

Abstract
Given the superior soft tissue contrasts obtained by MRI and the long residence times of magnetic nanoparticles (MNPs) in soft tissues, MNP-based theranostic systems are being developed for simultaneous imaging and treatment. However, development of such theranostic nanoformulations presents significant challenges of balancing the therapeutic and diagnostic functionalities in order to achieve optimum effect from both. Here we developed a simple theranostic nanoformulation based on magnetic nanoclusters (MNCs) stabilized by a bisphosphonate-modified poly(glutamic acid)-b-(ethylene glycol) block copolymer and complexed with cisplatin. The MNCs were decorated with luteinizing hormone releasing hormone (LHRH) to target LHRH receptors (LHRHr) overexpressed in ovarian cancer cells. The targeted MNCs significantly improved the uptake of the drug in cancer cells and decreased its IC50 compared to the nontargeted formulations. Also, the enhanced LHRHr-mediated uptake of the targeted MNCs resulted in enhancement in the T2-weighted negative contrast in cellular phantom gels. Taken together, the LHRH-conjugated MNCs show good potential as ovarian cancer theranostics.
Graphical Abstract
INTRODUCTION
Advances in material sciences and nanotechnology have enabled design of nanocarriers capable of releasing their payload in response to specific biological stimuli such as pH,1–3 temperature,1–3 and redox potential.4–6 Moreover, recent studies in nanomedicine have focused on spatiotemporal delivery of drugs, genes, and imaging agents as implementation of a “theranostics” platform. Over the last two decades, superparamagnetic iron oxide nanoparticles (MNPs) have been studied comprehensively and used in a wide variety of applications ranging from water purification to high capacity data storage7 to numerous biomedical applications such as protein and antibody detection,8 toxin separation,9 and magnetic resonance imaging (MRI) contrast agents.10 MNPs interact with protons in water molecules and shorten their transverse (T2) relaxation times thereby producing dark T2-weighted negative contrasts in the surrounding tissues. Because of this ability, the MNPs have been applied clinically as MRI contrast agents for diagnosis of several diseases such as blood pool agents in magnetic resonance angiography (MRA) of the aortoiliac region and coronary arteries.11–13
Several attempts were made to coformulate MNPs with therapeutic agents. Such theranostic systems can present a desirable improvement in cancer chemotherapy due to their ability to target the tumor and simultaneously allow non-invasive visualization and treatment.14,15 Development of MNP-based theranostic agents requires availability of a nanoformulation platform where the diagnostic and therapeutic components are properly balanced. Moreover, MNP-based formulations can provide an opportunity for remote control of theranostic agents by applied magnetic fields.16,17 For example, biodegradable polymersomes loaded with MNPs and doxorubicin displayed magnetic field-responsive drug release along with excellent MRI contrast.18 In another study, MNP aggregates stabilized by Pluronic F127 and loaded with doxorubicin exhibited sustained drug release in vivo while simultaneously enabling MRI of the target tumor tissue.19 However, formulation-related issues such as colloidal stability and systemic toxicity hinder the clinical development of such MNP-based theranostic systems.
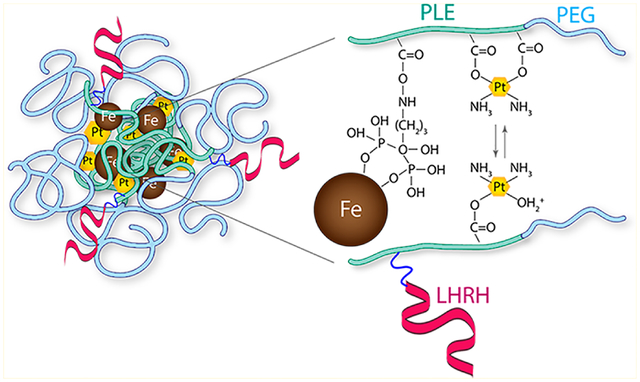
Here we report a facile process for theranostic nanoformulation through magnetic nanoclusters (MNC) for anticancer therapy and simultaneous MRI. These MNCs were prepared by reacting MNPs dispersed in an aqueous medium with anionic bisphosphonate-modified poly(glutamic acid-b-ethylene glycol) (PLE-b-PEG) block copolymer. The MNCs were loaded with cisplatin through coordination interactions with the remaining carboxylic groups of the block copolymer. A synthetic peptide analogue of the LHRH was conjugated to the MNCs to enable targeting the nanoformulation to ovarian cancer cells that overexpress the LHRHr. This work reports the synthesis of targeted MNCs, drug loading, drug release, and in vitro cytotoxicity in ovarian cancer cells and evaluates T2 relaxivity of the nanoformulations in LHRHr overexpressing wild type (A2780-WT) and cisplatin-resistant (A2780-CisR) ovarian cancer cells.
MATERIALS AND METHODS
Chemicals.
PLE-b-PEG block copolymers with different PLE block lengths, PLE10-b-PEG113, PLE50-b-PEG113, PLE100-b-PEG113, polyaspartic acid100-b-PEG113 (PLD100-b-PEG113), and the respective homopolymers were purchased from Alamanda Polymers (Huntsville, AL). The numbers in subscripts after the copolymer blocks define the average number of the repeating units (r.u.) in the respective block. The free end of the PEG had a methoxy end group. Alendronate sodium trihydrate (ALN) was purchased from Ultratech (Navi Mumbai, India). 1-Ethyl-3-(3-(dimethylamino)propyl)-carbodiimide (EDC), N-hydroxysuccinimide (NHS), iron(III) acetylacetonate (Fe(acac)3), benzyl alcohol (anhydrous), nitric acid (TraceSELECT), hydrochloric acid (TraceSELECT), inductively coupled plasma mass spectrometry (ICP-MS) grade standards for Fe, Pt, and P (Fluka), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO). Acetone (histology grade), N,N-dimethylformamide (DMF), cisplatin, pyridine, dimethyl sulfoxide (DMSO), and all other anhydrous HPLC grade organic solvents were purchased from Thermo Fisher Scientific (Waltham, MA). N-Hydroxysulfosuccinimide (S-NHS) was purchased from Covachem (Loves Park, IL). A LHRH peptide, d-Lys-6-LHRH (Glp–His–Trp–Ser–Tyr– DLys–Leu–Arg–Pro–Gly; 1253.4 Da) was purchased from American Peptide Company (Sunnyvale, CA). Alexa Fluor-647 hydrazide, Lysotracker Green, and Hoechst 33342 were purchased from Life Technologies (Carlsbad, CA).
Synthesis of MNPs.
MNPs were synthesized by thermal decomposition of iron(III) acetylacetonate (Fe(acac)3) in benzyl alcohol with minor modifications to the method developed by Pinna et al.20 Briefly, 6.2 μmoles of Fe(acac)3 was charged to a three-necked flask containing 45 mL of anhydrous benzyl alcohol. The reaction mixture was heated at 110 °C for 1 h to ensure complete evaporation of water. The size of the MNPs was tuned by subsequently varying the heating rates of the reaction. Four different heating profiles were employed as described below and depicted in Figure S1. (1) Profile S1-A: The reaction mixture was heated gradually at 0.4 °C/min to 205 °C. Once at reflux the temperature was kept constant for 40 h. (2) Profile S1-B: The mixture was heated to 150 °C at 4 °C/min and kept isothermally at 150 °C for 2 h, and then the temperature was raised to reflux at 4 °C/min and heated for 40 h at reflux. (3) Profile S1-C: The reaction mixture was heated to 180 °C at 4 °C/min and kept isothermally at 180 °C for 2 h, and then the temperature was raised to reflux at 4 °C/min and heated for 40 h at reflux. (4). Profile S1-D: The reaction mixture was heated quickly at 5 °C/min to reflux and heated at reflux for 40 h. All the reactions were conducted under an inert atmosphere. After cooling, the MNPs were precipitated and washed in acetone. The final product was collected by magnetic separation, dried under vacuum by rotary evaporation at 40 °C, and stored for further use under vacuum in a sealed vial filled with argon. The saturation magnetization (M; emu/g) of the smallest and the largest particles obtained was determined by superconducting quantum interference device (SQUID) analysis (Quantum Design Co.) at 300 K.
ALN Conjugation to PLE50-b-PEG113.
Prior to ALN conjugation, PLE50-b-PEG113 sodium salt was dissolved in deionized water and acidified by slow addition of 0.1 N HCl to restore the carboxylic acid functionality by removal of the sodium salt protecting the pendant carboxylate groups. The precipitate was dialyzed overnight against deionized water and lyophilized, and final deprotected polymer was stored under vacuum in a sealed vial filled with argon for further use. For the conjugation reaction, 60 mg of this polymer was dissolved in 3 mL of DMSO. In a separate vial, 30 mg of EDC and 85 mg of NHS were dissolved in 0.5 mL of DMSO and added to the polymer solution. The reaction mixture was stirred for 4h at room temperature (RT). ALN (145 mg) was dissolved in 4 mL of deionized water, and this was followed by adjustment of the pH to 9.0 with NaOH. The EDC-NHS-activated PLE50-b-PEG113 in DMSO was added dropwise to the aqueous solution of ALN, and the reaction mixture was stirred for 48 h. The unconjugated ALN as well as the reaction byproducts were removed by centrifugal filtration through 3000 MWCO PES centrifugal membrane filters (Sartorius, NY), and the resultant ALN-conjugated PLE50-b-PEG113 was lyophilized. The ALN conjugation was confirmed by 1H NMR and quantified by inductively coupled plasma mass spectrometry (ICP-MS). A standard curve was prepared from a commercial standard solution of phosphorus (Fluka) in the 50–500 ppb concentration range. The lyophilized polymer was dispersed in 1 mL of deionized (DI) water at a concentration of 1 mg/mL. Concentrated HCl ((50 μL; TraceSELECT) was added to it, and the mixture was incubated at 70 °C for 12 h. The sample was allowed to cool to RT, and then the volume was adjusted to 2 mL with DI water. The final solution was filtered through a 0.2 μm nylon syringe filter and analyzed by ICP-MS to quantify the bisphosphonate end groups in the polymer using argon and oxygen carrier gases.
Preparation of Polymer-Stabilized MNCs.
(1) MNCs prepared from unmodified PLE-b-PEG polymers were prepared as follows. Briefly, the polymer (40 mg) was dissolved in 10 mL of alkaline water (pH 9–9.5) and added to 10 mL of MNP dispersion (2 mg/mL) in alkaline water (pH 12) in a 2:1 (w/w) ratio. (2) MNCs prepared from ALN-modified PLE50-b-PEG113 polymers were prepared as follows. (a) The 30–40 nm MNCs were prepared by mixing the ALN-modified polymers and MNPs in the same ratio as that used for preparing MNCs from unmodified polymers. (b) The MNCs in 60–80 nm size ranges were obtained by dissolving the ALN modified A19-PLE50-b-PEG113 polymer (40 mg) and MNPs (20 mg) in 5 mL each of the respective solvents. The mixture was then stirred for 12–15 h followed by extensive dialysis for 72 h, with six water changes. To obtain the final product, the dialyzed mixture was further purified by filtration through 0.45 and 0.22 μm Whatman Anotop syringe filters, lyophilized, and then stored in a sealed vial filled with argon for further use.
Preparation of Cisplatin-Loaded MNCs.
The MNCs were dispersed in deionized water at 5–10 mg/mL, and the pH of the colloidal dispersion was adjusted to 9.5 with NaOH. Then 0.7 mol equiv of cisplatin relative to the available carboxylate groups of MNCs (each cisplatin molecule reacts with two carboxylates) was dissolved in DMF and added under stirring to the MNCs dispersion. (The number of carboxyl groups in MNCs was quantified by potentiometric titration beforehand.) The solution was stirred for 48 h in the dark. The unconjugated cisplatin was separated by centrifugal filtration, and the cisplatin-loaded MNCs were washed thrice with deionized water (2000-MWCO). The final product was recovered from the filter surface as a viscous colloid, lyophilized, and stored under vacuum at −20 °C for further use.
Preparation of LHRH-Conjugated MNCs.
(d-Lys6)-LHRH was conjugated to the free carboxylic groups of MNCs by an EDC/S-NHS coupling reaction via the d-lysine group on the peptide. Briefly, 10 mg of lyophilized unloaded or cisplatin loaded MNCs were dispersed separately in 0.5 mL of 0.05 M 2-(N-morpholino) ethanesulfonic acid (MES) buffer (pH 6.0). EDC/S-NHS (3 equiv/6 equiv) at RT was used to activate 5 mol % of the total available carboxylic groups (as determined by potentiometric titration) for 2 h. The excess of unreacted reagents was neutralized by 20 mM 2-mercaptoethanol. (d-Lys6)-LHRH (10 mg; 8 mM) was prepared in 1 mL of PBS (1×, pH 7.4). This solution (0.4 mL) was mixed with the activated MNC and cisplatin-loaded MNC formulations. The pH of the mixture was adjusted to 7.4, and the solutions were stirred for 7 h at RT. The dispersions were purified by centrifugal filtration (3000 MWCO; three washes) to remove any unreacted peptide and other reactants. The final formulations were lyophilized and stored under vacuum at −20 °C for further use.
Physicochemical Characterization of MNCs.
The z-average hydrodynamic diameter (Deff), polydispersity index (PDI), and ζ-potential were determined by dynamic light scattering (DLS) using a Malvern Zetasizer (Malvern Instruments, MA, U.S.A.) at a concentration of 1 mg/mL, Transmission electron microscopy (TEM) images of the MNCs were taken on a JEOL 2010F-FasTEM HRTEM (Peabody, MA). Briefly, MNPs and MNCs were diluted to approximately 0.2 mg/mL in alkaline water (pH 12) and DI water, respectively. A 10 μL sample was placed on a copper TEM grid coated with carbon support films (Ted Pella INC, Redding, CA) and allowed to settle for 2 min. The excess sample was carefully removed, and the grid was allowed to dry for another 10 min before placement in the microscope. The composition of the unloaded MNCs was determined by thermogravimetric analysis (TGA; Q50, TA Instruments, DE). Briefly, 5–10 mg of the lyophilized unloaded-formulation was placed on the tared TGA pan. The formulation was dried in the furnace at 110 °C in order to remove the moisture followed by steady heating at 5 °C/min to 1000 °C. The thermogram was analyzed using the Universal Analysis software (TA Instruments, DE) to deduce the total loss of the organic components and amount of iron oxide per mg of MNCs. The content of benzyl alcohol coating on the MNPs was determined by loss of organic components upon thermal decomposition by TGA. The loss on ignition (LOI) of MNCs was corrected by subtracting the content of benzyl alcohol in the MNPs to obtain the content of polymer in the MNCs. LHRH conjugation on the MNCs was determined by Fourier transform infrared spectroscopy (FTIR) using a Nicolet 380 FTIR equipped with a silvergate assembly (Thermo Fisher Scientific, Waltham, MA). The lyophilized sample was placed on the ZnSe crystal of the silvergate assembly and gently compressed with a flat-head anvil to form a pellet on the crystal. The spectrum was obtained by subtraction of the blank signal.
Relaxation Measurements.
A Bruker Biospec MRI and spectroscopy (MS) system (7T/21 cm, Bruker, Karlsruhe, Germany) was used to determine the longitudinal (r1) and transverse (r2) relaxivities of the various MNC formulations. The sequence used for T2 mapping was a Carr–Purcell–Meiboom–Gill (CPMG) phase cycled single slice multiecho sequence. One 1 mm thick coronal image was acquired with an acquisition matrix of 256 × 128 mm2, 40 × 40 mm2 field of view (FOV), 32 echoes at 10 ms first echo time and 10 ms echo spacing, repetition time of 3000 ms, one average, for a total acquisition time of 6.4 min. The sequence used for T1 mapping was a fast spin echo sequence with variable TR from 0.4 s to 10 s, TE = 7.0 ms, 1 coronal slice of thickness 1 mm, in-plane resolution 0.156 × 0.156 mm2, FOV = 40 × 40 mm2. The stabilized colloidal particle dispersions were diluted to 0, 0.018, 0.045, 0.09, 0.18, 0.27, and 0.36 mM Fe with distilled water and 20 mM PBS. T1 and T2 relaxation times were measured and converted to their corresponding relaxation rates (R1 and R2; 1/T; s−1). The r1 (longitudinal molar relaxivity) and r2 (transverse molar relaxivity) values (s−1·mM−1) were obtained as the slope of the plots of R1 or R2 versus the corresponding Fe concentrations.
