

Significance
Changes to pathogens’ ability to infect particular hosts (“host tropism”) often dictate modern infectious disease outbreaks. Lyme disease bacteria present a suitable model to study the molecular drivers of host tropism, as variants selectively survive in different hosts. To escape from killing by complement, part of the immune system, these bacteria produce the outer surface protein CspZ, which binds the complement inhibitor factor H (FH) to promote infection. Here, we applied multidisciplinary approaches to identify a small variable AA motif within the FH-binding site that drives host-specific FH-binding activity and infectivity. We further elucidated the evolutionary history of the recent CspZ divergence to provide new insights into the emergence of pathogen host tropism.
Keywords: Lyme disease, CspZ, factor H, complement, host tropism
Abstract
Modern infectious disease outbreaks often involve changes in host tropism, the preferential adaptation of pathogens to specific hosts. The Lyme disease-causing bacterium Borrelia burgdorferi (Bb) is an ideal model to investigate the molecular mechanisms of host tropism, because different variants of these tick-transmitted bacteria are distinctly maintained in rodents or bird reservoir hosts. To survive in hosts and escape complement-mediated immune clearance, Bb produces the outer surface protein CspZ that binds the complement inhibitor factor H (FH) to facilitate bacterial dissemination in vertebrates. Despite high sequence conservation, CspZ variants differ in human FH-binding ability. Together with the FH polymorphisms between vertebrate hosts, these findings suggest that minor sequence variation in this bacterial outer surface protein may confer dramatic differences in host-specific, FH-binding-mediated infectivity. We tested this hypothesis by determining the crystal structure of the CspZ–human FH complex, and identifying minor variation localized in the FH-binding interface yielding bird and rodent FH-specific binding activity that impacts infectivity. Swapping the divergent region in the FH-binding interface between rodent- and bird-associated CspZ variants alters the ability to promote rodent- and bird-specific early-onset dissemination. We further linked these loops and respective host-specific, complement-dependent phenotypes with distinct CspZ phylogenetic lineages, elucidating evolutionary mechanisms driving host tropism emergence. Our multidisciplinary work provides a novel molecular basis for how a single, short protein motif could greatly modulate pathogen host tropism.
The emergence of infectious disease outbreaks often involves changes in host tropism, the preferential adaptation of pathogens to selectively invade and persist in hosts (1). Such host tropism is often the result of ongoing host-pathogen interactions (2). Evolution theoretically favors the emergence of host-specializing pathogens, but host-generalist strategies can be advantageous in environments where pathogens have the potential to regularly interact with multiple hosts (3). Specialism vs. generalism can be attributed to polymorphisms within pathogen proteins that differentially interact with host ligands (2). Such polymorphism-mediated host ranges are often thought to require complex host-specific adaptive mechanisms (4 and 5), but actually can be conferred by a few variable amino acids (AAs) (6). Understanding how such minor differences impact diverse host-adapted phenotypes can elucidate the mechanistic insights into the emergence of modern infectious diseases, allowing for the development of earlier and more efficient public health interventions.
Species within the Borrelia (Borreliella) burgdorferi sensu lato genospecies complex cause Lyme disease (LD). These spirochetal bacteria are transmitted by ticks and maintained by several reservoir hosts, primarily rodents and birds (7). Lyme borreliae have been genotyped using different polymorphic loci, such as ospA, ospC, the 16S-23S rRNA intergenic spacer (RST [typing]), and multilocus sequence typing (8, 9). Laboratory and field studies have shown not only that Lyme borreliae species differ in the host species they infect, but that individual genotypes within single species display distinct preferential host associations, particularly within B. burgdorferi sensu stricto (hereafter B. burgdorferi, “Bb”), the primary LD agent in North America (10, 11). This species- and genotype-specific host selectivity distinguishes Lyme borreliae as a model system for studying the molecular basis and evolutionary history of host tropism (10, 11).
Lyme borreliae host associations require the spirochetes to transmit from infected ticks to hosts, establish infection at tick-biting sites, and disseminate hematogenously to persist in distal tissues (10, 11). The survival of Lyme borreliae during these discrete events necessitates evasion of multiple host immune responses, including complement, a first-line innate immune defense mechanism (12–14). Complement can be activated by the classical, lectin, and/or alternative pathways (15). The activation of these pathways results in the cascading cleavage and recombination of multiple complement components, ultimately resulting in phagocytic clearance, inflammation, and lytic pore formation on the pathogen surface by the C5b-9 protein complex. Complement is downregulated by diverse regulatory proteins that inhibit activation, preventing native cell damage in the absence of pathogens. For example, the alternative pathway is inhibited by factor H (FH), which is comprised of 20 individual short consensus repeat (SCR) domains (16).
Similar to many bloodborne pathogens that evolved mechanisms to circumvent complement-mediated clearance, Lyme borreliae produce several outer surface proteins that bind and recruit complement components and/or regulatory proteins on the spirochete surface to inactivate complement (12–14). One of these proteins is an FH-binding protein, CspA (also known as Complement Regulator Acquiring Surface Protein 1, CRASP-1, or BBA68 of pFam54-60 in Bb strain B31) that confers spirochete survival via complement evasion in the tick bloodmeal (17). However, this protein is downregulated after spirochetes infect hosts, suggesting other functionally redundant proteins may facilitate spirochete survival at this infection stage. In fact, CspZ (also known as CRASP-2 or BBH06 in Bb strain B31), which binds to the 6th and 7th domains of the SCR (SCR6-7), is upregulated when spirochetes reside in hosts (18, 19). This protein is highly conserved both between and within different Lyme borreliae species [>85% AA identity between species and 98% between Bb strains] (20–23). We and others have previously reported that the FH-binding activity of one CspZ variant promotes spirochete survival in vertebrate sera, and consequently promotes host infectivity using non-physiologically relevant infection routes (i.e., not tick-transmitted) (24, 25). However, CspZ variants differ in their human FH-binding activity (20, 21), and vertebrate complement components or regulatory proteins vary between host taxa (e.g., ~40% AA identity between mammalian and avian FH) (26). These findings raise several intriguing questions: Could this minor divergence among CspZ variants confer host-specific differences in FH-binding activity, resulting in varying host infectivity phenotypes? Further, how did such divergences evolve to impact Lyme borreliae host associations?
In this study, we solved the high-resolution structure of the CspZ-human FH complex, identified a polymorphic CspZ motif within the FH-binding interface, and defined its contribution to host-specific FH-binding activity. We then examined the role of this motif in dictating strain-specific, host-dependent dissemination, using mice and quail as rodent and avian models, respectively. Paired with evolutionary analyses of cspZ, we further elucidated how the evolutionary history behind a minor divergence in an immune evasion determinant can impact pathogen host tropism.
