerated, and in the study group receiving 4.5 mg/ week, a 10.8 % weight reduction was observed. In a phase-1 clinical trial in which AM833 was combined with the GLP-1-agonist Semaglutid, 20 weeks of treatment, participants lost an average of 17.1 % body weight. Overweight and obesity are risk factors for T2D and cardiovascular disease and the presented weight loss exceed reductions observed for GLP-1 and metformin. It should be noted that an IAPP derivative, pramlintide (symlin) is used, in addition to insulin, in patients with insulin-dependent T1D and T2D that have been difficult to regulate with insulin by a single drug.42
2.2. Cross-correlation with other diseases
Epidemiological studies link diabetes with neurodegenerative diseases, including Alzheimer’s disease43–46 and Parkinson’s disease.47–49 The link is unclear, but the three conditions are multifactorial and have protein aggregation in their pathophysiology in common. If protein aggregation constitutes the link between these diseases, however, still needs to be proven. The Rotterdam study50, published in 1999, showed that patients with T2D have an almost doubled risk of developing dementia and Alzheimer’s disease (AD). Data from the ULSAM study (Uppsala Longitudinal study of adult men) showed that already a moderate disturbance in the first-phase insulin release at the age of 55 increased the risk of developing AD thirty years later.51 This suggests that diabetes precedes AD. In fact, another type of diabetes has been proposed52–54, which is type 3 diabetes (T3DM) which is an AD associated insulin resistance, also described as “brain diabetes phenotype”. A considerable number of biophysical studies have shown a close link between the amyloid-forming proteins IAPP and Aβ.55–58 IAPP immunoreactivity is present in formic acid brain extracts from patients with AD59 and exhibited pattern on the western blot ranges from dimers to 16 mers.60 The laddering pattern suggests that IAPP is present in the Aβ amyloid deposit. Proximity ligation assay (PLA) using two primary antibodies can be applied to determine the co-deposition of proteins. The positive PLA signal obtained after the combination of anti-IAPP and anti-Aβ antibodies points to the co-deposition of IAPP and Aβ in vivo. To confirm that the peptides interact in vivo, hIAPP transgenic mice were injected with preformed fibrils of Aβ followed by high-fat feeding for ten months.60 Mice injected with preformed Aβ fibrils developed amyloid in 15% of the islets, which is comparable to mice injected with preformed fibrils of proIAPP. In mice injected with preformed IAPP fibrils, IAPP amyloid was found in 24 % of the islets. Control mice injected with fibrils produced from the proinsulin metabolite C-peptide/A-chain developed small amount of amyloid in 5% of the islets. The result confirms interaction between the peptides, but as found in vitro, homologous seeding is preferred over heterologous seeding.60 In the past, for many years, the hypothesis that the killer of neurons (AD) or beta cells (T2D) were the insoluble amyloid fibrils (amyloid hypothesis) has been supported.21,61–63 Subsequently, the hypothesis of toxic oligomers was advanced, where the toxicity of Aβs or hIAPP is due to the oligomers, transient species that precede the fibrillar state.61,64–68 Experimental data have shown that polyclonal antibodies prepared by Aβ40, can recognize not only the toxic oligomers of Aβ40 but also the oligomers of a-synuclein, lysozyme, insulin, IAPP, prion protein, and many other proteins62,63,69–72. The same result was obtained using IAPP oligomeric preparation, rather than Aβ mimic. De novo designed peptides derived from hIAPP interacting with Aβ form colloid-like oligomers having β-like structures73. From this, we can deduce that oligomers of different proteins may have the similar conformational characteristics. Alternatively, it has been proposed that the recognition between Aβ and hIAPP is due to the presence of hot regions that have a similar amino acid sequence. To identify this hot region membrane-bound peptide arrays of 10-amino acid residues covering full-length Aβ40 and positionally shifted by one residue were used.74 The results showed that self-association and cross-association of the two peptides depend exclusively on the polypeptide conformation. Using theoretical models, molecular dynamics simulations, and experimental measurements, it has been proposed that self-assembled polypeptides monomers having a backbone with substantial flexibility became unstable and evolved toward an alternating of partial ordered (β-sheet and α-helix) disorder (turn and random-coil) array. This information converges toward the concept of symmetry-breaking transitions. Symmetry-breaking transitions occurr when the energy difference between ordered and disordered structures became similar, and spatially modulate patterns spontaneously are formed75. This behavior was observed for a large number of intrinsically disordered proteins, including Aβ and hAPP mixtures, and is conformation-dependent and not dependent on the amino acid sequence. The results also qualitatively explain why amyloid aggregates escape a detailed structural characterization by X-ray analysis76. Surface Enhanced Raman Spectroscopy (SERS) imaging spectroscopic map of Aβ40 and hIAPP in equimolar mixtures have shown that the dimension of oligomeric aggregate ranges from 0.5 to 2 μm. The protein composition of oligomeric aggregate has a non-homogeneous distribution with an hIAPP rich core, an intermediate shell containing both peptides and a peripheral shell consisting of Aβ40. Also, the oligomer of pure Aβ40 and hIAPP was observed.77 Solid-state NMR experimental studies of fibril structures and molecular dynamic simulations of hybrid cross-seeding Aβ40–hIAPP assemblies with double-layer arrangements were also performed. These studies have explored the effects of interfacial variations in the U shaped β-sheet self-assembling of Aβ40 and hIAPP oligomers on the structural stability and interfacial interaction of Aβ40–hIAPP assemblies. All combinations N- and C-terminal residues of the two proteins were considered. Twelve polymorphic cross-seeding Aβ40–hIAPP assemblies were studied. Among investigated structures, only two configurations were identified by their high structural stability and favorable interfacial interaction, i.e., N-terminal of Aβ40-C-terminal of hIAPP and C-terminal of Aβ-N-terminal of hIAPP with a partially dehydrated interface. Besides, mutation studies also showed that salt bridges at interfaces reduced favorable interfacial interactions between Aβ40 and hIAPP78. Since Aβ42 is more toxic than Aβ40, another research group investigated the co-aggregation between the full-length Aβ42 oligomer and full-length hIAPP oligomers taking also into account that hIAPP has four variants79 that differ in the orientation of the residues along the β-strands and the turn region of the β-arch. The authors focused their attention on the parallel β-sheet structure of the Aβ42 oligomer interacting with four hIAPP variants. Simulations results indicate that all four variants of the full-length hIAPP oligomers preferred to interact with Aβ42 oligomers to form polymorphic single layer conformations. Yet, the interactions between the cross-seeding hIAPP and Aβ42 both in single and in double-layer conformations affect the structure differently. In particular, the main differences were found in the flexibility/rigidity of the turn region80. In the hIAPP-Aβ heterodimer formation, who between the two proteins is the driver? Molecular dynamics studies have proposed a plausible answer to this question. In the self-assembling process, strong inter-protein interaction is established. These forces induce a conformational transition from α-helix to β-sheet in the central amyloidogenic region of Aβ42. This transition contributes to lowering the aggregation free energy barrier. In other words, it was proposed that hIAPP induces Aβ42 aggregation and consequent conformational transition helix-sheet. The result of this conformational transition is the reduction of the free energy barrier, according to the symmetry-breaking theory81,82. The central role that hIAPP plays in hIAPP-Aβ cross-interaction has also been investigated by other authors using experimental methodologies. In particular, using a mixture of hIAPP and Aβ in the presence of anionic and raft model membranes, it has been observed that hIAPP drives the aggregation process. Furthermore, this was confirmed using hIAPP-GI (non-amyloidogenic mimic hIAPP) interacting with Aβ82. Other simulation studies confirm the central role of hIAPP in the interaction with Aβ in the presence of model membranes. Model membranes containing mixtures of zwitterionic and negatively charged lipids (POPC/POPG) have been used. It has been reported that the adsorption of Aβ-hIAPP cross-seeding on the membrane surface is strongly influenced by membrane electrostatic and the initial orientation of peptides. Aβ-hIAPP cross seeding interacts more effectively with the POPO/POPG bilayer rather than POPC. This is due to the strong electrostatic interaction between Aβ-hIAPP and POPG. Also, it adsorbs on both membranes on the N-terminal side of the Aβ on the surface of the bilayer83. The pathogenesis of both Alzheimer’s disease and T2D is also associated with inflammatory processes in the brain and pancreas, respectively. The cascade of inflammatory processes in related tissues is triggered by the internalization of Aβ or hIAPP oligomers into microglial cells or macrophages in T2D and thus their degradation via lysosomes. IAPP-GI, a non-amyloidogenic analog of hIAPP, has been shown to block the inflammatory processes mediated by Aβ or hIAPP. IAPP-GI forms highly soluble hetero-oligomers with both proteins, which are usually degraded84.
In adulthood, α-synuclein is expressed at high concentration in the brain, and only limited expression remains in peripheral tissues.85 α-synuclein has been ascribed to a variety of functions, and in beta cells α-synuclein binds to Kir6.2 on insulin secretory granules, and down-regulates insulin secretion.86 Less is known about the connection between hIAPP and α-synuclein and the development of Parkinson’s disease, but comparison of insulin and hIAPP secretion in patients with Parkinson and health controls revealed a significantly higher hIAPP:insulin ratio in the Parkinson group. Since insulin’s inhibitory effect on hIAPP aggregation is concentration dependent, this could give rise to a shift that facilitates hIAPP aggregation.87 It was recently reported that hIAPP could accelerate α-synuclein aggregation in vitro when mixed at a molar ratio of 1:10 hIAPP:α-synuclein88 indicating the underlying intermolecular interactions. In cynomolgus monkeys with spontaneously developed T2D, an increased accumulation of α-synuclein and phosphorylated α-synuclein appeared in the pancreatic β-cells.89 In the same study, increased accumulation of α-synuclein appeared in brain regions, including the cortical neurons and dopaminergic neurons in the substantia nigra. The degree of accumulation correlated with levels of fasting plasma glucose and circulation triglyceride. Much less is known about hIAPP and α-synuclein interactions and the development of Parkinson’s disease. Data collected hitherto suggest the co-presence of hIAPP and α-synuclein in the brain and the islets in patents with Parkinson’s disease. But it is unclear whether the in vivo deposits consist of peptides in amyloid form.
2.3. Effect of insulin on IAPP amyloid aggregation
Insulin is a polypeptide hormone containing 51 amino acids. Insulin has two chains: chain-A containing 21 amino acid and one intrachain disulfide bond (residue A6–A11), and chain-B containing 30 amino acids. Two disulfide bridges (residues A7 to B7, and A20 to B19) covalently tether the chains and the positions of these three disulfide bonds are invariant in mammalian forms of insulin90. Insulin and hIAPP are co-secreted by the β-cells in the Islets of Langerhans, consisting of hexamers (six insulin molecules grouped around two Zn2+ ions) as a result of interactions between hydrophobic surface. Insulin may also form dimers, but the active form is apparently a monomer. Insulin and hIAPP are stored into granule at 1–2:50 molar ratio91. It is known that in vitro aggregation of hIAPP disrupts the lipid membranes at μM concentrations68. hIAPP concentration in the β-cell granules is in mM without aggregation and no membrane damage has been observed in heathy individuals92. Thus, it is reasonable to think that the amyloid formation and membrane damage by hIAPP is inhibited by the elements contained in β-cell granules, such as low pH93, high Zn++ ions94,95, and insulin96. The concentration dependent effects of Zn2+ binding to hIAPP have been investigated. Zn2+ can suppress the fiber formation of hIAPP by forming an off-pathway complex97–99. A recent study reported that Zn2+ concentration determines the equilibrium of both hIAPP95 and insulin oligomers100 by forming both higher molecular-weight insulin oligomers and hIAPP homodimers. Therefore, zinc deficiency promotes the formation of zinc-free insulin monomers and dimers, which bind to hIAPP monomers versus zinc-related hexameters. The formation of the hetero-molecular complex prevents the self-assembling and thus subsequent aggregation of hIAPP. The role of negatively charged surface on hIAPP aggregation in the presence of insulin was also investigated. Using a negatively charged supported lipid bilayer, insulin has been shown to repress hIAPP self-assemby101,102, whereas a negatively charged Ta2O5 surface promotes co-assembled aggregation depending on the hIAPP/insulin molar ratio103. In aqueous solution, hIAPP has been shown to form first helical homo-dimers and then self-assemble into fibrils. On the other hand, the presence of insulin has been shown to inhibit fibril formation by hIAPP by forming heterodimers and the inhibition effect has been shown to increase with the increasing insulin concentration, suggesting that insulin is a competitive inhibitor of hIAPP5,104. Moreover, insulin B chain was found to be responsible for the heterodimer complex formation with residues 11 to 19 of chain-B of insulin and with residues 8 to 18 of insulin105,106. The effect of glycated insulin on hIAPP was explored107. Long time and high glucose concentration has been shown to cause about 10% of insulin glycation. An increased toxicity of hIAPP as monomer and dimer in the presence of glycated insulin has been observed. It is also remarkable that insulin inhibits hIAPP amyloid formation in the presence of lipid membrane.5 Interestingly, insulin does not prevent membrane pore formation by hIAPP. Additionally, fluorescence experiments demonstrated that insulin is capable of suppressing the fiber-dependent membrane disruption whereas the fiber-independent, pore-forming, mechanism is not altered by the presence of insulin.5 Therefore, insulin does not prevent the formation of small hIAPP oligomers on the membrane96. This means that to fully understand the co-existence of hIAPP-insulin and the implications in the pathology of the disease, other factors such as pH, ionic strength, lipid membrane composition and the role of free lipids should also be considered.
3. THE ISLET AMYLOID POLYPEPTIDE AND THE “AMYLOID HYPOTHESIS” OF T2D
3.1. Islet amyloid polypeptide and T2D
Occurrence of a hyaline alteration in islets of Langerhans constitutes one of the earliest described structural changes in diabetic pancreas.108 Its strong association with the form of diabetes later called T2D was found at an early date.109–111 The nature of the hyaline material was a matter of several studies and its resemblance to amyloid was noted109,112,113, but was denied by others. With the unified definition of amyloid on the molecular level, this discussion is now over.114
Islet amyloid is the most characteristic morphological islet feature of T2D, to some degree present in more than 90% of T2D pancreata.1,2 Its importance in the pathogenesis of β-cell failure is still not fully accepted and has been contradicted by the finding of similar alterations in non-diabetic subjects, albeit not to the same extent.115,116 With the finding that amyloid is a specific protein aggregation that result in characteristic β-sheet fibrils, it was natural to believe that insulin or proinsulin should be the origin of islet amyloid, particularly since insulin can be converted into fibrils.117 This assumption was further emphasized when electron microscopy investigations provided evidence for the production of islet amyloid fibrils from β-cells.118 However, it was only when the molecular composition of islet amyloid was directly elucidated by the amino acid sequence analysis that it was realized that the amyloid fibrils consist of a previously unknown β-cell product,119–121 today called islet amyloid polypeptide (IAPP) or amylin. More details regarding earlier studies of islet amyloid can be found in a recent review.122
3.2. IAPP aggregation in vitro and in vivo: pathways to β-cells toxicity
It has been well established that hIAPP is a highly aggregative polypeptide both in vitro and in vivo.123 Upon dissolving hIAPP in water or the dilution of concentrated hIAPP solution in organic solvents into aqueous buffer, there is a rapid and very efficient formation of amyloid fibrils as could be observed with various microscopy techniques including electron microscopy and atomic force microscopy. The fibrils that are formed possess classical amyloidal features including distinct β-sheet structure as observed by spectroscopy and strong typical birefringence upon staining with Congo red and examination using cross-polarised light.124 Furthermore, X-ray diffraction analysis of hIAPP fibrils show clear cross-β organization with a typical 4.7 Å meridional signal as observed with other amyloid fibrils.125 The formation of amyloid structures by hIAPP was not only observed in human patients but also in test tube, as well as animal models. When pancreatic samples from transgenic mice or islets transplanted into T1D patients were examined, there was a clear indication of the formation of IAPP deposits within the β-cells. Pancreas from patients with type 2 diabetes recovered during autopsy is autolytic, and the amyloid appears extracellular. The introduction of transgenic mice expressing human IAPP made it possible to recover islet material with preserved morphology. On sections from this material, it was possible to detect intracellular amyloid. The same is seen when normal human islets are transplanted into T1D patients.126 These deposits could be stained with the Congo red and show other features of amyloid fibrils. The β-cell mass loss in islets with hIAPP aggregates as observed in T2D suggested a role for IAPP aggregates in the progression of the multifactorial disorder as was previously suggested for neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease. From the mouse models, it was clear that IAPP amyloid appeared both intracellularly and extracellularly.127 hIAPP is synthesized as a proIAPP molecule and processed by the two prohormone convertases PC2 and PC1/3. This pH-dependent process takes place in the late Golgi and the secretory granules.128,129 Rising blood glucose levels result in a compensatory increase in hIAPP and insulin synthesis and secretion. During this stressed condition, there is an increased risk for aberrant pro-hormone processing, and the production of incomplete hormones, further increasing the demand on beta cells. TEM analysis of intracellular amyloid in mouse and human pancreas identified intragranular fibrils, and with specific antibodies, the presence of proIAPP in the amyloid was confirmed.130 Fibrils at this location suggest that pro-hormone processing may be affected and that proIAPP accumulates. Expression of proIAPP in the beta-cell containing PC2 and PC1/3, and where proIAPP is processed into IAPP, did not result in amyloid formation while the expression of proIAPP in GHC4 cells lacking both convertases resulted in amyloid composed of proIAPP.131 These results suggest that aberrant processing can result in the accumulation of proIAPP and proIAPP intermediates that are more amyloidogenic.131,132 During the passage, there is an opportunity for proIAPP to misfold, especially if the peptide is present at a high concentration. Isolated human islets can be used for transplantation and function as an enhance model system for islet amyloid. In human islets transplanted under the kidney capsule of nude mice islet amyloid developed rapidly and fibrillary aggregates were present intracellularly in compartments resembling organelles in the constitutive pathway (Figure1).133
Figure 1.

A) Human islet from a diabetic subject where most of the β-cells are replaced by amyloid. Congo red- Bar 50 μm. B) β-cell with intracellular amyloid-containing secretory granules. White arrows point to amyloid. C) Proposed sequence of events leading to islet amyloidosis: a) First, the processing of proIAPP is affected by factors such as high levels of NEFAs or glucose. Granules of amyloid-like fibrils fuse and form proIAPP amyloid deposits; b) Over time this aggregate enlarges and replaces most of the cell; c) the affected cell dies and the amyloid becomes extracellular and can act as a template for further amyloid formation; d) amyloid is now made up by IAPP secreted from neighboring β-cells. Formation of extracellular amyloid is preceded by the formation of cytotoxic intermediates, which can interact with cell membrane of surrounding cells and cause ion influx triggering apoptosis.130 This Figure is reproduced with copyright permission from Reference 130.
Despite the presence of hIAPP amyloid aggregates in vivo, there was a need to establish the mechanistic link between the fibrils and the observed toxicity. Indeed, the exposure of β-cells to hIAPP aggregates results in a clear cytotoxic effect as could be determined by cell viability assays such as 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. This assay is based on the reduction of yellow tetrazole to purple formazan in living cells that allows the quantitative assessment of level of viability using colorimetric tests. hIAPP aggregates, and to higher extent their early soluble oligomers, were found to be highly toxic to pancreatic cells in a similar manner as the toxicity of Alzheimer’s disease β-amyloid assemblies to various cells including neuronal ones. hIAPP samples were shown to interfere with the integrity of cellular membranes as well as model lipid membranes.134,135 One of the toxic species of hIAPP is reported to be the soluble oligomers which are transient in nature.136 This observation is similar perhaps to the studies of other amyloidal polypeptides and proteins including Alzheimer’s disease β-amyloid polypeptide and Parkinson’s disease α-synuclein protein. Also on the morphological level, the oligomeric assemblies that are formed by hIAPP, highly resembles those formed by Aβ-amyloid and α-synuclein when they are examined using electron microscopy.136
Membrane-bound biological macromolecules cover the cell surface, and their composition differs between cell types but include different compositions of cell surface proteoglycans. The association between amyloid and proteoglycans, particularly heparan sulfate proteoglycan (HSPG), is well known.137,138 When hIAPP amyloid deposits extracellularly, mainly β-cells are affected while α-cells appear to be less sensitive. hIAPP added to cultured islets, or β-cells interact with the cell surface causing invaginations of the cell membrane and pointing to direct interaction.139 The addition of heparin potentiates hIAPP aggregation in vitro, and a region at the N-terminal part of hIAPP has been identified as a binding site.140–142 Heparanase cleaves the carbohydrate chains of cell-surface HSPG present on beta cells.143 The culture of islets isolated from double transgenic mice overexpressing heparanase and hIAPP results in reduced islet amyloid deposits142 supporting cell-surface association driven by sulfated carbohydrates. IAPP interacts with the death receptors present on the β-cell surface.142,144 Through the intracellular domain, the procaspase 8 pathway is activated and, subsequently, the intrinsic pathway that results in procaspase 3 cleavage and induction of apoptosis.145
3.3. Molecular determinants of IAPP amyloid growth.
There are a number of physicochemical properties which govern the growth of hIAPP into amyloid fibrils. They can be divided into two groups: those intrinsic to the polypeptide chain and those of the environment in which the peptide self-assembly takes place, which are usually well-determined in in vitro assays and less well-determined in an in vivo context. Here we focus on the intrinsic properties of hIAPP. We first discuss the key regions of the IAPP sequence which promote the aggregation of the peptide, and then focus on how the amino acid sequence affects aggregation by collating the results from a large number of studies using sequence variants. Finally, we summarise the structural conformations of hIAPP and the possible intermediates that are on-pathway to amyloid fibril formation. We also highlight the need for new approaches and the integration of different techniques for the study of these complex amyloid assembly processes.
3.3.1. Aggregation-prone regions of hIAPP
The amyloidogenic behavior of proteins and peptides depends critically on the presence of stretches of contiguous amino acids in their sequences which have a tendency to aggregate (namely aggregation-prone regions, or APRs). These sequences can promote aggregation and the formation of the cross-β structures of amyloid fibrils.146,147 The sequence of IAPP is highly conserved across different species,148,149 yet crucial inter-species differences have been identified which play important roles in modulating the propensity of the peptide to aggregate. Figure 2 shows the amino acid sequences of IAPP from various species. Human, baboon and buffer fish IAPP have been reported to form amyloid fibrils.148,150,151 Praire vole, cat and porcine IAPP can form amyloid fibrils, but with lower aggregation propensity.148,152 Rat, mouse, bovine and dog IAPP do not from amyloid aggregates.148,153,154 Monkey, bear and horse IAPP have not been studied in vitro. The numbers at the bottom of each column in Figure 2 denote the degree of conservation according to the coloured scale under the alignment, determined by ConSurf.155 These alignments show how small sequence changes can have dramatic effects on aggregation. For example, hIAPP and rat-IAPP (rIAPP) have 83% sequence identity, but while hIAPP aggregates rapidly into amyloid fibrils in vitro, rIAPP does not form fibrils under the same experimental conditions.154,156 When comparing the sequences of these two peptidess, the variation is particularly obvious in the region spanning residue 20 to 29. Early work by Westermark and colleagues evaluated the aggregation capability of peptides containing this region and found that this segment can form amyloid fibrils in isolation in vitro.123 It was therefore suggested that residues 20–29 of hIAPP play a crucial role in its aggregation into amyloid and might be the key determinant which drives its amyloid growth.157
Figure 2. Amino acid sequence of IAPP from various species.
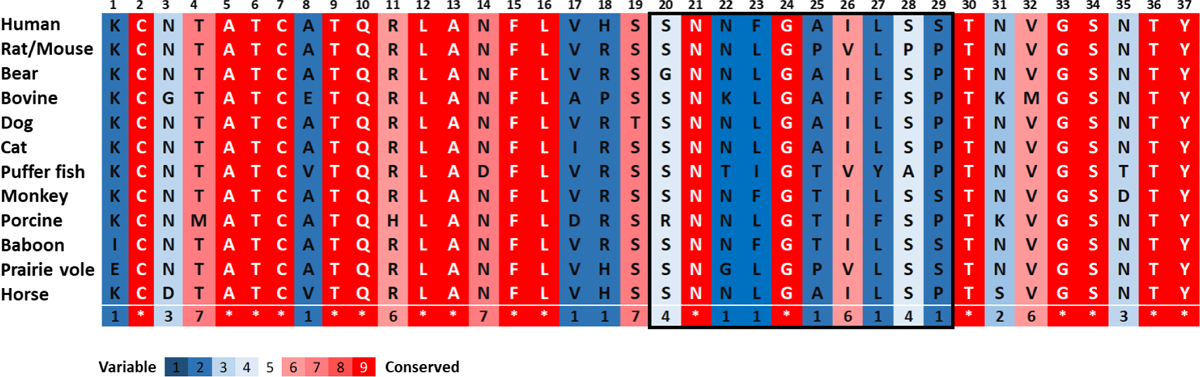
The amino acid sequence of IAPP is highly conserved with a disulfide bond between Cys-2 and Cys-7 residues and an amidated C-terminus. The peptide region spanning residues 20 to 29 shows the highest sequence variation among the species which is highlighted in the box.
However, later research showed that other peptide fragments from different regions of hIAPP can also self-assemble into amyloid fibril structures. These fragments of 5 to 30 residues in length covered peptides spanning residue 8 to the C-terminal residue of hIAPP.158–160 The work identified the shortest APR to be the pentapeptide FGAIL (residues 23–27) which in isolation was sufficient to form amyloid fibrils.158 Apart from this pentapeptide, Mazor and colleagues identified other two pentapeptides (FLVHS (residues 15–19) and NFLVH (residues 14–18)) which also formed amyloid fibrils in vitro.161 By developing a peptide array scanning approach, these authors also systematically mapped the regions of hIAPP which might contribute to the molecular self-recognition process during amyloid formation. Surprisingly, the fragment showing the highest binding affinity to full-length hIAPP was a ten-residue peptide spanning residues 11 to 20 (RLANFLVHSS), indicating that despite not being part of the major APR, this region of the polypeptide is important for inter-molecular interactions that could be relevant to amyloid formation. Therefore, the aggregation of this peptide hormone is not likely to be governed by a single molecular recognition motif in the polypeptide chain. Instead, its self-assembly process could be more complex and it is essential to consider this process in the context of the whole polypeptide chain, rather than as the behaviour of a series of short peptide fragments which are unlikely to mimic the behavior of the full-length polypeptide chain.
3.3.2. Residue-specific determinants of hIAPP aggregation
The intrinsic physicochemical properties of a polypeptide chain play a major role in determining the tendency of a protein/peptide to form amyloid fibrils, especially when aggregation is initiated from an unfolded or intrinsically disordered state.146,162,163 This is the case for hIAPP, for which aggregation commences from a disordered 37 amino acid peptide. The chemical properties of the amino acids, such as charge, hydrophobicity and aromaticity, are the key factors which affect aggregation property.146 Therefore, an understanding of the relationship between amino acid composition and amyloidogenicity is essential to define the molecular determinants of hIAPP amyloid growth. Studies of hIAPP sequence variants of both full-length hIAPP and its truncated variant, IAPP8–37, have been used to determine the role of different amino acids in hIAPP amyloidogenicity (Figure 3). Because the N-terminal region of the hIAPP sequence (residue 1 to 13) does not aggregate on in isolation164 and is hypothesised to be outside the fibril core165 (as confirmed by three recent cryo-EM structures of hIAPP amyloid formed in vitro166–168), most studies have focused on the central to C-terminal region of the sequence, which is known to contain IAPP’s APRs.165,169 Interestingly, the only known natural variation of the hIAPP sequence is a serine to glycine substitution at position 20.170 This residue is located in the second APR (residues 20–29), dramatically enhances the rate of hIAPP aggregation in vitro, and it has been linked to an early-onset form of T2D.171,172 However, the mechanism by which this single point mutation accelerates aggregation and disease onset remains unknown.
Figure 3. Studies of sequence alterations on the aggregation of full-length hIAPP and truncated IAPP8–37.
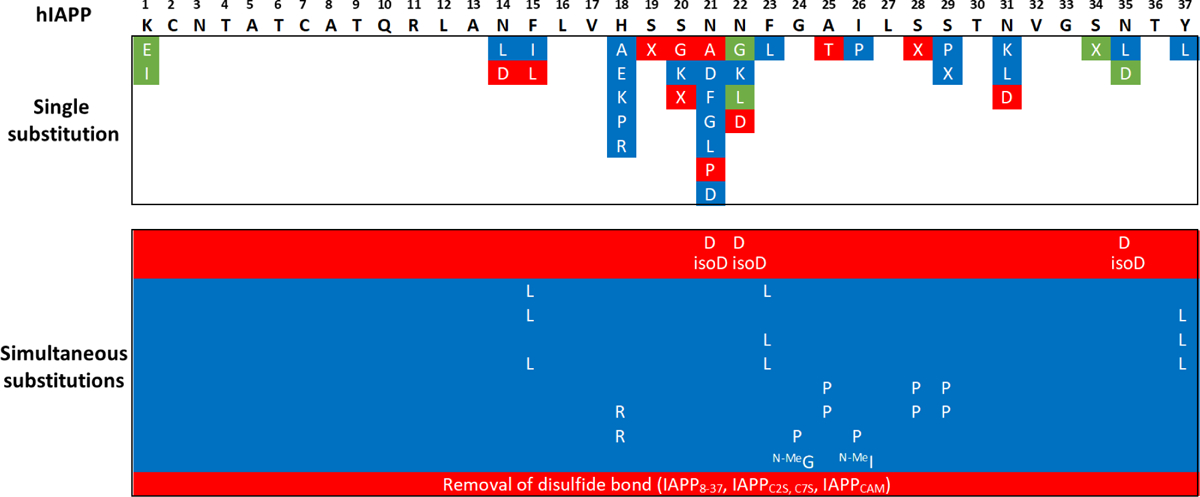
Amino acid substitutions in red show higher aggregation propensity as compared to wild-type hIAPP, with a shorter lag-time; substitutions in blue show a lower aggregation propensity with an extended lag-time or no aggregation; substitutions in green did not change the aggregation behavior as compared to that of the wild-type hIAPP. X is 2-aminobutyric acid (2-Abu). Cam is carboxyamidomethyl protecting group. N-MeG and N-MeI are N-methylation of the peptide bonds at Gly-24 and Ile-26. isoD is isoAsp.
There are three positively charged residues (Lys-1, Arg-11 and His-18) in the sequence of hIAPP, leading to a positively charged peptide at physiological pH (theoretical pI = 8.90). On the one hand, these positive charges contribute significantly to the interactions with negatively charged membranes, which are relevant for membrane-catalysed fibril formation as well as for peptide-induced cell toxicity.173–175 On the another hand, it is well established that an increase of the overall net charge of unfold proteins or polypeptides can decrease their aggregation propensity due to increased inter-molecular electrostatic repulsion.146 To determine whether positively charged Lys-1 affects the aggregation of hIAPP, as well as the importance of electrostatic repulsion in modulating peptide self-assembly, three variants have been studied (ΔLys-1,176 and the amino acid substitutions, K1E152 and K1I150). The aggregation kinetics of the three variants is similar to those of wild-type hIAPP in PBS buffer, suggesting that modification of Lys-1 has no dramatic influence on amyloid formation and is unlikely to be a key determinant of self-association under the reported experimental conditions. Interestingly, all these variants are less toxic to INS-1 β-cells, which suggests that the N-terminal residue is involved in modulating toxicity.150,152,176
Another amino acid which has been extensively studied is His-18. The protonation state of histidine is dependent on the pH of the buffer and it is speculated that the ionisation state of this residue plays a key role in the fibrillation of hIAPP.177 The importance of His-18 is also suggested by this residue being one of the six amino acids that differ between hIAPP and the non-amyloidogenic rIAPP which contains an arginine in this position (Figure 2). By generating a “humanizing” R18H amino acid substitution in rIAPP, Green and co-workers demonstrated that this variant containing only one amino acid substitution can form amyloid fibrils, highlighting the importance of His-18 in modulating hIAPP fibril formation.178 A follow-up study in which His-18 in hIAPP was replaced with four different amino acids that varied in polarity, charge and size showed that all the sequence variants could form fibril aggregates, but are less amyloidogenic with an extended lag time than the wild-type sequence, indicating that the size, charge and hydrophobicity of this residue position are important for inter- and/or intra-molecular interactions which are relevant to hIAPP amyloid growth.171,172
Another crucial chemical feature of amino acids is their aromaticity. There are three aromatic residues in the hIAPP sequence (Phe-15, Phe-23 and Tyr-37) (Figure 2 and 3). The role of these amino acids in facilitating hIAPP amyloid growth has been extensively explored,159,179–182 but their contributions to peptide self-assembly are still under debate. Early studies focused on fibrillogenic segments which contain Phe-15 or Phe-23, i.e. segments 12–18 (LANFLVH),159 20–29 (SNNFGAILSS)123,179 and 22–27 (NFGAIL)180. To dissect the effect of Phe-23 on fibril growth (relative to other amino acid in this region), an alanine scan was performed on the hexapeptide NFGAIL.180 The substitution of this phenylalanine in this peptide sequence to alanine abolished amyloid formation, demonstrating that aromatic interactions can serve as a key stabilising element in fibril growth, possibly through π-π interactions.183 Both the chemical and structural properties of alanine cannot fulfil this role, as alanine is not aromatic, is less hydrophobic and has lower β-sheet propensity.182 Amino acid substitutions of Phe-15 and Phe-23 were also made on peptides LANFLVH and SNNFGAILSS, respectively, and their aggregation propensity was determined by various biophysical techniques.159,179 Surprisingly, both studies showed that an aromatic residue at either of these two positions is not required for fibril formation, demonstrating that the requirement of an aromatic residue for aggregation is exquisitely sensitive to the precise sequence of peptide, including its length and hence the flanking residues surrounding the Phe in question. Nonetheless, the different substitutions of Phe-15 or Phe-23 did affect the rate of aggregation, again highlighting how a single amino acid substitution can alter the aggregation properties of the peptide sequence.
To further explore the effect of aromatic amino acids on hIAPP aggregation, substitution of Phe-15 and Phe-23 were also made in the context of the full-length peptide, the results showing that a Phe at these positions is not required for amyloid fibril formation.181,182,184 Similar to the peptide segments described above, replacement of these aromatic residues has an impact on the aggregation kinetics, leading to the suggestion that these residues are involved in forming early inter-molecular interactions at the onset of the aggregation pathway.181 In particular, the formation of hIAPP amyloid fibrils has been suggested to require long-range interactions between the C-terminal Tyr-37, with Phe-15 and Phe-23.165,184,185 A more comprehensive study on the role Phe-15 in hIAPP aggregation demonstrated the relationship between the local secondary structure of the peptide and its aggregation propensity.181 Specifically, amino acid substitutions in this position introducing residues with a high α-helical propensity and low β-sheet propensity resulted in peptides which formed fibrils more rapidly with shortened lag time, as monitored by thioflavin T (ThT) fluorescence assay.181 This observation supports a helix-mediated association model for hIAPP, in which the initial oligomerisation process of hIAPP involves the acquisition of an α-helix conformation which results in high local concentration of the aggregation-prone sequences and promotes amyloid formation.186
Although the studies described above led to the conclusion that aromaticity is not required for hIAPP amyloid growth, the variation in aggregation kinetics among the different peptide variants studied may result from the change in both hydrophobicity and β-sheet propensity of the corresponding peptide sequences. Indeed, several groups have demonstrated that the aggregation propensity of proteins or peptides is strongly correlated with hydrophobicity and β-sheet propensity.146,163,187 More importantly, these two properties of amino acids have been widely used as key predictors of amyloid propensity in computational studies.188,189 Nilsson et al. explored the relationship between hydrophobicity and self-assembly by using the hIAPP fragment (20SNNFGAILSS29).179 To cover a broad range of hydrophobicity values both natural and non-natural amino acids were introduced, of which cyclohexylalanine (Cha), α-naphthylalanine (1-Nap), and β-naphthylalanine (2-Nap) with hydrophobic values of 2.72, 3.08 and 3.12, respectively, are more hydrophobic than phenylalanine (1.79). These authors found that all sequence variants could form amyloid fibrils, with variants containing 1-Nap or 2-Nap exhibiting more rapid aggregation kinetics and more favourable thermodynamics of amyloid formation than wild-type peptide fragment. The propensity to form a β-sheet has also been suggested as another crucial molecular determinant of amyloid fibril formation. Replacement of amino acids in the β-sheet core regions of fibrils with β-sheet breaker amino acids (such as proline) can attenuate or abolish the aggregation of hIAPP, as exemplified by several single residue variants (Figure 3).172,190,191 In another study, N-methylation of the main-chain was exploited to inhibit peptide self-assembly by disfavouring β-sheet formation,192 with a double N-methyl modification in hIAPP at positions 24 (Gly) and 26 (Ile) (Figure 3) arresting fibril formation for up to 2 weeks, while the wild-type hIAPP sequence forms fibrils within 2.5 h.193 Moreover, hIAPP containing this modification is also able to inhibit aggregation of wild-type hIAPP into β-sheet rich structures by trapping the wild-type sequence in non-amyloidogenic conformations.193 Together, these studies shed light on the key roles of both hydrophobicity and β-sheet propensity in hIAPP aggregation.
The sequence of hIAPP contains a high percentage of asparagine residues (6 of its 37 amino acids) (Figure 3). There are several reasons for the study of asparagine residues in the context of amyloid formation. First, several of the asparagine residues in hIAPP have been proposed to contribute key interactions in oligomer formation.152,194,195 Second, deamidation of asparagine (forming aspartic acid) can occur spontaneously without enzymatic catalysis and can affect the aggregation of polypeptides, including hIAPP.196 Several studies have shown that deamidation of Asn residues can change the fibril formation of hIAPP.194,197–199 For example, O’Connor and colleagues used mass spectrometry to determine the sites of deamidation of hIAPP in both soluble species and insoluble fibrils.197 To determine the effect of deamidation on fibril growth, variants containing aspartic acid or iso-aspartic acid at the deamidation sites (Asn-21, Asn-22 and Asn-35) were synthesised and their aggregation behaviour was assessed using ThT fluorescence. The three variants formed fibrils with enhanced kinetics, suggesting that deamidation can indeed accelerate the aggregation of hIAPP, possible by enhancing favourable electrostatic interactions with positively charged residues.
Miranker and co-workers followed a different approach to evaluate the role of asparagine in hIAPP aggregation. They systematically substituted five of its six asparagine residues (omitting only Asn-3 for their study) to leucine in the truncated IAPP8–37 sequence.200 The substitution of Asn with Leu maintains a similar steric volume yet removes the possibility of hydrogen bond formation by the Asn amide side chain. Surprisingly, apart from N22L which had little effect on aggregation, the other four single Asn variants were found to behave slightly differently in terms of their aggregation behavior when compared with wild-type hIAPP: N31L and N35L slowed down aggregation, while N14L and N21L inhibited amyloid formation, highlighting the importance of inter- and/or intra-molecular interactions (e.g. hydrogen bond formation) by these residues in fibril growth and/or fibril stability. Other researchers have also studied the role of Asn-21 on hIAPP self-association.195 Strikingly, introduction of a proline residue at this position was found to accelerate the aggregation kinetics, which is contradictory to the expectation that proline substitutions usually inhibit the fibrillation process since Pro is incompatible with secondary structure formation. The authors speculated that a turn-inducing residue such as proline could constrain the peptide into conformations that facilitate self-assembly. In a different study Raleigh and co-workers evaluated the effect of Asn-22 on hIAPP amyloid formation and cytotoxicity, and showed that replacement of Asn-22 with Gly has only a modest effect on the rate of amyloid fibril formation.152 This observation was also reproduced in the context of the truncated variant IAPP8–37.200 Although there is no obvious difference in the rate of amyloid formation, the N22G substitution showed reduced toxicity towards β-cells, which was interpreted by the oligomeric intermediates of assembly being drastically different for the two peptides because of the amino acid substitution. In summary, results from these studies using both truncated and full-length hIAPP show that its asparagine residues, especially Asn-21, are involved in specific interactions such as hydrogen bond formation that mediate intra- and inter-molecular interactions and ultimately determine the conversion from soluble peptide into insoluble amyloid fibrils.
3.3.3. The presence of a disulfide bond affects hIAPP aggregation
IAPP from all species contains a conserved disulfide bond between Cys-2 and Cys-7 which is outside the fibril core structure (Figure 2).165 Although the self-assembly of proteins into amyloid fibrils usually depends critically on the sequence of the amyloid core regions or APRs, regions outside the core can also contribute significantly to the amyloidogenicity of proteins or peptides.201 The single conserved disulfide bond in hIAPP forms a six-residue loop linking Cys-2 and Cys-7 which could constrain the conformation of the peptide and prohibit the formation of a β-strand structure in this region. To evaluate how this intra-molecular disulfide bridge affects the aggregation of hIAPP, different variants have been generated and studied, including the well-characterised IAPP8–37 which lacks these two Cys residues, and also a reduced and carboxyamidomethyl (CAM) blocked full-length peptide (hIAPPCAM). The fibril growth kinetics of hIAPP variants lacking the disulphide bridge showed that the presence of the disulphide is not required for amyloid formation.202 Nonetheless, these variants were shown to aggregate more rapidly than the wild-type sequence, demonstrating that this peripheral region can influence the self-assembly kinetics of hIAPP, specifically by shortening the lag phase. Later on another study further discussed the role of this conserved disulfide bond.203 More variants were created, with all variants which lacked the disulfide bond-containing region resulting in enhanced amyloidogenicity, indicating that the disulfide bond is able to protect the peptide from aggregation into amyloid fibrils. This protective role of the disulfide bond was also observed in a NMR study conducted with both reduced and oxidised forms of hIAPP.204 The detailed structural characterisation revealed that the conserved disulfide bond stabilises the N-terminal region of hIAPP in an α-helical conformation, and hypothesized that this more structured monomeric state slows down self-association into amyloid. Interestingly, stabilising these pre-amyloid α-helical intermediates with α-helical mimics (foldamer) also inhibits the aggregation of hIAPP, further highlighting the finding that maintaining the α-helical conformation of the N-terminal domain of hIAPP can prevent its amyloidogenic aggregation.205,206 Despite the aggregation kinetics being highly dependent on the experimental conditions, such as buffer, pH and co-solvents among others, several groups using different experimental conditions have observed the same protective effect of the internal disulfide bond in disfavouring amyloid formation. Thus, despite being outside of the fibril core, this flanking region containing the disulfide bond protects against hIAPP aggregation into amyloid fibrils.
