

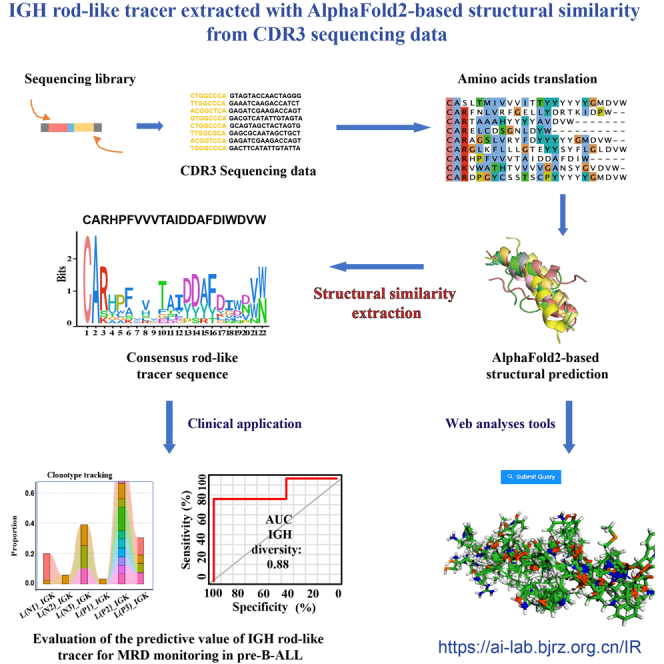
Summary
Sequence variation resulting from the evolution of IGH clones and immunophenotypic drift makes it difficult to track abnormal B cells in children with precursor B cell acute lymphoblastic leukemia (pre-B-ALL) by flow cytometry, qPCR, or next-generation sequencing (NGS). The V-(D)-J regions of immunoglobulin and T cell receptor of 47 pre-B-ALL samples were sequenced using the Illumina NovaSeq platform. The IGH rod-like tracer consensus sequence was extracted based on its rod-like alpha-helices structural similarity predicted by AlphaFold2. Additional data from published 203 pre-B-ALL samples were applied for validation. NGS-IGH (+) patients with pre-B-ALL had a poor prognosis. Consistent CDR3-coded protein structures in NGS-IGH (+) samples could be extracted as a potential follow-up marker for pre-B-ALL children during treatment. IGH rod-like tracer from quantitative immune repertoire sequencing may serve as a class of biomarker with significant predictive values for the dynamic monitoring of MRD in pre-B-ALL children.
Subject areas: Molecular physiology, Cancer, Genomics
Graphical abstract
Highlights
-
•
The structure of IGH CDR3 have been predicted based on AlphaFold2 for the first time
-
•
IGH rod-like tracer has predictive values for the dynamic monitoring of MRD
-
•
IGH rod-like tracer may serve as a class of biomarker in pre-B-ALL children
Molecular physiology; Cancer; Genomics
Introduction
Precursor B cell acute lymphoblastic leukemia (pre-B-ALL) is an aggressive immature B cell neoplasm, and its outcomes have been substantially improved in pediatric and adult patients.1,2 An important prognostic factor for patients with B-ALL is the initial response to therapy, which is determined by the level of “measurable” (also referred to as “minimal”) residual disease (MRD) at the end of induction chemotherapy.2,3,4 Detection of MRD in patients with pre-B-ALL helps to identify patients who may require treatment intensification.5 However, MRD assessment via flow cytometry can be challenging due to the difficulty in consistently interpreting complex multidimensional data, interfered with therapy-induced immunophenotypic drift and marrow regeneration. MRD assessment by qPCR for tracking the patient-specific clonal rearrangements of the immunoglobulin B cell receptor (BCR) and T cell receptor (TCR) requires significant laboratory and institutional commitment to maintain the continuity and quality of testing. Recently, the next-generation sequencing (NGS) has been introduced to assess MRD in patients with B-ALL.6 NGS universally amplifies antigen-receptor gene segments and identifies all clonal gene rearrangements at diagnosis, enabling the monitoring of disease progression and clonal evolution during therapy. Longitudinal BCR repertoire sequencing has also been used and revealed that the BCR underwent an unexpectedly high level of clonal diversification in B-ALL cells through both somatic hypermutation and secondary rearrangements.7 Furthermore, Gawad8 and Bashford-Rogers9 demonstrated massive clonal evolution in immunoglobulin heavy-chain genes (IGH). Clinical NGS has shown its superiority over flow cytometry and qPCR.
