

SUMMARY
Multiple myeloma remains an incurable disease, and the cellular and molecular evolution from precursor conditions including monoclonal gammopathy of undetermined significance and smoldering multiple myeloma is incompletely understood. Here, we combine single cell RNA- and B-cell receptor-sequencing from fifty-two patients with myeloma precursors in comparison with myeloma and normal donors. Our comprehensive analysis reveals early genomic drivers of malignant transformation, distinct transcriptional features, and divergent clonal expansion in hyperdiploid versus non-hyperdiploid samples. Additionally, we observe intra-patient heterogeneity with potential therapeutic implications and identify distinct patterns of evolution from myeloma precursor disease to myeloma. We also demonstrate distinctive characteristics of the microenvironment associated with specific genomic changes in myeloma cells. These findings add to our knowledge about myeloma precursor disease progression, providing valuable insights into patient risk stratification, biomarker discovery, and possible clinical applications.
Graphical Abstract
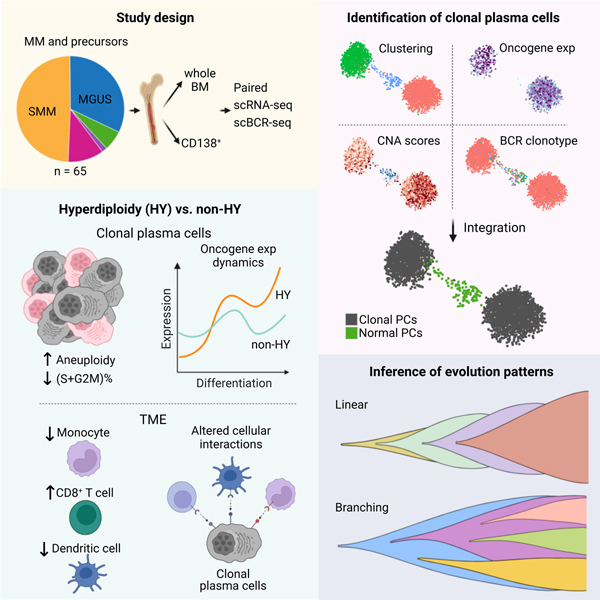
eTOC Blurb
Dang et al. present an in-depth analysis of the transcriptional landscapes in multiple myeloma precursors, revealing a high degree of transcriptional heterogeneity and distinct transcriptional and immunological characteristics of hyperdiploidy (HY) and non-HY subtypes. Integrating scRNA-seq and scBCR-seq, they elucidate transcriptional dynamics and evolutionary patterns in early disease.
INTRODUCTION
Multiple myeloma (MM) is a bone marrow cancer that originates in plasma cells (PCs). Despite great advances in treatment over the last two decades, myeloma remains mostly incurable1,2 and, contrary to many cancers, it is treated only after the tumor burden produces end-organ damage, or when this burden is high enough to increase this risk3. An opportunity exists for earlier intervention given the stepwise progression that myeloma follows from monoclonal gammopathy of undetermined significance (MGUS) to smoldering multiple myeloma (SMM), and then to multiple myeloma (MM) requiring treatment4. Despite this, not all MGUS and SMM patients progress to MM, and treatment is still mostly not recommended outside of clinical trials. This is due, in part, to the large range in the risk of progression of high versus low-risk monoclonal gammopathies, which varies from 70% vs 3%, respectively, at 2 years5. Moreover, different prognostic systems do not have a high level of concordance as to the risk of progression of each individual patient, further complicating choices as to who should be treated and who should not. Thus, further comprehensive cellular and molecular characterization is needed to advance our understanding of this evolutionary process and to identify biomarkers that can more accurately quantify individual risk of progression and set the stage for early intervention with novel therapies.
Cytogenetic abnormalities classify MM cases into two major subtypes: hyperdiploid (HY) and non-hyperdiploid (non-HY) which are almost mutually exclusive6–8. These two subtypes differ in the primary cytogenetic aberrancy that drives PCs towards the MGUS-MM pathway. HY in MGUS-MM includes a lack of IgH translocations and have a hyperdiploid chromosome number characterized by trisomies of chromosomes 3, 5, 7, 9, 11, 15, 19 and 21. The extra chromosomes carry genes the overexpression of which, may promote dysregulated cell replication and growth, transforming normal PCs (nPCs) into an MGUS-MM clone. In contrast, the non-HY subtype is mainly characterized by translocations of the immunoglobulin heavy chain (IgH) locus on 14q32, which juxtaposes an oncogene on the affected recipient chromosome to IgH enhancers. The etiology of acquisition of these primary events remains unknown, but genomic studies have shown that MGUS/SMM clones already harbor chromosomal alterations that define MM (both for HY or non-HY).
It is also recognized that tumors are part of complex ecosystems regulated through interactions with the tumor microenvironment (TME)9. For example, bulk transcriptomic analysis of MGUS/SMM has shown extensive changes in TME and tumor cell properties, both at baseline and during disease progression10,11. Despite this, bulk analysis of myeloma precursor diseases has been limited by its low tumor burden/purity and difficulty in tumor cell acquisition. Over the past few years, multiple single-cell RNA sequencing (scRNA-seq) studies have been published in myeloma12–21. These studies are either only focused on active myeloma or have limited sample size for precursor diseases. Importantly, the lack of single-cell B cell receptor sequencing (scBCR-seq) in those studies makes it difficult to accurately define malignant cells and infer tumor cells-of-origin, clonal architectures and evolution patterns. Yet, systematic characterization has to be done to better understand the impact of distinct genetic background (i.e., HY vs. non-HY) on tumor and TME cell properties18,19,21 and evolution. Here, we analyzed paired scRNA-seq and scBCR-seq data from whole bone marrow (BM) aspirate samples or enriched CD138+ PCs collected from 64 patients with monoclonal gammopathy at various stages. To our knowledge, this is the dataset that includes the largest number of patients for single cell analysis including scBCR-seq specifically for myeloma precursors. We interrogated the cellular and molecular programs that drive malignant transformation of precursors and characterized tumor evolution patterns and early immunological changes. This study provides valuable resource and can be leveraged to identify new targets and strategies for precursor diseases.
RESULTS
Single-cell analysis demonstrates a high degree of transcriptional heterogeneity in both precursor stages and NDMM
We performed paired scRNA-seq and scBCR-seq on 65 freshly collected BM aspirate samples from 64 patients including 21 MGUS, 32 SMM, 7 newly diagnosed multiple myeloma (NDMM), 1 refractory recurrent multiple myeloma (RRMM), and 4 normal BM samples from healthy donors (Fig. 1A, Table S1 and S2). Fluorescence in situ hybridization (FISH) was performed as part of the standard clinical work-up to define the genetic subtypes (Fig. 1B, Table S1). Single-cell data was processed in a manner as we previously described22–26. Following rigorous quality control (see Methods), we retained 183,928 cells which were further resolved into PCs (n = 64,078) and TME cells (n = 119,850) for subsequent analyses (Fig. 1C, Table S3). As expected, we observed increased fractions of PCs along the MGUS->SMM->MM axis (Fig. 1D). We obtained paired scBCR-seq data on 74,241 cells for subsequent phenotype and clonotype integration.
Figure 1. Workflow, sample information and single cell transcriptome map of this study.
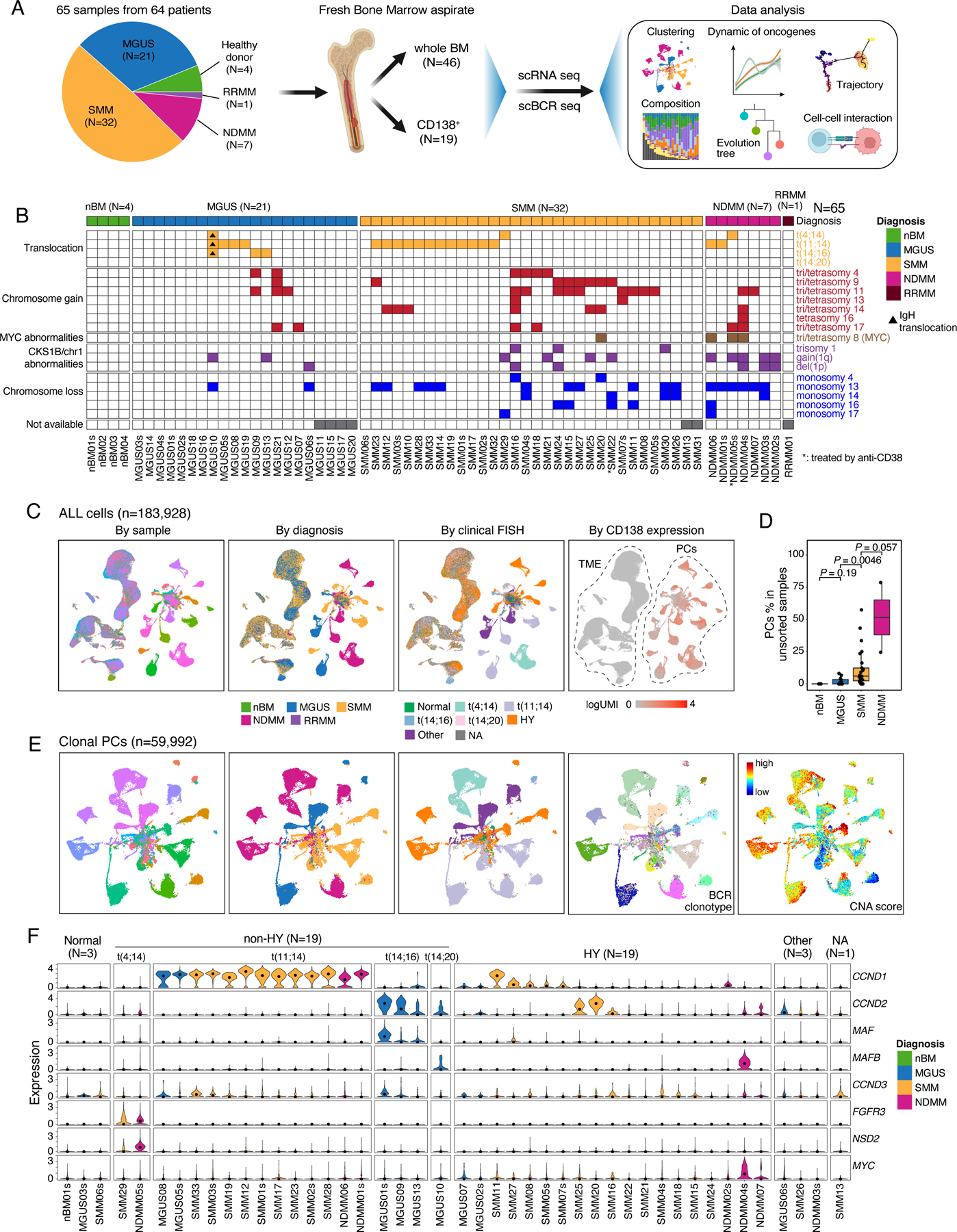
(A) A schema depicting the workflow of this study. (B) fluorescence in situ hybridization (FISH) results of 65 samples in this study. Samples were grouped by their diagnosis. (C) Uniform manifold approximation and projection (UMAP) visualization of unsupervised clustering analysis of all cells except erythrocytes (n = 183,928) that passed quality filtering. Cells were color coded for CD138 expression level (left), diagnosis (middle) and cytogenetic abnormalities determined by FISH (right). (D) Boxplots showing the comparisons of PC proportions in unsorted samples between nBM (N=3), MGUS (N=13), SMM (N=25) and NDMM (N=2). (E) UMAP visualization of unsupervised clustering analysis of all cPCs (n = 59,992) from 44 samples with ≥ 49 cPCs. Cells were color coded (from left to right) for their corresponding patient origins, diagnosis, cytogenetic abnormalities, BCR clonotypes and inferred CNA score. (F) Violin plots showing expression of translocation related genes in cPCs in 44 patients with ≥ 49 cPCs and nPCs in 1 healthy donor. Samples were grouped by their cytogenetic abnormalities and color coded by diagnosis. See also Figures S1, S2, Tables S1–S3.
