

Abstract
Co-exposure to tobacco and marijuana has become common in areas where recreational marijuana use is legal. To assist in the determination of the combined health risks of this co-exposure, an analytical method capable of simultaneously measuring tobacco and marijuana metabolites is needed to reduce laboratory costs and the required sample volume. So far, no such analytical method exists. Thus, we developed and validated a method to simultaneously quantify urinary levels of trans-3′-hydroxycotinine (3OH-COT), cotinine (COT), and 11-nor-9-carboxy-Δ9-tetrahydrocannabinol (COOH-THC) to assess co-exposure to tobacco and marijuana. Urine (200 μL) was spiked with labelled internal standards and enzymatically hydrolyzed to liberate the conjugated analytes before extraction using solid-supported liquid-liquid extraction (SLE) with ethyl acetate serving as an eluent. The target analytes were separated on a C18 (4.6 × 100 mm, 5 μm) analytical column with a gradient mobile phase elution and analyzed using tandem mass spectrometry with multiple reaction monitoring of target ion transitions. Positive electrospray ionization (ESI) was used for 3OH-COT and COT, while negative ESI was used for COOH-THC. The total run time was 13 min. The extraction recoveries were 18.4–23.9 % (3OH-COT), 65.1–96.8 % (COT), and 80.6–95.4 % (COOH-THC). The method limits of quantification were 5.0 ng/mL (3OH-COT) and 2.5 ng/mL (COT and COOH-THC). The method showed good accuracy (82.5–98.5 %) and precision (1.22–6.21 % within-day precision and 1.42–6.26 % between-day precision). The target analytes were stable for at least 144 h inside the autosampler (10 °C). The analyses of reference materials and 146 urine samples demonstrated good method performance. The use of a 96-well plate for preparation makes the method useful for the analysis of large numbers of samples.
Keywords: Trans-3’-hydroxycotinine, Cotinine, 11-nor-9-carboxy-delta 9-tetrahydrocannabinol, Supported liquid extraction, Tobacco metabolites, Marijuana metabolites
1. Introduction
Tobacco and cannabis (marijuana) are among the world’s most used psychoactive substances. A common method for consumption of these substances is smoking [1]. Their smoke contains hundreds of chemical constituents, including carbon monoxide, volatile organic chemicals, polycyclic aromatic hydrocarbons, and alkaloids or cannabinoids [2]. In addition to smoking, smokeless consumption of tobacco (e.g., chewing tobacco, snuff, and dissolvable tobacco products) and marijuana (e.g., marijuana edibles) is increasingly common [3,4]. Because alkaloids in tobacco plants and cannabinoids in cannabis plants are present in large amounts [3,5], regardless of how they are consumed, measuring alkaloid- or cannabinoid-derived chemical markers in biospecimens is a typical way to assess human exposure to tobacco and marijuana smoke or products.
Nicotine is the principal tobacco alkaloid, accounting for 1.5 % (wt) of tobacco in cigarettes from the United States (US) [6]. Approximately 95 % of the alkaloid fraction is nicotine. Upon entering the human body, 70–80 % of nicotine is metabolized to cotinine (COT) and then to trans-3′-hydroxycotinine (3OH-COT) [3]. Both compounds are conjugated and excreted as glucuronide-bound metabolites in urine. Typically, COT glucuronide and 3OH-COT glucuronide account for 12–17 % and 7–9 % of the nicotine dose, respectively. The free form COT and 3OH-COT accoimt for 10–15 % and 33–40 %, respectively [7]. The half-lives of 3OH-COT and COT in urine are about 6 and 16 h, respectively [3,8]. Although ethnic differences affect the clearance rate of cotinine [9], COT and 3OH-COT have commonly been used as biomarkers of tobacco smoke exposure [3,10]. Among marijuana cannabinoid compounds, Δ9-tetrahydrocannabinol (Δ9-THC) is a major constituent and provides psychoactive effects [11,12]. The average amount of Δ9-THC in dried marijuana has increased from 6.0 % in 2008 to 9.4 % in 2017 (wt) [5]. Δ9-THC is metabolized to the psychoactive 11-hydroxy-THC (OH-THC), which subsequently oxidizes to form the inactive 11-nor-9-carboxy-Δ9-tetrahydrocannabinol (COOH-THC). For a given dose, about 80–90 % of Δ9-THC is metabolized and excreted alongside its metabolites within 5 days in feces (>65 %) and urine (20 %) [13,14]. Among the major metabolites, OH-THC is found predominantly in feces, whereas COOH-THC glucuronide is a primary metabolite in urine. Δ9-THC has an elimination half-life of 1–3 days in occasional users and 5–13 days in chronic users [12,13,15]. COOH-THC is regarded commonly as the most suitable biomarker for determining human exposure to marijuana smoke [11,16].
Co-use of tobacco and marijuana products via smoking is common [17,18]. In the US, ~5.2 % of adults who participated in the 2011–2012 National Survey on Drug Use and Health (NSDUH) reported co-use of tobacco and marijuana. The US prevalence of co-use has increased, with a higher prevalence found in specific demographic groups (i.e., males 26–34 years, and African Americans) [18]. Among US youth (12–17 years old), 5.4 % were reported as tobacco and marijuana co-users in the 2013–2014 NSDUH [19]. Cohn, et al., reported ~ 21 % of US adult participants aged 18–24 years in their study were tobacco and marijuana co-users [20]. Co-use of tobacco and marijuana products increases the risk of addiction and mental illnesses [21]. In addition, co-use of tobacco and marijuana products during pregnancy was associated with smaller head circumference and higher occurrence of birth defects among newborns [22]. Another study found that infants born from co-using mothers had lower self-regulation and attention scores on the Neonatal Intensive Care Unit Network Neurobehavioral Scale [23]. Because tobacco smokers who increased their marijuana use had poorer smoking cessation outcomes than non-cannabis using smokers [24], co-users of tobacco and marijuana products are more likely to experience long-term health effects from both substances.
Many epidemiologic studies failed to assess the health effects of tobacco and marijuana co-exposure at the population level because they relied on self-reported exposure data [25]. To assess the exposure magnitude of tobacco-marijuana smoke accurately, specific biomarkers must be measured in the same biological matrices. Traditionally, this was accomplished by analyzing either tobacco or marijuana biomarkers separately using a different method in a given biospecimen. Several methods have been developed for the analysis of these biomarkers in urine and serum [10,16,26,27]. Currently, no reported analytical method exists that is capable of measuring tobacco and marijuana biomarkers together in any biological samples. Running separate analytical methods to measure tobacco and marijuana biomarkers has several disadvantages as it increases laboratory and labor costs, is time-consuming, and requires more sample volume. Thus, analytical methods that are able to measure tobacco- and marijuana- specific biomarkers simultaneously in one biospecimen are advantageous. Such methods can offer high-throughput analysis and will be useful for epidemiologic investigations where many samples must be analyzed in a short timeframe.
This study aimed to develop an analytical method for the simultaneous quantification of urinary 3OH-COT, COT, and COOH-THC and demonstrate its utility using samples from an African American maternal-child cohort in Atlanta. The method uses liquid chromatography coupled with electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) and utilizes a rapid and efficient extraction procedure for measuring these biomarkers in samples with good precision, accuracy, and efficiency.
2. Materials and methods
2.1. Chemicals and reagents
3OH-COT (1.0 mg/mL in methanol), COT (1.0 mg/mL in methanol), COOH-THC (1.0 mg/mL in methanol), 3OH-COT-D3 (100 μg/mL in methanol), and COOH-THC-D9 (100 μg/mL in methanol) were purchased from Cerilliant (Round Rock, Texas, USA). Isotopically labeled COT (2′,3′,4′−13C3) (100 μg/mL in water) was purchased from Cambridge Isotope Laboratories (Tewksbury, MA, USA).