However, the immune repertoire of bone marrow samples obtained at multiple recurrence time points in children with pre-B-ALL has not yet been evaluated. Moreover, the state-of-the-art computational methods showed its potential to facilitate the evaluation of the clonal evolution of IGH. However, these methods are complicated and lack standardization.8,9 Sequence variation resulting from the evolution of IGH clones and immunophenotypic drift makes it more difficult to track abnormal B cells by flow cytometry, qPCR, or NGS as pre-B-ALL biomarkers. Consequently, there is an urgent need to identify valuable biomarkers from mass next-generation sequencing data for dynamic MRD monitoring in patients with pre-B-ALL. In recent years, the predictive value of protein structure has been more accurate than the amino acid sequence in disease phenotyping.10 AlphaFold2 was a breakthrough in providing high-accuracy structural predictions, which facilitates unraveling previously unsolved questions from a brand-new structural perspective and as a next-generation biomarker.11
In this study, we used AlphaFold2 to generate protein structure predictions directly from NGS of bone marrow samples to characterize IGH, IGK, TRB, and TRG sequences during the treatment of children with pre-B-ALL. AlphaFold2 for the first was applied to extract the IGH rod-like tracer as a predictive biomarker of MRD in children with pre-B-ALL based on structural similarity analyses of IGH CDR3. We further investigated the dynamic MRD monitoring performance of IGH rod-like tracer in our sequencing data and published data, respectively.
Results
IGH clonotypes were characteristically different from IGK, TRB, and TRG
The principles of leukemia-associated immunophenotype and different from normal were used to distinguish abnormal tumor cells from normal differentiated cells by flow cytometry (FCM) (Figure 1A). Multiplex PCR and NGS were used to amplify and sequence rearrangements within the IGH, IGK, TRB, and TRG V-(D)-J regions (Figure 1B). Individual clonotypes with ≥5% frequency were designated disease clonotypes.8 Using this threshold, we identified 61 IGH disease clonotypes, 36 IGK disease clonotypes, 12 TRB disease clonotypes, and 2 TRG disease clonotypes in the bone marrow samples from children with pre-B-ALL (Table S1). Compared to those of IGK, TRB, and TRG, IGH CDR3 may better reflect the abnormal immune cell status in children with pre-B-ALL. IGH rod-like tracer was defined as the class of disease IGH CDR3 coding domain with rod-like alpha-helices structure in NGS-IGH (+) samples. The consensus rod-like tracer was extracted from our IGH sequencing data, and AlphaFold2 predicted the structure. The length of most IGH clonotypes was greater than 20 amino acids (Figure 1C). The length of clonotypes from other chains was mostly less than 20 amino acids long (medians: 10–15 amino acids) (Figures 1D, 1E, and 1F).
Figure 1.

Detection of MRD and distribution of clonotypes lengths
(A) Multicolor flow cytometry of pre-B-ALL samples.
(B) Schematic of the PCR primer strategy and sequencing assay.
(C) The length distribution of IGH clonotypes.
(D) The length distribution of IGK clonotypes.
(E) The length distribution of TRB clonotypes.
(F) The length distribution of TRG clonotypes.
NGS-IGH (+) predicted a poor prognosis for patients with pre-B-ALL
We evaluated the prognostic value of NGS-IGH. Patients were dichotomized into two groups based on their NGS-IGH: NGS-IGH (+) group includes patients with the proportion of hyperexpanded clonotypes in the bone marrow samples greater than 50% at the primary diagnosis; and NGS-IGH (−) group includes those with the proportion of hyperexpanded clonotypes in the bone marrow samples less than 50% at the primary diagnosis. We found that NGS-IGH (−) patients had significantly improved survival rates than NGS-IGH (+) patients (p < 0.05) (Figure 2A). We also used another dichotomization method (NGS-IGH >5% or NGS-IGH <5%). Progression-free survivalshowed no significant difference between patients with NGS-IGH >5% and those with NGS-IGH <5% (Figure 2B).
Figure 2.

Clinical relevance of NGS-MRD
(A) Kaplan–Meyer plot estimated PFS for patients between NGS-IGH (+) and NGS-IGH (−) groups.
(B) Kaplan–Meyer plot estimated PFS for patients between NGS-IGH (>5%) and NGS-IGH (<5%) groups.
