

Significance
Generation of memory CD8 T cells is the goal of many vaccination strategies to confer protection against intracellular infections and malignancies. While the depleting effects of ionizing radiation on immune cells are established, the long-term impact of sublethal ionizing radiation on preexisting memory CD8 T cells remains unclear. Here, we demonstrate that in addition to rapid loss of circulating memory CD8 T cells, ionizing radiation leads to a lasting lesion which prevents the surviving circulating memory CD8 T cells from numerically and functionally recovering. These findings are a critical step in the development of treatments that aim to curb radiation-induced immunosuppression with high efficacy.
Keywords: memory CD8 T cells, irradiation, impaired development, long-term dysfunction
Abstract
The increasing use of nuclear energy sources inevitably raises the risk of accidental or deliberate radiation exposure and associated immune dysfunction. However, the extent to which radiation exposure impacts memory CD8 T cells, potent mediators of immunity to recurring intracellular infections and malignancies, remains understudied. Using P14 CD8 T cell chimeric mice (P14 chimeras) with an lymphocytic choriomeningitis virus (LCMV) infection model, we observed that sublethal (5Gy) whole-body irradiation (WBI) induced a rapid decline in the number of naive (TN) and P14 circulating memory CD8 T cells (TCIRCM), with the former being more susceptible to radiation-induced numeric loss. While TN cell numbers rapidly recovered, as previously described, the number of P14 TCIRCM cells remained low at least 9 mo after radiation exposure. Additionally, the remaining P14 TCIRCM in irradiated hosts exhibited an inefficient transition to a central memory (CD62L+) phenotype compared to nonirradiated P14 chimeras. WBI also resulted in long-lasting T cell intrinsic deficits in memory CD8 T cells, including diminished cytokine and chemokine production along with impaired secondary expansion upon cognate Ag reencounter. Irradiated P14 chimeras displayed significantly higher bacterial burden after challenge with Listeria monocytogenes expressing the LCMV GP33-41 epitope relative to nonirradiated controls, likely due to radiation-induced numerical and functional impairments. Taken together, our findings suggest that sublethal radiation exposure caused a long-term numerical, impaired differentiation, and functional dysregulation in preexisting TCIRCM, rendering previously protected hosts susceptible to reinfection.
Memory CD8 T cells provide enhanced protection against intracellular pathogens and malignancies (1–3). Upon antigen reencounter, memory CD8 T cells respond by robust production of cytotoxic granules, effector cytokines, and chemokines as well as rapid secondary expansion in numbers that all act in concert to mediate pathogen clearance (4–7). The level of memory CD8 T cell–mediated protection depends on a multitude of factors including the number and subset composition of memory CD8 T cells, their localization within the host, and the nature of the insult (8–11). Based on their migration patterns, memory CD8 T cells consist of circulating memory CD8 T (TCIRCM) cells and tissue-resident memory CD8 T (TRM) cells (12–14). TCIRCM cells are further subdivided into CD62L- effector memory (TEM) cells and CD62L+ central memory (TCM) cells, each with preferential localization, phenotype, and function (9, 15–17). Nevertheless, presence of a numerically stable memory CD8 T cell pool with diverse but balanced subset composition is required to confer the most efficient recall responses to a wide range of pathogens. Multiple lines of investigation have demonstrated that either numeric loss or a shift in the subset makeup of the memory CD8 T cell pool negatively impacts the ability of immunized hosts to effectuate robust control during reinfection (18–22).
Nuclear energy ranks as the second-largest global source for low-carbon electricity (23). With the growing interest in replacing fossil fuels with clean and sustainable energy sources, the use of nuclear energy is expected to rise globally, with projections estimating a two-fold growth in the nuclear power capacity for electricity generation by 2050 (24). The increased availability of nuclear energy raises the chances of malicious use of nuclear energy or occurrence of large-scale incidents that expose the public to high doses of ionizing radiation, known to induce massive tissue damage and cell death (25–27). Although the immune ablative properties of whole-body irradiation (WBI) are used to condition patients for stem cell transplant (28), next to nothing is known about the impact of WBI on memory CD8 T cells. A previous study documented the increased sensitivity of naïve CD8 T (TN) cells compared to TCIRCM cells to radiation-induced cell death (29), but the long-term impacts of WBI on memory CD8 T cells remain unknown. Understanding the magnitude and the mechanisms by which ionizing radiation affects memory CD8 T cell biology is critical for formulation of the most effective therapeutic interventions to preserve the memory pool upon WBI exposure.
In agreement with previous literature (29), our results indicated that TCIRCM cells were lost to a lesser extent than TN cells shortly after WBI. However, longitudinal analysis revealed that in contrast to TN cells, the number of TCIRCM cells never recovered. Over time, TCIRCM cells from irradiated hosts showed altered subset composition due to hampered transition to a central memory phenotype. Additionally, WBI induced long-lasting impairment in the ability of TCIRCM cells to secrete cytokines and chemokines, and to proliferate upon antigen reencounter. These all collectively suggest that sublethal ionizing radiation inflicts irreversible numerical and functional damages on TCIRCM cells that cripple secondary responses.
Results
TCIRCM Cells, but Not TN Cells, Are Permanently Lost after Exposure to High Doses of Ionizing Radiation.