β-glucuronidase/sulfatase (enzyme) from Helix pomatia, H1 was purchased from Sigma Life Sciences (St. Louis, MO, USA). Ammonium acetate was purchased from Alfa Aesar (Haverhill, MA, USA). Glacial acetic acid was obtained from Mallinckrodt (United Kingdom). HPLC grade ethyl acetate and methanol were purchased from Thermo Fisher Scientific (Waltham, MA, USA). HPLC-grade acetonitrile was purchased from Honeywell Burdick & Jackson (Muskegon, MI, USA). Water was purified using an EMD Millipore Milli-Q Ultrapure water system (Burlington, MA, USA). Standard reference materials (SRM 3672, 3673, and 1507b) were purchased from the National Institute of Standards and Technology (NIST) (Gaithersburg, MD, USA). Proficiency testing materials were purchased from the German External Quality Assessment Scheme (G-EQUAS materials 18A and 18B) (Erlangen, Germany).
2.2. Enzymatic digestion procedure
Enzyme solution was prepared by adding enzyme to 0.5 M ammonium acetate buffer (pH 5.1) solution to yield a concentration of 20,000 units/mL. A 100 μL aliquot of this enzyme solution was added to the samples before incubating at 37 °C overnight to deconjugate the target analytes. The enzymatic digestion procedure was performed according to the method of McGuffey, et al. [28], with a slight modification of the enzyme concentration (2000 units in our study vs 1600 units per sample).
2.3. Preparation of calibration, quality control, and labeled internal standard solutions
Calibration solutions were prepared by mixing stock solutions of the target analytes and diluting with methanol:Milli-Q water (40:60, v/v) to yield standard solutions with the following concentrations: 20,000, 10,000, 5,000, 2,000, 1,000, 500, 200, 100, 50, 20,10, 5, and 2 ng/mL. Quality control (QC) solutions were prepared by mixing stock solutions of the target analytes and diluting with methanol:Milli-Q water (40:60, v/v) to yield four QC solutions at 8,000, 5,000, 500, and 50 ng/mL for 3OH-COT and COT and at 16,000, 5,000, 500, and 50 ng/mL for COOH-THC, respectively. Labeled internal standard (IS) solution was prepared by mixing labeled analogue stock solutions and diluting with methanol: Milli-Q water (40:60, v/v) to yield a labeled IS solution (concentration: 2,000 ng/mL).
2.4. Collection of non-smoker urine
Urine samples were anonymously collected front self-identified non-smoking individuals, pooled, and used as a matrix for calibrants, QCs, and matrix blanks. The pooled non-smoker urine was screened to confirm the absence of the target analytes using the developed method.
2.5. Preparation of blanks, calibrants, and quality control samples
To prepare the matrix blank sample, an aliquot of 100 μL pooled non-smoker urine was mixed with 50 μL of labeled IS solution and 150 μL of Milli-Q water. For the solvent blank sample, 250 μL of Milli-Q water was mixed with 50 μL of labeled IS solution. To both blank samples, 100 μL of enzyme solution was added and mixed, resulting in a final volume of 400 μL prior to the overnight incubation.
To prepare calibrants and QC samples, 100 μL of pooled non-smoker urine was spiked with 50 μL of labeled IS solution, 100 μL of the corresponding calibration or QC solution, and 50 μL of Milli-Q water. To these samples, 100 μL of enzyme solution was added and mixed well, resulting in a total volume of 400 μL. The amounts of target analytes for all calibrants that were used during method development were: 0.20, 0.50, 1.0, 2.0, 5.0, 10, 20, 50, 100, 200, 500, 1,000, and 2,000 ng (or 1.0–10,000 ng/1 mL urine). The amounts of 3OH-COT and COT in the QC samples were: 5.0, 50, 500, and 800 ng (or 25, 250, 2,500, and 4,000 ng/1 mL urine). The amount of COOH-THC in the QC samples were: 5.0, 50, 500, and 1,600 ng (or 25, 250, 2,500, and 8,000 ng/1 mL urine). The calibrants, QC, and blank samples underwent overnight incubation at 37 °C and extraction in a matter analogous to the unknown samples as described in Section 2.6. During quantification, 1/x weighted regression models were used for all calibration curves.
2.6. Preparation and extraction of unknown samples
Urine samples were stored at −20 °C and thawed to room temperature before analysis. Unknown urine (200 μL) was mixed with 50 μL of labeled IS solution and 50 μL of Milli-Q water. All samples underwent enzymatic digestion by adding 100 μL of enzyme solution (for a final volume of 400 μL) and mixing well before incubating at 37 °C overnight.
Next, the samples were loaded onto a Novum™ simplified liquid extraction Max 96-well (SLE) plate (Phenomenex, Torrance, CA, USA). Positive pressure (16.9 kPa) was applied for 10 sec using a Presston 100 positive pressure manifold (Phenomenex). The SLE plate was left for 5 min at room temperature. Next, samples were gravity fed with 900 μL of ethyl acetate, twice. Positive pressure (16.9 kPa) was applied for 10 sec to push the remaining eluate into a collecting plate (VWR, Radnor, PA, USA) placed on the bottom of the SLE plate. The eluates were evaporated to dryness using a Glas-Col evaporator (Terre Haute, IN, USA) set to 65 °C. The samples were reconstituted with 100 μL of methanol:Milli-Q water (40:60, v/v). A flow chart of the sample preparation procedure is given in Fig. S1.
During sample analysis, 10 calibrants, one solvent blank, one matrix blank, duplicates of low-, medium-, and high-level QC samples, 78 unknown samples, and additional quality assurance samples were prepared and analyzed concurrently. The calibrants covered the same quantification range (min–max concentrations) as those used during the method validation. A total of 96 samples were prepared in each batch. If the unknown samples had concentrations higher than the highest calibration point, they were diluted with Milli-Q water using either a 1:2 v/v or 1:5 v/v ratio and re-analyzed. Final concentrations were calculated based on the dilution factor used.
2.7. Chromatographic separation and mass spectrometric conditions
A 1260 Infinity LC (Agilent Technologies, Santa Clara, CA, USA) coupled with a triple quadrupole 6460 mass spectrometer (MS) (Agilent Technologies) equipped with a jet stream ESI source was used. The autosampler was set to 10 °C. The injection volume was 5 μL. The analytical column [Kinetex® EVO C18 (100 × 4.6 mm, 5 μm) (Phenomenex)] was kept at 40 °C inside the column compartment. The mobile phase consisted of (A) 6.5 mM ammonium acetate, pH 5.1, and (B) acetonitrile. The detailed, stepwise, gradient elution program is presented in Table 1. The total runtime was 13 min. The autosampler washing program was applied to eliminate absorption of COOH-THC to the external surfaces of the needle. The autosampler washing program involved rinsing the external needle surface with a mixture of isopropanol (70 %, v/v): Milli-Q water: methanol (25:25:50, v/v/v) for 20 sec between injections.
Table 1.
Multi-stepwise gradient elution program and multiple reaction monitoring (MRM) transitions and their associated mass spectrometric parameters for analysis of target analytes.
| Gradient elution program | MRM and mass spectrometric parameters | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||
| Time, min | (A), % | (B), % | Flow rate, mL/min | Analyte | RT, min | Ion type | Ion transition | CE, eV | Dwell time, ms | Fragmentor, V |
|
| ||||||||||
| 0.01 | 95 | 5 | 0.75 | 3OH-COT | 3.1 | Q | 193.1 → 80.0 | 28 | 175 | 115 |
| 3.00 | 85 | 15 | 0.75 | C | 193.1 → 58.1 | 36 | 175 | 115 | ||
| 5.00 | 70 | 30 | 0.75 | L | 196.1 → 80.1 | 28 | 175 | 115 | ||
| 6.00 | 15 | 85 | 0.75 | COT | 4.2 | Q | 177.1 → 80.1 | 20 | 175 | 115 |
| 8.00 | 15 | 85 | 0.75 | C | 177.1 → 98.0 | 20 | 175 | 115 | ||
| 9.00 | 0 | 100 | 1.00 | L | 180.1 → 80.1 | 28 | 175 | 120 | ||
| 11.00 | 0 | 100 | 1.00 | COOH-THC | 8.1 | Q | 343.4 → 299 | 17 | 175 | 204 |
| 12.00 | 95 | 5 | 1.00 | C | 343.4 → 245 | 29 | 175 | 204 | ||
| 13.00 | 95 | 5 | 1.00 | L | 352.5 → 308 | 21 | 175 | 204 | ||
Note: (A) = 6.5 mM ammonium acetate (pH 5.1), (B) = acetonitrile, RT = retention time, CE = collision energy, 3OH-COT = trans-3′-hydroxycotinine, COT = cotinine, COOH-THC = 11-nor-9-carboxy-Δ9-tetrahydrocannabinol, Q = quantification ion, C = confirmation ion, L = labeled ion. The mass spectrometer divert valve position was switched to waste during the following time segments: 0–2.6 min, 5.0–7.5 min, 8.5–13 min.