3.3.4. Conformational states of monomeric hIAPP and oligomeric structures related to hIAPP aggregation.
The self-assembly of hIAPP into amyloid fibrils occurs via classical nucleation-growth mechanism which shows sigmoidal aggregation kinetics characterised by a lag phase, growth phase and plateau phase.207 Primary nucleation occurs during the lag phase in which the soluble hIAPP monomers assemble into oligomers, followed by the generation of assembly nuclei. These nuclei can then further develop and elongate to form protofibrils and fibrils in the growth phase. Newly formed fibrils can accumulate and further catalyse the formation of new fibrils via secondary nucleation on the fibril surface and by fragmentation which generates more fibril ends. Eventually a steady-state phase is achieved wherein soluble peptide is in equilibrium with the fibrils.207 These highly dynamic aggregation processes include the formation of potentially a large number of different, transient oligomeric intermediates with different conformations, stability and potentially different roles in cytotoxicity and the fibril formation process. Owning to the complex nature of amyloid self-assembly processes, it remains a challenge to acquire detailed structural information of these transient and heterogeneous intermediates, although exciting recent progress has been made for some amyloidogenic systems.208–210
Recently, different techniques including solid-state and solution NMR,209 2D infrared spectroscopy (2D IR),211 native mass spectrometry (MS) coupled with ion mobility sepctrometry,212 hydrogen deuterium exchange (HDX) MS and fast photochemical oxidation of proteins (FPOP) MS213 have provided exciting possibilities to study oligomers and their structures, and to further understand their roles in amyloid fibril growth. Indeed, Buchanan and colleagues used 2D-IR spectroscopy and specific isotope (13C) labelling to characterise oligomers formed during the lag phase of hIAPP assembly. These experiments showed that structured prefibrillar oligomers of hIAPP contain a parallel β-sheet structure that includes residues 23 to 27 (FGAIL), and this region has long been identified as an APR.211 Detailed mutational studies and molecular dynamics simulations also indicated that this oligomeric state is likely to be on-pathway to fibril formation. Interestingly, this β-sheet structure is thought to be disrupted during aggregation, as eventually this sequence is thought to adopt a loop conformation in the final fibril structure.165 A different study used double isotopic labelling of Leu-12 and Ala-13 to monitor changes in the dihedral angle of these residues during aggregation, and demonstrated that this region displays helical features when hIAPP is in its monomeric state.214 It was hypothesized that these α-helices could seed hIAPP oligomer formation, consistent with the results using amino acid substitutions described above, but also able to stabilise small β-sheet oligomers reported previously.214
Most conventional biophysical approaches used for the study of amyloid growth are restricted in that they can only provide information that corresponds to a global, average structural property of the different species that co-exist during aggregation. Developments in MS techniques, however, have enabled the native structures of biological molecules to be maintained in the gas phase, allowing different oligomers formed during aggregation into amyloid to be individually identified and characterised without requiring prior separation (e.g. for β2-microglobulin,215 hIAPP and rIAPP,156 Aβ40216). When paralleled with ion mobility spectrometry (IM), nESI-IM-MS allows complex mixtures transiently populated oligomers to be separated and each species individually characterised based on their mass, shape, charge and size, and also allows the discrimination of species with the same mass-to-charge ratio.217,218 Using ESI-IM-MS two distinct structural families of hIAPP monomers were observed and these two conformations were identified as β-hairpin and helix-coil structural super families by replica exchange molecular dynamics (REMD) simulations.219 By comparing these species with those observed in the non-amyloidogenic rIAPP, the β-hairpin conformation was proposed to be the amyloidogenic precursor of hIAPP that may directly assemble into amyloid fibrils.
3.4. The role of metal ions: Ca++, Mg++, Zn++, Cu++
IAPP is co-secreted together with insulin and carried to the extracellular space in lipid-bearing vesicles through an endocytosis/exocytosis mechanism. The phospholipid composition of lipid vesicles is asymmetric and similar to the islet cell membrane: i.e. they contain negatively charged phospholipids in their inner lumen. An abnormal peptide/membrane interaction may negatively influence the availability of hIAPP to extracellular compartments. It is important to understand the role of adverse environmental factors such as the dyshomeostasis of some metal ions in influencing the properties of hIAPP and its interactions with lipid bilayers, which could provide further insights into the molecular mechanisms of T2D. It has been observed that a genetic disease known as hypercalcemia linked to the reduction in the expression of the calcium receptor gene is implicated in the development of diabetes in people with this genetic disease. This observation supports the idea that calcium dyshomeostasis may be linked to the development of diabetes220. From this observation, it has been suggested that high concentrations of calcium in the plasma may activate mechanisms involving hIAPP in the process of membrane destruction. Biophysical investigations have demonstrated that Ca++ and not Mg++ act as a switch in the mechanism of membrane damage induced by hIAPP on negatively charged (POPC:POPS 3:1) membranes, i.e., these metal ions repress ion-like pore formation and promote the detergent-like mechanism221. No influence of Ca++ or Mg++ was observed in the interaction of rIAPP and these membranes. It was also observed that hIAPP in the presence of Ca++ may penetrate into the hydrophobic core of the membrane, assuming a β-sheet conformation.222 The same behaviour was reported for Aβ40 interacting with model membranes. Other metal ions, such as zinc, copper, and iron, have also been associated with diabetes.223,224 Interestingly, the same behavior observed on hIAPP was found in the interaction of Aβ40 and model membrane,225 suggesting a common molecular mechanism in damage of model membranes. hIAPP and Zn++ play an essential role in regulating glucose concentration. Zn++ is stored in islet granules, where its concentration is of the order of mM.99 Notably Zn++ deficiency has been observed in patients with diabetes. NMR and AFM studies have indicated that Zn++ binds to prefibrillar oligomeric species of hIAPP during the lag-phase and promotes fibril formation by coordinating to His-18.95 Interestingly a high-resolution NMR structure revealed an off-pathway zinc-bound complex in which Zn++ is coordinated to His-18.97 Also, the interaction of hIAPP with Zn++ is strongly dependent on the concentration of the metal ion: during the lag-phase (tipically 40 min.) high concentrations of Zn++ (10 mM) favors the formation of large Zn++-hIAPP aggregates over the control sample. At a lower concentration (100 μM Zn++), the size of Zn++-hIAPP aggregates is even larger than those observed at a high concentration of Zn++. These studies also showed that Cu++ ions behave differently to Zn++ ions. Inthe presence of Cu++ ions, no hIAPP dimers are formed, and Cu++ inhibits the formation of fibrils94,226,227. Moreover, studies on hIAPP fragments have shown that the Cu++ ion prefers to follow interatct with region 18HSSNN22 as Zn++ ions. While the presence of His-18 is necessary for the formation of the hIAPP-Zn++ complex, Cu++ binds to hIAPP even in the absence of histidine. On the contrary, other studies have shown that Cu++ ions bind to His-18 and Ser-19 and Ser-20 residues of hIAPP228.
3.5. The role of membrane composition, raft, and cholesterol
The cell membrane is a very complex entity and plays the role of separating the highly ordered space inside the cell from the chaotic space outside the cell. It consists of phospholipids and cholesterol organized in a bilayer where proteins are also present. The phospholipid composition of the bilayer is asymmetric, containing zwitterionic and negatively charged229,230 phospholipids. Negatively charged phospholipids are mainly present on the inner side of the cell membrane. The bilayer contains 30 to 50 % cholesterol231 which has the role of making the membrane fluid and the order-disorder state of the bilayer,232 increase the bilayer mechanically rigid,233 regulate the mechanism controlling membrane phospholipid asymmetry234 and contributing to the integrity of the raft domains.235 Polymorphism of model membranes containing single-component phospholipids having saturated acyl chains (di-acyl-phosphatidylcholine) change with temperature: at low-temperature acyl chains are strongly packed in a state known as the gel-phase (Lβʹ), with increasing temperature first the ripple phase (Pβʹ) occurs and then the so-called liquid crystalline lamellar phase (Lα). Two micro-phases can be detected in a ternary system containing lipids with a high phase transition temperature, lipids with low phase transition temperature, and cholesterol in the liquid crystalline phase. These phases are called the liquid-ordered (lo) and liquid-disordered (ld) phases in reference to the lipid acyl chains order within the micro-each phase.236–240,240,241 These micro-domains are called lipid rafts. To explain the interaction between cholesterol and phospholipid acyl chains, the “push and pull” mechanism was proposed, where unsaturated acyl chains push cholesterol and the saturated acyl chains pull cholesterol, and the interactions that push cholesterol away are driven by enthalpy,242,243 It is known that cholesterol regulates glucose metabolism in adipocytes altering raft membrane properties244 and by activating several transcription factors.245 There is evidence that cholesterol is implicated in lipotoxicity of the β-cell dysfunction in T2D246. A recent study showed that an elevation in islet cholesterol promotes IAPP aggregation and islet amyloid formation in mice.247 The same results were obtained using a model membrane containing different amounts of cholesterol. In particular, in a model membrane containing lipid raft (1:2 mole ratio of POPC:DPPC), increasing cholesterol concentration enhanced ion-channel formation and repressed the “detergent-like” mechanism of membrane disruption. Conversely, increasing cholesterol in the non-raft-containing (7:3 mole ratio of POPC:POPS) membrane, ion-channel formation was repressed and ‘detergent-like’ mechanism was enhanced. Also, increasing cholesterol concentration in the raft increases the micro-domain in the membrane and decreases their size. Besides, hIAPP-penetrating model membranes (7:3 POPC:POPS) containing cholesterol is driven by three effects: the electrostatic interaction between membrane and hIAPP, the match between the hydrophobic domain of hIAPP and membrane thickness, and the stiffness effect that cholesterol induces on the bilayer. Conversely, in model membranes containing lipid raft, the small domains formed by DOPC lipids that have a small thickness (ld phase) suggests that they favor the formation of pores, while the large domains of DPPC (lo phase)248 that have a large thickness disfavor the interaction with hIAPP and the formation of aggregates249. The data on raft and non-raft model membranes agree with push and pull mechanism. It is interesting to note that recently reported fluorescence anisotropy studies on model membranes containing POPC, POPS, and several sterols have shown the close correlation between the packing of sterols in the bilayer matrix and the formation of fibrils and pores. Sterols that pack efficiently in the membrane slow down the formation of fibrils and decrease the formation of ion-channel like pores250. Other studies on pores and fibrils formation of hIAPP in the presence of DOPC, POPC, and sphingomyelin and mixture of negatively charged phospholipids (POPS) and cholesterol have been reported251–255 showing that zwitterionic phospholipids do not affect amyloid formation, while they affect pore formation (dye-leakage), with a downward trend in the following order DOPC>POPC>sfingomyelin. The presence of 40% or 20% cholesterol inhibits the formation of fibrils and pores. Also, the authors emphasize that there is an inverse correlation between the rate of fibril formation and dye loss from vesicles256. Notably, Aβ40 interacting with a model membrane containing cholesterol shows a similar behavior. Other authors link the distribution of cholesterol in the membrane with topological features of transmembrane Aβ oligomers and membrane permeability and deformability257,258. Asymmetric distribution of cholesterol into the membrane favors the formation of channel-like Aβ assemblies permeable to calcium ions, (e.g., in response to synaptic stimulation) would promote membrane vesiculation and Aβ endosomal re-uptake. The symmetric distribution of cholesterol and increased membrane rigidity associated with aging would promote instead, an increased Aβ aggregation into linear assemblies, triggering, in turn, their dismissal from the native membrane259.
3.6. Intracellular pathological event of oxidative stress and inflammation
One of the observed effects of hIAPP challenge to cells is the formation of reactive oxygen species (ROS) as noted also with other amyloid forming proteins and polypeptides. It was shown that the process of amyloid formation, but also its disassembly is associated with the production of ROS in a process that is methionine-independent.260 It may well be that beyond its multiple effects, including those on membrane integrity, the formation of hIAPP fibrils and oligomers results in an extensive oxidative stress on the pancreatic tissue. It is further demonstrated that the binding of copper ions to hIAPP generate dityrosine cross-links via ROS formation.261 The formation of ROS and their effect on the tissues may partially account for the loss in β-cell mass that is clear outcome in the advanced stages of T2D. Indeed, the modification of various cellular components by ROS serves as biomarkers for T2D and pre-diabetic states.262,263 The ability of various aromatic polyphenols to act both as potent antioxidant and inhibitors against the formation of amyloidal toxic assemblies, as will be discussed below, is an important feature of the potential treatment of the multifactorial T2D.
Another well-established response to the presence of amyloids and other crystalline materials is a significant inflammation.264 A major mechanism that was observed is the activation of the cytosolic receptor complex called the NACHT, LRR, and PYD domains–containing protein 3 (NLRP3) inflammasome. This results in a systemic response that could have additional effects on various tissues and organs. In the case of hIAPP, it has revealed that the protein can induce the expression of interleukin-1β which is the main cytokine involved in inflammation and T2D causing inflammation.265 The inflammation process appears to be an important part of various degenerative processes as well as other pathophysiological states.
3.7. Increased expression of RAGE and β-cells toxicity
Hyperglycemia is a central feature of diabetes. Therefore, it is reasonable to hypothesize that the known effects of hyperglycemia on endothelial malfunction and immune cells failure may be rooted in adverse biochemical mechanisms due to excessive blood glucose levels. High glucose blood levels promote the irreversible non-enzymatic glycation of lipids and proteins thus forming advanced glycation end products (AGEs).266 AGEs constitute a highly heterogeneous class of post-translationally modified species, including several adducts such as carboxy ethyl lysine (CEL), carboxy methyl lysine (CML), pyralline, and pentosidine to mention a few.267 An unbalanced accumulation of AGEs in tissues and their interaction with their principal cellular receptor (RAGE) plays a key role in endothelial and immune cells damage.
RAGE is a 35-kDa membrane protein which belongs to the superfamily of immunoglobulins.268 It is highly homologous to other neuronal cell adhesion proteins as axonin and NCAM.269,270 The structure of RAGE consists of five different domains: i) an extracellular N-terminal domain, which is needed for cell signaling, consists of a N-terminal V-type and two C-type Ig regions,271 ii) two separated constant domains named C1 and C2, iii) one transmembrane domain anchoring the protein to the membrane, iv) an intracellular C-terminal domain required for signal transduction.272,273 The N-terminal V domain of RAGE is the primary site involved in ligand binding. Albeit the mechanisms by which RAGE binds its ligands are mostly unknown, the cytoplasmic domains of the receptor are homo-dimerized by bridging several charged residues upon ligand binding. These movements activate downstream signaling that may induce cell proliferation/migration and inflammation274,275, which in turn are linked to diverse diseases including neurodegeneration,276 cancer,272 and diabetes273. Downstream signalling and the pathogenic pathways activated depend on the type and levels of ligands present in different cells. Hence, a detailed understanding of RAGE interactions with diverse range of ligands and the related signal pathways is essential for the development of effective treatments of RAGE-related diseases.277 Indeed, beyond the AGEs, RAGE is also known to be a receptor for several members of high mobility group box 1 (HMGB1), the S100/calgranulin class,278 lysophosphatidic acid (LPA), Mac-1 and phosphatidylserine266 amyloid-beta (Aβ) peptide and β-sheet fibrils.279 Therefore, both glucose- and lipid-modified species may bind RAGE and the diverse groups of RAGE ligands suggest that significant links do exist between RAGE activity and an altered metabolic state.280 Notably, inscreased RAGE expression is observed in those cells where high levels of its ligands are expressed such as endothelial cells, neurons and immune cells (i.e. lymphocytes and monocytes/macrophages).281 The observed co-localization of RAGE with many of its ligands suggests that additional RAGE-ligands/target cells may exist in diabetes. Based on all these findings, some authors have hypothesized that RAGE may play a significant role in the development of hIAPP mediated β-cells toxicity: indeed, RAGE is known to activate pathological pathways (such as inflammation, oxidative stress, and apoptosis) which results upregulated in hIAPP-induced toxicity.282 In turn, RAGE is upregulated by an abnormal accumulation of ligands and cannn activate pathogenic intracellular signalling which leads to β cell death.283,284 A recent report has demonstrated that toxic prefibrillar forms of exogenous hIAPP bind and upregulate RAGE in INS-1 β-cells.285 Interestingly, high levels of glucose induced a similar overexpression of RAGE in INS-1 β-cells which anticipated the effects of glucotoxicity and supported a role of RAGE in hIAPP-induced glucotoxic stress in β-cells. Moreover, the extracellular, soluble ligand-binding domain of RAGE (sRAGE) was shown to bind toxic prefibrillar forms of hIAPP, but not rIAPP, mature fibrils or non-toxic hIAPP aggregates. Consistent with its specific binding to toxic hIAPP oligomers, sRAGE was able to halt the formation of β-sheet-rich structures and amyloid growth when added to hIAPP solutions before the occurrence of the lag phase. Given that sRAGE has the same ligand-binding motifs as its membrane-bound cognate, it is likely to compete with RAGE for binding to toxic hIAPP species. Consistent with this hypothesis, toxic hIAPP oligomers considerably upregulated the RAGE encoding genes; however, a treatment with sRAGE during incubation with toxic hIAPP oligomers suppressed the expression of proinflammatory genes. Remarkably, neither hIAPP oligomers nor sRAGE or rIAPP alone were able to upregulate these inflammation mediators. The role of RAGE in mediating hIAPP-induced toxicity in β-cells was also confirmed through in vivo studies using hemyzigous hemi_h-IAPP mice expressing RAGE and hIAPP.286,287 These transgenic mice are known to develop islet amyloidosis and β-cells failure but not T2D. For this reason, this prediabetic in vivo model is used to disentangle the effects of RAGE in hIAPP-induced β-cell toxicity from other stressful factors including the glucose-activated formation of other RAGE ligands such as AGEs.287 In particular, pancreatic β-cells of hemi_h-IAPP mice exhibited an overexpression of inflammatory genes which was effectively reversed by treatment with sRAGE; next, sRAGE was also observed to reduce hIAPP deposition and β-cells apoptosis in hemi_h-IAPP mice. The importance of these findings to the development of T2D was also probed in human pancreas samples. In particular, hIAPP amyloid accumulation was paralleled by a significant increase in RAGE expression in human diabetic β-cells when compared to controls. In addition, RAGE colocalizes with insulin and hIAPP-rich areas in human diabetic β-cells. Therefore, mature amyloid hIAPP fibers and non toxic hIAPP aggregates do not bind RAGE. Blockade of RAGE i) reduces hIAPP-induced inflammation and apoptosis in β-cells and ii) ameliorates glucose intolerance in diabetes. sRAGE was also found to conteract toxic hIAPP amyloid aggregation in vivo (Figure 4).285 All these findings suggest that RAGE is a valuable target for the development of therapeutics for protecting β-cells in diabetes and metabolic disorders, and may also inspire further research focusing on the design of targeting RAGE/hIAPP interactions.
Figure 4.

Schematic diagram illustrating a RAGE-mediated mechanism of hIAPP cytotoxicity in β-cells.
Remarkably, beyond metabolic disorders and diabetes, similar considerations may be argued for many other diseases associated to adverse RAGE signalling including autoimmunity, cancer and Alzheimer’s disease: various ligands may specifically accumulate, bind and upregulate RAGE which, in turn, activates toxic cellular signals. For example, RAGE interaction with Aβ in neurons and microglia is known to foster and activate a number of deleterious effects in neuronal function and synaptic transmission thus contributing to the Aβ-mediated cognitive dysfunction observed in AD. Notably, in striking analogy to what observed for hIAPP-mediated toxicity in INS-1 β-cells, RAGE blockade by specific inhibitors. ameliorates Aβ-mediate neuronal failure.288
3.8. IAPP trafficking by extracellular vesicles and exosomes
Extracellular vesicles are released by cells as messengers in cell-to-cell communications. In recent years intense research was addressed in extracellular vesicles, especially on exosomes. Extracellular vesicles can be classified according to their size. They can be divided into three types: exosomes with size 50–200 nm, ectosomes with size 50–1000 nm, and apoptotic bodies with size 500–2000 nm. Exosomes are micro-vesicles that transport proteins and RNAs. Their bilayer has a thickness of about 5 nm and contains different types of lipid such as cholesterol, phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), lysophosphatidylcholine (lyPC), phosphatidylinositol (PI), ceramides (Cer) and sphingomyelin (Smy)289–291. Exosomes regulate many pathophysiological processes including inflammation and infection292,293. To date, their role in aberrant protein signalling is not clear. Few data are available in the literature and do not show a clear role of exosomes in the interaction with amyloidogenic proteins. To explore the relationship between Parkinson’s disease and exosomes it was reported that exosomes containing PE, PS, PI, PC, GM2, and GM3 gangliosides promote the formation of fibrils. The same authors also report that mimic model membranes SUVs containing cardiolipin, phosphatidylserine, phosphatidylcholine, GM1 or GM3 accelerate the formation of α-synuclein fibrils294. On the contrary, concerning Alzheimer’s disease, investigations on exosomes interacting with Aβ40 and Aβ42 have suggested that exosomes favor the clearance of amyloidogenic fragments. In particular, both exosomes and Aβs are released into extracellular space. Exosomes enhance Aβs fibril formation on its surface, and then Aβ fibrils are incorporated into microglia PS-mediated to degrade Aβ295. Recently, the interaction between hIAPP and exosomes was investigated using exosomes extracted from people affected of T2D, healthy people, and human plasma that was used as a control. It has been found that exosomes extracted from healthy individuals inhibit the formation of hIAPP fibrils, while exosomes extracted from patients having T2D and human plasma do not affect the formation of fibrils. Furthermore, the authors characterized the lipid composition of the three used extracts. The following lipids were found: steryl ester, triacylglycerol, cholesterol, phosphatidylcholine, and sphingomyelin. Surprisingly, no charged phospholipids were found, as in the case of neuronal exosomes296. Considering that negatively charged phospholipids accelerate fibrillogenesis processes as widely reported, the lack of the latter in the islet exosomes probably causes this apparent anomaly. However, there are currently few data available to make accurate models describing the role of exosomes in amyloidogenic protein-related diseases.
4. HIGH-RESOLUTION STRUCTURES OF IAPP
4.1. Computational studies
The molecular dynamics (MD) studies of IAPP have been reviewed recently by Moore et al.,297 and in a broad review article that discusses simulations of several amyloidogenic (poly)peptides.298 Furthermore, two review articles have focussed on the interactions of IAPP with the membrane299,300 and another review deals with experimental and computational methods for investigating cross-seeding of Aβ and IAPP.301 These review articles were published and/or submitted in 2018 so that we focus here on the papers published since 2018. A Web of Science search (performed on March 5, 2020) with the keywords “(amylin OR IAPP) AND simulation*” retrieved a total of 71 papers published in the past two years and two months. A total of 50 papers remained after filtering out the reviews, the papers with simulations of spectra, and the articles that did not present new simulation results but only compared with previous computational studies. About one third of these 50 papers deal with the simulation studies of IAPP oligomers in the presence of small molecule- or peptide-modulators of self-assembly.
We first discuss the simulation studies of monomeric IAPP followed by the investigations of multimeric systems, and the influence of self-assembly modulators. The computational models and protocols employed are heterogeneous with a prevalence of atomistic force field-based MD simulations with explicit or implicit solvent. In the following, the term “MD simulations” is used for classical force field-based models with explicit solvent (and membrane) and unbiased simulation protocols (e.g., canonical sampling). Protocols for enhanced sampling, e.g., replica exchange MD (REMD), metadynamics, and/or coarse-grained models are mentioned explicitly.
4.1.1. Simulations of monomeric IAPP and interactions with lipid membrane
A single molecule of IAPP has been simulated in the presence of a lipid bilayer302–304 or without membrane to compare the monomeric state of hIAPP and pIAPP305 or rIAPP.306 In μs-long MD simulations Qiao et al. observed transient α-helical and β-sheet structure of monomeric hIAPP during adsorption to the surface of a POPG (palmitoyloleoyl phosphatidylglycerol) bilayer.304 The hIAPP adsorption caused bending of the POPG bilayer and hydrophobic irregularities.
Scollo et al. investigated hIAPP insertion in a POPC (phosphatidylcholine) bilayer by unbiased MD, umbrella sampling MD simulations, and kinetics measurements of dye leakage from LUV (large unilammelar vesicles).302 The simulation and experimental results provide evidence that the membrane insertion of monomeric hIAPP is facilitated by its association to POPC molecule that are free in solution, i.e., in dynamic equilibrium with the POPC bilayer. The free energy profile of insertion, calculated from the umbrella sampling, shows that membrane insertion of a 1:1 hIAPP/POPC complex is more favorable than insertion of a 1:3 hIAPP/POPC complex.
Christensen and Schiott combined a coarse-grained model with umbrella sampling and MD simulations to study the binding of hIAPP on ganglioside-rich membranes.303 Their multi-scale simulations suggest that hIAPP peptides localize in the membrane regions with higher local concentration of gangliosides because electrostatic interactions favour the association of the cationic hIAPP peptides and the anionic headgroup of the gangliosides.
Alves and Frigori have compared the monomeric state of hIAPP and pIAPP in solution (without membrane) by REMD simulations.305 They observed a random-coil ensemble for the IAPP of both species. One major difference between hIAPP and pIAPP was the presence only in hIAPP of β-sheet propensity in the so-called core mutation region (segment 20–29).
Su et al. have analyzed the monomeric state of hIAPP and rIAPP in solution (without membrane) by metadynamics sampling.306 The enhanced sampling results were carried out separately with and without NMR chemical shift restraints, and compared to unbiased sampling. The simulations suggest different helical propensity for hIAPP and rIAPP but significant differences were observed for the same peptide depending on the sampling protocol and/or use of experimental restraints. Overall, hIAPP showed higher solvent-exposure of hydrophobic side chains than rIAPP.
4.1.2. Simulations of oligomerization and fibrillar aggregates
These simulation studies can be classified in three categories that correspond to the number of peptides in the self-assembly, namely, dimers, oligomers, and simulations started from (models of) fibrillar structures, respectively.
4.1.2.1. Dimerization
Guo et al. used metadynamics to enhance the sampling of the hIAPP dimeric state and the finite-temperature string method was employed for generating pathways of dimerization.307 The pathway analysis revealed a free energy barrier of approximately 7 kBT and nucleation of β-sheet structure in the segment 20–29 and also for Leu12-Ala13.
Qi et al. reported an application of REMD to study the dimerization of the hIAPP segment 11–25.308 This application was published together with an introduction to REMD simulations and it was supplemented by input files for REMD simulation protocols that the authors deemed useful for other researchers.
4.1.2.2. Oligomerization
Ilitchev et al. used ion-mobility mass spectrometry, AFM, circular dichroism (CD), and REMD simulations in implicit solvent to study the co-aggregation of hIAPP and the fragment 106–126 of the prion protein.309 The segment 106–126 connects the flexible tail (residues 23–123) and globular domain (124–231) of the prion protein and is not homologous to hIAPP. They proposed a new model in which the segment 106–126 of the prion protein induces the formation of β-hairpin structure in hIAPP as well as in rIAPP despite the presence of three β-sheet breaking proline substitutions. The prion-hIAPP heterodimer converts into a β-hairpin hIAPP dimer by ligand exchange in a process which is faster than hIAPP dimerization in the absence of the prion protein segment.
The dimeric, tetrameric, and hexameric states of hIAPP have been investigated by MD simulations and compared the results with the Aβ40 peptide, the ovine prion protein, and the SOD1 dimer.75 For all of these amyloidogenic polypeptides they observed oligomeric ensembles with alternate composition of partially structured and disordered monomers. The MD simulation results, as well as a theoretical model based on linear stability analysis, were congruent with surface enhanced Raman scattering (SERS) measurements of hIAPP.
Discrete MD (DMD) simulations and ThT fluorescence kinetics experiments have been performed to analyze the influence of hIAPP on the aggregation propensity of Aβ.310 DMD is an approximative methodin which discrete step functions instead of continuous potentials are used to model interatomic interactions in the solute in combination with an implicit treatment of the solvent. The DMD simulations, carried out with 2:2 and 4:4 peptide ratios, suggest that the formation of heterodimers is favoured by electrostatic attraction between the positively charged hIAPP peptide and negatively charged Aβ peptide. In the heterodimer the unfolding of the helical segment 16–22 of Aβ promotes Aβ aggregation. The ThT experiments confirmed previous reports on the lag phase of Aβ and IAPP coaggregation being shorter than that of Aβ alone and only slightly longer than IAPP. DMD was used also to simulate the oligomerization phase of hIAPP.311 Most oligomeric states had larger α-helical than β-sheet content, and in about 5% of the simulations β-barrel conformations were formed by pentamers and hexamers. Furthermore, the same research group reported that five fragments of amyloidogenic peptides, namely the cytotoxic hIAPP(19–29) and its Ser20Gly mutant as well as hIAPP(22–28), Aβ(16–22) and α-nuclein(68–78), formed oligomers with β-barrel content in DMD simulations while two non-toxic fragments (hIAPP(15–25) and its Ser20Gly mutant) polymerized without assuming β-barrel conformations.312
4.1.2.3. Formation and/or stability of fibrillar structures
Qian et al. carried out MD simulations of the early steps of insertion of hIAPP fibrillar oligomers in a DPPG (dipalmitoyl-phosphatidylglycerol) lipid bilayer or a mixed DPPG/DPPC (dipalmitoyl-phosphatidylcholine) (3:7) bilayer at acidic or neutral pH.313 The simulations indicate that lipid composition and pH conditions influence the tilt angles and insertion depth of the hIAPP fibrils. The pH dependence is due to the protonation of His18. The aromatic side chains of Phe15 and Phe23 seem to promote membrane insertion of fibrillar hIAPP which in turn has an influence on the bilayer thickness.
Davidson et al. employed a polarizable force field to investigate the electrostatic stabilization of fibrils of tau, Aβ, and IAPP.314 The MD simulations suggested that, irrespective of the peptide sequence and fibrillar structure, dipole moments in the side-chains, structured waters molecules in the fibril interior, and the surrounding of salt bridges contribute to the stability of fibrillar aggregates.
Li et al. approximated polarization effects by pre-calculating partial charges specific for a fibrillar structure of hIAPP using an iterative procedure based on quantum chemistry calculations in the gas phase and estimation of solvent effects by the Poisson-Boltzmann equation.315 The MD simulations indicated that electrostatic polarization, approximated by the pre-calculated partial charges, contributes significantly to the enthalpic gain during self-assembly.
Tofoleanu et al. analyzed the structural stability of the parallel β-sheet fibril arrangement of IAPP from multiple species by MD simulations.316 They also compared a model of a fibril of infinite length (using periodic boundary conditions) and a fibril segments solvated uniformly in all directions by explicit solvent. The simulation results on human, cat, rat, and pig IAPPs were consistent with experimentally measured aggregation propensity. The authors proposed that Asn31Lys is a mutation that destabilizes the fibril structure and increases its solvent accessibility.
Mahmoudinobar et al. used umbrella sampling and REMD to assess the structural stability of cross-β aggregates consisting of six identical peptides from the polar segment S28STNVG33 of hIAPP.317 A model structure of the parallel aggregate of hexameric hIAPP(28–33) was less stable at elevated temperature (up to 375 K) than at physiological temperature. Surprisingly, an antiparallel cross-β arrangement of the mainly nonpolar segment 16KLVFFA21 of Aβ was more stable in the high temperature range.
Baweja and Roche used a model that imposes the fibrillar structure as free energy minimum (called Go model from Nobuhiro Go, the scientist who originally proposed it) to study the growth of hIAPP.318 Their Go-model simulations reveal a dock-and-lock mechanism of elongation which is consistent with the stop-and-go behavior of Aβ42 elongation observed by AFM.319
Lu et al. developed a coarse-grained model of IAPP to analyze the structural stability and mechanical properties of different amyloid fibrils.320 In their simplified model, monomeric IAPP was approximated by as a patchy particle consisting of three spheres and four patches. They observed that the persistence length of IAPP fibrils increases together with their pitch length which indicates that, somewhat counterintuitively, the bending stiffness of the fibrillar structures is larger for less twisted fibrils.
4.1.3. Simulations of aggregation in the presence of self-assembly modulators
These simulation studies can be classified into three categories based on the chemical structures of the modulators, namely, small molecules, peptides, or others, respectively.
4.1.3.1. Small organic compounds
The influence of several small-molecule chemotypes on IAPP aggregation has been investigated by MD simulations and/or docking. Most of these small molecules feature multiple aromatic rings, e.g., polyphenols. They are natural products, dietary supplements and/or drug analogues, namely, tanshinone I and IIA (from the Chinese herb Danzhen also called red sage, Salvia miltiorrhiza),321 genistein (a major phytoestrogen in soybean),321 heparin fragments,322 naproxen analogues (also called pharmaco chaperones),323 the dietary supplements lipoic acid and ascorbic acid (vitamin C),324 epigallocatechin gallate which is the most abundant catechin in tea,325 diazenyl-derivatives of pyridazinylpyrazolone,326 fullerene C60 and polyhydroxylated derivatives of fullerene,327,328 azadirachtin (a secondary metabolite isolated from the traditional medicinal plant Neem, Azadirachta indica),329 a cationic polymethacrylate-copolymer,330 choline-O-sulfate, dopamine,331 and lithospermic acid (a polycyclic phenolic carboxylic acids isolated from plants of the genus Lithospermum).332 As it is difficult to assess the significance and robustness of the computational sampling of these studies we do not discuss them in detail considering also that the models and simulation protocols used were not novel. Some of these computational studies were combined with experimental results.
4.1.3.2. Peptides
Levine et al. carried out both simulations and experimental study of the influence of humanin, a 24-residue mitochondrial-derived peptide, on IAPP aggregation.333 REMD simulations of the S14G mutant of humanin (abbreviated as HNG14), which is known to be neuro- and cyto-protective, revealed that HNG14 is able to cap protofibrillar structures and thus hinder the elongation of amyloid oligomers. Furthermore, HNG14-IAPP heterodimers were about an order of magnitude more stable than IAPP heterodimers which together with the capping effect explains the substochiometric ability of humanin to hinder amyloid elongation. Kinetics measurements with ThT and electron microscopy showed that HNG14 does not break fibrillar structures of IAPP in agreement with the simulation results.333
Niu et al. investigated the inhibition of Aβ aggregation by derivatives of IAPP whose design was inspired by the IAPP/Aβ cross-amyloid interaction surface.334 The authors employed a combined experimental and computational approach based on fluorescence spectroscopy, solution-state and solid-state NMR spectroscopy, transmission electron microscopy (TEM), dynamic light scattering, and MD simulations. The multi-disciplinary study revealed that a peptide derived from the two segments of IAPP(8–18) and IAPP(22–28) (linked by an -RRR- tripeptide and with methylated backbone nitrogen on the Phe23 and Ala25) is disordered, has transient β-sheet content, and can oligomerize into colloid-like assemblies that can interact with Aβ40.334
Baram et al. have reported pi-pi interactions between Tyr16 in insulin and Phe15 in hIAPP by MD simulations of free insulin approaching model structures of hIAPP fibrils.335 These findings are relevant in vivo because insulin and hIAPP are both secreted by the pancreas and have been observed to form mixed aggregates.
4.1.3.3. Others: carbon nanotubes, graphene quantum dots and chiral silica nanoribbons
Mo et al. investigted the influence of hydroxylated single-walled carbon nanotubes (abbreviated as SWCNT-OHs) on the dimerization of hIAPP by REMD simulations.336
The REMD sampling provided evidence that SWCNT-OHs hinder interpeptide β-sheet formation which was consistent with a large body of experimental results, namely turbidity measurements, ThT fluorescence kinetics, CD, TEM, and AFM data.
Two studies, both from the Ke and Ding groups, used DMD to analyze the inhibition of IAPP aggregation by graphene quantum dots which is a biocompatible nanomaterial.337,338 In another study, the Ke and Ding groups studied the influence of chiral silica nanoribbons on IAPP fibrillization by a multi-disciplinary approach consistsing of ThT kinetics measurements, TEM, cell viability assays, zebrafish developmental and behavioral assays, and coarse-grained DMD.339 The DMD simulations were carried out with an 11-bead model that was inspired by a previously developed 10-bead model with two main states, an amyloid-prone structure and an amyloid-protected conformation.340,341 The multi-disciplinary study consistently revealed that the right-handed silica nanoribbons shortened the nucleation and elongation phases of IAPP. Furthermore, the accelerated kinetics of self-assembly resulted in a reduced toxicity in the cell viability assays and in vivo experiments with zebrafish embryos.339 It is interesting to note that the previous study with the coarse-grained 10-bead model had revealed already 15 years ago that fast kinetics of self-assembly are essential for functional amyloids.340 These cross-β fibrillar aggregates have a physiological role and their lack of toxicity is due to a tight control of their location and fast self-assembly.
4.2. Experimental characterization of small oligomeric species
Since the toxic oligomer hypothesis was suggested185,342–344, intense investigation has been focused on characterizing these transient species. Toxic oligomer hypothesis suggests that oligomers rather than fully matured fibrils damage the cell membrane. The fragment of sequence 20SNNFGAILSS29 has been shown to be responsible for the amyloidogenic propensities of the full length protein, by forming in a first stage trimer antiparallel β-sheet345 and in a second stage evolves in fibril as reporter167. To better understand the complex process of amyloid aggregation and oligomer formation induced membrane damage, in vitro biophysical studies used lipid membrane to probe the role of aggregation on the lipid bilayer structure and also on the catalytic role of the lipid membrane towards amyloid aggregation. Based on the experimental results, a broad two-step mechanism has been proposed. In step-1, an initial fiber-independent step in which the generation of stable transmembrane protein pores (poration). In step-2, membrane-assisted amyloid aggregation results in the removal of lipid components from the bilayer via a detergent-like mechanism173. In the fiber-dependent step-2, peptides initially associate with the lipid membrane like a carpet mechanism, a process that has been well studied for antimicrobial peptides, followed by amyloid aggregation. Whether these two steps are mutually exclusive or if (and how) they cooperate in triggering membrane damage remains to be established.
The molecular events underlying the fiber-dependent process are well investigated by solid-state NMR studies that detected the fragmentation of lipid bilayer and the presence of lipids in the fibril346,347. On the other hand, the step-1 is a challenge to most biophysical techniques as the lag-time for amyloid aggregation by hIAPP is quite fast (with in 2 hours). Nevertheless, helical intermediates are reported based on NMR and other biophysical studies348–350. Another study investigated ‘micelles-like’ oligomer formation by hIAPP but not by rIAPP and IAPP-1–19351. A recent study introduced hypothesis that free lipids in aqueous phase form a stable lipid-hIAPP complex and then transfer into lipid bilayer.302 In this framework oligomer formation plays a central role. Due to the transient nature of oligomeric species few experimental techniques are available and simulations continue to play an important role. Since oligomers are formed in the aqueous phase at a low concentration and are in non-equilibrium conditions, the ideal technique to investigate transient oligomeric species should be sensitive, fast to acquire data, probe-free and yield atomic-level structural details. NMR techniques can be cleverly utilized to probe the early events of IAPP aggregation.352 To investigate oligomeric species at nM concentration, surface enhanced Raman spectroscopy (SERS) using silver nano-particles has been used to probe the secondary structures of IAPP, Aβ and their mixture77. Pulsed field gradient NMR experiments provided evidence for the formation of toxic monomeric compact α-helix conformation of hIAPP, which was not found for the non-toxic and non-amyloidogenic rIAPP. Also, a large oligomeric species greater than 100 nm in diameter has been observed but it does not trigger the nucleation-dependent aggregation, indicating the large oligomeric species is likely to be an off-pathway intermediate.67 A mixture (1:1 molar ratio) of hIAPP and Aβ40 has been shown to form oligomeric aggregate having a diameter of about 500 nm.77 This study also tested two polyphenolic compounds, resveratrol and curcumin,353 to stabilize off-pathway oligomers in order to inhibit hIAPP toxicity.354,355 Moreover, using 2D-IR on solution containing 13C18O FGAIL labeled hIAPP and unlabeled hIAPP the evolution from monomer to fibril was investigated.356 By fitting the obtained data to a 3-state kinetic model, it was found that an activation free energy barrier of about 3 kJ mole−1 necessary for refolding the oligomeric intermediate into the structure of the amyloid fibril (oligomer activation). The barrier creates a transition state in the free energy landscape that slows the fibril formation and creates a stable population of oligomers during the lag-phase.356 The role of lipoproteins interacting with hIAPP has also been explored. It is known that the blood of diabetic patients contains low concentrations of high-density lipoproteins (HDLs) and high concentrations of very low-density lipoproteins (VLDLs), low-density lipoproteins (LDLs) and triglycerides (TGs). LDLs are spherical and non-fluorescent, whereas in the presence of hIAPP they were found to become intrinsically fluorescent characteristic of its interaction with hIAPP. This might indicate that β-sheets are already preformed in these oligomeric structures. hIAPP solubilized in plasma and LDL yielded high molecular weight oligomer complexes with an apparent molecular weight in the range of about 15 kDa to ≥100 kDa, and that in presence of glucose induced hIAPP oligomers which yielded an increased hemolytic activity as well as cellular toxicity.357 This finding suggests a link between diabetes and cardiovascular diseases. Molecular dynamics simulations agree with experimental evidences mentioned above; small oligomers are highly toxic to cell.358 Also membrane-bound small aggregates/oligomers are likely to be higly toxic as they can form ion-channel pores containing 4 o 5 molecules359,360 with consequent loss of ions homeostasis. Thus, the property of small oligomers is a common characteristic of many amyloid proteins such as hIAPP, Aβ and α-synulein.361 Recently it was suggested the toxic species are the smallest oligomers (monomer and dimer), since their concentration in solution strongly decrease with the dimension going from nM of monomer to 0.001 pM of trimer.362 Further studies investigating the formation, stability, toxicity, and high-resolution structures of a variety of small oligomers of hIAPP and its variants would be valuable to better understand the loss of islet cells in type-2 diabetes and could potentially aid in the development of compounds to suppress the toxicity.