Drug Release.
The drug release of the anionic formulations was studied in potassium free 20 mM phosphate buffered saline (PBS), pH 7.4, or 50 mM sodium acetate buffered saline (ABS), pH 5.5. Briefly, a formulation equivalent to 25 μg of cisplatin was placed in Float-alyzer© dialysis membrane tubes (3.5–5 kDa MWCO; 1 mL capacity; Spectrum Laboratories, USA). The volume in the tube membrane was adjusted to 1 mL, and the assembly was placed in 24 mL of the dissolution medium and stirred at approximately 125 rpm for 5 days at 37 °C. At specific time points during the study, 0.5 mL aliquots were removed from the dissolution medium outside the dialysis membrane and replaced with the same volume of fresh buffer. The cumulative release of cisplatin was quantified by ICP-MS and plotted against time.
Cell Culture.
The wild-type human ovarian cancer cells A2780-WT and cisplatin-resistant human ovarian cancer cells A2780-CisR were kindly provided by Dr. T.K. Bronich (UNMC, Omaha, NE). Both of the cell lines were cultured in RPMI complete growth medium (Gibco, Grand Island, NY) supplemented with 10% v/v fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO) and 1% v/v penicillin:streptomycin (Gibco, Grand Island, NY).
Cellular Uptake of MNCs.
A2780-WT and A2780-CisR cells (106 cells/flask) were plated in T25 cell culture flasks, were allowed to grow for 4 days until they achieved approximately 90% confluency, and then were treated with 7.5 mL of a colloidal dispersion of various MNCs (at Fe concentration of 5 μg/mL) in complete RPMI medium supplemented with 10% v/v FBS for up to 24 h. At various time points during the incubation the medium was removed and the cells were washed three times with ice-cold PBS and acid saline (pH 3.0). The cells were harvested and cell viability was determined by the trypan blue assay. The harvested cells were then centrifuged to form a pellet. The supernatant was removed completely, and 0.5 mL of 100 ppb solution of iridium in 2% v/v HNO3 was added to the pellets. The cells were then lysed mechanically by probe-sonication. Concentrated HNO3 (50 μL, TraCERT; Fluka) was added to the lysate, which was then treated overnight at 70 °C. The samples were then diluted to 5 mL with 2% HNO3. The cell debris was separated by ultra-centrifugation at 10000 rpm for 30 min. The supernatant was further filtered through 0.2 μm syringe filters and analyzed by Nexion 300-D ICP-MS equipped with collision cell and auto sampler (PerkinElmer, USA). The intensities of the 57Fe and 195Pt isotopes were considered for quantification of the uptake. The final Fe or Pt content was normalized to the total protein content of the cells as determined by standard BCA assay, and the uptake was expressed as μg Fe per mg of total cellular protein.
In parallel, live-cell confocal microscopy was performed to study the cellular localization pattern of the various fluorescent-labeled MNCs. For this purpose, MNCs were fluorescently labeled by an EDC/NHS coupling reaction with hydrazide functionalized Alexa-Fluor-647. For confocal imaging, A2780-WT and A2780-CisR cells were plated in 8-chamber confocal slides, allowed to grow for 24 h, and then incubated with Alexa Fluor-647 labeled MNCs (at Fe concentration of 5 μg/mL) in 0.5 mL of RPMI complete media for 1, 3, and 24 h. At the end of each time period the cells were washed thoroughly with sterile 1× PBS (10 mM, 0.14 M NaCl) and further incubated for 30 min with Lysotracker Green (504 nm/511 nm Ex/Em) and Hoechst nuclear stain followed by visualization under a LSM 710 (Zeiss, CA) confocal microscope equipped with a live-cell imaging stage.
In Vitro Cytotoxicity.
The A2780-WT and A2780-CisR cells (3000 cells/well) were seeded in 96-well plates and cultured in complete RPMI 1640 medium for 24 h before treatments with the drugs or drug-free MNCs for either 24 or 72 h. At the end of each time point the culture medium was replaced with complete RPMI, and the cells were further incubated for 12 h. The cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.21,22
In vitro MRI.
Approximately 10 million A2780-WT and A2780-CisR cells were incubated with LHRH-conjugated and unconjugated MNCs at Fe concentrations of 0.045, 0.09, and 0.18 mM in complete RPMI growth medium supplemented with 10% v/v FBS for 24 h. Following incubation, the cells were collected and cell viability was determined by the trypan blue assay. Nine million viable cells were redispersed in 0.5 mL of PBS and mixed carefully with an equal volume of hot 4% w/v agar solution. The samples were cooled to 4 °C to allow formation of a solid matrix. Nine million untreated cells were used as a control. Phantom gels were scanned in Siemens Magnetom TIM Trio 3T human magnetic resonance (MR) scanner. Images of the phantom gels were obtained by using similar parameters as mentioned previously for the relaxation measurements at a 1.64 ms/3000 ms TE/TR and 40° flip angle for T1 and 112.5 ms/3000 ms TE/TR for T2. The cellular phantoms were compared with phantoms of similar concentrations of formulations without any cells.
Statistical Analysis.
Statistical analysis was performed using PRISM and IBM SPSS statistical software. Experimental designs with two groups were analyzed by t test, and designs with more than two groups were analyzed by ANOVA followed by Bonferroni posthoc test.
RESULTS
Synthesis of MNPs.
At the onset of this work we concluded that it was important to control and fine-tune the sizes of the MNPs as these sizes could affect the properties of the resulting MNCs. In the modified one-pot solvent-controlled synthesis of magnetite nanocrystals20,23 benzyl alcohol acts as a solvent and reducing agent as well as a ligand or a mild surfactant. In this process the colloidal metal oxide nanocrystal nuclei of MNPs grow by consuming the surrounding Fe(acac)3 precursor while Ostwald’s ripening or oriented attachment mechanisms are not observed.24,25 Notably, the thermal decomposition of Fe(acac)3 in benzyl alcohol allows good control over the nucleation and growth stages of the colloidal nanocrystals of magnetite and yields MNPs with a narrow size distribution as compared to the widely used coprecipitation technique.20 The rate of decomposition of Fe(acac)3 and therefore the onset, rate, and extent of the nucleation phase in nanocrystallization of magnetite from the organometallic precursor were controlled by changing the heating rate and temperature of the reaction mixture. To determine the rate of decomposition of the precursor during the MNPs nucleation and growth phases, 5-μL aliquots of the reaction mixture were collected, diluted with HPLC-grade acetone, and centrifuged, and the supernatant was analyzed by UV spectroscopy in the 250–600 nm wavelength range. The broad peaks at 355 and 435 nm correspond to the precursor while the sharp peak at 330 nm corresponds to the colloidal nuclei in the reaction mixture.26 The Fe content in the supernatant was also quantified by ICP-MS. Several heating regimes were evaluated (Figure 1, Supporting Information Figure S1).
Figure 1.
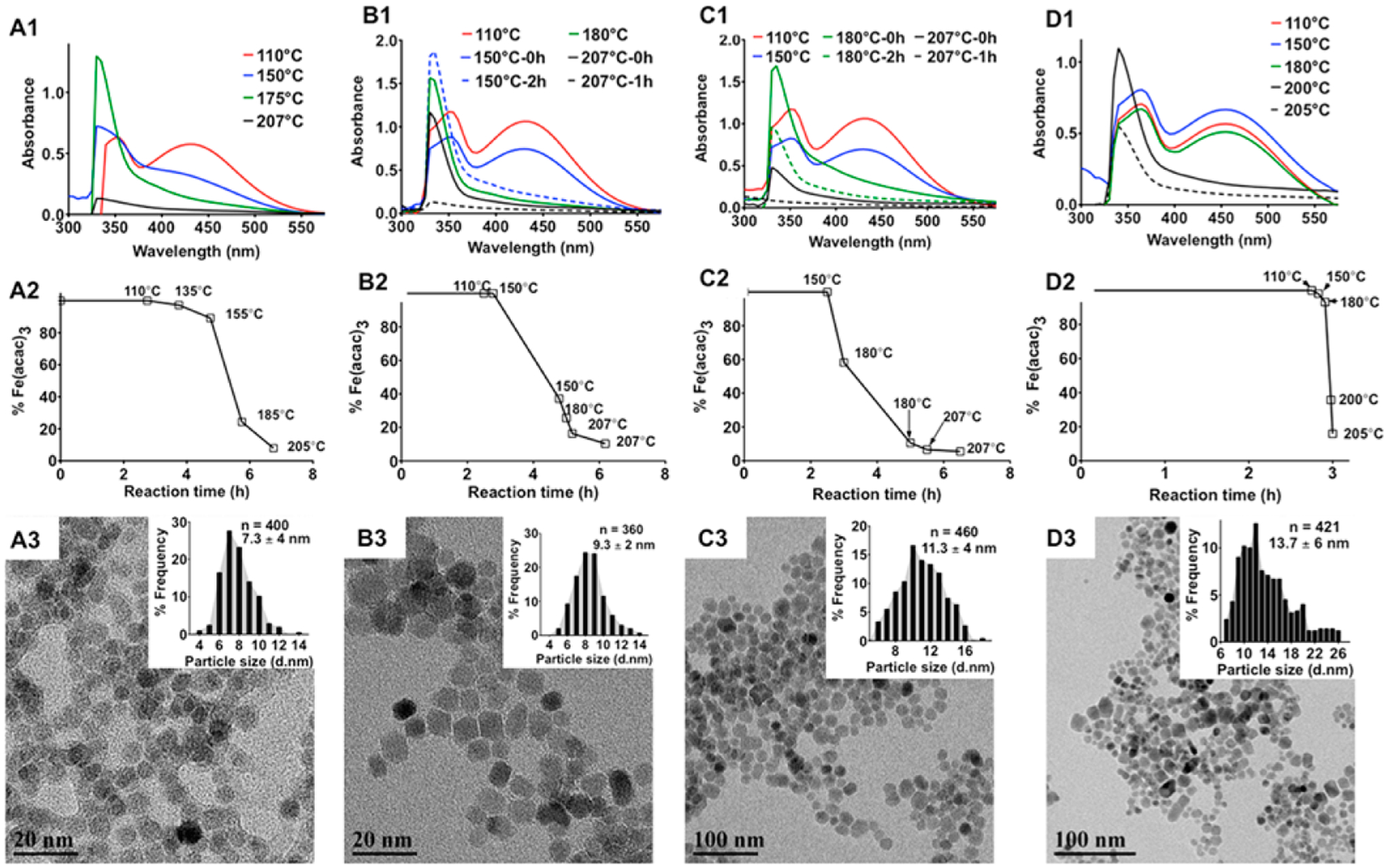
Effect of the heating patterns on the size of MNPs. A1-D1: UV-absorption spectra of the reaction mixtures upon different heating patterns as depicted in Supporting Information Figure S1. Aliquots of the reaction mixture were collected at specific time intervals, precipitated in acetone, and centrifuged to separate any formed MNPs, and the absorbance spectrum of the supernatant was measured to determine the nucleation and growth phases during the synthesis of MNPs. The solid lines correspond to measurement taken at the start of the incubation periods. The dashed lines are spectra taken 2 h after incubation of the system at a given temperature except for D1 where the spectra were taken at the start of reflux. A2-D2: Amount of unreacted Fe(acac)3 in the reaction mixture at the corresponding temperatures during the ramp phase of the heating process as measured by ICP-MS. A3-D3: TEM images of the MNPs synthesized by different heating patterns. Particle size distributions are shown in the respective insets. Diameters of the MNPs were calculated from the TEM images using ImageJ software.
1. Slow Heating.
As the temperature was raised slowly at 0.4 °C/min (Supporting Information Figure S1A), the nucleation stage was initiated very early (at ~150 °C), as indicated by the attenuation of the precursor peak intensities at 355 and 435 nm and elevation of the colloidal nuclei peak intensity at 330 nm (Figure 1A1). Approximately 75% of the precursor was converted into colloidal nuclei at around 160–180 °C (Figure 1A2). As the temperature approached reflux (205 °C), the remaining precursor in the reaction mixture was entirely consumed for the growth of the nuclei as shown by a complete loss of the absorbance (Figure 1A1). The mean size of the resultant MNPs was 7 ± 4 nm (Figure 1A3).
2. Step-Wise heating at 150 °C.
In this regime the reaction mixture was heated to 150 °C at 4 °C/min, and then the temperature was kept constant at 150 °C for 2 h to prolong the nucleation stage compared to the regime 1 by slowing down the precursor decomposition (Supporting Information Figure S1B). Under those conditions approximately 70 wt % of the precursor was consumed during the nucleation stage (Figure 1B1,B2) resulting in nearly monodisperse MNPs with a mean diameter of 9 ± 2 nm (Figure 1B3).
3. Step-Wise Heating at 180 °C.
In this regime the reaction mixture was heated at 4 °C/min to 180 °C, and then the temperature was kept constant for 2 h at 180 °C (Supporting Information Figure S1C). As a result, the nucleation phase was shortened compared to regimes 1 and 2. Under these conditions, at 180 °C and above, a large excess of the precursor was involved in the particles growth on the existing nuclei as demonstrated by the attenuation of the peak intensity at 330 nm (Figure 1C1). After 2 h at 180 °C, a significant amount of nuclei had undergone growth and precipitated as discrete MNPs (Figure 1C2). Larger MNPs with a mean diameter of 11 ± 4 nm and a broader size distribution were obtained using this regime (Figure 1C3).
4. Rapid Heating.
In the final regime the temperature was increased rapidly at a rate of 5 °C/min until the reaction mixture refluxed (205 °C), and this was followed by 40 h of annealing (Supporting Information Figure S1D). Rapid heating of the mixture resulted in an overlap of the nucleation and growth phases leading to availability of large excesses of the precursor to promote rapid growth of the formed nuclei (Figure 1D1,D2). This regime resulted in particles with a mean diameter of 14 ± 7 nm (Figure 1D3).
The rapid increase in temperature and prolonged high temperature annealing is known to provide a higher degree of control over the crystal size.27 Specifically, high temperature annealing was shown to enhance the saturation magnetization (Ms) of the superparamagnetic single domain MNPs synthesized by thermal decomposition.28,29 The magnetic properties of the 7 and 14 nm diameter MNPs were studied by measuring the saturation magnetization (Ms) at 300 K (Figure 2). The magnetization curves did not show any hysteresis which is consistent with the superparamagnetic properties of the MNPs.30 As the particle size increased from 7 to 14 nm, their Ms values nearly doubled from 36.2 emu/g to 65.5 emu/g while their superparamagnetic behavior was preserved. Larger MNPs, due to their lower surface–volume ratio, have smaller numbers of atoms at the surface thereby leading to decreased surface effects such as spin–spin canting and spin-glass-like behavior.31 As a result, the number of atoms contributing to the magnetic moment of the MNPs increases leading to higher Ms values.32 Increase in Ms also leads to an increased T2 relaxation capability of the MNPs.30,33 Therefore, from the theranostic perspective, the 14 nm MNPs were expected to have higher T2 relaxivity and darker negative contrast.
Figure 2.