Results
A Polymorphic CspZ Loop Structure Defines Human FH-Binding Activity.
We set to identify the regions driving the differences in human FH-binding activity among CspZ variants. We first pinpointed the AAs that are involved in FH-binding by co-crystalizing SCR6-7 of human FH and a recombinant CspZ variant with human FH-binding activity, from Bb strain B408 (“CspZB408”) (21). The resulting crystal structure (2.59 Å) showed an extensive binding interface between CspZB408 and both SCR6 and SCR7 (Fig. 1A). Specifically, Asp47, Tyr50, Asn51, Thr54, Asn58, and Thr62 in helix-B, Arg142 in helix-F, Asn183 from helix-G, and Tyr214 in helix-I of CspZB408 interacted with SCR7 (Fig. 1 B and C and SI Appendix, Table S1). Additionally, Asp71 from the loop between helix-B and -C, and Asp73 and Ser75 from the same region, bound to SCR7 and SCR6, respectively (Fig. 1 B and C and SI Appendix, Table S1).
Fig. 1.
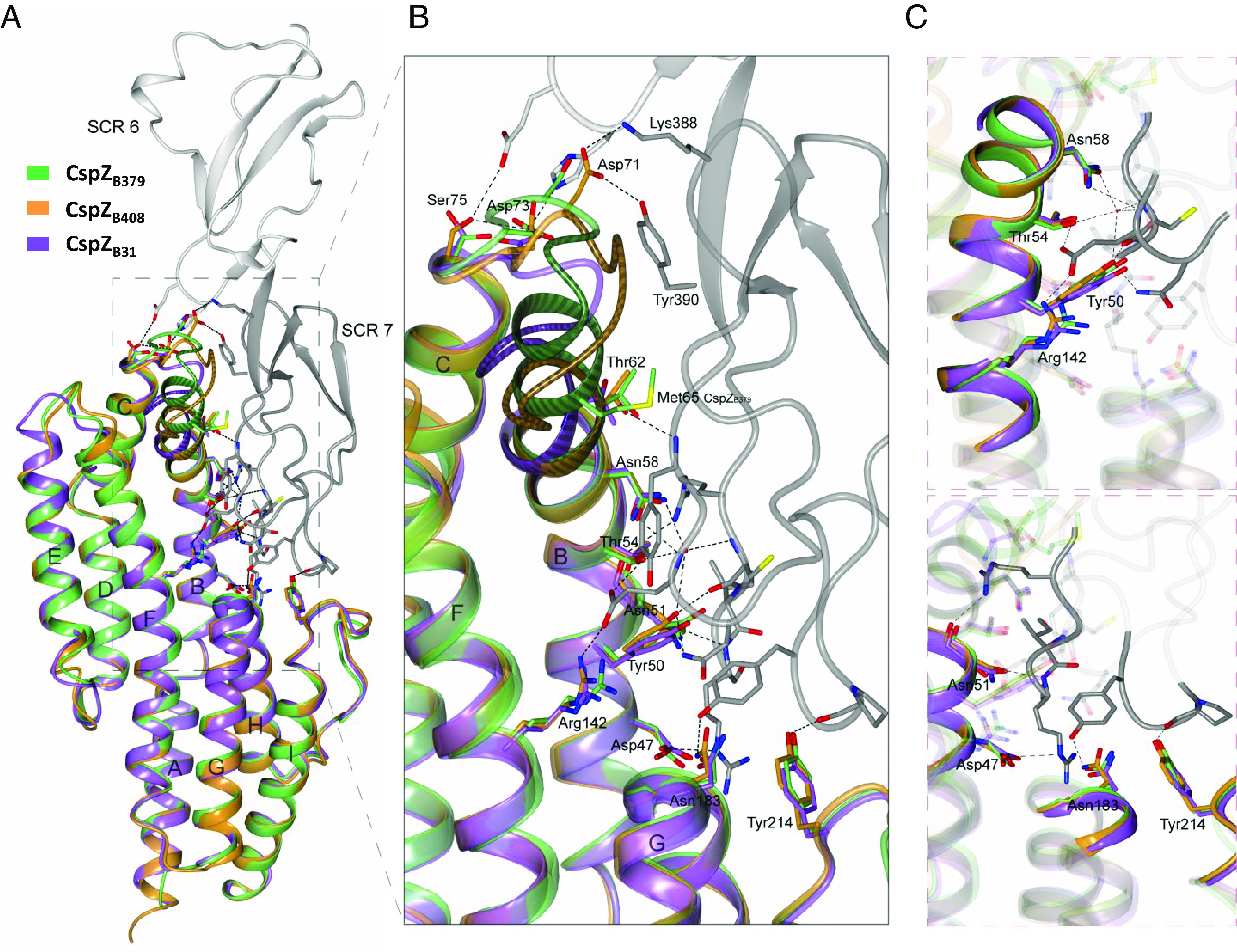
Superimposed crystal structures of the CspZB408-SCR6-7 complex, CspZB31, and CspZB379 reveal the FH-binding mechanisms of CspZ variants. (A) The crystal structure of human FH domains SCR6 (light gray) and SCR7 (dark gray) in complex with CspZB408 (orange) was superimposed with that of CspZB31 (purple) and CspZB379 (green). (B) The FH-binding interface with the location of the CspZB408 residues involved in human SCR6-7 binding, and the equivalent residues in CspZB31 and CspZB379. The numbering of the residues is given for CspZB408. α-helices in CspZ are labeled from A to I starting from the N terminus. The polymorphic loop region between α-helices B and C in CspZB408 (residues 60-IMTYSEVNNVTD-71), CspZB31 (residues 60-IMTYSEGT-67) and CspZB379 (residues 60-IMTYIMTYSEGT-71) are indicated. A more detailed illustration of the CspZB408-SCR6-7 complex interface involving the CspZB408 residues (C, Top) Tyr50, Thr54, Asn58 and Arg142 and (C, Bottom) the residues Asp47, Asn51, Asn183 and Tyr214 is shown.