3OH-COT and COT were analyzed in positive mode while COOH-THC was analyzed in negative mode. Polarity switching occurred at 5 min after the injection, where the ionization was changed from positive to negative mode. Multiple reaction monitoring (MRM) was used. The MRM transitions and MS parameters are summarized in Table 1. The MS conditions were set as follows: 350 °C gas temperature, 5 L/min gas flow rate, 400 °C sheath gas temperature, 10 L/min sheath gas flow rate, 310 kPa nebulizer pressure, 4,000 V capillary voltage, 800 V electron charge voltage, and 5 V collision cell voltage.
2.8. Determination of limits of quantitation
Following the US Food and Drug Administration (FDA)’s Bio-analytical Method Validation Guidance [29], the analyte limit of quantification (LOQ) was determined from repeat analysis (n = 5) of the four lowest calibrants. The lowest calibrant meeting the following criteria was set as the LOQ: the accuracy of 80–120 %, the precision of ± 20 %, and calculated concentration was ≥ 5 times than that of blank samples.
2.9. Accuracy and precision
The method accuracy and precision were assessed by analyzing replicates (n = 5) of QC samples for 5 consecutive days (n = 25/level). The accuracies were calculated by dividing the grand means of observed concentrations by the expected concentration [30] and then expressed as percentage values. Within-day and between-day precisions were calculated using the values generated from a one-way analysis of variation (ANOVA) table and expressed as relative standard deviations (% RSD). The procedure for calculating precision was based on the guidelines developed by the Scientific Working Group for Forensic Toxicology (SWGTOX) [30].
2.10. Extraction recoveries
Extraction recoveries were assessed using two sets of QC samples. The reference group was prepared by mixing pooled non-smoker urine (100 μL) with enzyme solution (100 μL) and Milli-Q water (200 μL) for a final volume of 400 μL. The reference group samples were extracted. Prior to evaporation, eluates were spiked with QC solution (100 μL) and labeled IS solution (50 μL). The experimental group was prepared by mixing pooled non-smoker urine, QC solution, and enzyme solution in the same volumes as above. Milli-Q water was added to yield a final volume of 400 μL. The experimental group samples were extracted. Before evaporation, the eluates were spiked with labeled IS solution (50 μL). All samples were analyzed together. In each set, three levels of QC samples were prepared in triplicate. The extraction recovery for each QC level was calculated by dividing the mean relative response ratio (RR) (the peak area of native analyte divided by the peak area of isotopically labeled analogue of the analyte) of the experimental group by the mean RR of the reference group and multiplying by 100.
2.11. Stability and carryover assessment
The autosampler stability of target analytes in urine extracts was evaluated. Replicates of extracted QC samples (low, medium, and high) (n = 5) were kept in the autosampler at 10 °C and re-injected using 0, 24, 50, 120, and 144 h time intervals. For each QC level, their RRs were plotted against the time intervals. The upper and lower control limits were calculated using the values at ± 15 % of the mean RR value observed at time zero.
To estimate the carryover, a solvent blank sample was injected immediately after the highest calibranl and 10 highly concentrated samples (concentration > 1000 ng/mL). The carryover was determined by dividing the concentration of the target analytes in the solvent blank sample with the concentration of the target analytes in the respective calibrant or samples and expressed as percentage values [30]. The percentage values were then averaged.
2.12. Matrix effects
The procedure for evaluation of matrix effects was based on the SWGTOX guideline [30]. To assess the matrix effects, two solvent-based QC samples (low and high levels) were prepared by mixing the corresponding QC solution (100 μL) and labeled IS solution (50 μL) together. The total volume was 150uL. These solvent-based QC samples were injected 6 times. The peak areas of the target analytes were assessed, averaged, and used as the reference for subsequent comparison with the matrix-based QC samples prepared from 10 individual urine samples. Duplicates of 10 matrix-based QC samples (total n = 20) were prepared by mixing 200 μL of urine, 100 μL of Milli-Q water, and 100 μL of enzyme solution. These samples were extracted in the manner described above (see Section 2.6). After evaporation, one set of 10 individual samples were reconstituted with the low-level QC solution (100 μL) and labeled IS solution (50 μL). Another set of 10 individual samples were reconstituted with the high- level QC solution (100 μL) and labeled IS solution (50 μL). The total volume for each matrix-based QC samples was 150 μL. All samples were injected individually. The matrix effects were calculated per target analytes using the following equation:
The same calculation was done using the mean peak area of the isotopically labeled analogue of the analyte. The matrix effects were presented as percentage values.
2.13. Dilution effects
To evaluate the effects of sample dilution, a pooled non-smoker urine sample was spiked with stock solutions of the target analytes to yield concentrations of 7,500 ng/mL for 3OH-COT and COT and 15,000 ng/mL for COOH-THC in a 10 mL solution. Then, the urine sample was diluted using a dilution ratio of 1:2 v/v and 1:5 v/v with Milli-Q water (i. e., 100 μL of urine mixed with 100 μL of Milli-Q water and 40 μL of urine mixed with 160 μL of Milli-Q water). Five replicates of diluted samples (in each dilution ratio) were analyzed for 3 consecutive days (n = 15/each dilution ratio). The accuracies and precisions of these diluted urine samples were calculated as described in Section 2.9.
2.14. Quality assurance
The method performance was assessed by repeat analysis of NIST SRM 3672 (organic contaminants in smokers’ urine), SRM 3673 (organic contaminants in non-smokers’ urine), and SRM 1507b (COOH-THC in urine, 3 levels). For COT, the method performance was also assessed by analysis of G-EQUAS materials 18A and 18B (round 64/2019, n = 4).
2.15. Method application
The method was used to analyze 146 unknown urine samples. Urine samples were collected from pregnant African American women who enrolled in the Atlanta African American Maternal-Child Cohort [31]. Participants were recruited from prenatal clinics affiliated with Grady Memorial Hospital and Emory University Hospital Midtown in Atlanta, Georgia. Urine samples were collected at clinic visits (8–14 weeks and 24–30 weeks gestation) and at home (20–24 weeks gestation) to assess environmental chemical exposures, including tobacco and marijuana smoke. The urine samples analyzed in this study were a random subset of those collected during the home visit. Socio-demographic, health survey, and clinical data were also collected. Subjects provided informed consent before data and biological sample collections. The study was approved by the Emory University Institutional Review Board and the Grady Memorial Hospital Research Oversight Committee (IRB00068441).
2.16. Data analysis
The data were processed and statistically analyzed using Microsoft Excel 2019 (Microsoft Corporation, Redmond, WA, USA). Prior to descriptive data analysis of the target analyte concentrations, the concentrations below the LOQ were assigned a value equal to the [32]. For the urinary data analysis of tobacco metabolites, the total number of samples was 146. The total number of samples for the urinary data analysis of marijuana metabolites was 145 because one subject was excluded due to missing self-report usage information. In addition, the sensitivity and specificity values of urinary 3-OH-COT, COT, and COOH-THC biomarkers were calculated using a 2×2 contingency table constructed using detection results (detected/not detected) (predictor) and self-report usage data (yes/no) (outcome).
3. Results
3.1. Sample extraction and chromatographic separation conditions
Ethyl acetate was used to extract the target analytes from the SLE sorbent. For all the concentrations tested, the extraction recoveries ranged from 18.4 to 23.9 % for 3OH-COT, 65.1 to 96.8 % for COT, and 80.6 to 95.4 % for COOH-THC (Fig. S2). Chromatographic separation was achieved. Typical chromatograms of the blanks, lowest calibrant, and unknown urine samples are shown in Fig. 1.
Fig. 1.