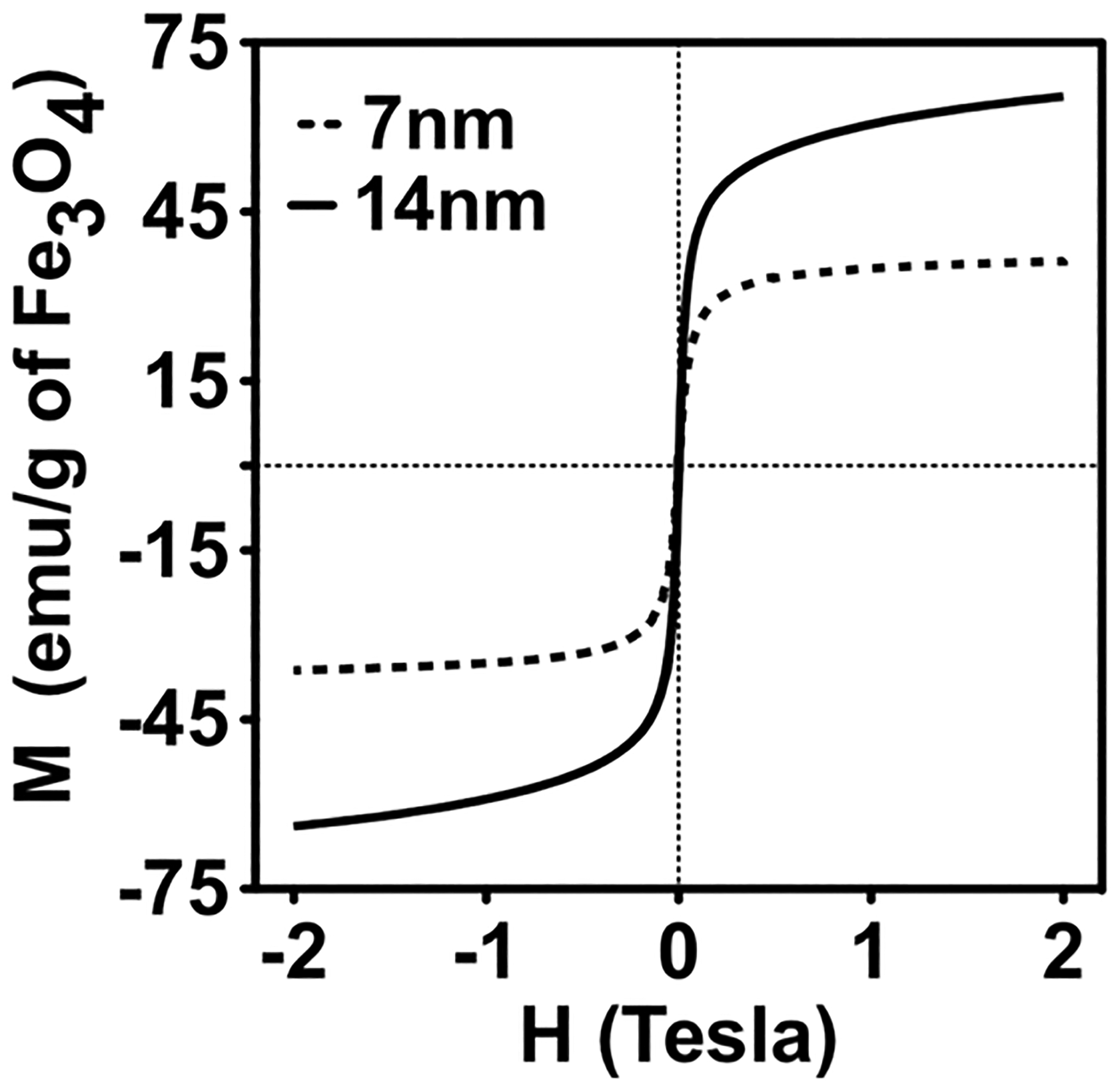
Saturation magnetization of MNPs with two different core sizes synthesized by thermal decomposition of Fe (acac)3.
Preparation and Characterization of the Polymer-Stabilized MNC.
MNCs stabilized by PLE-b-PEG block copolymers were formed via electrostatic binding of pendant carboxylate groups of the PLE blocks and the MNP surface (Scheme 1). In this study the length of the PEG block of the PLE-b-PEG was kept constant, while the PLE block length was varied. The hydrodynamic diameters of the resulting clusters (Supporting Information Figure S2) varied in the range of 40 to 70 nm depending on the length of the PLE block (Table 1). All MNCs were negatively charged as was evident from their ζ-potential values. In this study we also prepared MNCs stabilized by PLE100 homopolymer as well as PLD100-b-PEG113 block copolymer and PLD100 homopolymer. All these clusters in the deionized water had relatively small particle size (under 100 nm) and negative ζ-potential (Supporting Information Table S1). Pegylated polymers are preferable in our work because they impart stealth properties and enhance systemic circulation of nanoformulations.34,35 PLE-b-PEG was chosen because of more facile lysosomal biodegradation of PLE compared to PLD and the respective block copolymer.36,37 PLD100-b-PEG113 and the corresponding homopolymer based MNCs were anticipated to undergo degradation at a slower rate and hence were not developed further.
Scheme 1.

Formulation Process for Preparation of MNP Nanoclusters Stabilized by PLE-b-PEG or ALN-Modified PLE-b-PEG
Table 1.
Physicochemical Characteristics of MNCs Stabilized by Anionic PLE-b-PEG Block Copolymers Having Different PLE Block Lengths
| composition (wt %)b | |||||||
|---|---|---|---|---|---|---|---|
| organic content | DLS characteristicsc | ||||||
| formulationa | abbreviated designation | polymer | BAd | MNP | Deff (nm) | PDI | ζ-potential (mV) |
| PLE10-b-PEG113/MNP | E10-MNC | 19 | 6.7 | 74.3 | 51.4 ± 11.3 | 0.334 ± 0.01 | −20 |
| PLE50-b-PEG113/MNP | E50-MNC | 65.7 | 4.6 | 29.7 | 45.2 ± 4.3 | 0.171 ± 0.01 | −26 |
| PLE100-b-PEG113/MNP | E100-MNC | 56.2 | 5.5 | 38.3 | 68.8 ± 7.8 | 0.222 ± 0.04 | −36 |
The size of the MNPs in all formulations was 7.3 nm as quantified by ImageJ software from TEM images.
The composition of the MNCs was determined by TGA. A total of 5–8 mg of MNCs were heated to 110 °C at 5 °C/min. The temperature was maintained at 110 °C for 10 min and then elevated to 1000 °C at 5 °C/min. The polymer content was determined by subtracting the benzyl alcohol ligand content on the MNP from the overall organic fraction content.
DLS characteristics were measured in DI water at 0.5 mg/mL MNCs at 25 °C.
BA, benzyl alcohol.
For the PLE-b-PEG stabilized MNCs altering the PLE block length affected the polydispersity and composition of the clusters. Interestingly, the greatest MNPs content in the clusters, as determined by TGA, was observed in E10-MNC formed using copolymers with the shortest PLE chain (10 r.u.). As the ionic block length increased, the MNP content decreased to ~30–40 wt % in E50-MNCs and in E100-MNC compared to ~74% E10-MNC. Irrespective of the length of the PLE block in the copolymer used for cluster formation, the MNCs showed considerable sensitivity to the environmental pH, as well as the concentration and/or composition of the elementary salts in the solution. For example, E100-MNC appeared to be less stable in magnesium sulfate, pH 6.0, than in sodium acetate, pH 5.0, sodium bicarbonate, pH 7.2, or disodium phosphate, pH 7.4, solutions (Figure 3A). This is possibly explained by the coordination of the divalent magnesium cation with the PLE block38 resulting in detachment of this block from the MNPs surface. In the phosphate buffer (pH 7.4) the E100-MNC stability strongly depended on the concentration of the disodium phosphate (Figure 3B) or NaCl (Figure 3C). This was attributed to a relatively weak attachment of the pendant carboxylate groups of the PLE to the MNP surface that can be substituted by the phosphate ions or disrupted by increasing the ionic strength.
Figure 3.
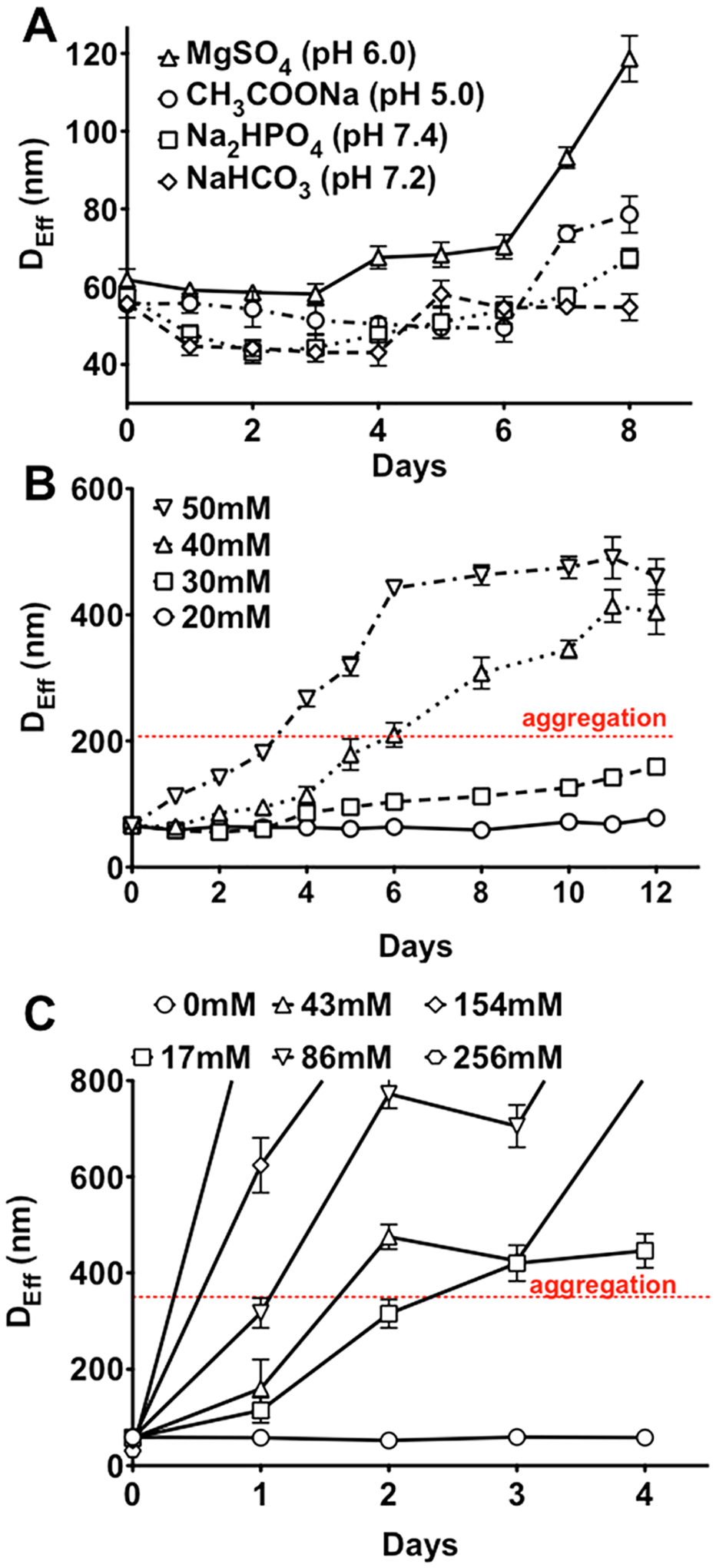
Colloidal stability of E100-MNC as a function of (A) pH and salt composition (20 mM each), (B) concentrations of phosphate buffer, pH 7.4, and (C) NaCl concentration in 10 mM phosphate buffer, pH 7.4. All samples were prepared at a concentration of 0.5 mg/mL. Data are mean ± SD (n = 3).
Introducing anchor groups such as dopamine39–41 or bisphosphonate-containing compounds that form very strong coordination complexes with the MNPs23,42 can enhance binding of the block copolymer to the MNP surface and increase stability of the resulting MNCs under physiological conditions. Toward this goal we conjugated ALN to the PLE chain of PLE50-b-PEG113 by EDC/NHS chemistry in a DMSO–water system (Supporting Information Scheme S1). The product of the reaction was confirmed by 1H NMR (Supporting Information Figure S3) and 31P NMR (Supporting Information Figure S4). The number of ALN groups linked to the PLE chain, termed here “degree of conjugation” (Dconj), was quantified by ICP-MS as well as NMR (Table 2, Supporting Information Table S2).
Table 2.
Physicochemical Characteristics of MNCs Stabilized by ALN-Conjugated PLE50-b-PEG113 Copolymers
| composition (wt %)d | DLS characteristicsd | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| organic content | DI water | PBS pH 7.4 | ||||||||||
| formulation | abbreviated designation | ALN groups per PLE chaina | COOH groups (μmole per mg MNC)b | MNP diameter (nm)c | polymer | BA | MNP | Deff (nm) | PDI | ζ-potential (mV) | Deff (nm) | PDI |
| ALN6-PLE50-b-PEG113/MNP | A6-MNC | 6* | 8.5 | 9 | 76 | 2.5 | 21.5 | 33.4 ± 6 | 0.21 ± 0.02 | −24.6 | 39.6 ± 11 | 0.31 ± 0.03 |
| ALN12-PLE50-b-PEG113/MNP | A12-MNC | 12 | 11.5 | 9 | 67.3 | 3.1 | 29.6 | 35.4 ± 8.4 | 0.25 ± 0.01 | −33.1 | 34.6 ± 9 | 0.25 ± 0.02 |
| ALN19-PLE50-b-PEG113/MNP-1 | A19-MNC-1 | 19 | 16.5 | 9 | 53.7 | 5.8 | 40.7 | 69.5 ± 5.4 | 0.22 ± 0.05 | −29.7 | 67.4 ± 10 | 0.24 ± 0.01 |
| ALN19-PLE50-b-PEG113/MNP-2 | A19-MNC-2 | 19 | 26.7 | 12 | 68.4 | 4.8 | 26.8 | 78 ± 7.2 | 0.20 ± 0.01 | −20.4 | 79.8 ± 7 | 0.22 ± 0.02 |
Number of ALN molecules per polymer chain was quantified by ICP-MS except for A6-MNC (*) wherein the number of ALN units per polymer chain were estimated from 1H NMR.
Carboxylic acid (COOH) groups concentration was determined by potentiometric acid–base titration on formulation diluted to 0.1 mg/mL in DI water.
The average MNP sizes were estimated from TEM images by measuring a minimum of 400 core MNP sizes with ImageJ software.
The composition and DLS characteristics of the MNCs were determined as explained in Table 1, except for the last two columns where the DLS measurements were done in PBS, pH 7.4.
The cluster size was altered by increasing the concentration of the polymer and MNP solution by twofold. The number of ALN groups also strongly influenced the size of the resulting MNCs (Table 2). The A6-MNC and A12-MNC formed using copolymers with Dconj equal to 6 and 12 respectively were relatively small (~33–35 nm). As Dconj increased to 19 in A19-MNC-1 the particle size increased nearly twofold. Interestingly, TEM images suggest that A19-MNC-1 particles appear to be more elongated and display greater proportion of chain-like structures compared to A12-MNC (Figure 4). Using the copolymer with Dconj = 19 and MNPs with different particle diameter (9 and 12 nm) we also produced two types of clusters, A19-MNC-1 and A19-MNC-2. Despite the difference in their MNPs sizes the resulting clusters had practically the same overall particle size. Similar to the above-described non-stabilized clusters, the MNCs obtained using the ALN copolymers were negatively charged as was evident from the high absolute values of their negative ζ-potential (Table 2). Perhaps the greatest difference between these clusters was in their stability in physiological ionic strength conditions. Thus, all ALN-stabilized clusters had nearly the same particle size in PBS, pH 7.4, as in the DI water. When compared with each other the A6-MNC and A12-MNC having a smaller number of ALN groups per copolymer were relatively less stable upon storage and aggregated over time (Supporting Information Figure S5). In contrast, A19-MNC-1 was stable in both DI water and PBS at RT and 37 °C displaying little if any change in particles size upon incubation for at least 30 days (Figure 5).
Figure 4.