Subclustering analysis of PCs by patient identified a total of 153 subclusters from 48 samples (with ≥ 50 PCs) (Fig. S1A, Table S4, Methods). We observed a high degree of intra-tumoral heterogeneity (ITH) in their transcriptome profiles in both precursor diseases and NDMMs. BCR clonotypes were defined using scBCR-seq data (Methods), and copy number alterations (CNAs) were inferred from scRNA-seq data, based on which, CNA scores24,25 were computed for each individual cell to profile CNAs across all samples (Fig. S1B). By integrating information from inferred CNAs, the presence of a dominant B cell clone based on scBCR-seq, and high expression of known oncogenes such as translocation-related genes, we identified 59,992 clonal plasma cells (cPCs, Fig. 1E). When projecting the cPCs back to the UMAP containing all PCs, we observed that the cPCs were largely separated from normal PCs (nPCs), which clustered together at the center (Fig. S2A). Additionally, the proportion of cPCs inferred from scRNA-seq data was overall correlated with that measured by flow cytometric analysis (Fig. S2B), suggesting that our integrative method can accurately identify cPCs and estimate the relative proportions of cell populations. Moreover, we compared our integrative method to two other commonly used approaches – a kNN-based classifier16 and a clustering-based method21 - which do not utilize BCR data, to define cPCs (Fig. S2C). Our results suggest that our integrative method could improve the accuracy of cPCs identification compared to approaches rely solely on scRNA-seq data. This is supported by a significant number of cells (13,811 cells in kNN-based classifier and 6,679 cells in clustering-based method) that exhibited strong abnormal features and could potentially be cPCs but were likely misclassified as nPCs in these two methods.
Most of the cPCs from MGUS, SMM, and NDMM patients formed sample-specific clusters independent of disease stages or cytogenetic abnormalities (Fig. 1E). Clusters having the same diagnosis (e.g., SMM) and/or cytogenetic changes such as t(11;14) and t(4;14) were closer, indicating overall more similar transcriptome profiles. Consistently across these multiple modalities, all sample-specific clusters were driven by dominant BCR clonotypes, suggesting cells of each cluster were clonally expanded from a single ancestor cell (Fig. 1E). Notably, despite the same origin of all cells in a sample-specific cluster, we observed substantial variations in their CNA scores across most of the clusters (Fig. 1E), demonstrating a high degree of genomic ITH.
scRNA-seq is synergistic with FISH in defining the genetic subtypes of MM
Next generation sequencing (NGS)-based FISH (Seq-FISH) has demonstrated improved sensitivity and similar specificity relative to clinical FISH at detecting relevant chromosomal abnormalities in MM27. We sought to examine whether scRNA-seq could also be useful in confirming the transcriptional consequence of translocations or improve clinical FISH detection of subclonal events. Expression of translocation-related genes and inferred CNAs using scRNA-seq were examined in cPCs (Fig. S2D, Fig. S1B). Although scRNA-seq was not designed to detect genomic structure variations or CNAs, we observed an overall good consistence with the clinical FISH (Fig. S2E). Approximately, 63.7% (65/102) of chromosome aneuploidy events detected by FISH were confirmed using scRNA-seq data (e.g., tri/tetrasomy chr11). Consistently, CCND1 upregulation was predominantly observed in samples with t(11;14) and FGFR3 and NSD2 were highly expressed in samples with t(4;14), although less frequent. However, in 3 patients with normal clinical FISH results (Fig. S2F), we observed high expression of translocation-related genes like CCND2, MAF and CCND3 (Fig. S2D), and chromosomal abnormalities such as gains of chrs 9, 11 and losses 14q, 22p in cPCs in patient MGUS01s; gains of chrs 5, 9, 11, 19 and losses of chrs 13, 14q, 16q in MGUS02s; and loss of 1p, gain of 1q and additional CNAs in NDMM02s (Fig. S2F). This shows improved sensitivity by scRNA-seq relative to clinical FISH at detecting chromosomal abnormalities. The information from scRNA-seq was therefore integrated with clinical FISH to refine the genetic stratification of patients into two subtypes, HY and non-HY (Fig. 1F & S2G).
Genomic abnormalities in cPCs contribute to transcriptional heterogeneity
The evidence that clusters having the same diagnosis and/or cytogenetic changes were closer in the low-dimensional embeddings (Fig. 1E) motivated us to interrogate the overall transcriptome similarity and to dissect factors that contribute to heterogeneity of cPCs. We performed unsupervised clustering of cPCs at the sample level (Methods). As observed in previous studies16,21, the clustering results were primarily driven by genomic alterations, with samples from the same cytogenetic groups being grouped together (Fig. 2A). Additionally, we measured sample transcriptome similarities using Bhattacharyya pairwise distance (BPD) calculated from the top-ranking Principal Components (Fig. 2B and Fig. S3A, methods). Overall, samples with translocations and/or SMM were transcriptionally more similar displaying smaller BPD. Unexpectedly, we observed larger distances between MGUS samples especially with HY and others, indicating that, in the MGUS stage, cPCs already showed high degree of transcriptome ITH comparable to that in late stage, whereas in the SMM stage, the heterogeneity decreased. What’s more, HY contributed more heterogeneity than translocations in different stages of MM (Fig. 2B and Fig. S3A). However, due to the small sample size, these observations need to be carefully interpreted and validated in a larger cohort.
Figure 2. Characterization of cPCs.
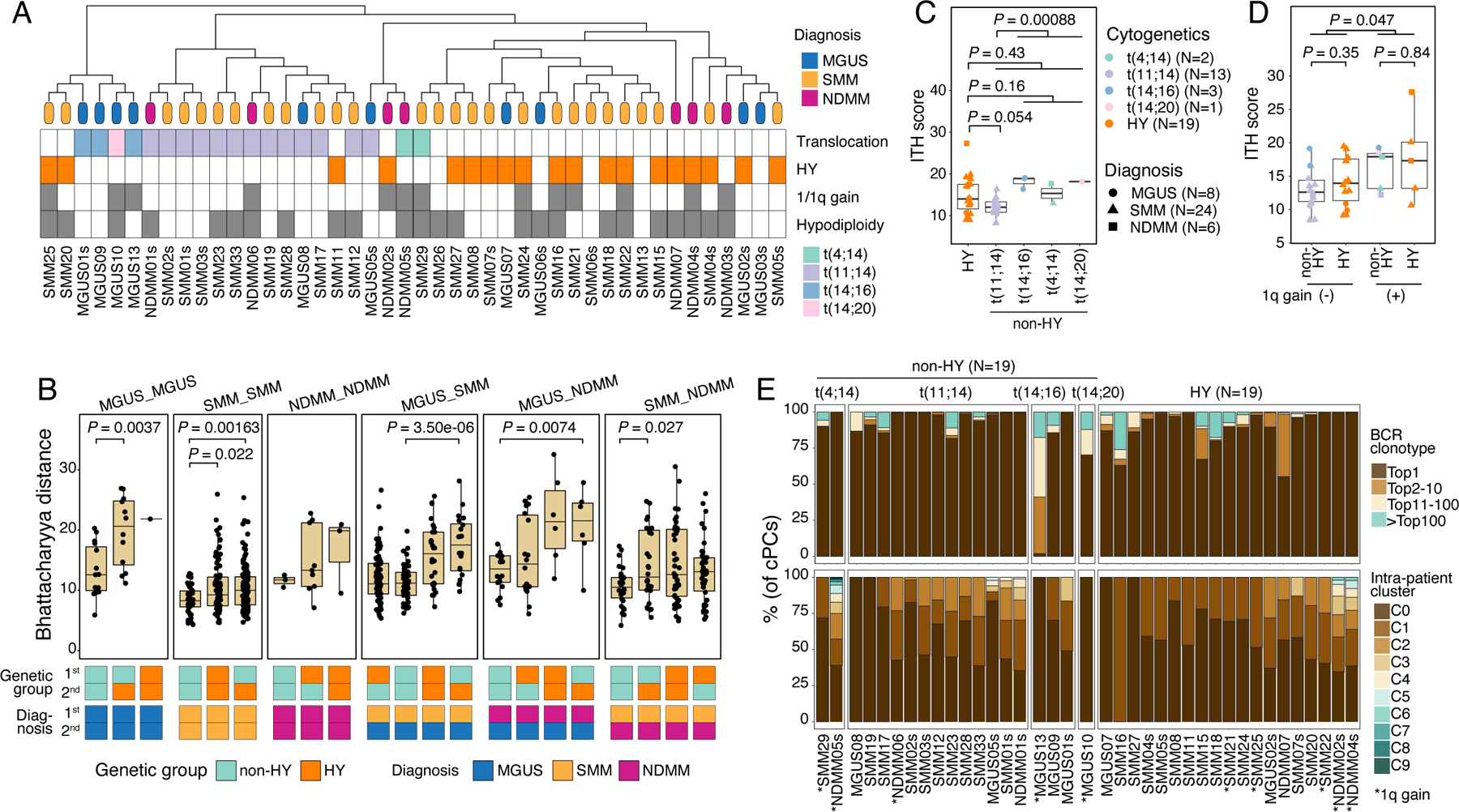
(A) Hierarchical clustering analysis (top) showing similarities of transcriptome of cPCs across patients (N=44). Leaves of the phylogenetic tree were color coded by their diagnosis. Annotations (bottom) showing the states of cytogenetic abnormalities of each patient. (B) Boxplot showing the comparisons of Bhattacharyya distance between different diagnosis-cytogenetics groups. Comparisons were ordered by their median values in each group. Annotation tracks at the bottom showed the diagnosis and cytogenetic abnormalities of the two samples between which Bhattacharyya distance was calculated. (C) Boxplots showing the comparison of ITH score between samples in HY and different translocation subgroups. Dots are color-coded by cytogenetic abnormalities and shape-coded by disease stages. (D) Boxplots showing the comparisons of ITH score between samples with 1q gain (N=10) and those without (N=28). Dots are color-coded according to cytogenetics abnormalities and shape-coded based on disease stages. (E) Stacked bar charts showing BCR clonotype (upper) and intra-sample subcluster (lower) distributions in each non-HY (N=19) and HY (N=19) patient. Clonotypes were grouped and colored by clonotype frequency categories. See also Figure S3.
We next compared sample-level ITH score, aneuploidy level and cell proliferative properties and found no significant difference of these properties between different diagnoses, though an increasing trend was shown. When comparing these properties between the two genetic subtypes, non-HY and HY, we observed higher degree of ITH as measured by Euclidean distance in high-risk translocations such as t(4;14), t(14;16) compared to that in low/intermediate risk translocation t(11;14) (Fig. 2C). Meanwhile, samples in the HY subtype showed an intermediate level of ITH when compared to the different translocation subgroups. As expected, we observed higher CNA scores and lower fractions of proliferating cells in samples of HY subtype, while the opposite was true in the non-HY patients, with different translocation subgroups showing a similar level of these properties (Fig. S3B). We also investigated the impact of 1q gain, a genomic alteration associated with high risk, on ITH levels (Fig. 2D). Our analysis revealed that samples with 1q gain had significantly higher levels of ITH, indicating a more heterogeneous nature. Notably, non-HY and HY samples with the same 1q status showed very similar levels of ITH, suggesting that 1q gain has a substantial contribution to ITH in both genetic subtypes. However, we observed no significant difference in aneuploidy levels and cell proliferative properties between samples with and without 1q gain in both subtypes (Fig. S3C). BCR clonality and intra-sample subclustering demonstrated an overall similar degree of clonal complexity between non-HY and HY (Fig. 2E). Notably, over a third of MGUS and SMM samples already exhibited a high degree of transcriptomic ITH at a level that is parallel to that of NDMMs (Fig. S3D). These results suggested that despite being initiated from different driver events and having distinct properties, myeloma cells acquired similar degree of clonal complexity during their evolution and progression and the acquisition of high-risk genomic events such as 1q gain would increase the transcriptomic ITH.
Intra-sample subcluster heterogeneity in expression of key oncogenic features
Comparative analyses between cPCs from non-HY and HY samples at both single-cell (Fig. S3E) and sample (Fig. S3F) levels identified significantly upregulated CCND1, CKS1B, SDF2L1, LY6E, DUSP1, CD99, LIME1 and CTHRC1 in cPCs of non-HY samples, which is consistent with the previous studies28,29. In contrast, expression levels of LAMP5, SULF2, SLAMF1, FAM30A, ITM2C were significantly elevated in cPCs of HY samples. In addition to unsupervised DEG analysis as described above, we also examined the expression of translocation related genes, other biologically important genes in myeloma cells16,30–39, as well as MHC class I genes in cPCs. We found MYC, SDC1, TNFRSF17, GPRC5D, FCRL5, FRZB and ASS1 increased as disease progressed (Fig. S3G) but they showed no difference between non-HY and HY subtypes. Likewise, expression of MHC class I genes showed no differences between non-HY and HY samples or by disease stage.