(C) The diagnostic value of IGH clonotype diversity for disease risk classification.
(D) The diagnostic value of IGH clonotype diversity for the number of relapses of patients.
(E) The diagnostic value of the proportion of hyperexpanded IGH clonotypes for disease risk classification.
(F) The diagnostic value of the proportion of hyperexpanded IGH clonotypes for the number of relapses.
(G) The diagnostic value of the number of IGH/TRB clonotypes for disease risk classification.
(H) The relationship between the number of IGH/TRB clonotypes and disease risk classification. Group mean values and standard errors are shown. ∗ = p<0.05. ∗∗ = p < 0.01. ∗∗∗ = p<0.001.
Next, we investigated the diagnostic value of IGH diversity, the proportion of hyperexpanded IGH for disease risk classification, and the number of relapses. IGH diversity had great diagnostic value in disease risk classification (Figure 2C) (AUC = 0.88, p < 0.05) but had no significant value in the number of relapses (Figure 2D) (ROC area = 0.67, p > 0.05) of patients. The proportion of hyperexpanded IGH also had a great diagnostic value in disease risk classification (Figure 2E) (ROC area = 0.86, p < 0.05) and the number of relapses (Figure 2F) (ROC area = 0.70, p < 0.05) of patients. Moreover, the ratio of IGH/TRB clonotypes was associated with disease risk classification (p < 0.001) (Figure 2G) and also had a great diagnostic value in disease risk classification (Figure 2H) (ROC area = 0.80, p < 0.05). These results showed that NGS-IGH (+) patients with pre-B-ALL had a poor prognosis.
The CDR3 protein structures were consistent across NGS-IGH (+) samples
AlphaFold2 was used to predict the protein structure of CDR3. Figure 3A showed that the CDR3 structure with alpha helices in the NGS-IGH (+) group had 23 corresponding atoms between protein chains, with the value of root mean squared deviation (RMSD) being 1.181. The CDR3 structure with alpha helices in the NGS-IGH (−) sample only had three corresponding atoms between protein chains (Figure 3B). In both NGS-IGH (+) and NGS-IGH (−) groups, the CDR3 with beta folds all showed dissimilar structures (RMSD = 4.179, 3.292) (Figures 3C and 3D). In both NGS-IGH (+) and NGS-IGH (−) groups, the CDR3 with irregular curls only had three corresponding atoms between protein chains (Figures 3E and 3F). The CDR3 structures in the NGS-IGH (+) group were more similar, while the CDR3 structures in the NGS-IGH (−) group were more diverse. On the other hand, multiple sequence alignment showed that the different CDR3 sequences were not similar, while the protein structures of the NGS-IGH (+) group were highly similar (Figure 3A). These results suggested that consistent CDR3 rod-like protein structures in NGS-IGH (+) samples could be used as a potential follow-up marker in patients with pre-B-ALL during treatment.
Figure 3.

The protein structure of IGH CDR3 in NGS-IGH (+) and NGS-IGH (−) groups
(A)The multiple sequence alignment (MSA) plot and CDR3 structure with alpha helices in the NGS-IGH (+) group.
(B) The MSA plot and CDR3 structure with beta folds in the NGS-IGH (+) group.
(C) The MSA plot and CDR3 structure with irregular curls in the NGS-IGH (+) group.
(D) The MSA plot and CDR3 structure with alpha helices in the NGS-IGH (−) group.
(E) The MSA plot and CDR3 structure with beta folds in the NGS-IGH (−) group.
(F) The MSA plot and CDR3 structure with irregular curls in the NGS-IGH (−) group.
The predictive value of IGH rod-like tracer identified from the structure of IGH CDR3 with alpha helices
The CDR3 structure with alpha helices was similar across the NGS-IGH (+) group. According to the proportion of primary diagnosis samples in each patient, we attempted to evaluate the predictive value of molecular tracers identified from the structure of IGH CDR3 with alpha helices. For example, in patients L and J, the top 10 CDR3s that dominated the primary diagnosis sample were not tracked in IGH (Figures 4A and 4B). The IGH CDR3 structures with alpha helices were similar at different time points (Figures 4E and 4F) (RMSD = 0.036, 2.031), while the sequences at different time points were not similar (Figures 4C and 4D). On the other hand, the top 10 CDR3s tracked their distribution at other time points in patients L and J (Figures 4G and 4H). The IGH rod-like tracer can better reflect the dynamic changes of abnormal B cells in patients. These data suggested that IGH rod-like tracer provided excellent dynamic MRD monitoring performance in patients with pre-B-ALL.