To locate the residues that impact the variable human FH-binding activity, we aligned the sequences of CspZB408 with one CspZ variant that binds to human FH (CspZ from Bb B31, “CspZB31”), and one that lacks human FH-binding ability from Bb strain B379 (“CspZB379”) (21). All the aforementioned residues involved in human FH-binding are conserved among these variants, except Asn51 and Asp71 in CspZB408 (SI Appendix, Fig. S1A). We also determined the crystal structures of CspZB379 and CspZB408 (2.10 and 2.45 Å, respectively). The previously crystalized CspZB31 superimposes very well with both CspZB379 (RMSD: 0.88 Å over 190 aligned Cα atoms) and CspZB408 (RMSD: 0.90 Å over 195 aligned Cα atoms), reflecting the high conservation between the proteins (23) (Fig. 1A). Moreover, the crystal structure of CspZB408 shows very high similarity to CspZB408 from the complex structure (RMSD: 0.93 Å over 201 aligned Cα atoms). For the two aforementioned nonconserved AAs involved in human FH-binding: in helix-B, Asn51 in CspZB408 is substituted by a Ser51 in CspZB379. The superimposed structure suggests that the relatively small side chain of serine would not inhibit human FH-binding in CspZB379 because the position of helix-B is identical in both proteins (RMSD: 0.15 Å over 16 aligned Cα atoms) (Fig. 1 B and C). The second nonconserved AA, Asp71 in CspZB408, is part of an insertion that is not present in CspZB31 and CspZB379 (Fig. 1B), and CspZB31 binds human FH. Therefore, these two non-conserved residues alone cannot explain the differential human FH-binding ability of these CspZ variants. However, the C-terminus of helix-B and the subsequent loop region between helix-B and helix-C harbor polymorphisms (Fig. 1A): CspZB379 has a unique duplication of the last residues in helix-B (Ile60, Met61, Thr62, and Tyr63) (SI Appendix, Fig. S1A), resulting in an extended helix-B in this variant (Fig. 1B). This structural extension leads to steric hindrance between CspZB379 and human FH to selectively prevent human FH-binding activity by CspZB379. The abovementioned regions are well defined in the electron density map and are not influenced by crystal packing (SI Appendix, Fig. S2). Thus, our results suggest variant-specific CspZ structural differences in a loop and the adjacent helix-B (hereafter, “loop structures”), providing mechanistic insights into the CspZ-mediated, polymorphic human FH-binding activity.
CspZ Loop Structures Determine Host-Specific FH-Binding Activity.
FH SCR6-7 is divergent among vertebrate species (SI Appendix, Fig. S1B), suggesting the CspZ loop structures may dictate host-specific FH-binding activity. We found that CspZB31 and CspZB408, but not CspZB379, bound to mouse FH. CspZB31 and CspZB379, but not CspZB408, bound to quail FH (Fig. 2A and SI Appendix, Fig. S3 and Table S2). We then searched for structural evidence of this FH-binding activity by superimposing the complex structure of CspZB408-human FH with CspZB31, CspZB379, the previously resolved structures of mouse SCR6-7, and the AlphaFold-predicted structure of quail SCR6-7 (pLDDT > 70) (Fig. 2C). There are similar tertiary structures between mouse and human SCR6-7 (RMSD: 2.47 Å over 108 aligned Cα atoms), including segments facing the protein complex interface which is consistent with the ability of both FH variants to selectively bind CspZB31 and CspZB408 (Fig. 2A and SI Appendix, Fig. S3 and Table S2) (21). The CspZB379-specific duplication showed potential structural hindrance in the mouse FH-binding interface, similar to human FH (Fig. 2D). However, the equivalent region in quail SCR7 (median pLDDT: 92) is positioned away from the CspZ-FH-binding interface, allowing sufficient space for the extended helix-B of CspZB379 to interact with quail FH (Fig. 2D). Further, the crystal structure of CspZB408-SCR6-7 superimposed with the predicted structure of quail SCR6-7 showed that Asp71 of CspZB408 may collide with quail FH, while the equivalent region in mouse SCR7 would not interfere with the FH-binding of CspZB408 (Fig. 2D).
Fig. 2.
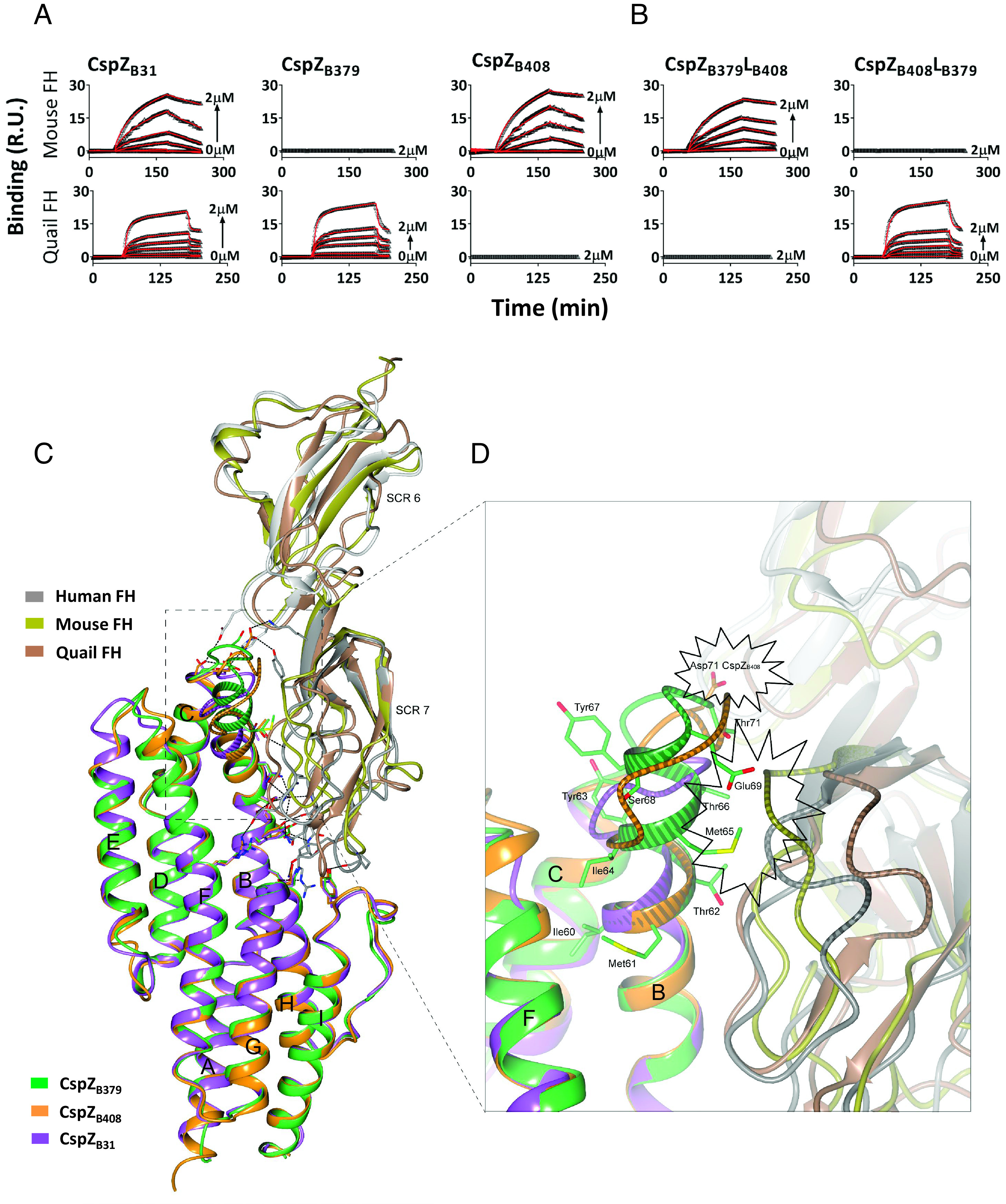
The polymorphic loop region in CspZ promotes host-specific FH-binding ability. (A and B) Untagged CspZ variants or mutants were flowed over the chip surface, conjugated with indicated FH variants. Binding was measured in response units (R.U.) by SPR. The kon, koff, and KD values were determined from the average of these three experiments (SI Appendix, Table S2). (C) The crystal structure of CspZB408-SCR6-7 where human SCR6-7 (gray) is superimposed with mouse SCR6-7 (gold, PDB: 2YBY), and the predicted structure of quail SCR6-7 (brown), while CspZB408 (orange) is superimposed with CspZB31 (purple) and CspZB379 (green). (D) The loop region in CspZ proteins located between α-helices B and C. The Asp71 in CspZB408, and the residues 60-IMTYIMTYSEGT-71 in CspZB379 are highlighted. The polymorphic loop region between α-helices B and C in CspZ variants, as well as the loop region in human, mouse and quail SCR6-7 located at the interface with helix-B in CspZ, is showed with a striped pattern. The potential structural hindrance region between CspZB408 and quail FH, as well as the structural hindrance between CspZB379 and both mouse and human FH, are indicated with the starbursts.