To better understand the transcriptional ITH across these cPC subclusters especially in expression of key oncogenic features, we quantified expression levels of key genes mentioned above across 130 cPC subclusters as well as in nPCs (Fig. 3A). Overall, we observed substantial ITH across cPC subclusters of individual sample (Figs. 3A and S4A) in both precursors and NDMM. For example, LAMP5 was differentially expressed across subclusters of MGUS06s, SMM08, SMM15, SMM22, NDMM03s, NDMM04s, NDMM05s et al. CKS1B was differentially expressed across subclusters of MGUS05s, MGUS06s, SMM01, SMM02, NDMM02s, NDMM03s, NDMM05s et al. One of the t(4;14) related genes, NSD2, was highly expressed in C1 but not C0 in SMM29; this gene was also differentially expressed in subclusters of NDMM05s. MYC was expressed in C3, C6, C7, C8 but not the other clusters in MGUS06s. Intra-sample subcluster heterogeneity may have therapeutic implications as different subclusters may be more or less sensitive to targeted treatments and may shape tumor evolution further.
Figure 3. Intra-sample subcluster heterogeneity of myeloma related key genes is associated with genomic abnormalities and tumor evolution.
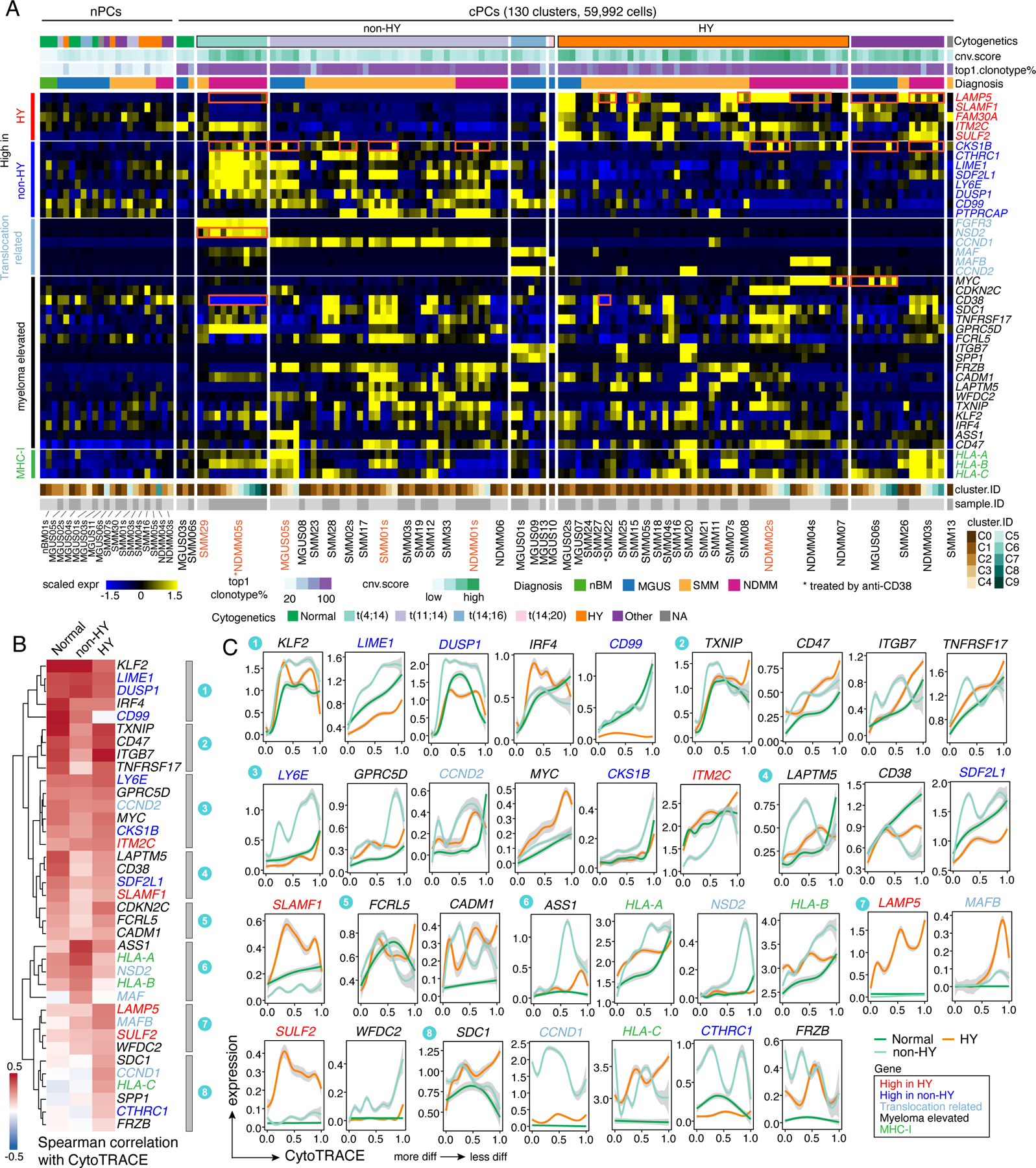
(A) Heatmap showing the scaled average expression of myeloma related key genes in PCs at intra-sample subcluster level. Clusters were firstly grouped by normal or clonal. Within the cPCs, clusters were ordered by cytogenetic abnormalities of corresponding patient origins. The top annotation tracks show (from top to bottom): the cytogenetic abnormalities of corresponding patient, the cluster averaged CNA score, proportion of cells harboring the most abundant clonotype in the cluster and diagnosis of corresponding patient. (B) Heatmap with hierarchical clustering by row showing spearman correlations of gene expression levels with CytoTRACE score separately in normal, non-HY and HY cells. Genes were color coded as shown in (A). (C) Two-dimensional plots showing the dynamic changes in expression levels of genes as shown in (A) along the CytoTRACE score. Genes were grouped by their cluster as shown in (B) and color coded as shown in (A). Shaded band, 95th confidence interval. See also Figure S4, Table S4.
To investigate the expression dynamics of key genes along the cell differentiation trajectory, we used CytoTRACE40 to infer cellular differentiation states of all PCs (Fig. S4B). Overall, we observed substantial variations of CytoTRACE scores in nPCs as well as cPCs. The increasing trend of CytoTRACE score from nPCs to cPCs in MGUS and NDMM (but not SMM) (Fig. S4C) suggested a less differentiated state in more advanced disease stage. Intriguingly, we noted that myeloma cells from non-HY and HY samples had distinct CytoTRACE score distributions (Fig. S4C) with HY having a higher score (less differentiated) and non-HY a lower score compared with normal. We then calculated the spearman correlations of gene expression level with CytoTRACE score separately in normal, non-HY and HY PCs, based on which the unsupervised clustering identified shared and unique patterns in these three groups (Fig. 3B). While most of those key genes had positive correlations with CytoTRACE score, we found some genes with different trends (Fig. 3B and 3C). For example, TNFRSF17 (BCMA) in cluster 2 exhibited strong correlation with CytoTRACE scores in normal and HY but not in non-HY PCs. LAPTM5 and CD38 in cluster 4 only showed strong correlations in nPCs but not in cPCs. SDC1 in cluster 8 only showed positive correlation in HY. HLA-A and HLA-B in cluster 6 showed a higher correlation in non-HY, while HLA-C in cluster 8 only showed positive correlation in HY. The genes mentioned above also enriched at the top (negative correlation) or bottom (positive correlation) in the corresponding ranked gene list when analyzing the expression dynamics of the top 2,000 variable genes along the CytoTRACE score (Fig. S4E). These results suggest that the expression of those myeloma related genes is not evenly distributed but dynamically changed along with tumor evolution and strongly associated with genomic abnormalities and cell differentiation states.
Pseudo-time reconstruction of malignant transformation reveals distinct evolution patterns
To better understand the trajectory of malignant transformation from normal B cells, we performed pseudo-time reconstruction of B lineage cells for each sample and annotated the cells with defined major cell types (B cells, nPCs and cPCs) by mapping them to the new embeddings from which the trajectories were reconstructed using Monocle241–43. We observed two different clonal evolution patterns: linear and branching (Fig. S5A–B), which were independent of cytogenetic abnormalities (fisher’s exact test, p=0.70) but marginally associated with disease stages (NDMM vs MGUS+SMM, fisher’s exact test, p=0.057). These observations suggest that during the early stage of tumorigenesis, tumor cells might favor linear evolution, while in the late stage, as the tumor expands and cells become more adaptable, branching evolution could potentially gain prominence.
MGUS01s is a representative case (among 16 cases shown in Fig. S5A) of the linear evolution pattern (Fig. 4). Unsupervised clustering analysis of all B and PCs in MGUS01s identified 3 B-cell clusters (i.e., CD34+ Pro B, MKI67+ proliferating Pre-B, and Pre/Immature B) and 5 PC clusters (Fig. 4A, Table S5). Pre/Immature B cells were identified based on their expression of CD19/CD24/CD81/IGHM and downregulation of rearrangement related genes such as DNTT and VPREB1. The five PC clusters (C0-C4, Fig. 4A) highly expressed PC signature genes such as SDC1 (CD138) and MZB1 plus cluster-specific expression features (Table S5). Pseudo-time reconstruction suggested a path from pro B to Pre B, Immature B, as well as plasma cells C0, C1, C2, C3 and C4 (Fig. 4A), with gradually increased levels of aneuploidy (Fig. 4B) and BCR clonal expansion (Figs. 4C) along the cell differentiation trajectory inferred by Monocle341–43. Notably, cells of C0 and C1 were polyclonal with diverse BCR clonotypes while cells of C2, C3, and C4 were clonal and shared the same (top 1) BCR clonotype (Fig. 4A), implying that cells of these 3 clusters were expanded from the same ancestor cell. However, distinct CNAs (Fig. 4D) and expression profiles (Fig. 4E) were observed among these 3 clusters. For example, C3 shared similar CNAs with C4, and cells of both clusters acquired additional CNAs when compared to C2 (Fig. 4D). Consistently, C4 shared more cluster specific differentially expressed genes (DEGs) with C3 than with C2 (Fig. 4E). DEG analysis between C4 and C3 identified upregulated genes like ASS1, PTP4A3 and PRPSAP2 in the ‘terminally’ differenced cluster C4 (Fig. 4F, Table S5), and these 3 genes were also upregulated in the late stage in our independent cohort (Fig. 4G). Analysis of expression dynamics along the cell differentiation trajectory revealed interesting cluster specific expression features (Figs. 4H–J). For example, ITGB7 and CCND2 were highly enriched in cells of C2, C3, and C4, KLF2 was highly expressed in cells of C3 and C4, while ASS1 was specifically enriched in C4 cells (Fig. 4I). In addition, we found highly enriched pathways in cells of C3 and C4 (vs. C2) including hypoxia, apoptosis, glycolysis, TGF-B signaling, NF-kB and P53 pathways (Fig. 4K).
Figure 4. Intra-patient heterogeneity and evolution of B/plasma subpopulations for patient MGUS01s.