To determine the extent to which sublethal ionizing radiation impacts the number of preexisting memory CD8 T cells, we first adoptively transferred 104 congenically distinct (Thy1.1) naive P14 CD8 T cells into Thy1.2 recipients followed by lymphocytic choriomeningitis virus-Armstrong (LCMV-Arm) infection. At an early memory timepoint (D45), the P14 chimeric mice were either exposed to mock (0Gy) or sublethal WBI (5Gy) and 24 h later, the memory P14 CD8 T cells were enumerated in lymphoid and nonlymphoid tissues (Fig. 1A). Comparing the number of memory P14 cells in 5Gy mice with the P14 number in mock-treated 0Gy mice revealed significant tissue-wide decline in the number of memory CD8 T cells shortly after WBI (Fig. 1B). To investigate whether either TEM or TCM cells are differentially susceptible to WBI, the frequency of CD62L+ memory P14 cells was measured prior to and 1 d post-WBI in blood (PBL) and spleens. No differences were identified in the frequency of CD62L+ memory P14 cells in either tissue at this timepoint. (Fig. 1C). These results indicated that the number of preexisting TCIRCM cells systemically declined as early as 1 d after WBI, with no preferential loss of either TEM or TCM cells in this period.
Fig. 1.

WBI substantially decreases the number of pathogen-specific circulating memory CD8+ T cells. (A) Experimental design: 104 naive Thy1.1+ P14 CD8+ T cells were adoptively transferred into Thy1.2+ naïve hosts, followed by LCMV-Arm infection to generate memory P14 CD8 T cells. Forty-five days later, the memory P14 chimeric mice were either exposed to mock (0Gy) or 5Gy WBI and 1 d later, the mice were killed for further analysis. (B) The number of memory P14 CD8 T cells was determined in blood a day prior and after WBI and the number of memory P14 CD8 T cells was determined in the indicated organs 1 d after WBI (C) Frequency of CD62L+ P14 memory CD8 T cells was assessed in blood a day prior and after WBI and frequency of CD62L+ P14 memory CD8 T cells was assessed in the spleen 1 d after WBI. All data are representative of at least two independent experiments with 4 to 10 mice per group. *=P < 0.05, **=P < 0.01, ***=P < 0.001. Error bars represent SEM.
We next sought to investigate the long-term impact of ionizing radiation on the number of preexisting TCIRCM cells. To this end, after generating P14 chimeric mice and exposing them to mock or WBI 45 d postinfection, we monitored the number of memory P14 cells as well as TN cells in PBL for 270 d after WBI (Fig. 2A). At 1D post-WBI, both the number of memory P14 cells and TN cells declined, with the naive compartment exhibiting a much greater fold-loss as compared with memory P14 cells, which is in line with the previous literature (Fig. 2 B–D) (29). In contrast to TN cells, the number of memory P14 cells appeared to be further reduced beyond D1 timepoint as the loss of memory P14 cells was continued, implying that radiation-induced memory CD8 T cell loss is gradual (Fig. 2 D and E). Next, we examined the kinetics of recovery for the TN and TCIRCM cells. WBI creates a lymphopenic environment, known to trigger an antigen-independent expansion of TN cells regarded as homeostatic proliferation (30, 31). This is a compensatory mechanism which allows the body to replenish the physiologic number of CD8 T cells and has been documented in both naive and memory CD8 T cells following sepsis-induced lymphopenia (21, 22, 32). As expected, the number of TN cells started repopulating the irradiated hosts as early as D21 after WBI, and in time, the number of TN cells returned to pre-WBI baseline (Fig. 2C). In sharp contrast, the number of memory P14 cells never recovered, suggesting a permanent numerical decline following WBI (Fig. 2D). The lack of recovery of circulatory memory P14 cells was also observed in other lymphoid and nonlymphoid tissues as well as in endogenous GP33-41-specific memory CD8 T cells (SI Appendix, Fig. S1), ruling out the possibility of preferential localization of memory P14 cells in inflamed tissues. Similarly, the number of memory P14 and CD69+ CD103+ tissue–resident memory (TRM) P14 cells in salivary glands (SG) of 5Gy hosts is diminished and remained significantly lower following WBI compared to nonirradiated control mice (SI Appendix, Fig. S2). These data collectively suggest that although TCIRCM cells were less susceptible to WBI than TN cells, TCIRCM cells displayed long-lasting impairment in numerical recovery following WBI while TN cells were restored to pre-WBI numbers (Fig. 2E).
Fig. 2.

Long-lasting decline in the number of memory CD8+ T cells following WBI in contrast to numerical recovery of naïve CD8+ T cells. (A) Experimental design: 104 naive Thy1.1+ P14 CD8+ T cells were adoptively transferred into Thy1.2+ naïve hosts, followed by LCMV-Arm infection to generate memory P14 CD8 T cells. Forty-five days later, the memory P14 chimeric mice were either exposed to mock or 5Gy WBI. Analysis was performed on the indicated timepoints after irradiation. The number of (B) total CD8+ T cells, (C) naïve CD8+ T cells, and (D) P14 memory CD8 T cells was assessed in the blood up to D270. (E) Ratio of the number of indicated cells from 5Gy hosts to 0Gy hosts in blood following WBI. All data are representative of at least two independent experiments with 4 to 10 mice per group. *=P < 0.05, **=P < 0.01, ***=P < 0.001. Error bars represent SEM.
Radiation Induces Alterations in the Subset Composition of TCIRCM Cells.
TEM cells are the dominant subset of TCIRCM cells at an early memory timepoint after clearance of LCMV infection. However, with time, TCM cells slowly become the predominant subset due to the direct conversion of TEM cells to TCM and/or enhanced homeostatic maintenance of TCM cells (33–35). This gradual increase in the frequency of TCM cells is associated with enhanced proliferation and IL-2 production of the TCIRCM pool (36, 37). The lasting inability of those TCIRCM cells that survive irradiation to repopulate the lymphopenic space post-WBI suggested that WBI may impose long-lasting functional defects in memory CD8 T cells. This prompted us to examine whether WBI influences the ability of surviving TCIRCM cells to progress to a central memory phenotype over time given that WBI exposure occurs at an early memory timepoint in our model (Fig. 3 A and B). The inability of the memory P14 cells from irradiated hosts to differentiate into central memory was observed as early as 31D and 50D post-WBI (Fig. 3C). Although memory P14 cells from 0Gy hosts showed increasing frequency of CD62L+ subset with time, the frequency of CD62L+ memory P14 cells in 5Gy mice remained low, suggesting that CD62L− memory CD8 T cells are enriched in WBI-exposed hosts (Fig. 3C). In fact, this lack of transition to central memory continued to 270D post-WBI, with CD62L− CD27− memory P14 cells being overrepresented in 5Gy mice compared with 0Gy mice (Fig. 3 B–D). In addition, memory P14 cells from 5Gy hosts exhibited reduced expression of CD122 and CD127, markers associated with long-term memory and memory maintenance, up to 270 d post-WBI (Fig. 3E). These data indicated that WBI impeded the differentiation of TCIRCM cells toward central memory phenotype, resulting in long-lasting enrichment of CD62L− CD27− memory CD8 T cells.