These structural differences in mouse/human vs. quail FH, paired with the respective impactful AAs from CspZ variants, suggest the loop structures of CspZ may cause host-specific FH-binding activity. To test this, we swapped the loop regions of the two CspZ variants with distinct host-specific FH-binding ability to generate two chimeric proteins: CspZB379LB408 has the backbone of the strictly quail FH-binder (CspZB379), and the loop structures of the strictly mouse FH-binder (CspZB408), whereas CspZB408LB379 has the backbone of CspZB408 and the loop structures of CspZB379 (SI Appendix, Fig. S1A). We found that CspZB379LB408 and CspZB408LB379 selectively bound to mouse and quail FH, respectively, demonstrating the CspZ loop structures are a determinant of host-specific FH-binding activity (Fig. 2B and SI Appendix, Fig. S3 and Table S2).
The CspZ Loop Structures Dictate Spirochete Strain- and Host-Specific Complement Inactivation.
We next examined the host-dependent complement inactivation of these CspZ variants produced on the spirochete surface. We thus obtained a wild-type Bb strain B31-A3 (referred to hereafter as “WT B31-A3”) and a cspZ-deficient mutant strain in this background harboring an empty vector (“ΔcspZ/Vector”). We complemented this mutant with a plasmid encoding cspZB31 (“pCspZB31”) (25), cspZB379 (“pCspZB379”), cspZB408 (“pCspZB408”), or the loop-swapped mutants (“pCspZB379LB408” and “pCspZB408LB379”). We verified that these strains had similar generation times (SI Appendix, Table S3), no differences in the CspZ surface production level (SI Appendix, Fig. S4), and that surface-produced CspZ bound FH in the same manner as the recombinant proteins (SI Appendix, Fig. S5). We then used flow cytometry to determine the ability of these CspZ variants and mutants to inactivate mouse and quail complement by measuring the deposition levels of mouse C5b-9 and quail C8, respectively, after incubation with sera from these animals. All strains had levels of mouse C5b-9 or quail C8 deposition significantly lower than the control strain: high passage, noninfectious, and mouse and quail complement-susceptible Bb strain B313 (Fig. 3 A and B and SI Appendix, Fig. S6 A and B) (19). ΔcspZ/Vector had significantly greater levels of mouse C5b-9 and quail C8 deposition than WT B31-A3 or pCspZB31 (Fig. 3 A and B and SI Appendix, Fig. S6 A and B) (25, 27). Compared to ΔcspZ/Vector, pCspZB379 had indistinguishable levels of mouse C5b-9 deposition but significantly lower levels of quail C8, whereas pCspZB408 harbored significantly lower levels of mouse C5b-9 but indistinguishable levels of quail C8 (Fig. 3 A and B and SI Appendix, Fig. S6 A and B). Further, pCspZB379LB408 recruited significantly lower levels of mouse C5b-9 but indistinguishable levels of quail C8 than ΔcspZ/Vector, whereas pCspZB408LB379 bound indistinguishable levels of mouse C5b-9 but significantly lower levels of quail C8 than ΔcspZ/Vector (Fig. 3 A and B and SI Appendix, Fig. S6 A and B). These results correspond with these variants’ host-specific FH-binding activity, suggesting the loop structure-driven host-specific FH-binding activity confers host-specific complement inactivation.
Fig. 3.

The CspZ loop-driven, variable FH-binding activity confers host-specific complement inactivation and early dissemination. (A and B) Indicated Bb strains were incubated with mouse or quail sera (20%) followed by straining with antibodies against mouse C5b-9 or quail C8. The deposition levels of (A) mouse C5b-9 or (B) quail C8 on the surface of the indicated strains were measured by flow cytometry. Each bar represents the mean of three independent experiments ±SEM. Significant differences in the deposition levels relative to B313 (“Φ”), ΔcspZ/Vector (“*”), or between two strains relative to each other (“#”), are indicated. (C and D) The B. burgdorferi strains were incubated with (C) white-footed mouse sera or (D) quail sera (40%). The number of live spirochetes was assessed microscopically using live/dead staining. The percent survival of the strains was calculated using the number of live spirochetes at 4-h post incubation normalized to that prior to incubation with sera. Each bar represents the mean of three independent experiments (n = 3) ±SEM. Significant differences in the percent survival of spirochetes relative to the ΔcspZ/Vector (“*”) or between two strains relative to each other (“#”) are indicated. (E–M) I. scapularis nymphs carrying indicated Bb strains were allowed to feed until repletion on indicated animals. Shown are the geometric mean of recA copies in animal tissues, normalized to 104 copies of (E–I) mouse nidogen or (J–M) quail β-actin ±SEM of five animals per group, except for the BALB/c blood from the B31-A3 and ΔcspZ/Vector groups, which have six and 10 mice per group, respectively. Significant differences in the spirochete burdens relative to the ΔcspZ/Vector (“*”) or between two strains relative to each other (“#”) are indicated.