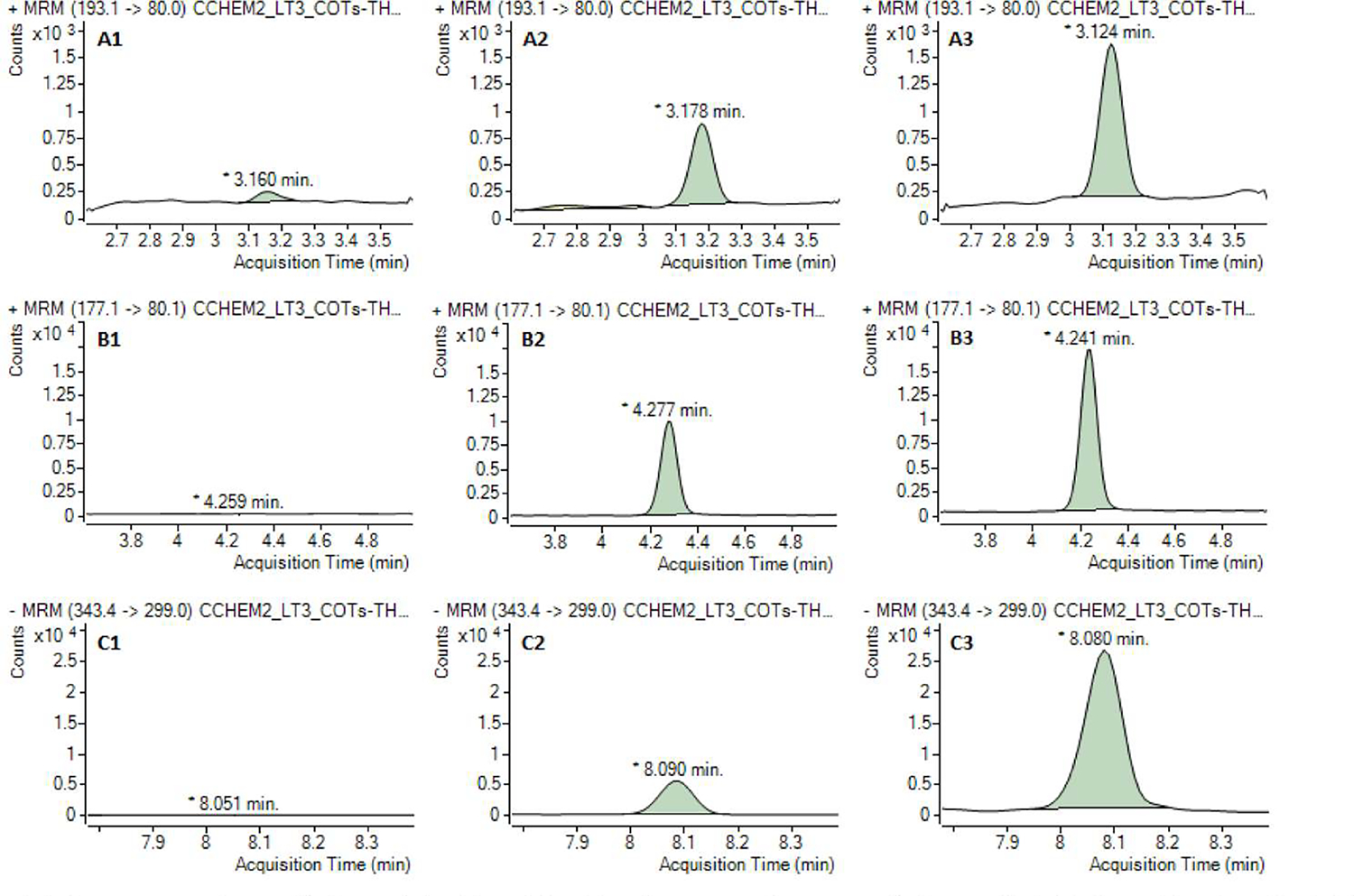
Typical chromatograms of trans-3′-hydroxycotinine (A), cotinine (B), and 11-nor-9-caiboxy-Δ9-tetrahydrocannabinol (C) obseived in solvent blank (1), lowest calibrant (2), and unknown mine samples (3). Note: The concentrations of trans-3′-hydroxycotinine, cotinine, and 11-nor-9-carboxy-Δ9-tetrahydrocannabinol in unknown samples are 22.2 (A3), 10.5 (B3), and 11.3 ng/mL (C3), respectively.
3.2. Calibration curves, quantification ranges, and LOQs
The calibration curves for 3OH-COT and COT consisted of 12 calibrants and covered a quantification range of 5.0–5,000 ng/mL and 2.5–5,000 ng/mL, respectively. Both curves were best fitted with a quadratic regression equation, with average correlation coefficient values (R2) greater than 0.999 (n = 5 curves). The average agreement between the observed and expected concentrations across the calibration curve was 95.9 % and 94.6 % for 3OH-COT and COT, respectively.
Two calibration curves were used for COOH-THC quantification. A nine-point calibration curve fitted with a linear regression equation, covering a concentration range of 2.5–500 ng/mL was used to quantify low and medium COOH-THC levels. The average R2 was greater than 0.996 (n = 5). Another six-point calibration curve fitted with a quadratic regression equation, covering a range of 250–10,000 ng/mL was used to quantify high COOH-THC levels (above 500 ng/mL) (Fig. S3). The average R2 was greater than 0.991 (n = 5). The average agreement between the observed and expected concentrations across the calibration curve was 93.8 % and 99.2 % for the low and medium COOH-THC levels and high COOH-THC levels, respectively.
For all target analytes, the background concentrations were at least 5 times lower than the LOQ and did not greatly affect the calibration curve accuracies. The LOQs for 3OH-COT, COT, and COOH-THC were 5.0 ng/mL, 2.5 ng/mL, and 2.5 ng/mL, respectively.
3.3. Method performance
For all target analytes, the accuracies of the method ranged from 82.5 to 98.5 %. The within-day %RSDs were 1.22–6.21 %, while the between-day %RSDs were 1.42–6.26 %. For NIST material analysis, the reported concentrations were in a range of 86.3–95.3 % of the certified values for all target analytes. For COT, the reported concentrations in G-EQUAS 18A and 18B materials were 392 ng/mL and 909 ng/mL, respectively, which were well within the acceptable ranges (18A acceptable range: 355.0–540.4 ng/mL and 18B acceptable range: 840.2–1165.4 ng/mL). Table 2 shows the accuracies, precisions, and results of NIST and G-EQUAS material analyses.
Table 2.
The method accuracies, precisions, and concentrations (cone.) of trans-3′-hydroxycotinine (3OH-COT), cotinine (COT), and 11-nor-9-carboxy-Δ9-tetrahydrocannabinol (COOH-THC) in control materials.
| Analyte | Accuracies and precisions | Quality assurance materials conc., ng/mL | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| QC level | Expected conc., ng/200 μL | Accuracy, % (n = 25) | Precision, % RSD | NIST SRM (n = 10) | G-EQUAS (n = 4) | ||||
|
|
|
|
|||||||
| Within-day (n = 25) | Between-day (n = 25) | Reported conc. (%)A | Reference conc. | Reported conc. (%)A | Reference conc. | ||||
|
| |||||||||
| 3OH-COT | Low | 5.00 | 82.5 | 3.76 | 3.62 | 3030 (87.6) 1) | 3460 | ||
| Medium-1 | 50.0 | 86.9 | 1.94 | 2.15 | |||||
| Medium-2 | 500 | 96.4 | 2.81 | 2.65 | |||||
| High | 800 | 98.5 | 1.60 | 1.78 | |||||
| COT | Low | 5.00 | 89.0 | 5.16 | 5.00 | 20.8 (86.7) 2) | 24 | 392 (87.6) 6) | 447.7 |
| Medium-1 | 50.0 | 93.8 | 1.58 | 1.71 | 1039 (95.3) 1) | 1090 | 909 (90.6) 7) | 1002.8 | |
| Medium-2 | 500 | 97.1 | 3.50 | 3.29 | |||||
| High | 800 | 93.9 | 1.22 | 1.42 | |||||
| COOH-THC | Low | 5.00 | 86.6 | 6.21 | 5.77 | <LOQ 3) | <1 | ||
| Medium-1 | 50.0 | 95.6 | 1.59 | 2.53 | 10.1 (86.3) 4) | 11.7 | |||
| Medium-2 | 500 | 96.7 | 5.54 | 6.26 | 21.9 (90.9) 5) | 24.1 | |||
| High | 1600 | 95.0 | 2.76 | 5.13 | |||||
Note: QC = quality control, NIST SRM = National Institute for Standard and Technology standard reference material, G-EQUAS = German External Quality Assessment Scheme
= the percentage was calculated by dividing the repotted concentration with the reference concentration and multiplied by 100, LOQ = limit of quantification. The number of reference materials analyzed:
NIST SRM 3672 (n = 2)
NIST SRM 3673 (n = 2)
NIST SRM 1507b-0 (n = 2)
NIST SRM 1507b-1 (n = 2)
NIST SRM 1507b-2 (n = 2)
G-EQUAS 18A of round 64/2019 (n = 2)
G-EQUAS 18B of round 64/2019 (n = 2).