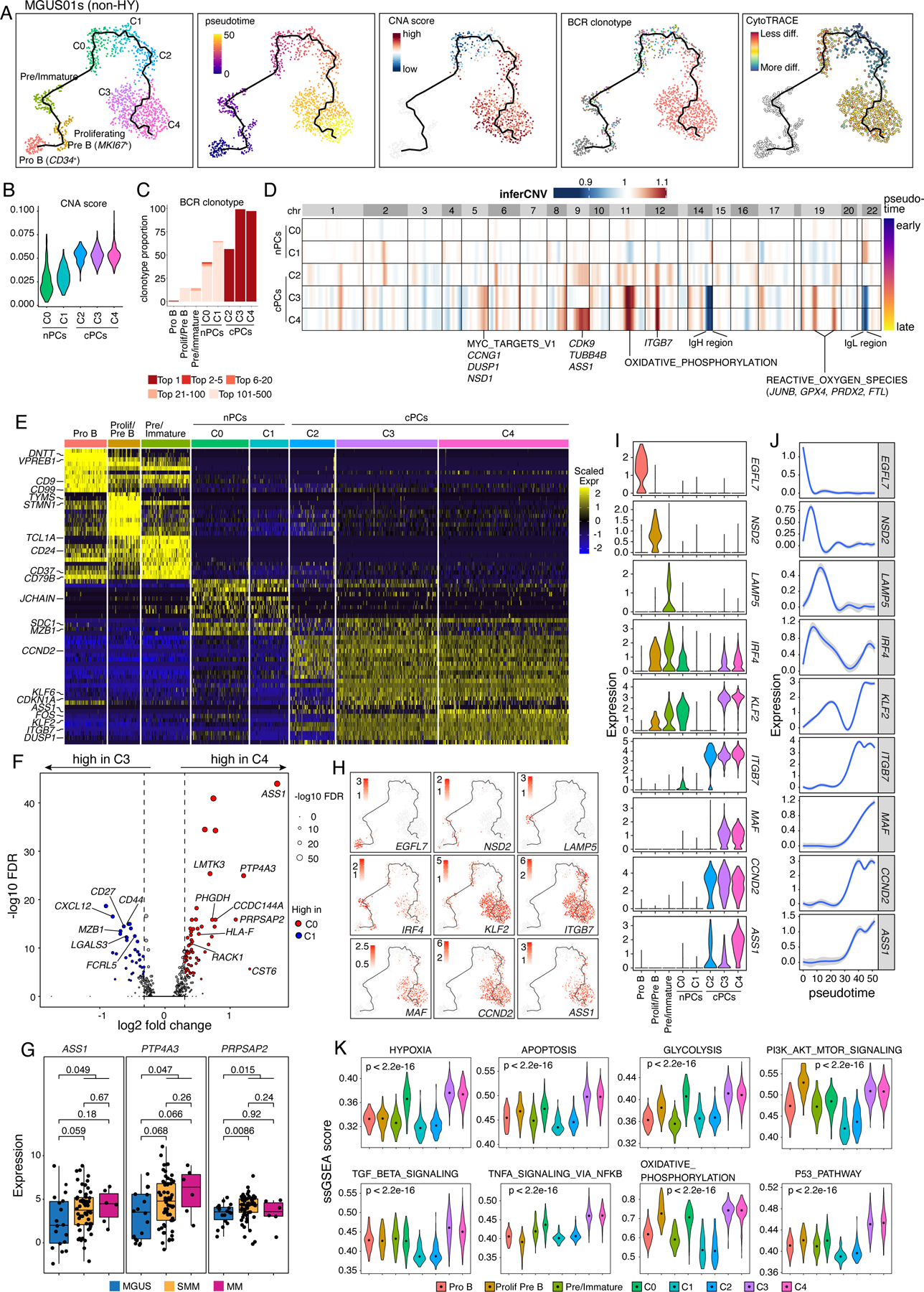
(A) Uniform manifold approximation and projection (UMAP) visualization of unsupervised clustering analysis of B cells and PCs from MGUS01s (n=1,210) with inferred trajectory and cluster label, pseudotime, CNA score, BCR clonotype and CytoTRACE score (from left to right) mapped on. Pro B cell cluster was selected as the root of trajectory. (B) Violin plots showing the differences of CNA scores in 5 PC subclusters. Clusters were ordered from early stage to late stage based on inferred pseudotime. (C) Stacked bar chart showing BCR clonotype distributions in each B and PC subclusters. Clonotypes were grouped and colored by clonotype frequency category. (D) Heatmap on the top showing genome wide inferred CNA profiles for 5 PC clusters of patient MGUS01s. Clusters were ordered by inferred pseudotime as shown on the right-hand side. Enriched hallmark pathways or important genes related to the most striking differences of CNAs between 5 clusters were labelled at the bottom. (E) Heatmap showing scaled expression of top 10 DEGs of B and PC clusters, with selected cluster specific DEGs labeled on the left. (F) Volcano plots of differentially expressed genes between PC cluster C3 and C4. FDR, two-sided Wilcoxon rank sum test with Bonferroni correction. Dashed line, log2FC > 0.3 or < −0.3. Labels, biologically important genes. (G) Boxplot showing the comparisons of expression levels of ASS1, PTP4A3 and PRPSAP2 between MGUS, SMM and MM in our validation cohort. (H) UMAP as in (A) showing the expression of representative genes. (I) The violin plots showing expression of representative genes across different B and PC clusters. Clusters were ordered from early stage to late stage based on inferred pseudotime. (J) Two-dimensional plots showing the dynamic changes in expression levels of representative genes as shown in (H&I) along the pseudotime. Shaded band, 95th confidence interval. (K) The violin plots showing gene set enrichment as measured by ssGSEA score of 10 representative hallmark pathways across different B and PC clusters. P values were calculated by one-way Kruskal-Wallis rank-sum test. See also Figure S5, Table S5.
However, we are still able to observe branching evolution patterns at early stages like MGUS and SMM (Fig. S5). MGUS06s and SMM07s are representative cases (Fig. 5). In MGUS06s, we identified 9 PC clusters, each of which was characterized by unique transcriptome and/or CNA profiles (Figs. 5A–C, Table S5). C5 had no detectable CNAs while the other 8 clusters were cPCs. C2 cells are polyclonal and serve as the root of the branching evolution model. Six cPC clusters were expanded from the same ancestor cell but were split into 3 major branches (C1, C0/C4, C3/C6/C8) (Fig. 5A) and further diverged into 6 clones with distinct CNAs and expression profiles (Figs. 5B–D). Cells of all 3 branches highly expressed CCND2 but only the branch of C3/C6/C8 showed unique expression of LAMP5. Likewise, sample SMM07s showed a similar evolution pattern (Fig. 5E–H, Table S5). Among 4 PC clusters, C2 was a normal-like and serves as a root of the branch evolution of clusters C1, C0 and C3. Although they carried the same BCR clonotype, cells of C0 showed distinct CNAs from C1 indicating divergent evolution.
Figure 5. Intra-patient heterogeneity and evolution of B/plasma subpopulations for patient MGUS06s and SMM07s.
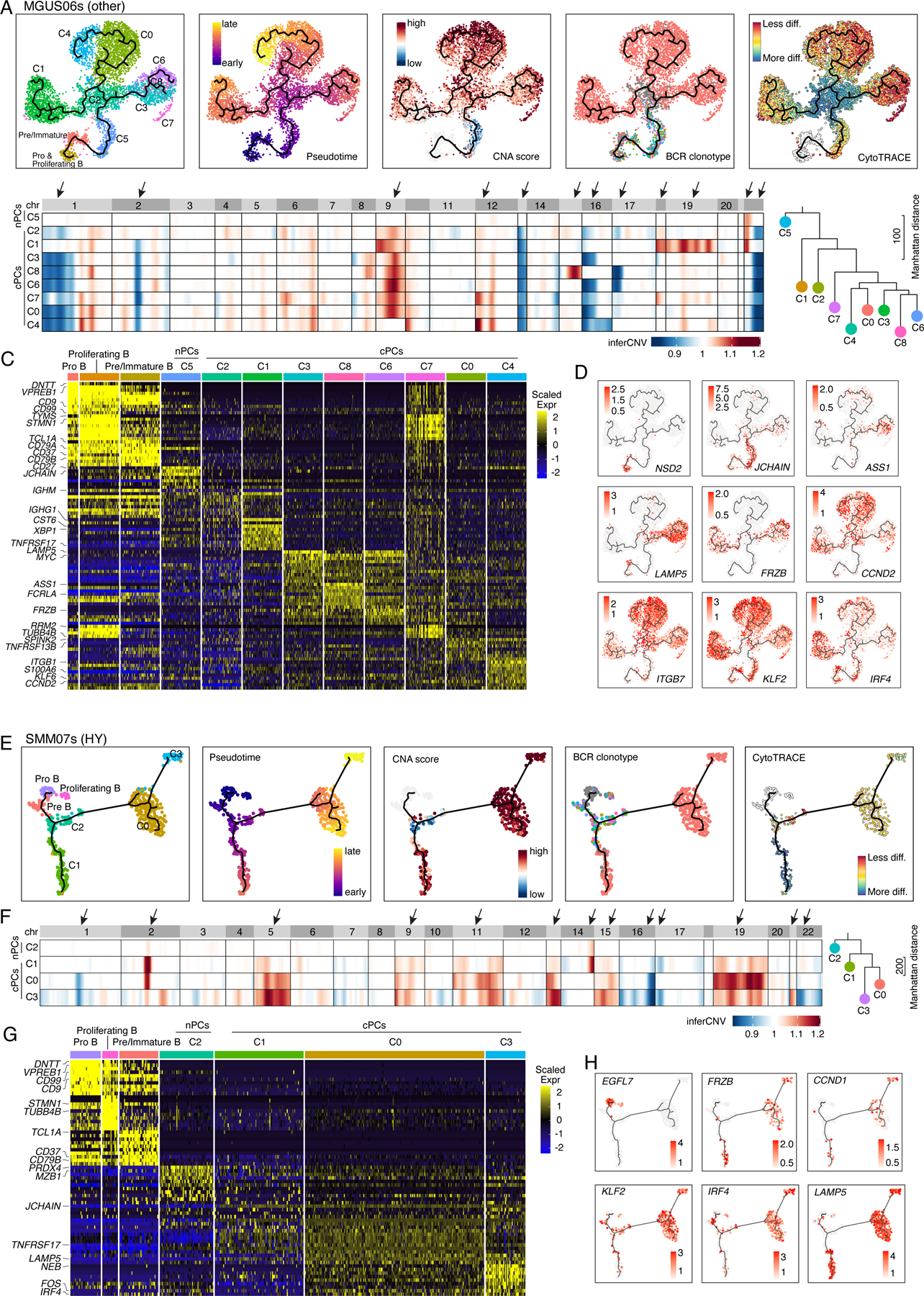
(A) Uniform manifold approximation and projection (UMAP) visualization of unsupervised clustering analysis of B cells and PCs from MGUS06s (n=5,660) with inferred trajectory and cluster label, pseudotime, CNA score, BCR clonotype and CytoTRACE score (from left to right) mapped on. Pro B cell cluster was selected as the root of trajectory. (B) Heatmap on the left showing genome wide inferred CNA profiles for 9 PC clusters of patient MGUS06s. Clusters were organized by inferred trajectories. Black arrows indicated the most striking differences of CNAs between 9 clusters. Phylogenetic tree on the right showing minimum evolution trees generated using cluster-averaged/consensus CNA profiles of subclusters for MGUS06s and rooted by a neutral node. (C) Heatmap showing scaled expression of top 10 DEGs of B and PC clusters, with selected cluster specific DEGs labeled on the left. (D) UMAP as in (A) showing the expression of representative genes. (E) UMAP visualization of unsupervised clustering analysis of B cells and PCs from SMM07s (n=483) with inferred trajectory and cluster label, pseudotime, CNA score, BCR clonotype and CytoTRACE score (from left to right) mapped on. Pro B cell cluster was selected as the root of trajectory. (F) Heatmap on the left showing genome wide inferred CNA profiles for 4 PC clusters of patient SMM07s. Clusters were organized by inferred trajectories. Black arrows indicated the most striking differences of CNAs between 4 clusters. Phylogenetic tree on the right showing minimum evolution trees generated using cluster-averaged/consensus CNA profiles of subclusters for SMM07s and rooted by a neutral node. (G) Heatmap showing scaled expression of top 10 DEGs of B cell and PC clusters, with selected cluster specific DEGs labeled on the left. (H) UMAP as in (A) showing the expression of representative genes. See also Figure S5, Table S5.
Differential remodeling of TME in non-HY and HY samples
We then analyzed TME cells and identified 15 major cell types (Fig. 6A, Fig S6A–B, Table S6). To explore the role of disease burden in TME dysregulation, we stratified SMM patients by IMWG 20/2/20 score44. We observed great variations in cellular compositions of these major cell types across different disease stages (Fig. 6B and Fig. S6C) and within sample groups that carried the same genomic alterations (Fig. 6C and Fig. S6D). Partially consistent with previous studies14,18,21,45, we observed trends toward decreased proportions of mature B cell, NK cell, CD14+ monocyte, and increased proportions of CD8+ T cell, CD16+ monocytes, TAMs, endothelial and fibroblasts when disease burden increased (Fig. S6C). Notably, we noted substantial differences in TME landscapes between samples with distinct genomic abnormalities (Fig. 6C and Fig. S6D). Despite the great variations between different translocation subgroups, we observed significantly decreased proportions of CD8+ T cells and increased proportions of CD14+ and CD16+ monocytes and DCs in non-HY samples (Fig. 6C and Fig. S6D) compared to HY samples. Such differences were unlikely related to disease stages based on our stratified analyses (Fig. S6E). We also observed significantly decreased proportions of rare cell types like TAM, Endothelial and Fibroblast in non-HY SMM samples (Fig. S6F). Furthermore, we explored the correlations of TME cellular compositions with tumor properties (Fig. S6G) mentioned in Fig. 2C as well as CytoTRACE score within each subtype and found differential remodeling of TME between non-HY and HY samples. For example, the fractions of CD14+ monocytes were negatively correlated with myeloma cell proliferative properties in HY samples while the correlation was not significant in non-HY samples. The fractions of cDC2 were positively correlated with levels of aneuploidy in HY samples but in non-HY sample, a reversed trend was observed. Our results suggested that differential adaptations in the TME between non-HY and HY samples might be driven by distinct genomic alterations in the myeloma cells. These findings are hypothesis-generating and require confirmation through follow-up functional studies.