4.3. Structures of IAPP in lipid membrane
The interaction of model membranes and IAPP was studied using a variety of phospholipids (pureor in mixture) having different acyl chains length and polar head groups. In general, a complete understanding of the transfer of an amyloidogenic protein from the aqueous phase to the membrane, and the determination of the structure of the various intermediates, is a key requisite to describe the cytotoxicity of IDPs. However, the achievement of this objective is very complex due to the transient nature of the various aggregates that are formed before the final equilibrium state is reached363. For example, it has been observed that hIAPP interacts immediately with supported lipid bilayer (SBL), and the transfer from the aqueous phase to the bilayer occurs immediately and continues over about eight hours.256,364,365 It was hypothesized from data obtained in different laboratories and a variety of experimental techniques, that small IAPP unstructured aggregates (oligomers), rather than mature fibril, are cytotoxic366–370 and the toxicity was associated with membrane permeabilization371. Membrane permeabilization was linked to the self-assembling of hIAPP within hydrocarbon core of the bilayer by forming ion channel-like pores. The insertion of IAPP into the lipid bilayer occurs fast as monomeric α-helix and the N-terminal part of hIAPP is largely responsible for insertion61,372–377. Multiscale simulations and statistical thermodynamics have shown some different dynamics of oligomerization behavior of hIAPP and rIAPP into the bilayer. Starting from 36 monomer embedded into the bilayer, aggregation occurs in the first stage by way of dimer and trimer, and in the second stage dimer and trimer merge to form a pentamer having a pentagonal topology. Fused pentagons can be formed by adding (one-by-one) three peptides to a pentagonal assembly; aggregates contain 5, 8,… (3n+2) hIAPP molecules.378 The formation of pentamers is favored by the electrostatic distribution along hIAPP in α-helical conformation where an amphipathic structure can be see with one side is rich in hydrophobic residues and the other side contain hydrophilic residues. This asymmetric charge distribution in the protein in α-helix conformation drives the pentamer formation having the topology of an ion-channel. This finding agrees with AFM measurements previously reported.66,359 The growth of pentameric aggregates leads to the formation of transient large non-linear aggregates containing 26 hIAPP molecules. These aggregates lead to the curvature of the membrane according to AFM measurements.377–381 The curvature of the membrane is due to the truncated-cone shape of the aggregates. It is interesting to note that a curvature of the membrane was also observed for the Aβ40.360
Molecular dynamics simulations have shown that hIAPP adsorbs on the membrane surface without penetrating the bilayer. The failure to observe the insertion of hIAPP into the bilayer core is principally due to the high viscosity of the bilayer and, therefore, the enormous simulation time required to observe the protein insertion. The current computational power does not allow this type of problem to be solved. Therefore, it has been chosen to simulate the transfer of the protein from the aqueous phase to the membrane surface. Simulating the adsorption of protein on the membrane surface has been reported that electrostatic interaction between hIAPP and negatively charge lipid bilayer plays a major role in peptide-lipid interaction.382,383 Lys1 and Arg11 act as anchor on the bilayer surface and pH act as a switch for the poly-peptides aggregation:384–386 acidic environment represses aggregation as a consequence of His protonation. These results agree with neutron scattering measurement, where a preferential interaction between hIAPP and negatively charged phospholipid is more favorable instead of zwitterionic lipids.387 Moreover, hIAPP inserts into the bilayer containing zwitterionic lipids from C-terminal, whereas in the presence of negatively charged phospholipid, the insertion occurs from N-terminal.358 The Cys2-Cys7 disulfide bridge does not appreciably influence the interaction between hIAPP and the membrane388, while in the adsorption of hIAPP on the membrane surface, disulfide bridge favours the formation of beta sheets as opposed to the rat isoform389.
From the data available in the literature, hIAPP structure on the membrane surface depends on the electrostatic property of the phospholipid i.e., zwitterionic phospholipids induce the formation of α-helices, while negatively charged phospholipids favor β-sheet structures. While if the protein is in the phospholipid bilayer, the most stable conformation is the α-helix, which later self-aggregates to form ion-channel like pores.363 Since membranes catalyse the amyloid aggregation of IAPP, micelles have been used to solve the high-resolution structures of full-length hIAPP and rIAPP and the non-fibril forming 1–19 segments. These helical structures support the low-resolution structural models for membrane insertion and membrane disruption by IAPP peptides390–392.
4.3.1. Are the fibril growths on the membrane surface and in solution correlated?
Biophysical studies have shown that the amyloid aggregation kinetics is significantly altered by the lipid membrane. The lipid charge, lipid composition, rafts, and bilayer thickness affect the aggregation process. In general, the toxicity of amyloidogenic proteins is associated with membrane damage: the amyloid protein from the aqueous phase is transferred into the bilayer then in a first step forms small pores (ion channel-like mechanism) during the lag phase and in a second step very large pores are formed with a mechanism called “detergent-like” mechanism. The detergent-like mechanism involves the growth of fibrils in the bilayer, which acts as a detergent by tearing lipids from the bilayer (Figure 5). This model of membrane damage, called the two-step mechanism, has been deduced from experimental dye-leakage measurements using fluorescent probes.393 In parallel, fluorescent measurements were conducted using ThT, a probe that selectively binds to fibrils, thus highlighting the kinetics of amyloid fibril formation. By overlapping these kinetics data, it was observed that ion-channel and detergent-like mechanisms occur before the fibril formation. From these data, the following questions arise: since ThT assay cannot distinguish whether amyloid fibrils grow in the aqueous phase or on the membrane, can we somehow separate the two phenomena? Furthermore, are the two processes related? Several strategies have been developed to answer these questions.
Figure 5.
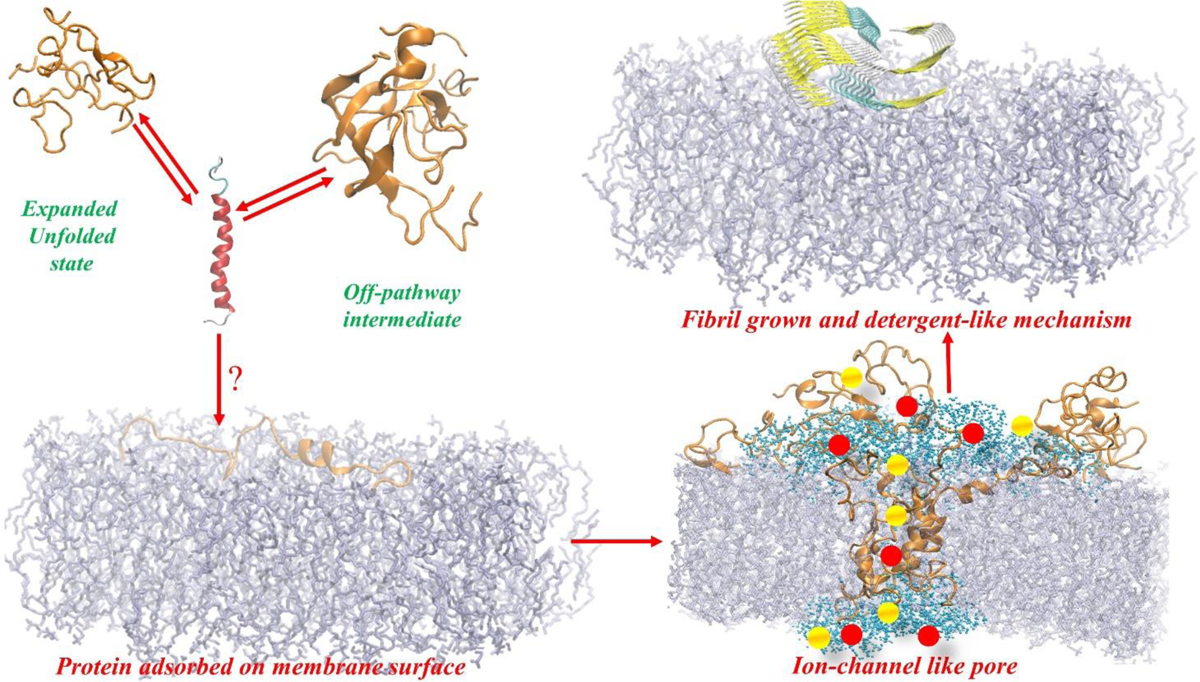
Schematic illustration of a two-step membrane disruption mechanism. In the aqueous phase, the oligomers, formed on-pathway and off-pathway, are in chemical equilibrium with the monomers existing in α-helix conformation. In the first step hIAPP insert as α-helix within the lipid bilayer, then the peptides self-assemble to form a pentameric aggregate having an ion-channel like structure which can enable ions to cross the bilayer resulting in an electrolytic imbalance. In the second step, monomers from the solution can associate with the lipid bilayer surface which can catalyze the self-assembly process to form fibrils; the fibril forming process can remove lipids from one of the leaflets of the bilayer and then ripping the other leaflet causing membrane damage like a “detergent-like” carpet mechanism.
A biophysical study showed that the N-terminal1–19 region of hIAPP is responsible for the membrane insertion, while the 20–29 region is responsible for the fibril formation.364 The interaction between the hIAPP1–19 fragment with negatively charged model membranes containing POPG was studied. It is shown that hIAPP1–19 at low concentrations damages the membrane similar to that of the full-length hIAPP. At high concentrations, the hIAPP1–19 has a more significant action in the membrane destruction rather than the full-length peptide. Also, the hIAPP1–19 fragment, like the full-length protein in the aqueous phase, has a random-coil conformation that evolves into α-helix in the presence of POPG vesicles. It has also been shown that the hIAPP1–19 in the presence of POPG bilayer does not form fibrils as opposed to the full-length protein. From these data, it can be deduced that membrane destruction and fibril formation are two independent phenomena.364 The same conclusion has been reached by other authors, starting from theoretical models prediction of amyloidogenic properties from the amino acid sequence of the selected protein,394,395 which focused on the regions hIAPP12–18 (LANFLVH) and hIAPP21–27 (NNFGAIL).396 Besides, it has been reported that His and Phe do not affect the amyloidogenic and membrane interaction properties.386 Another indication of no correlation between the growth of fibrils on membrane surface and in the aqueous phase was observed in the interaction of hIAPP with model membranes in the presence of resveratrol and their derivatives 4′-O-Phosphorylresveratrol (4′-O-PR) and 4′-O-(1,2-di-O-myristoyl-sn-glycero-3-phosphoryl) resveratrol.397–399
4.3.2. Model membranes for experimental studies
In 1972 S.J. Singer and G.L. Nicolson400, based on the thermodynamic principles of lipid organization in the aqueous phase, proposed the model of biological membranes called fluid mosaic, where proteins are embedded in an asymmetric fluid bilayer consisting of phospholipids and cholesterol. Since 1972 our knowledge of biological membranes has been enriched with many important details, but the basic concepts from the initial model remain valid. From this, it can be deduced that cell membranes are very complex systems, and therefore this study contemplates a high number of dependent and independent variables such that the in vivo study becomes almost impossible. To bypass this difficulty, the bottom-up strategy i.e., to build simple systems with few variables, has been applied. Of these model membranes, there are different types, and the choice of the model to use depends mainly on what one wants to investigate. In this review, we describe the most popular ones for studying the structure and dynamics of the bilayer itself and those used in protein-membrane investigations. Historically, the first phospholipidic vesicles have been the multi-lamellar vesicles (MLVs). From an experimental point of view, the preparation of MLVs is simple. A certain amount of lipid is dissolved in an organic solvent, by evaporation of the solvent in a tube a thin film is formed that then by hydration and agitation results in the formation of MLVs.401 MLVs are multi-state vesicles having an onion-like structure. They have an average size between 1 μm and 5 μm and broad size distribution402. MLVs containing zwitterionic phospholipids show polymorphism as temperature changes. At low temperature, lipids exist in the gel phase where the hydrocarbon tails of phospholipids have a large order parameter, S and a slow diffusion coefficient403 DT (approximately 10−3 μm2s−1). Increasing the temperature leads to a phase transition, the ripple phase is formed in which S and DT do not vary appreciably, while the membrane surface becomes wavy. If the temperature increases again, a phase transition (called the main transition) is observed where the lipid-crystalline phase is formed. The crystalline liquid phase (liquid disordered) is characterized by a low S and a fast DT (about 1 μm2s−1); on the other hand, the liquid order “raft”, formed in the presence the of cholesterol, coexist with liquid disordered and shows a high S and a fast DT (about 1 μm2s−1)404,405. Also, phases transitions show a positive value of enthalpy, entropy and volume406. By extrusion of MLVs, large unilamellar vesicles (LUVs) are obtained through a polycarbonate membrane with appropriately sized pores. The LUVs are mono lamellar and have average dimensions ranging from 100 to 400 nm with a very narrow size distribution. The polymorphism, DT, and S are equal to those of the MLVs. Another model membrane having spherical shape is the small unilamellar vesicles (SUVs) that are obtained by sonication of LUVs, and they have dimensions ranging from 10 to 100 nm407. Some physicochemical characteristics of SUVs are identical to those of LUVs, excluding the transition temperatures and cooperativity that are lower due to the small radius of curvature defined as reported in ref360 that decreases from giant unilamellar vesicles (GUVs) to SUVs. GUVs have dimensions higher than 5 μm and are prepared by evaporation408 or electroforming409, and they have a curvature radius close to zero. A better membrane mimic of the eukaryotic plasma membrane should consider the asymmetric lipid distribution of two leaflets of the bilayer. GUVs, LUVs, and SUVs can be assembled having asymmetric bilayer using methyl-β-cyclodextrin-induced lipid exchange410,411.
Since the model membrane is used to explore the molecular mechanism of amyloidogenic protein-membrane interaction, the choice of model membrane selection in these investigations is crucial. In this framework, some physicochemical properties of the above-mentioned model membranes must be considered. The strain due to curvature radii strongly affects stability of SUVs over time. To decrease curvature elastic free energy412, SUVs fuse spontaneously to form larger unilamellar vesicles413. Supported lipid bilayer is formed just from SUVs. Dye-leakage experiments require to load the inner space of the model membrane with dye fluorescent probes, which induce osmotic stress. A model membrane having a radius of >100 nm are more resistant to deformation by osmotic stress, thus making them more suited to applications involving osmotic pressure gradients414. It was reported that membrane curvature properties strongly affect negatively charged membrane-IAPP and α-synuclein interaction381. Planar lipid bilayer (PLB) is another useful platform to investigate the physicochemical properties of model membranes and their interaction with amyloidogenic proteins. Some of the most commonly used PLBs to study amyloid proteins are: black lipid membrane (BLM), planar or spherical supported lipid bilayer (SLB), bicelles, and nanodiscs. BLM consists of phospholipid bilayer across about 1 mm hole in a solid plate between two solution compartments415. The membrane becomes opaque due to destructive interference of reflected light from the front and back of the film, hence the name black lipid membrane. The main application of BLMs is to study the membrane pore formation by measuring the conductance of solution in the two compartments. SLB mimetic membrane is formed by deposition of a drop of aqueous solution on a solid surface; usually, SiO2, mica, or SiO2 coated with Au or other transition metal. An useful technique to study the interaction of membrane and amyloidogenic proteins is quartz crystal microbalance with dissipation, QCM-D416. QCM-D detects the weight deposition on the sensor (crystal solid support) by revealing the change in the oscillation frequency of crystal using the Sauerbrey relationship417. QCM-D has been used to character the interaction between hIAPP and POPC SBL that occurs immediately without any lag phase418. Bicelles are attractive membrane mimetics as they can be prepared easily with varying lipid composition and size, and have been successfully used in membrane protein structure biology419. Bicelles are typically prepared by mixing the desired ratio of short-chain and long-chain phospholipids in organic solvents, and then lyophilization to remove the solvents and hydration followed by a series of freeze-thaw-vortex cycles. An exciting feature of bicelles is the presence of high curvature region and flat lipid bilayer region. Structure and properties of bicelles are influenced by factors including concentration and phospholipid composition, the ratio of long-chain phospholipids to short-chain phospholipids, metal ions, and processing conditions420. Isotropic bicelles are frequently used for studies using solution NMR while large bicelles that align magnetically are used in solid-state NMR experiments. Static solid-state NMR experiments probing the interaction of hIAPP with the lipid bilayer in magnetically-aligned bicelles evidenced that non-toxic rIAPP prefer the flat region of bicelles whereas the toxic hIAPP bind to the high curvature region. This finding is in agreement with the pore formation of hIAPP on LUVs by inducing excessive membrane curvature377, as shown in other findings using simulations and AFM measurements378. SLB fabrication is typically used as the adsorbing and rupture of phospholipid SUVs on the solid support, bicelles represent and useful alternative method to this purpose since bicelles avoid the rupture step. AFM and neutron reflectometry investigation shown that bicelles aligned parallel to the solid surface of SiO2, whereas unoriented phospholipid bilayers were formed on Si if it is covered with the amine group. Thus, the solid surface is covered strongly by phospholipid depending on the chemistry of the surface421. Nanodiscs are disk-shaped nanoparticles containing phospholipid bilayer surrounded by amphipathic belt protein/polypeptides422 or synthetic amphiphilic polymers423–425. Molecular details of the nanodiscs formation were obtained by using coarse-grained molecular dynamics revealing large membrane defects, including small, water-filled pores. Polymers bind to the lipid bilayer interface, driven by the hydrophobic effect426. Particularly, polymers have the significant advantage of being able to extract membrane proteins from their native environment423. The macro-nanodiscs (diameter > 20 nm) that magnetically-align can be used for the measurement of dipolar couplings using high-resolution NMR are an effective, alignment medium to obtain structural information on a wide of biomolecules such as natural products, proteins, and nucleic acids. Besides, it was shown that the degree of alignment could be scaled, by changing the lipid-nanodiscs concentration or temperature427. Other model membranes used in solid-state NMR studies include mechanically-aligned bilayers, which are prepared on glass-plates or by trapping the lipid bilayer in the aluminium oxide nanopores. They are used in solid-state NMR experiments to reveal the membrane fragmentation induced by hIAPP and suppression by the addition of congored.346,347
4.3.3. Lipid-assisted transfer of IAPP from solution to the membrane
Studies have reported the toxicity of amyloid aggregation related to the pathology of amyloid diseases such as T2D and Alzheimer’s disease. While mapping the molecular events that define the toxicity would be valuable in better understanding the pathology of the amyloid diseases and also for the development of compounds to potentially treat the diseases, the process is way too complex to be investigated by biophysical and biochemical approaches. Nevertheless, several biophysical studies have reported the molecular events driving the aggregation in solution and in the membrane-water interface352,428,429. In this section, we focus on the important steps underlying the membrane interaction of soluble amyloid species.
Amyloidogenic IDPs are toxic to cells which are associated with pore formation on the membrane, and many studies reported the molecular details underlying the mechanisms of toxicity. Available data suggest that protein transfers from the aqueous phase directly to the surface of the membrane, which influences its structure and enables the premeation into the bilayer. There is no information available on the transfer process from the surface to the core of the bilayer. The lack of this last step has been taken into consideration recently. The most popular model membrane is vesicles suspension in water. These systems are heterogeneous where phospholipid assembled into bilayer are in chemical equilibrium with free lipids in the aqueous phase. The concentration of lipids in the solution is named the critical micellar concentration (CMC), and its value is linked to the number of acyl chains carbon atoms and polar head of the lipid430,431. CMC of a lipid that is commonly used in IDPs-membrane interaction ranges from μM to nM, whereas protein concentration has the order of magnitude of μM, thus the interaction between free lipid and protein should be considered. In this framework, a thermodynamic non-equilibrium model of the diffusion-reaction to investigate a concerted mechanism coupling lipid and protein fluxes was developed. The main issue is that several phenomena occur with different rates such as: the formation of lipid-protein complexes in water, reassembling to form complexes with lipids or other membrane components in solution, migration and merging of the complexes with the lipid membrane, and desorption of lipid monomers from the bilayer to replace those bound to translocating proteins. A vital point of this intricate multi-scale behaviour is the formation of the lipid-protein complex. Molecular dynamics simulation of hIAPP and DMPC was performed, since the time scale of the lipid-complex formation is very short. Theoretical development is based on two main assumptions: lipid-protein formation is the fast step, whereas lipid escape from the bilayer and penetration of the lipid-protein complex within the bilayer core is the slow step. The main conclusions are that amyloidogenic proteins insert into hydrocarbon core of the bilayer if the complex lipid-protein is formed i.e., lipids having low CMC do not favor complex formation whereas lipids having high CMC act as chaperone enabling protein insertion into the bilayer. The theoretical model was tested by dye-leakage experiments on hIAPP or Aβ40 interacting with phospholipid model membranes having different CMC432. To validate the concept of lipid-assisted protein transport, a systematic study on hIAPP interacting with LUVs containing di-acyl-phosphatidylcholine having different CMC was carried out. In particular, umbrella sampling molecular dynamics simulation, dye leakage, and ThT assay were performed to investigate the membrane damage and fibril formation vs. CMC. The results are in good agreement with the outcome using a theoretical model. Extensive molecular dynamics simulation confirms that the lipid-complex is formed, and 1:1 stoichiometry was the most favoured and protein assume an α-helix secondary structure433. Moreover, results from ThT assay and dye-leakage experiments concluded that lipids having low CMC favor the fibril formation and repress ion-channel like pores, whereas lipids having high CMC induce ion-channel like pores and repress the fibril formation. It is concluded that free lipids in dynamic equilibrium with the lipid bilayer might play an essential role in modulating the interaction between amyloidogenic proteins and membrane, according to the theoretical model discussed above and the lipid-protein transport inside the bilayer is independent of the bilayer thickness.302 Previously, this finding reported for hIAPP was also observed for Aβ434 in which the role of free DLPC lipids (with a CMC of ~100 nM) in equilibrium with LUVs has been demonstrated to have significant effect on the amyloid aggregation of Aβ. There are also other studies that supported the idea of the important role played by the free lipids on the fibril formation and membrane damage (Figure 6).225,435,436 Of course, additional investigations are needed to validate this mechanism such as its extension to other IDPs437, since a recent study has shown that IDPs in the oligomeric state share a common structures which are independent of its amino acid sequence, and depend on a balance of intramolecular energy vs. intermolecular energy438. Besides, the role of cholesterol, pH, metal ions, the fluidity of bilayer, and the investigation of mixed phospholipids should also shed light on their influence on the CMC of the complex membrane. Extending these in vitro biophysical observations using simple lipid membrane system to in vivo conditions is not easy as numerous factors associated with the crowded molecular environment complicate the problem.
Figure 6.

Schematic illustration of lipid-assisted bilayer penetration. Chemical equilibrium between dispersed lipid monomers (or free lipids) in solution and their supramolecular assemblies (LUVs) is always established and is characterized by the Critical Micellar Concentration (CMC). For lipid molecules having short hydrocarbon tails or charged head-groups, the concentration of free lipids in equilibrium with LUVs may reach values up to the μM range, while the CMC of long-tails containing lipids the CMC value drops to few nM. In the aqueous phase, three simultaneous and competing equilibria should be taken into account: lipid-lipid, protein-protein and lipid-protein with the monomeric species. Lipids having a high CMC value favour the formation of a lipid-protein complex. The lipid-protein complex transfers into the lipid bilayer spontaneously due to the low chemical potential of the lipid bilayer when compared to that of the aqueous phase. Thus, lipids with a high CMC value would favour ion-channel like pores formation whereas lipids having a low CMC value would favour a detergent-like mechanism and fibril formation.
5. CLEARANCE OF MISFOLDED IAPP
5.1. Pathophysiological degradation of IAPP by proteases
Aggregation of hIAPP into amyloid fibrils is toxic to β-cells.174,439–442 Therefore, a comprehensive understanding of the mechanisms by which hIAPP self-assembly is reduced or inhibited may be highly beneficial for the prevention of β-cell loss in T2D. These mechanisms include the inhibition of hIAPP amyloid aggregation, the reduction of hIAPP production, and the degradation of hIAPP. Several enzymes are known to play a role in hIAPP clearance.442,443 Two zinc dependent matrix metalloproteinases, MMP-2 and MMP-9 have been reported to degrade the Aβ peptide thus reducing its amyloidogenic potential.444,445 However, these two enzymes are also expressed in islet β-cells446 and are located in the extracellular space where hIAPP amyloids accumulate.447 Notably, albeit MMP-2 is expressed in islet β-cells at lower levels than MMP-9, the two enzymes have been reported to cleave hIAPP at the same sites, since as the digestion products from MMP-2 cleavage of hIAPP matched those from MMP-9 peptide hydrolysis.448 In particular, mass spectrometry (ESI-MS) assessment of molecular weights of the fragmented peptides allowed determining the putative cleavage sites to be located between the A25 and I26 of hIAPP (Fig. 7). It is known that MMP-2 and MMP-9 preferentially hydrolyze peptides immediately before Ser, Gly or Ala residues and after hydrophobic residues as Leu or Ile.449,450 Therefore, MMP-2 and MMP-9 putative cleavage sites of hIAPP are consistent with their preferential target residues for all peptide substrates. Intriguingly, this cleavage site falls within the hIAPP region (NFGAIL) which is known to play a key role in amyloid growth.123 Furthermore, the fragmented hydrolysis products i.e. hIAPP(1–25) amd hIAPP(26–37) do not have any propensity to aggregate into amyloid fibrils.451,452 It is thus likely that peptide fragments released after hIAPP cleavage being unable to form amyloid thus explaining why the action of MMP-2 and MMP-9 may limit hIAPP amyloid growth and toxicity. However, hIAPP is not a substrate for all MMP. MMP-7, for example, cannot hydrolyze hIAPP.448 More interestingly, it was also reported that the non amyloidogenic mouse IAPP (mIAPP) could not be hydrolyzed by MMP-2 and MMP-9 suggesting that the amino acidic sequence is critical for the activity of both proteolytic enzymes. Actually, the residues at position 25 and 26 in hIAPP (Ala and Ile, respectively) are different from those present in mIAPP in the same positions (Pro and Val) and do not match with the preferred cleavage sites of MMP-2 and MMP-9.449
Figure 7.

Human-IAPP degradation by proteases. Solid lines represent the cleavage sites targeted by the proteases. The yellow highlighted residues 22–27 form the amyloidogenic core of hIAPP.
Besides MMP-2 and MMP-9, other amyloid-degrading proteases are present in pancreatic islets and may counteract hIAPP amyloid aggregation.453 Neprilysin (NEP) is a 100 kDa zinc metalloprotease characterized by a hydrophobic trasmembrane domain, a short cytoplasmic N-terminus and an active site located in a large extracellular domain. NEP may degrade a wide range of substrates depending on its tissue localization. As an example it has been reported that in neuronal tissues NEP may hydrolyze the amyloid Aβ peptide, the main component of amyloid plaques found in the AD brain.454 A decrease of NEP levels has been shown to increase hIAPP amyloid formation in vivo. Moreover, NEP may inhibit hIAPP amyloid aggregation in vitro.442 In addition to its anti-aggregating effect, some reports have demonstrated that NEP may degrade hIAPP albeit 30 times slower that Aβ.443 HIAPP cleavage sites were identified by incubating the peptide with NEP for 60 min at 37 °C. Next, the digested fragments were separated by HPLC and analyzed by mass spectrometry. Consistently with the known substrate specificity of NEP which mainly degrades non polar residues, cleavage of hIAPP was observed at the following sites: Arg11- Leu12, Leu12-Ala13, Asn14-Phe15, Phe15-Leu16 and Ala25-Ile26 (Fig. 7).
Beta-site APP-cleaving enzyme 2 (BACE2) is a membrane-bound aspartyl protease able to hydrolyze the amyloid protein precursor (APP).455 Besides its well established role in APP processing, BACE2 has been also shown to cleave Aβ peptide at positions Phe19 and Phe20.456 Notably, BACE2 may be also relevant to T2D since as it expressed in pancreas at higher levels than the brain.457 There is evidence that hIAPP is a substrate for BACE2 and that, consequently to this proteolysis, it can modulate hIAPP fibril growth.458 Cleavage sites of hIAPP by BACE2 are located at positions Phe-15 and Phe-23. HIAPP is also a substrate for beta-site APP-cleaving enzyme 1 (BACE1) proteolysis albeit only at position Phe-15. The BACE1/BACE2 cleavage of hIAPP at positions 15 and 23 are consistent with the observed effects of both proteases on hIAPP fibril fromation and morphology albeit the additional cleavage site at position 23 by BACE2 was shown to interfere with hIAPP fibrillogenesis at a greater extent. The ability of BACE2 to modulate hIAPP fibril growth in test tubes suggested that its modulation may represent a convenient therapeutic target to treat T2D. Inspired by this observation several in vivo studies were performed using transgenic mice carrying the hIAPP transcript (hIAPP-Tg) and BACE2-deficient mice (BACE2-ko).459 HIAPP-Tg mice exhibited hyperglycemia and lowered insulin levels, but when crossed with BACE2-ko mice, animals exhibited improvements in insulin levels and in glucose tolerance. Although in apparent contrast with the observed effects of BACE2 on hIAPP degradation, these experiments suggest that BACE2 modulation may be potentially considered as a convenient target for T2D treatment, albeit further studies are needed to decode all the intertwined factors involved in the regulation of these proteases and the pathogenic mechanisms occurring in T2D.
5.1.1. The Insulin Degrading Enzyme and IAPP
The Insulin degrading enzyme (IDE) is a zinc metallopeptidase known to be a member of a class of proteases named “inverzicins” characterized by the inverted zinc binding motif HxxEH which has also been found in NEP and thermolysin.460 The 3D structure of monomeric IDE (M.W. 114 kDa) is made up by four structurally similar αβ coil domains encompassing residues 43–285 (domain I), 286–515 (domain II), 542–768 (domain III) and 796–1016 (domain IV).461 The first two domains (IDE-N) are connected by a 28-residues extended loop to the last two domains (IDE-C) and enclose the catalytic pocket. In particular, the Zn++ ion, which is responsible for the catalytic activity of IDE is placed in domain 1 and is coordinated by H108, H112 and E189 residues. Another residue, E111, although not directly coordinated to the Zn++ ion, still actively participates to proteolysis by bridging a water molecule that mediates hydrolysis of short peptides.461 For degradation of substrates larger than 12–15 residues, however, it is required the assistance of an exosite located in domain II and distant about 30 Å from the Zn++ ion which provide additional interaction surface with the enzyme.462
Within the last two decades, IDE has long been recognized as associated to insulin degradation and turnover463 and, as such, to play a key role in the age-related impaired glucose metabolism in TD2.464,465 More recently, IDE has been found to degrade other hormones and peptides such has IAPP or glucagon, thus suggesting a multifaceted contribution of IDE to glucose metabolism.466 Notably, IDE may also degrade the Aβ peptide and its levels are negatively related to amyloid load in AD brains, thus suggesting an intriguing association between AD and T2D.467 Although IDE is expressed in many different tissues and can be thus considered an ubiquitous enzyme, its levels in liver and kidneys are maximal thus indicating a specific role in insulin, insulin growth factors (IGF) and glucagon metabolism.468 IDE expression is closely associated to insulin signalling since as insulin resistance results in lower levels of IDE and, in turn, in impaired insulin turnover and T2D progression.469 Abnormal IDE levels and/or activity was observed in IDE-ko Tg mice also concomitantly with neurodegeneration.470 Notably, IDE levels result significantly lowered in diabetic patients if compared to healthy individuals, thus suggesting that a self-perpetuating vicious cycle of pathogenic processes occurs in diabetes pathogenesis.471 Albeit most of its known substrates are extracellular, IDE is mainly localized into the cytosol.472 In particular, Aβ proteolysis by IDE occurs extracellular in the outer leaflet of the plasma membrane where it is generated after cleavage of APP by secretases.473 During its enzymatic activity, IDE degrades Aβ peptide mainly at positions 13–15 and 18–21, leading to the formation of non-amyloidogenic fragments474 thus demonstrating the significant role played by IDE in AD. IDE may also degrade hIAPP at position L12-A13 and F15-L16.475 Notably, hIAPP cleavage by IDE is strictly dependent on the experimental conditions adopted.461,476 As an example, in a more recent work some previously neglected cleavage sites were identified in H18-S19, N23-F24 and, albeit to a lesser extent, S28-S29, S29-T30 and S34-N35.475.
5.1.2. The role of the ubiquitin proteasome pathway in IAPP turn over
The ubiquitin proteasome system (UPS) is a major cytosolic proteolytic pathway that targets and degrade abnormal/misfolded proteins.477,478 Ubiquitin (Ub) is a small (76 residues) protein, first described as a covalent moiety for post-translational processing of intracellular proteins. It is characterized by a compact structure in which a parallel/anti-parallel β-sheet assembly groups against an α-helix thus creating a hydrophobic core.479 Albeit Ub is principally known as a prologue to proteasomal degradation, it regulates a range of diverse biological processes, including protein trafficking, gene transcription, autophagy and apoptosis.480 Ub is particularly versatile due to the diversity of post-translational modifications that it can form with its susbstrate. The COOH-terminal glycine of Ub covalently binds the ε-amino group lysines of the substrate by an isopeptide bond. However, seven lysine residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, Lys63) are present in Ub and this enables the formation of a large variety of polyubiquitin chains and each one may encode diverse biochemical pathways. Ub tagging to a protein substrate is the result of a complex sequential process which involves three enzymes i.e. E1 (Ub activating enzyme), E2 (Ub conjugating enzyme) and E3 (Ub ligase).481 In the first step of the process, Ub forms an ATP-dependent high-energy thioester link with an E1 enzyme.482 Second, Ub is transferred from the E1-activating enzyme to the E2 Ub-conjugating enzymes (Ubc), which, by linking to different Ub Lys residues, confers specificity to protein ubiquitination. Ub-tagged protein susbtrates are then transferred to the proteasome, a multi subunit, multifunctional 2500 kDa enzyme assembly.483 The catalytic chamber of the proteasome is a cylinder-shaped multisubunit protein complex, named the 20S proteasome or the core particle (CP). Each end of the CP is capped by one (26S) or two (30S) regulatory particles RPs (19S). The 19S particle recognizes, de-ubiquitinates, unfolds and transfers protein substrates into the catalytic chamber of the 20S particle.484 The 20S proteasome is a multimeric enzyme (MW ~ 700 kDa) formed by 28 subunits assembled in four stacked α/β heptameric rings where the two outer α-rings (antechamber) rim the two central β-rings (catalytic chamber).485,486 The 20S proteasome exhibits at least three different proteolyitic activities termed as chymotrypsin-like (ChT-L), trypsin-like (T-L) and peptidylglutamyl-peptide hydrolyzing (PGPH) which occur in β1, β2 and β5 subunits respectively.487
At this time, our knowledge of the cellular factors that regulate hIAPP toxicity is still incomplete, but several reports suggest that an impaired turnover and clearance of the accumulating protein may play a primary role in T2D pathogenesis.488 In particular, an age-related derangement of the UPS has been suggested to be linked to the development of the disease.489 Biochemical methods coupled with confocal microscopy studies showed that, upon internalization into pancreatic islet cells, hIAPP mainly localizes in the nucleus, lysosomes and mithochondria.490 It has been also demonstrated that hIAPP may physically interact with proteasome subunits α4 of the 20S and RPn8 of the 19S in pancreatic islets and that, in response to hIAPP treatment, proteasome activity results inhibited. Notably, further biochemical assays evidenced that Lactacystin, a proteasome inhibitor, induced a significant intracellular accumulation of hIAPP, thus pointing to the proteasome as a crucial hub for hIAPP clearance in pancreatic islets. In the same work, and in accordance with previous studies,139 experiments performed on Rat insulinoma (RIN-m5F) β-cells, evidenced 20S proteasome inhibition in response to either amyloidogenic hIAPP or non-amyloidogenic mIAPP treatment thus suggesting that amyloid aggregation is not required for IAPP-mediated proteasome inhibition. Other experiments carried out at low temperatures and in the absence of Ub and ATP suggested that neither the ubiquitinated nor the aggregated form of hIAPP interact with the 20S proteasome. These findings reconcile with many reports suggesting that intrinsically disordered proteins (IDPs) interact with the 20S proteasome without the assistance of the recognitions step regulated by the 19S.491 However, a concomitant ATP-dependent degradation of ubiquitinated hIAPP cannot be totally ruled out. In fact, ubiquitinated intracellular forms of hIAPP have been observed492 as well as the existence of hIAPP/19S complexes thus suggesting that Ub-dependent proteasomal degradation may still play a role in hIAPP clearance. Although the role played by the 20S proteasome in the proteolysis of internalized hIAPP is firmly established, its contribution to endogenous hIAPP synthesis, secretion and trafficking is still incomplete. Hyperglycemia has been associated with an impaired proteasome activity and an abnormal biosynthesis of both insulin and hIAPP.493 In particular, intracellular levels of hIAPP decreases in T2D human islets when compared to age-matched healthy controls.494 Furthermore, Western Blot analysis performed in the same cells, showed that polyubiquitinated substrates were significantly increased when compared to healthy cells, thus confirming an impaired UPS efficiency. Consistently with this result, a 3-fold decrease of proteasome peptidase activity was observed in islets from a diabetic donor, and this decline was equivalent with that measured in the presence of lactacystin, a known 20S proteasome inhibitor. Even more significantly, the gene expression analysis of islets from diabetic donors evidenced a decrease in the levels of FOXA2, a key hIAPP transcription factor, thus confirming a causal relationship between proteasome activity and hIAPP transcription.494 In accordance with this double-edged view of the role of proteasome in hIAPP regulation, it has been evidenced that a modest proteasome inhibition (35%) resulted in an increase of hIAPP levels in islet cells thus indicating a direct role of proteasome in hIAPP degradation.494
5.2. Role of metal ions in IAPP degradation
The toxic effects of hIAPP aggregates toward islet β-cells are widely demonstrated, and intense efforts have been devoted to determine what factors may influence this proteotoxic event. Accordingly, metal ions and lipid membranes have been identified as key effective modulators of hIAPP aggregation and their effects on its conformational, aggregation and biological properties are subject of intense debate (see Sections 3.5, 4.3, 4.3.3 of this review). On the other hand, IAPP proteostasis is strictly controlled by a number of efficient sentinels, including metalloproteases, proteasome and lysosomes/autophagy that both directly hydrolyse the polypeptide or, as an alternative, regulate its biosynthesis acting on their transcription factors. However, dysregulation of metal ions, besides its effects on amyloid growth, may also have an impact on the proteolytic activity of hIAPP degrading proteases. As an example, it has been recently demonstrated that Cu++ ions may inhibit IDE activity and that Zn++ may also modify the preferential cleavage sites of several substrates.495 Cu++ and Zn++ ions were found to inhibit IAPP degradation by IDE albeit with some specific differences.475 In particular the presence of Cu++ ions abolished hIAPP cleavage by IDE at positions N22-F23 and L27-S28 suggesting that a metal-dependent change in the hIAPP conformation significantly affects enzyme activity. Indeed, this finding reconciles with the known Cu++ coordination site of hIAPP which involve the segment encompassing residues between N22 and I26.496 Notably a lower inhibitory effect of Cu++ was observed when mIAPP was used as a substrate. Besides N22-F23 and L27-S28, Zn(II) ions inhibit IDE activity on F15-L16 and S29-T30 hIAPP cleavage sites. Zn++ ions are known to induce structural changes in hIAPP residues H18, and this may explain the observed effects on the F15-L16 cleavage site.497 However, besides the metal-induced conformational changes on protease substrates, there are other ways by which metal ions may have an effect on IDE peptidase activity. For example, metal ions may bind directly to IDE and thus influence the activity of the enzyme. This effect may be due either to the coordination of exogenous ion to the active site or replacement of the catalytic metal ion.498 Moreover, it cannot be ruled out that abnormal levels of metal ions may also regulate localization and expression of IDE in the cell. Of course, these different metal-related alterations of hIAPP degradation by IDE may be closely interconnected but thorough descriptions of their mechanisms remain elusive. In an attempt to describe the effects of metal ion on the peptidase activity of IDE some authors have investigated the cleavage of a synthetic peptide (B20–30) unable to bind metal ions.499 According to that report, IDE activity is irreversibly compromised on Cu++ binding. On the contrary, IDE is still able to process substrates when incubated with excess Cu++ ions, although with a lower degradation rate.
Besides IDE, other Zn++-dependent endopeptidase like NEP previously described as hIAPP-degrading enzymes have been shown to be inhibited by excess Cu++ ions.500 Indeed, Zn++ ions present in metallopeptidases are known to significantly contribute to enzyme folding and stability. Moreover, the distorted tetrahedral geometry adopted by Zinc centres in metalloprotease enhances the Lewis acidity of the catalytic site and of the coordinated water molecules as well which are both essential for the peptidase activity. Therefore, if a metal ion such as Cu++ characterized by a higher affinity for the amino acid residues and a different coordination sphere should replace Zn++ in metalloprotease catalytic center, the metal protein site would result perturbed,501 as reported for the many Zinc-proteases as thermolysin,502 endopeptidase from Lactococcus lactis503 carboxypeptidase A504, or aminopeptidase B505 which are all inactivated upon Zn(II) replacement by Cu++. In some cases, it has been shown that Cu++ ions may significantly inhibit hydrolytic activity of some specific Zinc proteases (e.g NEP) toward Aβ peptides, but their effects on hIAPP degradation are unknown. Dyshomeostasis of metal ions, including Cu++ and Zn++, may also impact UPS function. It has been reported that Cu++ ions may bind to Ub and promote its aggregation. Conversely, other cations as Ni++, Al3+ and Cd++ do not alter Ub stability suggesting a specific effect of Cu++ ions.506,507 Furthermore, both Cu++ ions and Zn++ ions, over a 40–70 μM concentration range, inhibit Lys63 and Lys48-linked polyubiquitination reaction.508 Besides these effects on the upstream events of UPS, Cu++ ions have been shown to significantly impair proteasome activity. Enzyme assays conducted on isolated 20S have shown that Cu(II) ions inhibit (IC50 ~ 1 μM) the three different ChT-L, C-L, and T-L peptidase activities of the core particle implicitly suggesting that the inactivation mechanism could be ascribed to altered gating dynamics.509 All these findings do suggest that metal dyshomeostasis may, on one hand, affect hIAPP conformation and aggregation propensity and, on the other hand, significantly interfere with the protease-mediated digestion of excess hIAPP. However, the effects of metal ions on hIAPP degradation by proteasome and/other proteases are poorly investigated.