3.4. Autosampler stability, matrix effects, dilution effects, and carry-over
The target analytes were stable in the extracts for up to 144 h when stored inside the autosampler at 10 °C. The RR of each analyte per concentration tested was ± 15 % of the average RR obtained at time zero (Fig. S4).
Matrix effects were observed for all analytes. The calculated ion suppression percentages for the native 3OH-COT, COT, and COOH-THC were in a range of −2.4 to 0.0, −1.6 to −0.4, and −18.3 to −11.7, respectively. The calculated ion suppression percentages for the labeled 3OH-COT, COT, and COOH-THC were in a range of −1.4 to −0.2, −1.3 to −0.3, and −20.0 to −11.6, respectively. The calculated percent coefficient of variation (%CVs) of the suppression for all analytes were in a range of 0.4–7.9 %.
In the dilution effects study, the accuracies for the 1:5 v/v diluted samples were 102.2 % for 3-OH-COT, 92.0 % for COT, and 97.9 % for 3OH-COT. The 1:5 v/v diluted samples had precisions ranging from 1.79 to 2.83 % (within-day) and 2.35 to 4.67 % (between-day). For the samples with the dilution ratio of 1:2 v/v, the accuracies were 106.9 % for 3OH-COT, 96.9 % for COT, and 91.6 % for 3OH-COT. The precisions for the 1:2 v/v diluted samples were in the range of 1.85–4.32 % (within-day) and 2.00–8.22 % (between-day) for all compounds.
Initially, carryover was observed for COOH-THC and was calculated to be approximately 0.25 %. To eliminate the carryover likely caused by absorption of COOH-THC to the external surfaces of the needle, an autosampler washing program was developed and applied. We found that rinsing the external needle surface with a mixture of isopropanol (70 %, v/v): Milli-Q water: methanol (25:25:50, v/v/v) for 20 sec between injections was able to reduce the carryover concentration of COOH-THC to negligible concentrations (<LOQ).
3.5. Sample analysis
Of the total urine samples analyzed, detection frequencies for 3OH-COT, COT, and COOH-THC were 63.7 % (93 samples), 64.4 % (94 samples), and 36.6 % (53 samples), respectively. All three target analytes were detected in 51 samples (34.9 %). The concentration ranges of 3OH-COT, COT, and COOH-THC were < LOQ-30,194 ng/mL (geometric mean, 35.8 ng/mL), <LOQ-9,448 ng/mL (geometric mean, 15.1 ng/mL), and < LOQ-4,615 ng/mL (geometric mean, 6.9 ng/mL), respectively. When the data were analyzed based on self-reported usage information, among the self-identified non-tobacco smokers, 57.9 % had detectable levels of urinary 3OH-COT and COT with geometric means values of 19.0 and 8.3 ng/mL, respectively. Additionally, among the individuals reported as non-marijuana users, 22.0 % of these subjects had detectable levels of urinary COOH-THC with a geometric mean value of 4.1 ng/mL. Urinary 3OH-COT and COT were detected in 92 and 96 % (23 of 25 and 24 of 25) of subjects who reported smoking tobacco, respectively. The geometric mean values of urinary 3OH-COT and COT concentrations from tobacco users were 754.1 and 270.8 ng/mL, respectively. Urinary COOH-THC was detected in 80.6 % (29 of 36) of subjects who reported using marijuana sometime during the last month. Their urinary COOH-THC geometric mean concentration was 34.2 ng/mL. Additional data are shown in Table 3. In addition, the sensitivity values for 3-OH-COT, COT, and COOH-THC were 92, 96, and 81 %, respectively. The specificity values for 3-OH-COT, COT, and COOH-THC were 42, 42, and 78 %, respectively.
Table 3.
Descriptive data for trans-3′-hydroxycotinine (3OH-COT), cotinine (COT), and 11-nor-9-carboxy-Δ9-tetrahydrocannabinol (COOH-THC) concentrations measured in urine samples collected from pregnant African American women participants in the Atlanta area.
| Analyte | Descriptive item | All subjects | Self-report user | Self-report non-user |
|---|---|---|---|---|
|
| ||||
| 3OH-COT | n (%) | 146 (100.0) | 25 (17.1) | 121 (82.9) |
| Positive samples (%) | 93 (63.7) | 23 (92.0) | 70 (57.9) | |
| Geometric mean, ng/mL | 35.8 | 754.1 | 19.0 | |
| 50th percentile, ng/mL | 16.0 | 1393.4 | 10.0 | |
| Range (min–max), ng/mL | <LOQ-30194 | <LOQ-21068 | <LOQ-30194 | |
| COT | n (%) | 146 (100.0) | 25 (17.1) | 121 (82.9) |
| Positive samples (%) | 94 (64.4) | 24 (96.0) | 70 (57.9) | |
| Geometric mean, ng/mL | 15.1 | 270.8 | 8.3 | |
| 50th percentile, ng/mL | 6.7 | 480.2 | 3.9 | |
| Range (min–max), ng/mL | <LOQ-9448 | <LOQ-9448 | <LOQ-1957 | |
| COOH-THC | n (%) | 145 (100.0) | 36 (24.8) | 109 (75.2) |
| Positive samples (%) | 53 (36.6) | 29 (80.6) | 24 (22.0) | |
| Geometric mean, ng/mL | 6.9 | 34.2 | 4.1 | |
| 50th percentile, ng/mL | <LOQ | 29.3 | <LOQ | |
| Range (min–max), ng/mL | <LOQ-4615 | <LOQ-4615 | <LOQ-771 | |
Note: LOQ = limit of quantification: 5 ng/mL for 3OH-COT, 2.5 ng/rnL for COT and COOH-THC. For the urinary data analysis of tobacco metabolites, the total number of samples was 146. The total number of samples for the urinary data analysis of marijuana metabolites was 145 because one subject was excluded due to missing self-report usage information. Self-report user information was collected based on the following questions: 1) Have you smoked cigarettes or cigars?, 2) During the last month, have you smoked marijuana?
4. Discussion
We developed a high-throughput method to quantify urinary 3OH-COT, COT, and COOH-THC simultaneously to assess co-exposure to tobacco and marijuana products. Traditionally, these urinary metabolites have been measured using separate methods because they contain distinctive functional groups that dictated how efficiently they were extracted from samples and how they ionized during MS analysis. 3OH-COT and COT are highly polar basic compounds. They are usually extracted and analyzed in one method using positive mode ESI [26]. Conversely, COOH-THC is typically extracted and analyzed separately using a method with negative mode ESI [27]. Therefore, the advantages of using this method are reduced sample volume, personnel and supply costs, and sample preparation and analysis time. This method can be utilized in laboratories to support high-throughput, large-scale studies where available sample volume is limited.
Our results demonstrate that it is practical to use ethyl acetate to extract all analytes in urine using SLE. Urine is typically regarded as the preferred biospecimen for assessing tobacco or marijuana use primarily because 3OH-COT, COT, and COOH-THC are present in higher concentrations in urine than in other biological matrices [10,11]. In addition, urine sample collection is relatively easy and is not invasive. During the analysis of the target compounds, the MS was operated alternatively in positive and negative ESI mode. Polarity switching can only be applied when the target analytes (of a different ionization mode) are eluted from the column at least one minute apart to allow ionization stabilization. This requires proper selection of analytical columns and mobile phase. To our knowledge, this is the first method reported to measure 3OH-COT, COT, and COOH-THC simultaneously using SLE-LC-MS/MS.