Fig. 3.

WBI impedes the differentiation of memory CD8+ T cells to central memory phenotype. (A) Experimental design: 104 naive Thy1.1+ P14 CD8+ T cells were adoptively transferred into Thy1.2+ naïve hosts, followed by LCMV-Arm infection to generate memory P14 CD8 T cells. Forty-five days later, the memory P14 chimeric mice were either exposed to mock or 5Gy WBI. Analysis was performed on the indicated timepoints after irradiation. (B) Representative gating of CD62L and CD27 (Left) and geometric mean fluorescence intensity (gMFI) of CD122 and CD127 (Right) of memory P14 cells in blood at D270. (C) Frequency of CD62L+ memory P14 CD8 T cells in blood up to D50. (D) Frequency of TCM (CD62L+ CD27+) memory P14 CD8 T cells (Left), CD62L− CD27+ (Middle), and CD62L− CD27− (Right) memory P14 CD8 T cells at indicated timepoints after WBI in blood. (E) Quantification of gMFI of CD127 and CD122 at D190 (Left) and D270 (Right) on memory P14 CD8 T cells from blood. All data are representative of at least two independent experiments with 4 to 10 mice per group. *=P < 0.05, **=P < 0.01, ***=P < 0.001. Error bars represent SEM.
Antigen-Driven Cytokine Production of TCIRCM Cells Remains Impaired after Radiation Exposure for Long Time.
We next studied the impact of WBI on antigen-dependent functions of TCIRCM cells. In response to antigen stimulation, memory CD8 T cells not only secrete proinflammatory cytokines such as IFN-γ, TNF-α, and IL-2, but they also release chemokines such as CCL4, CCL5, and XCL-1 (38–40). To evaluate how ionizing radiation alters the functional avidity of TCIRCM cells, as a measure of T cell responsiveness to antigen (41), splenocytes from 0Gy or 5Gy P14 chimeric mice were stimulated ex-vivo with graded doses of GP33-41 peptide on 12D and 58D post-WBI, followed by intracellular staining (ICS) to measure IFN-γ production (Fig. 4A). At 12D post-WBI, memory P14 cells from 5Gy hosts had diminished ability to produce IFN-γ at all GP33-41 peptide concentrations. Reduced IFN-γ production at 0.2 nM [GP33-41], a physiologically relevant concentration of the antigen (42), was still detected at 58D post WBI (Fig. 4 B and C). Calculating the EC50 of maximum IFN-γ production indicated decreased antigen sensitivity of memory P14 cells that does not fully recover up to 58D post-WBI (Fig. 4D). Further analyses showed long-lasting impairment in the production of additional effector cytokines including TNF-α and IL-2 (Fig. 4E) as well as chemokines such as CCL5 and XCL-1 (SI Appendix, Fig. S3). Overall, these results illustrated that WBI causes long-term reduction in antigen-dependent cytokine and chemokine production of TCIRCM cells.
Fig. 4.

WBI results in long-lasting decreased antigen sensitivity and the ability of memory CD8 T cells to secrete cytokines upon cognate antigen stimulation. (A) Experimental design: 104 naive Thy1.1+ P14 CD8+ T cells were adoptively transferred into Thy1.2+ naïve hosts, followed by LCMV-Arm infection to generate memory P14 CD8 T cells. Forty-five days later, the memory P14 chimeric mice were either exposed to mock or 5Gy WBI. At D12 and D58, splenocytes from both groups were harvested and were either left unstimulated or stimulated with different concentrations of GP33-41 peptide for 5 h. (B) Representative gating of IFN-γ, TNF-α, and IL-2-producing memory P14 T cells from 0Gy (Top) and 5Gy (Bottom) hosts after stimulating with 200 nM of GP33-41 at D12. (C) Frequency of IFN-γ+ memory P14 CD8 T cells at D12 (Left) and D58 (Right). (D) EC50 graph (Left) and summary of EC50 (Right) of memory 0Gy and 5Gy memory P14 cells at D12 and D58. (E) Frequency of IFN-γ+, TNF-α+, and IL-2+ memory P14 CD8 T cells at D12 (Left) and D58 (Right). All data are representative of at least two independent experiments with four to five mice per group. *=P < 0.05, **=P < 0.01, ***=P < 0.001. Error bars represent SEM.
These lasting defects raised the question as to whether T cell–intrinsic or –extrinsic factors lead to defective cytokine and chemokine responses. To normalize cell-extrinsic factors, splenocytes from 0Gy and 5Gy P14 chimeric mice were differentially labeled with CFSE and coincubated in the presence of GP33-41 peptide ex-vivo (SI Appendix, Fig. S4A). We still observed compromised cytokine production by memory P14 cells from irradiated hosts, suggesting that exposure to sublethal ionizing radiation causes cell-intrinsic defects as normalizing the environment does not alleviate the function of TCIRCM cells (SI Appendix, Fig. S4 B and C). To determine whether T cell–intrinsic defects could be attributed to impairments in T cell receptor (TCR) signaling, we bypassed TCR signaling with PMA/ionomycin and tested whether this restored the magnitude of cytokine response (SI Appendix, Fig. S5A). Interestingly, memory P14 cells from 5Gy hosts still exhibited long-lasting decreased effector cytokine production (SI Appendix, Fig. S5 B and C). This suggested that WBI impairs signaling events distal to the TCR but does not formally rule out the possibility of WBI-induced TCR signaling impairments. Thus, these findings indicated that WBI instigates T cell–intrinsic defects that lead to diminished capacity of TCIRCM cells to secrete cytokines and chemokines upon antigen rechallenge.