5.3. Removal of IAPP by autophagy
Autophagy is multicomponent regulating cell systems that digest large cytosolic components, such as organelles or proteins, by transporting them to the proteolytic assemblies termed lysosomes. To date, three different categories of autophagy have been described i.e. i) macroautophagy (henceforth termed as autophagy), ii) microautophagy, a bulk lysosomal hydrolytic process initiated by the lysosomal membrane wich incorporates portions of the cytosol for degradation and removal510; and iii) the chaperone mediated autophagy (CMA), in which the chaperone protein Heat shock cognate 70 (hsc70) recognizes and binds the cytosolic substrates onto the lysosomal membrane, thus supporting the release of the chaperone-substrate complex into the lysosome511. It has been reported that hIAPP, besides proteasome, is degraded via autophagic processes. In particular, β-cells loss was observed in hIAPP expressing mice with defective autophagy thus suggesting an active role of this process in counteracting islets damage.492 It is therefore reasonable that internalized hIAPP may be, in part, degraded also by lysosome. However, recent studies have shown that lysosomal inhibition is not directly related to significant hIAPP accumulation490 in contrast with other reports suggesting a key role in autophagy-related mithochondrial dysfunction in hIAPP toxicity.512 However, it should be taken into account that different hIAPP trafficking and localization observed in different cell lines, may account for these apparent discrepancies. In particular, in vitro experiments have demonstrated higher exogenous hIAPP β-cells toxicity in the presence of autophagy inhibitors.492,513 Whereas autophagy impairment may be correlated with hIAPP aggregation and islet cell damage, on the other hand, hIAPP aggregation into toxic oligomers also inhibit autophagy, as shown by the concomitant increase of P62, a known autophagy substrate. HIAPP accumulation is also associated to abnormal mitochondrial function and autophagic digestion (mitophagy),514 thus suggesting a vicious cycle in which hIAPP accumulation resulting from a decreased autophagy may further inhibit autophagic processes. Autophagy and UPS are the two major degradative systems in cell catabolism and their carefully orcherstrated cross-talk is generally considered a key factor in protein homeostasis maintenance.515 It has also been observed that a derangement of the UPS is always counterbalanced by an increase in the efficiency of the autophagic clearance processes and that this compensatory mechanism is fundamental to ensure the remove the excessive load of proteotoxic aggregates.516 However, recent studies have suggested that the regulation of hIAPP level by UPS is independent from autophagy.494
6. ANTIAGGREGANTS OF hIAPP
6.1. Inhibitors of IAPP amyloid growth
Peptide concentration is not the only, but an essential determinant for hIAPP aggregation and contrary to increased synthesis, reduced degradation and clearance results in increasing hIAPP concentration that is a potential risk factor for aggregation. At a given time point, a single human β-cell contains 5–6 thousand IAPP-insulin granules designated for exocytosis via the regulated pathway. Secretory granules have a best before date, and if not released, the granules are degraded via crinophagy. Crinophagy is selective autophagy that acts as quality control and regulator of the β-cell granular homeostasis.517–519
In vitro, hIAPP is highly amyloidogenic, while in vivo, hIAPP remains in solution for many days under normal conditions. This difference suggests the existence of endogenous inhibitors. In the mature secretory granule, insulin is packed together with Zn2+ forming the dense core while hIAPP and C-peptide localize to the halo region. Being in close association with hIAPP, insulin made a putative candidate as an hIAPP inhibitor. Therefore, insulin was added to hIAPP during aggregation and shown to be a concentration-dependent inhibitor of hIAPP aggregation.520–522 During posttranslational processing, a mixture of prohormones, mature hIAPP and insulin, together with the processing intermediates, are expected to be present in the immature secretory granule. ProIAPP’s two processing intermediates are N-terminal flanking peptide+IAPP (NIAPP) and IAPP+the C-terminal flanking peptide (IAPPC). At pH 7.0, NIAPP and IAPP form amyloid-like fibrils to the same extent, and aggregation is as expected prevented by insulin. At pH 5.2, insulin’s activity as an inhibitor is reduced, and at this low pH, N-IAPP is more prone to form amyloid.523 Therefore, intragranular peptide composition may be relevant, but not only to peptide concentration.
Molecular chaperones such as heat-shock proteins have been evolved to assist folding and counteract misfolding of proteins. A different group of molecules that possess chaperone activity contains the domain BRICHOS. BRICHOS is about 100-residue long domain found in 10 distantly related protein families (>300 different proteins). Integral transmembrane (ITM) is one of these families, and ITMB2, also known as Bri2 is recognized for its association with Familial British and Danish dementias.524 Bri2 molecule is 266-residue long and contains 4 regions with distinct properties where BRICHOS constitutes the third domain of the protein. During synthesis, Bri2 is inserted into the ER membrane with the C-terminal part of the protein facing the lumen of ER/golgi. Cleaved of the luminal part of Bri2 by Furin and ADAM10 releases the 100-residue long BRICHOS domain.525–527
Bri2 has a role in the generation of Aβ which originates from cleavage of the Amyloid precursor protein (APP) by β-secretase followed by γ-secretase cleavage. Gamma secretase is a multi-protein complex, and Bri2 regulates the activity of this complex and, thereby, the generation of Aβ.528 When BRICHOS was added to monomeric Aβ in solution, it decreases the aggregation process of Aβ with increased lag phase.529
Transgene expression of Aβ driven to the NLVs neuron in Drosophila melanogaster results in neuronal death, but this was counteracted by simultaneous expression of BRICHOS.530 Bri2 is also expressed in peripheral tissues and to a moderate extent, in the islets of Langerhans. Bri2 synthesized by β-cells co-localizes with hIAPP in secretory granules and is present in extracellular deposits of hIAPP amyloid. BRICHOS also had an inhibitory effect on hIAPP aggregation and could prevent hIAPP fibrillation if added to monomeric IAPP and prevent secondary nucleation. Down-regulation of Bri2 in β-cells made the cells more sensitive to metabolic stress, but the simultaneous up-regulation of BRICHOS expression ameliorated this.531 The BRICHOS domain is an endogenous inhibitor of hIAPP aggregation and is therefore interesting and putative drug target.
One of the interesting directions toward the development of therapeutic means to treat T2D is the development of inhibitors that modulate the formation of aberrant hIAPP assemblies. As hIAPP appears to exert its toxic effect in specific aggregated forms, the ability to interfere the assembly process of hIAPP or to divert assembly into the formation of non-toxic species is highly attractive. There are multiple approaches to achieve these goals. These include the use of small molecules, peptide fragments, full-length polypeptides, antibodies and chelation agents. Here, we provide details on several of the approaches that are currently studied or under development as novel therapeutic means.
6.2. Natural compounds as IAPP anti-aggregating and anti-oxidant agents
It is well-established that several natural compounds, most notable polyphenols, can control the aggregation of amyloid assemblies. In pioneering work, Ono et al., studied a collection of natural polyphenols and determined their effect on amyloidal formation.532 Notable examples of such compounds include Epigallocatechin gallate (EGCG)533 that is found in green tea and other plant-based sources, rosmarinic acid that is found in a variety of plants, and curcumin of turmeric534. Intriguingly, these compounds are used as traditional medicine in treatment of T2D. The molecular mechanism of inhibition by these compounds is not fully understood. The vast majority of the natural inhibitors are aromatic in nature and they may be able to interfere with the formation of aromatic interactions known to be import for hIAPP amyloid formation (see above). Another intriguing direction is the activity of many of these compound as antioxidants.535 There are three important steps in which an successful anti-aggregation compound can interfere: (i) nucleus formation which is the thermodynamically unfavourable event in which monomers form a specific conformation or assemble into a nucleus of critical size, (ii) fibril elongation, thermodynamically favourable event in which (usually) monomers are sequentially added to the pre-formed seed to form fibrils of increased size, and (iii) a compound could energetically destabilize a mature fibrils and promote its disassembly536. In a detailed analysis, in which the chemical character and the antioxidative potential of the compounds were studies, it was found that the hydrophobic and/or aromatic nature of the polyphenols inhibits the formation and extension of the amyloid fibril seeds, while the antioxidative potency relates mostly to the promotion of destabilization of the fibrillary assemblies. Another molecule that is clearly associated with hIAPP is insulin. Insulin is known to bind to hIAPP and thus can modulate its self-assembly.521 Therefore the use of insulin or its analogues appears as an attractive way to modulate the assembly of hIAPP into toxic architectures. It was established that the B-chain of insulin is the active element in this inhibitory activity.537 Peptide arrays have also been used to map the interacting domains within the B-chain and hIAPP that mediate the co-assembly of the two polypeptides, which showed what regions to be important?.537 This work served as a basis for the design of small molecules that could alter the aggregation propensity of hIAPP.
6.3. Peptide inhibitors of IAPP aggregation
Following the identification of sequence 20–29 as a critical region in hIAPP assembly (see section 3.3.2 above) attempts have been made to design peptides that could modulate the aggregation of hIAPP into amyloid. There were several approaches including β-sheet breaker and hydrogen-bonds modulators. One of the straightforward directions is the modulation of recognition modules with elements that do not allow further growth of the amyloid fibrils. The main concept of peptide inhibition is based on two main properties of the active biomolecules. The first is the ability to recognize and bind to amyloid-forming modules at high affinity. As the on-rate kinetics of the binding (kon) is mostly diffusion-limited, a strong binding affinity and thus low thermodynamic dissociation constant (Kd) is translated to slower off-rate kinetics of binding (koff). A high affinity maybe obtained by a combination of several non-covalent interactions between the ligand and hIAPP, such as hydrogen boding, aromatic interactions, or electrostatic interactions. The second key element for the inhibitory molecules is their ability to affect further growth of amyloid bysteric hindrance, electrostatic repulsion and/or competition with H-bonding donors and acceptors or aromatic moieties that are involved in the interactions between the amyloid-forming proteins and polypeptides.
The identification of recognition modules was achieved by either minimalistic approach to indemnity the shortest sequence element that can form fibrils or by systematic screening of the entire peptide sequence. In a pioneering work, Kapurniotu and co-workers identified the NFGAIL hexpeptide as the smallest element of IAPP that can form canonical amyloid assemblies.158 A follow up work had determined the key role of the aromatic phenylalanine in this recognition event. Later studies using peptide arrays, in which labelled-IAPP was incubated with a series of partially overlapping fragments of IAPP mobilized to a solid support, it was confirmed that this region is important for the interaction as well as an additional aromatic module within the peptide sequence.161 These peptide motif served as recognition template for the design of further inhibitors by the addition of β-breaker elements, modulators of hydrogen bonding and steric hindrance elements.538 Experimental osbservation suggest the critical role played by oligomers. Miranker and co-worker206 suggest tetraquinoline as inhibitors. Tetraquinoline is charged dianionic molecule soluble in water and crosses biological membranes without cellular assistance. Tetraquinoline stabilizes a pre-amyloid, α-helical conformation of IAPP and docks specifically with intracellular IAPP.
6.4. Antibody inhibitors of IAPP aggregation
Another direction for the control of IAPP aggregation is the development of antibodies that specifically recognizes the soluble oligomers that are formed during amyloid assembly.539,540 Methods that were developed to prepare stable oligomers of Aβ were used to make stable IAPP oligomers. It was shown the vaccination of transgenic mice model, which expresses hIAPP and has high fasting glucose levels, could be brought back to normal levels by vaccination with these stable oligomer preparations. The antibodies could thus be used in either active or passive immunization. Antibodies that were purified from the immunized mice were also shown to reduce IAPP cytotoxicity towards pancreatic β-cells in a dose-dependent manner. The immunization of transgenic mice with the oligomer preparation also resulted in an increase IAPP-antibody serum titer, lower blood glucose levels, higher insulin levels, and lower hIAPP accumulation. The study thus highlighted the significance of targeting the early amyloid self-assembly events for potential disease management and demonstrated that α-helical oligomers conformers are valid epitope for the development of future immunization therapy.
6.5. Metal chelators
Metal ions are known to play a role in amyloid formation. This is most likely by the intermolecular coordination of residues (most notably the imidazole ring of His-18 in hIAPP; see Section 3.3.2 above) thus allowing energetic barriers in the nucleation step to be reduced, enhacing the rate of amyloid assemblies formation.541 Therefore, the use of metal ion chelators appear an attractive possibility to delay of the onset of various amyloid-associated disorders500,542,543. Bush and colleagues made pioneering contributions in the study of the ability of metal ion chelators to interfere with amyloid formation.544 Although these metal chelators were promising potential drugs to fight Alzheimer’s, they were abandoned due to their side effects. One interesting direction is the combination of aggregation inhibitors with metal ion chelators. A recently introduced candidate is Gallic acid, a naturally occurring hydroxybenzoic acid found in red fruits, onions and tea that was shown to be both an excellent modulator of protein aggregation as well as a metal ion chelator.545 Also in the case of hIAPP, it was shown that the addition of metal ions, most notably Cu++, increase the toxic activity of the polypeptide.546 To rule out the specific role of reactive oxygen sepcies (ROS) in the induction of this toxicity (see 3.6), the researchers added Ni++, which is a redox inactive but has similar protein binding affinity as compared Cu++. The discovery that Ni++ also caused an increase in toxicity suggested that metal ion-mediated aggregation, rather that the formation of oxidative species may account for the mechanism of these metal ions, and thus supported the use of metal chelation to treat T2D. Other studies have investigated the role of Zn++in fibrillization of Aβ547 and IAPP.548 Indeed, the use of Zn++ chelators was shown to inhibit metal ion-associated IAPP aggregation and its induction of apoptosis of cultured cells.262,547
7. RESCUING IAPP PROTEOSTASIS BY SMALL MOLECULES
7.1. hIAPP proteostasis machinery
Proteasome malfunction plays a key role in hIAPP toxic aggregation. As an example, treatment of Min6 and RIN-m5F cells with proteasome inhibitors as lacatcystin or epoxomicin resulted in an accumulation of exogenous hIAPP aggregates. On the contrary, treatment with the proteasome activation particle PA28 protected the cells from the toxic effects of hIAPP.139 Based on this evidence, it is assumed that stimulation of proteasome activity by small molecules may represent a promising strategy for the treatment of T2D. However, the question arises pf whether hIAPP may be digested by the 20S proteasome or by the mature 26S proteasome after its ubiquitination. Evidences of colocalization of hIAPP and ubquitin in human islet cells suggest that ubquitin-dependent hIAPP degradation may indeed be possible, although in vitro studies have also clearly demonstrated that hIAPP may be degraded by the 20S proteasome without the assistance of ubiquitin.490,492 This evidence reconciles with the notion that the 26S proteasome degrades mainly globular proteins with the assistance of the 19S particles which bind, deubiquitynate, unfold and transfer the substrate into the proteasome core catalytic sites549, whereas unstructured proteins are most likely digested by the 20S proteasome via an ubquitin-independent process.550 Moreover, the 20S/26S molar ratio is 3:1 in healthy cells,491 but aging and pathogenic conditions which are known to prompt accumulation of oxidized/misfolded proteins, favour disassembly of the capped proteasome and a subsequent increase of the 20S form.551 Based on all these considerations, the 20S proteasome may be considered a major target for therapies focusing on restoring UPS function in T2D.
In principle, small molecules may activate the 20S proteasome by two different mechanisms: i) the induction of gate opening which facilitates substrate entrance into the catalytic chamber or ii) the stimulation of substrate binding to the catalytic sites through allosteric interactions. However, proteasome activity may be altered by small molecules by indirect mechanisms. As an example, it is known that 20S proteasome activity is sensitive to oxidative stress.552 Therefore, small molecule antioxidants may activate the proteasome by counteracting the inactivating effects due to unbalanced oxidation. In general, UPS activity may also be enhanced by increasing the level of free ubiquitin,553 or overexpressing ubquitin ligases.554 Currently, an increasingly large subset of UPS-enhancing small molecule compounds have been reported as promising candidates to treat protein misfolding diseases including T2D. However, for most of them the mechanism of action is complex and not completely understood.555
If UPS or autophagy impairment may cause hIAPP accumulation and β-cells loss, UPS or autophagy enhancers could provide a novel route to search for new drugs to prevent or treat T2D. However, it may be difficult to single out the effect of a given drug candidate on the two degradative systems. In fact, we know that the two pathways target different substrates since UPS degrades short-lived globular proteins or soluble misfolded proteins, whereas auotphagy removes insoluble protein aggregates, or whole organelles. Nonetheless, in addition to an indirect relationship between the two systems provided by ubiquitin (both UPS and autophagy recognize their targets by specific ubiquitin tags), there is a plenty of significant connections and reciprocal compensatory mechanisms between the two systems.556,557
Within this complex scenario, it could be convenient to propose a mulitarget strategy aimed at selecting/designing molecules able to interfere synergistically with the diverse pathways involved in abnormal hIAPP accumulation. To achieve this goal and overcome current limitations of drug discovery strategies focusing solely to the design of molecules targeting pathogenic hIAPP self assembly, it is important to select those molecular entities able to rescue the entire hIAPP proteostasis machinery which is impaired in T2D.
Here, we have selected some small molecules that are known to be involved in different hIAPP proteostasis pathways and may thus considered prototypal for future investigations in this direction (Figure 8).
Figure 8.
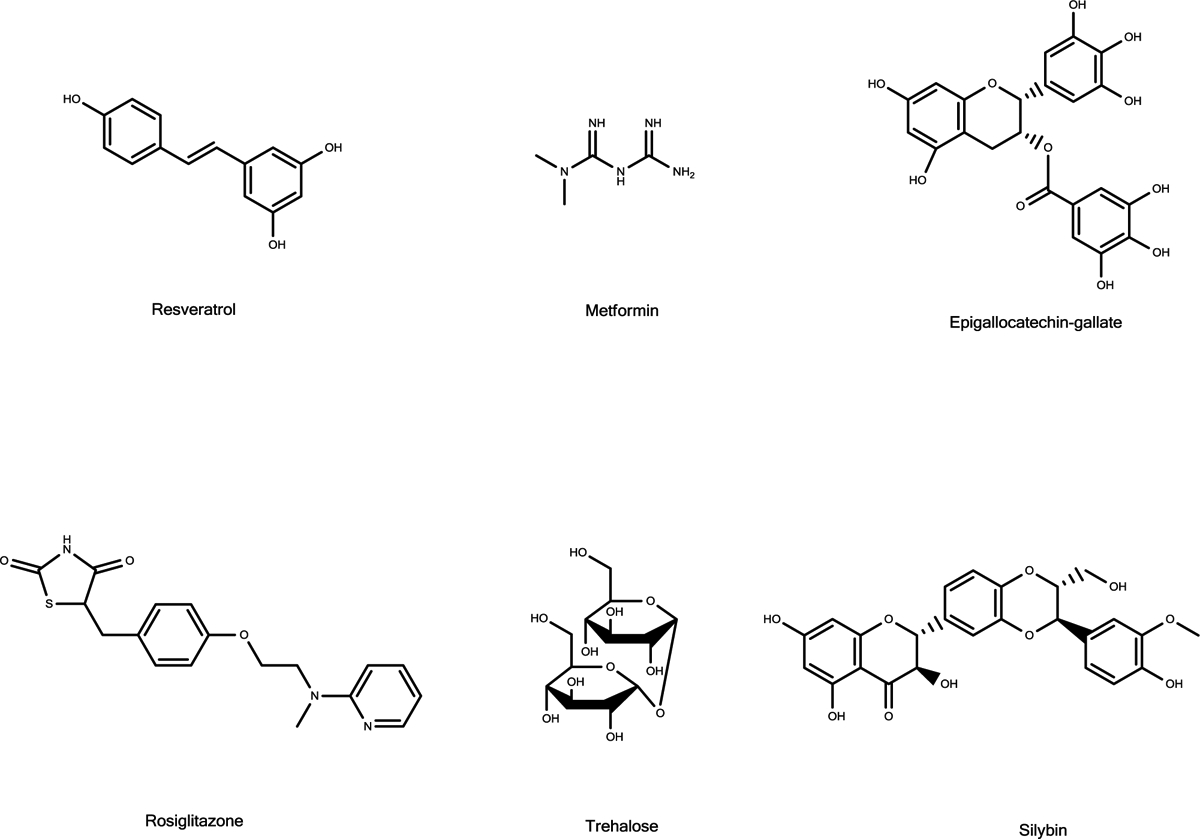
Chemical structures of the multifunctional compounds discussed in the text.
7.2. Metformin
Metformin is a small molecule widely used as a first-line treatment for T2D since the late 1950s.558 Albeit metformin is known to regulate glucose metabolism by suppressing hepatic glucose productions and increasing the rate of glucose utilization our knowledge of its roles in regulating insulin sensitivity, glucose homeostasis, and promoting β-cell survival is far from complete. As an example, metformin reduced islet amyloid accumulation in the pancreas of mice expressing hIAPP.559 However, in vitro studies have demonstrated that metformin does not inhibit hIAPP aggregation in vitro94 thus indicating that other mechanisms are likely involved in hIAPP clearance in vivo. Unfortunately, studies addressing the effect of Metformin on isolated proteasomes are not currently available albeit some reports performed on human liver cancer cells suggest that it may suppress proteasome activity.560 On the other hand, Metformin has been reported to augment autophagy in pancreatic β-cells561, suggesting that activation of this pathway may play a role in the inhibition of toxic hIAPP accumulation by this molecule.
7.3. Rosiglitazone
Rosiglitazone is a member of a class of hypoglycemic agents known as thiazolidinediones.559 Albeit with some controversy,562 some studies have demonstrated that rosiglitazone, when administered to transgenic mice expressing hIAPP, reduces hIAPP accumulation and decreases β-cells loss, when compared to untreated controls.559 It has been also proposed that β-cell protection associated with rosiglitazone treatment may be related directly to its ability to inhibit hIAPP fibrillation. However, biophysical assays demonstrated that rosiglitazone has only minoreffect on hIAPP aggregation and did not protect INS-1 cells from proteotoxicity of exogenous hIAPP.563 Thus, it makes sense that rosiglitazone may partly exert its anti-diabetic effects by increasing the cellular degradative mechanisms such as UPS and autophagy. Our knowledge about the effects of rosiglitazone on UPS is limited. An early work reported a decreased UPS activity in the carotid plaques of diabetic patients when matched to non diabetic people564, but detailed informations about the effect of this molecule on purified proteasomes is missing. Rosiglitazone is also known to be an autophagy enhancer, and this effect may explain its role in the improvement of metabolism and response to excess amyloid load in diabetic patients.565
7.4. Trehalose
Trehalose, a non-reducing disaccharide present in non-mammalian species, is known to protect cells against a range of adverse environmental factors. Due to its osmolyte properties, trehalose has been considered as a small “chaperone” ie. a molecule capable of converting β-sheet rich conformations into less fibrillogenic protein structures. There is evidence that low doses (<100 mM) of trehalose may inhibit hIAPP α-helix to β-sheet rich conformational transitions and delay amyloid growth. By contrast, high doses (>500 mM) of trehalose induce conformational transitions towards β-sheet rich hIAPP structures that seed hIAPP fibril growth566. This behaviour has been observed for other osmolytes567,568 and is in accordance with the hypothesis that low concentrations of trehalose may replace water molecules close to hIAPP, bind the peptide chain through a hydrogen bond network, reduce protein-protein interactions, and eventually delay fibril growth. By contrast, at high concentrations trehalose molecules may self assemble to form clusters that, acting as crowding agents, could seed hIAPP fibrillation. Although some of the trehalose protective effects may be explained by its chemical chaperone properties, the biochemical mechanisms at the root of its effects on pancreatic β-cells remain largely unknown. When treated with trehalose transgenic mice expressing hIAPP showed an increased clearance of hIAPP amyloid oligomers in pancreatic islets and increased autophagy.569 Moreover, these mice exhibited a better metabolic profile, glucose consumption and a higher β-cell viability compared with their untreated counterparts.570 While the role of trehalose as an autophagy enhancer is widely recognized, its effects in modulating proteasome activity are still controversial. Administration of trehalose to neuroblastoma cells was reported to revert the accumulation of polyubiquitinated proteins induced by epoxomicin, a known proteasome inhibitor.571 In vitro studies showed that the protective effects of trehalose against toxic protein aggregation in neuroblastoma cells were turned off by treatment with the proteasome inhibitor MG132.572 Similar effects were also observed in liver derived cultured cells, where trehalose reduced apoptosis in samples treated with a proteasome inhibitor.572 However, further studies are needed to confirm the ability of trehalose to activate the isolated 20S proteasome.
7.5. Ghrelin
Ghrelin is 28 residues peptide hormone (sequence: FLSPEHQRVQQRKESKKPPAKLQPR) produced in X/A cells from the stomach. Ghrelin plays a role in modifying insulin and glucose metabolism, blood pressure lipid homeostasis, and phlogistic processes.573 Notably, ghrelin levels are lowered in individuals with T2D. Moreover, ghrelin has a multifaceted impact in metabolism regulation, influencing gastric emptying, vascular resistance and memory persistence.574 Notably, recent work conducted on neuroblastoma cells has demonstrated that ghrelin has neuroprotective and anti-apopototic properties. In the same work, it was also demonstatred that ghrelin activates the proteasome and modulate the UPS/autophagy crosstalk.575 Finally, exogenous ghrelin stimulates autophagy in rat cortical neurons.576
7.6. Epigallocatechin gallate
Epigallocatechin gallate (EGCG) is natural compound present in green tea (7380 mg per 100 g of dried leaves). The multifaceted roles of EGCG in i) decreasing glucose uptake, ii) regulating lipid metabolism, iii) mitochondrial failure, iv) oxidative stress and v) inflammation are widely recognized.577 EGCG is known to interfere with the toxic hIAPP aggregation pathway. In particular, upon interacting with EGCG, hIAPP undergoes off-pathway aggregation causing the formation of non toxic, amorphous aggregates differently from mature fibrils which exhibit a twisted fibrillar morphology.325 Moreover, biophysical assays conducted on different amyloidogenic proteins including hIAPP, α-Synuclein, prions, huntingtin, and Aβ peptide, suggest that the inhibitory effect of EGCG may be ascribed to a common mechanism in which the ligand binds aggregation-competetnt intermediates characterized by cross-β sheet structures.578 The role played by EGCG in modulating proteasome activity is controversial. Some authors have reported that EGCG is a proteasome inhibitor in vitro, and has the ability to activate aopototic processes by promoting the accumulation of polyubiquitinated substrates in carcinoma cell lines.579 Moreover, molecular simulations have also shown that the ester carbon in EGCG plays a major role in driving interactions with the 20S proteasome.580 More recently, EGCG was found to effectively counteract the ability of bortezomib to inhibit the 20S proteasome, thus suggesting a complex effect of EGCG on proteasome activity.581 These apparently controversial results may be explained by the poor EGCG stability under physiological conditions. Indeed, a biologically active derivative of EGCG was observed to form in neutral and neutral-alkaline conditions with different activities on several cellular processes although further investigations are needed to better understand EGCG/proteasome interactions at a molecular level.579 By contrast, the role of EGCG in activating autophagy is firmly established. EGCG promotes the formation of autophagosomes also by regulating the cytosolic levels of two key proteins involved in autophagy (LC-3 and beclin).582
7.7. Resveratrol
Resveratrol (trans-3,5,4 trihydroxy-trans-stilbene) is a member of a family of natural small molecules (polyphenols) known for their antioxidant and cytoprotective effects which are known to inhibit amyloid formation.583 Resveratrol, in particular, has been shown to have remarkable beneficial effects on patients affected by T2D584, and a large number of studies have confirmed that resveratrol inhibits hIAPP aggregation in buffer solution in vitro397 and in the presence of model membranes.585 Molecular simulations have also shown that resveratrol binds the segment of hIAPP encompassing residues 22–27 and inhibits amyloid aggregation by interfering with inter-peptide β-sheet interactions driven by stacking of its aromatic rings.586 Resveratrol is also known to be a proteasome inhibitor, similar to other polyphenols,587 butits role in regulating amyloid clearance is poorly understood. In a recent report, the beneficial effects of resveratrol in mitigating ischemia or myocardial damage in streptozotocin-induced diabetic rats was ascribed to enhanced autophagy.588 A recent study also addressed the effect of resveratrol in promoting autophagy-mediated hIAPP degradation, using a pancreatic cell line (hIAPP-INS1) obtained by transfecting INS1 cells with a hIAPP overexpressing lentivirus.589 hIAPP amyloid deposition was significantly decreased in hIAPP-INS1 cells after treatment with resveratrol. Concomitantly, insulin secretion and glucose metabolism were restored. Of note, if autophagy was inhibited by 3-methyladenine, resveratrol was ineffective in alleviating hIAPP amyloid deposition and did not restore insulin secretion, indicating the role of autophage for removal of hIAPP aggregates.
7.8. Silybins
Flavonolignans are a family of natural compounds with attractive pharmacological properties.590 Silybins are likely the most known members of this class of compounds. A 1:1 mixture of two diastereoisomers, Silybin A and Silybin B, collectively known as silibinin, are the major components of silymarin, an extract from the milk thistle of Silybum Marianum. Silibinin has been recently shown to ameliorate glucose metabolism pathways and its use has been proposed as a promising treatment for patients affected by T2D.591 In particular, silibinin protected INS1 cells from hIAPP toxicity by a mechanism involving an enhanced expression of apoptosis-associated signals.592 Ion mobility mass spectrometry (ESI-IMS-MS) experiments demonstrated that Silybin may bind most effectively to a highly extended population of hIAPP peptides, altering the aggregation/fibrillization process.156 Moreover, in vitro studies have also shown the ability of Silybin to retard hIAPP aggregation and to modulate the structure of the monomeric peptide.563 The anti-aggregation and anti-amyloidogenic effects of Silybin were also observed for Aβ, reinformcing the similarities in the aggregation of these peptides.593 There are few papers regarding the effect of sylibins on proteasome activity. Some biochemical assays have demonstrated that silibinin may inhibit Platelet-Derived Growth Factor-Driven (PDGF) cell proliferation in human fibroblasts by enhancing the UPS activity, albeit information about a direct silibyn/proteasome interactions is largely incomplete.594 Silibinin is also known as an effective autophagy enhancer, since as silibinin treatment rescued glucose metabolism in streptozotocin-induced diabetic mice.595
6.2. Natural compounds as IAPP anti-aggregation and anti-oxidant agents
Chemical structures of the multifunctional compounds discussed in the main text.
8. CONCLUSION AND PERSPECTIVES
Why is it difficult to develop drugs for pathological amyloids?
Evidence has emerged from simulations with atomistic detail596 and coarse-grained models597 that amyloid self-assembly is kinetically controlled, with the final amyloid fibril structure not necessarily being at the global minimum of the free energy.598 In contrast to protein folding in which the same native structure is usually found irrespective of solution conditions and how folding is controlled, amyloid fibrils can form from the same, or very similar sequence, with an array of different structures dependenting on the growth conditions. Single-point mutations and/or minor changes in external factors, e.g., temperature, pH, buffer composition, growth under agitated or quiescent conditions, can thus result in different fibrillar structures, i.e., polymorphism, which has been observed in simulations597 and experiments,599 including for hIAPP. In a cellular context, fibril formation can also depend on the cell type, cell age and subcellular localization of the aggregation process. In contrast, for most globular proteins the folding process and the proteins’ final state, i.e., the folded structure, are usually robust to sequence changes and external/cellular conditions. It is plausible, therefore, that a small molecule modulator of amyloid aggregation could shift the free energy barriers along the self-assembly process, which could result in a new fibril form, potentially formed via a new pathway in which more or less toxic oligomeric intermediates are formed. As an example, a small molecule or peptide that hinders the formation of fibrillar aggregates might promote the self-assembly of long-lived and toxic oligomeric species. The failures in clinical studies of antibodies and small molecules, that were developed to prevent the formation of pathological amyloids, may have resulted, at least in part, from the lack of knowledge on the nature of these toxic species and the kinetics of their formation. The recent cryoEM structures of hIAPP fibrils166,167,168 and nanodisc-trapped oligomer349 will open the door to the design of small molecules that target amylin fibrils and oligomers, respectively, allowing clarification of the contribution of fibrils to the pathogenesis associated with T2D.
The relevance of the kinetics of fibrillogenesis calls for more detailed time-resolved experiments, backed up with the atomistic resolution of simulation, to determine how fibrils are formed and how the solution conditions determine the mechanism of fibril formation. Time-resolved biophysical measurements and concomitant cellular assays have been employed to shed light on some toxic species.600 Importantly, these experiments have provided evidence that cytotoxic hIAPP oligomers contain a modest β-sheet content, and thus anti-amyloid agents, i.e. molecules developed to hinder cross-β fibril formation, could potentially extend the lifetime of toxic aggregation intermediates of hIAPP amyloid assembly. Conversely, a small molecule (or antibody) that stabilizes fibrillar structures might reduce the population of toxic oligomeric species, but would increase the amyloid burden. One example of small molecule stabilizer of amyloid fibrils was designed by MD simulations and validated in vivo.601 Specifically, using umbrella sampling MD for a series of polythiophene derivatives functionalized with negatively charged carboxyl groups, the calculated free energy of binding of the compounds to amyloid fibrils showed a remarkable correlation with their efficacy in a mouse model of the prion disease.601
9. OUTLOOK
In this review, we have presented the molecular determinants of hIAPP amyloid growth by dissecting the sequence composition of the hIAPP polypeptide chain, the properties of the various hIAPP sequence substitutions and the specific interactions they establish, and the conformational features of monomeric, oligomeric and fibrillar hIAPP. Of course, there are other factors that are likely to modulate the growth of the hIAPP into amyloid, including glycosylation602 and fragmentation of hIAPP fibrils.603 hIAPP amyloid formation can also be influenced by interactions with other molecules, including the possible co-aggregation of hIAPP with amyloid-β, with which hIAPP shares about 50% sequence similarlity.60,604–607 Finally, it should be noted that almost all the experiments described above were conducted in vitro, leaving open the question of how hIAPP aggregates in vivo. It is likely that cellular and physiological factors that are difficult to reproduce in vitro could play a significant role in the growth of hIAPP into amyloid in vivo under physiological and pathological conditions. For example, insulin is known to inhibit hIAPP amyloid formation.520,521,608 There are also data indicating that hIAPP amyloid formation initially takes place intracellularly in endoplasmic reticulum, golgi or secretory vesicles126,130, and involved both the mature hIAPP sequence, as well as proIAPP131 (a 64 amino acid peptide in which IAPP is flanked by two short propeptides).609,610 Consequently, the properties of the amylin precursor and its possible interaction with mature hIAPP need also to be studied in the future.
10. FUTURE DIRECTIONS
The future directions for hIAPP research and its relevance to T2DM are many-fold. Key areas for future research endeavors could include: (a) Stabilizing hIAPP aggregation intermediates: Studies focusing on developing approaches to stabilize hIAPP oligomers under various conditions and solving their high-resolution structures would be highly valuable in the development of potent amyloid inhibitors that can be used to suppress the cell toxicity and potentially to contribute to therapeutic strategies for T2DM. (b) Solving structures of hIAPP formed at the membrane: The use of lipid-nanodiscs to obtain high-resolution structural insights of various intermediates and pore-forming structures could provide insights into the toxic mechanism of hIAPP. (c) Structural studies on hIAPP aggregates extracted from islet cells to explore the variety of amyloid fibril structures formed in a cellular setting. (d) Elucidating the mechanism(s) of hIAPP aggregation by combining biophysical techniques including cryoEM, solid-state NMR, EPR and MD simulations. (e) In-cell studies to probe hIAPP aggregation and the use of animal models to better understand the molecular origins of islet toxicity and the effect of small molecule compounds on hIAPP-induced cell loss. (f) Studies probing the roles of cofactors such as metabolites, carbohydrates, small molecules, metals, insulin, chaperones, free lipids and other crowding agents. (g) Probing cross-seeding effects of other amyloid proteins including amyloid-β and insulin, and the effects of antimicrobial peptides such as LL-37 and defensins. (h) Systematic screening of small molecule compounds, nanoparticles, and enzymes to inhibit hIAPP aggregation and also clear pre-existing aggregates. (i) Develop better understanding of the mechanisms of hIAPP degradation by proteolysis, including IDE, proteasome and autophagy. (j) Design and characterize small molecules that could rescue an impaired hIAPP proteostasis by enhancing the activity of proteolytic systems. (k) A cross-disease analysis of apparently unrelated pathologies (including neurodegenerative disorders) to provide hints for a deeper understanding of the factors affecting hIAPP accumulation and suggest what drugs may be repurposed to treat T2DM. What emerges from all of this, and we hope clearly annotated in this review, is that there is still much to be learned about hIAPP as a small, but crucial peptide hormone, that if left unchecked, can contribute to one of the most serious of diseases facing the mankind today.
Acknowledgments
Research in A.C. lab is supported by an Excellence Grant of the Swiss National Science Foundation (310030B-189363 to A.C.). S.E.R., Y.X. and R.G. are supported by funds from Wellcome (no. 204963). Research in G.W. and P.W. labs is supported by Novo Nordisk (grant number: NNF 180c0034256), the Swedish Diabetes Foundation (grant number: DIA 2017–296) and Family Ernfors Fund. Research in A.R. lab is supported by funds from NIH (AG048934 to A.R.).
Biographies
Danilo Milardi received his Ph.D. in Physical Chemistry from the University of Catania (Italy) in 1996. After completing his postdoctoral position at the University of Catania, in 2001 he was appointed Researcher at the National Research Council of Italy (CNR). His current research activity focuses on the molecular mechanisms regulating protein misfolding and amyloid assembly, protein-membrane interactions, and protein clearance by proteasome and proteases.
Ehud Gazit is a Professor and Endowed Chair at the Shmunis School of Biomedicine and Cancer Research, Faculty of Life Sciences and the Department of Materials Science and Engineering, Faculty of Engineering at Tel Aviv University. He received his B.Sc. (summa cum laude) after completing his studies at the Special Program for Outstanding Students of Tel Aviv University and his Ph.D. (with highest distinction) at the Department of Membrane Research and Biophysics, Weizmann Institute of Science in 1997. He has been a faculty member at Tel Aviv University since 2000, after completing his postdoctoral studies at the Massachusetts Institute of Technology where he also had held a visiting appointment (2002–2011). He also had a visiting appointment at St John’s College, Cambridge University, UK (2016). Gazit had received numerous awards and honors including Landau Research Award, Dan David Scholarship Award for Young Researchers, Hestrin Award for Young Scientists, Research Prize Award Named After “Teva” Founders, Rappaport Prize for Excellence in the Field of Biomedical Research and Kadar Family Award for Outstanding Research. He is a fellow of the Royal Society of Chemistry, a member of the European Molecular Biology Organization, and a Foreign Fellow of the National Academy of Sciences, India. In 2015, he was knighted by the Italian Republic for his service to science and society.
Sheena Radford is Astbury Professor for Biophysics at the University of Leeds and Director of the Astbury Centre for Structural Molecular Biology which unites chemists, physicist, biologists and medics, working together to understand life in molecular detail. She has worked in the field of protein folding and misfolding using different biophysical and structural methods for more than 25 years.
Yong Xu is a medicinal chemist and postdoctoral research fellow in the Astbury Centre for Structural Molecular Biology at the University of Leeds. He completed his PhD at King’s College London in 2017 and he received his Master’s degree from Sichuan University in 2013. His current interest focuses on the development of novel small molecules to modulate amyloid formation.
Rodrigo Gallardo is a biophysicist and structural biologist, currently working as postdoctoral research fellow in the Astbury Centre for Structural Molecular Biology at the University of Leeds. He worked previously as a postdoc in the group of Joost Schymkowitz and Frederic Rousseau at the VIB Switch Laboratory, KU Leuven, developing the Targeted-Protein-Aggregation technology and synthetic amyloids. His current work focuses on using cryo-EM to analyse amyloid structures formed in vitro and in vivo, making full use of the two Titan Krios EMs in the Astbury Biostructure Laboratory.
Amedeo Caflisch studied physics at the ETH in Zurich. During 1992–1994 he was a postdoctoral fellow in the group of Martin Karplus at Harvard University. In 1996 he was offered an assistant professorship position at the Department of Biochemistry of the University of Zurich, and promoted to full professor in 2001. His research team is known for the development of methods for enhanced sampling molecular dynamics and data-driven analysis of simulations of protein folding, aggregation, and ligand binding. The group has also developed fragment-based docking protocols, which have been applied to proteases, kinases, protein-protein, and protein-RNA recognition domains. In 2013, at the age of 50, he decided to start experimental activities in his group (from gene to structure), which have resulted in the crystal structures of nearly 300 protein/small-molecule ligand complexes. Most of the ligands in these complexes originate from docking.
Gunilla T Westermark, Ph.D., is a Professor of Medical Cell Biology at Uppsala University. Her research interest is in islet amyloid and its implication in beta cell death and diabetes development. She is also interested in IAPP’s possible role in Alzheimer’s and Parkinson’s diseases. Furthermore, she studies the transmission of amyloid using in model systems such as mice, Drosophila melanogaster, and Caenorhabditis elegans.
Per Westermark, MD, PhD is a Professor (em) of Pathology, Uppsala University. Consultant of Clinical Pathology, Uppsala University Hospital. Editor-in-Chief, Amyloid, the Official Journal of the International Society of Amyloidosis (ISA). Chairman of the Nomenclature Committee of ISA.
Carmelo La Rosa received a master’s degree in Chemistry and PhD in Physical-Chemistry from the University of Catania (Italy) working on lyotropic liquid crystals. After completing postdoctoral training on thermodynamics and kinetics of proteins unfolding at the University of Catania and Leiden University (The Netherland), he joined the department of Chemical Sciences, University of Catania. His current research focuses on the biophysics of amyloidogenic proteins and their interaction with model membrane.
Ayyalusamy Ramamoorthy is the Robert W. Parry Collegiate Professor of Chemistry and Biophysics, and Faculty of Macromolecular Science and Engineering, and Biomedical Engineering, at the University of Michigan, Ann Arbor. He joined the University of Michigan after getting his PhD in Chemistry from the Indian Institute of Technology (Kanpur, India), working in the Central Leather Research Institute (Chennai/Madras, India) and JEOL Ltd (Tokyo, Japan), and completing postdoctoral training at the University of Pennsylvania, Philadelphia. His current research focuses on the structural biophysics of amyloid proteins and membrane proteins and on the development and applications of solid-state NMR spectroscopy.
Contributor Information
Danilo Milardi, Consiglio Nazionale delle Ricerche, Istituto di Cristallografia, Via P. Gaifami 18, 95126 Catania, Italy..
Ehud Gazit, Department of Molecular Microbiology and Biotechnology, George S. Wise Faculty of Life Sciences, Tel Aviv University, Tel Aviv 6997801, Israel..
Rodrigo U. Gallardo, Astbury Centre for Structural Molecular Biology, School of Molecular and Cellular Biology, University of Leeds, Leeds LS2 9JT, UK.
Amedeo Caflisch, Department of Biochemistry, University of Zürich, Zürich CH-8057, Switzerland..
Gunilla T. Westermark, Department of Medical Cell Biology and Immunology, Uppsala University, SE-751 23 Uppsala, Sweden.
Per Westermark, Department of Immunology, Genetics and Pathology, Uppsala University, SE-751 85 Uppsala, Sweden..
Carmelo La Rosa, Università degli Studi di Catania, Dipartimento di Scienze Chimiche, Viale Andrea Doria 6, 95125 Catania, Italy..