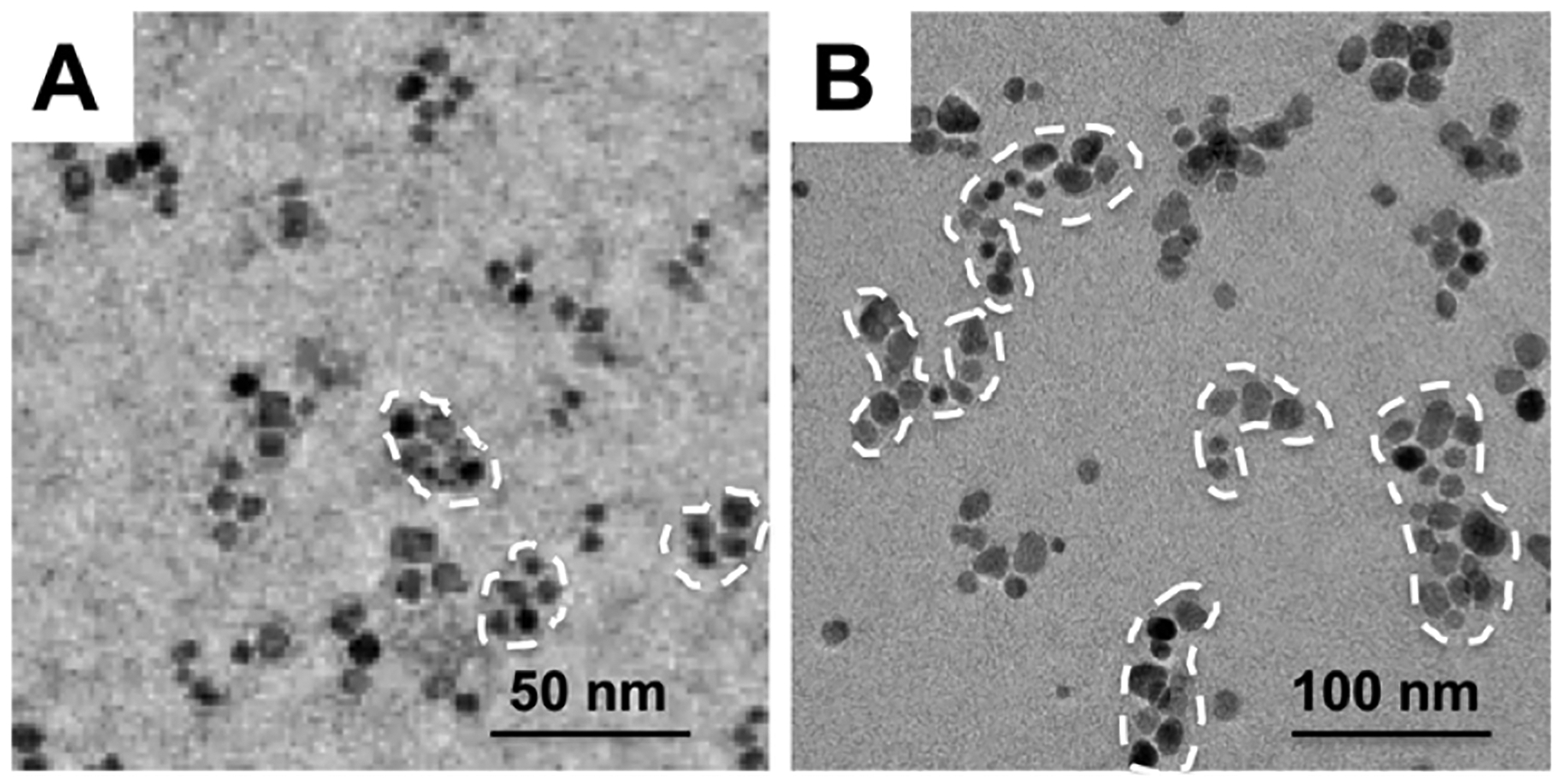
TEM images of (A) A12-MNC and (B) A19-MNC-1 formulations.
Figure 5.
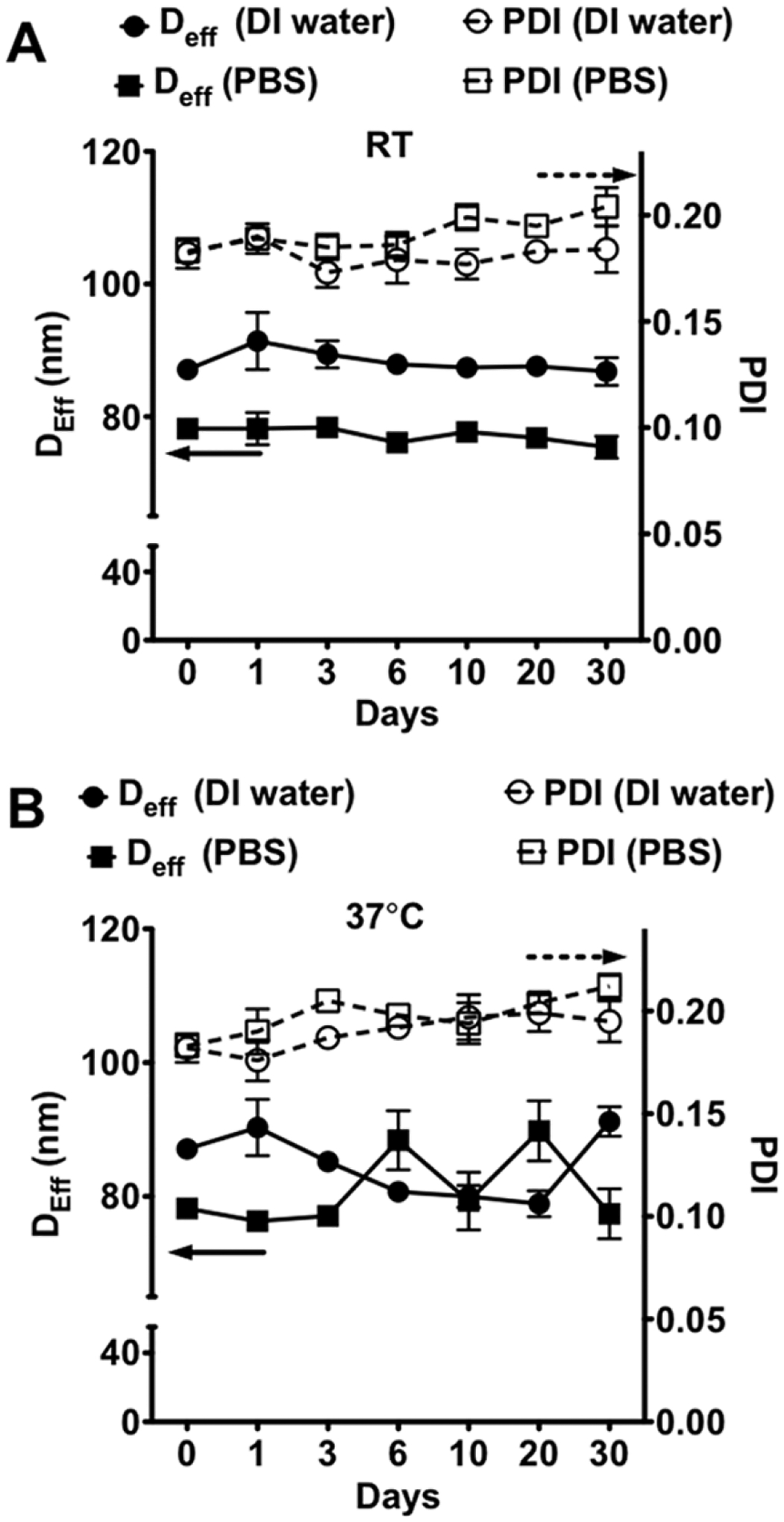
Colloidal stability of A19-MNC-1 in DI water and potassium free PBS pH 7.4 at (A) RT and (B) 37 °C.
Loading of Cisplatin in MNCs.
Cisplatin was loaded into the MNCs via the formation of a coordination complex of drug molecules with the carboxylate groups of PLE chains at pH 9.5. The technique employed for the drug loading was similar to that previously described by us for the loading of the cross-linked anionic nanogels.43,44 In addition, the bisphosphonate groups introduced on the polymer chain are also likely to have a higher binding affinity toward cisplatin than the carboxylates.45 We targeted 70 mol % of the total carboxylate groups available in MNCs, assuming that each mole of cisplatin reacts with two moles of the carboxylate groups. Four different types of ALN-stabilized MNCs described in the previous section were used in these experiments. The characteristics of the resulting cisplatin-loaded MNCs are presented in Table 3. In all cases from 24 to 40% of the drug used for the loading was actually incorporated in the MNCs. Both the loading efficiency (LE) and loading capacity (LC) appeared to increase as the number of ALN moieties elevated in the following order A6-MNC < A12-MNC < A19-MNC-1. As mentioned earlier, increase in the bisphosphonate groups (two bisphosphonates introduced per each carboxylate group attached) is also very likely to contribute to this trend in LE and LC due to higher affinity for cisplatin. However, in A19-MNC-2, as the size of the MNP grains increased, there was nearly twofold increase in the LC compared to A19-MNC-1. Although these two unloaded clusters had similar particle size, the greater phosphonate and carboxylate groups content in A19-MNC-2 compared to A19-MNC-1 probably explained the difference in the LC between them (Table 2). Notably, the particle size of all clusters in DI water noticeably did not change upon loading (Tables 2 and 3). However, and quite interestingly in all cases, the ζ-potentials of the loaded clusters appeared to become more negative after loading, which might seem counterintuitive as the ionic groups are partially consumed by cisplatin coordination. However, we believe that this effect may be explained by the thickening of the effective Stern layer that, as previously discussed43 in the nanogel-like particles, can span throughout the entire particle volume. Coordination of the drug is likely to displace the counterions from the interior of the MNCs to the more “hydrodynamically mobile” surface and periphery layers, resulting in an effective increase in the density of the uncompensated negative charge and absolute ζ-potential value of the particles.
Table 3.
Physicochemical Characteristics of Cisplatin-Loaded MNCs
| DLS characteristicsb | ||||||||
|---|---|---|---|---|---|---|---|---|
| DI water | PBS pH 7.4 | |||||||
| MNC used to prepare cisplatin-containing formulationa | abbreviated designation | Deff (nm) | PDI | ζ-potential (mV) | Deff (nm) | PDI | LE (%)c | LC (%)d |
| A6E50-PEG113/MNP | Pt-A6-MNC | 38.6 ± 8.2 | 0.261 ± 0.03 | −28.3 | 36.4 ± 11.4 | 0.348 ± 0.03 | 24 | 3.8 |
| A12E50-PEG113/MNP | Pt-A12-MNC | 41.7 ± 11.3 | 0.243 ± 0.03 | −36.1 | 43.3 ± 8.2 | 0.267 ± 0.03 | 32.7 | 5.2 |
| A19E50-PEG113/MNP | Pt-A19-MNC-1 | 69.3 ± 9.3 | 0.255 ± 0.02 | −35.4 | 70.2 ± 7 | 0.203 ± 0.01 | 37.3 | 8.4 |
| A19E50-PEG113/MNP | Pt-A19-MNC-2 | 86 ± 5.1 | 0.218 ± 0.02 | −43.4 | 83.8 ± 7.2 | 0.211 ± 0.01 | 40 | 17.6 |
The loading procedure is described in Materials and Methods.
DLS characteristics were measured in DI water or PBS, pH 7.4, at 0.5 mg/mL MNCs at 25 °C.
The loading efficiency (LE) was determined by ICP-MS as the percent of the drug incorporated in MNCs vs the drug added during loading.
The loading capacity (LC) was determined as the weight percent of cisplatin per the dispersed phase. Briefly, the drug-loaded MNCs were lyophilized, and the amount of cisplatin per mg of the formulation was determined by ICP-MS.
Release of Cisplatin from MNCs.
Cisplatin release was studied at 37 °C at pH 7.4 (PBS) and pH 5.5 (50 mM; ABS), which mimic conditions in extracellular media and endosomal compartments, respectively. As shown in Figure 6 both pH and Dconj of the MNCs affected the drug release. In all cases the fastest release kinetics was observed in Pt-A6-MNC having the smallest number of ALN anchor groups in the copolymer and lowest LC. As the Dconj increased the release rate decreased, the lowest being in Pt-A19-MNC-1. For all systems the release was faster at pH 5.5 than pH 7.4 although this difference was the smallest in Pt-A6-MNC and the greatest in Pt-A19-MNC. The pKa of the protons on the phosphonate groups of ALN may also play an important role in the drug release. The third pKa of alendronic acid is 6.4.46 Hence, at pH 5.5, the respective phosphonate groups of ALN can become protonated, and if these groups participate in binding cisplatin this could also cause increase in the drug release rate.
Figure 6.
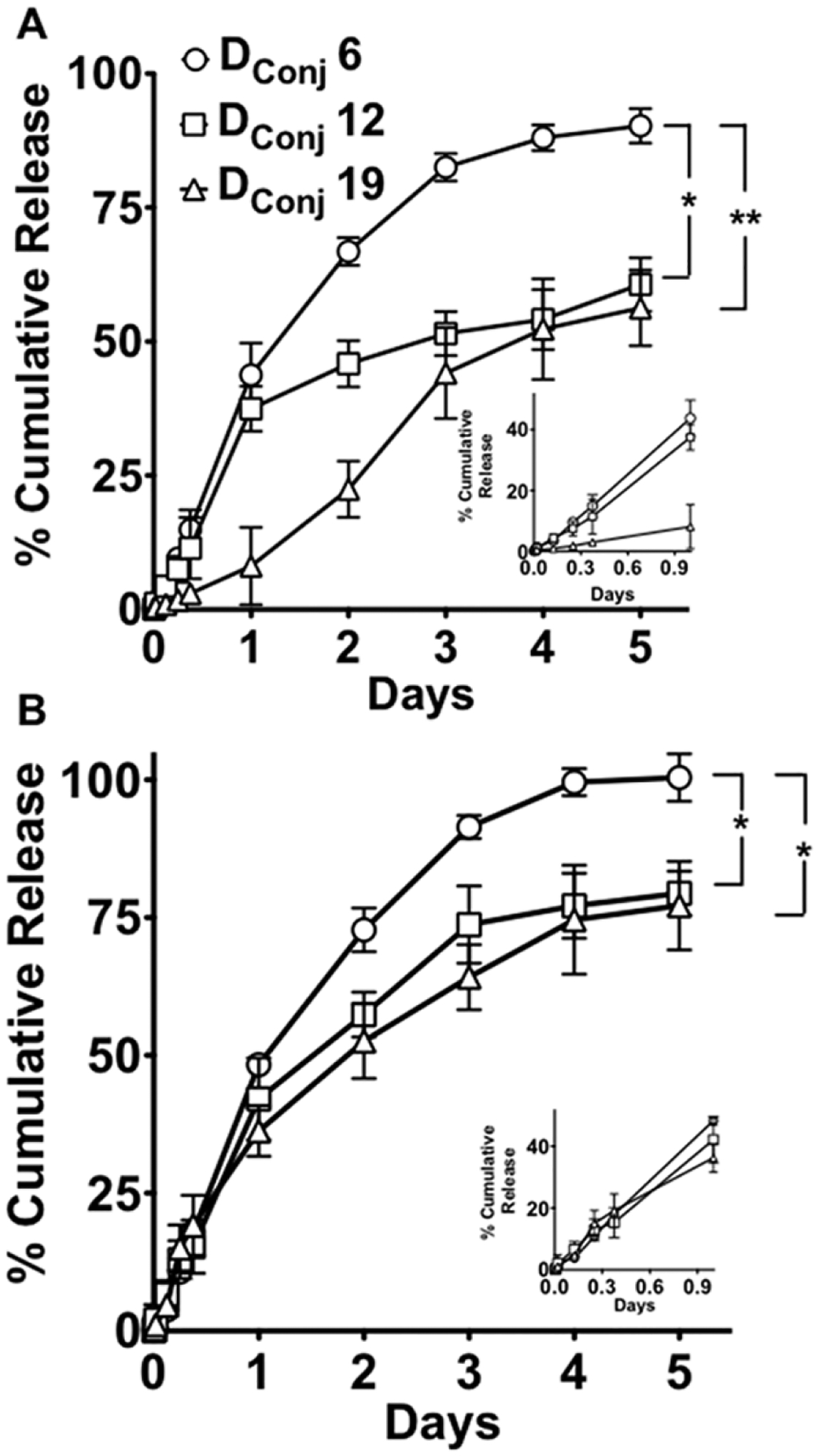
Release of cisplatin from Pt-A6-MNC, Pt-A12-MNC and Pt-A19-MNC-1 at (A) pH 7.4 and (B) pH 5.5. Data are mean ± SD (n = 3); statistical analysis performed by ANOVA using SPSS statistical software, *p < 0.05; **p < 0.01. The graphs in the insets of both the plots show the release patterns in the first 24 h.