Diminished Recall Responses Long after Radiation Exposure.
We next interrogated the impact of WBI on the ability of TCIRCM cells to undergo secondary expansion in numbers after antigen reencounter. Splenocytes were isolated from 0Gy and 5Gy P14 chimeric mice 23D post-WBI and an equal number of memory P14 cells were adoptively transferred into naive recipients. One day later, the recipient mice were challenged with attenuated Listeria monocytogenes expressing GP33-41 (att. LM-GP33) and the number of P14 cells was analyzed in PBL 4 and 5 d later (Fig. 5A). The hosts that received memory P14 cells from 5Gy mice contained a substantially (~100×) smaller number of memory P14–derived secondary effector cells than the hosts that had received memory P14 cells from 0Gy mice. These data revealed a severely diminished capacity of memory P14 cells to undergo secondary expansion, suggesting that WBI markedly reduced the antigen-induced expansion potential of memory CD8 T cells (Fig. 5 B and C).
Fig. 5.
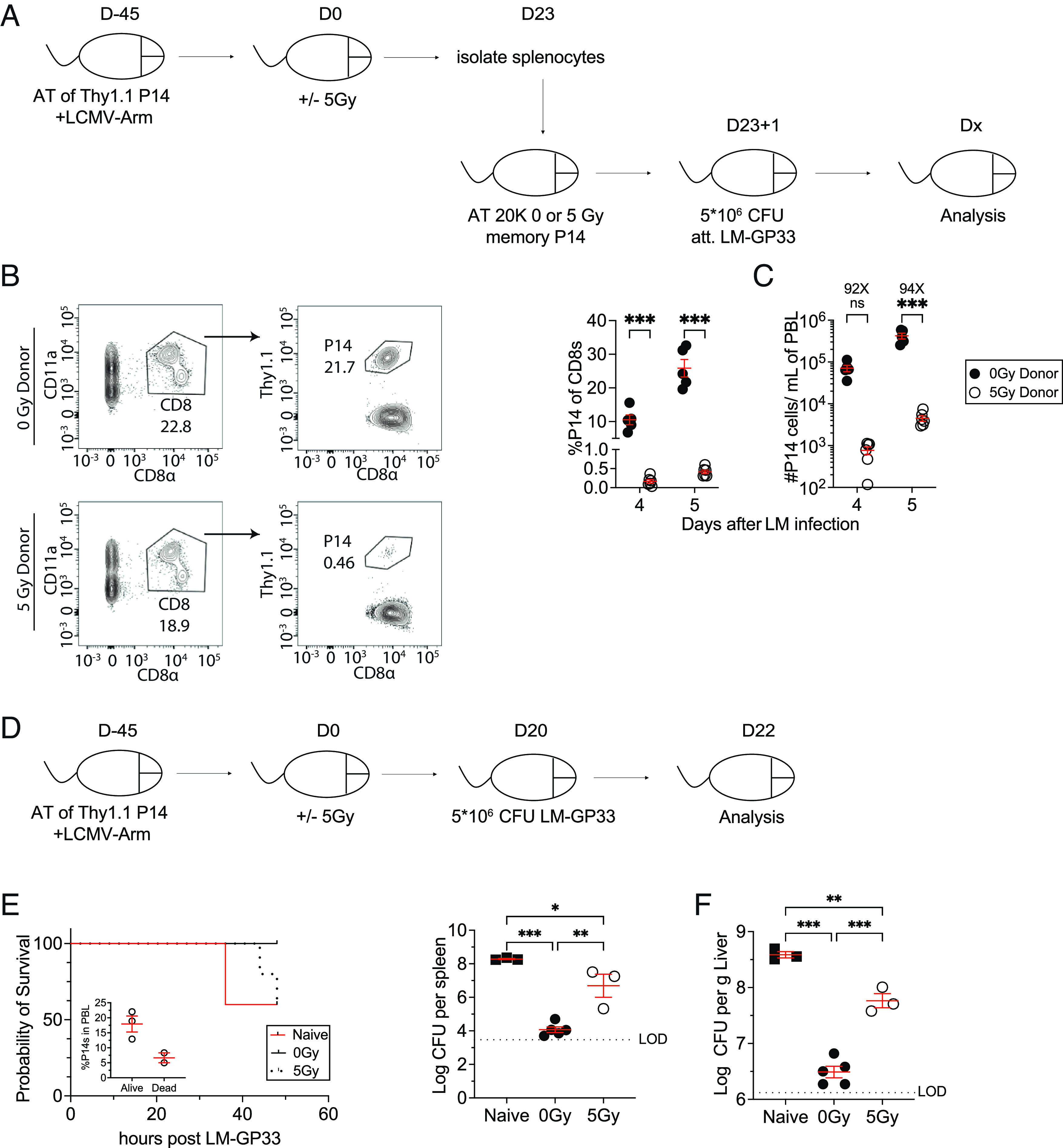
WBI substantially impairs the ability of memory CD8 T cells and immunized hosts to respond to rechallenges. (A) Experimental design: 104 naive Thy1.1+ P14 CD8+ T cells were adoptively transferred into Thy1.2+ naïve hosts, followed by LCMV-Arm infection to generate memory P14 CD8 T cells. Forty-five days later, the memory P14 chimeric mice were either exposed to mock or 5Gy WBI. Twenty-three days after mock or 5Gy WBI, splenocytes from 0Gy and 5Gy mice were isolated and 2 × 104 memory P14 CD8 T cells were then transferred into naïve mice. The P14-recipient mice were then infected with attenuated L. monocytogenes expressing GP33-41 (LM-GP33). (B) Representative gating of 0Gy donor (Top) and 5Gy donor (Bottom) P14 CD8 T cells in recipient mice 5 d after attenuated LM-GP33 challenge. (C) Frequency of donor P14 CD8 T cells of total recipient CD8 T cells (Left) and the number of donor P14 CD8 T cells (Right) 4 and 5 d after attenuated LM-GP33 challenge assessed in blood. (D) 104 naive Thy1.1+ P14 CD8+ T cells were adoptively transferred into Thy1.2+ naïve hosts, followed by LCMV-Arm infection to generate memory P14 CD8 T cells. Forty-five days later, the memory P14 chimeric mice were either exposed to mock or 5Gy WBI. These mice as well as a group of naïve mice with no previous pathogen exposure were then infected with 5 × 106 CFU of virulent L. monocytogenes expressing GP33-41 (LM-GP33) 20 d after mock or 5Gy WBI. (E) Mortality of mice after infection. (F) CFUs of LM-GP33 per gram of the liver (Left) and spleen (Right) 2 d after infection. All data are representative of at least two independent experiments with four to five mice per group. *=P < 0.05, **=P < 0.01, ***=P < 0.001. Error bars represent SEM.