Ayyalusamy Ramamoorthy, Biophysics, Department of Chemistry, Biomedical Engineering, Macromolecular Science and Engineering, University of Michigan, Ann Arbor, MI 41809-1055, USA..
References
- (1).Schwartz P New patho-anatomic observations on amyloidosis in the aged. Fluorescence microscopic investigations. In Amyloidosis; Mandema E, Ruinen L, Scholten JH, Cohen AS, Eds.; Excerpta Medica: Amsterdam, 1968; pp 400–15. [Google Scholar]
- (2).Westermark P Islet Amyloid Polypeptide and Amyloid in the Islets of Langerhans; Leslie RDG, Robbins D, Eds.; Cambridge University Press: Cambridge, UK, 1995. [Google Scholar]
- (3).Sengupta U; Nilson AN; Kayed R The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Scollo F; La Rosa C Amyloidogenic Intrinsically Disordered Proteins: New Insights into Their Self-Assembly and Their Interaction with Membranes. Life 2020, 10, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Klaips CL; Jayaraj GG; Hartl FU Pathways of Cellular Proteostasis in Aging and Disease. J. Cell Biol 2018, 217, 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Singal DP; Blajchman MA Histocompatibility (HL-A) Antigens, Lymphocytotoxic Antibodies and Tissue Antibodies in Patients with Diabetes Mellitus. Diabetes 1973, 22, 429–432. [DOI] [PubMed] [Google Scholar]
- (7).Nerup J; Platz P; Andersen OO; Christy M; Lyngsoe J; Poulsen JE; Ryder LP; Nielsen LS; Thomsen M; Svejgaard A HL-A Antigens and Diabetes Mellitus. Lancet 1974, 2, 864–866. [DOI] [PubMed] [Google Scholar]
- (8).Aly TA; Ide A; Jahromi MM; Barker JM; Fernando MS; Babu SR; Yu L; Miao D; Erlich HA; Fain PR; Barriga KJ; Norris JM; Rewers MJ; Eisenbarth GS Extreme Genetic Risk for Type 1A diabetes. Proc. Natl. Acad. Sci. USA 2006, 103, 14074–14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Erlich H; Valdes AM; Noble J; Carlson JA; Varney M; Concannon P; Mychaleckyj JC; Todd JA; Bonella P; Fear AL; Lavant E; Louey A; Moonsamy P; Consortium., T. D. G. HLA DR-DQ Haplotypes and Genotypes and Type 1 Diabetes Risk: Analysis of the Type 1 Diabetes Genetics Consortium Families. Diabetes 2008, 57, 1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bishay RH; Greenfield JR A Review of Maturity Onset Diabetes of the Young (MODY) and Challenges in the Management of Glucokinase-MODY. Med. J. Aus.t 2016, 205, 480–485. [DOI] [PubMed] [Google Scholar]
- (11).Stoffel M; Duncan SA The Maturity-Onset Diabetes of the Young (MODY1) Transcription Factor HNF4alpha Regulates Expression of Genes Required for Glucose Transport and Metabolism. Proc. Natl. Acad. Sci. USA 1997, 94, 13209–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Stoffel M; Froguel P; Takeda J; Zouali H; Vionnet N; Nishi S; Weber IT; Harrison RW; Pilkis SJ; Lesage S Human Glucokinase Gene: Isolation, Characterization, and Identification of Two Missense Mutations Linked to Early-Onset Non-Insulin-Dependent (Type 2) Diabetes Mellitus. Proc. Natl. Acad. Sci. USA 1992, 89, 7698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Yamagata K; Oda N; Kaisaki PJ; Menzel S; Furuta H; Vaxillaire M; Southam L; Cox RD; Lathrop GM; Boriraj VV; et al. Mutations in the Hepatocyte Nuclear Factor-1alpha Gene in Maturity-Onset Diabetes of the Young (MODY3. Nature 1996, 384, 455–458. [DOI] [PubMed] [Google Scholar]
- (14).www.diabetesatlas.org, IDF Diabetes Atlas; 2019.
- (15).Schwartz MW; Porte DJ Diabetes, Obesity, and the Brain. Science 2005, 307, 375–379. [DOI] [PubMed] [Google Scholar]
- (16).Tuomi T; Santoro N; Caprio S; Cai M; Weng J; L., G. The Many Faces of Diabetes: A Disease with Increasing Heterogeneity. Lancet 2014, 383, 1084–1094. [DOI] [PubMed] [Google Scholar]
- (17).Kahn SE; Prigeon RL; Schwartz RS; Fujimoto WY; Knopp RH; Brunzell JD; Porte DJ Obesity, Body Fat Distribution, Insulin Sensitivity and Islet Beta-Cell Function as Explanations for Metabolic Diversity. J. Nutr 2001, 131, 354–360. [DOI] [PubMed] [Google Scholar]
- (18).Novel Subgroups of Adult-Onset Diabetes and Their Association with Outcomes: A Data-Driven Cluster Analysis of Six Variables. Lancet Diabetes Endocrinol 2018, 6, 361–369. [DOI] [PubMed] [Google Scholar]
- (19).Prasad RB; Groop L Precision Medicine in Type 2 Diabetes. J. Intern. Med 2019, 285, 40–48. [DOI] [PubMed] [Google Scholar]
- (20).Skyler JS; Bakris GL; Bonifacio E; Darsow T; Eckel RH; Groop L; Groop PH; Handelsman Y; Insel RA; Mathieu C; McElvaine AT; Palmer JP; Pugliese A; Schatz DA; Sosenko JM; Wilding JP; Ratner RE Differentiation of Diabetes by Pathophysiology, Natural History, and Prognosis. Diabetes 2017, 66, 241–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Westermark P; Grimelius L The Pancreatic Islet Cells in Insular Amyloidosis in Human Diabetic and Non-Diabetic Adults. Acta Path. Microbiol. Scand. A 1973, 81, 291–300. [DOI] [PubMed] [Google Scholar]
- (22).Clark A; Wells CA; Buley ID; Cruickshank JK; Vanhegan RI; Matthews DR; Cooper GJS; Holman RR; Turner RC Islet Amyloid, Increased A-Cells, Reduced β-Cells and Exocrine Fibrosis: Quantitative Changes in the Pancreas in Type 2 Diabetes. Diab. Res 1988, 9, 151–159. [PubMed] [Google Scholar]
- (23).Butler AE; Janson J; Bonner-Weir S; Ritzel R; Rizza RA; Butler PC B-Cell Deficit and Increase B-Cell Apoptosis in Humans with Type 2 Diabetes. Diabetes 2003, 52, 102–110. [DOI] [PubMed] [Google Scholar]
- (24).Haeften TW; Pimenta W; Mitrakou A; Korytkowski M; Jenssen T; Yki-Jarvinen H; Gerich JE Relative Conributions of Beta-Cell Function and Tissue Insulin Sensitivity to Fasting and Postglucose-Load Glycemia. Metabolism 2000, 49, 1318–1325. [DOI] [PubMed] [Google Scholar]
- (25).Yoshioka N; Kuzuya T; Matsuda A; Taniguchi M; Iwamoto Y Serum Proinsulin Levels at Fasting and after Oral Glucose Load in Patients with Type 2 (Non-Insulin-Dependent) Diabetes Mellitus. Diabetologia 1988, 31, 355–360. [DOI] [PubMed] [Google Scholar]
- (26).Bell GI; Kayano T; Buse JB; Burant CF; Takeda J; Lin D; Fukumoto H; Seino S Molecular Biology of Mammalian Glucose Transporters. Diabetes Care 1990, 13, 198–208. [DOI] [PubMed] [Google Scholar]
- (27).Zierath JR; Houseknecht KL; Gnudi L; Kahn BB High-Fat Feeding Impairs Insulin-Stimulated GLUT4 Recruitment via an Early Insulin-Signaling Defect. Diabetes 1997, 46, 215–223. [DOI] [PubMed] [Google Scholar]
- (28).Johnson JH; Newgard CB; Milburn JL; Lodish HF; Thorens B The High Km Glucose Transporter of Islets of Langerhans Is Functionally Similar to the Low Affinity Transporter of Liver and Has an Identical Primary Sequence. J. Biol. Chem 1990, 265, 6548–6551. [PubMed] [Google Scholar]
- (29).Papatheodorou K; M B; Bekiari E; Rizzo M; Edmonds M Complications of Diabetes 2017. J. Diabetes Res 2018, 11, 3086167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hanefeld M; Fleischmann H; Siegmund T; Seufert J Rationale for Timely Insulin Therapy in Type 2 Diabetes within the Framework of Individualised Treatment: 2020 Update. Diabetes Ther 2020. 11, pages 1645–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Rizos CV; Kei A; Elisaf MS The Current Role of Thiazolidinediones in Diabetes Management. Arch. Toxicol 2016, 90, 1861–1881. [DOI] [PubMed] [Google Scholar]
- (32).Wang S; Dougherty EJ; Danner RL PPARγ Signaling and Emerging Opportunities for Improved Therapeutics. Pharmacol Res 2016, 111, 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nesto RW; Bell D; Bonow RO; Fonseca V; Grundy SM; Horton ES; Le Winter M; Porte D; Semenkovich CF; Smith S; Young LH; Kahn R Thiazolidinedione Use, Fluid Retention, and Congestive Heart Failure: A Consensus Statement from The American Heart Association and American Diabetes Association; Diabetes Care, 2004, 27, 256–263. [DOI] [PubMed] [Google Scholar]
- (34).MacDonald PE; El-kholy W; Riedel MJ; Salapatek AMF; Light PE; Wheeler MB The Multiple Actions of GLP-1 on the Process of Glucose-Stimulated Insulin Secretion. Diabetes 2002, 51, S434–S442. [DOI] [PubMed] [Google Scholar]
- (35).Deacon CF Circulation and Degradation of GIP and GLP-1. Horm. Metab. Res 2004, 36, 761–765. [DOI] [PubMed] [Google Scholar]
- (36).Garber AJ Long-Acting Glucagon-like Peptide 1 Receptor Agonists: A Review of Their Efficacy and Tolerability. Diabetes Care 2011, 2, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bahne E; Sun EWL; Young RL; Hansen M; Sonne DP; Hansen JS; Rohde U; Liou AP; Jackson ML; Fontgalland, et al. Metformin-Induced Glucagon-like Peptide-1 Secretion Contributes to the Actions of Metformin in Type 2 Diabetes. JCI Insight 2018, 6;3, e93936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Brown E; Rajeev SP; Cuthbertson DJ; Wilding JPH A Review of the Mechanism of Action, Metabolic Profile and Haemodynamic Effects of Sodium-Glucose Co-Transporter-2 Inhibitors. Diabetes Obes. Metab 2019, 21, 9–18. [DOI] [PubMed] [Google Scholar]
- (39).Lam CSP; Chandramouli C; Ahooja V; Verma S SGLT-2 Inhibitors in Heart Failure: Current Management, Unmet Needs, and Therapeutic Prospects. J. Am. Heart Assoc 2019, 8, e013389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Goncalves E; Bell DSH Combination Treatment of SGLT2 Inhibitors and GLP-1 Receptor Agonists: Symbiotic Effects on Metabolism and Cardiorenal Risk. Diabetes Ther 2018, 9, 919–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Nordisk. Novo Nordisk successfully completes AM833 phase 2 trial and phase 1 combination trial with AM833 and semaglutide in obesity https://ml-eu.globenewswire.com/Resource/Download/265c351d-c721-43e0-ab7d-ab841a44d72a.
- (42).Nally LM; Sherr JL; Van Name MA; Patel AD; Tamborlane WV Pharmacologic Treatment Options for Type 1 Diabetes: What’s New? Expert. Rev. Clin. Pharmacol 2019, 12, 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ott A; Stolk RP; Hofman A; Harskamp F; Grobbee DE; Breteler MM Association of Diabetes Mellitus and Dementia: the Rotterdam Study. Diabetologia 1996, 39, 1392–1397. [DOI] [PubMed] [Google Scholar]
- (44).Leibson CL; Rocca WA; Hanson VA; Cha R; Kokmen E; O’Brien PC Risk of Dementia among Persons with Diabetes Mellitus: a Population-Based Cohort Study. Am. J. Epidemiol 1997, 145, 301–308. [DOI] [PubMed] [Google Scholar]
- (45).Peila R; Rodriguez BL; Launer LJ Type 2 Diabetes, APOE Gene, and the Risk for Dementia and Related Pathologies: The Honolulu-Asia Aging Study. Diabetes 2002, 51, 1256–1262. [DOI] [PubMed] [Google Scholar]
- (46).Janson J; Laedtke T; Parisi JE; O’Brien P; Petersen RC; Butler PC Increased Risk of Type 2 Diabetes in Alzheimer Disease. Diabetes 2004, 53, 474–481. [DOI] [PubMed] [Google Scholar]
- (47).Hu G; Jousilahti P; Bidel S; Antikainen R; Tuomilehto J Type 2 Diabetes and the Risk of Parkinson’s Disease. Diabetes Care 2007, 30, 842–847. [DOI] [PubMed] [Google Scholar]
- (48).Schernhammer E; Hansen J; Rugbjerg K; Wermuth L; Ritz B Diabetes and the Risk of Developing Parkinson’s Disease in Denmark. Diabetes Care 2011, 34, 1102–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).De Pablo-Fernandez E; Goldacre R; Pakpoor J; Noyce AJ; Warner TT Association between Diabetes and Subsequent Parkinson Disease. Neurology 2018, 91, e139. [DOI] [PubMed] [Google Scholar]
- (50).Ott A; Stolk RP; Van Harskamp F; Pols HAP; Hofman A; Breteler MMB Diabetes Mellitus and the Risk of Dementia the Rotterdam Study. Neurology 1999, 53, 1937–1937. [DOI] [PubMed] [Google Scholar]
- (51).Rönnemaa E; Zethelius B; Sundelöf J; Sundström J; Degerman-Gunnarsson M; Berne C; Lannfelt L; Kilander L Impaired Insulin Secretion Increases the Risk of Alzheimer Disease. Neurology 2008, 71, 1065–1071. [DOI] [PubMed] [Google Scholar]
- (52).Raimundo AF; Ferreira S; Martins IC; Menezes R Islet Amyloid Polypeptide: A Partner in Crime with Aβ in the Pathology of Alzheimer’s Disease. Front. Mol. Neurosci 2020, 13, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Stanciu GD; Bild V; Ababei DC; Rusu RN; Cobzaru A; Paduraru L; Bulea D Link between Diabetes and Alzheimer’s Disease Due to the Shared Amyloid Aggregation and Deposition Involving Both Neurodegenerative Changes and Neurovascular Damages. J. Clin. Med 2020, 9, 1713–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Hanyu H Diabetes-Related Dementia. Adv. Exp. Med. Biol 2019, 1128, 147–160. [DOI] [PubMed] [Google Scholar]
- (55).O’Nuallian B; Williams AD; Westermark P; Wetzel R Seeding Specificity in Amyloid Growth Induced by Heterologous Fibrils. J. Biol. Chem 2004, 279, 17490–17499. [DOI] [PubMed] [Google Scholar]
- (56).Bharadwaj P; Solomon T; Sahoo BR; Ignasiak K; Gaskin S; Rowles J; Verdile G; Howard MJ; Bond CS; Ramamoorthy A; Martins RN; Newsholme P Amylin and Beta Amyloid Proteins Interact to Form Amorphous Heterocomplexes with Enhanced Toxicity in Neuronal Cells. Sci. Rep 2020, 10, 10356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ivanova MI; Lin Y; Lee Y-H; Zheng J; Ramamoorthy A Biophysical Processes Underlying Cross-Seeding in Amyloid Aggregation and Implications in Amyloid Pathology. Biophys. Chem 2020, in press, doi: 10.1016/j.bpc.2020.106507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Yan L-M; Velkova A; Tatarek‐Nossol M; Andreetto E; Kapurniotu A IAPP Mimic Blocks Aβ Cytotoxic Self-Assembly: Cross-Suppression of Amyloid Toxicity of Aβ and IAPP Suggests a Molecular Link between Alzheimer’s Disease and Type II Diabetes. Angew. Chem. Int. Ed 2007, 46, 1246–1252. [DOI] [PubMed] [Google Scholar]
- (59).Jackson K; Barisone GA; Diaz E; Jin LW; DeCarli C; Despa F Amylin Deposition in the Brain: A Second Amyloid in Alzheimer Disease? Ann Neurol 2013, 74, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Oskarsson ME; Paulsson JF; Schultz SW; Ingelsson M; Westermark P; Westermark GT In Vivo Seeding and Cross-Seeding of Localized Amyloidosis: A Molecular Link between Type 2 Diabetes and Alzheimer Disease. Am. J. Pathol 2015, 185, 834–846. [DOI] [PubMed] [Google Scholar]
- (61).Abedini A; Raleigh DP A Critical Assessment of the Role of Helical Intermediates in Amyloid Formation by Natively Unfolded Proteins and Polypeptides. Protein Eng. Des. Sel 2009, 22, 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Guerrero-Muñoz MJ; Castillo-Carranza DL; Sengupta U; White MA; Kayed R Design of Metastable β-Sheet Oligomers from Natively Unstructured Peptide. ACS Chem. Neurosci 2013, 4, 1520–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Kayed R Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [DOI] [PubMed] [Google Scholar]
- (64).Baglioni S; Casamenti F; Bucciantini M; Luheshi LM; Taddei N; Chiti F; Dobson CM; Stefani M Prefibrillar Amyloid Aggregates Could Be Generic Toxins in Higher Organisms. J. Neurosci 2006, 26, 8160–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Hardy J The Amyloid Hypothesis for Alzheimer’s Disease: A Critical Reappraisal: Amyloid Hypothesis for Alzheimer’s Disease. J. Neurochem 2009, 110, 1129–1134. [DOI] [PubMed] [Google Scholar]
- (66).Scalisi S; Sciacca MFM; Zhavnerko G; Grasso DM; Marletta G; La Rosa C Self-Assembling Pathway of HiApp Fibrils within Lipid Bilayers. ChemBioChem 2010, 11, 1856–1859. [DOI] [PubMed] [Google Scholar]
- (67).Soong R; Brender JR; Macdonald PM; Ramamoorthy A Association of Highly Compact Type II Diabetes Related Islet Amyloid Polypeptide Intermediate Species at Physiological Temperature Revealed by Diffusion NMR Spectroscopy. J. Am. Chem. Soc 2009, 131, 7079–7085. [DOI] [PubMed] [Google Scholar]
- (68).Padrick SB; Miranker AD Islet Amyloid: Phase Partitioning and Secondary Nucleation Are Central to the Mechanism of Fibrillogenesis. Biochemistry 2002, 41, 4694–4703. [DOI] [PubMed] [Google Scholar]
- (69).Evans ML; Schmidt JC; Ilbert M; Doyle SM; Quan S; Bardwell JCA; Jakob U; Wickner S; Chapman MRE Coli Chaperones DnaK, Hsp33 and Spy Inhibit Bacterial Functional Amyloid Assembly. Prion 2011, 5, 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Lasagna-Reeves CA; Clos AL; Castillo-Carranza D; Sengupta U; Guerrero-Muñoz M; Kelly B; Wagner R; Kayed R Dual Role of P53 Amyloid Formation in Cancer; Loss of Function and Gain of Toxicity. Biochem. Biophys. Res. Commun 2013, 430, 963–968. [DOI] [PubMed] [Google Scholar]
- (71).Aidt FH; Hasholt LF; Christiansen M; Laursen H Localization of A11-Reactive Oligomeric Species in Prion Diseases. Histopathology 2013, 62, 994–1001. [DOI] [PubMed] [Google Scholar]
- (72).Shin TM; Isas JM; Hsieh C-L; Kayed R; Glabe CG; Langen R; Chen J Formation of Soluble Amyloid Oligomers and Amyloid Fibrils by the Multifunctional Protein Vitronectin. Mol. Neurodegener 2008, 3, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Niu Z; Prade E; Malideli E; Hille K; Jussupow A; Mideksa YG; Yan L-M; Qian C; Fleisch M; Messias AC; Sarkar R; Sattler M; Lamb DC; Feige MJ; Camilloni C; Kapurniotu A; Reif B Structural Insight into IAPP-Derived Amyloid Inhibitors and Their Mechanism of Action. Angew. Chem. Int. Ed Engl 2020, 59, 5771–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Andreetto E; Yan L-M; Tatarek-Nossol M; Velkova A; Frank R; Kapurniotu A Identification of Hot Regions of the Aβ–IAPP Interaction Interface as High-Affinity Binding Sites in Both Cross- and Self-Association. Angew. Chem. Int. Ed 2010, 49, 3081–3085. [DOI] [PubMed] [Google Scholar]
- (75).La Rosa C; La Rosa C; Condorelli M; Compagnini G; Lolicato F; Milardi D; Do TN; Karttunen M; Pannuzzo M; Ramamoorthy A; Fraternali F; et al. Symmetry-Breaking Transitions in the Early Steps of Protein Self-Assembly. Eur. Biophys. J 2020, 49, 175–191. [DOI] [PubMed] [Google Scholar]
- (76).Toyama BH; Weissman JS Amyloid Structure: Conformational Diversity and Consequences. Annu. Rev. Biochem 2011, 80, 557–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).D’Urso L; Condorelli M; Puglisi O; Tempra C; Lolicato F; Compagnini G; Rosa CL Detection and Characterization at NM Concentration of Oligomers Formed by HIAPP, Aβ(1–40) and Their Equimolar Mixture Using SERS and MD Simulations. Phys. Chem. Chem. Phys 2018, 20, 20588–20596. [DOI] [PubMed] [Google Scholar]
- (78).Zhang M; Hu R; Chen H; Chang Y; Ma J; Liang G; Mi J; Wang Y; Zheng J Polymorphic Cross-Seeding Amyloid Assemblies of Amyloid-β and Human Islet Amyloid Polypeptide. Phys. Chem. Chem. Phys 2015, 17, 23245–23256. [DOI] [PubMed] [Google Scholar]
- (79).Wineman-Fisher V; Atsmon-Raz Y; Miller Y Orientations of Residues along the β-Arch of Self-Assembled Amylin Fibril-Like Structures Lead to Polymorphism. Biomacromolecules 2015, 16, 156–165. [DOI] [PubMed] [Google Scholar]
- (80).Baram M; Atsmon-Raz Y; Ma B; Nussinov R; Miller Y Amylin–Aβ Oligomers at Atomic Resolution Using Molecular Dynamics Simulations: A Link between Type 2 Diabetes and Alzheimer’s Disease. Phys. Chem. Chem. Phys 2016, 18, 2330–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Ge X; Yang Y; Sun Y; Cao W; Ding F Islet Amyloid Polypeptide Promotes Amyloid-Beta Aggregation by Binding-Induced Helix-Unfolding of the Amyloidogenic Core. ACS Chem. Neurosci 2018, 9, 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Seeliger J; Evers F; Jeworrek C; Kapoor S; Weise K; Andreetto E; Tolan M; Kapurniotu A; Winter R Cross-Amyloid Interaction of Aβ and IAPP at Lipid Membranes. Angew. Chem. Int. Ed 2012, 51, 679–683. [DOI] [PubMed] [Google Scholar]
- (83).Zhang M; Hu R; Ren B; Chen H; Jiang B; Ma J; Zheng J Molecular Understanding of Aβ-HIAPP Cross-Seeding Assemblies on Lipid Membranes. ACS Chem. Neurosci 2017, 8, 524–537. [DOI] [PubMed] [Google Scholar]
- (84).Aftabizadeh M; Tatarek-Nossol M; Andreetto E; El Bounkari O; Kipp M; Beyer C; Latz E; Bernhagen J; Kapurniotu A Blocking Inflammasome Activation Caused by β-Amyloid Peptide (Aβ) and Islet Amyloid Polypeptide (IAPP) through an IAPP Mimic. ACS Chem. Neurosci 2019, 10, 3703–3717. [DOI] [PubMed] [Google Scholar]
- (85).Ltic S; Perovic M; Mladenovic A; Raicevic N; Ruzdijic S; Rakic L Alpha-Synuclein Is Expressed in Different Tissues during Human Fetal Development. J. Mol. Neurosci 2004, 22, 199–204. [DOI] [PubMed] [Google Scholar]
- (86).Geng X; Lou H; Wang J; Li L; Swanson AL; Sun M α-Synuclein Binds the K(ATP) Channel at Insulin-Secretory Granules and Inhibits Insulin Secretion. Am. J. Physiol. Endocrinol. Metab 2011, 300, E276–E286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Westermark P; Li Z-C; Westermark GT; Leckström A; Steiner DF Effects of Beta Cell Granule Components on Human Islet Amyloid Polypeptide Fibril Formation. FEBS Lett 1996, 379, 203–206. [DOI] [PubMed] [Google Scholar]
- (88).Horvath I; Wittung-Stafshede P Cross-Talk between Amyloidogenic Proteins in Type-2 Diabetes and Parkinson’s Disease. Proc. Natl. Acad. Sci. US A 2016, 113, 12473–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Sun Y; Guo C; Yuan L; Li W; Wang ZY; Yue F Cynomolgus Monkeys with Spontaneous Type-2-Diabetes-Mellitus-like Pathology Develop Alpha-Synuclein Alterations Reminiscent of Prodromal Parkinson’s Disease and Related Diseases. Front. Neurosci 2020, 14, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Hua Q-X; Gozani SN; Chance RE; Hoffmann JA; Frank BH; Weiss MA Structure of a Protein in a Kinetic Trap. Nat. Struct. Mol. Biol 1995, 2, 129–138. [DOI] [PubMed] [Google Scholar]
- (91).Westermark P; Andersson A; Westermark GT Islet Amyloid Polypeptide, Islet Amyloid, and Diabetes Mellitus. Physiol. Rev 2011, 91, 795–826. [DOI] [PubMed] [Google Scholar]
- (92).Hutton JC The Insulin Secretory Granule. Diabetologia 1989, 32, 271–281. [DOI] [PubMed] [Google Scholar]
- (93).Charge SBP; de Koning EJP; Clark A Effect of pH and Insulin on Fibrillogenesis of Islet Amyloid Polypeptide in Vitro. Biochemistry 1995, 34, 14588–14593. [DOI] [PubMed] [Google Scholar]
- (94).Salamekh S; Brender JR; Hyung S-J; Nanga RPR; Vivekanandan S; Ruotolo BT; Ramamoorthy A A Two Site Mechanism for the Inhibition of IAPP Amyloidogenesis by Zinc. J. Mol. Biol 2011, 410, 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Brender JR; Krishnamoorthy J; Messina GML; Deb A; Vivekanandan S; Rosa CL; Penner-Hahn JE; Ramamoorthy A Zinc Stabilization of Prefibrillar Oligomers of Human Islet Amyloid Polypeptide. Chem. Commun 2013, 49, 3339–3341. [DOI] [PubMed] [Google Scholar]
- (96).Brender JR; Lee EL; Hartman K; Wong PT; Ramamoorthy A; Steel DG; Gafni A Biphasic Effects of Insulin on Islet Amyloid Polypeptide Membrane Disruption. Biophys. J 2011, 100, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Brender JR; Hartman K; Nanga RPR; Popovych N; de la Salud Bea R; Vivekanandan S; Marsh ENG; Ramamoorthy A Role of Zinc in Human Islet Amyloid Polypeptide Aggregation. J. Am. Chem. Soc 2010, 132, 8973–8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Wineman-Fisher V; Miller Y Insight into a New Binding Site of Zinc Ions in Fibrillar Amylin. ACS Chem. Neurosci 2017, 8, 2078–2087. [DOI] [PubMed] [Google Scholar]
- (99).Wineman-Fisher V; Miller Y Effect of Zn2+ Ions on the Assembly of Amylin Oligomers: Insight into the Molecular Mechanisms. Phys. Chem. Chem. Phys 2016, 18, 21590–21599. [DOI] [PubMed] [Google Scholar]
- (100).Nedumpully-Govindan P; Ding F Inhibition of IAPP Aggregation by Insulin Depends on the Insulin Oligomeric State Regulated by Zinc Ion Concentration. Sci. Rep 2015, 5, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Sellin D; Yan L-M; Kapurniotu A; Winter R Suppression of IAPP Fibrillation at Anionic Lipid Membranes via IAPP-Derived Amyloid Inhibitors and Insulin. Biophys. Chem 2010, 150, 73–79. [DOI] [PubMed] [Google Scholar]
- (102).Velkova A; Tatarek‐Nossol M; Andreetto E; Kapurniotu A Exploiting Cross-Amyloid Interactions to Inhibit Protein Aggregation but not Function: Nanomolar Affinity Inhibition of Insulin Aggregation by an IAPP Mimic. Angew. Chem. Int. Ed 2008, 47, 7114–7118. [DOI] [PubMed] [Google Scholar]
- (103).Liu P; Zhang S; Chen M; Liu Q; Wang C; Wang C; Li Y-M; Besenbacher F; Dong M Co-Assembly of Human Islet Amyloid Polypeptide (HIAPP)/Insulin. Chem. Commun 2012, 48, 191–193. [DOI] [PubMed] [Google Scholar]
- (104).Wiltzius JJW; Sievers SA; Sawaya MR; Eisenberg D Atomic Structures of IAPP (Amylin) Fusions Suggest a Mechanism for Fibrillation and the Role of Insulin in the Process. Protein Sci 2009, 18, 1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Susa AC; Wu C; Bernstein SL; Dupuis NF; Wang H; Raleigh DP; Shea J-E; Bowers MT Defining the Molecular Basis of Amyloid Inhibitors: Human Islet Amyloid Polypeptide–Insulin Interactions. J. Am. Chem. Soc 2014, 136, 12912–12919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Gilead S; Wolfenson H; Gazit E Molecular Mapping of the Recognition Interface between the Islet Amyloid Polypeptide and Insulin. Angew. Chem. Int. Ed 2006, 45, 6476–6480. [DOI] [PubMed] [Google Scholar]
- (107).Ma L; Yang C; Huang L; Chen Y; Li Y; Cheng C; Cheng B; Zheng L; Huang K Glycated Insulin Exacerbates the Cytotoxicity of Human Islet Amyloid Polypeptides: A Vicious Cycle in Type 2 Diabetes. ACS Chem. Biol 2019, 14, 486–496. [DOI] [PubMed] [Google Scholar]
- (108).Opie EL On Relation of Chronic Interstitial Pancreatitis to the Islands of Langerhans and to Diabetes Mellitus. J. Exp. Med 1901, 5, 397–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Gellerstedt N Die elektive, insuläre (Para-)Amyloidose der Bauchspeicheldrüse. Zugleich En Beitrag Zur Kennt. Senilen Amyloidose Beitr Path Anat 1938, 101, 1–13. [Google Scholar]
- (110).Ahronheim JH The Nature of the Hyaline Material in the Pancreatic Islands in Diabetes Mellitus. Am. J. Path 1943, 19, 873–82. [PMC free article] [PubMed] [Google Scholar]
- (111).Bell ET Hyalinization of the Islets of Langerhans in Diabetes Mellitus. Diabetes 1952, 1, 341–344. [DOI] [PubMed] [Google Scholar]
- (112).Ehrlich JC; Ratner IM Amyloidosis of the Islets of Langerhans. A Restudy of Islet Hyalin in Diabetic and Nondiabetic Individuals. Am. J. Path 1961, 38, 49–59. [PMC free article] [PubMed] [Google Scholar]
- (113).Warren S; LeCompte PM; Legg MA The Pathology of Diabetes Mellitus, Fourth.; Lea & Febiger: Philadelphia, 1966. [Google Scholar]
- (114).Glenner GG Amyloid Deposits and Amyloidosis. B-Fibrilloses N. Engl. J. Med 1980, 302, 333–343. [DOI] [PubMed] [Google Scholar]
- (115).Bell ET Hyalinization of the Islets of Langerhans in Nondiabetic Individuals. Am. J. Path 1959, 35, 801–805. [PMC free article] [PubMed] [Google Scholar]
- (116).Westermark P; Wilander E The Influence of Amyloid Deposits on the Islet Volume in Maturity Onset Diabetes Mellitus. Diabetologia 1978, 15, 417–21. [DOI] [PubMed] [Google Scholar]
- (117).Waugh DF A Fibrous Modification of Insulin. I. The Heat Precipitate of Insulin. J. Am. Chem. Soc 1946, 68, 247–250. [Google Scholar]
- (118).Westermark P Fine Structure of Islets of Langerhans in Insular Amyloidosis. Virchows Arch. A 1973, 359, 1–18. [DOI] [PubMed] [Google Scholar]
- (119).Westermark P; Wernstedt C; Wilander E; Sletten K A Novel Peptide in the Calcitonin Gene Related Peptide Family as an Amyloid Fibril Protein in the Endocrine Pancreas. Biochem. Biophys. Res. Commun 1986, 140, 827–31. [DOI] [PubMed] [Google Scholar]
- (120).Westermark P; Wernstedt C; Wilander E; Hayden DW; O’Brien TD; Johnson KH Amyloid Fibrils in Human Insulinoma and Islets of Langerhans of the Diabetic Cat Are Derived from a Neuropeptide-like Protein Also Present in Normal Islet Cells. Proc. Natl. Acad. Sci. USA 1987, 84, 3881–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Cooper GJ; Willis AC; Clark A; Turner RC; Sim RB; Reid KBM Purification and Characterization of a Peptide from Amyloid-Rich Pancreases of Type 2 Diabetic Patients. Proc. Natl. Acad. Sci. USA 1987, 84, 8628–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Westermark P; Andersson A; Westermark GT Islet Amyloid Polypeptide, Islet Amyloid and Diabetes Mellitus. Physiol. Rev 2011, 91, 795–826. [DOI] [PubMed] [Google Scholar]
- (123).Westermark P; Engström U; Johnson KH; Westermark GT; Betsholtz C Islet Amyloid Polypeptide: Pinpointing Amino Acid Residues Linked to Amyloid Fibril Formation. Proc. Natl. Acad. Sci. USA, 1990, 87, 5036–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Charge SB; Koning EJ; Clark A Effect of PH and Insulin on Fibrillogenesis of Islet Amyloid Polypeptide in Vitro. Biochemistry 1995, 34, 14588–14593. [DOI] [PubMed] [Google Scholar]
- (125).Makin OS; Serpell LC Structural Characterisation of Islet Amyloid Polypeptide Fibrils. J. Mol. Biol 2004, 335, 1279–1288. [DOI] [PubMed] [Google Scholar]
- (126).Westermark GT, Westermark P, Berne C, Korsgren O Widespread Amyloid Deposition in Transplanted Human Pancreatic Islets. N. Engl. J. Med 2008, 359, 977–979. [DOI] [PubMed] [Google Scholar]
- (127).Westermark GT; Gebre-Medhin S; Steiner DF; Westermark P Islet Amyloid Development in a Mouse Strain Lacking Endogenous Islet Amyloid Polypeptide (IAPP) but Expressing Human IAPP. Mol. Med 2000, 6, 998–1007. [PMC free article] [PubMed] [Google Scholar]
- (128).Marzban L; Trigo-Gonzalez G; Zhu X; Rhodes CJ; Halban PA; Steiner DF Role of Beta-Cell Prohormone Convertase (PC)1/3 in Processing of pro-Islet Amyloid Polypeptide. Diabetes 2004, 53, 414–418. [DOI] [PubMed] [Google Scholar]
- (129).Marzban L; Soukhatcheva G; Verchere CB Role of Carboxypeptidase E in Processing of Pro-Islet Amyloid Polypeptide in Beta-Cells. Endocrinology 2005, 146, 1808–1817. [DOI] [PubMed] [Google Scholar]
- (130).Paulsson JF; Andersson A; Westermark P; Westermark GT Intracellular Amyloid-Like Deposits Contain Unprocessed Pro Islet Amyloid Polypeptide (proIAPP) in Beta-Cells of Transgenic Mice Overexpressing Human IAPP and Transplanted Human Islets. Diabetologia 2006, 49, 1237–1246. [DOI] [PubMed] [Google Scholar]
- (131).Paulsson JF; Westermark GT Aberrant Processing of Human Proislet Amyloid Polypeptide Results in Increased Amyloid Production. Diabetes 2005, 54, 2117–2125. [DOI] [PubMed] [Google Scholar]
- (132).Marzban L; Rhodes CJ; Steiner DF; Haataja L; Halban PA; Verchere CB Impaired NH2-Terminal Processing of Human Proislet Amyloid Polypeptide by the Prohormone Convertase PC2 Leads to Amyloid Formation and Cell Death. Diabetes 2006, 55, 2192–2201. [DOI] [PubMed] [Google Scholar]
- (133).Westermark P; Eizirik DL; Pipeleers DG; Hellerström C; Andersson A Rapid Deposition of Amyloid in Human Islets Transplanted into Nude Mice. Diabetologia 1995, 38, 543–549. [DOI] [PubMed] [Google Scholar]
- (134).Anguiano M; Nowak RJ; Lansbury PT Protofibrillar Islet Amyloid Polypeptide Permeabilizes Synthetic Vesicles by a Pore-Like Mechanism that may be Relevant to Type II diabetes. Biochemistry 2002, 41, 11338–11343. [DOI] [PubMed] [Google Scholar]
- (135).Sasahara K Membrane-Mediated Amyloid Deposition of Human Islet Amyloid Polypeptide. Biophys. Rev 2018, 10, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Porat Y; Kolusheva S; Jelinek R; Gazit E The Human Islet Amyloid Polypeptide Forms Transient Membrane-Active Prefibrillar Assemblies. Biochemistry 2003, 42, 10971–10977. [DOI] [PubMed] [Google Scholar]
- (137).Kisilevsky R; Fraser P Proteoglycans and amyloid fibrillogenesis. Ciba Found Symp 1996, 1999, 58–67. [DOI] [PubMed] [Google Scholar]
- (138).Zhang X; Li JP Heparan Sulfate Proteoglycans in Amyloidosis. Prog. Mol. Biol. Transl. Sci 2010, 93, 309–34. [DOI] [PubMed] [Google Scholar]
- (139).Casas S; Gomis R; Gribble FM; Altirriba J; Knuutila S; Novials A Impairment of the Ubiquitin-Proteasome Pathway Is a Downstream Endoplasmic Reticulum Stress Response Induced by Extracellular Human Islet Amyloid Polypeptide and Contributes to Pancreatic Beta-Cell Apoptosis. Diabetes 2007, 56, 2284–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Park K; Verchere CB Identification of a Heparin Binding Domain in the N-Terminal Cleavage Site of Pro-Islet Amyloid Polypeptide. Implication Islet Amyloid Formation. J. Biol. Chem 2001, 276, 16611–16616. [DOI] [PubMed] [Google Scholar]
- (141).Abedini A; Tracz SM; Cho JH; Raleigh DP Characterization of the Heparin Binding Site in the N-Terminus of Human pro-Islet Amyloid Polypeptide: Implications for Amyloid Formation. Biochemistry 2006, 45, 9228–9237. [DOI] [PubMed] [Google Scholar]
- (142).Oskarsson ME; Singh K; Wang J; Vlodavsky I; Li JP; Westermark GT Heparan Sulfate Proteoglycans are Important for Islet Amyloid Formation and Islet Amyloid Polypeptide-Induced Apoptosis. J. Biol. Chem 2015, 290, 15121–15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Pikas DS; Li JP; Vlodavsky I; Lindahl U Substrate Specificity of Heparanases from Human Hepatoma and Platelets. J. Biol. Chem 1998, 273, 18770–18777. [DOI] [PubMed] [Google Scholar]
- (144).Park YJ; Lee S; Kieffer TJ; Warnock GL; Safikhan N; Speck M Deletion of Fas Protects Islet Beta Cells from Cytotoxic Effects of Human Islet Amyloid Polypeptide. Diabetologia 2012, 38, 543–549. [DOI] [PubMed] [Google Scholar]
- (145).Guicciardi ME; Gores GJ Life and Death by Death Receptors. FASEB J 2009, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Bemporad F; Calloni G; Campioni S; Plakoutsi G; Taddei N; Chiti F Sequence and Structural Determinants of Amyloid Fibril Formation. Acc. Chem. Res 2006, 39, 620–627. [DOI] [PubMed] [Google Scholar]
- (147).Paz ML de la; Serrano, L. Sequence Determinants of Amyloid Fibril Formation. Proc. Natl. Acad. Sci. USA 2004, 101, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Akter R; Cao P; Noor H; Ridgway Z; Tu L-H; Wang H; Wong A; Zhang X; Abedini A; Schmidt A; Raleigh D Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J. Diabetes Res 2016, 2798269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Rodriguez Camargo DC; Tripsianes K; Kapp TG; Mendes J; Schubert J; Cordes B; Reif B Cloning, Expression and Purification of the Human Islet Amyloid Polypeptide (HIAPP) from Escherichia Coli. Protein Expr. Purif 2015, 106, 49–56. [DOI] [PubMed] [Google Scholar]
- (150).Ridgway Z; Lee K-H; Zhyvoloup A; Wong A; Eldrid C; Hannaberry E; Thalassinos K; Abedini A; Raleigh DP Analysis of Baboon IAPP Provides Insight into Amyloidogenicity and Cytotoxicity of Human IAPP. Biophys. J 2020, 118, 1142–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Wong AG; Wu C; Hannaberry E; Watson MD; Shea J-E; Raleigh DP Analysis of the Amyloidogenic Potential of Pufferfish (Takifugu Rubripes) Islet Amyloid Polypeptide Highlights the Limitations of Thioflavin-T Assays and the Difficulties in Defining Amyloidogenicity. Biochemistry 2016, 55, 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Lee K-H; Noh D; Zhyvoloup A; Raleigh D Analysis of Prairie Vole Amylin Reveals the Importance of the N-Terminus and Residue 22 in Amyloidogenicity and Cytotoxicity. Biochemistry 2020, 59, 471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Cao P; Meng F; Abedini A; Raleigh DP The Ability of Rodent Islet Amyloid Polypeptide to Inhibit Amyloid Formation by Human Islet Amyloid Polypeptide has Important Implications for the Mechanism of Amyloid Formation and the Design of Inhibitors. Biochemistry 2010, 49, 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Akter R; Bower RL; Abedini A; Schmidt AM; Hay DL; Raleigh DP Amyloidogenicity, Cytotoxicity, and Receptor Activity of Bovine Amylin: Implications for Xenobiotic Transplantation and the Design of Nontoxic Amylin Variants. ACS Chem. Biol 2018, 13, 2747–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Goldenberg O; Erez E; Nimrod G; Ben-Tal N The ConSurf-DB: Pre-Calculated Evolutionary Conservation Profiles of Protein Structures. Nucleic Acids Res 2009, 37, 323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Young LM; Cao P; Raleigh DP; Ashcroft AE; Radford SE Ion Mobility Spectrometry–Mass Spectrometry Defines the Oligomeric Intermediates in Amylin Amyloid Formation and the Mode of Action of Inhibitors. J. Am. Chem. Soc 2014, 136, 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Betsholtz C; Christmanson L; Engström U; Rorsman F; Jordan K; O’Brien TD Structure of Cat Islet Amyloid Polypeptide and Identification of Amino Acid Residues of Potential Significance for Islet Amyloid Formation. Diabetes 1990, 39, 118–22. [DOI] [PubMed] [Google Scholar]