We also measured the survival of these strains in rodent and quail sera by counting the number of live bacteria using both live/dead staining and the number of colony-forming units (CFUs) after plating. Note that sera from white-footed mice (Peromyscus leucopus) rather than house mice (Mus musculus) were used to represent rodent sera. House mouse complement is highly labile in vitro, which leads to inconsistent results (28). We found that WT B31-A3 survived in white-footed mouse and quail sera more efficiently than ΔcspZ/Vector (Fig. 3 C and D and SI Appendix, Fig. S7). Because the percent survival yielded from live/dead staining and CFU counting were indistinguishable (SI Appendix, Fig. S7), counting the number of live bacteria via live/dead staining was used to determine the survival of the rest of the strains. We found that pCspZB31 also survived in white-footed mouse and quail sera at greater levels than ΔcspZ/Vector (Fig. 3 C and D) (25). Compared to ΔcspZ/Vector, pCspZB379 survived at significantly greater levels in quail but not white-footed mouse sera, whereas pCspZB408 survived at significantly higher levels in white-footed mouse but not quail sera (Fig. 3 C and D). Relative to ΔcspZ/Vector, pCspZB379LB408 survived significantly higher in white-footed mouse but not in quail sera, whereas pCspZB408LB379 survived significantly higher in quail but not in white-footed mouse sera (Fig. 3 C and D). However, these differences were not observed in the presence of sera treated with Cobra Venom factor or Ornithodorus moubata complement inhibitor (OmCI) that deplete functional rodent and quail complement, respectively (SI Appendix, Fig. S8) (29, 30). This suggests that CspZ loop structures determine spirochete host-specific serum survival.
The CspZ Loop Structures Define Host-Specific, Complement-Dependent, Early-Onset Dissemination.
We further evaluated whether the CspZ loop structure variants determine host infectivity by allowing Ixodes scapularis nymphs carrying similar burdens of each of the aforementioned Bb strains to feed on mice and quail (SI Appendix, Fig. S9 A and D). The burdens of ΔcspZ/Vector were significantly lower than WT B31-A3 at 7- and 10-days postfeeding (dpf) in mouse blood and at 10 dpf in distal tissues (joints, heart, and bladder), and at 9 dpf in quail blood and distal tissues (brain and heart) (SI Appendix, Fig. S10). There were no differences in burdens by a later time point (i.e., 14 dpf, SI Appendix, Fig. S10). We thus performed subsequent experiments using our entire Bb strain cohort at 10 dpf and 9 dpf in mouse and quail, respectively. In mice, we found indistinguishable spirochete burdens at initial infection sites (tick bite sites) between all seven strains tested at 10 dpf (Fig. 3E). pCspZB31, pCspZB408, and pCspZB379LB408, but neither pCspZB379 nor pCspZB408LB379, colonized mouse blood and distal tissues at significantly higher levels than ΔcspZ/Vector (Fig. 3 F–I). At 9 dpf in quail, we found no significantly different spirochete burdens at tick bite sites among strains. However, pCspZB31, pCspZB379, and pCspZB408LB379, but neither pCspZB408 nor pCspZB379LB408, colonized quail blood and distal tissues at significantly greater levels than ΔcspZ/Vector (Fig. 3 J–M). There were no significantly different burdens of any strains in any tissue from complement-deficient mice and quail (C3−/− mice or OmCI-treated quail) (SI Appendix, Fig. S11). These results demonstrate that the CspZ loop structures define complement-dependent, spirochete strain- and host-specific early onset dissemination.
The Population-Wide CspZ Loop Structures Evolved from a Variant with Versatile FH-Binding Ability.
To investigate the evolutionary history behind the CspZ loop structures, we mined Bb cspZ from NCBI GenBank and Sequence Read Archive (totaling 174 high-quality cspZ isolates originating from ticks, reservoir hosts, and patients, across North America, Europe, and Asia). A haplotype-based phylogenetic network distinctly separated three individual lineages, each of which contained one of the above-tested alleles: cspZB31, cspZB379, or cspZB408 (SI Appendix, Fig. S12). Examining the entire dataset, isolates within the same lineage had over 99% overall sequence identity and the same loop structure as cspZB408, cspZB379, or cspZB31 (defined as the unique insertion, duplication, or neither, respectively) (Fig. 4). We pinpointed single AA polymorphisms (SAPs) in the isolates and found none of the SAPs within each lineage were in the FH-binding interface, except for one SAP in one isolate in the CspZB379 lineage (SI Appendix, Tables S1 and S4). The majority of the SAPs were between lineages, located in the loop structures driving the FH-binding abilities. While this present study has been the only one to investigate the abilities of CspZ variants to bind house mouse and quail FH, the human FH-binding abilities of 13% of these 174 isolates have been evaluated (20, 21, 25). Incorporating these findings with the phylogeny, we found a 100% correlation of known host-specific FH-binding ability with lineages (Fig. 4). These results suggest each CspZ lineage with distinct loop structures can be linked to CspZ-specific, host-dependent FH-binding ability.
Fig. 4.

Phylogeny and sequence comparisons support polymorphic CspZ loop arising from host-specific adaptation of Lyme borreliae. Unrooted likelihood phylogenetic tree of 174 Bb cspZ isolates generated with IQ-tree and visualized in iTOL. The names of the isolates are highlighted based on their CspZ loop types. The percent sequence identity of each isolate relative to CspZB31 (purple), CspZB408 (orange), or CspZB379 (green) is indicated on the outer ring. The known FH-binding activity is marked: black or gray stick figures, mice, and quail indicate FH binding or lack thereof, respectively.
We explored the evolutionary mechanisms that could have led to this minor loop structure-dependent FH-binding specificity. No evidence for recombination was found (GARD method: 38 potential and one inferred breakpoints) or past changes in effective population size (Bayesian skyline analysis: all predicted mean population sizes are within the lowest and highest HPD95% of all timepoints). The evidence for episodic diversifying selection along cspZ was weak (BUSTED method including site-to-site synonymous rate variation and double/triple nucleotide substitutions: p = 0.025) while excluding terminal branches on the gene phylogeny. Several codons appear to deviate from expectations of neutrality (SI Appendix, Fig. S1A). We reconstructed the possible last common ancestor states (“LCAS”) of the entire Bb CspZ dataset using the previously estimated chromosome mutation rate (31). The diversification between the three lineages was dated at 261 to 784 years B.P. (YBP) (SI Appendix, Table S5), whereas the diversification of the Bb CspZ LCAS as a whole likely occurred 2166 YBP (HPD95% 896 to 5,718). The LCAS variants possess a loop region resembling that of CspZB31 both in primary and tertiary structure (SI Appendix, Figs. S1A and S13), and bound to human, mouse, and quail FH, similar to CspZB31 (SI Appendix, Fig. S14 and Table S6). These results suggest that modern CspZ variants arose relatively recently from a generalist variant with versatile FH-binding features.