Thus far, our results have demonstrated that WBI induces lasting damage to both the number and the function of TCIRCM cells. This led us to examine how the reduced number and function of TCIRCM cells in WBI-exposed hosts influenced pathogen control in a secondary challenge. At D20 post-WBI, 0Gy, and 5Gy, P14 chimeric mice along with naive mice with no history of pathogen exposure were challenged with a high dose of virulent LM-GP33. Mortality and bacterial clearance from the spleen and liver were determined at 2 d after the challenge (Fig. 5D). Presence of memory P14 cells was associated with protection from early death as a fraction of naïve mice (2/5) succumbed to LM-GP33 by 36 h postinfection (Fig. 5E). In addition, prechallenge PBL analysis indicated that the 5Gy mice that died by 48 h after LM-GP33 challenge had lower frequency of memory P14 cells than the ones that survived (Fig. 5 E, Inset). Of the surviving mice, P14 chimeric mice regardless of their status of WBI exposure exhibited better pathogen clearance than that of naive mice due to the presence of memory P14 cells (Fig. 5 E and F). Additionally, 5Gy P14 chimeric mice exhibited significantly higher pathogen burden in the spleen and liver than that of 0Gy P14 chimeric mice (Fig. 5F). These results cumulatively suggested that WBI-induced permanent numerical decline and per-cell dysfunction of TCIRCM cells significantly impaire pathogen clearance upon re-infection.
Discussion
The radiosensitivity of lymphocytes, endowed with high proliferative capacity, is well established in a dose-dependent manner and often exploited in treatments that require near-perfect eradication of lymphocytes (43–45). Despite the increased prevalence of nuclear energy utilization and instances of nuclear accidents that expose the public to large-scale ionizing radiation, the long-term impact of ionizing radiation on subsets of lymphocytes remains largely unknown. This knowledge gap is a barrier for the development of effective medical counter measures against radiation-induced immunosuppression. A prior study demonstrated the increased susceptibility of TN cells to radiation-induced cell death compared to preexisting TCIRCM cells (29). While this study confirms the previous findings, it also provides unique and unexpected evidence for the long-lasting detrimental effects of WBI on the number, differentiation, and function that lead to subpar secondary responses by TCIRCM cells.
The factors underlying differential susceptibility of TN and TCIRCM to radiation-induced cell loss are subject to further investigation. While there is an extensive list of transcriptomic and epigenetic differences between TN and TCIRCM cells, one interesting area of exploration is to compare the DNA damage response (DDR) elicited by each subset in response to WBI. Heylmann et al. have previously documented that T cell stimulation leads to downregulation of ATM, major kinase in response to DNA double-strand breaks (DSB), which results in reduced sensitivity of stimulated T cells to radiation-induced cell death compared to unstimulated T cells (46). In addition, one of the phosphorylation targets of ATM is Ser-139 residue of the histone variant H2AX to form γH2AX, a key step in initiating DDR and DSB repair. Pugh et al. have also shown that TN and TCIRCM cells demonstrate different kinetic of γH2AX upregulation following ionizing radiation (47). These notions imply that the effectiveness of DNA damage response by each subset is different and, hence, may be the basis for the differential initial susceptibility of TN and TCIRCM cell death mediated by ionizing radiation.
In addition to differences in the timing and magnitude of loss, longitudinal tracking of the number of CD8 T cells after WBI pointed to the distinct kinetic of recovery between TN and TCIRCM cells. In sharp contrast to TN cells, which recovered their numbers completely, the number of TCIRCM cells did not recover for at least 9 mo after WBI. Whether robust proliferation of the surviving TN cells in response to homeostatic cues and/or maturation of new TN cells from thymus results in gradual TN recovery remains to be determined. Additionally, TCIRCM cells from WBI hosts also failed to efficiently differentiate into TCM phenotype, resulting in the sustained enrichment of effector-like TCIRCM cells. These results are divergent from how sepsis, another lymphopenia-inducing event, shapes CD8 T cells. In contrast to WBI, TN and TCIRCM cells both show the same degree of susceptibility to sepsis-induced apoptosis (20, 48) and shortly after sepsis, both TN and TCIRCM cells numerically recover. Additionally, the homeostatic cues from the postseptic environment favor the rapid proliferation of TCM cells, leading to an overrepresentation of central memory phenotype (21, 49). Of note, WBI induces cell death through irreversible DNA damage, whereas cytokine-mediated apoptosis is responsible for lymphopenia associated with sepsis. Thus, one could postulate that the differences between the WBI and sepsis-induced TCIRCM cell loss and recovery are due to the varying nature of each lymphopenic event and their specific influences on CD8 T cell biology.