Figure 6. Landscape of tumor microenvironment.
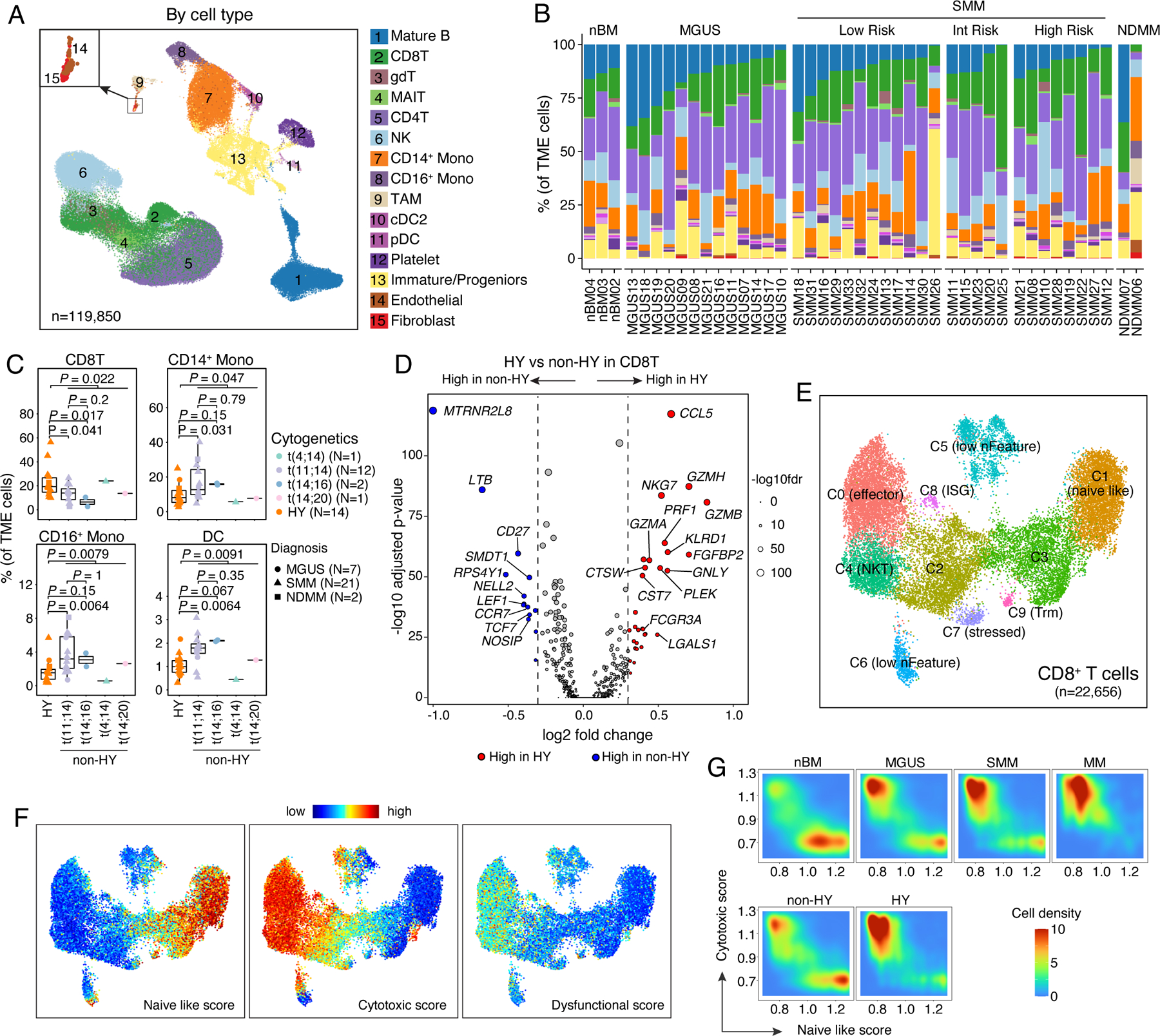
(A) Uniform manifold approximation and projection (UMAP) visualization of unsupervised clustering analysis of all TME cells (n = 119,850). Cells were color coded for their identified cell types. (B) Stacked bar plots showing corresponding % cell type composition in each unsorted sample grouped by diagnosis. (C) Boxplots showing the comparisons of CD8 T cell, CD14+ monocyte, CD16+ monocyte and DC proportion in tumor microenvironment between samples in HY and different translocation subgroups. Dots are color-coded by cytogenetic abnormalities and shape-coded by disease stages. (D) Volcano plots of differentially expressed genes in CD8 T cells between non-HY and HY samples. FDR, two-sided Wilcoxon rank sum test with Bonferroni correction. Dashed line, log2FC > 0.3 or < −0.3. Labels, biologically important genes. (E) UMAP visualization of unsupervised sub-clustering analysis of CD8 T cells (n = 22,656). (F) UMAP as shown in (E) with cells colored by naïve-like score, cytotoxic score and dysfunctional score (from left to right) calculated using ssGSEA method. (G) Distribution of CD8 T cells based on their naïve like score and cytotoxic score in patient at different stages of disease (first row) or with different cytogenetic abnormalities (second row). See also Figure S6, Tables S3, S6.
For each major cell type, different cell states were identified based on unsupervised clustering (Table S6). DEG analysis in CD8+ T cells between non-HY and HY identified upregulated cytolytic activity (e.g., Granzymes, NKG7, PRF1 and GNLY) in HY samples (Fig. 6D). Unsupervised clustering analysis identified ten CD8+ T cell clusters (Fig. 6E, Table S6) but none of them showed an exhausted phenotype (Fig. 6F). Consistently, CD8+ T cell in HY samples trended to have higher cytotoxic scores and lower naïve like scores (Fig. 6G) and we noted significantly increased proportions of C0 and decreased proportions of C3 and C9 in HY samples (Fig. S6H).
Similar analysis was performed on CD14+ monocytes. We found upregulation of pro-inflammatory cytokines and chemokines such as IL1B, CCL3, CXCL8, interferon-induced genes such as IFITM3, ISG15, IFI6, NFKB, inhibitor genes such as NFKBIA, NFKBIZ, TNFAIP3 in HY samples (Fig. S6I), suggesting an immunoactivating microenvironment. Further analysis of CD14+ monocytes identified six clusters (Fig. S6I and Table S6), but we found no differences in their cellular abundance/compositions with advanced stages or different genomic abnormalities, the same for the other major cell types including the regulatory T cells (CD4T_C3 in Table S6) in the comparative analysis.
Lastly, we inferred cellular interactions between cPCs and TME cells based on ligand-receptor (L-R) co-expression using CellPhoneDB (v2.0)46. Consistent with previous reports21, cPCs showed the strongest interactions with myeloid cells including monocytes and DCs (Fig. S7A), particularly in HY samples (Fig. 7A). Further investigation identified elevated interactions between cPCs and TME cells via CD74-MIF, ICAM1-AREG, TNFRSF1A/B-GRN, CCL3-CCR1, LGALS9-CD47 (Fig. 7B). We also observed potential reciprocal interactions between MIF and TNFRSF14, both of which were highly expressed in cPCs and a variety of TME cells (Fig. S7B). Intriguingly, we found most of the differential interactions between non-HY and HY were contributed by cell adhesion molecules (ICAMs, F11R) and CD74 from cPCs and their receptors from TME (Fig. 7B, Fig. S7B). ICAM1 was differentially expressed in cPCs in this study (Fig. 7C) as well as in a larger validation cohort (Fig. S7C) between non-HY and HY samples while this difference was observed at the relatively later stage of tumor cell differentiation (Fig. 7D). Interactions enhanced in non-HY samples includes CD94:NKG2A_HLA-E in γδ T, CD99_PILRA in CD16+ monocytes, both of which potentially convey inhibitory signals to corresponding immune cells; and CCL3_CCR1/5, FCER2_aM/Xb2 complex, which may be involved in recruiting, migration and adhesion of NK and myeloid cells to myeloma cells. Further investigation is needed to determine whether differential cellular interactions is associated with distinct oncogenic or immunological features we observed between these two genetic subtypes.
Figure 7. Cellular interactions between cPCs and TME cells.
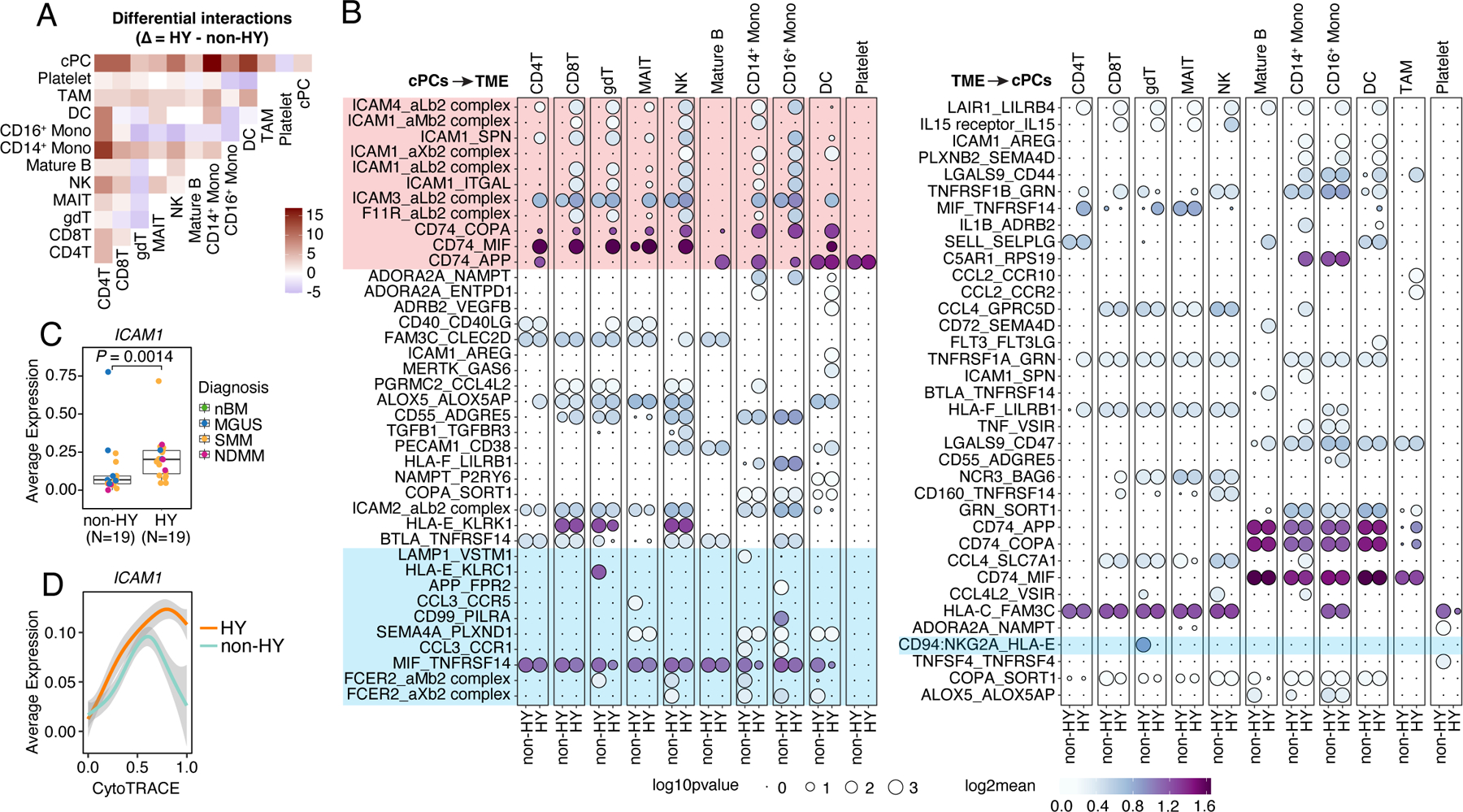
(A) Heatmap showing the different number of interactions between each major cell types (#cells > 1000) inferred by CellphoneDB between non-HY and HY samples. Positive number means increasing in HY. (B) Bubble plot showing the mean expression (color key) and significance (size key) of ligand-receptor pairs between cPCs and TME cells. Only interactions that showed differences between non-HY and HY are shown. (C) Boxplots showing the comparisons of averaged expression of ICAM1 in cPCs between non-HY (N=19) and HY (N=19) at sample level. Dots were color coded by diagnosis. (D) Two-dimensional plots showing the dynamic changes in expression level of ICAM1 along the CytoTRACE score. Shaded band, 95th confidence interval. See also Figure S7.