Discussion
Host tropism is an outcome of host-pathogen interactions (11). In vector-borne zoonotic pathogens, such interactions include vector-to-host transmission, pathogen dissemination and persistence, and host-to-vector acquisition (32). For Lyme borreliae to survive these steps, spirochetes need to overcome numerous host immunological mechanisms, including complement-mediated killing. In fact, Lyme borreliae inactivate complement in the tick blood meal for tick-to-host transmission, and in the host bloodstream for dissemination and persistence (25, 33, 34). In this study, we examined this inactivation conferred by the FH-binding protein, CspZ. We showed reduced burdens of a tick-introduced, cspZ-deficient mutant at the early stages of bloodstream survival and distal tissue colonization, defining this polymorphic protein as a contributor to survival at these early stages. Superimposing the crystal structures of the three polymorphic CspZ variants revealed a short, variable AA motif consisting of duplication and insertion/deletion events (23). Our newly-resolved and software-predicted complex structures of CspZ with human, mouse, and quail SCR6-7 further linked these variable residues to distinct host-specific FH-binding activity, in a unique manner compared to previously reported pathogenic FH-binding proteins (SI Appendix, Fig. S15) (35). These results are congruent with the ability of CspZ variants and their associated mutants with swapped loop structures to promote host-specific FH-binding activity, complement evasion, and dissemination, defining this CspZ loop structure as a determinant of host tropism. Our findings thus demonstrate the concept of how minor variation functionally impacts host-adapted phenotypes, providing a new mechanism for host tropism modulation by pathogens (36–38).
We integrated an evolutionary approach to investigate the emergence of such host tropism determinants, an approach whose importance has been recently demonstrated to understand and track modern disease emergence (39). Our findings demonstrate that modern CspZ variants emerged from a last common ancestor that could bind human, mouse, and quail FH. This diversification occurred relatively recently compared to the origin of Bb 60,000 YBP, and the diversification of another FH-binding protein, CspA, about 25,000 YBP (31, 33). The lack of CspZ orthologs also supports the predicted young age of this protein, and as such it may be undergoing widespread neutral evolution (23, 40). We detected diversifying selection using multiple models on three codons, all of which are located in helices. No evidence for positive selection was found in the loop structures driving FH-binding phenotypes, despite those loops harboring most of the variation and insertions/deletions across CspZ. Alignment columns with gaps are excluded from most selection scans, rendering the detection of selective pressure more challenging and less trustworthy.
Other polymorphic anticomplement loci, particularly ospC, are undergoing balancing selection (41, 42). Negative frequency-dependent selection (NFDS) and multiple-niche polymorphism (MNP) have been proposed as mechanisms driving ospC evolution. NFDS allows for high allele diversity to emerge through the negative correlation between pathogen fitness and the frequency of any alleles in the pathogen population (43). In Lyme borreliae, NFDS may explain the greater antibody-mediated clearance of spirochete strains harboring higher-frequency ospC alleles (44, 45). MNP maintains elevated levels of genetic diversity by varying pathogen fitness across reservoir hosts (42). As a systemically redundant anticomplement protein (46), CspZ may also be subjected to balancing selection. It is unlikely, but the lack of detectable selective pressure on the loop could also mean this locus is undergoing neutral selection. Balancing selection on one locus can increase diversity in another genetically-linked locus, and there is genetic linkage between cspZ and ospC types (20, 21, 27, 47). This raises the intriguing possibility that ospC-mediated balancing selection may promote diversification through drift in other functionally redundant genes such as cspZ without deleterious effects on the pathogen, warranting further investigations.
In this study, we used isogenic strains producing different CspZ variants under the control of an identical cspZ promoter to facilitate the equal production levels of each CspZ variant. While the strength of this approach can directly attribute phenotypes to the variation of coding sequences of CspZ (48), it does not consider the possibility that each cspZ variant is regulated differently in their parental strains. However, CspZB379 and CspZB408 appear to be produced at similar levels in their parental strains (21), which may alleviate the abovementioned limitation. Additionally, it should be noted that other anticomplement proteins from ticks or spirochetes, or noncomplement-mediated mechanisms may (and likely do) contribute to the overall strain-specific, host-adapted phenotypes. This highlights the potential of polygenic host-specific phenotypes, which would need to be further considered in defining drivers of host tropism (49–51). Overall, the information and the platform established in this study would provide greater insights into pathogen–host interactions, facilitating the understanding of host tropism as a cause of infectious diseases.
Materials and Methods
Ethics Statement.
All animal experiments were performed in strict accordance with all provisions of guidelines detailed in SI Appendix.
Bacteria, Mice, Quail, Ticks, Animal Sera, and Proteins.
The Escherichia coli, Pichia pastoris, and Borrelia strains, the sources of ticks, and animal sera, along with the FH and OmCI used in this study and their verification are detailed in the SI Appendix.
Structure Determination by Crystallization and Prediction by AlphaFold.
CspZB408 (4 mg/mL) and human SCR6-7 (3 mg/mL) were mixed at a molar ratio of 1:2 and loaded on a pre-equilibrated HiLoad 16/600 Superdex 200 prep grade column (GE Healthcare, Chicago, IL, USA), followed by size exclusion chromatography and SDS PAGE. Crystals of this complex and those of CspZB379 and CspZB408, the diffraction data for these proteins and their complex, are detailed in Supplementary Information. AlphaFold v2.0 (52) was used to predict the 3D structure for quail FH SCR6-7 extrapolated from the sequences of Coturnix japonica complement FH (GenBank: XM_015869474.2), detailed in the SI Appendix.
ELISAs and SPR.
Enzyme-linked immunosorbent assay (ELISA) was used to determine FH-binding by CspZ proteins, detailed in the SI Appendix. For Surface Plasmon Resonance (SPR), interactions of CspZ proteins with FH were analyzed by SPR using a Biacore T200 (Cytiva, Marlborough, MA), detailed in SI Appendix.
Flow Cytometry and Serum Resistance Assays.
CspZ production, FH-binding, complement deposition on spirochetes were determined using flow cytometry, detailed in SI Appendix. The serum resistance of Bb was measured as outlined in the SI Appendix.
Mouse and Quail Infection.
The generation of flat, infected I. scapularis nymphs, the feeding of nymphs on mice and quail, and the determination of bacterial burdens at animal tissues and ticks are detailed in the SI Appendix.
Genomic Analyses.