An interesting area of future investigation is to characterize the factors that lead to the enrichment and longevity of CD62L− CD27− TCIRCM cells in the irradiated hosts. This subset remains enriched in irradiated hosts for a much longer time than that in control hosts despite the lack of expression of markers associated with the maintenance and survival of long-term memory such as CD127 and CD122. Two of the many explanations for this observation include: 1) This subset may possess enhanced DNA repair capacity compared with TCM cells and over time CD62L− CD27− TCIRCM cells gradually become overrepresented in the irradiated hosts. This explanation is in line with a previous report where TEM (CD44hi CD62L−) cells were found to be more radioresistant than TCM (CD44hi CD62L+) cells in-vitro (47). 2) Post-WBI environment may provide survival cues and/or induce lasting transcriptional and epigenetic changes that allow for enrichment of this subset.
Our results demonstrate that the effect of WBI is not only limited to the maintenance and differentiation of TCIRCM cells in the resting state. TCIRCM cells from irradiated hosts also showed impaired recall responses. T cell–intrinsic factors appeared to play a critical role in driving the long-lasting dysfunction of TCIRCM cells after WBI; however, these factors remain elusive. Interestingly, recent studies have suggested that unlike TN cells (50), memory CD8 T cells that have undergone CRISPR/Cas9 genetic modifications fail to expand in response to cytokines (IL-7 and IL-15) and cognate antigen stimulation. This notion was attributed to p53, a sensor of genetic stress that orchestrates cellular response to DNA damage, as deletion of p53 restored the ability of memory CD8 T cells with CRISPR/Cas9 genomic alterations to numerically expand upon antigen encounter (51). Similar to CRISPR/CAS9 genome editing, ionizing radiation induces DNA DSB but most likely at a much larger scale. Therefore, p53-mediated DNA damage response may contribute to the debilitated capacity of irradiated TCIRCM cells to undergo proliferation.
Future investigations should interrogate the long-term impacts of ionizing radiation on other subsets of the memory CD8 T cell pool. For example, the effect of ionizing radiation on TRM cells, patrollers of nonlymphoid tissue for invading pathogens and at the forefront of secondary responses to many reinfections (12–14), remains unknown. In addition, the human population is seeded with memory CD8 T cells with a history of repeated cognate antigen encounters through recurring infections or reimmunizations. It will be important to characterize the differences that may exist between primary and higher-order memory CD8 T cells as each round of antigen stimulation induces lasting changes to the gene expression patterns of memory CD8 T cells (52, 53). It is also important to note that an X-ray tube is utilized to expose the subjects to sublethal WBI, with an exposure time of under 10 min. Given higher penetrating power of γ-rays and the widespread industrial use of γ-irradiators, it would also be of interest to investigate whether γ-rays and other forms of genotoxic stress would induce similar deficits on TCIRCM cells. Additionally, how exposure to a lower dose of ionizing radiation over a longer period impacts TCIRCM cells needs to be studied further. In conclusion, the findings of this study provide target cells for design of the most effective therapeutic measures to alleviate the severe immunosuppression associated with exposure to high doses of ionizing radiation.
Materials and Methods
Mice, Infections, and Memory CD8 T Cell Generation.
Experimental procedures using mice were approved by the University of Iowa Animal Care and Use Committee under protocol number #9101915. Inbred C57BL/6 (Thy 1.2/1.2) mice were purchased from Charles River and maintained in the animal facilities at the University of Iowa at the appropriate biosafety level. P14 TCR-transgenic mice (Thy1.1/1.1) were bred and maintained at the University of Iowa (Iowa City, IA).
To generate memory CD8 T cells, 104 naïve P14 TCR-Tg CD8 T cells were adoptively transferred into C57BL/six mice, followed by infection with 2 × 105 plaque-forming units (PFU) of LCMV-Arm by intraperitoneal (i.p.) injection a day later. In rechallenge studies, the mice were infected with 5 × 106 colony forming units (CFU) of either attenuated actA-deficient GP33-expressing L. monocytogenes [Att. LM-GP33] or virulent 10403s GP33-expressing L. monocytogenes strain [LM-GP33] by intravascular (I.V.) injection.
Irradiation.
WBI was performed by placing the mice in a 225-kv rotating X-ray tube (Small Animal Radiation Research platform; Xstrahl, Atlanta, GA) available at the Radiation Core at the University of Iowa. 5Gy was the radiation dose used in this study. For mock-treated mice, the mice were placed into the same mouse pie cages as 5Gy-treated were and remained there for the length of a 5Gy exposure (~8 min) without WBI exposure.
Cell Isolation.
Peripheral blood was collected by retroorbital bleeding. Single-cell suspensions from the spleen, liver, kidney, and lymph nodes were generated after mashing tissue through a 70-μm cell strainer without enzymatic digestion. For bone marrow, both femurs were collected and crushed using a mortar and pestle prior to mashing tissue through 70 μm cell strainer. Salivary glands were minced and incubated in 37 °C in the presence of collagenase II and DNAse I prior to mashing tissue through 70 μm cell strainer. Liver and kidney cells were subsequently run on a 35% Percoll gradient. ACK lysis buffer was used for red blood cell lysis of PBL, spleen, bone marrow, kidney, and liver samples.
Flow Cytometry, Peptides, and Cytokine Detection.