- (158).Tenidis K; Waldner M; Bernhagen J; Fischle W; Bergmann M; Weber M; Merkle ML; Voelter W; Brunner H; Kapurniotu A Identification of a Penta-and Hexapeptide of Islet Amyloid Polypeptide (IAPP) with Amyloidogenic and Cytotoxic Properties. J. Mol. Biol 2000, 295, 1055–1071. [DOI] [PubMed] [Google Scholar]
- (159).Milardi D; Sciacca MFM; Pappalardo M; Grasso DM; La Rosa C The Role of Aromatic Side-Chains in Amyloid Growth and Membrane Interaction of the Islet Amyloid Polypeptide Fragment LANFLVH. Eur. Biophys. J 2011, 40, 1–12. [DOI] [PubMed] [Google Scholar]
- (160).Krotee P; Rodriguez JA; Sawaya MR; Cascio D; Reyes FE; Shi D; Hattne J; Nannenga BL; Oskarsson ME; Philipp S; Griner S; Jiang L; Glabe CG; Westermark GT; Gonen T; Eisenberg DS Atomic Structures of Fibrillar Segments of HIAPP Suggest Tightly Mated β-Sheets Are Important for Cytotoxicity. eLife 2017, 6, e19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Mazor Y; Gilead S; Benhar I; Gazit E Identification and Characterization of a Novel Molecular-Recognition and Self-Assembly Domain within the Islet Amyloid Polypeptide. J. Mol. Biol 2002, 322, 1013–1024. [DOI] [PubMed] [Google Scholar]
- (162).Chiti F; Stefani M; Taddei N; Ramponi G; Dobson CM Rationalization of the Effects of Mutations on Peptide Andprotein Aggregation Rates. Nature 2003, 424, 805–808. [DOI] [PubMed] [Google Scholar]
- (163).Groot NS; Aviles FX; Vendrell J; Ventura S Mutagenesis of the Central Hydrophobic Cluster in Aβ42 Alzheimer’s Peptide: Side‐chain Properties Correlate with Aggregation Propensities. FEBS J 2006, 273, 658–668. [DOI] [PubMed] [Google Scholar]
- (164).Jaikaran ET; Higham CE; Serpell LC; Zurdo J; Gross M; Clark A; Fraser PE Identification of a Novel Human Islet Amyloid Polypeptide β-Sheet Domain and Factors Influencing Fibrillogenesis. J. Am. Chem. Soc 2001, 308, 515–525. [DOI] [PubMed] [Google Scholar]
- (165).Luca S; Yau W-M; Leapman R; Tycko R Peptide Conformation and Supramolecular Organization in Amylin Fibrils: Constraints from Solid-State NMR. Biochemistry 2007, 46, 13505–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Röder C; Kupreichyk T; Gremer L; Schäfer LU; Pothula KR; Ravelli RBG; Willbold D; Hoyer W; Schröder GF Cryo-EM Structure of Islet Amyloid Polypeptide Fibrils Reveals Similarities with Amyloid-β Fibrils. Nat. Struct. Mol. Biol 2020, 27, 660–667. [DOI] [PubMed] [Google Scholar]
- (167).Cao Q; Boyer DR; Sawaya MR; Ge P; Eisenberg DS Cryo-EM Structure and Inhibitor Design of Human IAPP (Amylin) Fibrils. Nat. Struct. Mol. Biol 2020, 27, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Gallardo R; Iadanza MG; Xu Y; Heath GR; Foster R; Radford SE; Ranson NA Fibril Structures of Diabetes-Related Amylin Variants Reveal a Basis for Surface Templated Assembly. Nat. Struct. Mol. Biol 2020, 27, 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Jaikaran ET; Clark A Islet Amyloid and Type 2 Diabetes: from Molecular Misfolding to Islet Pathophysiology. Biochim. Biophys. Acta 2001, 179–203. [DOI] [PubMed] [Google Scholar]
- (170).Sakagashira S; Sanke T; Hanabusa T; Shimomura H; Ohagi S; Kumagaye KY; Nakajima K; Nanjo K Missense Mutation of Amylin Gene (S20G) in Japanese NIDDM Patients. Diabetes 1996, 45, 1279–1281. [DOI] [PubMed] [Google Scholar]
- (171).Khemtemourian L; Guillemain G; Foufelle F; Killian JA Residue Specific Effects of Human Islet Polypeptide Amyloid on Self-Assembly and on Cell Toxicity. Biochimie 2017, 142, 22–30. [DOI] [PubMed] [Google Scholar]
- (172).Hoffmann ARF; Saravanan MS; Lequin O; Killian JA; Khemtemourian L A Single Mutation on the Human Amyloid Polypeptide Modulates Fibril Growth and Affects the Mechanism of Amyloid-Induced Membrane Damage. Biochim. Biophys. Acta BBA - Biomembr 2018, 1783–1792. [DOI] [PubMed] [Google Scholar]
- (173).Brender JR; Salamekh S; Ramamoorthy A Membrane Disruption and Early Events in the Aggregation of the Diabetes Related Peptide IAPP from a Molecular Perspective. Acc. Chem. Res 2012, 45, 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Janson J; Ashley RH; Harrison D; McIntyre S; Butler PC The Mechanism of Islet Amyloid Polypeptide Toxicity Is Membrane Disruption by Intermediate-Sized Toxic Amyloid Particles. Diabetes 1999, 48, 491–498. [DOI] [PubMed] [Google Scholar]
- (175).Kumar S; Schlamadinger DE; Brown MA; Dunn JM; Mercado B; Hebda JA; Saraogi I; Rhoades E; Hamilton AD; Miranker AD Islet Amyloid-Induced Cell Death and Bilayer Integrity Loss Share a Molecular Origin Targetable with Oligopyridylamide-Based α-Helical Mimetics. Chem. Biol 2015, 22, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Kyung-Hoon Lee AZ; Raleigh D Amyloidogenicity and Cytotoxicity of Des-Lys-1 Human Amylin Provides Insight into Amylin Self-Assembly and Highlights the Difficulties of Defining Amyloidogenicity. Protein Eng. Des. Sel. PEDS 2019, 32, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Jha S; Snell JM; Sheftic SR; Patil SM; Daniels SB; Kolling FW; Alexandrescu AT PH Dependence of Amylin Fibrillization. Biochemistry 2014, 53, 300–310. [DOI] [PubMed] [Google Scholar]
- (178).Green J; Goldsbury C; Mini T; Sunderji S; Frey P; Kistler J; Cooper G; Aebi U Full-Length Rat Amylin Forms Fibrils Following Substitution of Single Residues from Human Amylin. J. Mol. Biol 2003, 326, 1147–1156. [DOI] [PubMed] [Google Scholar]
- (179).Doran T; Kamens A; Byrnes N; Nilsson B Role of Amino Acid Hydrophobicity, Aromaticity, and Molecular Volume on IAPP(20–29) Amyloid Self-Assembly. Proteins 2012, 80, 1053–1065. [DOI] [PubMed] [Google Scholar]
- (180).Azriel R; Gazit E Analysis of the Minimal Amyloid-Forming Fragment of the Islet Amyloid Polypeptide an Experimental Support for the Key Role of the Phenylalanine Residue in Amyloid Formation. J. Biol. Chem 2001, 276, 34156–34161. [DOI] [PubMed] [Google Scholar]
- (181).Tu L-H; Raleigh DP Role of Aromatic Interactions in Amyloid Formation by Islet Amyloid Polypeptide. Biochemistry 2013, 52, 333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Marek P; Abedini A; Song B; Kanungo M; Johnson ME; Gupta R; Zaman W; Wong SS; Raleigh DP Aromatic Interactions Are Not Required for Amyloid Fibril Formation by Islet Amyloid Polypeptide but Do Influence the Rate of Fibril Formation and Fibril Morphology. Biochemistry 2007, 46, 3255–3261. [DOI] [PubMed] [Google Scholar]
- (183).Gazit E A Possible Role for π-Stacking in the Self-Assembly of Amyloid Fibrils. FASEB J 2002, 16, 77–83. [DOI] [PubMed] [Google Scholar]
- (184).Padrick SB; Miranker AD Islet Amyloid Polypeptide: Identification of Long-Range Contacts and Local Order on the Fibrillogenesis Pathway. J. Biol. Chem 2001, 308, 783–794. [DOI] [PubMed] [Google Scholar]
- (185).Abedini A; Plesner A; Cao P; Ridgway Z; Zhang J; Tu L-H; Middleton CT; Chao B; Sartori DJ; Meng, et al. Time-Resolved Studies Define the Nature of Toxic IAPP Intermediates, Providing Insight for Anti-Amyloidosis Therapeutics. eLife 2016, 5, e12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Abedini A; Raleigh DP A Critical Assessment of the Role of Helical Intermediates in Amyloid Formation by Natively Unfolded Proteins and Polypeptides. Protein Eng. Des. Sel 2009, 22, 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Bemporad F; Taddei N; Stefani M; Chiti F Assessing the Role of Aromatic Residues in the Amyloid Aggregation of Human Muscle Acylphosphatase. Protein Sci 2006, 15, 862–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).DuBay KF; Pawar AP; Chiti F; Zurdo J; Dobson CM; Vendruscolo M Prediction of the Absolute Aggregation Rates of Amyloidogenic Polypeptide Chains. J. Mol. Biol 2004, 341, 1317–1326. [DOI] [PubMed] [Google Scholar]
- (189).Família C; Dennison SR; Quintas A; Phoenix DA Prediction of Peptide and Protein Propensity for Amyloid Formation. PLOS One 2015, 10, 0134679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Moriarty DF; Raleigh DP Effects of Sequential Proline Substitutions on Amyloid Formation by Human Amylin20–29. Biochemistry 1999, 38, 1811–1818. [DOI] [PubMed] [Google Scholar]
- (191).Abedini A; Meng F; Raleigh DP A Single-Point Mutation Converts the Highly Amyloidogenic Human Islet Amyloid Polypeptide into a Potent Fibrillization Inhibitor. J. Am. Chem. Soc 2007, 129, 11300–11301. [DOI] [PubMed] [Google Scholar]
- (192).Kang L; Moriarty GM; Woods LA; Ashcroft AE; Radford SE; Baum J N-Terminal Acetylation of α-Synuclein Induces Increased Transient Helical Propensity and Decreased Aggregation Rates in the Intrinsically Disordered Monomer. Protein Sci 2012, 21, 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Yan L-M; Tatarek-Nossol M; Velkova A; Kazantzis A; Kapurniotu A Design of a Mimic of Nonamyloidogenic and Bioactive Human Islet Amyloid Polypeptide (IAPP) as Nanomolar Affinity Inhibitor of IAPP Cytotoxic Fibrillogenesis. In Proc. Natl. Acad. Sci. USA; 2006, 103, 2046–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Nguyen PT; Zottig X; Sebastiao M; Bourgault S Role of Site-Specific Asparagine Deamidation in Islet Amyloid Polypeptide Amyloidogenesis: Key Contributions of Residues 14 and 21. Biochemistry 2017, 56, 3808–3817. [DOI] [PubMed] [Google Scholar]
- (195).Godin E; Nguyen P; Zottig X; Bourgault S Identification of a Hinge Residue Controlling Islet Amyloid Polypeptide Self-Assembly and Cytotoxicity. J. Biol. Chem 2019, 294, 8452–8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Abedini A; Gupta R; Marek P; Meng F; Raleigh DP; Taskent H; Tracz S Role of Posttranslational Modifications in Amyloid Formation; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- (197).Lam YPY; Wootton CA; Hands-Portman I; Wei J; Chiu CKC; Romero-Canelon I; Lermyte F; Barrow MP; O’Connor PB Does Deamidation of Islet Amyloid Polypeptide Accelerate Amyloid Fibril Formation? Chem. Commun 2018, 54, 13853–13856. [DOI] [PubMed] [Google Scholar]
- (198).Dunkelberger EB; Buchanan LE; Marek P; Cao P; Raleigh DP; Zanni MT Deamidation Accelerates Amyloid Formation and Alters Amylin Fiber Structure. J. Am. Chem. Soc 2012, 134, 12658–12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Nilsson MR; Driscoll M; Raleigh DP Low Levels of Asparagine Deamidation Can Have a Dramatic Effect on Aggregation of Amyloidogenic Peptides: Implications for the Study of Amyloid Formation. Protein Sci 2002, 11, 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Koo BW; Hebda JA; Miranker AD Amide Inequivalence in the Fibrillar Assembly of Islet Amyloid Polypeptide. Protein Eng. Des. Sel 2008, 21, 147–154. [DOI] [PubMed] [Google Scholar]
- (201).Doherty CPA; Ulamec SM; Maya-Martinez R; Good SC; Makepeace J; Khan GN; Oosten-Hawle P; Radford SE; Brockwell DJ A Short Motif in the N-Terminal Region of α-Synuclein Is Critical for Both Aggregation and Function. Nat. Struct. Mol. Biol 2020, 27, 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Koo BW; Miranker AD Contribution of the Intrinsic Disulfide to the Assembly Mechanism of Islet Amyloid. Protein Sci 2005, 14, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Ridgway Z; Zhang X; Wong AG; Abedini A; Schmidt AM; Raleigh DP Analysis of the Role of the Conserved Disulfide in Amyloid Formation by Human Islet Amyloid Polypeptide in Homogeneous and Heterogeneous Environments. Biochemistry 2018, 57, 3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Rodriguez Camargo DC; Tripsianes K; Buday K; Franko A; Göbl C; Hartlmüller C; Sarkar R; Aichler M; Mettenleiter G; Schulz M et al. The Redox Environment Triggers Conformational Changes and Aggregation of HIAPP in Type II Diabetes. Sci. Rep 2017, 7, 44041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Kumar S; Brown MA; Nath A; Miranker AD Folded Small Molecule Manipulation of Islet Amyloid Polypeptide. Chem. Biol 2014, 21, 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Kumar S; Birol M; Schlamadinger DE; Wojcik SP; Rhoades E; Miranker AD Foldamer-Mediated Manipulation of a Pre-Amyloid Toxin. Nat. Commun 2016, 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Iadanza MG; Jackson MP; Hewitt EW; Ranson NA; Radford SE A New Era for Understanding Amyloid Structures and Disease. Nat. Rev. Mol. Cell Biol 2018, 19, 755–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Karamanos TK; Jackson MP; Calabrese AN; Goodchild SC; Cawood EE; Thompson GS; Kalverda AP; Hewitt EW; Radford SE Structural Mapping of Oligomeric Intermediates in an Amyloid Assembly Pathway. eLife, 2016, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Fusco G; Chen SW; Williamson PT; Cascella R; Perni M; Jarvis JA; Cecchi C; Vendruscolo M; Chiti F; Cremades N Structural Basis of Membrane Disruption and Cellular Toxicity by α-Synuclein Oligomers. Science 2017, 358, 1440–1443. [DOI] [PubMed] [Google Scholar]
- (210).Sandberg A; Luheshi LM; Söllvander S; Barros TP de; Macao B; Knowles TPJ; Biverstål H; Lendel C; Ekholm-Petterson F; Dubnovitsky A; et al. Stabilization of Neurotoxic Alzheimer Amyloid-β Oligomers by Protein Engineering. In Proc. Natl. Acad. Sci. USA 2010, 107, 15595–15600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Buchanan LE; Dunkelberger EB; Tran HQ; Cheng P-N; Chiu C-C; Cao P; Raleigh DP; De Pablo JJ; Nowick JS; Zanni MT Mechanism of IAPP Amyloid Fibril Formation Involves an Intermediate with a Transient β-Sheet. Proc. Natl. Acad. Sci. USA 2013, 110, 19285–19290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Young LM; Tu L-H; Raleigh DP; Ashcroft AE; Radford SE Understanding Co-Polymerization in Amyloid Formation by Direct Observation of Mixed Oligomers. Chem. Sci 2017, 8, 5030–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Cornwell O; Radford SE; Ashcroft AE; Ault JR Comparing Hydrogen Deuterium Exchange and Fast Photochemical Oxidation of Proteins: A Structural Characterisation of Wild-Type and ΔN6 B2-Microglobulin. J. Am. Soc. Mass Spectrom 2018, 29, 2413–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Maj M; Lomont JP; Rich KL; Alperstein AM; Zanni MT Site-Specific Detection of Protein Secondary Structure Using 2D IR Dihedral Indexing: A Proposed Assembly Mechanism of Oligomeric HIAPP. Chem. Sci 2018, 9, 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Woods LA; Platt GW; Hellewell AL; Hewitt EW; Homans SW; Ashcroft AE; Radford SE Ligand Binding to Distinct States Diverts Aggregation of an Amyloid-Forming Protein. Nat. Chem. Biol 2011, 7, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Bernstein SL; Dupuis NF; Lazo ND; Wyttenbach T; Condron MM; Bitan G; Teplow DB; Shea J-E; Ruotolo BT; Robinson CV Amyloid-β Protein Oligomerization and the Importance of Tetramers and Dodecamers in the Aetiology of Alzheimer’s Disease. Nat. Chem 2009, 1, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Woods L; Radford S; Ashcroft A Advances in Ion Mobility Spectrometry–Mass Spectrometry Reveal Key Insights into Amyloid Assembly. Biochim. Biophys. Acta 2013, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Williams DM; Pukala TL Novel Insights into Protein Misfolding Diseases Revealed by Ion Mobility‐mass Spectrometry. Mass Spectrom. Rev 2013, 32, 169–187. [DOI] [PubMed] [Google Scholar]
- (219).Dupuis NF; Wu C; Shea J-E; Bowers MT Human Islet Amyloid Polypeptide Monomers Form Ordered β-Hairpins: A Possible Direct Amyloidogenic Precursor. J. Am. Chem. Soc 2009, 131, 18283–18292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Milardi D; Sciacca MFM; Randazzo L; Raudino A; La Rosa C The Role of Calcium, Lipid Membranes and Islet Amyloid Polypeptide in the Onset of Type 2 Diabetes: Innocent Bystanders or Partners in a Crime? Front. Endocrinol 2014, 5, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Sciacca MF; Milardi D; Messina GM; Marletta G; Brender JR; Ramamoorthy A; La Rosa C Cations as Switches of Amyloid-Mediated Membrane Disruption Mechanisms: Calcium and IAPP. Biophys. J 2013, 104, 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Sciacca MF; Pappalardo M; Milardi D; Grasso DM; La Rosa C Calcium-Activated Membrane Interaction of the Islet Amyloid Polypeptide: Implications in the Pathogenesis of Type II Diabetes Mellitus. Arch. Biochem. Biophys 2008, 477, 291–298. [DOI] [PubMed] [Google Scholar]
- (223).Alghrably M; Czaban I; Jaremko Ł; Jaremko M Interaction of Amylin Species with Transition Metals and Membranes. J. Inorg. Biochem 2019, 191, 69–76. [DOI] [PubMed] [Google Scholar]
- (224).Wineman-Fisher V; Bloch DN; Miller Y Challenges in Studying the Structures of Metal-Amyloid Oligomers Related to Type 2 Diabetes, Parkinson’s Disease, and Alzheimer’s Disease. Coord. Chem. Rev 2016, 327–328, 20–26. [Google Scholar]
- (225).Sciacca MFM; Monaco I; La Rosa C; Milardi D The Active Role of Ca2+ Ions in Aβ-Mediated Membrane Damage. Chem. Commun 2018, 54, 3629–3631. [DOI] [PubMed] [Google Scholar]
- (226).Li H; Ha E; Donaldson RP; Jeremic AM; Vertes A Rapid Assessment of Human Amylin Aggregation and Its Inhibition by Copper(II) Ions by Laser Ablation Electrospray Ionization Mass Spectrometry with Ion Mobility Separation. Anal. Chem 2015, 87, 9829–9837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Nedumpully-Govindan P; Yang Y; Andorfer R; Cao W; Ding F Promotion or Inhibition of Islet Amyloid Polypeptide Aggregation by Zinc Coordination Depends on Its Relative Concentration. Biochemistry 2015, 54, 7335–7344. [DOI] [PubMed] [Google Scholar]
- (228).Atrián-Blasco E; Gonzalez P; Santoro A; Alies B; Faller P; Hureau C Cu and Zn Coordination to Amyloid Peptides: From Fascinating Chemistry to Debated Pathological Relevance. Coord. Chem. Rev 2018, 375, 38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Marrink SJ; de Vries AH; Tieleman DP Lipids on the Move: Simulations of Membrane Pores, Domains, Stalks and Curves. Biochim. Biophys. Acta 2009, 1788, 149–168. [DOI] [PubMed] [Google Scholar]
- (230).Ding W; Palaiokostas M; Wang W; Orsi M Effects of Lipid Composition on Bilayer Membranes Quantified by All-Atom Molecular Dynamics. J. Phys. Chem. B 2015, 119, 15263–15274. [DOI] [PubMed] [Google Scholar]
- (231).Hao M, Lin SX, Karylowski OJ, Wüstner D, McGraw TE, Maxfield FR Vesicular and Non-Vesicular Sterol Transport in Living Cells. The Endocytic Recycling Compartment is a Major Sterol Storance Organelle. J. Biol. Chem 2002, 277, 609–617. [DOI] [PubMed] [Google Scholar]
- (232).Fantini J; Garmy N; Mahfoud R; Yahi N Lipid Rafts: Structure, Function and Role in HIV, Alzheimer’s and Prion Diseases. Expert Rev. Mol. Med 2002, 4, 1–22. [DOI] [PubMed] [Google Scholar]
- (233).Henriksen J; Rowat AC; Ipsen JH Vesicle Fluctuation Analysis of the Effects of Sterols on Membrane Bending Rigidity. Eur. Biophys. J 2004, 33, 732–741. [DOI] [PubMed] [Google Scholar]
- (234).Arashiki N; Saito M; Koshino I; Kamata K; Hale J; Mohandas N; Manno S; Takakuwa Y An Unrecognized Function of Cholesterol: Regulating the Mechanism Controlling Membrane Phospholipid Asymmetry. Biochemistry 2016, 55, 3504–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Jacobson K; Mouritsen OG; Anderson RGW Lipid Rafts: At a Crossroad between Cell Biology and Physics. Nat. Cell Biol 2007, 9, 7–14. [DOI] [PubMed] [Google Scholar]
- (236).Hjort Ipsen J; Karlström G; Mourtisen OG; Wennerström H; Zuckermann MJ Phase Equilibria in the Phosphatidylcholine-Cholesterol System. Biochim. Biophys. Acta BBA - Biomembr 1987, 905, 162–172. [DOI] [PubMed] [Google Scholar]
- (237).Marsh D Liquid-Ordered Phases Induced by Cholesterol: A Compendium of Binary Phase Diagrams. Biochim. Biophys. Acta 2010, 1798, 688–699. [DOI] [PubMed] [Google Scholar]
- (238).Mouritsen OG The Liquid-Ordered State Comes of Age. Biochim. Biophys. Acta BBA - Biomembr 2010, 1798, 1286–1288. [DOI] [PubMed] [Google Scholar]
- (239).Semrau S; Schmidt T Membrane Heterogeneity – from Lipid Domains to Curvature Effects. Soft Matter 2009, 5, 3174–3186. [Google Scholar]
- (240).Schmidt ML; Davis JH Liquid Disordered–Liquid Ordered Phase Coexistence in Lipid/Cholesterol Mixtures: A Deuterium 2D NMR Exchange Study. Langmuir 2017, 33, 1881–1890. [DOI] [PubMed] [Google Scholar]
- (241).Ghysels A; Krämer A; Venable RM; Teague WE; Lyman E; Gawrisch K; Pastor RW Permeability of Membranes in the Liquid Ordered and Liquid Disordered Phases. Nat. Commun 2019, 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Krause MR; Daly TA; Almeida PF; Regen SL Push–Pull Mechanism for Lipid Raft Formation. Langmuir 2014, 30, 3285–3289. [DOI] [PubMed] [Google Scholar]
- (243).McHenry AJ; Sciacca MFM; Brender JR; Ramamoorthy A Does Cholesterol Suppress the Antimicrobial Peptide Induced Disruption of Lipid Raft Containing Membranes? Biochim. Biophys. Acta BBA - Biomembr 2012, 1818, 3019–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Lay SL; Krief S; Farnier C; Lefrère I; Liepvre XL; Bazin R; Ferré P; Dugail I Cholesterol, a Cell Size-Dependent Signal That Regulates Glucose Metabolism and Gene Expression in Adipocytes. J. Biol. Chem 2001, 276, 16904–16910. [DOI] [PubMed] [Google Scholar]
- (245).Brown MS; Goldstein JL The SREBP Pathway: Regulation of Cholesterol Metabolism by Proteolysis of a Membrane-Bound Transcription Factor. Cell 1997, 89, 331–340. [DOI] [PubMed] [Google Scholar]
- (246).Brunham LR; Kruit JK; Hayden MR; Verchere CB Cholesterol in β-Cell Dysfunction: The Emerging Connection Between HDL Cholesterol and Type 2 Diabetes. Curr. Diab. Rep 2010, 10, 55–60. [DOI] [PubMed] [Google Scholar]
- (247).Brunham LR; Kruit JK; Pape TD; Timmins JM; Reuwer AQ; Vasanji Z; Marsh BJ; Rodrigues B; Johnson JD; Parks JS; Verchere CB; Hayden MR Beta-Cell ABCA1 Influences Insulin Secretion, Glucose Homeostasis and Response to Thiazolidinedione Treatment. Nat. Med 2007, 13, 340–347. [DOI] [PubMed] [Google Scholar]
- (248).Ge M; Gidwani A; Brown HA; Holowka D; Baird B; Freed JH Ordered and Disordered Phases Coexist in Plasma Membrane Vesicles of RBL-2H3 Mast Cells. An ESR Study. Biophys. J 2003, 85, 1278–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Sciacca MF; Lolicato F; Di Mauro G; Milardi D; D’Urso L; Satriano C; Ramamoorthy A; La Rosa C The Role of Cholesterol in Driving IAPP-Membrane Interactions. Biophys. J 2016, 111, 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Zhang X; London E; Raleigh DP Sterol Structure Strongly Modulates Membrane–Islet Amyloid Polypeptide Interactions. Biochemistry 2018, 57, 1868–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Xu W; Wei G; Su H; Nordenskiöld L; Mu Y Effects of Cholesterol on Pore Formation in Lipid Bilayers Induced by Human Islet Amyloid Polypeptide Fragments: A Coarse-Grained Molecular Dynamics Study. Phys. Rev. E 2011, 84, 051922. [DOI] [PubMed] [Google Scholar]
- (252).Caillon L; Lequin O; Khemtémourian L Evaluation of Membrane Models and Their Composition for Islet Amyloid Polypeptide-Membrane Aggregation. Biochim. Biophys. Acta BBA - Biomembr 2013, 1828, 2091–2098. [DOI] [PubMed] [Google Scholar]
- (253).Caillon L; Duma L; Lequin O; Khemtemourian L Cholesterol Modulates the Interaction of the Islet Amyloid Polypeptide with Membranes. Mol. Membr. Biol 2014, 31, 239–249. [DOI] [PubMed] [Google Scholar]
- (254).Wakabayashi M; Matsuzaki K Ganglioside-Induced Amyloid Formation by Human Islet Amyloid Polypeptide in Lipid Rafts. FEBS Lett 2009, 583, 2854–2858. [DOI] [PubMed] [Google Scholar]
- (255).Lozano MM; Hovis JS; Moss FR; Boxer SG Dynamic Reorganization and Correlation among Lipid Raft Components. J. Am. Chem. Soc 2016, 138, 9996–10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Zhang X; St. Clair JR; London E; Raleigh DP Islet Amyloid Polypeptide Membrane Interactions: Effects of Membrane Composition. Biochemistry 2017, 56, 376–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Fabiani C; Antollini SS Alzheimer’s Disease as a Membrane Disorder: Spatial Cross-Talk Among Beta-Amyloid Peptides, Nicotinic Acetylcholine Receptors and Lipid Rafts. Front. Cell. Neurosci 2019, 13, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Ungureanu A-A; Benilova I; Krylychkina O; Braeken D; De Strooper B; Van Haesendonck C; Dotti CG; Bartic C Amyloid Beta Oligomers Induce Neuronal Elasticity Changes in Age-Dependent Manner: A Force Spectroscopy Study on Living Hippocampal Neurons. Sci. Rep 2016, 6, 25841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Pannuzzo M On the Physiological/Pathological Link between Aβ Peptide, Cholesterol, Calcium Ions and Membrane Deformation: A Molecular Dynamics Study. Biochim. Biophys. Acta BBA - Biomembr 2016, 1858, 1380–1389. [DOI] [PubMed] [Google Scholar]
- (260).Shoval H; Weiner L; Gazit E; Levy M; Pinchuk I; Lichtenberg D Polyphenol-Induced Dissociation of Various Amyloid Fibrils Results in a Methionine-Independent Formation of ROS. Biochim. Biophys. Acta BBA-Proteins Proteomics 2008, 1784, 1570–1577. [DOI] [PubMed] [Google Scholar]
- (261).Dong X; Svantesson T; Sholts SB; Wallin C; Jarvet J; Gräslund A; Wärmländer SK Copper Ions Induce Dityrosine-Linked Dimers in Human but Not in Murine Islet Amyloid Polypeptide (IAPP/Amylin. Biochem. Biophys. Res. Commun 2019, 510, 520–524. [DOI] [PubMed] [Google Scholar]
- (262).Zeng H; Tong R; Tong W; Yang Q; Qiu M; Xiong A; Sun S; Ding L; Zhang H; Yang L; Tian J Metabolic Biomarkers for Prognostic Prediction of Pre-Diabetes: Results from a Longitudinal Cohort Study. Sci. Rep 2017, 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Chiu CJ; Rabbani N; Rowan S; Chang ML; Sawyer S; Hu FB; Willett W; Thornalley PJ; Anwar A; Bar L; Kang JH Studies of Advanced Glycation End Products and Oxidation Biomarkers for Type 2 Diabetes. Biofactors 2018, 44, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Franklin BS; Mangan MS; Latz E Crystal Formation in Inflammation. Annu. Rev. Immunol 2016, 34, 173–202. [DOI] [PubMed] [Google Scholar]
- (265).Montane J; Novials A The Role of Human IAPP in Stress and Inflammatory Processes in Type 2 Diabetes. Explor. New Find. Amyloidosis 2016. [Google Scholar]
- (266).Egaña-Gorroño L; López-Díez R; Yepuri G; Ramirez LS; Reverdatto S; Gugger PF; Shekhtman A; Ramasamy R; Schmidt AM Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights from Human Subjects and Animal Models. Front. Cardiovasc. Med 2020, 7, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Kislinger T; Fu C; Huber B; Qu W; Taguchi A; Du Yan S; Hofmann M; Yan SF; Pischetsrieder M; Stern D; Schmidt AM N(Epsilon)-(Carboxymethyl)Lysine Adducts of Proteins Are Ligands for Receptor for Advanced Glycation End Products That Activate Cell Signaling Pathways and Modulate Gene Expression. J. Biol. Chem 1999, 274, 31740–31749. [DOI] [PubMed] [Google Scholar]
- (268).Schmidt AM; Stern DM RAGE: A New Target for the Prevention and Treatment of the Vascular and Inflammatory Complications of Diabetes. Trends Endocrinol. Metab. TEM 2000, 11, 368–375. [DOI] [PubMed] [Google Scholar]
- (269).Freigang J; Proba K; Leder L; Diederichs K; Sonderegger P; Welte W The Crystal Structure of the Ligand Binding Module of Axonin-1/TAG-1 Suggests a Zipper Mechanism for Neural Cell Adhesion. Cell 2000, 101, 425–433. [DOI] [PubMed] [Google Scholar]
- (270).Soroka V; Kolkova K; Kastrup JS; Diederichs K; Breed J; Kiselyov VV; Poulsen FM; Larsen IK; Welte W; Berezin V; Bock E; Kasper C Structure and Interactions of NCAM Ig1-2-3 Suggest a Novel Zipper Mechanism for Homophilic Adhesion. Struct. Lond. Engl. 1993 2003, 11, 1291–1301. [DOI] [PubMed] [Google Scholar]
- (271).Ramasamy R; Vannucci SJ; Yan SSD; Herold K; Yan SF; Schmidt AM Advanced Glycation End Products and RAGE: A Common Thread in Aging, Diabetes, Neurodegeneration, and Inflammation. Glycobiology 2005, 15, 16R–28R. [DOI] [PubMed] [Google Scholar]
- (272).Dattilo BM; Fritz G; Leclerc E; Kooi CWV; Heizmann CW; Chazin WJ The Extracellular Region of the Receptor for Advanced Glycation End Products Is Composed of Two Independent Structural Units. Biochemistry 2007, 46, 6957–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Khan Md. I.; Su Y-K; Zou J; Yang L-W; Chou R-H; Yu C S100B as an Antagonist to Block the Interaction between S100A1 and the RAGE V Domain. PLoS ONE 2018, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Leclerc E; Vetter SW The Role of S100 Proteins and Their Receptor RAGE in Pancreatic Cancer. Biochim. Biophys. Acta BBA - Mol. Basis Dis 2015, 1852, 2706–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Tesarova P; Kalousova M; Zima T; Tesar V HMGB1, S100 Proteins and Other RAGE Ligands in Cancer - Markers, Mediators and Putative Therapeutic Targets. Biomed. Pap 2016, 160, 1–10. [DOI] [PubMed] [Google Scholar]
- (276).Yan SD; Bierhaus A; Nawroth PP; Stern DM RAGE and Alzheimer’s Disease: A Progression Factor for Amyloid-Beta-Induced Cellular Perturbation? J. Alzheimers Dis. JAD 2009, 16, 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Wright NT; Cannon BR; Zimmer DB; Weber DJ S100A1: Structure, Function, and Therapeutic Potential. Curr. Chem. Biol 2009, 3, 138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Hofmann MA; Drury S; Fu C; Qu W; Taguchi A; Lu Y; Avila C; Kambham N; Bierhaus A; Nawroth P; Neurath MF; Slattery T; Beach D; McClary J; Nagashima M; Morser J; Stern D; Schmidt AM RAGE Mediates a Novel Proinflammatory Axis: A Central Cell Surface Receptor for S100/Calgranulin Polypeptides. Cell 1999, 97, 889–901. [DOI] [PubMed] [Google Scholar]
- (279).Yan SD; Chen X; Fu J; Chen M; Zhu H; Roher A; Slattery T; Zhao L; Nagashima M; Morser J; Migheli A; Nawroth P; Stern D; Schmidt AM RAGE and Amyloid-Beta Peptide Neurotoxicity in Alzheimer’s Disease. Nature 1996, 382, 685–691. [DOI] [PubMed] [Google Scholar]
- (280).Ramasamy R; Shekhtman A; Schmidt AM The Multiple Faces of RAGE - Opportunities for Therapeutic Intervention in Aging and Chronic Disease. Expert Opin. Ther. Targets 2016, 20, 431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Litwinoff E; Hurtado Del Pozo C; Ramasamy R; Schmidt AM Emerging Targets for Therapeutic Development in Diabetes and Its Complications: The RAGE Signaling Pathway. Clin. Pharmacol. Ther 2015, 98, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Wautier MP; Chappey O; Corda S; Stern DM; Schmidt AM; Wautier JL Activation of NADPH Oxidase by AGE Links Oxidant Stress to Altered Gene Expression via RAGE. Am. J. Physiol. Endocrinol. Metab 2001, 280, E685–694. [DOI] [PubMed] [Google Scholar]
- (283).Zhu Y; Shu T; Lin Y; Wang H; Yang J; Shi Y; Han X Inhibition of the Receptor for Advanced Glycation Endproducts (RAGE) Protects Pancreatic β-Cells. Biochem. Biophys. Res. Commun 2011, 404, 159–165. [DOI] [PubMed] [Google Scholar]
- (284).Lee B-W; Chae HY; Kwon SJ; Park SY; Ihm J; Ihm S-H RAGE Ligands Induce Apoptotic Cell Death of Pancreatic β-Cells via Oxidative Stress. Int. J. Mol. Med 2010, 26, 813–818. [PubMed] [Google Scholar]
- (285).Abedini A; Cao P; Plesner A; Zhang J; He M; Derk J; Patil SA; Rosario R; Lonier J; Song F; Koh H; Li H; Raleigh DP; Schmidt AM RAGE Binds Preamyloid IAPP Intermediates and Mediates Pancreatic β Cell Proteotoxicity. J. Clin. Invest 128, 682–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Matveyenko AV; Butler PC Islet Amyloid Polypeptide (IAPP) Transgenic Rodents as Models for Type 2 Diabetes. ILAR J 2006, 47, 225–233. [DOI] [PubMed] [Google Scholar]
- (287).Costal F; Oliveira E; Raposo A; Machado-Lima A; Peixoto E; Roma L; Santos L; Lopes Faria JB; Carpinelli AR; Giannella-Neto D; Passarelli M; Correa-Giannella ML Dual Effect of Advanced Glycation End Products in Pancreatic Islet Apoptosis. Diabetes Metab. Res. Rev 2013, 29, 296–307. [DOI] [PubMed] [Google Scholar]
- (288).Yan SS; Chen D; Yan S; Guo L; Chen JX RAGE Is a Key Cellular Target for Aβ-Induced Perturbation in Alzheimer’s Disease. Front. Biosci. Sch. Ed 2012, 4, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Vlassov AV; Magdaleno S; Setterquist R; Conrad R Exosomes: Current Knowledge of Their Composition, Biological Functions, and Diagnostic and Therapeutic Potentials. Biochim. Biophys. Acta 2012, 1820, 940–948. [DOI] [PubMed] [Google Scholar]
- (290).Vella LJ; Hill AF; Cheng L Focus on Extracellular Vesicles: Exosomes and Their Role in Protein Trafficking and Biomarker Potential in Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci 2016, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Kalra H; Drummen GPC; Mathivanan S Focus on Extracellular Vesicles: Introducing the Next Small Big Thing. Int. J. Mol. Sci 2016, 17, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Sarko DK; McKinney CE Exosomes: Origins and Therapeutic Potential for Neurodegenerative Disease. Front. Neurosci 2017, 11, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Barile L; Vassalli G Exosomes: Therapy Delivery Tools and Biomarkers of Diseases. Pharmacol. Ther 2017, 174, 63–78. [DOI] [PubMed] [Google Scholar]
- (294).Grey M; Dunning CJ; Gaspar R; Grey C; Brundin P; Sparr E; Linse S Acceleration of α-Synuclein Aggregation by Exosomes. J. Biol. Chem 2015, 30, 2969–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).Yuyama K; Sun H; Mitsutake S; Igarashi Y Sphingolipid-Modulated Exosome Secretion Promotes the Clearance of Amyloid-β by Microglia. J. Biol. Chem 2012, 287, 10977–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Ribeiro D; Horvath I; Heath N; Hicks R; Forslöw A; Wittung-Stafshede P Extracellular Vesicles from Human Pancreatic Islets Suppress Human Islet Amyloid Polypeptide Amyloid Formation. Proc. Natl. Acad. Sci. USA 2017, 114, 11127–11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Moore SJ Characterisation of the Structure and Oligomerisation of Islet Amyloid Polypeptides (IAPP): A Review of Molecular Dynamics Simulation Studies. Molecules 2018, 23, 2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (298).Ilie IM; Caflisch A Simulation Studies of Amyloidogenic Polypeptides and Their Aggregates. Chem. Rev 2019, 119, 6956–6993. [DOI] [PubMed] [Google Scholar]
- (299).Dong X Recent Computational Studies of Membrane Interaction and Disruption of Human Islet Amyloid Polypeptide: Monomers, Oligomers and Protofibrils. Biochim. Biophys. Acta-Biomembr 2018, 1860, 1826–1839. [DOI] [PubMed] [Google Scholar]
- (300).Press-Sandler O; Miller Y Molecular Mechanisms of Membrane-Associated Amyloid Aggregation: Computational Perspective and Challenges. Biochim. Biophys. Acta-Biomembr 2018, 1860, 1889–1905. [DOI] [PubMed] [Google Scholar]
- (301).Ren B Experimental and Computational Protocols for Studies of Cross-Seeding Amyloid Assemblies. Peptide Self-Assembly. Methods and Protocols Ed. Nilsson B Doran TM 2018, 1777, 429–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Scollo F Phospholipids Critical Micellar Concentrations Trigger Different Mechanisms of Intrinsically Disordered Proteins Interaction with Model Membranes. J. Phys. Chem. Lett 2018, 9, 5125–5129. [DOI] [PubMed] [Google Scholar]
- (303).Christensen MH; Schiott B Membrane Interactions of IAPP. Biophys. J 2019, 116, 491–491. [Google Scholar]
- (304).Qiao Q Formation of Alpha-Helical and Beta-Sheet Structures in Membrane-Bound Human IAPP Monomer and the Resulting Membrane Deformation. Phys. Chem. Chem. Phys 2019, 21, 20239–20251. [DOI] [PubMed] [Google Scholar]
- (305).Alves NA; Frigori RB In Silico Comparative Study of Human and Porcine Amylin. J. Phys. Chem. B 2018, 122, 10714–10721. [DOI] [PubMed] [Google Scholar]
- (306).Su X All-Atom Structure Ensembles of Islet Amyloid Polypeptides Determined by Enhanced Sampling and Experiment Data Restraints. Proteins-Struct. Funct. Bioinforma 2019, 87, 541–550. [DOI] [PubMed] [Google Scholar]
- (307).Guo AZ Early-Stage Human Islet Amyloid Polypeptide Aggregation: Mechanisms behind Dimer Formation. J. Chem. Phys 2018, 149. [DOI] [PubMed] [Google Scholar]
- (308).Qi RX Replica Exchange Molecular Dynamics: A Practical Application Protocol with Solutions to Common Problems and a Peptide Aggregation and Self-Assembly Example. Pept. Self-Assem. Methods Protoc 2018, 1777, 101–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (309).Ilitchev AI Hetero-Oligomeric Amyloid Assembly and Mechanism: Prion Fragment PrP(106–126) Catalyzes the Islet Amyloid Polypeptide Beta-Hairpin. J. Am. Chem. Soc 2018, 140, 9685–9695. [DOI] [PubMed] [Google Scholar]