Sequences of both FH and CspZ were aligned and visualized, as detailed in SI Appendix. To generate the cspZ phylogenetic trees, we mined all publicly available cspZ sequences on NCBI as of September 2021. All resulting sequences were analyzed as outlined in the SI Appendix. The ancestor state of CspZ was reconstructed, described in the SI Appendix.
Statistical Analysis.
Samples were compared using the Mann–Whitney U test or the Kruskal–Wallis test with the two-stage step-up method of Benjamini, Krieger, and Yekutieli (53).
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Simon Starkey, Quinton Smith, Deirdre Torrisi, and Joey Anderson from the Wadsworth Veterinary Sciences facility for animal husbandry; Richard Marconi, Utpal Pal, Patricia Rosa, Gary Wormser, and Ira Schwartz for providing the Bb strains; David Vance and Nicholas Mantis for sharing the E. coli strain; Susan Madison-Antonucci to allow us using her fluorescence microscope; Timothy Czajka for the assistance of SPR analysis; Nikhat Parveen, Klemen Strle, and Grace Chen for insightful comments; Carly Fernandes for assistance with making media; and Laurel Lown for assistance with running PCRs. We also appreciate Leslie Eisele and Renjie Song of Wadsworth Biochemistry and Immunology Core for CD spectroscopy, SPR, and flow cytometry; and Karen Chave of the Wadsworth Protein Expression Core for the help of the materials to purify FH. We thank the Wadsworth Center ATGC core for plasmid sequencing. Diffraction data for B. burgdorferi CspZ proteins and CspZ-human FH were collected on BL14.1 at the BESSY II electron storage ring operated by the Helmholtz-Zentrum, Berlin. We would particularly like to acknowledge the help and support of Manfred S. Weiss and Jan Wollenhaupt during the diffraction data collection. This work was supported by NSF IOS1754995 (S.-O.K.), NSF IOS1755286 (Y.-P.L., A.L.M., T.A.N., A.P.D., J.L.S., and A.T.C.), DoD TB170111, NIHR21AI144891, NIH R21AI146381, New York State Department of Health Wadsworth Center Start-Up Grant (A.L.M., T.A.N., and Y.-P.L.), and LOEWE Center DRUID Novel Drug Targets against Poverty-Related and Neglected Tropical Infectious Diseases, project C3 (P.K.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
A.L.M., K.B., S.-O.K., and Y.-P.L. designed research; A.L.M., K.B., A.P.D., T.M.H., M.Z.-C., T.A.N., J.L.S., I.A., A.K., J.B., A.T.C., S.-O.K., and Y.-P.L. performed research; A.L.M., K.B., and Y.-P.L. contributed new reagents/analytic tools; A.L.M., K.B., S.-O.K., and Y.-P.L. analyzed data; S.-O.K., K.B., A.T.C. and Y.-P.L. supervise the work; and A.L.M., K.B., A.P.D., A.T.C., P.K., S.-O.K., and Y.-P.L. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Kalvis Brangulis, Email: kalvis@biomed.lu.lv.
Sergios-Orestis Kolokotronis, Email: sok@downstate.edu.
Yi-Pin Lin, Email: Yi-Pin.Lin@health.ny.gov.
Data, Materials, and Software Availability
The coordinates and the structure factors for CspZB379, CspZB408, and human SCR-CspZB408 have been deposited in the Protein Data Bank with accession codes 7ZJJ (54), 7ZJK (55), and 7ZJM (56), respectively. All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Trovato M., Sartorius R., D’Apice L., Manco R., De Berardinis P., Viral emerging diseases: Challenges in developing vaccination strategies. Front. Immunol. 11, 2130 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Douam F., et al. , Genetic dissection of the host tropism of human-tropic pathogens. Annu. Rev. Genet. 49, 21–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kurtenbach K., et al. , Fundamental processes in the evolutionary ecology of Lyme borreliosis. Nat. Rev. Microbiol. 4, 660–669 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Longdon B., Brockhurst M. A., Russell C. A., Welch J. J., Jiggins F. M., The evolution and genetics of virus host shifts. PLoS Pathog. 10, e1004395 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin Y. P., et al. , Cellular and immunological mechanisms influence host-adapted phenotypes in a vector-borne microparasite. Proc. Biol. Sci. 289, 20212087 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dean A. M., Thornton J. W., Mechanistic approaches to the study of evolution: The functional synthesis. Nat. Rev. Genet. 8, 675–688 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radolf J. D., Strle K., Lemieux J. E., Strle F., Lyme disease in humans. Curr. Issues Mol. Biol. 42, 333–384 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz I., Margos G., Casjens S. R., Qiu W. G., Eggers C. H., Multipartite genome of Lyme disease Borrelia: Structure, variation and prophages. Curr. Issues Mol. Biol. 42, 409–454 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Margos G., et al. , MLST of housekeeping genes captures geographic population structure and suggests a European origin of Borrelia burgdorferi. Proc. Natl. Acad. Sci. U.S.A. 105, 8730–8735 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolcott K. A., Margos G., Fingerle V., Becker N. S., Host association of Borrelia burgdorferi sensu lato: A review. Ticks Tick Borne Dis. 12, 101766 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Tufts D. M., et al. , Outer surface protein polymorphisms linked to host-spirochete association in Lyme borreliae. Mol. Microbiol. 111, 868–882 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skare J. T., Garcia B. L., Complement evasion by Lyme disease spirochetes. Trends Microbiol. 28, 889–899 (2020), 10.1016/j.tim.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Y. P., Diuk-Wasser M. A., Stevenson B., Kraiczy P., Complement evasion contributes to Lyme borreliae-host associations. Trends Parasitol. 36, 634–645 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dulipati V., Meri S., Panelius J., Complement evasion strategies of Borrelia burgdorferi sensu lato. FEBS Lett. 594, 2645–2656 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Reis E. S., Mastellos D. C., Hajishengallis G., Lambris J. D., New insights into the immune functions of complement. Nat. Rev. Immunol. 19, 503–516 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jozsi M., Factor H family proteins in complement evasion of microorganisms. Front. Immunol. 8, 571 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hart T., et al. , Polymorphic factor H-binding activity of CspA protects Lyme borreliae from the host complement in feeding ticks to facilitate tick-to-host transmission. PLoS Pathog. 14, e1007106 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bykowski T., et al. , Coordinated expression of Borrelia burgdorferi complement regulator-acquiring surface proteins during the Lyme disease spirochete’s mammal-tick infection cycle. Infect. Immun. 75, 4227–4236 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartmann K., et al. , Functional characterization of BbCRASP-2, a distinct outer membrane protein of Borrelia burgdorferi that binds host complement regulators factor H and FHL-1. Mol. Microbiol. 61, 1220–1236 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Rogers E. A., Marconi R. T., Delineation of species-specific binding properties of the CspZ protein (BBH06) of Lyme disease spirochetes: Evidence for new contributions to the pathogenesis of Borrelia spp. Infect. Immun. 