Flow cytometry data were acquired on a FACSCanto or LSRII (BD Biosciences) and analyzed with FlowJo software (Tree Star). To determine the expression of cell surface proteins, monoclonal antibodies (mAb) were incubated at 4 °C for 20 to 30 min and cells were fixed using BD Cytofix (BD Biosciences). To stain intracellular proteins, after staining cell surface proteins, the cells were fixed using Cytofix/Cytoperm (BD Biosciences), followed by incubation with mAb for an additional 20 to 30 min. The following mAb clones were used to stain the samples: CD8a (53 to 6.7; eBioscience), CD11a (M17/4; BioLegend), Thy1.1 (HIS51; eBioscience), CD127 (eBioSB/199; eBioscience), CD62L (MEL-14; eBioscience), CD27 (LG.7F9; eBioscience), CD122 (TM-b1; eBioscience), CD69 (H1.2F3; BioLegend). CD103 (2E7; BioLegend), IFN-γ (XMG1.2; eBioscience), IL-2 (JES6-5H4; eBioscience), TNF-α (MP6-XT22; BioLegend), CCL4 (polyclonal goat; R&D Systems), and XCL-1 (polyclonal goat; R&D Systems)
Peptide Stimulation.
For cytokine detection, splenocytes were incubated for 5 h with the indicated concentration of GP33-41 peptide or with 5 ng/mL PMA and 500 ng/mL ionomycin in the presence of brefeldin A. To differentially label splenocytes in the coincubation experiment, splenocytes (107/mL) from 0Gy and 5Gy hosts were disparately labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE; eBioscience) by incubating the cells at room temperature for 15 min with either 1 μM or 0.1 μM CFSE, respectively. Labeled cells were then incubated for 5 min with 1 mL FCS on ice to remove any free CFSE and washed three times with RPMI prior to stimulation. [EC50] was calculated as previously described (54) by measuring the GP33-41 concentration required for obtaining 50% of the maximum IFN-γ production.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank members of our laboratories for technical assistance and helpful discussions. This study was supported by NIH Grants GM134880 (V.P.B.), AI114543 (V.P.B. and J.T.H.), T32AI007485 and T32AI007511 (I.J.J.), AI042767 and AI167847 (J.T.H.), T32AI007260 and 1F32AI174382 to (M. Hassert), AI121080, AI139874, and AI112579 (H.-H.X.), The Holden Comprehensive Cancer Center at The University of Iowa and its National Cancer Institute Award P30CA086862. V.P.B. is a University of Iowa-Distinguished Scholar.
Author contributions
M. Heidarian, I.J.J., H.-H.X., J.T.H., and V.P.B. designed research; M. Heidarian, I.J.J., and M. Hassert performed research; S.K.K., S.P., and H.-H.X. contributed to conceptualization of experiments; L.L.P. contributed new reagents/analytic tools; M. Heidarian, I.J.J., S.K.K., J.T.H., and V.P.B. analyzed data; I.J.J., S.S.K., M. Hassert, and H-H.X. edited the paper; and M. Heidarian, J.T.H., and V.P.B. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
John T. Harty, Email: john-harty@uiowa.edu.
Vladimir P. Badovinac, Email: vladimir-badovinac@uiowa.edu.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Adamo S., et al. , Signature of long-lived memory CD8+ T cells in acute SARS-CoV-2 infection. Nature 602, 148–155 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt M. E., Varga S. M., The CD8 T cell response to respiratory virus infections. Front Immunol. 9, 678 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sahin U., et al. , Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Lalvani A., et al. , Rapid effector function in CD8+ memory T cells. J. Exp. Med. 186, 859–865 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barber D. L., Wherry E. J., Ahmed R., Cutting edge: Rapid in vivo killing by memory CD8 T cells1. J. Immunol. 171, 27–31 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Veiga-Fernandes H., Walter U., Bourgeois C., McLean A., Rocha B., Response of naïve and memory CD8+ T cells to antigen stimulation in vivo. Nat. Immunol. 1, 47–53 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Davenport B., et al. , Chemokine signatures of pathogen-specific t cells II: Memory T cells in acute and chronic infection. J. Immunol. 205, 2188–2206 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seder R. A., et al. , Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science 341, 1359–1365 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Wherry E. J., et al. , Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 4, 225–234 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Bachmann M. F., Wolint P., Schwarz K., Jager P., Oxenius A., Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of IL-7 receptor alpha and CD62L. J. Immunol. 175, 4686–4696 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Nolz J. C., Harty J. T., Protective capacity of memory CD8(+) T cells is dictated by antigen exposure history and nature of the infection. Immunity 34, 781–793 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin M. D., Badovinac V. P., Defining memory CD8 T cell. Front. Immunol. 9, 2692 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jameson S. C., Masopust D., Understanding subset diversity in t cell memory. Immunity 48, 214–226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mueller S. N., Gebhardt T., Carbone F. R., Heath W. R., Memory T cell subsets, migration patterns, and tissue residence. Annu. Rev. Immunol. 31, 137–161 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Jung Y. W., Rutishauser R. L., Joshi N. S., Haberman A. M., Kaech S. M., Differential localization of effector and memory CD8 T cell subsets in lymphoid organs during acute viral infection. J. Immunol. 185, 5315–5325 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sallusto F., Lenig D., Forster R., Lipp M., Lanzavecchia A., Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Masopust D., Vezys V., Marzo A. L., Lefrancois L., Preferential localization of effector memory cells in nonlymphoid tissue. Science 291, 2413–2417 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Wu T., et al. , Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J. Leukoc. Biol. 95, 215–224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slütter B., et al. , Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci. Immunol. 2, eaag2031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duong S., et al. , Polymicrobial sepsis alters antigen-dependent and -independent memory CD8 T cell functions. J. Immunol. 192, 3618–3625 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jensen I. J., et al. , Sepsis leads to lasting changes in phenotype and function of memory CD8 T cells. Elife 10, e70989 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Condotta S. A., Rai D., James B. R., Griffith T. S., Badovinac V. P., Sustained and incomplete recovery of naive CD8+ T cell precursors after sepsis contributes to impaired CD8+ T cell responses to infection. J. Immunol. 190, 1991–2000 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.IEA, Nuclear Power in a Clean Energy System (Paris) (2019).