DISCUSSION
The most common cancer affecting PCs is multiple myeloma. Although revolutionary gains in the biological understanding and treatment of MM have been accomplished in the first part of the twentieth century, much remains incompletely understood, including the cellular and molecular drivers of malignant transformation, the patterns of tumor evolution and the immunological changes in the TME in the context of different genetic background. Importantly, we know little about the earliest changes in precursor disease. A better understanding of these would help to identify potential biomarkers of interest that could be clinically translated for patient benefit. To this end, we analyzed, to our knowledge, the largest cohort of myeloma precursors using a paired scRNA-seq and scBCR-seq approach derived from fresh bone marrow aspirates.
In this study, we show a high degree of genomic (e.g., CNA scores and aneuploidy profiles) and transcriptional ITH (e.g., expression profiles and transcriptional states as well as cell differentiation status) in both precursor diseases and NDMM, despite the same origin of all cells in each individual sample based on BCR clonotype analysis. We note that both disease stages and genomic abnormalities can influence phenotypic ITH of myeloma, and their relative contribution may vary across individuals. Notably, we observed substantial transcriptome ITH in cPCs in MGUS, the very early stage of MM. It is worth mentioning that our deep analysis of intra-sample subcluster heterogeneity in expression profiles of key myeloma related genes16,30–39 in the very early stage of MM as well as NDMMs may help better understand disease progression and tumor evolution in the context of therapy and have important therapeutic implications.
Importantly, the addition of tumor genomic scRNA-seq results to standard of care clinical FISH data refines and synergistically improves the identification of the different genetic subtypes of myeloma. It is well established that low tumor purity in myeloma precursor disease can severely limit the sensitivity of FISH in identifying known genomic abnormalities in myeloma. The combination of scRNA-seq and scBCR-seq overcomes this limitation and allows us to study the transcriptomic make up of even the earliest precursors of myeloma, such as MGUS.
The generation of paired scBCR-seq data allows phenotypic and genotypic integration and importantly, it can be used to trace the genomic and phenotypic evolution of myeloma cells at very early stage of the disease course. We observed two main evolution patterns in our cohort: linear and branching, which were independent of cytogenetic abnormalities but marginally associated with disease progression. It’s possible that this difference represents an underlying propensity for stable versus progressive disease, where precursors with branching evolution tend to be more progressive. However, due to the lack of longitudinal samples, we cannot determine the plasticity of evolution patterns over time. Our observation of different evolution patterns was in line with earlier reports by Morgan et al. using whole-exome sequencing and single-cell DNA sequencing47, indicating that paired scBCR-seq and scRNA-seq can be used to accurately infer myeloma evolution. Notable, our analysis provided important information on also clonotypic and phenotypic evolution of myeloma cells as well as cell differentiation states and gene expression dynamics along the evolution trajectory, which are missing from DNA-based platforms. Such information may help identify the cellular and molecular drivers promoting disease evolution and discover unique targets in the “terminally” differentiated myeloma cells for the development of new treatment. For example, ASS1 was specifically enriched in the ‘terminally’ differenced cluster C0 in MGUS01s. ASS1 has been reported to be critical for the growth of human cancers as it involves in the biosynthesis pathway of arginine which plays an important role in tumor metabolism and the immune system38. ASS1 overexpression is associated with an increased proliferation property of the plasma cells38 and lack of ASS1 sensitizes CD138+ myeloma cells to arginine depletion/analogue treatment48,49. Consistently, ASS1 has been reported highly expressed in non-responders to a quadruple treatment in primary refractory MM17. Nevertheless, it would be of great interest to better understand how therapy shape the patterns of myeloma cell evolution and how different evolution patterns contribute to the survival benefit of tumor cells.
Lastly, we further delineated TME landscape to better understand the immunological changes during disease progression. Our analysis revealed that TME changes may occur at very early stage during the disease course and TME landscape might be influenced by distinct genomic abnormalities. Differences in the states of CD8+ T cells and myeloid cells and their interaction networks between HY and non-HY samples are potentially interesting but would need further investigation to better understand their immunological consequences.
Our manuscript has some limitations including the small number of NDMM samples and the absence of longitudinally collected samples along the disease course. The insufficient number of progressors hampers further comparisons between progressors and non-progressors, which would be more clinically significant. The analysis with different translocation subgroups separated is limited by small sample size and need to be validated in larger patient cohorts. Despite this, it has numerous strengths including the prospective nature of sample acquisition, the large number of precursor myeloma samples, the generation of paired scBCR-seq data for accurate inference of clonal evolution, and integration with genomic subtypes. Some of our discoveries may have important clinical and therapeutic implications as this study provides an invaluable resource to the community and can be further leveraged to identify new targets and develop new strategies for myeloma precursor disease.
STAR METHODS
KEY RESOURCES TABLE
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Elisabet E. Manasanch (eemanasanch@mdanderson.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All single-cell RNA-sequencing data generated by this study have been deposited in the European Genome-Phenome Archive (EGA, https://ega-archive.org/). The data can be accessed under the accession number EGAS00001006694. Bulk mRNA-seq expression data (normalized) generated by Multiple Myeloma Research Foundation (MMRF) CoMMpass study (MMRF-COMMPASS) were downloaded from NCI Cancer Genomic Data Commons (NCI-GDC: https://gdc.cancer.gov). SeqFISH results of MMRF-COMMPASS samples were downloaded from the MMRF researcher gateway portal (https://research.themmrf.org). All other data supporting the findings of this study and Code used for all processing and analysis are available from the corresponding author upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human samples
Primary bone marrow samples were collected from normal donors and patients with MGUS, SMM or MM at MD Anderson Cancer Center. Patients were enrolled in studies LAB01–473, PA15–0575, 2005–0656 and 2015–0148 which were approved by the MD Anderson Cancer Center Institutional Review Board (Appendix 1). All the patients and healthy donors signed an informed consent in accordance with the Declaration of Helsinki protocol.
METHOD DETAILS
Patient samples and cell preparation
Whole bone marrow (n=46) was collected and processed by density gradient centrifugation (StemCell Technology Lymphoprep Catalog #07851). For patients with available CD138+ selected samples (n=19), the EasySep CD138 positive selection kit (CAT#:17887A) was used for CD138+ enrichment through magnetic bead separation. After processing, samples were transferred to the MD Anderson Genomics Core for library construction and sequencing.
Sequencing library construction using the 10x Genomics platform
10X Genomics 5’ scRNA and BCR enrichment, and next generation sequencing was performed by the Advanced Technology Genomics Core (ATGC) facility. Briefly, cell counts and viabilities were determined from investigator submitted cell suspensions in 1X PBS, using trypan blue exclusion and the Countess 3 FL automated cell counter (Life Technologies). Sample processing was performed following 10X Genomics’ 5’ scRNA-seq (version 1.1) and BCR enrichment guidelines (referencing user guide: Chromium Next GEM Single Cell V(D)J Reagent Kits v1.1). QC steps after cDNA amplification, BCR enrichment, and library preparation steps were carried out by utilizing both ThermoFisher Qubit HS dsDNA Assay along with Agilent (Santa Clara, CA) HS DNA Bioanalyzer for concentration and quality assessments, respectively. Sample libraries were normalized to 5 nM for pooling. Sample Library concentrations were verified using qPCR using a KAPA Biosystems KAPA Library Quantification Kit (KAPA/Roche Diagnostics). The gene expression libraries and BCR libraries were pooled in a ratio of 5 volumes gene expression library to 1 volume BCR library. The pool was sequenced using a NovaSeq6000 S4–200 cycle flow cell. The run parameters used were 26 cycles for read 1, 91 cycles for read2, 8 cycles for index1, and 0 cycles for index2 as stipulated in the protocol mentioned above.
Raw sequencing data processing, quality check and data filtering
The raw scRNA-seq data were pre-processed (demultiplex cellular barcodes, read alignment to human reference genome build 38, and generation of gene count matrix) using Cell Ranger Single Cell Software Suite (v5.0.0) provided by 10x Genomics. Detailed QC metrics were generated and evaluated. Cells with low complexity libraries or likely cellular debris (in which detected transcripts are aligned to less than 200 genes) were filtered out and excluded from subsequent analyses. Low-quality cells where >15% of transcripts derived from the mitochondria genome were considered apoptotic and also excluded. In addition, cells with number of detected genes >6,500 were discarded to remove likely doublet or multiplet captures.
Data normalization, unsupervised cell clustering and dimensionality reduction
Library size normalization was performed using the function NormalizeData in Seurat v4 (version 4.0.3)50 on the filtered gene-cell matrix to obtain the normalized UMI count. FindVariableFeatures of Seurat was applied to the normalized gene-cell matrix to identify highly variable genes (HVGs) for unsupervised cell clustering. Principal component analysis (PCA) was performed on the top 2000 HVGs. For plasma cells immunoglobulin genes were masked when performing PCA. For TME cells, Harmony51 was used to remove batch effects in the PCA space with the default parameters. The elbow plot was generated with the ElbowPlot function of Seurat and based on which, the number of significant principal components were determined. The FindNeighbors function of Seurat was used to construct the Shared Nearest Neighbor (SNN) Graph, based on which the unsupervised clustering was done with Seurat function FindClusters. Different resolution parameters for unsupervised clustering were then examined in order to determine the optimal number of clusters. For visualization, the dimensionality was further reduced using Uniform Manifold Approximation and Projection (UMAP)52 method with Seurat function RunUMAP. The principal components used to calculate the embedding were as the same as those used for clustering.
Doublets removal and determination of major cell types and cell states
This was achieved simultaneously by several rounds of hierarchical subclustering. In each round, unsupervised cell clustering and dimensionality reduction was performed as mentioned above. Differentially expressed genes (DEGs) were identified for each cluster using the FindAllMarkers function in Seurat R package. Top-ranked DEGs in each cell cluster were carefully reviewed and cell clusters were annotated according to the enrichment of canonical marker genes. Clusters with hybrid cell lineage markers were identified as doublets and thus removed. The first round of clustering on all cells identified TME cells and PCs. The second round of clustering was performed on TME cells and further dissected them into T and NK cells, B cells, myeloid cells and progenitor cells. Non-B/plasma cells with productive BCRs were removed. Several other rounds of subclustering were performed on each of these major cell types hierarchically and finally we identified 15 major cell types including mature B cells, 4 T cell subsets (CD4+ T, CD8+ T, γδ T, and MAIT cells), NK cells, 5 myeloid cell subsets (CD14+ and CD16+ monocytes, tumor associate macrophages (TAMs), cDC2 and pDCs), as well as endothelial cells, fibroblasts, platelets and immature/progenitor cells. We also identified multiple cell states of B cells, CD4+ and CD8+ T cells, NK cells and CD14+ monocytes by this way.
Identification of cPCs
To identify cPCs from nPCs, we performed subclustering analysis of PCs by sample. The resolution of each subclustering was manually adjusted so that the resulted intra-sample subclusters had either distinct differentially expressed genes or unique CNA profiles. CPCs were identified at intra-sample subcluster level based on the clonality of BCR as well as the CNA profiles inferred from single cell RNA sequencing data using inferCNV (v1.5.0)53.
Unsupervised Hierarchical clustering of cPCs
Pairwise Spearman correlations were calculated based on average expression level (Seurat function AverageExpression) of top 2000 highly variable genes (HVGs) with immunoglobulin variable genes excluded in cPCs from 44 samples which have more than 50 cPCs. HVGs were selected at the sample level by first generating pseudo-bulk for cPCs in each sample using the single-cell data and then selecting genes with top standardized variance. Pairwise Euclidean distances between samples were calculated from the correlation matrix. Hierarchical cluster analysis was performed by R function hclust on distance matrix and the dendrogram was drawn using R package ggtree.
Quantification of transcriptomic similarities of cPCs between patients
The Bhattacharyya distance metric (a distance metric that is effective at comparing pairwise probability distributions) was used to measure the similarity of gene expression distributions for cPCs of all pairs of patients. We embedded cPCs into PCA space using the highly variable genes and retained the top 40 principal components for computing the Bhattacharyya pairwise distance between patients using a similar approach as described previously54,55 as follows
where and are the mean vectors of each distribution, and .