75, 5272–5281 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rogers E. A., Abdunnur S. V., McDowell J. V., Marconi R. T., Comparative analysis of the properties and ligand binding characteristics of CspZ, a factor H binding protein, derived from Borrelia burgdorferi isolates of human origin. Infect. Immun. 77, 4396–4405 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marcinkiewicz A. L., et al. , The factor H-binding site of CspZ as a protective target against multistrain, tick-transmitted Lyme disease. Infect. Immun. 88, e00956-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brangulis K., et al. , Structural characterization of CspZ, a complement regulator factor H and FHL-1 binding protein from Borrelia burgdorferi. FEBS J. 281, 2613–2622 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Lin T., et al. , Analysis of an ordered, comprehensive STM mutant library in infectious Borrelia burgdorferi: Insights into the genes required for mouse infectivity. PLoS One 7, e47532 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marcinkiewicz A. L., et al. , Blood treatment of Lyme borreliae demonstrates the mechanism of CspZ-mediated complement evasion to promote systemic infection in vertebrate hosts. Cell. Microbiol. 21, e12998 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakao M., Somamoto T., The Evolution of Complement System Functions and Pathways in Vetebrates, Malagoli D., Ed. (Academic Press, 2016), 10.1016/C2014-0-01270-1. [DOI] [Google Scholar]
- 27.Combs M., et al. , Phylogenomic diversity elucidates mechanistic insights into Lyme borreliae-host association. mSystems 7, e0048822 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caine J. A., Coburn J., A short-term Borrelia burgdorferi infection model identifies tissue tropisms and bloodstream survival conferred by adhesion proteins. Infect. Immun. 83, 3184–3194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frye A. M., et al. , A soft tick Ornithodoros moubata salivary protein OmCI is a potent inhibitor to prevent avian complement activation. Ticks Tick Borne Dis. 11, 101354 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finnie J. A., Stewart R. B., Aston W. P., A comparison of cobra venom factor-induced depletion of serum C3 in eight different strains of mice. Dev. Comp. Immunol. 5, 697–701 (1981). [DOI] [PubMed] [Google Scholar]
- 31.Walter K. S., Carpi G., Caccone A., Diuk-Wasser M. A., Genomic insights into the ancient spread of Lyme disease across North America. Nat. Ecol. Evol. 1, 1569–1576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coburn J., et al. , Lyme disease pathogenesis. Curr. Issues Mol. Biol. 42, 473–518 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hart T. M., et al. , Host tropism determination by convergent evolution of immunological evasion in the Lyme disease system. PLoS Pathog. 17, e1009801 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawrenz M. B., et al. , Effect of complement component C3 deficiency on experimental Lyme borreliosis in mice. Infect. Immun. 71, 4432–4440 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneider M. C., et al. , Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature 458, 890–893 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wollert T., et al. , Extending the host range of Listeria monocytogenes by rational protein design. Cell 129, 891–902 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Viana D., et al. , A single natural nucleotide mutation alters bacterial pathogen host tropism. Nat. Genet. 47, 361–366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sabrina Pankey M., et al. , Host-selected mutations converging on a global regulator drive an adaptive leap towards symbiosis in bacteria. Elife 6, e24414 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh D., Yi S. V., On the origin and evolution of SARS-CoV-2. Exp. Mol. Med. 53, 537–547 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vishnoi A., Kryazhimskiy S., Bazykin G. A., Hannenhalli S., Plotkin J. B., Young proteins experience more variable selection pressures than old proteins. Genome Res. 20, 1574–1581 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang I. N., et al. , Genetic diversity of ospC in a local population of Borrelia burgdorferi sensu stricto. Genetics 151, 15–30 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brisson D., Dykhuizen D. E., ospC diversity in Borrelia burgdorferi: Different hosts are different niches. Genetics 168, 713–722 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haven J., et al. , Pervasive recombination and sympatric genome diversification driven by frequency-dependent selection in Borrelia burgdorferi, the Lyme disease bacterium. Genetics 189, 951–966 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haven J., Magori K., Park A. W., Ecological and inhost factors promoting distinct parasite life-history strategies in Lyme borreliosis. Epidemics 4, 152–157 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Qiu W. G., Martin C. L., Evolutionary genomics of Borrelia burgdorferi sensu lato: Findings, hypotheses, and the rise of hybrids. Infect. Genet. Evol. 27, 576–593 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Charlesworth D., Balancing selection and its effects on sequences in nearby genome regions. PLoSGenet. 2, e64 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Charlesworth D., Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet. 2, e64 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin Y. P., et al. , Strain-specific variation of the decorin-binding adhesin DbpA influences the tissue tropism of the lyme disease spirochete. PLoS Pathog. 10, e1004238 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia B. L., Zhi H., Wager B., Hook M., Skare J. T., Borrelia burgdorferi BBK32 inhibits the classical pathway by blocking activation of the C1 complement complex. PLoS Pathog. 12, e1005404 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caine J. A., et al. , Borrelia burgdorferi outer surface protein C (OspC) binds complement component C4b and confers bloodstream survival. Cell. Microbiol. 19, e12786 (2017), 10.1111/cmi.12786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pereira M. J., et al. , Lipoproteome screening of the Lyme disease agent identifies inhibitors of antibody-mediated complement killing. Proc. Natl. Acad. Sci. U.S.A. 119, e2117770119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jumper J., et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benjamini Y., Krieger A. M., Yekutieli D., Adaptive linear step-up procedures that control the false discovery rate. Biometrika 93, 491–507 (2006). [Google Scholar]
- 54.Brangulis K., et al. , CspZ (BbCRASP-2) from Borrelia burgdorferi strain B379. RCSB Protein Data Bank. 10.2210/pdb7ZJJ/pdb. Deposited 11 April 2022. [DOI]
- 55.Brangulis K., et al. , CspZ (BbCRASP-2) from Borrelia burgdorferi strain B408. RCSB Protein Data Bank. 10.2210/pdb7ZJK/pdb. Deposited 11 April 2022. [DOI]
- 56.Brangulis K., et al. , Crystal structure of a complex between CspZ from Borrelia burgdorferi strain B408 and human FH SCR domains 6-7. RCSB Protein Data Bank. 10.2210/pdb7ZJM/pdb. Deposited 11 April 2022. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
The coordinates and the structure factors for CspZB379, CspZB408, and human SCR-CspZB408 have been deposited in the Protein Data Bank with accession codes 7ZJJ (54), 7ZJK (55), and 7ZJM (56), respectively. All study data are included in the article and/or SI Appendix.