Interestingly, Pt-A19-MNC-1 at pH 7.4 displayed a complex release kinetics with very slow release of the drug during the first 2 days followed its by acceleration thereafter. The slow phase could be possibly attributed to the initial compact morphology of the cluster, which becomes looser as the drug loading decreases, thereby facilitating drug diffusion out of the particle. Notably, the slow release phase was not seen at pH 5.5 with the drug release being overall much faster and monotonic. It is noteworthy that the formulations did not aggregate or lose colloidal stability over the entire period of the study in either buffer systems (Supporting Information Figure S6).
Relaxometry Measurements of MNC Formulations.
The use of contrast agents for MRI depends on their ability to shorten the relaxation times of the surrounding protons.47,48 Positive contrast agents reduce T1 relaxation times to give a brighter signal while negative contrast agents reduce T2 resulting in a darker signal. The reciprocals of the relaxation times are called the relaxation rates, R1 and R2, with the effectiveness of contrast agents expressed as relaxivities, r1 and r2, which are determined from the slope of the relaxation rate curves expressed as a function of total Fe concentration (mM). In all cases the r1 relaxivity values of MNCs in PBS pH 7.4 values were relatively low and did not change or decrease upon drug loading (Table 4).
Table 4.
Longitudinal (r1) and Transverse (r2) Relaxivity of Various MNC Formulations
| formulation | r1 (s−1·mM−1)a | r2 (s−1·mM−1)a |
|---|---|---|
| A6-MNC | 0.694 ± 0.07 | 31.3 ± 2 |
| Pt-A6-MNC | 0.66 ± 0.04 | 56 ± 4.7 |
| A12-MNC | 1.91 ± 0.11 | 48.3 ± 2.2 |
| Pt-A12-MNC | 1.02 ± 0.06 | 86.5 ± 2.8 |
| A19-MNC-1 | 1.23 ± 0.05 | 87.8 ± 4.4 |
| Pt-A19-MNC-1 | 1.02 ± 0.16 | 154.8 ± 11.5 |
| A19-MNC-1b | 1.12 ± 0.1 | 72.2 ± 4.02 |
| Pt-A19-MNC-1b | 0.9 ± 0.04 | 120.8 ± 3 |
| A19-MNC-2 | 1.34 ± 0.08 | 81.4 ± 3.3 |
| Pt-A19-MNC-2 | 1.18 ± 0.07 | 140.3 ± 12 |
Data are mean ± SD (n = 3).
Relaxivity measurements of phantom gels in 2% v/v agar. All other measurements were conducted in PBS, pH 7.4.
In contrast the r2 values of MNCs increased progressively as the Dconj value increased A6-MNC < A12-MNC < A19-MNC-1 (Table 4). Notably for each empty and drug loaded MNCs the r2 value appeared to be higher as the particle size was larger. But quite unexpectedly for all MNCs the r2 value increased considerably, ~1.6 to 1.8 times after the drug loading although the particle size (Tables 2 and 3) did not change that significantly. For example, Pt-A19-MNC-1 had r2 values of 154.8 s−1·mM−1 vs 87.8 s−1·mM−1 for the unloaded A19-MNC-1. The T1 and T2 relaxivities of A19-MNC-1 and Pt-A19-MNC-1 were further measured in buffered (PBS pH 7.4) 2% w/w agar gel. Phantom agar gel is commonly used as reference material to mimic biological tissues due to similarities of its T1 and T2 values to those of biological tissues.49 In the gel the r1 and r2 values of both the loaded and unloaded MNCs showed only a minor decline compared to these values in PBS dispersion suggesting good potential for negative contrast in biological tissues.
Synthesis of LHRH-Targeted MNCs.
Unloaded A19-MNC-1 and Pt-A19-MNC-1 (41 wt % MNP and 8.4 wt % cisplatin) were used in this work as representative MNCs for LHRH conjugation. Similar to our previous publication on cisplatin containing nanogels50 (d-Lys6)-LHRH was conjugated through its lysine primary amino group to the free carboxyl groups of the A19-MNC-1 and Pt-A19-MNC-1 using EDC/S-NHS coupling chemistry. The conjugation was confirmed by an HPLC assay51 by measuring the amount of the free (d-Lys6)-LHRH in the filtrate after purification of the modified clusters by centrifugal filtration (3000 MWCO). Only 13.3% and 28.7% of the total reagent added in the reaction mixtures was present in the filtrates of A19-MNC-1 and Pt-A19-MNC-1 samples, respectively. The apparent greater yield of conjugation of the peptide to A19-MNC-1 compared to Pt-A19-MNC-1 was explained by availability of a greater number of free carboxylate groups prior to loading of the drug. The FTIR spectra (Supporting Information Figure S7) of the LHRH-conjugated Pt-A19-MNC-1 (LH-Pt-A19-MNC-1) suggested formation of a secondary amide bond between the primary amino group of the (d-Lys6)-LHRH and the carboxylic acid groups of the PLE.
Uptake of LHRH-Targeted Cisplatin-Loaded MNCs in Ovarian Cancer Cells.
The cell uptake of the unloaded LHRHr-targeted (LH-A19-MNC-1) and untargeted (A19-MNC-1) MNCs was studied by confocal microscopy in LHRHr overexpressing A2780-WT and A2780-CisR human ovarian cancer cells. A2780 human ovarian cancer cells are known for LHRHr overexpression. Numerous studies have characterized LHRHr expression by Western Blot analysis.50,52,53 As seen in Figure 7, untargeted MNCs showed very low cellular uptake even after 24 h in both wild type and cisplatin-resistant cells. The internalized MNCs were localized in the lysosomal compartments of the cells visualized by the bright yellow punctate regions in the overlay images. Conjugation of LHRH significantly enhanced the LH-A19-MNC-1 cellular uptake. In the A2780-WT cells the LH-A19-MNC-1 were seen localized along the cell membrane (red) and within the lysosomes (yellow punctate regions) as early as at 1 h post-incubation. Although the uptake of LH-A19-MNC-1 in A2780-CisR cells was slower, a significant red fluorescence of the labeled clusters was observed in the cells at as early as 3 h. Interestingly, literature reports suggest that certain cisplatin-resistant cancer cells (e.g., ovarian epidermal carcinoma) exhibit significant reduction in nonreceptor mediated fluid-phase endocytosis compared to their wild-type counterparts while the receptor-mediated endocytosis remains unaffected.54 The observed increased uptake of LH-A19-MNC-1 in A2780-CisR cells specifically is in line with these reports and strongly suggests that receptor-mediated targeted therapy is an effective strategy for achieving a faster onset and selective enhancement of cellular uptake in drug resistant tumors as it circumvents the defective mechanisms of uptake in drug resistant cancers.
Figure 7.
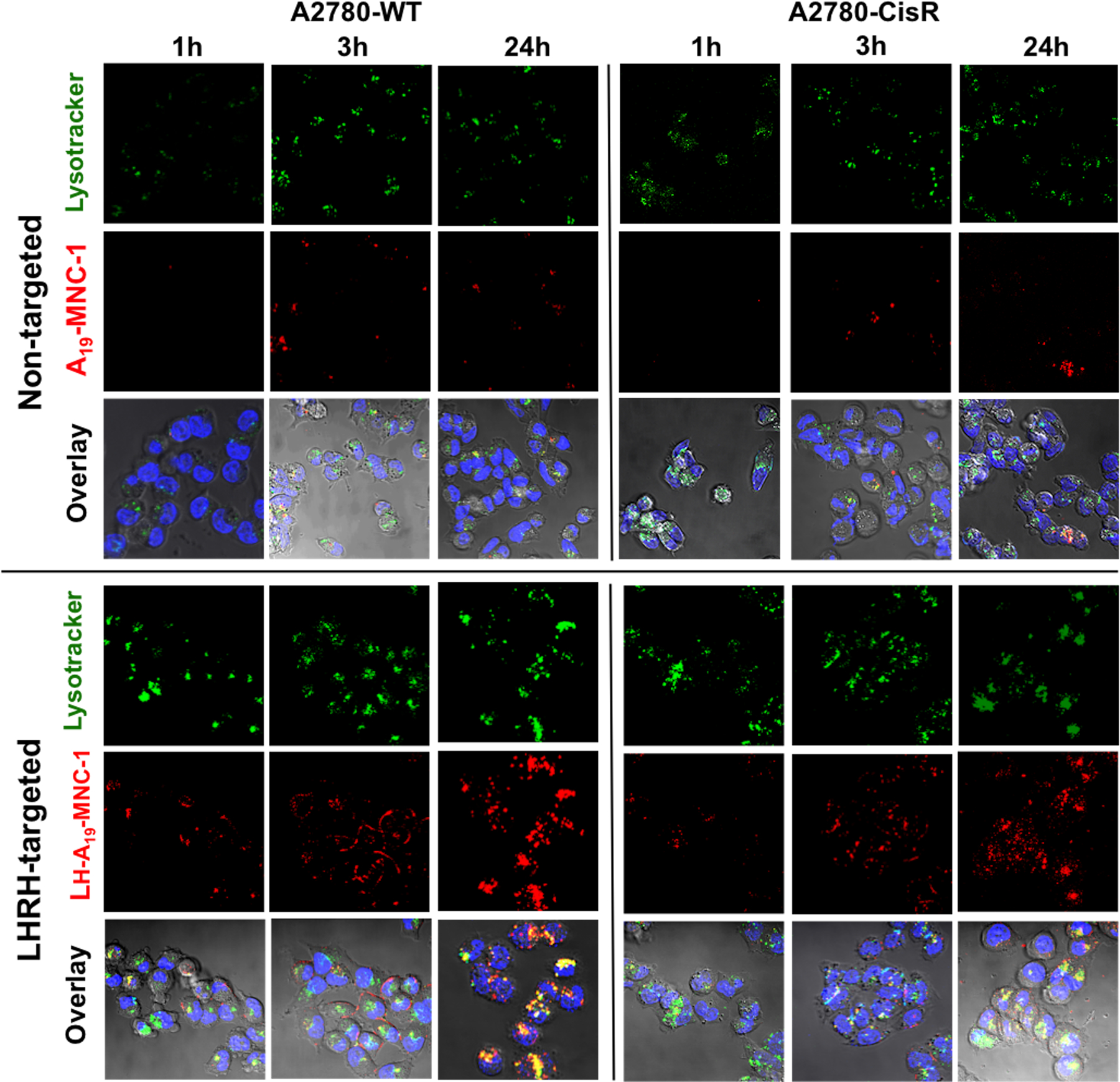
Confocal microscopy of the wild-type A2780-WT and cisplatin resistant A2780-CisR human ovarian cancer cells at different time points during their incubation with nontargeted A19-MNC-1 and LHRH-conjugated A19-MNC-1. Live cells were exposed to the said Alexa-Fluor 647 labeled (red) MNCs and stained with the lysotracker green (green) and Hoechst nuclear stain (blue) for 30 min. The colocalization of the labeled MNCs in lysosomes is seen in the overlay (yellow punctate regions) (magnification 63×).
The uptake was further quantified by ICP-MS. In this case the cells were incubated with Pt-A19-MNC-1 and LH-Pt-A19-MNC-1 for up to 24 h, and the Fe and Pt content in the cells was normalized per mg of cellular protein and plotted over time (Figure 8). Similar to the trend observed in the confocal images, LHRH conjugation resulted in an almost twofold increase in intracellular Fe concentration in the A2780-WT cells (Figure 8A). In the A2780-CisR cells there was also a significant increase in the uptake of targeted LH-Pt-A19-MNC-1 compared to untargeted Pt-A19-MNC-1, although as expected, the overall uptake was slower than that in the WT cells. A similar pattern was observed for the intracellular Pt content also (Figure 8B). It is important to note that LHRH targeting significantly improved the rate of uptake of the cisplatin-loaded MNCs during the first few hours, thereby decreasing the effect of the release of free cisplatin in the culture medium. This was apparent in the analysis of the ratios of the cisplatin/Fe cell levels (Figure 8C). In the case of the nontargeted Pt-A19-MNC formulation the cisplatin/Fe ratio in both cell lines decreased significantly after 3 h over time indicating that a significant amount of drug was released into the media during the incubation time. For the targeted LH-Pt-A19-MNC-1 formulation this ratio steadily decreased over time but remained much higher than that for Pt-A19-MNC-1. This was consistent with more rapid internalization of the drug-loaded particles prior to a considerable release of the drug into the media.
Figure 8.
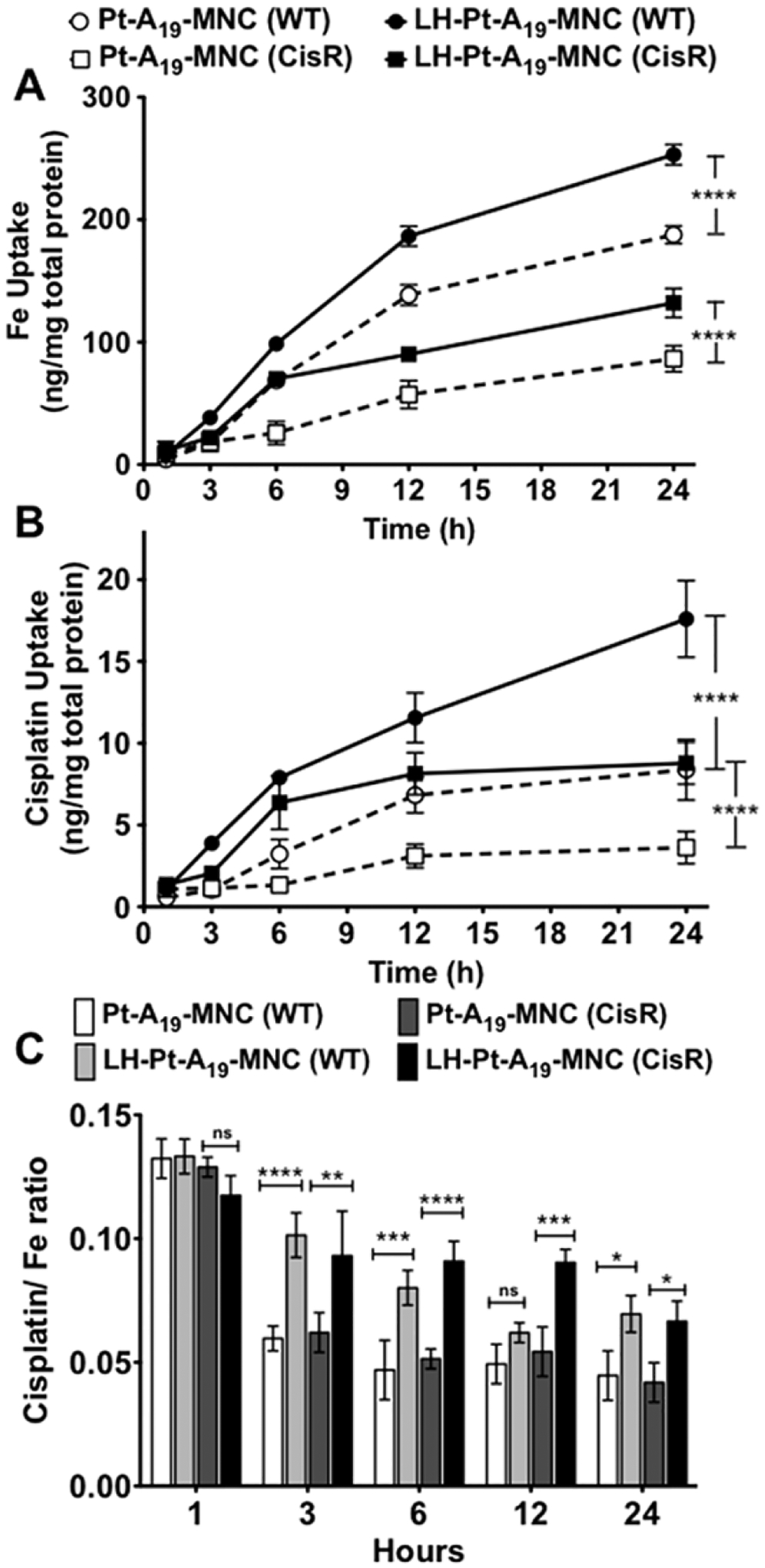
Uptake of LHRH-targeted and untargeted cisplatin-loaded MNCs in A2780-WT and A2780-CisR cells as quantified by ICP-MS and presented as (A) Fe levels, (B) Pt (cisplatin) levels, and (C) cisplatin/Fe ratio over time. Data are mean ± SD. Statistical analysis done by ANOVA using SPSS software. Statistical significance denoted as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Finally, preincubating both cell lines with 100 nM free d-Lys-6-LHRH peptide for 1 h before adding the MNCs greatly diminished the uptake of LH-Pt-A19-MNC-1 and had little if any effect on Pt-A19-MNC-1 (Supporting Information Figure S8). This suggests that the free peptide competitively inhibited the uptake of LHRH-targeted LH-Pt-A19-MNC-1 and reinforces the receptor-mediated mechanism of its internalization in cancer cells.