In our method, SLE was used instead of traditional liquid-liquid or solid-phase extraction. In addition, the method used isotope dilution quantification, which corrects for extraction recovery in each sample. SLE permits the use of small volumes of urine (200 μL) and organic solvents (1.8 mL). The 96-well plate format allowed 78 unknown samples to be processed at a time given our QC protocol. Typically, SLE works best when targeted analytes are in their neutral forms. However, we found that sample pH adjustment using acetic acid or ammonium hydroxide led to unsatisfactory recoveries. Therefore, we chose to use the initial pH (5.1) after enzymatic digestion as a compromise, which resulted in better extraction recoveries for both COT and COOH-THC. SLE was found to be more suitable for COOH-THC and COT, which are less polar compounds. For the most polar analyte, 3OH-COT, extraction recoveries were low. Low extraction recoveries of 3OH-COT in urine were observed in previous studies [33,34]. Despite low extraction recovery of 3OH-COT, the method demonstrated sufficient sensitivity during MS analysis and the use of isotope dilution allows for the automatic correction for recovery thus preserving the integrity of the quantitative value. Although assessing tobacco exposure can be adequately done using the concentrations of COT alone, concentrations of 3OH-COT will permit the evaluation of the associated toxicity of tobacco modified by the individual genetic polymorphisms [3,35]. Thus, inclusion of 3OH-COT as one of the target analytes in the method has advantages for epidemiological studies.
The LOQ, method accuracy and precision, matrix effects, dilution effects, and autosampler stability were evaluated during the method validation. All of the studied parameters yielded satisfactory results. Within-day precisions and between-day precisions for all analytes were within ± 15 and ± 20 % RSD, as suggested by FDA and SWGTOX guidelines [29,30]. The accuracies for all analytes were acceptable according to the SWGTOX guidelines (±20 % of nominal concentrations). Minimal matrix effects were observed for 3OH-COT and COT. For COOH-THC, a higher degree of ion suppression was observed. It should be noted that each mine sample is unique and contains different concentrations of matrix compounds, thus the results may not reflect the matrix effects possibly happening in all unknown samples. However, because the method used the isotope dilution technique, matrix effects were adequately controlled for. The average ionization suppression did not exceed 25 % and the calculated %CVs of the suppression were lower than 15 % for all target analytes [30]. Thus, no further modification was required to overcome these matrix effects. In addition, the dilution integrity results were acceptable for all target analytes, with accuracy values ranging within ± 15 % of nominal concentration and precision values ranging within ± 15 % RSD [29].
The analysis of NIST samples indicates that the method has good performance and can produce accurate results (±20 % of the reference values). The analysis of G-EQUAS samples demonstrated that the method produced results comparable to laboratories that participated in the same proficiency testing program.
3OH-COT, COT, and COOH-THC were stable for at least 144 h in the autosampler at 10 °C. Long-term and freeze–thaw storage stabilities were not assessed because they have been investigated by other laboratories. In general, total COOH-THC and COT are stable in urine up to 1 year of storage at −20 °C [36,37]. COT and 3OH-COT and their glucuronides are stable in urine through 6 freeze–thaw cycles [38]. In addition, urinary COOH-THC and COOH-THC-glucuronide are stable through 3 freeze–thaw cycles [39].
In this method, the LOQs for 3OH-COT, COT, and COOH-THC were 5.0, 2.5, and 2.5 ng/mL, respectively. To analyze the target compounds in a wide range of concentrations, the calibration curves had dynamic concentration ranges greater than 1000-fold, resulting in lower accuracies for the low concentration calibrants. Accordingly, the LOQs were high for 3OH-COT and COT. However, lower LOQs could be achieved for 3OH-COT and COT by narrowing the concentration range of their calibration curves. Splitting the calibration curve for COT into two curves (1–100 ng/mL and 100–5000 ng/mL) would have resulted in an LOQ of 1 ng/mL for COT (Fig. S5). According to the literature, the LOQs for 3OH-COT, COT, and COOH-THC were in the range of 0.5–9.5, 0.2–5.0, and 1.0–10 ng/mL, respectively [26,27,40,41]. These LOQs are applicable for analyzing urine samples obtained from active and passive tobacco and marijuana users. According to the Substance Abuse and Mental Health Services Administration, the cut-off value in confirmatory tests of COOH-THC is set to 15 ng/mL in urine [42]. The cut-off value for urinary COT, proposed by different studies, varies from 40.5 to 91.7 ng/mL [43,44]. The cutoff for urinary 3OH-COT was reported to be 128.0 ng/mL for the U.S. population [43]. Thus, based on the current LOQ, our method is sensitive enough to assess exposure to tobacco smoke among active smokers and passive-smokers as well as to assess co-exposure to tobacco and marijuana products via smoking.
This method used quadratic calibration models for quantification of 3OH-COT, COT, and higher concentrations of COOH-THC. Although linear calibration models are often used for quantification of urinary tobacco and marijuana metabolites in previous methods, quadratic calibration models are also applicable for quantitative analysis of urinary biomarkers [30,45]. When using the quadratic calibration models, urine samples must be diluted and re-extracted for analysis if the concentration of the target analytes in analyzed sample exceeded the highest calibrants.
The method was used to analyze urine samples obtained from pregnant African American women residing in the Atlanta metropolitan area. This method was developed specifically to evaluate this population because of their high self-report of marijuana use. Tobacco and marijuana metabolites were detected in 35 % of the samples, suggesting coexposures to tobacco and marijuana smoke. The observed wide range of metabolite concentrations indicate that these pregnant women are exposed to tobacco and marijuana products as active and passive users. Based on previous studies, pregnant women who were active tobacco users had urinary COT in a range of 19 to 28,020 ng/mL or greater than 100 ng/mL [46,47]. In one study, active marijuana users had measured urinary COOH-THC levels in a range of 34 to 6,330 ng/mL [48]. These values noted in our investigation were comparable to concentrations of those who reported themselves as active users. Thus, it is evident that our method can be used to assess tobacco-marijuana exposures by measuring the concentrations of both tobacco and marijuana biomarkers. Based on our results, urinary COT and COOH-THC are excellent biomarkers that can be used to indicate exposure to tobacco and marijuana products or smoke. Urinary 3-OH-COT, COT, and COOH-THC have high sensitivity (>81 %) in indicating active users. Although poor specificity (42 %) was noted for 3OH-COT and COT in our study, it can easily be explained. Poor specificity was likely a result of either passive exposure or non-concordant self-report of tobacco use. Therefore, to assess exposures to tobacco and marijuana accurately and reliably, urinary biomarker measurements should be used, instead of self-report data collection. Moreover, recent studies among pregnant women suggest that tobacco and marijuana use are under-reported [49,50]. Thus, a method capable of detecting urinary tobacco and marijuana metabolites has important epidemiologic applications.
5. Conclusion
We developed a robust LC-MS/MS method that allows the simultaneous quantification of the principal tobacco and marijuana metabolites: 3OH-COT, COT, and COOH-THC. The method was fully validated and is suitable for the analysis of urine samples for assessing co-exposure to tobacco and marijuana products.
Supplementary Material
Acknowledgements
We are grateful to our colleagues Nathan Mutic, Estefani Ignacio Gallegos, Nikolay Patrushev, Kristi Maxwell Logue, Castalia Thome, Shirleta Reid, Cassandra Hall, Mumu Rahman, and the staff at the recruiting sites for data and sample collection and logistics.
Funding information
This work was funded by the National Institutes of Health (NIH-R01NR014800, NIH-R01MD009064, NIH-R24ES029490, NIH-R01MD009746 NIH-P50ES02607, NIH-P30ES019776, NIH-UH3OD023318, NIH-U2CES026560, NIH-U2CES026542) and the US Environmental Protection Agency (EPA-83615301).