- (310).Ge X Islet Amyloid Polypeptide Promotes Amyloid-Beta Aggregation by Binding-Induced Helix-Unfolding of the Amyloidogenic Core. ACS Chem. Neurosci 2018, 9, 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (311).Sun Y Nucleation of Beta-Rich Oligomers and Beta-Barrels in the Early Aggregation of Human Islet Amyloid Polypeptide. Biochim. Biophys. Acta-Mol. Basis Dis 2019, 1865, 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (312).Sun YX Beta-Barrel Oligomers as Common Intermediates of Peptides Self-Assembling into Cross-Beta Aggregates. Sci. Rep 2018, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (313).Qian Z Atomistic-Level Study of the Interactions between HIAPP Protofibrils and Membranes: Influence of PH and Lipid Composition. Biochim. Biophys. Acta-Biomembr 2018, 1860, 1818–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (314).Davidson DS; Brown AM; Lemkul JA Insights into Stabilizing Forces in Amyloid Fibrils of Differing Sizes from Polarizable Molecular Dynamics Simulations. J. Mol. Biol 2018, 430, 3819–3834. [DOI] [PubMed] [Google Scholar]
- (315).Li Y; Wang X; Ren L; Cao X; Ji C; Xia F; Zhang JZH Electrostatic Polarization Effect on Cooperative Aggregation of Full Length Human Islet Amyloid. J. Chem. Inf. Model 2018, 58, 1587–1595. [DOI] [PubMed] [Google Scholar]
- (316).Tofoleanu F; Yuan Y; Pickard FC; Tywoniuk B; Brooks BR; Buchete N-V Structural Modulation of Human Amylin Protofilaments by Naturally Occurring Mutations. J. Phys. Chem. B 2018, 122, 5657–5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (317).Mahmoudinobar F; Urban JM; Su Z; Nilsson BL; Dias CL Thermodynamic Stability of Polar and Nonpolar Amyloid Fibrils. J. Chem. Theory Comput 2019, 15, 3868–3874. [DOI] [PubMed] [Google Scholar]
- (318).Baweja L; Roche J Pushing the Limits of Structure-Based Models: Prediction of Nonglobular Protein Folding and Fibrils Formation with Go-Model Simulations. J. Phys. Chem. B 2018, 122, 2525–2535. [DOI] [PubMed] [Google Scholar]
- (319).Watanabe-Nakayama T; Ono K; Itami M; Takahashi R; Teplow DB; Yamada M High-Speed Atomic Force Microscopy Reveals Structural Dynamics of Amyloid B1–42 Aggregates. Proc. Natl. Acad. Sci. USA 2016, 113, 5835–5840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (320).Lu L; Deng Y; Li X; Li H; Karniadakis GE Understanding the Twisted Structure of Amyloid Fibrils via Molecular Simulations. J. Phys. Chem. B 2018, 122, 11302–11310. [DOI] [PubMed] [Google Scholar]
- (321).Ren B Tanshinones Inhibit HIAPP Aggregation, Disaggregate Preformed HIAPP Fibrils, and Protect Cultured Cells. J. Mater. Chem. B 2018, 6, 56–67. [DOI] [PubMed] [Google Scholar]
- (322).Asthana S; Sahu M; Nayak PS; Mallick B; Jha S The Smaller Heparin Fragments Bind Non-Specifically through the IAPP Sequence: An in Silico Study. Int. J. Biol. Macromol 2018, 113, 1092–1104. [DOI] [PubMed] [Google Scholar]
- (323).Fernández-Gómez I; Sablón-Carrazana M; Bencomo-Martínez A; Domínguez G; Lara-Martínez R; Altamirano-Bustamante NF; Jiménez-García LF; Pasten-Hidalgo K; Castillo-Rodríguez RA; Altamirano, et al. Diabetes Drug Discovery: HIAPP1–37 Polymorphic Amyloid Structures as Novel Therapeutic Targets. Molecules 2018, 23, 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Azzam SK; Jang H; Choi MC; Alsafar H; Lukman S; Lee S Inhibition of Human Amylin Aggregation and Cellular Toxicity by Lipoic Acid and Ascorbic Acid. Mol. Pharm 2018, 15, 2098–2106. [DOI] [PubMed] [Google Scholar]
- (325).Kakinen A; Adamcik J; Wang B; Ge X; Mezzenga R; Davis TP; Ding F; Ke PC Nanoscale Inhibition of Polymorphic and Ambidextrous IAPP Amyloid Aggregation with Small Molecules. Nano Res 2018, 11, 3636–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (326).Farrukh SUB; Javed I; Ather AQ; Emwas A-H; Alazmi M; Gao X; Chotana GA; Davis TP; Ke PC; Saleem RSZ Synthesis and Identification of Novel Pyridazinylpyrazolone Based Diazo Compounds as Inhibitors of Human Islet Amyloid Polypeptide Aggregation. Bioorganic Chem 2019, 84, 339–346. [DOI] [PubMed] [Google Scholar]
- (327).Bai C; Lin D; Mo Y; Lei J; Sun Y; Xie L; Yang X; Wei G Influence of Fullerenol on HIAPP Aggregation: Amyloid Inhibition and Mechanistic Aspects. Phys. Chem. Chem. Phys 2019, 21, 4022–4031. [DOI] [PubMed] [Google Scholar]
- (328).Bai C; Lao Z; Chen Y; Tang Y; Wei G Pristine and Hydroxylated Fullerenes Prevent the Aggregation of Human Islet Amyloid Polypeptide and Display Different Inhibitory Mechanisms. Front. Chem 2020, 8, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (329).Dubey R; Patil K; Dantu SC; Sardesai DM; Bhatia P; Malik N; Acharya JD; Sarkar S; Ghosh S; Chakrabarti R; Sharma S; Kumar A Azadirachtin Inhibits Amyloid Formation, Disaggregates Pre-Formed Fibrils and Protects Pancreatic β-Cells from Human Islet Amyloid Polypeptide/Amylin-Induced Cytotoxicity. Biochem. J 2019, 476, 889–907. [DOI] [PubMed] [Google Scholar]
- (330).Sahoo BR; Genjo T; Nakayama TW; Stoddard AK; Ando T; Yasuhara K; Fierke CA; Ramamoorthy A A Cationic Polymethacrylate-Copolymer Acts as an Agonist for β-Amyloid and an Antagonist for Amylin Fibrillation. Chem. Sci 2019, 10, 3976–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (331).Lao Z; Chen Y; Tang Y; Wei G Molecular Dynamics Simulations Reveal the Inhibitory Mechanism of Dopamine against Human Islet Amyloid Polypeptide (HIAPP) Aggregation and Its Destabilization Effect on HIAPP Protofibrils. ACS Chem. Neurosci 2019, 10, 4151–4159. [DOI] [PubMed] [Google Scholar]
- (332).Pang B; Bian X; Xing J; Liu S; Liu Z; Song F Effects of Lithospermic Acid on HIAPP Aggregation and Amyloid-Induced Cytotoxicity by Multiple Analytical Methods. Biochim. Biophys. Acta BBA - Proteins Proteomics 2020, 1868, 140283. [DOI] [PubMed] [Google Scholar]
- (333).Levine ZA The Mitochondrial Peptide Humanin Targets but Does Not Denature Amyloid Oligomers in Type II Diabetes. J. Am. Chem. Soc 2019, 141, 14168–14179. [DOI] [PubMed] [Google Scholar]
- (334).Niu Z Structural Insight into IAPP-Derived Amyloid Inhibitors and Their Mechanism of Action; Angew. Chem. Int. Ed, 2020, 132, 5820–5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (335).Baram M; Gilead S; Gazit E; Miller Y Mechanistic Perspective and Functional Activity of Insulin in Amylin Aggregation. Chem. Sci 2018, 9, 4244–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (336).Mo Y; Brahmachari S; Lei J; Gilead S; Tang Y; Gazit E; Wei G The Inhibitory Effect of Hydroxylated Carbon Nanotubes on the Aggregation of Human Islet Amyloid Polypeptide Revealed by a Combined Computational and Experimental Study. ACS Chem. Neurosci 2018, 9, 2741–2752. [DOI] [PubMed] [Google Scholar]
- (337).Wang M; Sun Y; Cao X; Peng G; Javed I; Kakinen A; Davis TP; Lin S; Liu J; Ding F; Ke PC Graphene Quantum Dots against Human IAPP Aggregation and Toxicity in Vivo. Nanoscale 2018, 10, 19995–20006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (338).Faridi A; Sun Y; Mortimer M; Aranha RR; Nandakumar A; Li Y; Javed I; Kakinen A; Fan Q; Purcell AW; Davis TP; Ding F; Faridi P; Ke PC Graphene Quantum Dots Rescue Protein Dysregulation of Pancreatic β-Cells Exposed to Human Islet Amyloid Polypeptide. Nano Res 2019, 12, 2827–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (339).Faridi A Mitigating Human IAPP Amyloidogenesis In Vivo with Chiral Silica Nanoribbons. Small 2018, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (340).Pellarin R; Caflisch A Interpreting the Aggregation Kinetics of Amyloid Peptides. J. Mol. Biol 2006, 360, 882–892. [DOI] [PubMed] [Google Scholar]
- (341).Pellarin R Pathways and Intermediates of Amyloid Fibril Formation. J. Mol. Biol 2007, 374, 917–924. [DOI] [PubMed] [Google Scholar]
- (342).Gurlo T; Ryazantsev S; Huang C; Yeh MW; Reber HA; Hines OJ; O’Brien TD; Glabe CG; Butler PC Evidence for Proteotoxicity in Beta Cells in Type 2 Diabetes: Toxic Islet Amyloid Polypeptide Oligomers Form Intracellularly in the Secretory Pathway. Am. J. Pathol 2010, 176, 861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (343).Bram Y; Frydman-Marom A; Yanai I; Gilead S; Shaltiel-Karyo R; Amdursky N; Gazit E Apoptosis Induced by Islet Amyloid Polypeptide Soluble Oligomers Is Neutralized by Diabetes-Associated Specific Antibodies. Sci. Rep 2015, 4, 4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (344).Haataja L; Gurlo T; Huang CJ; Butler PC Islet Amyloid in Type 2 Diabetes, and the Toxic Oligomer Hypothesis. Endocr. Rev 2008, 29, 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (345).Mo Y; Lu Y; Wei G; Derreumaux P Structural Diversity of the Soluble Trimers of the Human Amylin(20–29) Peptide Revealed by Molecular Dynamics Simulations. J. Chem. Phys 2009, 130, 125101. [DOI] [PubMed] [Google Scholar]
- (346).Brender JR; Dürr UHN; Heyl D; Budarapu MB; Ramamoorthy A Membrane Fragmentation by an Amyloidogenic Fragment of Human Islet Amyloid Polypeptide Detected by Solid-State NMR Spectroscopy of Membrane Nanotubes. Biochim. Biophys. Acta 2007, 1768, 2026–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (347).Brender JR; Heyl DL; Samisetti S; Kotler SA; Osborne JM; Pesaru RR; Ramamoorthy A Membrane Disordering Is Not Sufficient for Membrane Permeabilization by Islet Amyloid Polypeptide: Studies of IAPP(20–29) Fragments. Phys. Chem. Chem. Phys 2013, 15, 8908–8915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (348).Nanga RPR; Brender JR; Vivekanandan S; Ramamoorthy A Structure and Membrane Orientation of IAPP in Its Natively Amidated Form at Physiological PH in a Membrane Environment. Biochim. Biophys. Acta BBA - Biomembr 2011, 1808, 2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (349).Rodriguez Camargo DC; Korshavn KJ; Jussupow A; Raltchev K; Goricanec D; Fleisch M; Sarkar R; Xue K; Aichler M; Mettenleiter G; Walch AK; Camilloni C; Hagn F; Reif B; Ramamoorthy A Stabilization and Structural Analysis of a Membrane-Associated HIAPP Aggregation Intermediate. eLife 2017, 6, e31226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (350).Birol M; Kumar S; Rhoades E; Miranker AD Conformational Switching within Dynamic Oligomers Underpins Toxic Gain-of-Function by Diabetes-Associated Amyloid. Nat. Commun 2018, 9, 1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Brender JR; Krishnamoorthy J; Sciacca MFM; Vivekanandan S; D’Urso L; Chen J; La Rosa C; Ramamoorthy A Probing the Sources of the Apparent Irreproducibility of Amyloid Formation: Drastic Changes in Kinetics and a Switch in Mechanism Due to Micellelike Oligomer Formation at Critical Concentrations of IAPP. J. Phys. Chem. B 2015, 119, 2886–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Patel HR; Pithadia AS; Brender JR; Fierke CA; Ramamoorthy A In Search of Aggregation Pathways of IAPP and Other Amyloidogenic Proteins: Finding Answers through NMR Spectroscopy. J. Phys. Chem. Lett 2014, 5, 1864–1870. [DOI] [PubMed] [Google Scholar]
- (353).Pithadia AS; Bhunia A; Sribalan R; Padmini V; Fierke CA; Ramamoorthy A Influence of a Curcumin Derivative on HIAPP Aggregation in the Absence and Presence of Lipid Membranes. Chem. Commun 2016, 52, 942–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (354).Nedumpully-Govindan P; Kakinen A; Pilkington EH; Davis TP; Chun Ke P; Ding F Stabilizing Off-Pathway Oligomers by Polyphenol Nanoassemblies for IAPP Aggregation Inhibition. Sci. Rep 2016, 6, 19463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (355).Lolicato F; Raudino A; Milardi D; La Rosa C Resveratrol Interferes with the Aggregation of Membrane-Bound Human-IAPP: A Molecular Dynamics Study. Eur. J. Med. Chem 2015, 92, 876–881. [DOI] [PubMed] [Google Scholar]
- (356).Serrano AL; Lomont JP; Tu L-H; Raleigh DP; Zanni MT A Free Energy Barrier Caused by the Refolding of an Oligomeric Intermediate Controls the Lag Time of Amyloid Formation by HIAPP. J. Am. Chem. Soc 2017, 139, 16748–16758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (357).Rodriguez Camargo DC; Garg D; Buday K; Franko A; Rodriguez Camargo A; Schmidt F; Cox SJ; Suladze S; Haslbeck M; Mideksa YG; et al. HIAPP Forms Toxic Oligomers in Plasma. Chem. Commun 2018, 54, 5426–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (358).Dong X; Qiao Q; Qian Z; Wei G Recent Computational Studies of Membrane Interaction and Disruption of Human Islet Amyloid Polypeptide: Monomers, Oligomers and Protofibrils. Biochim. Biophys. Acta BBA - Biomembr 2018, 1860, 1826–1839. [DOI] [PubMed] [Google Scholar]
- (359).Quist A; Doudevski I; Lin H; Azimova R; Ng D; Frangione B; Kagan B; Ghiso J; Lal R Amyloid Ion Channels: A Common Structural Link for Protein-Misfolding Disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (360).Pannuzzo M; Milardi D; Raudino A; Karttunen M; La Rosa C Analytical Model and Multiscale Simulations of Aβ Peptide Aggregation in Lipid Membranes: Towards a Unifying Description of Conformational Transitions, Oligomerization and Membrane Damage. Phys. Chem. Chem. Phys 2013, 15, 8940–8951. [DOI] [PubMed] [Google Scholar]
- (361).Bengoa-Vergniory N; Roberts RF; Wade-Martins R; Alegre-Abarrategui J Alpha-Synuclein Oligomers: A New Hope. Acta Neuropathol 2017, 134, 819–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (362).Iljina M; Garcia GA; Dear AJ; Flint J; Narayan P; Michaels TCT; Dobson CM; Frenkel D; Knowles TPJ; Klenerman D Quantitative Analysis of Co-Oligomer Formation by Amyloid-Beta Peptide Isoforms. Sci. Rep 2016, 6, 28658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (363).Sciacca MFM; Tempra C; Scollo F; Milardi D; La Rosa C Amyloid Growth and Membrane Damage: Current Themes and Emerging Perspectives from Theory and Experiments on Aβ and HIAPP. Biochim. Biophys. Acta Biomembr 2018, 1860, 1625–1638. [DOI] [PubMed] [Google Scholar]
- (364).Brender JR; Lee EL; Cavitt MA; Gafni A; Steel DG; Ramamoorthy A Amyloid Fiber Formation and Membrane Disruption Are Separate Processes Localized in Two Distinct Regions of IAPP, the Type-2-Diabetes-Related Peptide. J. Am. Chem. Soc 2008, 130, 6424–6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).Hebda JA; Miranker AD The Interplay of Catalysis and Toxicity by Amyloid Intermediates on Lipid Bilayers: Insights from Type II Diabetes. Annu. Rev. Biophys 2009, 38, 125–152. [DOI] [PubMed] [Google Scholar]
- (366).Janson J; Ashley RH; Harrison D; McIntyre S; Butler PC The Mechanism of Islet Amyloid Polypeptide Toxicity Is Membrane Disruption by Intermediate-Sized Toxic Amyloid Particles. Diabetes 1999, 48, 491–498. [DOI] [PubMed] [Google Scholar]
- (367).Anguiano M; Nowak RJ; Lansbury PT Protofibrillar Islet Amyloid Polypeptide Permeabilizes Synthetic Vesicles by a Pore-like Mechanism That May Be Relevant to Type II Diabetes. Biochemistry 2002, 41, 11338–11343. [DOI] [PubMed] [Google Scholar]
- (368).Ashburn TT; Lansbury PT Interspecies Sequence Variations Affect the Kinetics and Thermodynamics of Amyloid Formation: Peptide Models of Pancreatic Amyloid. J. Am. Chem. Soc 1993, 115, 11012–11013. [Google Scholar]
- (369).Knight JD; Miranker AD Phospholipid Catalysis of Diabetic Amyloid Assembly. J. Mol. Biol 2004, 341, 1175–1187. [DOI] [PubMed] [Google Scholar]
- (370).Sparr E; Engel MFM; Sakharov DV; Sprong M; Jacobs J; Kruijff B. de; Höppener JWM; Killian JA Islet Amyloid Polypeptide-Induced Membrane Leakage Involves Uptake of Lipids by Forming Amyloid Fibers. FEBS Lett 2004, 577, 117–120. [DOI] [PubMed] [Google Scholar]
- (371).Brender JR; Salamekh S; Ramamoorthy A Membrane Disruption and Early Events in the Aggregation of the Diabetes Related Peptide IAPP from a Molecular Perspective. Acc. Chem. Res 2012, 45, 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (372).Engel MFM; Yigittop H; Elgersma RC; Rijkers DTS; Liskamp RMJ; de Kruijff B; Höppener JWM; Antoinette Killian J Islet Amyloid Polypeptide Inserts into Phospholipid Monolayers as Monomer. J. Mol. Biol 2006, 356, 783–789. [DOI] [PubMed] [Google Scholar]
- (373).Lopes DHJ; Meister A; Gohlke A; Hauser A; Blume A; Winter R Mechanism of Islet Amyloid Polypeptide Fibrillation at Lipid Interfaces Studied by Infrared Reflection Absorption Spectroscopy. Biophys. J 2007, 93, 3132–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (374).Williamson JA; Loria JP; Miranker AD Helix Stabilization Precedes Aqueous and Bilayer-Catalyzed Fiber Formation in Islet Amyloid Polypeptide. J. Mol. Biol 2009, 393, 383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).Jayasinghe SA; Langen R Membrane Interaction of Islet Amyloid Polypeptide. Biochim. Biophys. Acta BBA - Biomembr 2007, 1768, 2002–2009. [DOI] [PubMed] [Google Scholar]
- (376).Martel A; Antony L; Gerelli Y; Porcar L; Fluitt A; Hoffmann K; Kiesel I; Vivaudou M; Fragneto G; Pablo JJ Membrane Permeation versus Amyloidogenicity: A Multitechnique Study of Islet Amyloid Polypeptide Interaction with Model Membranes. J. Am. Chem. Soc 2017, 139, 137–148. [DOI] [PubMed] [Google Scholar]
- (377).Smith PES; Brender JR; Ramamoorthy A Induction of Negative Curvature as a Mechanism of Cell Toxicity by Amyloidogenic Peptides: The Case of Islet Amyloid Polypeptide. J. Am. Chem. Soc 2009, 131, 4470–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (378).Pannuzzo M; Raudino A; Milardi D; La Rosa C; Karttunen M α-Helical Structures Drive Early Stages of Self-Assembly of Amyloidogenic Amyloid Polypeptide Aggregate Formation in Membranes. Sci. Rep 2013, 3, 2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (379).Terakawa MS; Lin Y; Kinoshita M; Kanemura S; Itoh D; Sugiki T; Okumura M; Ramamoorthy A; Lee Y-H Impact of Membrane Curvature on Amyloid Aggregation. Biochim. Biophys. Acta Biomembr 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (380).Qiao Q; Wei G; Yao D; Song Z Formation of α-Helical and β-Sheet Structures in Membrane-Bound Human IAPP Monomer and the Resulting Membrane Deformation. Phys. Chem. Chem. Phys 2019, 21, 20239–20251. [DOI] [PubMed] [Google Scholar]
- (381).Kegulian NC; Sankhagowit S; Apostolidou M; Jayasinghe SA; Malmstadt N; Butler PC; Langen R Membrane Curvature-Sensing and -Inducing Activity of Islet Amyloid Polypeptide and Its Implications for Membrane Disruption. J. Biol. Chem 2015, jbc.M115.659797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (382).Zhang Y; Luo Y; Deng Y; Mu Y; Wei G Lipid Interaction and Membrane Perturbation of Human Islet Amyloid Polypeptide Monomer and Dimer by Molecular Dynamics Simulations. PLOS ONE 2012, 7, e38191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (383).Dignon GL; Zerze GH; Mittal J Interplay Between Membrane Composition and Structural Stability of Membrane-Bound HIAPP. J. Phys. Chem. B 2017, 121, 8661–8668. [DOI] [PubMed] [Google Scholar]
- (384).Christensen M; Skeby KK; Schiøtt B Identification of Key Interactions in the Initial Self-Assembly of Amylin in a Membrane Environment. Biochemistry 2017, 56, 4884–4894. [DOI] [PubMed] [Google Scholar]
- (385).Skeby KK; Andersen OJ; Pogorelov TV; Tajkhorshid E; Schiøtt B Conformational Dynamics of the Human Islet Amyloid Polypeptide in a Membrane Environment: Toward the Aggregation Prone Form. Biochemistry 2016, 55, 2031–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (386).Pappalardo G; Milardi D; Magrì A; Attanasio F; Impellizzeri G; La Rosa C; Grasso D; Rizzarelli E Environmental Factors Differently Affect Human and Rat IAPP: Conformational Preferences and Membrane Interactions of IAPP17–29 Peptide Derivatives. Chem. Eur. J 2007, 13, 10204–10215. [DOI] [PubMed] [Google Scholar]
- (387).Junghans A; Watkins EB; Majewski J; Miranker A; Stroe I Influence of the Human and Rat Islet Amyloid Polypeptides on Structure of Phospholipid Bilayers: Neutron Reflectometry and Fluorescence Microscopy Studies. Langmuir 2016, 32, 4382–4391. [DOI] [PubMed] [Google Scholar]
- (388).Khemtémourian L; Engel MFM; Kruijtzer JAW; Höppener JWM; Liskamp RMJ; Killian JA The Role of the Disulfide Bond in the Interaction of Islet Amyloid Polypeptide with Membranes. Eur. Biophys. J 2010, 39, 1359–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (389).Milardi D; Pappalardo M; Pannuzzo M; Grasso DM; La Rosa C The Role of the Cys2-Cys7 Disulfide Bridge in the Early Steps of Islet Amyloid Polypeptide Aggregation: A Molecular Dynamics Study. Chem. Phys. Lett 2008, 463, 396–399. [Google Scholar]
- (390).Nanga RPR; Brender JR; Xu J; Hartman K; Subramanian V; Ramamoorthy A Three-Dimensional Structure and Orientation of Rat Islet Amyloid Polypeptide Protein in a Membrane Environment by Solution NMR Spectroscopy. J. Am. Chem. Soc 2009, 131, 8252–8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (391).Nanga RPR; Brender JR; Vivekanandan S; Ramamoorthy A Structure and Membrane Orientation of IAPP in Its Natively Amidated Form at Physiological pH in a Membrane Environment. Biochim. Biophys. Acta 2011, 1808, 2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (392).Nanga RPR; Brender JR; Xu J; Veglia G; Ramamoorthy A Structures of Rat and Human Islet Amyloid Polypeptide IAPP(1–19) in Micelles by NMR Spectroscopy. Biochemistry 2008, 47, 12689–12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (393).Sciacca MFM; Kotler SA; Brender JR; Chen J; Lee D; Ramamoorthy A Two-Step Mechanism of Membrane Disruption by Aβ through Membrane Fragmentation and Pore Formation. Biophys. J 2012, 103, 702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Cecchini M; Curcio R; Pappalardo M; Melki R; Caflisch A A Molecular Dynamics Approach to the Structural Characterization of Amyloid Aggregation. J. Mol. Biol 2006, 357, 1306–1321. [DOI] [PubMed] [Google Scholar]
- (395).Caflisch A Computational Models for the Prediction of Polypeptide Aggregation Propensity. Curr. Opin. Chem. Biol 2006, 10, 437–444. [DOI] [PubMed] [Google Scholar]
- (396).Sciacca MFM; Pappalardo M; Attanasio F; Milardi D; Rosa CL; Grasso DM Are Fibril Growth and Membrane Damage Linked Processes? An Experimental and Computational Study of IAPP12–18 and IAPP21–27 Peptides. New J. Chem 2010, 34, 200–207. [Google Scholar]
- (397).Sciacca MFM; Chillemi R; Sciuto S; Greco V; Messineo C; Kotler SA; Lee D-K; Brender JR; Ramamoorthy A; La Rosa C; Milardi D A Blend of Two Resveratrol Derivatives Abolishes HIAPP Amyloid Growth and Membrane Damage. Biochim. Biophys. Acta - Biomembr 2018, 1860, 1793–1802. [DOI] [PubMed] [Google Scholar]
- (398).Sciacca MF; Chillemi R; Sciuto S; Pappalardo M; Rosa CL; Grasso D; Milardi D Interactions of Two< I> O-Phosphorylresveratrol Derivatives with Model Membranes. Arch. Biochem. Biophys 2012, 521, 111–116. [DOI] [PubMed] [Google Scholar]
- (399).Lolicato F; Raudino A; Milardi D; La Rosa C Resveratrol Interferes with the Aggregation of Membrane-Bound Human-IAPP: A Molecular Dynamics Study. Eur. J. Med. Chem 2015, 92, 876–881. [DOI] [PubMed] [Google Scholar]
- (400).Singer SJ; Nicolson GL The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. [DOI] [PubMed] [Google Scholar]
- (401).Hope MJ; Bally MB; Mayer LD; Janoff AS; Cullis PR Generation of Multilamellar and Unilamellar Phospholipid Vesicles. Chem. Phys. Lipids 1986, 40, 89–107. [Google Scholar]
- (402).Kim S; Jacobs RE; White SH Preparation of Multilamellar Vesicles of Defined Size-Distribution by Solvent-Spherule Evaporation. Biochim. Biophys. Acta - Biomembr 1985, 812, 793–801. [DOI] [PubMed] [Google Scholar]
- (403).Gruner SM; Lenk RP; Janoff AS; Ostro NJ Novel Multilayered Lipid Vesicles: Comparison of Physical Characteristics of Multilamellar Liposomes and Stable Plurilamellar Vesicles. Biochemistry 1985, 24, 2833–2842. [DOI] [PubMed] [Google Scholar]
- (404).van Meer G; Voelker DR; Feigenson GW Membrane Lipids: Where They Are and How They Behave. Nat. Rev. Mol. Cell Biol 2008, 9, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (405).Pappalardo M; Milardi D; Grasso D; La Rosa C Phase Behaviour of Polymer-Grafted DPPC Membranes for Drug Delivery Systems Design. J. Therm. Anal. Calorim 2005, 80, 413–418. [Google Scholar]
- (406).Raudino A; Zuccarello F; La Rosa C; Buemi G Thermal Expansion and Compressibility Coefficients of Phospholipid Vesicles: Experimental Determination and Theoretical Modeling. J. Phys. Chem 1990, 94, 4217–4223. [Google Scholar]
- (407).Drazenovic J; Wang H; Roth K; Zhang J; Ahmed S; Chen Y; Bothun G; Wunder SL Effect of Lamellarity and Size on Calorimetric Phase Transitions in Single Component Phosphatidylcholine Vesicles. Biochim. Biophys. Acta BBA - Biomembr 2015, 1848, 532–543. [DOI] [PubMed] [Google Scholar]
- (408).Moscho A; Orwar O; Chiu DT; Modi BP; Zare RN Rapid Preparation of Giant Unilamellar Vesicles. Proc. Natl. Acad. Sci. USA 1996, 93, 11443–11447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (409).Patil YP; Ahluwalia AK; Jadhav S Isolation of Giant Unilamellar Vesicles from Electroformed Vesicle Suspensions and Their Extrusion through Nano-Pores. Chem. Phys. Lipids 2013, 167–168, 1–8. [DOI] [PubMed] [Google Scholar]
- (410).Chiantia S; Schwille P; Klymchenko AS; London E Asymmetric GUVs Prepared by MβCD-Mediated Lipid Exchange: An FCS Study. Biophys. J 2011, 100, L1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (411).Cheng H-T; London E Preparation and Properties of Asymmetric Large Unilamellar Vesicles: Interleaflet Coupling in Asymmetric Vesicles Is Dependent on Temperature but Not Curvature. Biophys. J 2011, 100, 2671–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Lin C-M; Li C-S; Sheng Y-J; Wu DT; Tsao H-K Size-Dependent Properties of Small Unilamellar Vesicles Formed by Model Lipids. Langmuir 2012, 28, 689–700. [DOI] [PubMed] [Google Scholar]
- (413).Lichtenberg D; Freire E; Schmidt CF; Barenholz Y; Felgner PL; Thompson TE Effect of Surface Curvature on Stability, Thermodynamic Behavior, and Osmotic Activity of Dipalmitoylphosphatidylcholine Single Lamellar Vesicles. Biochemistry 1981, 20, 3462–3467. [DOI] [PubMed] [Google Scholar]
- (414).Tayebi L; Vashaee D; Parikh AN Stability of Uni- and Multillamellar Spherical Vesicles. ChemPhysChem 2012, 13, 314–322. [DOI] [PubMed] [Google Scholar]
- (415).Castellana ET; Cremer PS Solid Supported Lipid Bilayers: From Biophysical Studies to Sensor Design. Surf. Sci. Rep 2006, 61, 429–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (416).Keller CA; Kasemo B Surface Specific Kinetics of Lipid Vesicle Adsorption Measured with a Quartz Crystal Microbalance. Biophys. J 1998, 75, 1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (417).Cho N-J; Frank CW; Kasemo B; Höök F Quartz Crystal Microbalance with Dissipation Monitoring of Supported Lipid Bilayers on Various Substrates. Nat. Protoc 2010, 5, 1096–1106. [DOI] [PubMed] [Google Scholar]
- (418).Scalisi S; Sciacca MFM; Zhavnerko G; Grasso DM; Marletta G; La Rosa C Self-Assembling Pathway of HiApp Fibrils within Lipid Bilayers. ChemBioChem 2010, 11, 1856–1859. [DOI] [PubMed] [Google Scholar]
- (419).Dürr UHN; Gildenberg M; Ramamoorthy A The Magic of Bicelles Lights Up Membrane Protein Structure. Chem. Rev 2012, 112, 6054–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (420).Glover KJ; Whiles JA; Wu G; Yu N; Deems R; Struppe JO; Stark RE; Komives EA; Vold RR Structural Evaluation of Phospholipid Bicelles for Solution-State Studies of Membrane-Associated Biomolecules. Biophys. J 2001, 81, 2163–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (421).Yamada NL; Sferrazza M; Fujinami S In-Situ Measurement of Phospholipid Nanodisk Adhesion on a Solid Substrate Using Neutron Reflectometry and Atomic Force Microscopy. Phys. B Condens. Matter 2018, 551, 222–226. [Google Scholar]
- (422).Bayburt TH; Grinkova YV; Sligar SG Self-Assembly of Discoidal Phospholipid Bilayer Nanoparticles with Membrane Scaffold Proteins. Nano Lett 2002, 2, 853–856. [Google Scholar]
- (423).Knowles TJ; Finka R; Smith C; Lin Y-P; Dafforn T; Overduin M Membrane Proteins Solubilized Intact in Lipid Containing Nanoparticles Bounded by Styrene Maleic Acid Copolymer. J. Am. Chem. Soc 2009, 131, 7484–7485. [DOI] [PubMed] [Google Scholar]
- (424).Hardin NZ; Kocman V; Mauro GMD; Ravula T; Ramamoorthy A Metal-Chelated Polymer Nanodiscs for NMR Studies. Angew. Chem. Int. Ed 2019, 58, 17246–17250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (425).Hardin NZ; Ravula T; Mauro GD; Ramamoorthy A Hydrophobic Functionalization of Polyacrylic Acid as a Versatile Platform for the Development of Polymer Lipid Nanodisks. Small 2019, 15, 1804813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Xue M; Cheng L; Faustino I; Guo W; Marrink SJ Molecular Mechanism of Lipid Nanodisk Formation by Styrene-Maleic Acid Copolymers. Biophys. J 2018, 115, 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (427).Ravula T; Ramamoorthy A Magnetic Alignment of Polymer Macro-Nanodiscs Enables Residual-Dipolar-Coupling-Based High-Resolution Structural Studies by NMR Spectroscopy. Angew. Chem. Int. Ed 2019, 58, 14925–14928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (428).Brender JR; Krishnamoorthy J; Sciacca MFM; Vivekanandan S; D’Urso L; Chen J; La Rosa C; Ramamoorthy A Probing the Sources of the Apparent Irreproducibility of Amyloid Formation: Drastic Changes in Kinetics and a Switch in Mechanism Due to Micellelike Oligomer Formation at Critical Concentrations of IAPP. J. Phys. Chem. B 2015, 119, 2886–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (429).Brender JR; Salamekh S; Ramamoorthy A Membrane Disruption and Early Events in the Aggregation of the Diabetes Related Peptide IAPP from a Molecular Perspective. Acc. Chem. Res 2012, 45, 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (430).Marsh D Thermodynamics of Phospholipid Self-Assembly. Biophys. J 2012, 102, 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (431).Marsh D Equation of State for Phospholipid Self-Assembly. Biophys. J 2016, 110, 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (432).La Rosa C; Scalisi S; Lolicato F; Pannuzzo M; Raudino A Lipid-Assisted Protein Transport: A Diffusion-Reaction Model Supported by Kinetic Experiments and Molecular Dynamics Simulations. J. Chem. Phys 2016, 144, 184901. [DOI] [PubMed] [Google Scholar]
- (433).Tempra C; La Rosa C; Lolicato F The Role of Alpha-Helix on the Structure-Targeting Drug Design of Amyloidogenic Proteins. bioRxiv 2020, doi:2020.11.20.391409. [DOI] [PubMed] [Google Scholar]
- (434).Korshavn KJ; Satriano C; Lin Y; Zhang R; Dulchavsky M; Bhunia A; Ivanova MI; Lee Y-H; La Rosa C; Lim MH; Ramamoorthy A Reduced Lipid Bilayer Thickness Regulates the Aggregation and Cytotoxicity of Amyloid-β. J. Biol. Chem 2017, 292, 4638–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (435).Galvagnion C; Brown JWP; Ouberai MM; Flagmeier P; Vendruscolo M; Buell AK; Sparr E; Dobson CM Chemical Properties of Lipids Strongly Affect the Kinetics of the Membrane-Induced Aggregation of α-Synuclein. Proc. Natl. Acad. Sci.USA 2016, 113, 7065–7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (436).Dignon GL; Zerze GH; Mittal J Interplay Between Membrane Composition and Structural Stability of Membrane-Bound HIAPP. J. Phys. Chem. B 2017, 121, 8661–8668. [DOI] [PubMed] [Google Scholar]
- (437).Sciacca MFM; Lolicato F; Tempra C; Scollo F; Sahoo BR; Watson MD; García-Viñuales S; Milardi D; Raudino A; Lee JC; Ramamoorthy A; La Rosa C The Lipid-Chaperon Hypothesis: A Common Molecular Mechanism of Membrane Disruption by Intrinsically Disordered Proteins. ACS Chem Neurosci 2020, Doi: 10.1021/acschemneuro.0c00588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (438).La Rosa C; Condorelli M; Compagnini G; Lolicato F; Milardi D; Do TN; Karttunen M; Pannuzzo M; Ramamoorthy A; Fraternali F; et al. Symmetry-Breaking Transitions in the Early Steps of Protein Self-Assembly. Eur. Biophys. J 2020, 49, 175–191. [DOI] [PubMed] [Google Scholar]
- (439).Lorenzo A; Razzaboni B; Weir GC; Yankner BA Pancreatic Islet Cell Toxicity of Amylin Associated with Type-2 Diabetes Mellitus. Nature 1994, 368, 756–760. [DOI] [PubMed] [Google Scholar]
- (440).Bai JZ; Saafi EL; Zhang S; Cooper GJ Role of Ca2+ in Apoptosis Evoked by Human Amylin in Pancreatic Islet β-Cells. Biochem. J 1999, 343, 53–61. [PMC free article] [PubMed] [Google Scholar]
- (441).Zraika S; Hull RL; Udayasankar J; Aston-Mourney K; Subramanian SL; Kisilevsky R; Szarek WA; Kahn SE Oxidative Stress Is Induced by Islet Amyloid Formation and Time-Dependently Mediates Amyloid-Induced Beta Cell Apoptosis. Diabetologia 2009, 52, 626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (442).Zraika S; Aston-Mourney K; Marek P; Hull RL; Green PS; Udayasankar J; Subramanian SL; Raleigh DP; Kahn SE Neprilysin Impedes Islet Amyloid Formation by Inhibition of Fibril Formation Rather than Peptide Degradation. J. Biol. Chem 2010, 285, 18177–18183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (443).Guan H; Chow KM; Shah R; Rhodes CJ; Hersh LB Degradation of Islet Amyloid Polypeptide by Neprilysin. Diabetologia 2012, 55, 2989–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (444).Backstrom JR; Lim GP; Cullen MJ; Tökés ZA Matrix Metalloproteinase-9 (MMP-9) Is Synthesized in Neurons of the Human Hippocampus and Is Capable of Degrading the Amyloid-β Peptide (1–40). J. Neurosci 1996, 16, 7910–7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (445).Roher AE; Kasunic TC; Woods AS; Cotter RJ; Ball MJ; Fridman R Proteolysis of Aβ Peptide from Alzheimer Disease Brain by Gelatinase A. Biochem. Biophys. Res. Commun 1994, 205, 1755–1761. [DOI] [PubMed] [Google Scholar]
- (446).Tomita T; Iwata K Gelatinases and Inhibitors of Gelatinases in Pancreatic Islets and Islet Cell Tumors. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc 1997, 10, 47–54. [PubMed] [Google Scholar]
- (447).Sternlicht MD; Werb Z How Matrix Metalloproteinases Regulate Cell Behavior. Annu. Rev. Cell Dev. Biol 2001, 17, 463–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (448).Aston-Mourney K; Zraika S; Udayasankar J; Subramanian SL; Green PS; Kahn SE; Hull RL Matrix Metalloproteinase-9 Reduces Islet Amyloid Formation by Degrading Islet Amyloid Polypeptide. J. Biol. Chem 2013, 288, 3553–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (449).Netzel-Arnett S; Sang QX; Moore WG; Navre M; Birkedal-Hansen H; Van Wart HE Comparative Sequence Specificities of Human 72- and 92-KDa Gelatinases (Type IV Collagenases) and PUMP (Matrilysin). Biochemistry 1993, 32, 6427–6432. [DOI] [PubMed] [Google Scholar]
- (450).Turk BE; Huang LL; Piro ET; Cantley LC Determination of Protease Cleavage Site Motifs Using Mixture-Based Oriented Peptide Libraries. Nat. Biotechnol 2001, 19, 661–667. [DOI] [PubMed] [Google Scholar]
- (451).Levy M; Garmy N; Gazit E; Fantini J The Minimal Amyloid-Forming Fragment of the Islet Amyloid Polypeptide Is a Glycolipid-Binding Domain. FEBS J 2006, 273, 5724–5735. [DOI] [PubMed] [Google Scholar]
- (452).Nilsson MR; Raleigh DP Analysis of Amylin Cleavage Products Provides New Insights into the Amyloidogenic Region of Human Amylin. J. Mol. Biol 1999, 294, 1375–1385. [DOI] [PubMed] [Google Scholar]
- (453).Zraika S; Hull RL; Udayasankar J; Clark A; Utzschneider KM; Tong J; Gerchman F; Kahn SE Identification of the Amyloid-Degrading Enzyme Neprilysin in Mouse Islets and Potential Role in Islet Amyloidogenesis. Diabetes 2007, 56, 304–310. [DOI] [PubMed] [Google Scholar]
- (454).Eckman EA; Adams SK; Troendle FJ; Stodola BA; Kahn MA; Fauq AH; Xiao HD; Bernstein KE; Eckman CB Regulation of Steady-State β-Amyloid Levels in the Brain by Neprilysin and Endothelin-Converting Enzyme but Not Angiotensin-Converting Enzyme. J. Biol. Chem 2006, 281, 30471–30478. [DOI] [PubMed] [Google Scholar]
- (455).Farzan M; Schnitzler CE; Vasilieva N; Leung D; Choe H BACE2, a β-Secretase Homolog, Cleaves at the β Site and within the Amyloid-β Region of the Amyloid-β Precursor Protein. Proc. Natl. Acad. Sci. USA 2000, 97, 9712–9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (456).Hussain I; Powell DJ; Howlett DR; Chapman GA; Gilmour L; Murdock PR; Tew DG; Meek TD; Chapman C; Schneider K ASP1 (BACE2) Cleaves the Amyloid Precursor Protein at the β-Secretase Site. Mol. Cell. Neurosci 2000, 16, 609–619. [DOI] [PubMed] [Google Scholar]
- (457).Bennett B; Babu-Khan S; Loeloff R; Louis J; Curran E; Citron M; Vassar R Expression Analysis of BACE2 in Brain and Peripheral Tissues. J. Biol. Chem 2000, 275, 20647–20651. [DOI] [PubMed] [Google Scholar]