Anticancer of LHRH-Targeted Cisplatin-Loaded MNCs in Ovarian Cancer Cells.
Prior to evaluating the anticancer activity of drug formulations the toxicity of unloaded MNCs was determined in A2780-WT and A278-CisR human ovarian cancer cells. The cells were incubated with various concentrations of A19-MNC-1 and LH-A19-MNC-1 for 24 and 72 h, and cell viability was measured by an MTT assay. These MNCs did not show toxicity in a broad range of concentrations (Supporting Information Figure S9), and they were further used to assess the anticancer activity of the drug loaded MNCs.
The anticancer activity of LHRH-targeted and untargeted cisplatin-loaded MNCs was determined in A2780-WT and A278-CisR cells after 24 and 72-h treatments. The two time points were used initially to exclude possible effects of the release of the drug into the extracellular media from the untargeted and targeted MNCs. However, the observed trends in cytotoxicity were essentially the same for both exposure times. As expected, the untargeted Pt-A19-MNC-1 was several-fold less active compared to the free cisplatin in both A2780-WT and A2780-CisR cells (Table 5 and Supporting Information Figure S10). LH-Pt-A19-MNC-1 was more active compared to untargeted Pt-A19-MNC-1 in both cell lines. Although LH-Pt-A19-MNC-1 was still less active than the free drug in the resistant cells, its IC50 was close to that of cisplatin (10.9 μM vs 7.9 μM; 72 h). This trend in activity of MNC formulations is consistent with the slow and low cellular uptake of the untargeted MNCs and somewhat faster uptake of the LHRH targeted particles. Since, as discussed above, during the cell exposure time the free drug may release from nanoparticles into the extracellular media and contribute to cytotoxicity, the “actual” difference in the activity of the targeted and untargeted MNCs could be even greater than revealed herein.
Table 5.
IC50 Values of Different Treatment Groups in A2780-WT and Cisplatin Resistant A2780-CisR Ovarian Cancer Cell Lines
| IC50 values (μM) | |||
|---|---|---|---|
| exposure timea | treatment | A2780-WT | A2780-CisR (μM) |
| 24 h | cisplatin | 1.1 | 9.1 |
| Pt-A19-MNC-1 | 6.8 | 28.6 | |
| LH-Pt-A19-MNC-1 | 3.2 | 18.3 | |
| 72 h | cisplatin | 0.9 | 7.9 |
| Pt-A19-MNC-1 | 4.4 | 16.7 | |
| LH-Pt-A19-MNC-1 | 2.3 | 10.9 | |
Exposure time is the total duration of incubation of cells with respective treatments.
In Vitro Evaluation of the MRI Potential of MNCs.
The effect of LHRHr targeting on the MRI capability of the formulations was studied in both cell lines. Following 24 h of incubation with different unloaded and cisplatin loaded MNCs A19-MNC-1, LH-A19-MNC-1, Pt-A19-MNC-1, and LH-Pt-A19-MNC-1 at various Fe concentrations, the cells were washed with 1× PBS and harvested, and 9 × 106 live cells per group were molded into agar and scanned in the Siemens 3T human MR scanner to obtain T1 and T2-weighted contrast images (Figure 9). MNPs are predominantly used as negative (T2) contrast agents. The negative contrast was indeed more prominent and showed a dose-dependent increase. LHRH conjugation promoted cellular uptake of LH-Pt-A19-MNC-1 and resulted in greater T2 contrast and this was more profound in the A2780-WT cells that took up more material than A2780-CisR. In line with the relaxivity values, the presence of cisplatin resulted in greater contrasts irrespective of the presence of LHRH. This was more obvious in the case of A2780-CisR cells than in A2780-WT cells due to overexposure in the latter. Overall, when compared to the contrast obtained with the phantom gels, the targeted MNCs provided significant T2 contrast even after internalization within the cells.
Figure 9.
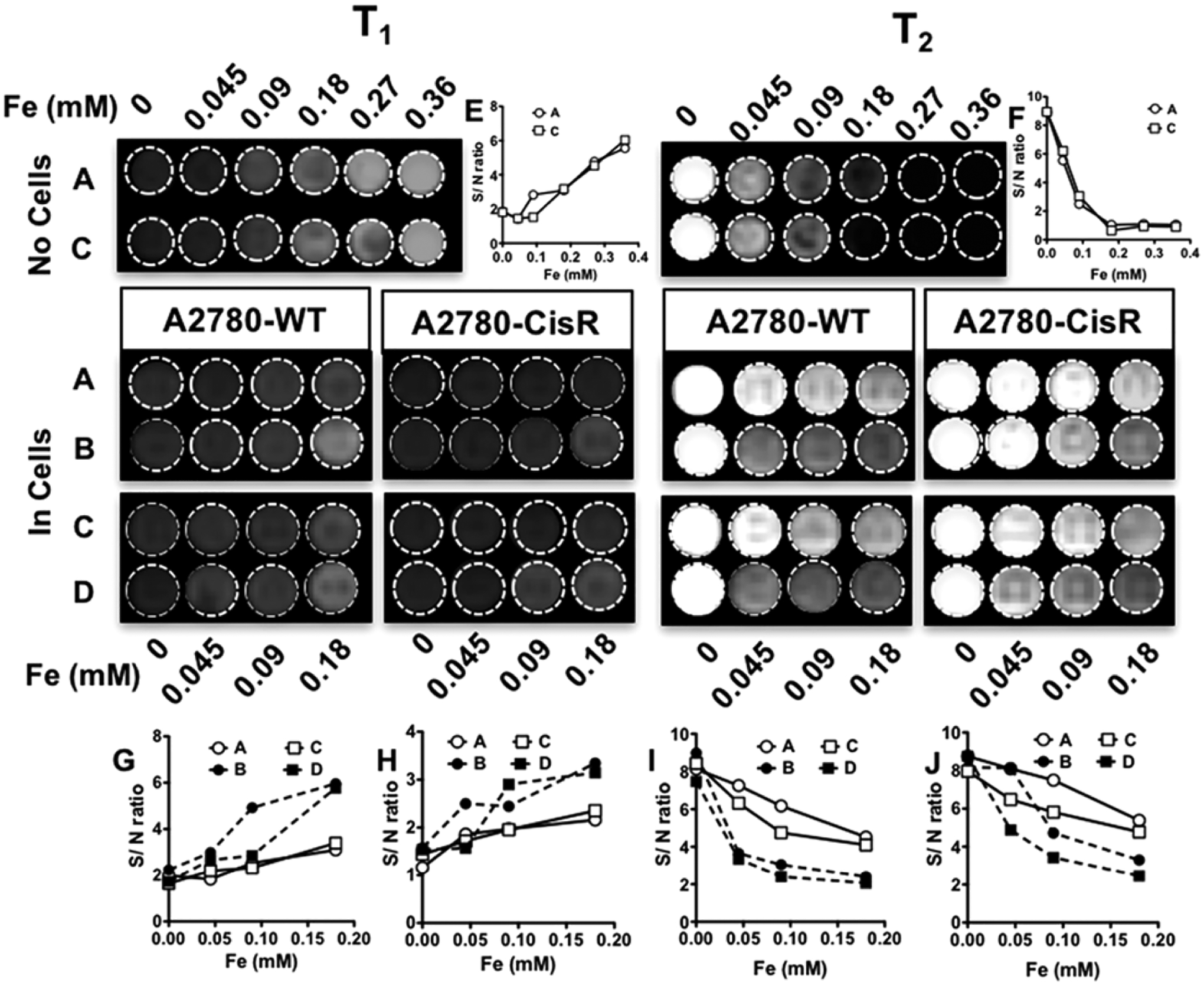
In vitro evaluation of MRI potential of different formulations. T1 and T2 images of phantom gels of A2780-WT and A2780-CisR cells incubated with different concentrations of (A) A19-MNC-1, (B) LH-A19-MNC-1, (C) Pt-A19-MNC-1, and (D) LH-Pt-A19-MNC-1. The image contrasts are compared with those obtained from serial dilutions of A19-MNC-1 and Pt-A19-MNC-1 in the absence of cells (top). The signal-to-noise (S/N) ratios for the different sets of contrasts were plotted against the corresponding Fe concentrations to quantitatively compare the contrasts obtained by the four different formulations A–D. The comparisons of S/N ratios of T1 and T2-weighted contrasts of formulations A and C in phantoms with no cells are represented in parts E and F respectively. parts G and I represent the S/N ratios of T1 and T2-weighted contrasts in phantoms of A2870-WT cells while parts H and J represent the same of A2780-CisR cells incubated with different concentrations of formulations A–D.
DISCUSSION
We have developed the new superparamagnetic nanoformulations for drug delivery and MRI and demonstrated (1) their use as carriers for a chemotherapeutic drug, cisplatin, (2) their targeted receptor mediated delivery into drug resistant and sensitive ovarian cancer cells, and (3) and their potential use as MR contrast agents for future theranostic applications. Structurally, these systems are distant relatives of previously described nanogels or polymeric micelles with cross-linked polyion cores and PEG shells that have been synthesized using the anionic block copolymers, including PLE-b-PEG.43,50,55,56 However, instead of chemical cross-links the polyion chains of the block copolymers are physically cross-linked through ionic interactions with the magnetite MNPs that bind these chains to each other.
To synthesize the MNPs we employed a well-established method of thermal decomposition of Fe(acac)3, which is a simple one-pot technique allowing access to relatively uniform MNPs having good magnetization properties. In this method the solvent serves as a mild surfactant as well as a reducing agent.20 The sizes of the MNPs are governed by the rate of heating while magnetic properties depend on the annealing temperature and duration of the reaction.57,58 High boiling point solvents such as benzyl ether (298 °C) are known to allow for shortening of the duration of synthesis but require additional surfactants and reducing agents.59 We show herein that, in spite of a relatively lower reflux temperature in benzyl alcohol, the nucleation and growth phases could be effectively controlled in this solvent by altering the rate of reaction thereby enabling control over the core size.
Formation of nanoclusters of MNPs with unmodified PLE-b-PEG block copolymers was governed by complex interplay between 1) the charge coordination and electrostatic interactions of the carboxylate groups of PLE with the magnetite particle surface, 2) the magnetite particle–particle interactions, and 3) the steric repulsion of the PEG blocks. From this standpoint it is interesting that PLE10-PEG113 having the shortest anionic PLE block formed the clusters that were enriched by magnetite compared to other clusters. One possible reason for such behavior was that the short PLE block could not prevent coalescence of MNPs with each other. Moreover, given the PLE and PEG block length ratio the complete coverage of the MNP surface by the copolymer might have been prevented by the PEG steric repulsion that is particularly high in the case of the shortest PLE10-PEG113 compared to two other block copolymers. As a result while E10-MNC showed the highest capacity to incorporate MNPs these clusters were also the least stable compared to E50-MNC and E100-MNC. An increase in the number of anchoring groups was shown to improve surface coating and reduce aggregation of the MNPs.60
Colloidal stabilization of the MNCs in aqueous dispersion was achieved using biodegradable ALN-modified PLE-b-PEG block copolymer.55 Notably, for the ALN-modified PLE-b-PEG block copolymer, both the phosphate and the carboxylic groups interacted with the MNPs surface during clusters formation. Although the interactions of carboxylic groups with the MNPs were disturbed in PBS, the structures were stabilized by phosphate groups of the alendronate. Hydrogels of carboxylated polymers are known to exhibit significant swelling behavior at physiological pH due to the presence of charged carboxylate groups.61–63 In the case of cross-linked nanogels formed by anionic block copolymers such as PLE-b-PEG, swelling results in significant increase in the particle size that is varied by changing the cross-linking degree. Moreover, cross-linking can significantly enhance the colloidal stability of nanogels and define the rate of the release of the encapsulated payload.55,64 In this work we have demonstrated that MNCs can be successfully stabilized by introducing sufficient amount of ALN bis-phosphonate groups onto the PLE block that tightly anchor the copolymer to the MNP surface. Cisplatin loading in the MNCs was successfully achieved by coordinating the drug with a fraction of the remaining carboxylic and phosphonate groups of the PLE chains. The presence of MNPs did not hinder the loading as was evident from the relatively high LC values. The drug loaded MNCs exhibited sustained drug release profiles that were altered by changing the number of bis-phosphonate “cross-links” between the polymer and the MNPs. The utilization of a few carboxylate groups for cisplatin loading did not hamper the colloidal stability of the formulations. The samples showed only a marginal pH-dependent increase if Deff and did not undergo any aggregation throughout the period of the cisplatin release study. In addition, we believe that cisplatin loading can, in fact, further increase the stability and integrity of the formulation by simultaneously cross-linking the polymer chains within the cluster.
Recently, Pothayee et al.65 developed theranostic MNCs by adsorbing the polyacrylate block of an amino functional poly(ethylene glycol-b-acrylate) (H2N-PEG-b-PAA) copolymer onto magnetite MNP and loaded these MNCs with a cationic antibacterial drug, gentamycin, through electrostatic interactions. Despite some similarities that can be found with this prior work there were major differences with our nanoformulations. First, in contrast to H2N-PEG-b-PAA the PLE-b-PEG used here has a biodegradable PLE block, which is essential for the use of this nanoformulation for systemic drug delivery. Second, H2N-PEG-b-PAA chains were covalently cross-linked via nondegradable PEG diacrylate oligomer to form the nanoclusters. In this work we introduced controlled levels of bisphosphonate groups onto the PLE block via ALN addition. The bisphosphonates form stronger links to both the magnetite and the Pt from cisplatin than having only carboxylates present. These interactions allow for facile noncovalent self-assembly of MNCs. Such clusters based on ALN-modified PLE have stable colloidal sizes in physiological pH and ionic strength. Selection of an appropriate polymer coating is critical to ensure colloidal stability, high drug loading capacity, biocompatibility and biodegradability, and long circulation time of the MNP-based nanoformulations. Block copolymers comprising biodegradable charged polyamino acid chains can serve as ideal stabilizing agents for formation of MNCs. In our case, the resulting MNCs contained an excess of carboxyl and bisphosphonate groups that were used to load an anticancer drug, cisplatin. Since the loading proceeded through coordination of the drug with the ionic groups, it increased as the amount of these groups was increased and achieved a maximum of 17.6%, which is an excellent LC value for a nanoformulation used in cancer therapy.