BCR V(D)J sequence assembly, paired clonotype calling and integration with scRNA-seq data
Cell Ranger v5.0.0 for V(D)J sequence assembly was applied for BCR reconstruction and paired BCR clonotype calling. The CDR3 motif was located and the productivity was determined for each cell. The clonotype landscape was then assessed and the clonal fraction of each identified clonotype was calculated. The BCR clonotype data was then integrated with the B/plasma-cell phenotype data inferred from single cell gene expression analysis based on the shared cell barcodes.
Correlation of expression dynamics with differentiation states
To investigate the overall relationship of expression of myeloma related key genes with cell differentiation states, CytoTRACE40 was applied with default parameters to infer cellular differentiation states of all PCs. To show the dynamics of gene expression with cell differentiation states, gene expression values were plotted along the CytoTRACE score axis and the smoothed lines was generated using the geom_smooth function in ggplot2, with smoothing methods automatically determined by the size of data. We further checked the ranks of these key genes among top 2,000 highly variable genes regarding the correlation of expression level with CytoTRACE score. Downsampling was performed to avoid the domination of samples with large cell numbers.
Reconstruction of cPCs differentiation trajectories
To reconstruct the differentiation trajectory of cPCs and identify the evolution pattern, we firstly applied Monocle 241–43 with default parameters to infer the pseudotemporal ordering of cells according to their transcriptome similarity for samples with more than 50 PCs. In addition, to provide a better visualization, Monocle3 (version 0.2.0)41–43 was also applied to infer the differentiation states of B cells and PCs for MGUS01s, MGUS06s and SMM07s. For the pseudotime analysis, we selected the B cells at earliest stage as the root of the trajectory.
Statistical analysis
In addition to the algorithms described above, all other basic statistical analysis was performed in the R statistical environment (v4.0.0). All statistical tests performed in this study were two-sided. Statistical significance of differences observed between tumor and normal was determined by non-parametric Mann-Whitney U test when comparing continuous variables like gene expression level. The Spearman’s correlation coefficient was calculated to assess the association between two continuous variables (such as expression levels of two genes, proportions of two cell types) at sample level. To control the false discovery rate (FDR) and correct p-values for multiple testing, we apply Benjamini-Hochberg method56 and an FDR adjusted p-value (or q-value) < 0.05 is considered as statistically significant.
Supplementary Material
Table S1- Summarized and detailed clinical characterization of each individual patient, related to Figure 1.
Table S2- scRNA and scBCR sequencing matrix, related to Figure 1.
Table S3- Number of cells per cell type per sample and number of cells with BCR per sample, related to Figures 1 and 6.
Table S4- Cell counts and sequencing matrix per intra-sample subcluster of PCs, related to Figures 3 and S1.
Table S5- Top 50 DEGs between B lineage cell subsets of MGUS01s, MGUS06s and SMM07s (related to Figures 4 and 5).
Table S6- Top 50 DEGs between subclusters of major TME cell types including CD4+ T cells, CD8+ T cells, NK cells, B cells, Myeloid cells, CD14+ and CD16+ monocytes (related to Figures 6 and S6).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Normal donor human bone marrow samples | University of Texas Lymphoma/Myeloma Department Myeloma Tissue Bank | https://www.mdanderson.org/research/departments-labs-institutes/departments-divisions/lymphoma-myeloma.html |
| Myeloma precursor disease and multiple myeloma patient bone marrow samples | University of Texas Lymphoma/Myeloma Department Myeloma Tissue Bank | https://www.mdanderson.org/research/departments-labs-institutes/departments-divisions/lymphoma-myeloma.html |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 5’ Library and Gel Bead kit v1.1, 16 rxns. | 10x Genomics | 1000165 |
| Chromium Single Cell V(D)J Enrichment Reagent Kits Human Bcells | 10x Genomics | 1000016 |
| Chromium Next GEM Chip G Single Cell kit | 10X Genomics | 1000120 |
| Qubit HS dsDNA Assay | ThermoFisher | Q32854 |
| HS DNA Bioanalyzer | Agilent | 5067–4626 |
| KAPA Biosystems KAPA Library Quantification Kit | KAPA/Roche Diagnostics | KK4835/079602040 01 |
| EasySep CD138 Positive Selection Kit | StemCell Technology | Cat#: 17887A |
| 40-micron sterile cell strainer | VWR | Cat#: 21008–949 |
| Density gradient centrifugation, LymphoPrep | StemCell Technology LymphoPrep | Cat#: 07851 |
| Deposited data | ||
| Aligned scRNA-seq and scBCR-seq data of myeloma and precursors | This paper | EGAS00001006694 |
| Bulk mRNA-seq expression data | NCI Cancer Genomic Data Commons | MMRF-COMMPASS |
| Software and algorithms | ||
| Cell Ranger-5.0.0 | 10x Genomics | https://www.10xgenomics.com/ |
| R-4.0.0 | R Core Team | https://www.r-project.org/ |
| Seurat-4.0.3 | Stuart et al.50 | https://satijalab.org/seurat/ |
| Harmony-0.1.0 | Korsunsky et al.51 | https://portals.broadinstitute.org/harmony |
| inferCNV-1.5.0 | Tickle et al.53 | https://github.com/broadinstitute/inferCNV |
| CytoTRACE | Gunsagar et al.40 | https://cytotrace.stanford.edu/ |
| Monocle2 | Trapnell et al. (2014)41 Qiu et al. (2017)42,43 |
http://cole-trapnell-lab.github.io/monocle-release/ |
| Monocle3–0.2.0 | Trapnell et al. (2014)41 Qiu et al. (2017)42,43 |
http://cole-trapnell-lab.github.io/monocle-release/ |
Highlights.
Multiple myeloma precursors exhibit high degree of transcriptional heterogeneity
The HY and non-HY subtypes are transcriptionally and immunologically distinct
Early-stage tumors favor linear evolution; branching evolution arise with expansion
ACKNOWLEDGEMENTS
This work was supported in part by The MD Anderson Cancer Center Support Grant (P30 CA016672), Core Grant CA016672 (ATGC) and NIH 1S10OD024977–01 award to the Advanced Technology Genomics Core Facility, the Leukemia and Lymphoma Society Specialized Center of Research (SCOR-12206–17), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the Paula and Rodger Riney Foundation’s Riney Family Multiple Myeloma Research Fund at MD Anderson, and the University of Texas MD Anderson Moon Shot Program. We would like to thank participating patients and their families.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
H.L. has received consulting fees from Adaptive Biotechnologies, Celgene, Pimera and Takeda and research support from Amgen, Daiichi Sankyo, Janssen and Takeda. S.N. has received research support from Kite/Gilead, Celgene, Cellectis, Poseida, Merck, Acerta, Karus, and BMS; served as consultant and advisory board member for Kite/Gilead, Celgene, Novartis, Unum Therapeutics, Pfizer, CellMedica, and Merck. R.O. has received consulting fees from Amgen, Inc., Bristol-Myers Squibb, Celgene, GSK Biologicals, Ionis Pharmaceuticals, Inc., Janssen Biotech, Karyopharm Therapeutics, Molecular Partners, Neoleukin Corporation, Oncopeptides AB, Regeneron Pharmaceuticals, Sanofi-Aventis, Servier, and Takeda Pharmaceuticals North America, Inc. Clinical research support has come from CARsgen Therapeutics, Celgene, Exelixis, Janssen Biotech, Sanofi-Aventis, Takeda Pharmaceuticals North America, Inc., while laboratory research support has come from Asylia Therapeutics, Inc., BioTheryX, and Heidelberg Pharma. E.M. has received research support from Sanofi, Quest Diagnostics, Novartis, JW Pharma, Merck, GSK; consultant fees from Takeda, Celgene, Sanofi, Seattle Genetics, BMS, GSK. The other authors report no conflicts of interest.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Alexanian R, Delasalle K, Wang M, Thomas S, and Weber D (2012). Curability of multiple myeloma. Bone Marrow Res 2012, 916479. 10.1155/2012/916479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barlogie B, Mitchell A, van Rhee F, Epstein J, Morgan GJ, and Crowley J (2014). Curing myeloma at last: defining criteria and providing the evidence. Blood 124, 3043–3051. 10.1182/blood-2014-07-552059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, Kumar S, Hillengass J, Kastritis E, Richardson P, et al. (2014). International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 15, e538–548. 10.1016/S1470-2045(14)70442-5. [DOI] [PubMed] [Google Scholar]
- 4.Kunacheewa C, and Manasanch EE (2020). High-risk smoldering myeloma versus early detection of multiple myeloma: Current models, goals of therapy, and clinical implications. Best Pract Res Clin Haematol 33, 101152. 10.1016/j.beha.2020.101152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dhodapkar MV, Sexton R, Waheed S, Usmani S, Papanikolaou X, Nair B, Petty N, Shaughnessy JD Jr., Hoering A, Crowley J, et al. (2014). Clinical, genomic, and imaging predictors of myeloma progression from asymptomatic monoclonal gammopathies (SWOG S0120). Blood 123, 78–85. 10.1182/blood-2013-07-515239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pawlyn C, and Morgan GJ (2017). Evolutionary biology of high-risk multiple myeloma. Nat Rev Cancer 17, 543–556. 10.1038/nrc.2017.63. [DOI] [PubMed] [Google Scholar]
- 7.Morgan GJ, Walker BA, and Davies FE (2012). The genetic architecture of multiple myeloma. Nat Rev Cancer 12, 335–348. 10.1038/nrc3257. [DOI] [PubMed] [Google Scholar]
- 8.Manier S, Salem KZ, Park J, Landau DA, Getz G, and Ghobrial IM (2017). Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol 14, 100–113. 10.1038/nrclinonc.2016.122. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Ortiz A, Rodriguez-Garcia Y, Encinas J, Maroto-Martin E, Castellano E, Teixido J, and Martinez-Lopez J (2021). The Role of Tumor Microenvironment in Multiple Myeloma Development and Progression. Cancers (Basel) 13. 10.3390/cancers13020217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manasanch EE, Han G, Zhang Z, Dang M, Singh M, Khan M, Singh S, Lee HC, Patel KK, Kaufman GP, et al. (2020). Extensive Changes of the Immune Microenvironment Are Associated with Progression from Precursor Stages to Multiple Myeloma. Blood Abstract 485. ASH Annual Meeting 2020. [Google Scholar]
- 11.Isola I, Braso-Maristany F, Moreno DF, Mena MP, Oliver-Calders A, Pare L, Rodriguez-Lobato LG, Martin-Antonio B, Cibeira MT, Blade J, et al. (2021). Gene Expression Analysis of the Bone Marrow Microenvironment Reveals Distinct Immunotypes in Smoldering Multiple Myeloma Associated to Progression to Symptomatic Disease. Front Immunol 12, 792609. 10.3389/fimmu.2021.792609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geng S, Wang J, Zhang X, Zhang JJ, Wu F, Pang Y, Zhong Y, Wang J, Wang W, Lyu X, et al. (2020). Single-cell RNA sequencing reveals chemokine self-feeding of myeloma cells promotes extramedullary metastasis. FEBS Lett 594, 452–465. 10.1002/1873-3468.13623. [DOI] [PubMed] [Google Scholar]
- 13.Zavidij O, Haradhvala NJ, Mouhieddine TH, Sklavenitis-Pistofidis R, Cai S, Reidy M, Rahmat M, Flaifel A, Ferland B, Su NK, et al. (2020). Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat Cancer 1, 493–506. 10.1038/s43018-020-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryu D, Kim SJ, Hong Y, Jo A, Kim N, Kim HJ, Lee HO, Kim K, and Park WY (2020). Alterations in the Transcriptional Programs of Myeloma Cells and the Microenvironment during Extramedullary Progression Affect Proliferation and Immune Evasion. Clin Cancer Res 26, 935–944. 10.1158/1078-0432.CCR-19-0694. [DOI] [PubMed] [Google Scholar]
- 15.Jang JS, Li Y, Mitra AK, Bi L, Abyzov A, van Wijnen AJ, Baughn LB, Van Ness B, Rajkumar V, Kumar S, and Jen J (2019). Molecular signatures of multiple myeloma progression through single cell RNA-Seq. Blood Cancer J 9, 2. 10.1038/s41408-018-0160-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ledergor G, Weiner A, Zada M, Wang SY, Cohen YC, Gatt ME, Snir N, Magen H, Koren-Michowitz M, Herzog-Tzarfati K, et al. (2018). Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat Med 24, 1867–1876. 10.1038/s41591-018-0269-2. [DOI] [PubMed] [Google Scholar]