- (458).Rulifson IC; Cao P; Miao L; Kopecky D; Huang L; White RD; Samayoa K; Gardner J; Wu X; Chen K et al. Identification of Human Islet Amyloid Polypeptide as a BACE2 Substrate. PLOS ONE 2016, 11, e0147254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (459).Alcarraz-Vizán G; Castaño C; Visa M; Montane J; Servitja J-M; Novials A BACE2 Suppression Promotes β-Cell Survival and Function in a Model of Type 2 Diabetes Induced by Human Islet Amyloid Polypeptide Overexpression. Cell. Mol. Life Sci 2017, 74, 2827–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (460).Sbardella D; Tundo GR; Sciandra F; Bozzi M; Gioia M; Ciaccio C; Tarantino U; Brancaccio A; Coletta M; Marini S Proteasome Activity Is Affected by Fluctuations in Insulin-Degrading Enzyme Distribution. PLOS ONE 2015, 10, e0132455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (461).Shen Y; Joachimiak A; Rich Rosner M; Tang W-J Structures of Human Insulin-Degrading Enzyme Reveal a New Substrate Recognition Mechanism. Nature 2006, 443, 870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (462).Im H; Manolopoulou M; Malito E; Shen Y; Zhao J; Neant-Fery M; Sun C-Y; Meredith SC; Sisodia SS; Leissring MA; Tang W-J Structure of Substrate-Free Human Insulin-Degrading Enzyme (IDE) and Biophysical Analysis of ATP-Induced Conformational Switch of IDE. J. Biol. Chem 2007, 282, 25453–25463. [DOI] [PubMed] [Google Scholar]
- (463).Mirsky IA; Broh-Kahn RH The Inactivation of Insulin by Tissue Extracts; the Distribution and Properties of Insulin Inactivating Extracts. Arch. Biochem 1949, 20, 1–9. [PubMed] [Google Scholar]
- (464).Duckworth WC Insulin Degradation: Mechanisms, Products, and Significance. Endocr. Rev 1988, 9, 319–345. [DOI] [PubMed] [Google Scholar]
- (465).Maianti JP; McFedries A; Foda ZH; Kleiner RE; Du XQ; Leissring MA; Tang W-J; Charron MJ; Seeliger MA; Saghatelian A; Liu DR Anti-Diabetic Activity of Insulin-Degrading Enzyme Inhibitors Mediated by Multiple Hormones. Nature 2014, 511, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (466).Tundo GR; Sbardella D; Ciaccio C; Grasso G; Gioia M; Coletta A; Polticelli F; Di Pierro D; Milardi D; Van Endert P Multiple Functions of Insulin-Degrading Enzyme: A Metabolic Crosslight? Crit. Rev. Biochem. Mol. Biol 2017, 52, 554–582. [DOI] [PubMed] [Google Scholar]
- (467).Vekrellis K; Ye Z; Qiu WQ; Walsh D; Hartley D; Chesneau V; Rosner MR; Selkoe DJ Neurons Regulate Extracellular Levels of Amyloid β-Protein via Proteolysis by Insulin-Degrading Enzyme. J. Neurosci 2000, 20, 1657–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (468).Bondy CA; Zhou J; Chin E; Reinhardt RR; Ding L; Roth RA Cellular Distribution of Insulin-Degrading Enzyme Gene Expression. Comparison with Insulin and Insulin-like Growth Factor Receptors. J. Clin. Invest 1994, 93, 966–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (469).Rezende LF; Camargo RL; Branco RCS; Cappelli APG; Boschero AC; Carneiro EM Reduced Insulin Clearance and Lower Insulin-Degrading Enzyme Expression in the Liver Might Contribute to the Thrifty Phenotype of Protein-Restricted Mice. Br. J. Nutr 2014, 112, 900–907. [DOI] [PubMed] [Google Scholar]
- (470).Deprez-Poulain R; Hennuyer N, Bosc D; Liang WG, Enée E, Marechal X; Charton J, Totobenazara J, Berte G, Jahklal J, et al. Catalytic Site Inhibition of Insulin-Degrading Enzyme by a Small Molecule Induces Glucose Intolerance in Mice. Nat. Commun 2015, 6, 8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (471).Fernández-Gamba A; Leal MC; Morelli L; Castaño EM Insulin-Degrading Enzyme: Structure-Function Relationship and Its Possible Roles in Health and Disease. Curr. Pharm. Des 2009, 15, 3644–3655. [DOI] [PubMed] [Google Scholar]
- (472).Hersh LB The Insulysin (Insulin Degrading Enzyme) Enigma. Cell. Mol. Life Sci 2006, 63, 2432–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (473).McDermott JR; Gibson AM Degradation of Alzheimer’s Beta-Amyloid Protein by Human and Rat Brain Peptidases: Involvement of Insulin-Degrading Enzyme. Neurochem. Res 1997, 22, 49–56. [DOI] [PubMed] [Google Scholar]
- (474).Chesneau V; Vekrellis K; Rosner MR; Selkoe DJ Purified Recombinant Insulin-Degrading Enzyme Degrades Amyloid Beta-Protein but Does Not Promote Its Oligomerization. Biochem. J 2000, 351, 509–516. [PMC free article] [PubMed] [Google Scholar]
- (475).Bellia F; Grasso G The Role of Copper (II) and Zinc (II) in the Degradation of Human and Murine IAPP by Insulin-Degrading Enzyme. J. Mass Spectrom 2014, 49, 274–279. [DOI] [PubMed] [Google Scholar]
- (476).Guo Q; Manolopoulou M; Bian Y; Schilling AB; Tang W-J Molecular Basis for the Recognition and Cleavages of IGF-II, TGF-α, and Amylin by Human Insulin-Degrading Enzyme. J. Mol. Biol 2010, 395, 430–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (477).Goldberg AL Protein Degradation and Protection against Misfolded or Damaged Proteins. Nature 2003, 426, 895–899. [DOI] [PubMed] [Google Scholar]
- (478).Varshavsky A Regulated Protein Degradation. Trends Biochem. Sci 2005, 30, 283–286. [DOI] [PubMed] [Google Scholar]
- (479).Vijay-Kumar S; Bugg CE; Cook WJ Structure of Ubiquitin Refined at 1.8 A Resolution. J. Mol. Biol 1987, 194, 531–544. [DOI] [PubMed] [Google Scholar]
- (480).Mukhopadhyay D; Riezman H Proteasome-Independent Functions of Ubiquitin in Endocytosis and Signaling. Science 2007, 315, 201–205. [DOI] [PubMed] [Google Scholar]
- (481).Pickart CM; Rose IA Functional Heterogeneity of Ubiquitin Carrier Proteins. J. Biol. Chem 1985, 260, 1573–1581. [PubMed] [Google Scholar]
- (482).Pickart CM Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem 2001, 70, 503–533. [DOI] [PubMed] [Google Scholar]
- (483).Hough R; Pratt G; Rechsteiner M Ubiquitin-Lysozyme Conjugates. Identification and Characterization of an ATP-Dependent Protease from Rabbit Reticulocyte Lysates. J. Biol. Chem 1986, 261, 2400–2408. [PubMed] [Google Scholar]
- (484).Voges D; Zwickl P; Baumeister W The 26S Proteasome: A Molecular Machine Designed for Controlled Proteolysis. Annu. Rev. Biochem 1999, 68, 1015–1068. [DOI] [PubMed] [Google Scholar]
- (485).Harris JR Release of a Macromolecular Protein Component from Human Erythrocyte Ghosts. Biochim. Biophys. Acta 1968, 150, 534–537. [DOI] [PubMed] [Google Scholar]
- (486).Hegerl R; Pfeifer G; Pühler G; Dahlmann B; Baumeister W The Three-Dimensional Structure of Proteasomes from Thermoplasma Acidophilum as Determined by Electron Microscopy Using Random Conical Tilting. FEBS Lett 1991, 283, 117–121. [DOI] [PubMed] [Google Scholar]
- (487).Orlowski M; Wilk S Catalytic Activities of the 20 S Proteasome, a Multicatalytic Proteinase Complex. Arch. Biochem. Biophys 2000, 383, 1–16. [DOI] [PubMed] [Google Scholar]
- (488).Mukherjee A; Morales-Scheihing D; Butler PC; Soto C Type 2 Diabetes as a Protein Misfolding Disease. Trends Mol. Med 2015, 21, 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (489).Jaisson S; Gillery P Impaired Proteostasis: Role in the Pathogenesis of Diabetes Mellitus. Diabetologia 2014, 57, 1517–1527. [DOI] [PubMed] [Google Scholar]
- (490).Singh S; Trikha S; Sarkar A; Jeremic AM Proteasome Regulates Turnover of Toxic Human Amylin in Pancreatic Cells. Biochem. J 2016, 473, 2655–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (491).Erales J; Coffino P Ubiquitin-Independent Proteasomal Degradation. Biochim. Biophys. Acta 2014, 1843, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (492).Rivera JF; Costes S; Gurlo T; Glabe CG; Butler PC Autophagy Defends Pancreatic β Cells from Human Islet Amyloid Polypeptide-Induced Toxicity. J. Clin. Invest 2014, 124, 3489–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (493).Broca C; Varin E; Armanet M; Tourrel-Cuzin C; Bosco D; Dalle S; Wojtusciszyn A Proteasome Dysfunction Mediates High Glucose-Induced Apoptosis in Rodent Beta Cells and Human Islets. PLOS ONE 2014, 9, e92066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (494).Chatterjee Bhowmick D; Jeremic A Functional Proteasome Complex Is Required for Turnover of Islet Amyloid Polypeptide in Pancreatic β-Cells. J. Biol. Chem 2018, 293, 14210–14223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (495).Bellia F; Pietropaolo A; Grasso G Formation of Insulin Fragments by Insulin-Degrading Enzyme: The Role of Zinc (II) and Cystine Bridges. J. Mass Spectrom 2013, 48, 135–140. [DOI] [PubMed] [Google Scholar]
- (496).Kim M-J; Kim H-T Investigation of the Copper Binding Site on the Human Islet Amyloid Polypeptide Hormone. Eur. J. Mass Spectrom 2012, 18, 51–58. [DOI] [PubMed] [Google Scholar]
- (497).Salamekh S; Brender JR; Hyung S-J; Nanga RPR; Vivekanandan S; Ruotolo BT; Ramamoorthy A A Two-Site Mechanism for the Inhibition of IAPP Amyloidogenesis by Zinc. J. Mol. Biol 2011, 410, 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (498).Maric S; Donnelly SM; Robinson MW; Skinner-Adams T; Trenholme KR; Gardiner DL; Dalton JP; Stack CM; Lowther J The M17 Leucine Aminopeptidase of the Malaria Parasite Plasmodium Falciparum: Importance of Active Site Metal Ions in the Binding of Substrates and Inhibitors. Biochemistry 2009, 48, 5435–5439. [DOI] [PubMed] [Google Scholar]
- (499).Grasso G; Salomone F; Tundo GR; Pappalardo G; Ciaccio C; Spoto G; Pietropaolo A; Coletta M; Rizzarelli E Metal Ions Affect Insulin-Degrading Enzyme Activity. J. Inorg. Biochem 2012, 117, 351–358. [DOI] [PubMed] [Google Scholar]
- (500).Mital M; Bal W; Frączyk T; Drew SC Interplay between Copper, Neprilysin, and N-Truncation of β-Amyloid. Inorg. Chem 2018, 57, 6193–6197. [DOI] [PubMed] [Google Scholar]
- (501).Malgieri G; Grasso G The Clearance of Misfolded Proteins in Neurodegenerative Diseases by Zinc Metalloproteases: An Inorganic Perspective. Coord. Chem. Rev 2014, 260, 139–155. [Google Scholar]
- (502).Bertini I; Canti G; Kozłowski H; Scozzafava A Spectroscopic Characterization of Copper (II) Thermolysin. J. Chem. Soc. Dalton Trans 1979, 0, 1270–1273. [Google Scholar]
- (503).Tan PS; Pos KM; Konings WN Purification and Characterization of an Endopeptidase from Lactococcus Lactis Subsp. Cremoris Wg2. Appl. Environ. Microbiol 1991, 57, 3593–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (504).Rosenberg RC; Root CA; Bernstein PK; Gray HB Spectral Studies of Copper(II) Carboxypeptidase A and Related Model Complexes. J. Am. Chem. Soc 1975, 97, 2092–2096. [DOI] [PubMed] [Google Scholar]
- (505).Hirose J; Ohsaki T; Nishimoto N; Matuoka S; Hiromoto T; Yoshida T; Minoura T; Iwamoto H; Fukasawa KM Characterization of the Metal-Binding Site in Aminopeptidase B. Biol. Pharm. Bull 2006, 29, 2378–2382. [DOI] [PubMed] [Google Scholar]
- (506).Milardi D; Arnesano F; Grasso G; Magrì A; Tabbì G; Scintilla S; Natile G; Rizzarelli E Ubiquitin Stability and the Lys63-Linked Polyubiquitination Site Are Compromised on Copper Binding. Angew. Chem. Int. Ed Engl 2007, 46, 7993–7995. [DOI] [PubMed] [Google Scholar]
- (507).Arena G; Fattorusso R; Grasso G; Grasso GI; Isernia C; Malgieri G; Milardi D; Rizzarelli E Zinc(II) Complexes of Ubiquitin: Speciation, Affinity and Binding Features. Chem. Weinh. Bergstr. Ger 2011, 17, 11596–11603. [DOI] [PubMed] [Google Scholar]
- (508).Ecker DJ; Butt TR; Marsh J; Sternberg EJ; Margolis N; Monia BP; Jonnalagadda S; Khan MI; Weber PL; Mueller L Gene Synthesis, Expression, Structures, and Functional Activities of Site-Specific Mutants of Ubiquitin. J. Biol. Chem 1987, 262, 14213–14221. [PubMed] [Google Scholar]
- (509).Santoro AM; Monaco I; Attanasio F; Lanza V; Pappalardo G; Tomasello MF; Cunsolo A; Rizzarelli E; De Luigi A; Salmona M; Milardi D Copper(II) Ions Affect the Gating Dynamics of the 20S Proteasome: A Molecular and in Cell Study. Sci. Rep 2016, 6, 33444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (510).Lee J-A Neuronal Autophagy: A Housekeeper or a Fighter in Neuronal Cell Survival? Exp. Neurobiol 2012, 21, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (511).Kaushik S; Cuervo AM Chaperone-Mediated Autophagy: A Unique Way to Enter the Lysosome World. Trends Cell Biol 2012, 22, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (512).Lim Y-A; Rhein V; Baysang G; Meier F; Poljak A; Raftery MJ; Guilhaus M; Ittner LM; Eckert A; Götz J Abeta and Human Amylin Share a Common Toxicity Pathway via Mitochondrial Dysfunction. Proteomics 2010, 10, 1621–1633. [DOI] [PubMed] [Google Scholar]
- (513).Rivera JF; Gurlo T; Daval M; Huang CJ; Matveyenko AV; Butler PC; Costes S Human-IAPP Disrupts the Autophagy/Lysosomal Pathway in Pancreatic β -Cells: Protective Role of P62-Positive Cytoplasmic Inclusions. Cell Death Differ 2011, 18, 415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (514).Hernández MG; Aguilar AG; Burillo J; Oca RG; Manca MA; Novials A; Alcarraz-Vizan G; Guillén C; Benito M Pancreatic β Cells Overexpressing HIAPP Impaired Mitophagy and Unbalanced Mitochondrial Dynamics. Cell. Death Dis 2018, 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (515).Korolchuk VI; Menzies FM; Rubinsztein DC Mechanisms of Cross-Talk between the Ubiquitin-Proteasome and Autophagy-Lysosome Systems. FEBS Lett 2010, 584, 1393–1398. [DOI] [PubMed] [Google Scholar]
- (516).Pandey UB; Nie Z; Batlevi Y; McCray BA; Ritson GP; Nedelsky NB; Schwartz SL; DiProspero NA; Knight MA; Schuldiner O; et al. HDAC6 Rescues Neurodegeneration and Provides an Essential Link between Autophagy and the UPS. Nature 2007, 447, 859–863. [DOI] [PubMed] [Google Scholar]
- (517).Hutton JC The insulin secretory granule. Diabetologia 1989, 32. [DOI] [PubMed] [Google Scholar]
- (518).Hou JC; Min L; Pessin JE Insulin Granule Biogenesis, Trafficking and Exocytosis. Vitam. Horm 2009, 80, 473–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (519).Marsh BJ; Soden C; Alarcón C; Wicksteed BL; Yaekura K; Costin AJ Regulated autophagy controls hormone content in secretory-deficient pancreatic endocrine beta-cells. Mol. Endocrinol 2007, 21, 2255–2269. [DOI] [PubMed] [Google Scholar]
- (520).Westermark P; Li ZC; Westermark GT; Leckstrom A; Steiner DF Effects of Beta Cell Granule Components on Human Islet Amyloid Polypeptide Fibril Formation. FEBS Lett 1996, 379, 203–206. [DOI] [PubMed] [Google Scholar]
- (521).Jaikaran ET; Nilsson MR; Clark A Pancreatic Beta-Cell Granule Peptides Form Heteromolecular Complexes Which Inhibit Islet Amyloid Polypeptide Fibril Formation. Biochem. J 2004, 377, 709–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (522).Knight JD; Williamson JA; Miranker AD Interaction of Membrane-Bound Islet Amyloid Polypeptide with Soluble and Crystalline Insulin. Protein Sci 2008, 17, 1850–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (523).Andersson A; Bohman S; Borg LA; Paulsson JF; Schultz SW; Westermark GT Amyloid Deposition in Transplanted Human Pancreatic Islets: A Conceivable Cause of Their Long-Term Failure. Exp. Diabetes Res 2008, 2008, 562985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (524).Lashley T; Revesz T; Plant G; Bandopadhyay R; Lees AJ; Frangione B Expression of BRI2 mRNA and Protein in Normal Human Brain and Familial British Dementia: Its Relevance to the Pathogenesis of Disease. Neuropathol. Appl. Neurobiol 2008, 34. 492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (525).Del Campo M; Hoozemans JJM; Dekkers L-L; Rozemuller AJ; Korth C; Müller-Schiffmann A; Scheltens P; Blankenstein MA; Jimenez CR; Veerhuis R; Teunissen CE BRI2-BRICHOS Is Increased in Human Amyloid Plaques in Early Stages of Alzheimer’s Disease. Neurobiol. Aging 2014, 35, 1596–1604. [DOI] [PubMed] [Google Scholar]
- (526).Del Campo M; Oliveira CR; Scheper W; Zwart R; Korth C; Müller-Schiffmann A; Kostallas G; Biverstal H; Presto J; Johansson J; Hoozemans JJ; Pereira CF; Teunissen CE BRI2 Ectodomain Affects Aβ42 Fibrillation and Tau Truncation in Human Neuroblastoma Cells. Cell. Mol. Life Sci 2015, 72, 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (527).Garringer HJ; Sammeta N; Oblak A; Ghetti B; Vidal R Amyloid and Intracellular Accumulation of BRI2. Neurobiol. Aging 2017, 52, 90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (528).Matsuda S; Giliberto L; Matsuda Y; McGowan EM; D’Adamio L BRI2 Inhibits Amyloid Beta-Peptide Precursor Protein Processing by Interfering with the Docking of Secretases to the Substrate. J. Neurosci 2008, 28, 8668–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (529).Willander H; Presto J, Askarieh G, Biverstål H, Frohm B, D Knight SD, Johansson J, Linse S BRICHOS Domains Efficiently Delay Fibrillation of Amyloid β-Peptide. J. Biol. Chem 2012, 287, 31608–31617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (530).Hermansson E; Schultz S; Crowther D; Linse S; Winblad B; Westermark G The Chaperone Domain BRICHOS Prevents CNS Toxicity of Amyloid-β Peptide in Drosophila Melanogaster. Model Mech 2014, 7, 659–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (531).Oskarsson ME; Hermansson E; Wang Y; Welsh N; Presto J; Johansson J BRICHOS Domain of Bri2 Inhibits Islet Amyloid Polypeptide (IAPP) Fibril Formation and Toxicity in Human Beta Cells. Proc. Natl. Acad. Sci. USA 2018, 115, E2752–E2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (532).Ono K; Yoshiike Y; Takashima A; Hasegawa K; Naiki H; Yamada M Potent Anti‐amyloidogenic and Fibril‐Destabilizing Effects of Polyphenols in Vitro: Implications for the Prevention and Therapeutics of Alzheimer’s Disease. J. Neurochem 2003, 87, 172–181. [DOI] [PubMed] [Google Scholar]
- (533).Lee Y-H; Lin Y; Cox SJ; Kinoshita M; Sahoo BR; Ivanova M; Ramamoorthy A Zinc Boosts EGCG’s HIAPP Amyloid Inhibition Both in Solution and Membrane. Biochim. Biophys. Acta - Proteins Proteomics 2019, 1867, 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (534).Pithadia A; Brender JR; Fierke CA; Ramamoorthy A Inhibition of IAPP Aggregation and Toxicity by Natural Products and Derivatives. J. Diabetes Res 2016, 2016, 2046327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (535).Shoval H; Lichtenberg D; Gazit E The Molecular Mechanisms of the Anti-Amyloid Effects of Phenols. Amyloid 2007, 14, 73–87. [DOI] [PubMed] [Google Scholar]
- (536).Cox SJ; Camargo DCR; Lee Y-H; Dubini RCA; Rovó P; Ivanova MI; Padmini V; Reif B; Ramamoorthy A Small Molecule Induced Toxic Human-IAPP Species Characterized by NMR. Chem. Commun 2020, 56, 13129–13132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (537).Gilead S; Wolfenson H; Gazit E Molecular Mapping of the Recognition Interface between the Islet Amyloid Polypeptide and Insulin. Angew. Chem. Int. Ed 2006, 118, 6626–6630. [DOI] [PubMed] [Google Scholar]
- (538).Gilead S; Gazit E Inhibition of Amyloid Fibril Formation by Peptide Analogues Modified with A-aminoisobutyric Acid. Angew. Chem. Int. Ed 2004, 43, 4041–4044. [DOI] [PubMed] [Google Scholar]
- (539).Bram Y; Frydman-Marom A; Yanai I; Gilead S; Shaltiel-Karyo R; Amdursky N; Gazit E Apoptosis Induced by Islet Amyloid Polypeptide Soluble Oligomers Is Neutralized by Diabetes-Associated Specific Antibodies. Sci. Rep 2014, 4, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (540).Bram Y; Peled S; Brahmachari S; Harlev M; Gazit E Active Immunization Against HIAPP Oligomers Ameliorates the Diabetes-Associated Phenotype in a Transgenic Mice Model. Sci. Rep 2017, 7, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (541).Santos MA; Chand K; Chaves S Recent Progress in Multifunctional Metal Chelators as Potential Drugs for Alzheimer’s Disease. Coord. Chem. Rev 2016, 327–328, 287–303. [Google Scholar]
- (542).Budimir A Metal Ions, Alzheimer’s Disease and Chelation Therapy. Acta Pharm. Zagreb Croat 2011, 61, 1–14. [DOI] [PubMed] [Google Scholar]
- (543).Sales TA; Prandi IG; de Castro AA; Leal DHS; da Cunha EFF; Kuca K; Ramalho TC Recent Developments in Metal-Based Drugs and Chelating Agents for Neurodegenerative Diseases Treatments. Int. J. Mol. Sci 2019, 20, 1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (544).Cuajungco MP; Faget KY; Huang X; Tanzi RE; Bush AI Metal Chelation as a Potential Therapy for Alzheimer’s Disease. Ann. N. Y. Acad. Sci. USA 2000, 920, 292–304. [DOI] [PubMed] [Google Scholar]
- (545).Khan AN; Hassan MN; Khan RH Gallic Acid: A Naturally Occurring Bifunctional Inhibitor of Amyloid and Metal Induced Aggregation with Possible Implication in Metal-Based Therapy. J. Mol. Liq 2019, 285, 27–37. [Google Scholar]
- (546).Yu Y-P; Lei P; Hu J; Wu W-H; Zhao Y-F; Li Y-M Copper-Induced Cytotoxicity: Reactive Oxygen Species or Islet Amyloid Polypeptide Oligomer Formation. Chem. Commun 2010, 46, 6909–6911. [DOI] [PubMed] [Google Scholar]
- (547).Ben-Shushan S; Hecel A; Rowinska-Zyrek M; Kozlowski H; Miller Y Zinc Binding Sites Conserved in Short Neuropeptides Containing a Diphenylalanine Motif. Inorg. Chem 2020, 59, 925–929. [DOI] [PubMed] [Google Scholar]
- (548).Wineman-Fisher V; Miller Y Insight into a New Binding Site of Zinc Ions in Fibrillar Amylin. ACS Chem. Neurosci 2017, 8, 2078–2087. [DOI] [PubMed] [Google Scholar]
- (549).Husnjak K; Elsasser S; Zhang N; Chen X; Randles L; Shi Y; Hofmann K; Walters KJ; Finley D; Dikic I Proteasome Subunit Rpn13 Is a Novel Ubiquitin Receptor. Nature 2008, 453, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (550).Ben-Nissan G; Sharon M Regulating the 20S Proteasome Ubiquitin-Independent Degradation Pathway. Biomolecules 2014, 4, 862–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (551).Wang X; Yen J; Kaiser P; Huang L Regulation of the 26S Proteasome Complex During Oxidative Stress. Sci. Signal 2010, 3, ra88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (552).Farout L; Mary J; Vinh J; Szweda LI; Friguet B Inactivation of the Proteasome by 4-Hydroxy-2-Nonenal Is Site Specific and Dependant on 20S Proteasome Subtypes. Arch. Biochem. Biophys 2006, 453, 135–142. [DOI] [PubMed] [Google Scholar]
- (553).Lee FKM; Wong AKY; Lee YW; Wan OW; Chan HYE; Chung KKK The Role of Ubiquitin Linkages on α-Synuclein Induced-Toxicity in a Drosophila Model of Parkinson’s Disease. J. Neurochem 2009, 110, 208–219. [DOI] [PubMed] [Google Scholar]
- (554).Al-Ramahi I; Lam YC; Chen H-K; De Gouyon B; Zhang M; Pérez AM; Branco J; De Haro M; Patterson C; Zoghbi HY; Botas J CHIP Protects from the Neurotoxicity of Expanded and Wild-Type Ataxin-1 and Promotes Their Ubiquitination and Degradation. J. Biol. Chem 2006, 281, 26714–26724. [DOI] [PubMed] [Google Scholar]
- (555).Lee B-H; Lee MJ; Park S; Oh D-C; Elsasser S; Chen P-C; Gartner C; Dimova N; Hanna J; Gygi SP; Wilson SM; King RW; Finley D Enhancement of Proteasome Activity by a Small-Molecule Inhibitor of USP14. Nature 2010, 467, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (556).Chen J-W; Wang B-Z; Gao X-X; Zhang Y-Y Crosstalk between Autophagy and Proteasome Systems. Prog. Biochem. Biophys 2013, 40, 199–208. [Google Scholar]
- (557).Ji CH; Kwon YT Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Mol. Cells 2017, 40, 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (558).Viollet B; Guigas B; Sanz Garcia N; Leclerc J; Foretz M; Andreelli F Cellular and Molecular Mechanisms of Metformin: An Overview. Clin. Sci. Lond. Engl. 1979 2012, 122, 253–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (559).Hull RL; Shen Z-P; Watts MR; Kodama K; Carr DB; Utzschneider KM; Zraika S; Wang F; Kahn SE Long-Term Treatment with Rosiglitazone and Metformin Reduces the Extent of, but Does Not Prevent, Islet Amyloid Deposition in Mice Expressing the Gene for Human Islet Amyloid Polypeptide. Diabetes 2005, 54, 2235–2244. [DOI] [PubMed] [Google Scholar]
- (560).Obara A; Fujita Y; Abudukadier A; Fukushima T; Oguri Y; Ogura M; Harashima S-I; Hosokawa M; Inagaki N DEPTOR-Related MTOR Suppression Is Involved in Metformin’s Anti-Cancer Action in Human Liver Cancer Cells. Biochem. Biophys. Res. Commun 2015, 460, 1047–1052. [DOI] [PubMed] [Google Scholar]
- (561).Jiang Y; Huang W; Wang J; Xu Z; He J; Lin X; Zhou Z; Zhang J Metformin Plays a Dual Role in MIN6 Pancreatic β Cell Function through AMPK-Dependent Autophagy. Int. J. Biol. Sci 2014, 10, 268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (562).Udayasankar J; Zraika S; Aston-Mourney K; Subramanian SL; Brooks-Worrell BM; Taborsky GJ; Hull RL Rosiglitazone Treatment Does Not Decrease Amyloid Deposition in Transplanted Islets from Transgenic Mice Expressing Human Islet Amyloid Polypeptide. Transplant. Proc 2013, 45, 574–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (563).Fortin JS; Benoit-Biancamano M-O In Vitro Evaluation of Hypoglycemic Agents to Target Human Islet Amyloid Polypeptide: A Key Protein Involved in Amyloid Deposition and Beta-Cell Loss. Can. J. Diabetes 2015, 39, 373–382. [DOI] [PubMed] [Google Scholar]
- (564).Hwang JS; Ham SA; Yoo T; Lee WJ; Paek KS; Kim J-H; Lee C-H; Seo HG Upregulation of MKP-7 in Response to Rosiglitazone Treatment Ameliorates Lipopolysaccharide-Induced Destabilization of SIRT1 by Inactivating JNK. Pharmacol. Res 2016, 114, 47–55. [DOI] [PubMed] [Google Scholar]
- (565).Lee Y; Kim J; Park K; Lee M-S β-Cell Autophagy: Mechanism and Role in β-Cell Dysfunction. Mol. Metab 2019, 27, S92–S103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (566).Chen C-H; Yao T; Zhang Q; He Y-M; Xu L-H; Zheng M; Zhou G-R; Zhang Y; Yang H-J; Zhou P Influence of Trehalose on Human Islet Amyloid Polypeptide Fibrillation and Aggregation. RSC Adv 2016, 6, 15240–15246. [Google Scholar]
- (567).Reddy G; Muttathukattil AN; Mondal B Cosolvent Effects on the Growth of Amyloid Fibrils. Curr. Opin. Struct. Biol 2020, 60, 101–109. [DOI] [PubMed] [Google Scholar]
- (568).Kushwah N; Jain V; Yadav D Osmolytes: A Possible Therapeutic Molecule for Ameliorating the Neurodegeneration Caused by Protein Misfolding and Aggregation. Biomolecules 2020, 10, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (569).Sarkar S; Davies JE; Huang Z; Tunnacliffe A; Rubinsztein DC Trehalose, a Novel MTOR-Independent Autophagy Enhancer, Accelerates the Clearance of Mutant Huntingtin and α-Synuclein. J. Biol. Chem 2007, 282, 5641–5652. [DOI] [PubMed] [Google Scholar]
- (570).Kim J; Cheon H; Jeong YT; Quan W; Kim KH; Cho JM; Lim Y-M; Oh SH; Jin S-M; Kim JH; Lee M-K; Kim S; Komatsu M; Kang S-W; Lee M-S Amyloidogenic Peptide Oligomer Accumulation in Autophagy-Deficient β Cells Induces Diabetes. J. Clin. Invest 2014, 124, 3311–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (571).Casarejos MJ; Solano RM; Gómez A; Perucho J; De Yébenes JG; Mena MA The Accumulation of Neurotoxic Proteins, Induced by Proteasome Inhibition, Is Reverted by Trehalose, an Enhancer of Autophagy, in Human Neuroblastoma Cells. Neurochem. Int 2011, 58, 512–520. [DOI] [PubMed] [Google Scholar]
- (572).Honma Y; Sato-Morita M; Katsuki Y; Mihara H; Baba R; Harada M Trehalose Activates Autophagy and Decreases Proteasome Inhibitor-Induced Endoplasmic Reticulum Stress and Oxidative Stress-Mediated Cytotoxicity in Hepatocytes. Hepatol. Res 2018, 48, 94–105. [DOI] [PubMed] [Google Scholar]
- (573).Ukkola O Ghrelin in Type 2 Diabetes Mellitus and Metabolic Syndrome. Mol. Cell. Endocrinol 2011, 340, 26–28. [DOI] [PubMed] [Google Scholar]
- (574).Olszewski W; Głuszek J Ghrelin Antagonists in Type 2 Diabetes Mellitus Therapy - Is It a Safe Path? Przeglad Kardiodiabetologinczny 2010, 5, 98–105. [Google Scholar]
- (575).Cecarini V; Bonfili L; Cuccioloni M; Keller JN; Bruce-Keller AJ; Eleuteri AM Effects of Ghrelin on the Proteolytic Pathways of Alzheimer’s Disease Neuronal Cells. Mol. Neurobiol 2016, 53, 3168–3178. [DOI] [PubMed] [Google Scholar]
- (576).Ferreira-Marques M; Aveleira CA; Carmo-Silva S; Botelho M; de Almeida LP; Cavadas C Caloric Restriction Stimulates Autophagy in Rat Cortical Neurons through Neuropeptide Y and Ghrelin Receptors Activation. Aging 2016, 8, 1470–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (577).Casanova E; Salvadó J; Crescenti A; Gibert-Ramos A Epigallocatechin Gallate Modulates Muscle Homeostasis in Type 2 Diabetes and Obesity by Targeting Energetic and Redox Pathways: A Narrative Review. Int. J. Mol. Sci 2019, 20, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (578).Andrich K; Bieschke J The Effect of (−)-Epigallo-Catechin-(3)-Gallate on Amyloidogenic Proteins Suggests a Common Mechanism. Adv. Exp. Med. Biol 2015, 863, 139–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (579).Bonfili L; Cuccioloni M; Mozzicafreddo M; Cecarini V; Angeletti M; Eleuteri AM Identification of an EGCG Oxidation Derivative with Proteasome Modulatory Activity. Biochimie 2011, 93, 931–940. [DOI] [PubMed] [Google Scholar]
- (580).Yang H; Landis-Piwowar K; Chan TH; Dou QP Green Tea Polyphenols as Proteasome Inhibitors: Implication in Chemoprevention. Curr. Cancer Drug Targets 2011, 11, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (581).Glynn SJ; Gaffney KJ; Sainz MA; Louie SG; Petasis NA Molecular Characterization of the Boron Adducts of the Proteasome Inhibitor Bortezomib with Epigallocatechin-3-Gallate and Related Polyphenols. Org. Biomol. Chem 2015, 13, 3887–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (582).Kim H-S; Montana V; Jang H-J; Parpura V; Kim J-A Epigallocatechin Gallate (EGCG) Stimulates Autophagy in Vascular Endothelial Cells: A Potential Role for Reducing Lipid Accumulation. J. Biol. Chem 2013, 288, 22693–22705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (583).Porat Y; Abramowitz A; Gazit E Inhibition of Amyloid Fibril Formation by Polyphenols: Structural Similarity and Aromatic Interactions as a Common Inhibition Mechanism. Chem. Biol. Drug Des 2006, 67, 27–37. [DOI] [PubMed] [Google Scholar]
- (584).Goh KP; Lee HY; Lau DP; Supaat W; Chan YH; Koh AFY Effects of Resveratrol in Patients with Type 2 Diabetes Mellitus on Skeletal Muscle SIRT1 Expression and Energy Expenditure. Int. J. Sport Nutr. Exerc. Metab 2014, 24, 2–13. [DOI] [PubMed] [Google Scholar]
- (585).Sciacca MFM; Chillemi R; Sciuto S; Pappalardo M; La Rosa C; Grasso D; Milardi D Interactions of Two O-Phosphorylresveratrol Derivatives with Model Membranes. Arch. Biochem. Biophys 2012, 521, 111–116. [DOI] [PubMed] [Google Scholar]
- (586).Jiang P; Li W; Shea J-E; Mu Y Resveratrol Inhibits the Formation of Multiple-Layered β-Sheet Oligomers of the Human Islet Amyloid Polypeptide Segment 22–27. Biophys. J 2011, 100, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (587).Silswal N; Reddy NS; Qureshi AA; Qureshi N Resveratrol Downregulates Biomarkers of Sepsis Via Inhibition of Proteasomeʼs Proteases: SHOCK 2018, 50, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (588).Qu X; Chen X; Shi Q; Wang X; Wang D; Yang L Resveratrol Alleviates Ischemia/Reperfusion Injury of Diabetic Myocardium Via Inducing Autophagy. Exp. Ther. Med 2019, 18, 2719–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (589).Lv W; Zhang J; Jiao A; Wang B; Chen B; Lin J Resveratrol Attenuates HIAPP Amyloid Formation and Restores the Insulin Secretion Ability in HIAPP-INS1 Cell Line via Enhancing Autophagy. Can. J. Physiol. Pharmacol 2019, 97, 82–89. [DOI] [PubMed] [Google Scholar]
- (590).Fraschini F; Demartini G; Esposti D Pharmacology of Silymarin. Clin. Drug Investig 2002, 22, 51–65. [Google Scholar]
- (591).Das S; Roy P; Pal R; Auddy RG; Chakraborti AS; Mukherjee A Engineered Silybin Nanoparticles Educe Efficient Control in Experimental Diabetes. PLOS ONE 2014, 9, e101818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (592).Yang J; Sun Y; Xu F; Liu W; Mai Y; Hayashi T; Hattori S; Ushiki-Kaku Y; Onodera S; Tashiro S; Ikejima T Silibinin Ameliorates Amylin-Induced Pancreatic β-Cell Apoptosis Partly via Upregulation of GLP-1R/PKA Pathway. Mol. Cell. Biochem 2019, 452, 83–94. [DOI] [PubMed] [Google Scholar]
- (593).Sciacca MF; Romanucci V; Zarrelli A; Monaco I; Lolicato F; Spinella N; Galati C; Grasso G; D’Urso L; Romeo M et al. Inhibition of Aβ Amyloid Growth and Toxicity by Silybins: The Crucial Role of Stereochemistry. ACS Chem. Neurosci 2017, 8, 1767–1778. [DOI] [PubMed] [Google Scholar]
- (594).Chen Y-H; Chen C-L; Lu D-W; Liang C-M; Tai M-C; Chen J-T Silibinin Inhibits Platelet-Derived Growth Factor-Driven Cell Proliferation via Downregulation of N-Glycosylation in Human Tenon’s Fibroblasts in a Proteasome-Dependent Manner. PLOS ONE 2016, 11, e0168765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (595).Wang Q; Liu M; Liu W-W; Hao W-B; Tashiro S; Onodera S; Ikejima T In Vivo Recovery Effect of Silibinin Treatment on Streptozotocin-Induced Diabetic Mice Is Associated with the Modulations of Sirt-1 Expression and Autophagy in Pancreatic β-Cell. J. Asian Nat. Prod. Res 2012, 14, 413–423. [DOI] [PubMed] [Google Scholar]
- (596).Hwang W; Karplus M Kinetic Control of Dimer Structure Formation in Amyloid Fibrillogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 12916–12921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (597).Pellarin R; Schuetz P, Guarnera E, Caflisch A Amyloid Fibril Polymorphism Is under Kinetic Control. J. Am. Chem. Soc 2010, 132, 14960–14970. [DOI] [PubMed] [Google Scholar]
- (598).Caflisch A Kinetic Control of Amyloidogenesis Calls for Unconventional Drugs to Fight Alzheimer’s Disease. ACS Chem. Neurosci 2020, 11, 103–104. [DOI] [PubMed] [Google Scholar]
- (599).Kollmer M Cryo-EM Structure and Polymorphism of Aβ Amyloid Fibrils Purified from Alzheimer’s Brain Tissue. Nat. Commun 2019, 10, 4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (600).Abedini A Time-Resolved Studies Define the Nature of Toxic IAPP Intermediates, Providing Insight for Anti-Amyloidosis Therapeutics. Elife 2016, 5, e12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (601).Herrmann US; Schütz AK, Shirani H, Huang D, Saban D, Nuvolone N, Li B, Ballmer B, Åslund AKO, Mason JJ et al. Structure-Based Drug Design Identifies Polythiophenes as Antiprion Compounds. Sci. Transl. Med 2015, 7, 299ra123. [DOI] [PubMed] [Google Scholar]
- (602).Kapurniotu A; Bernhagen J; Greenfield N; Al-abed Y; Teichberg S; Frank R; Voelter W; Bucala R Contribution of Advanced Glycosylation to the Amyloidogenicity of Islet Amyloid Polypeptide. Eur. J. Biochem 1998, 251, 208–216. [DOI] [PubMed] [Google Scholar]
- (603).Xue W-F; Hellewell AL; Hewitt EW; Radford SE Fibril Fragmentation in Amyloid Assembly and Cytotoxicity. Prion 2010, 4, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (604).Young LM; Mahood RA; Saunders JC; Tu L-H; Raleigh DP; Radford SE; Ashcroft AE Insights into the Consequences of Co-Polymerisation in the Early Stages of IAPP and Aβ Peptide Assembly from Mass Spectrometry. Analyst 2015, 140, 6990–6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (605).Andreetto E; Yan L-M; Tatarek-Nossol M; Velkova A; Frank R; Kapurniotu A Identification of Hot Regions of the Aβ–IAPP Interaction Interface as High-Affinity Binding Sites in Both Cross- and Self-Association. Angew. Chem. Int. Ed 2010, 49, 3081–3085. [DOI] [PubMed] [Google Scholar]
- (606).Westermark P Amyloid and Polypeptide Hormones: What Is Their Inter-Relationship? Amyloid 1994, 1, 47–60. [Google Scholar]
- (607).Lee S; Pioszak AA Molecular interaction of an antagonistic amylin analog with the extracellular domain of receptor activity-modifying protein 2 assessed by fluorescence polarization. Biophys. Chem 2020, 267, 106477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (608).Janciauskiene S; Eriksson S; Carlemalm E; Ahrén B β-Cell Granule Peptides Affect Human Islet Amyloid Polypeptide (IAPP) Fibril Formation in Vitro. Biochem. Biophys. Res. Commun 1997, 236, 580–5. [DOI] [PubMed] [Google Scholar]
- (609).Sanke T; Bell GI; Sample C; Rubenstein AH; Steiner DF An Islet Amyloid Peptide Is Derived from an 89-Amino Acid Precursor by Proteolytic Processing. J. Biol. Chem 1988, 263, 17243–17246. [PubMed] [Google Scholar]
- (610).Betsholtz C; Svensson V; Rorsman F; Engström U; Westermark GT; Wilander E Islet Amyloid Polypeptide (IAPP): CDNA Cloning and Identification of an Amyloidogenic Region Associated with Species-Specific Occurrence of Age-Related Diabetes Mellitus. Exp. Cell. Res 1989, 183, 484–93. [DOI] [PubMed] [Google Scholar]