A key element in our nanoformulation design is the decoration of MNCs with a targeting moiety to increase accumulation of the MNCs and their payload in ovarian cancer cells. Ovarian cancer represents a major health problem accounting for about 22 000 new cases and over 14 000 deaths annually in the US alone.66 In spite of the dose limiting nephrotoxicity and other severe side effects,67 cisplatin still remains the first line of treatment for ovarian cancer. To address the toxicity and delivery issues of cisplatin the drug has previously been loaded into various polymeric nanocarriers.56 Attempts of targeted delivery of cisplatin have also been reported. For example, cisplatin has been incorporated within the folate-decorated cross-linked PEG-b-polymethacrylate (PEG-b-PMA) nanogels to treat the folate receptor positive ovarian cancer tumor in mice.43 Here we targeted LHRHr that is overexpressed in more than 70% of human ovarian cancers as well as several other cancers.68–70 These receptors are not present in most visceral organs, which could help in reducing the off-target delivery of the nanoformulations to normal tissues and decrease side effects. The LHRH peptide, a synthetic analogue of the natural hormone, has successfully been used to improve delivery of chemotherapeutic agents to LHRHr overexpressing tumors.74 We also previously used this approach with LHRH-decorated PEG-b-PMA nanogels loaded with cisplatin to target LHRHr-positive ovarian cancer in mice.50 In a separate study MNPs were conjugated directly with LHRH-peptide in order to improve the uptake of such MNPs in LHRH overexpressing breast cancer cells.71
As expected, decoration of the MNCs with LHRH resulted in the increased uptake of both MNPs and the drug presumably due to the LHRHr-mediated endocytosis of the targeted nanoformulation.72–74 This translated to increased cytotoxic effects of the targeted nanoformulated drug in both cisplatin-sensitive and resistant cancer cells. Notably, cisplatin loaded in untargeted nanoformulations was also active albeit much less than the free drug. The differences between the free and nanoformulated drug were minimal in the drug resistant cell line. Interestingly, our targeted nanoformulations were somewhat more active in A2780 cells in comparison to previously reported LHRH-decorated PEG-b-PMA nanogels loaded with cisplatin (24 h IC50 ≈ 30 μM).50 We attribute the enhanced efficacy of our new nanofomulation to the biodegradability of PLE-b-PEG in the lysosomal compartments, which can enhance the drug release.56 As MNCs accumulate in endosomal-lysosomal organelles, the drug release is triggered by acidic pH in these organelles. Hydrolysis of the neutral cisplatin molecule inside the cells results in aquation of the molecule involving displacement of the chlorine ligand with water. LHRH targeting further increases influx of the drug-loaded MNCs in the targeted cancer cells where the paucity of chloride ions facilitates drug activation. The activated cationic mono- and di-aqua complexes of cisplatin are thereby released and trapped within the target cells where they intercalate with the N7 atoms of the purines in the DNA base pairs, which prevents DNA repair and DNA replication and leads to cell death.56,75,76 By incorporating cisplatin in the targeted MNCs one can protect the drug from premature aquation in the blood that is responsible for undesired side effects and more selectively affect tumor cells.
Interestingly we also show here that targeting can improve the MRI imaging of cancer cells, which is consistent with increased uptake of LHRH-decorated MNCs in these cells. This result reinforces the importance of tissue specific targeting in designing successful theranostic nanosystems, which can enable diagnosis as well continuous monitoring of therapy. The MNP-based MR contrast agents have been in commercial clinical use in several countries for almost a decade.77,78 To be clinically relevant, negative contrast agents should have higher transverse (T2) relaxivities. Transverse relaxivity is governed by several factors such as MNP core size, chemical nature, oxidation state, applied magnetic field, site-specific uptake, and proximity of the metal ions to the water.79–82 A formulation that enables deeper penetration of the water molecules toward the magnetic core allows for faster relaxation of the protons and a higher T2-relaxivity.82 Clusters of magnetic nanoparticles experience a greater force and magnetic moment than single, well separated MNPs in a magnetic field gradient resulting in an increase in saturation magnetization.83 Clustering of several MNP cores into compact agglomerates has been shown to be beneficial especially in the case of T2 relaxivity.84,85 It was previously reported that aggregates comprising few closely held MNPs measured with long pulse-echo times, can result in greatly improved T2-relaxivity.86,87 Berret et al.88 have shown that ionic block copolymers having an opposite charge to that of the MNPs can be successfully used to formulate stable MNCs wherein the aggregation number of the MNPs can be controlled by altering the copolymer block length. The MNCs discussed herein exhibited a similar trend, where T2 relaxivity was governed by the cluster size and MNP loading in the clusters. In contrast the T1 relaxivity was only marginally affected either by cluster formation or increased uptake since the primary factor governing T1-weighted positive contrast is the MNP core size. Only ultrasmall superparamagnetic iron oxide nanoparticles (less than 5–6 nm in size) are known to exhibit positive contrast enhancement as well.89 Hence, the 9 nm MNP cores were expected to produce only marginal changes in T1 images.
Unexpectedly, we discovered that cisplatin loading in the MNCs considerably increased the r2 relaxivities for all of our nanoformulations. It is well-known that FePt nanoparticles have considerably faster r2 relaxivities as compared to iron oxide nanoparticles with similar sizes.90,91 This can be attributed to the much higher magnetization of FePt as compared to the iron oxides. It is also established that T2 relaxivities of magnetic nanoparticles increase significantly as the size of the nanoparticle is increased, and this is attributed to the diffusion rates of proximal water molecules around the particulate contrast enhancement agents.92 There are also some examples of small clusters of iron oxide nanoparticles in core–shell arrangements where the core is relatively hydrophilic that can display very high r2 relaxivities,93 but this is not well understood theoretically. This is the case with the cisplatin-loaded MNCs reported herein. It is believed that access of water molecules around the iron oxide nanoparticles in the cores of these MNC clusters has been changed as the cisplatin was added, and that the change results in the observed increased r2 values in a rather uniform manner. Such alterations would require considerably more work to understand these structure–property relationships. This effect, nonetheless, could play a positive role in theranostic applications using cisplatin-loaded MNCs.
CONCLUSION
We have produced stable MNCs by reacting magnetite MNPs with the biodegradable PLE50-b-PEG113 block copolymer having ALN anchor groups attached to some of the carboxylic groups of the PLE via polymer-analogous modification. The resulting MNCs were loaded with cisplatin using remaining carboxylic and bisphosphonate groups of the PLE. The MNCs were decorated with the LHRH as the targeting groups to enhance delivery of the nanoformulations to ovarian cancer cells overexpressing LHRHr. The LHRH modification enhanced uptake of the MNCs, and their drug and MNP payload in the wild-type as well as cisplatin-resistant ovarian cancer cells. Enhanced uptake further translated into superior MRI contrast and significant enhancement in cytotoxicity of the nanoformulated drug. Thus, theranostic nanoformulations were developed here that can be applied for simultaneous MR imaging and targeted drug delivery in cancer cells. The encouraging in vitro results certainly warrant further investigation of the in vivo efficacy of the proposed theranostic nanosystem.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the innovative Research in Cancer Nanotechnology (IRCN) Grant (U01CA198910-01) of the National Cancer Institute Alliance for Nanotechnology in Cancer and the Carolina Partnership, a strategic partnership between the UNC Eshelman School of Pharmacy and The University Cancer Research Fund through the Lineberger Comprehensive Cancer Center.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemmater.6b00197.
Heating patterns for Figure 1, physicochemical characterization of MNCs stabilized by homopolymers, synthetic scheme for ALN conjugation, 1H NMR and 31P NMR of ALN-modified PLE50-b-PEG113, quantification of ALN conjugation by NMR, colloidal stability of A6-MNC and A12-MNC, Relaxation measurements at 7T in water, FTIR spectra of LHRH-conjugated MNCs, inhibition of in vitro intracellular uptake in A2780-WT and A2780-CisR cells, and formulation toxicity and anticancer activity of targeted and untargeted cisplatin loaded MNCs (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Sershen S; Westcott S; Halas N; West J Temperature-sensitive polymer–nanoshell composites for photothermally modulated drug delivery. J. Biomed. Mater. Res 2000, 51, 293–298. [DOI] [PubMed] [Google Scholar]
- (2).Na K; Lee KH; Lee DH; Bae YH Biodegradable thermo-sensitive nanoparticles from poly (L-lactic acid)/poly (ethylene glycol) alternating multi-block copolymer for potential anti-cancer drug carrier. Eur. J. Pharm. Sci 2006, 27, 115–122. [DOI] [PubMed] [Google Scholar]
- (3).Nuopponen M; Tenhu H Gold nanoparticles protected with pH and temperature-sensitive diblock copolymers. Langmuir 2007, 23, 5352–5357. [DOI] [PubMed] [Google Scholar]
- (4).Cheng R; Meng F; Deng C; Klok H-A; Zhong Z Dual and multi-stimuli responsive polymeric nanoparticles for programmed site-specific drug delivery. Biomaterials 2013, 34, 3647–3657. [DOI] [PubMed] [Google Scholar]
- (5).Li J; Huo M; Wang J; Zhou J; Mohammad JM; Zhang Y; Zhu Q; Waddad AY; Zhang Q Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials 2012, 33, 2310–2320. [DOI] [PubMed] [Google Scholar]
- (6).Nguyen DH; Choi JH; Joung YK; Park KD Disulfide-crosslinked heparin-pluronic nanogels as a redox-sensitive nanocarrier for intracellular protein delivery. J. Bioact. Compat. Polym 2011, 26, 287–300. [Google Scholar]
- (7).Reiss G; Hütten A Magnetic nanoparticles: applications beyond data storage. Nat. Mater 2005, 4, 725–726. [DOI] [PubMed] [Google Scholar]
- (8).Perez JM; Simeone FJ; Saeki Y; Josephson L; Weissleder R Viral-induced self-assembly of magnetic nanoparticles allows the detection of viral particles in biological media. J. Am. Chem. Soc 2003, 125, 10192–10193. [DOI] [PubMed] [Google Scholar]
- (9).Wang L; Yang Z; Gao J; Xu K; Gu H; Zhang B; Zhang X; Xu B A biocompatible method of decorporation: bisphosphonate-modified magnetite nanoparticles to remove uranyl ions from blood. J. Am. Chem. Soc 2006, 128, 13358–13359. [DOI] [PubMed] [Google Scholar]
- (10).Gu H; Ho P-L; Tsang KW; Wang L; Xu B Using biofunctional magnetic nanoparticles to capture vancomycin-resistant enterococci and other gram-positive bacteria at ultralow concentration. J. Am. Chem. Soc 2003, 125, 15702–15703. [DOI] [PubMed] [Google Scholar]
- (11).Tombach B; Reimer P; Bremer C; Allkemper T; Engelhardt M; Mahler M; Ebert W; Heindel W First-pass and equilibrium-MRA of the aortoiliac region with a superparamagnetic iron oxide blood pool MR contrast agent (SH U 555 C): results of a human pilot study. NMR Biomed 2004, 17, 500–506. [DOI] [PubMed] [Google Scholar]
- (12).Allkemper T; Bremer C; Matuszewski L; Ebert W; Reimer P Contrast-enhanced Blood-Pool MR Angiography with Optimized Iron Oxides: Effect of Size and Dose on Vascular Contrast Enhancement in Rabbits. Radiology 2002, 223, 432–438. [DOI] [PubMed] [Google Scholar]
- (13).Taylor AM; Panting JR; Keegan J; Gatehouse PD; Amin D; Jhooti P; Yang GZ; McGill S; Burman ED; Francis JM; Firmin DN; Pennell DJ Safety and preliminary findings with the intravascular contrast agent NC100150 injection for MR coronary angiography. J. Magn. Reson. Imaging 1999, 9, 220–227. [DOI] [PubMed] [Google Scholar]
- (14).Enochs WS; Harsh G; Hochberg F; Weissleder R Improved delineation of human brain tumors on MR images using a long-circulating, superparamagnetic iron oxide agent. J. Magn. Reson. Imaging 1999, 9, 228–232. [DOI] [PubMed] [Google Scholar]
- (15).Yu MK; Jeong YY; Park J; Park S; Kim JW; Min JJ; Kim K; Jon S Drug-Loaded Superparamagnetic Iron Oxide Nanoparticles for Combined Cancer Imaging and Therapy In Vivo. Angew. Chem., Int. Ed 2008, 47, 5362–5365. [DOI] [PubMed] [Google Scholar]
- (16).Golovin YI; Gribanovsky SL; Golovin DY; Klyachko NL; Majouga AG; Master AM; Sokolsky M; Kabanov AV Towards nanomedicines of the future: Remote magneto-mechanical actuation of nanomedicines by alternating magnetic fields. J. Controlled Release 2015, 219, 43–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Singh D; McMillan JM; Liu X-M; Vishwasrao HM; Kabanov AV; Sokolsky-Papkov M; Gendelman HE Formulation design facilitates magnetic nanoparticle delivery to diseased cells and tissues. Nanomedicine 2014, 9, 469–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sanson C; Diou O; Thevenot J; Ibarboure E; Soum A; Brûlet A; Miraux S; Thiaudière E; Tan S; Brisson A; et al. Doxorubicin loaded magnetic polymersomes: theranostic nanocarriers for MR imaging and magneto-chemotherapy. ACS Nano 2011, 5, 1122–1140. [DOI] [PubMed] [Google Scholar]
- (19).Jain TK; Richey J; Strand M; Leslie-Pelecky DL; Flask CA; Labhasetwar V Magnetic nanoparticles with dual functional properties: drug delivery and magnetic resonance imaging. Biomaterials 2008, 29, 4012–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pinna N; Grancharov S; Beato P; Bonville P; Antonietti M; Niederberger M Magnetite Nanocrystals: Nonaqueous Synthesis, Characterization, and Solubility†. Chem. Mater 2005, 17, 3044–3049. [Google Scholar]
- (21).Edmondson JM; Armstrong LS; Martinez AO A rapid and simple MTT-based spectrophotometric assay for determining drug sensitivity in monolayer cultures. J. Tissue Cult. Methods 1988, 11, 15–17. [Google Scholar]
- (22).Cole S Rapid chemosensitivity testing of human lung tumor cells using the MTT assay. Cancer Chemother. Pharmacol 1986, 17, 259–263. [DOI] [PubMed] [Google Scholar]
- (23).Goff J; Huffstetler P; Miles W; Pothayee N; Reinholz C; Ball S; Davis R; Riffle J Novel phosphonate-functional poly (ethylene oxide)-magnetite nanoparticles form stable colloidal dispersions in phosphate-buffered saline. Chem. Mater 2009, 21, 4784–4795. [Google Scholar]
- (24).Sun S; Murray C; Weller D; Folks L; Moser A Monodisperse FePt nanoparticles and ferromagnetic FePt nanocrystal superlattices. Science 2000, 287, 1989–1992. [DOI] [PubMed] [Google Scholar]
- (25).Shevchenko EV; Talapin DV; Rogach AL; Kornowski A; Haase M; Weller H Colloidal Synthesis and Self-Assembly of CoPt3 Nanocrystals. J. Am. Chem. Soc 2002, 124, 13958–13958. [DOI] [PubMed] [Google Scholar]