Abbreviations:
- US
the United States
- COT
cotinine
- 3OH-COT
trans-3′-hydroxycotinine
- Δ9-THC
Δ9-tetrahydrocannabinol
- COOH-THC
11-nor-9-carboxy-Δ9-tetrahydrocannabino
- NSDUH
National Survey on Drug Use and Health
- LC
liquid chromatography
- ESI
electrospray ionization
- MS/MS
tandem mass spectrometry
- SRM
standard reference material
- NIST
the National Institute of Standards and Technology
- G-EQUAS
the German External Quality Assessment Scheme
- QC
quality control
- IS
internal standard
- SLE
solid-supported liquid-liquid extraction
- MS
mass spectrometer
- MRM
multiple reaction monitoring
- FDA
Food and Drug Administration
- LOQ
limit of quantification
- ANOVA
analysis of variance
- %RSD
relative standard deviations
- SWGTOX
Scientific Working Group for Forensic Toxicology
- RR
relative response
- R2
correlation coefficient
- %CV
percent coefficient of variation
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Volha Yakimavets: Investigation, Validation, Writing – original draft, Visualization. Tian Qiu: Methodology, Investigation, Writing – original draft, Visualization. Parinya Panuwet: Methodology, Conceptualization, Investigation, Validation, Formal analysis, Writing – review & editing, Supervision, Project administration. Priya E. D’Souza: Resources, Writing – review & editing. Patricia A. Brennan: Resources, Writing – review & editing, Funding acquisition. Anne L. Dunlop: Resources, Writing – review & editing, Data curation, Funding acquisition. P. Barry Ryan: Resources, Writing – review & editing, Supervision, Project administration, Funding acquisition. Dana Boyd Barr: Conceptualization, Resources, Writing – review & editing, Supervision, Project administration, Funding acquisition.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jchromb.2022.123378.
References
- [1].Hindocha C, Freeman TP, Ferris JA, Lynskey MT, Winstock AR, No smoke without tobacco: a global overview of cannabis and tobacco routes of administration and their association with intention to quit, Front. Psychiatry 7 (2016) 104, 10.3389/fpsyt.2016.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Graves BM, Johnson TJ, Nishida RT, Dias RP, Savareear B, Harynuk JJ, Kazemimanesh M, Olfert JS, Boies AM, Comprehensive characterization of mainstream marijuana and tobacco smoke, Scientific Rep. 10 (2020) 1–12, 10.1038/s41598-020-63120-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Benowitz NL, Hukkanen J, Jacob P, Nicotine chemistry, metabolism, kinetics and biomarkers, Nicotine Psychopharmacology 192 (2009) 29–60, 10.1007/978-3-540-69248-5_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Spindle TR, Cone EJ, Herrmann ES, Mitchell JM, Flegel R, LoDico C, Bigelow GE, Vandrey R, Pharmacokinetics of cannabis brownies: a controlled examination of Δ9-tetrahydrocannabinol and metabolites in blood and oral fluid of healthy adult males and females, J. Anal. Toxicol. 44 (2020) 661–671, 10.1093/jat/bkaa067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chandra S, Radwan MM, Majumdar CG, Church JC, Freeman TP, ElSohly MA, New trends in cannabis potency in USA and Europe during the last decade (2008–2017), Eur. Arch. Psychiatry Clin. Neurosci. 269 (2019) 5–15, 10.1007/s00406-019-00983-5. [DOI] [PubMed] [Google Scholar]
- [6].Kozlowski LT, Mehta NY, Sweeney CT, Schwartz SS, Vogler GP, Jarvis MJ, West RJ, Filter ventilation and nicotine content of tobacco in cigarettes from Canada, the United Kingdom, and the United States, Tob. Control. 7 (4) (1998) 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hukkanen J, Jacob P, Benowitz NL, Metabolism and disposition kinetics of nicotine, Pharmacol. Rev. 57 (1) (2005) 79–115. [DOI] [PubMed] [Google Scholar]
- [8].Benowitz NL, Jacob III P, Trans-3′-hydroxycotinine: disposition kinetics, effects and plasma levels during cigarette smoking, Br. J. Clin. Pharmacol. 51 (2001) 53–59, 10.1046/j.1365-2125.2001.01309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Benowitz NL, Perez-Stable EJ, Fong I, Modin G, Herrera B, Jacob P, Ethnic differences in N-glucuronidation of nicotine and cotinine, J. Pharmacol. Exp. Therapeutics 291 (1999) 1196–1203. [PubMed] [Google Scholar]
- [10].El-Khoury JM, Wang S, Recent advances in MS methods for nicotine and metabolite analysis in human matrices: clinical perspectives, Bioanalysis. 6 (2014) 2171–2183, 10.4155/bio.14.176. [DOI] [PubMed] [Google Scholar]
- [11].Battista N, Sergi M, Montesano C, Napoletano S, Compagnone D, Maccarrone M, Analytical approaches for the determination of phytocannabinoids and endocannabinoids in human matrices, Drug Test, and Anal. 6 (2014) 7–16, 10.1002/dta.1574. [DOI] [PubMed] [Google Scholar]
- [12].Huestis MA, Pharmacokinetics and metabolism of the plant cannabinoids, Δ 9-tetrahydrocannibinol, cannabidiol and cannabinol, in: Barret JE (Ed.), Handbook of Experimental Pharmacology, Springer, Germany, 2005, pp. 657–690. [DOI] [PubMed] [Google Scholar]
- [13].Sharma P, Murthy P, Bharath MS, Chemistry, metabolism, and toxicology of cannabis: clinical implications, Iranian J. Psychiatry 7 (2012) 149. [PMC free article] [PubMed] [Google Scholar]
- [14].Huestis M, Human cannabinoid pharmacokinetics, Chem. & Biodivers. 4 (8) (2007) 1770–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Huestis MA, Henningfield JE, Cone EJ, Blood cannabinoids. I. Absorption of THC and formation of 11-OH-THC and THCCOOH during and after smoking marijuana, J. Anal. Toxicol. 16 (1992) 276–282. 10.1093/jat/16.5.276. [DOI] [PubMed] [Google Scholar]
- [16].Citti C, Braghiroli D, Vandelli MA, Cannazza G, Pharmaceutical and biomedical analysis of cannabinoids: A critical review, J. Pharm. Biomed. Anal. 147 (2018) 565–579, 10.1016/j.jpba.2017.06.003. [DOI] [PubMed] [Google Scholar]
- [17].Ramo DE, Liu H, Prochaska JJ, Tobacco and marijuana use among adolescents and young adults: a systematic review of their co-use, Clin. Psychol. Rev. 32 (2012) 105–121, 10.1016/j.cpr.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schauer GL, Berg CJ, Kegler MC, Donovan DM, Windle M, Assessing the overlap between tobacco and marijuana: Trends in patterns of co-use of tobacco and marijuana in adults from 2003–2012, Addictive Behav. 49 (2015) 26–32, 10.1016/j.addbeh.2015.05.012. [DOI] [PubMed] [Google Scholar]
- [19].Schauer GL, Peters EN, Correlates and trends in youth co-use of marijuana and tobacco in the United States, 2005–2014, Drug Alcohol Depend. 185 (2018) 238–244, 10.1016/j.drugalcdep.2017.12.007. [DOI] [PubMed] [Google Scholar]
- [20].Cohn AM, Abudayyeh H, Perreras L, Peters EN, Patterns and correlates of the co-use of marijuana with any tobacco and individual tobacco products in young adults from Wave 2 of the PATH Study, Addictive Behav. 92 (2019) 122–127, 10.1016/j.addbeh.2018.12.025. [DOI] [PubMed] [Google Scholar]
- [21].Benowitz N, Nardone N, Helen GS, Addo N, Jacob P 3rd, Liakoni E, Jain S, Hooshfar S, Lynch K, Quantitative biochemical screening for marijuana use and concordance with tobacco use in urban adolescents, Drug Alcohol Depend. 205 (2019), 107583, 10.1016/j.drugalcdep.2019.107583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Coleman-Cowger VH, Oga EA, Peters EN, Mark K, Prevalence and associated birth outcomes of co-use of Cannabis and tobacco cigarettes during pregnancy, Neurotoxicol. Teratol. 68 (2018) 84–90, 10.1016/j.ntt.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Stroud LR, Papandonatos GD, McCallum M, Kehoe T, Salisbury AL, Huestis MA, Prenatal tobacco and marijuana co-use: Impact on newborn neurobehavior, Neurotoxicol. Teratol. 70 (2018) 28–39, 10.1016/j.ntt.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Driezen P, Gravely S, Wadsworth E, Smith DM, Loewen R, Hammond D, Li L, Abramovici H, McNeill A, Borland R, Increasing cannabis use is associated with poorer cigarette smoking cessation outcomes: Findings from the ITC Four Country Smoking and Vaping Surveys, 2016–2018, Nicotine & Tob. Res. (2021). 10.1093/ntr/ntab122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hindocha C, McClure EA, Unknown population-level harms of cannabis and tobacco co-use: if you don’t measure it, you can’t manage it, Addiction. 116 (2021) 1622–1630, 10.1111/add.15290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wei B, Feng J, Rehmani IJ, Miller S, McGuffey JE, Blount BC, Wang L, A high-throughput robotic sample preparation system and HPLC-MS/MS for measuring urinary anatabine, anabasine, nicotine and major nicotine metabolites, Clin. Chim. Acta 436 (2014) 290–297, 10.1016/j.cca.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Scheidweiler KB, Desrosiers NA, Huestis MA, Simultaneous quantification of free and glucuronidated cannabinoids in human urine by liquid chromatography tandem mass spectrometry, Clin. Chim. Acta 413 (2012) 1839–1847, 10.1016/j.cca.2012.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].McGuffey JE, Wei B, Bernert JT, Morrow JC, Xia B, Wang L, Blount BC, Taffe M, Validation of a LC-MS/MS method for quantifying urinary nicotine, six nicotine metabolites and the minor tobacco alkaloids -anatabine and anabasine - in smokers’ urine, PLoS ONE 9 (7) (2014) e101816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].US Department of Health Human Services, Bioanalytical method validation guidance for industry, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry, 2018. (accessed 2 June 2022).