- 17.Cohen YC, Zada M, Wang SY, Bornstein C, David E, Moshe A, Li BG, Shlomi-Loubaton S, Gatt ME, Gur C, et al. (2021). Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single-cell sequencing. Nature Medicine 27, 491-+. 10.1038/s41591-021-01232-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Jong MME, Kellermayer Z, Papazian N, Tahri S, Bruinink DHO, Hoogenboezem R, Sanders MA, van de Woestijne PC, Bos PK, Khandanpour C, et al. (2021). The multiple myeloma microenvironment is defined by an inflammatory stromal cell landscape. Nat Immunol 22, 769-+. 10.1038/s41590-021-00931-3. [DOI] [PubMed] [Google Scholar]
- 19.Liu RY, Gao QS, Foltz SM, Fowles JS, Yao LJ, Wang JT, Cao S, Sun H, Wendl MC, Sethuraman S, et al. (2021). Co-evolution of tumor and immune cells during progression of multiple myeloma. Nature Communications 12. ARTN 2559 10.1038/s41467-021-22804-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merz M, Merz AMA, Wang J, Wei L, Hu Q, Hutson N, Rondeau C, Celotto K, Belal A, Alberico R, et al. (2022). Deciphering spatial genomic heterogeneity at a single cell resolution in multiple myeloma. Nature Communications 13. ARTN 807 10.1038/s41467-022-28266-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tirier SM, Mallm JP, Steiger S, Poos AM, Awwad MHS, Giesen N, Casiraghi N, Susak H, Bauer K, Baumann A, et al. (2021). Subclone-specific microenvironmental impact and drug response in refractory multiple myeloma revealed by single-cell transcriptomics. Nature Communications 12. ARTN 6960 10.1038/s41467-021-26951-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abbas HA, Hao D, Tomczak K, Barrodia P, Im JS, Reville PK, Alaniz Z, Wang W, Wang R, Wang F, et al. (2021). Single cell T cell landscape and T cell receptor repertoire profiling of AML in context of PD-1 blockade therapy. Nat Commun 12, 6071. 10.1038/s41467-021-26282-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng Q, Han G, Puebla-Osorio N, Ma MCJ, Strati P, Chasen B, Dai E, Dang M, Jain N, Yang H, et al. (2020). Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat Med 26, 1878–1887. 10.1038/s41591-020-1061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinjab A, Han G, Treekitkarnmongkol W, Hara K, Brennan PM, Dang M, Hao D, Wang R, Dai E, Dejima H, et al. (2021). Resolving the Spatial and Cellular Architecture of Lung Adenocarcinoma by Multiregion Single-Cell Sequencing. Cancer Discov 11, 2506–2523. 10.1158/2159-8290.CD-20-1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang R, Dang M, Harada K, Han G, Wang F, Pool Pizzi M, Zhao M, Tatlonghari G, Zhang S, Hao D, et al. (2021). Single-cell dissection of intratumoral heterogeneity and lineage diversity in metastatic gastric adenocarcinoma. Nat Med 27, 141–151. 10.1038/s41591-020-1125-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang S, Jiang VC, Han G, Hao D, Lian J, Liu Y, Zhang R, McIntosh J, Wang R, Dang M, et al. (2021). Longitudinal single-cell profiling reveals molecular heterogeneity and tumor-immune evolution in refractory mantle cell lymphoma. Nat Commun 12, 2877. 10.1038/s41467-021-22872-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller C, Yesil J, Derome M, Donnelly A, Marrian J, McBride K, Auclair D, Keats JJ, and Commpass NM (2016). A Comparison of Clinical FISH and Sequencing Based FISH Estimates in Multiple Myeloma: An Mmrf Commpass Analysis. Blood 128. DOI 10.1182/blood.V128.22.374.374. [DOI] [Google Scholar]
- 28.Van Wier S, Braggio E, Baker A, Ahmann G, Levy J, Carpten JD, and Fonseca R (2013). Hypodiploid multiple myeloma is characterized by more aggressive molecular markers than non-hyperdiploid multiple myeloma. Haematologica 98, 1586–1592. 10.3324/haematol.2012.081083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chng WJ, Kumar S, Vanwier S, Ahmann G, Price-Troska T, Henderson K, Chung TH, Kim S, Mulligan G, Bryant B, et al. (2007). Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res 67, 2982–2989. 10.1158/0008-5472.CAN-06-4046. [DOI] [PubMed] [Google Scholar]
- 30.Sanchez E, Li MJ, Kitto A, Li J, Wang CS, Kirk DT, Yellin O, Nichols CM, Dreyer MP, Ahles CP, et al. (2012). Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Brit J Haematol 158, 727–738. 10.1111/j.1365-2141.2012.09241.x. [DOI] [PubMed] [Google Scholar]
- 31.Cohen Y, Gutwein O, Garach-Jehoshua O, Bar-Haim A, and Kornberg A (2013). GPRC5D is a promising marker for monitoring the tumor load and to target multiple myeloma cells. Hematology 18, 347–350. 10.1179/1607845413y.0000000079. [DOI] [PubMed] [Google Scholar]
- 32.Elkins K, Zheng B, Go M, Slaga D, Du CC, Scales SJ, Yu SF, McBride J, de Tute R, Rawstron A, et al. (2012). FcRL5 as a Target of Antibody-Drug Conjugates for the Treatment of Multiple Myeloma. Mol Cancer Ther 11, 2222–2232. 10.1158/1535-7163.Mct-12-0087. [DOI] [PubMed] [Google Scholar]
- 33.Neri P, Ren L, Azab AK, Brentnall M, Gratton K, Klimowicz AC, Lin C, Duggan P, Tassone P, Mansoor A, et al. (2011). Integrin beta7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood 117, 6202–6213. 10.1182/blood-2010-06-292243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robbiani DF, Colon K, Ely S, Ely S, Chesi M, and Bergsagel PL (2007). Osteopontin dysregulation and lytic bone lesions in multiple myeloma. Hematol Oncol 25, 16–20. 10.1002/hon.803. [DOI] [PubMed] [Google Scholar]
- 35.De Vos J, Couderc G, Tarte K, Jourdan M, Requirand G, Delteil MC, Rossi JF, Mechti N, and Klein B (2001). Identifying intercellular signaling genes expressed in malignant plasma cells by using complementary DNA arrays. Blood 98, 771–780. DOI 10.1182/blood.V98.3.771. [DOI] [PubMed] [Google Scholar]
- 36.Bret C, Hose D, Reme T, Sprynski AC, Mahtouk K, Schved JF, Quittet P, Rossi JF, Goldschmidt H, and Klein B (2009). Expression of genes encoding for proteins involved in heparan sulphate and chondroitin sulphate chain synthesis and modification in normal and malignant plasma cells. Brit J Haematol 145, 350–368. 10.1111/j.1365-2141.2009.07633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohguchi H, Hideshima T, Bhasin MK, Gorgun GT, Santo L, Cea M, Samur MK, Mimura N, Suzuki R, Tai YT, et al. (2016). The KDM3A-KLF2-IRF4 axis maintains myeloma cell survival. Nature Communications 7. ARTN 10258 10.1038/ncomms10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rojas EA, Corchete LA, Mateos MV, Garcia-Sanz R, Misiewicz-Krzeminska I, and Gutierrez NC (2019). Transcriptome analysis reveals significant differences between primary plasma cell leukemia and multiple myeloma even when sharing a similar genetic background. Blood Cancer J 9. ARTN 90 10.1038/s41408-019-0253-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Danielsen JMR, Knudsen LM, Dahl IM, Lodahl M, and Rasmussen T (2007). Dysregulation of CD47 and the ligands thrombospondin 1 and 2 in multiple myeloma. Brit J Haematol 138, 756–760. 10.1111/j.1365-2141.2007.06729.x. [DOI] [PubMed] [Google Scholar]
- 40.Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, Ilagan F, Kuo AH, Hsieh RW, Cai S, et al. (2020). Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405-+. 10.1126/science.aax0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386. 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14, 979–982. 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu XJ, Hill A, Packer J, Lin DJ, Ma YA, and Trapnell C (2017). Single-cell mRNA quantification and differential analysis with Census. Nature Methods 14, 309-+. 10.1038/Nmeth.4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lakshman A, Rajkumar SV, Buadi FK, Binder M, Gertz MA, Lacy MQ, Dispenzieri A, Dingli D, Fonder AL, Hayman SR, et al. (2018). Risk stratification of smoldering multiple myeloma incorporating revised IMWG diagnostic criteria. Blood Cancer J 8, 59. 10.1038/s41408-018-0077-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zavidij O, Haradhvala NJ, Mouhieddine TH, Sklavenitis-Pistofidis R, Cai SJ, Reidy M, Rahmat M, Flaifel A, Ferland B, Su NK, et al. (2020). Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat Cancer 1. 10.1038/s43018-020-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc 15, 1484–1506. 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 47.Melchor L, Brioli A, Wardell CP, Murison A, Potter NE, Kaiser MF, Fryer RA, Johnson DC, Begum DB, Hulkki Wilson S, et al. (2014). Single-cell genetic analysis reveals the composition of initiating clones and phylogenetic patterns of branching and parallel evolution in myeloma. Leukemia 28, 1705–1715. 10.1038/leu.2014.13. [DOI] [PubMed] [Google Scholar]
- 48.Jacobi B, Stroeher L, Leuchtner N, Echchannaoui H, Desuki A, Kuerzer L, Habermeier A, Antunes E, Amann E, Bomalaski J, et al. (2015). Interfering with Arginine Metabolism As a New Treatment Strategy for Multiple Myeloma. Blood 126, 3005–3005. 10.1182/blood.V126.23.3005.3005. [DOI] [Google Scholar]
- 49.Windschmitt J, Jacobi B, Bülbül Y, Sester L, Tappe J, Hiebel C, Behl C, Theobald M, and Munder M (2018). Arginine Depletion in Combination with Canavanine Supplementation Induces Massive Cell Death in Myeloma Cells By Interfering with Their Protein Metabolism and Bypassing Potential Rescue Mechanisms. Blood 132, 3205–3205. 10.1182/blood-2018-99-113396. [DOI] [Google Scholar]
- 50.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2019). Dimensionality reduction for visualizing single-cell data using UMAP. Nature Biotechnology 37, 38-+. 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 53.Tickle T TI, Georgescu C, Brown M, Haas B (2019). inferCNV of the Trinity CTAT Project https://github.com/broadinstitute/inferCNV.
- 54.Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, et al. (2018). Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 174, 1293–1308 e1236. 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cillo AR, Kurten CHL, Tabib T, Qi Z, Onkar S, Wang T, Liu A, Duvvuri U, Kim S, Soose RJ, et al. (2020). Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity 52, 183–199 e189. 10.1016/j.immuni.2019.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Benjamini Y, Drai D, Elmer G, Kafkafi N, and Golani I (2001). Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125, 279–284. 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1- Summarized and detailed clinical characterization of each individual patient, related to Figure 1.
Table S2- scRNA and scBCR sequencing matrix, related to Figure 1.
Table S3- Number of cells per cell type per sample and number of cells with BCR per sample, related to Figures 1 and 6.
Table S4- Cell counts and sequencing matrix per intra-sample subcluster of PCs, related to Figures 3 and S1.
Table S5- Top 50 DEGs between B lineage cell subsets of MGUS01s, MGUS06s and SMM07s (related to Figures 4 and 5).
Table S6- Top 50 DEGs between subclusters of major TME cell types including CD4+ T cells, CD8+ T cells, NK cells, B cells, Myeloid cells, CD14+ and CD16+ monocytes (related to Figures 6 and S6).
Data Availability Statement
All single-cell RNA-sequencing data generated by this study have been deposited in the European Genome-Phenome Archive (EGA, https://ega-archive.org/). The data can be accessed under the accession number EGAS00001006694. Bulk mRNA-seq expression data (normalized) generated by Multiple Myeloma Research Foundation (MMRF) CoMMpass study (MMRF-COMMPASS) were downloaded from NCI Cancer Genomic Data Commons (NCI-GDC: https://gdc.cancer.gov). SeqFISH results of MMRF-COMMPASS samples were downloaded from the MMRF researcher gateway portal (https://research.themmrf.org). All other data supporting the findings of this study and Code used for all processing and analysis are available from the corresponding author upon reasonable request.