- 24.IAEA, Nuclear Energy for a Net Zero World (Vienna) (2021).
- 25.Cytlak U. M., et al. , Immunomodulation by radiotherapy in tumour control and normal tissue toxicity. Nat. Rev. Immunol. 22, 124–138 (2022). [DOI] [PubMed] [Google Scholar]
- 26.Giuranno L., Ient J., De Ruysscher D., Vooijs M. A., Radiation-Induced Lung Injury (RILI). Front Oncol. 9, 877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andreyev J., Gastrointestinal symptoms after pelvic radiotherapy: A new understanding to improve management of symptomatic patients. Lancet Oncol. 8, 1007–1017 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Slavin S., Total lymphoid irradiation. Immunol. Today 8, 88–92 (1987). [DOI] [PubMed] [Google Scholar]
- 29.Grayson J. M., Harrington L. E., Lanier J. G., Wherry E. J., Ahmed R., Differential sensitivity of naive and memory CD8+ T cells to apoptosis in vivo. J. Immunol. 169, 3760–3770 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Cheung K. P., Yang E., Goldrath A. W., Memory-like CD8+ T cells generated during homeostatic proliferation defer to antigen-experienced memory cells1. J. Immunol. 183, 3364–3372 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldrath A. W., Bevan M. J., Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity 11, 183–190 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Unsinger J., Kazama H., McDonough J. S., Hotchkiss R. S., Ferguson T. A., Differential lymphopenia-induced homeostatic proliferation for CD4+ and CD8+ T cells following septic injury. J. Leukocyte Biol. 85, 382–390 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaech S. M., Cui W., Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 12, 749–761 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou X., et al. , Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 33, 229–240 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harty J. T., Badovinac V. P., Shaping and reshaping CD8+ T-cell memory. Nat. Rev. Immunol. 8, 107–119 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Martin M. D., et al. , Phenotypic and functional alterations in circulating memory CD8 T cells with time after primary infection. PLoS Pathog 11, e1005219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eberlein J., et al. , Aging promotes acquisition of naive-like CD8+ memory T cell traits and enhanced functionalities. J. Clin. Invest. 126, 3942–3960 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho B. K., Wang C., Sugawa S., Eisen H. N., Chen J., Functional differences between memory and naive CD8 T cells. Proc. Natl. Acad. Sci. U.S.A. 96, 2976–2981 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Badovinac V. P., Corbin G. A., Harty J. T., Cutting edge: Off cycling of TNF production by antigen-specific CD8+ T cells is antigen independent1. J. Immunol. 165, 5387–5391 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Boutet M., et al. , Memory CD8<sup>+</sup> T cells mediate early pathogen-specific protection via localized delivery of chemokines and IFNγ to clusters of monocytes. Sci. Adv. 7, eabf9975 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slifka M. K., Whitton J. L., Functional avidity maturation of CD8+ T cells without selection of higher affinity TCR. Nat. Immunol. 2, 711–717 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Garcia V., Richter K., Graw F., Oxenius A., Regoes R. R., Estimating the in vivo killing efficacy of cytotoxic T lymphocytes across different peptide-MHC complex densities. PLOS Comput. Biol. 11, e1004178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trowell O., The sensitivity of lymphocytes to ionising radiation. J. Pathol. Bacteriol. 64, 687–704 (1952). [DOI] [PubMed] [Google Scholar]
- 44.Seki H., et al. , Ionizing radiation induces apoptotic cell death in human TcR-γ/δ+ T and natural killer cells without detectable p53 protein. Eur. J. Immunol. 24, 2914–2917 (1994). [DOI] [PubMed] [Google Scholar]
- 45.Wilkins R. C., et al. , Differential apoptotic response to ionizing radiation in subpopulations of human white blood cells. Mutat. Res./Genet. Toxicol. Environ. Mutagenesis 513, 27–36 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Heylmann D., Badura J., Becker H., Fahrer J., Kaina B., Sensitivity of CD3/CD28-stimulated versus non-stimulated lymphocytes to ionizing radiation and genotoxic anticancer drugs: Key role of ATM in the differential radiation response. Cell Death Disease 9, 1053 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pugh J. L., et al. , Histone deacetylation critically determines T cell subset radiosensitivity. J. Immunol. 193, 1451–1458 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jensen I. J., Sjaastad F. V., Griffith T. S., Badovinac V. P., Sepsis-Induced T cell immunoparalysis: The Ins and Outs of impaired T cell immunity. J. Immunol. 200, 1543–1553 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heidarian M., Griffith T. S., Badovinac V. P., Sepsis-induced changes in differentiation, maintenance, and function of memory CD8 T cell subsets. Front Immunol. 14 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nüssing S., et al. , Efficient CRISPR/Cas9 gene editing in uncultured naive mouse T cells for in vivo studies. J. Immunol. 204, 2308–2315 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Kurup S. P., Moioffer S. J., Pewe L. L., Harty J. T., p53 hinders CRISPR/Cas9-mediated targeted gene disruption in memory CD8 T cells in vivo. J. Immunol. 205, 2222–2230 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Masopust D., Ha S. J., Vezys V., Ahmed R., Stimulation history dictates memory CD8 T cell phenotype: Implications for prime-boost vaccination. J. Immunol. 177, 831–839 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Wirth T. C., et al. , Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity 33, 128–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Richer Martin J., Nolz Jeffrey C., Harty John T., Pathogen-specific inflammatory milieux tune the antigen sensitivity of CD8+ T cells by enhancing T cell receptor signaling. Immunity 38, 140–152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.