- (26).Jang J-H; Lee E; Park J; Kim G; Hong S; Kwon Y-U Rational syntheses of core-shell Fex@Pt nanoparticles for the study of electrocatalytic oxygen reduction reaction. Sci. Rep 2013, 3, 2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kanczler JM; Sura HS; Magnay J; Green D; Oreffo RO; Dobson JP; El Haj AJ Controlled differentiation of human bone marrow stromal cells using magnetic nanoparticle technology. Tissue Eng., Part A 2010, 16, 3241–3250. [DOI] [PubMed] [Google Scholar]
- (28).Herzer G Grain structure and magnetism of nanocrystalline ferromagnets. IEEE Trans. Magn 1989, 25, 3327–3329. [Google Scholar]
- (29).Yoon TJ; Lee H; Shao H; Weissleder R Highly magnetic core–shell nanoparticles with a unique magnetization mechanism. Angew. Chem., Int. Ed 2011, 50, 4663–4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lu AH; Salabas E. e. L.; Schüth F Magnetic nanoparticles: synthesis, protection, functionalization, and application. Angew. Chem., Int. Ed 2007, 46, 1222–1244. [DOI] [PubMed] [Google Scholar]
- (31).Taqaddas A Use of Magnetic Nanoparticles in Cancer Detection with MRI. Int. J. Med.l, Health, Biomed.l, Bioeng. Pharm. Eng 2014, 8, 616–624. [Google Scholar]
- (32).Koo Y-EL; Reddy GR; Bhojani M; Schneider R; Philbert MA; Rehemtulla A; Ross BD; Kopelman R Brain cancer diagnosis and therapy with nanoplatforms. Adv. Drug Delivery Rev 2006, 58, 1556–1577. [DOI] [PubMed] [Google Scholar]
- (33).Stephen ZR; Kievit FM; Zhang M Magnetite nanoparticles for medical MR imaging. Mater. Today 2011, 14, 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Owens DE; Peppas NA Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm 2006, 307, 93–102. [DOI] [PubMed] [Google Scholar]
- (35).Otsuka H; Nagasaki Y; Kataoka K PEGylated nanoparticles for biological and pharmaceutical applications. Adv. Drug Delivery Rev 2012, 64, 246–255. [DOI] [PubMed] [Google Scholar]
- (36).Li C Poly(l-glutamic acid)–anticancer drug conjugates. Adv. Drug Delivery Rev 2002, 54, 695–713. [DOI] [PubMed] [Google Scholar]
- (37).Kishore B; Lambricht P; Laurent G; Maldague P; Wagner R; Tulkens PM Mechanism of protection afforded by polyaspartic acid against gentamicin-induced phospholipidosis. II. Comparative in vitro and in vivo studies with poly-L-aspartic, poly-L-glutamic and poly-D-glutamic acids. J. Pharmacol. Exp. Ther 1990, 255, 875–885. [PubMed] [Google Scholar]
- (38).Sajadi S Metal ion-binding properties of L-glutamic acid and L-aspartic acid, a comparative investigation. Nat. Sci 2010, 2, 85. [Google Scholar]
- (39).Xu C; Xu K; Gu H; Zheng R; Liu H; Zhang X; Guo Z; Xu B Dopamine as a robust anchor to immobilize functional molecules on the iron oxide shell of magnetic nanoparticles. J. Am. Chem. Soc 2004, 126, 9938–9939. [DOI] [PubMed] [Google Scholar]
- (40).Frey NA; Peng S; Cheng K; Sun S Magnetic nanoparticles: synthesis, functionalization, and applications in bioimaging and magnetic energy storage. Chem. Soc. Rev 2009, 38, 2532–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Xie J; Liu G; Eden HS; Ai H; Chen X Surface-engineered magnetic nanoparticle platforms for cancer imaging and therapy. Acc. Chem. Res 2011, 44, 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Pothayee N; Balasubramaniam S; Davis RM; Riffle JS; Carroll MRJ; Woodward RC; St. Pierre TG Synthesis of ‘ready-to-adsorb’ polymeric nanoshells for magnetic iron oxide nanoparticles via atom transfer radical polymerization. Polymer 2011, 52, 1356–1366. [Google Scholar]
- (43).Nukolova NV; Oberoi HS; Cohen SM; Kabanov AV; Bronich TK Folate-decorated nanogels for targeted therapy of ovarian cancer. Biomaterials 2011, 32, 5417–5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kim JO; Kabanov AV; Bronich TK Polymer micelles with cross-linked polyanion core for delivery of a cationic drug doxorubicin. J. Controlled Release 2009, 138, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Xue Z; Lin M; Zhu J; Zhang J; Li Y; Guo Z Platinum (II) compounds bearing bone-targeting group: synthesis, crystal structure and antitumor activity. Chem. Commun 2010, 46, 1212–1214. [DOI] [PubMed] [Google Scholar]
- (46).Markovsky E; Koroukhov N; Golomb G Additive-free albumin nanoparticles of alendronate for attenuating inflammation through monocyte inhibition. Nanomedicine 2007, 2, 545–553. [DOI] [PubMed] [Google Scholar]
- (47).Na HB; Song IC; Hyeon T Inorganic nanoparticles for MRI contrast agents. Adv. Mater 2009, 21, 2133–2148. [Google Scholar]
- (48).Strijkers GJ; Mulder M; Willem J; van Tilborg F; Geralda A; Nicolay K MRI contrast agents: current status and future perspectives. Anti-Cancer Agents Med. Chem 2007, 7, 291–305. [DOI] [PubMed] [Google Scholar]
- (49).Vre RM-D; Grimee R; Parmentier F; Binet J The use of agar gel as a basic reference material for calibrating relaxation times and imaging parameters. Magn. Reson. Med 1985, 2, 176–179. [DOI] [PubMed] [Google Scholar]
- (50).Nukolova NV; Oberoi HS; Zhao Y; Chekhonin VP; Kabanov AV; Bronich TK LHRH-Targeted Nanogels as a Delivery System for Cisplatin to Ovarian Cancer. Mol. Pharmaceutics 2013, 10, 3913–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Bi X; Shi X; Baker JR Synthesis, characterization and stability of a luteinizing hormone-releasing hormone (LHRH)-functionalized poly (amidoamine) dendrimer conjugate. J. Biomater. Sci., Polym. Ed 2008, 19, 131–142. [DOI] [PubMed] [Google Scholar]
- (52).Dharap S; Wang Y; Chandna P; Khandare J; Qiu B; Gunaseelan S; Sinko P; Stein S; Farmanfarmaian A; Minko T Tumor-specific targeting of an anticancer drug delivery system by LHRH peptide. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 12962–12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Dharap S; Qiu B; Williams G; Sinko P; Stein S; Minko T Molecular targeting of drug delivery systems to ovarian cancer by BH3 and LHRH peptides. J. Controlled Release 2003, 91, 61–73. [DOI] [PubMed] [Google Scholar]
- (54).Chauhan S; Liang X; Su A; Pai-Panandiker A; Shen D; Hanover J; Gottesman M Reduced endocytosis and altered lysosome function in cisplatin-resistant cell lines. Br. J. Cancer 2003, 88, 1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Desale SS; Cohen SM; Zhao Y; Kabanov AV; Bronich TK Biodegradable hybrid polymer micelles for combination drug therapy in ovarian cancer. J. Controlled Release 2013, 171, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Oberoi HS; Nukolova NV; Kabanov AV; Bronich TK Nanocarriers for delivery of platinum anticancer drugs. Adv. Drug Delivery Rev 2013, 65, 1667–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Maaz K; Mumtaz A; Hasanain S; Ceylan A Synthesis and magnetic properties of cobalt ferrite (CoFe 2 O 4) nanoparticles prepared by wet chemical route. J. Magn. Magn. Mater 2007, 308, 289–295. [Google Scholar]
- (58).Tung LD; Kolesnichenko V; Caruntu G; Caruntu D; Remond Y; Golub V; O’connor C; Spinu L Annealing effects on the magnetic properties of nanocrystalline zinc ferrite. Phys. B 2002, 319, 116–121. [Google Scholar]
- (59).Sun S; Zeng H Size-controlled synthesis of magnetite nanoparticles. J. Am. Chem. Soc 2002, 124, 8204–8205. [DOI] [PubMed] [Google Scholar]
- (60).Miles WC; Huffstetler PP; Goff JD; Chen AY; Riffle J; Davis RM Design of Stable Polyether– Magnetite Complexes in Aqueous Media: Effects of the Anchor Group, Molecular Weight, and Chain Density. Langmuir 2011, 27, 5456–5463. [DOI] [PubMed] [Google Scholar]
- (61).Gyenes T; Torma V; Gyarmati B; Zrínyi M Synthesis and swelling properties of novel pH-sensitive poly (aspartic acid) gels. Acta Biomater 2008, 4, 733–744. [DOI] [PubMed] [Google Scholar]
- (62).Gonzales D; Fan K; Sevoian M Synthesis and swelling characterizations of a poly (gamma-glutamic acid) hydrogel. J. Polym. Sci., Part A: Polym. Chem 1996, 34, 2019–2027. [Google Scholar]
- (63).Markland P; Zhang Y; Amidon GL; Yang VC A pH-and ionic strength-responsive polypeptide hydrogel: synthesis, characterization, and preliminary protein release studies. J. Biomed. Mater. Res 1999, 47, 595–602. [DOI] [PubMed] [Google Scholar]
- (64).Desale SS; Soni KS; Romanova S; Cohen SM; Bronich TK Targeted delivery of platinum-taxane combination therapy in ovarian cancer. J. Controlled Release 2015, 220, 651–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Pothayee N; Pothayee N; Jain N; Hu N; Balasubramaniam S; Johnson LM; Davis RM; Sriranganathan N; Riffle J Magnetic block ionomer complexes for potential dual imaging and therapeutic agents. Chem. Mater 2012, 24, 2056–2063. [Google Scholar]
- (66).Jayson GC; Kohn EC; Kitchener HC; Ledermann JA Ovarian cancer. Lancet 2014, 384, 1376–1388. [DOI] [PubMed] [Google Scholar]
- (67).Florea A-M; Büsselberg D Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Emons G; Pahwa GS; Brack C; Sturm R; Oberheuser F; Knuppen R Gonadotropin releasing hormone binding sites in human epithelial ovarian carcinomata. Eur. J. Cancer Clin. Oncol 1989, 25, 215–221. [DOI] [PubMed] [Google Scholar]
- (69).Ohno T; Imai A; Furui T; Takahashi K; Tamaya T Presence of gonadotropin-releasing hormone and its messenger ribonucleic acid in human ovarian epithelial carcinoma. Am. J. Obstet. Gynecol 1993, 169, 605–610. [DOI] [PubMed] [Google Scholar]
- (70).Srkalovic G; Schally A; Wittliff J; Day T; Jenison E Presence and characteristics of receptors for [D-Trp6] luteinizing hormone releasing hormone and epidermal growth factor in human ovarian cancer. Int. J. Oncol 1998, 12, 489–587. [DOI] [PubMed] [Google Scholar]
- (71).Kumar CS; Leuschner C; Urbina M; Ozkaya T; Hormes J Glutaric acid as a spacer facilitates improved intracellular uptake of LHRH–SPION into human breast cancer cells. Int. J. Nanomed 2007, 2, 175. [PMC free article] [PubMed] [Google Scholar]
- (72).Meng J; Fan J; Galiana G; Branca R; Clasen P; Ma S; Zhou J; Leuschner C; Kumar C; Hormes J; et al. LHRH-functionalized superparamagnetic iron oxide nanoparticles for breast cancer targeting and contrast enhancement in MRI. Mater. Sci. Eng., C 2009, 29, 1467–1479. [Google Scholar]
- (73).Minko T; Patil ML; Zhang M; Khandare JJ; Saad M; Chandna P; Taratula O LHRH-targeted nanoparticles for cancer therapeutics. In Cancer Nanotechnology; Springer: 2010; pp 281–294. [DOI] [PubMed] [Google Scholar]
- (74).Taheri A; Dinarvand R; Atyabi F; Ahadi F; Nouri FS; Ghahremani MH; Ostad SN; Borougeni AT; Mansoori P Enhanced anti-tumoral activity of methotrexate-human serum albumin conjugated nanoparticles by targeting with luteinizing hormone-releasing hormone (LHRH) peptide. Int. J. Mol. Sci 2011, 12, 4591–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Pinto AL; Lippard SJ Binding of the antitumor drug cis-diamminedichloroplatinum(II) (cisplatin) to DNA. Biochim. Biophys. Acta, Rev. Cancer 1985, 780, 167–180. [DOI] [PubMed] [Google Scholar]
- (76).Jung Y; Lippard SJ Direct Cellular Responses to Platinum-Induced DNA Damage. Chem. Rev 2007, 107, 1387–1407. [DOI] [PubMed] [Google Scholar]
- (77).Cole AJ; Yang VC; David AE Cancer theranostics: the rise of targeted magnetic nanoparticles. Trends Biotechnol 2011, 29, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Singh A; Sahoo SK Magnetic nanoparticles: a novel platform for cancer theranostics. Drug Discovery Today 2014, 19, 474–481. [DOI] [PubMed] [Google Scholar]
- (79).Carroll MR; Woodward RC; House MJ; Teoh WY; Amal R; Hanley TL; St Pierre TG Experimental validation of proton transverse relaxivity models for superparamagnetic nanoparticle MRI contrast agents. Nanotechnology 2010, 21, 035103. [DOI] [PubMed] [Google Scholar]
- (80).Carroll MR; Huffstetler PP; Miles WC; Goff JD; Davis RM; Riffle JS; House MJ; Woodward RC; St Pierre TG The effect of polymer coatings on proton transverse relaxivities of aqueous suspensions of magnetic nanoparticles. Nanotechnology 2011, 22, 325702. [DOI] [PubMed] [Google Scholar]
- (81).Roch A; Gossuin Y; Muller RN; Gillis P Superparamagnetic colloid suspensions: water magnetic relaxation and clustering. J. Magn. Magn. Mater 2005, 293, 532–539. [Google Scholar]
- (82).Matsumoto Y; Jasanoff AT 2 relaxation induced by clusters of superparamagnetic nanoparticles: Monte Carlo simulations. Magn. Reson. Imaging 2008, 26, 994–998. [DOI] [PubMed] [Google Scholar]
- (83).Zhang Q; Thompson MS; Carmichael-Baranauskas AY; Caba BL; Zalich MA; Lin Y-N; Mefford OT; Davis RM; Riffle JS Aqueous Dispersions of Magnetite Nanoparticles Complexed with Copolyether Dispersants: Experiments and Theory. Langmuir 2007, 23, 6927–6936. [DOI] [PubMed] [Google Scholar]
- (84).Ge J; Hu Y; Biasini M; Beyermann WP; Yin Y Superparamagnetic Magnetite Colloidal Nanocrystal Clusters. Angew. Chem., Int. Ed 2007, 46, 4342–4345. [DOI] [PubMed] [Google Scholar]
- (85).Lee N; Choi Y; Lee Y; Park M; Moon WK; Choi SH; Hyeon T Water-Dispersible Ferrimagnetic Iron Oxide Nanocubes with Extremely High r2 Relaxivity for Highly Sensitive in Vivo MRI of Tumors. Nano Lett 2012, 12, 3127–3131. [DOI] [PubMed] [Google Scholar]
- (86).Koppolu B; Bhavsar Z; Wadajkar AS; Nattama S; Rahimi M; Nwariaku F; Nguyen KT Temperature-sensitive polymer-coated magnetic nanoparticles as a potential drug delivery system for targeted therapy of thyroid cancer. J. Biomed. Nanotechnol 2012, 8, 983–990. [DOI] [PubMed] [Google Scholar]
- (87).Brown KA; Vassiliou CC; Issadore D; Berezovsky J; Cima MJ; Westervelt RM Scaling of transverse nuclear magnetic relaxation due to magnetic nanoparticle aggregation. J. Magn. Magn. Mater 2010, 322, 3122–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Berret J-F; Schonbeck N; Gazeau F; El Kharrat D; Sandre O; Vacher A; Airiau M Controlled clustering of superparamagnetic nanoparticles using block copolymers: design of new contrast agents for magnetic resonance imaging. J. Am. Chem. Soc 2006, 128, 1755–1761. [DOI] [PubMed] [Google Scholar]
- (89).Neuwelt EA; Hamilton BE; Varallyay CG; Rooney WR; Edelman RD; Jacobs PM; Watnick SG Ultrasmall superparamagnetic iron oxides (USPIOs): a future alternative magnetic resonance (MR) contrast agent for patients at risk for nephrogenic systemic fibrosis (NSF)? Kidney Int 2009, 75, 465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Chen S; Wang L; Duce SL; Brown S; Lee S; Melzer A; Cuschieri SA; André P Engineered biocompatible nanoparticles for in vivo imaging applications. J. Am. Chem. Soc 2010, 132, 15022–15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Taylor RM; Huber DL; Monson TC; Esch V; Sillerud LO Structural and magnetic characterization of superparamagnetic iron platinum nanoparticle contrast agents for magnetic resonance imaging. J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom 2012, 30, 02C101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Balasubramaniam S; Kayandan S; Lin Y-N; Kelly DF; House MJ; Woodward RC; St. Pierre TG; Riffle JS; Davis RM Toward design of magnetic nanoparticle clusters stabilized by biocompatible diblock copolymers for T 2-weighted MRI contrast. Langmuir 2014, 30, 1580–1587. [DOI] [PubMed] [Google Scholar]
- (93).Pothayee N; Balasubramaniam S; Pothayee N; Jain N; Hu N; Lin Y; Davis RM; Sriranganathan N; Koretsky AP; Riffle J Magnetic nanoclusters with hydrophilic spacing for dual drug delivery and sensitive magnetic resonance imaging. J. Mater. Chem. B 2013, 1, 1142–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.