- [30].Scientific Working Group for Forensic Toxicology (SWGTOX) standard practices for method validation in forensic toxicology, J. Anal. Toxicol. 37 (2013) 452–474. 10.1093/jat/bkt054. [DOI] [PubMed] [Google Scholar]
- [31].Corwin EJ, Hogue CJ, Pearce B, Hill CC, Read TD, Mulle J, Dunlop AL, Protocol for the Emory University African American vaginal, oral, and gut microbiome in pregnancy cohort study, BMC Pregnancy and Childbirth. 17 (2017) 1–8, 10.1186/s12884-017-1357-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hornung RW, Reed LD, Estimation of average concentration in the presence of nondetectable values, Appl. Occup. Environ. Hyg. 5 (1990) 46–51, 10.1080/1047322X.1990.10389587. [DOI] [Google Scholar]
- [33].Feldhammer M, Menasco D, Zhang W, Ritchie J, Validation of a liquid chromatography-tandem mass spectrometry method for nicotine and its metabolites reveals a low clinical utility for the tobacco alkaloid anabasine, Am. J. Clin. Pathol. 147 (2017) S159–S159. 10.1093/ajcp/aqw191. [DOI] [Google Scholar]
- [34].Tuomi T, Johnsson T, Reijula K, Analysis of nicotine, 3-hydroxycotinine, cotinine, and caffeine in urine of passive smokers by HPLC-tandem mass spectrometry, Clin. Chem. 45 (1999) 2164–2172, 10.1093/clinchem/45.12.2164. [DOI] [PubMed] [Google Scholar]
- [35].Nakajima M, Yokoi T, Interindividual variability in nicotine metabolism: C-oxidation and glucuronidation, Drug Metab. Pharmacokinet. 20 (2005) 227–235, 10.2133/dmpk.20.227. [DOI] [PubMed] [Google Scholar]
- [36].Church TR, Anderson KE, Le C, Zhang Y, Kampa DM, Benoit AR, Yoder AR, Carmella SG, Hecht SS, Temporal stability of urinary and plasma biomarkers of tobacco smoke exposure among cigarette smokers, Biomark. 15 (2010) 345–352, 10.3109/13547501003753881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Desrosiers NA, Lee D, Scheidweiler KB, Concheiro-Guisan M, Gorelick DA, Huestis MA, In vitro stability of free and glucuronidated cannabinoids in urine following controlled smoked cannabis, Anal. Bioanal. Chem. 406 (2014) 785–792, 10.1007/s00216-013-7524-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Piller M, Gilch G, Scherer G, Scherer M, Simple, fast and sensitive LC-MS/MS analysis for the simultaneous quantification of nicotine and 10 of its major metabolites, J. Chromatography B. 951 (2014) 7–15, 10.1016/j.jchromb.2014.01.025. [DOI] [PubMed] [Google Scholar]
- [39].Dong X, Li L, Ye Y, Zheng L, Jiang Y, Simultaneous determination of major phytocannabinoids, their main metabolites, and common synthetic cannabinoids in urine samples by LC- MS/MS, J. Chromatography B. 1033 (2016) 55–64, 10.1016/j.jchromb.2016.08.002. [DOI] [PubMed] [Google Scholar]
- [40].Scheidweiler KB, Shakleya DM, Huestis MA, Simultaneous quantification of nicotine, cotinine, trans-3′-hydroxycotinine, norcotinine and mecamylamine in human urine by liquid chromatography–tandem mass spectrometry, Clin. Chim. Acta 413 (2012) 978–984, 10.1016/j.cca.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rumpler MJ, Quantitative analysis of ll-nor-9-carboxy-tetrahydrocannbinol (THC–COOH) in urine by LC-MS/MS following a simple filtration, J. Chromatography B. 957 (2014) 77–83, 10.1016/j.jchromb.2014.02.056. [DOI] [PubMed] [Google Scholar]
- [42].The Substance Abuse and Mental Health Services Administration, U.S. Department of Health and Human Services, Mandatory guidelines for federal workplace drug testing programs. https://www.federalregister.gov/d/2017-00979/p-37, 2017. (accessed 2 June 2022).
- [43].Jain RB, Estimates of cutoffs with specificities and sensitivities for urine cotinine and hydroxycotinine for US adults aged≥ 20 years to classify smokers and nonsmokers, Environ. Sci. Pollut. Res. 27 (2020) 10882–10887, 10.1007/S11356-020-07710-X. [DOI] [PubMed] [Google Scholar]
- [44].Edwards KC, Naz T, Stanton CA, Goniewicz ML, Hatsukami DK, Smith DM, Wang L, Villanti A, Pearson J, Blount BC, Urinary cotinine and cotinine+ trans-3′-hydroxycotinine (TNE-2) cut-points for distinguishing tobacco use from nonuse in the United States: PATH study (2013–2014), Cancer Epidemiology and Prevention Biomark. 30 (2021) 1175–1184. 10.1158/1055-9965.EPI-20-0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Moos RK, Angerer J, Wittsiepe J, Wilhelm M, Brüning T, Koch HM, Rapid determination of nine parabens and seven other environmental phenols in urine samples of German children and adults, Int. J. Hyg. Environ. Health 217 (8) (2014) 845–853. [DOI] [PubMed] [Google Scholar]
- [46].Pickett KE, Rathouz PJ, Kasza K, Wakschlag LS, Wright R, Self-reported smoking, cotinine levels, and patterns of smoking in pregnancy, Paediatric and Perinat, Epidemiology. 19 (2005) 368–376, 10.1111/j.1365-3016.2005.00660.x. [DOI] [PubMed] [Google Scholar]
- [47].Jhun H-J, Seo H-G, Lee D-H, Sung M-W, Kang Y-D, Syn HC, Jun JK, Self-reported smoking and urinary cotinine levels among pregnant women in Korea and factors associated with smoking during pregnancy, J. Korean Med. Sci. 25 (2010) 752–757, 10.3346/jkms.2010.25.5.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Odell MS, Frei MY, Gerostamoulos D, Chu M, Lubman DI, Residual cannabis levels in blood, urine and oral fluid following heavy cannabis use, Forensic Sci. Int. 249 (2015) 173–180, 10.1016/j.forsciint.2015.01.026. [DOI] [PubMed] [Google Scholar]
- [49].Garg M, Garrison L, Leeman L, Hamidovic A, Borrego M, Rayburn WF, Bakhireva L, Validity of self-reported drug use information among pregnant women, Maternal and Child Health J. 20 (2016) 41–47, 10.1007/S10995-015-1799-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ashford K, Wiggins A, Rayens E, Assef S, Fallin A, Rayens MK, Perinatal biochemical confirmation of smoking status by trimester, Nicotine & Tob. Res. 19 (2017) 631–635, 10.1093/ntr/ntw332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.