Figure 4.

Predictive value of the IGH rod-like tracer identified from IGH CDR3 structure with alpha helices
In patient L, the top 10 CDR3s that dominate the primary diagnosis sample were not tracked in IGH (A) and tracked in IGK (G). The IGH CDR3 structure with alpha helices at different times points was similar (E). The IGH CDR3 sequences with alpha helices at different times were not similar (C). In patient J, the top 10 CDR3s that dominate the primary diagnosis sample were not tracked in IGH (B) and tracked in IGK (H). The IGH CDR3 structure with alpha helices at different time points was similar (F). The IGH CDR3 sequences with alpha helices at different times were not similar (D).
Evaluate the predictive value of IGH rod-like tracer in tracking and monitoring abnormal B cells in patients with pre-B-ALL in published IGH sequencing data
To further evaluate the predictive value of IGH rod-like tracer in tracking and monitoring abnormal B cells in patients with pre-B-ALL, we downloaded the published IGH sequencing data of 152 pre-treatment and 57 post-treatment patients with pre-B-ALL. In pre-treatment samples, 213 IGH CDR3 sequences were defined as disease clonotypes, in which 147 clonotypes were identified with alpha-helices structures. One hundred nineteen sequences (81%) had high similarity with the consensus IGH CDR3 structures. In post-treatment samples, only 33 IGH CDR3 sequences were defined as disease clonotypes, in which only 22 clonotypes were identified with alpha-helices structures, and 10 sequences (45%) had high similarity with the consensus IGH CDR3 structures. The consensus IGH rod-like tracer had great predictive value in tracking and monitoring abnormal B cells in pre-treatment patients with pre-B-ALL. We constructed the IGH rod-like tracer website (https://ai-lab.bjrz.org.cn/IR) and uploaded IGH sequences and PDB files of the consensus IGH rod-like tracer identified from our sequencing data. This website can predict the three-dimensional structure of IGH CDR3 online and compare the similarity with the structure of the consensus IGH rod-like tracer. These results verified the dynamic MRD monitoring performance of IGH rod-like tracer in published data.
Relapsed samples had higher relative abundance, lower diversity of IGH, and the different V-J gene usage and related motifs
The samples were grouped according to the number of relapses, FCM-MRD status, gene mutation status, and disease risk level. Groups were divided as hyperexpanded (0.01 < X<=1), large (0.001 < X<=0.01), medium (1e-04 < X<=0.001), and small (0 < X<=1e-04) according to the percentage of relative abundance of IGH clonotypes. The samples of the third relapse had the highest relative abundance, and the samples of the first relapse had the lowest relative abundance (p = 0.03) in the hyperexpanded group (Figure 5A), indicating that hyperexpanded IGH clonotypes (0.01 < X<=1) could reveal the number of relapses. There were no significant differences across different clonotypes in terms of gene mutation status, FCM-MRD status, and disease risk level (Figures 5B, 5C, and 5D). This result may be biased by the number of our patients. The diversity of IGH clonotypes, the different V-J gene usage, and related motifs were also evaluated among the samples with different number of relapses (Figures S1 and S2).
Figure 5.

Summary of clonotypes with specific frequencies
(A–D) The relationship between IGH clonotypes and relapse frequency (A), gene mutation status (B), FCM-MRD status (C), and disease risk classification (D). Group mean values and standard errors are shown.
Discussion
The prognostic value of MRD during treatment has been demonstrated in numerous studies performed in patients with newly diagnosed childhood and adult ALL3,12 or relapsed ALL,13,14,15 and in patients undergoing hematopoietic stem cell transplantation.16,17 Despite these encouraging findings, MRD monitoring showed limited utility after induction in some studies, probably due to the sequence variations from the evolution of IGH clones and the immunophenotypic drift. Consequently, there is an urgent need to identify valuable predictive biomarkers from mass next-generation sequencing data for MRD monitoring. In our current study, the IGH rod-like tracer consensus sequence was extracted based on its rod-like alpha-helices structure predicted by AlphaFold2. Our findings underscored the prediction value of the biomarker-IGH rod-like tracer in children with pre-B-ALL, showing great potential for improving MRD monitoring.
Cross-lineage TCR gene rearrangements frequently occurred in immature B cell malignancies, especially in pre-B-ALL (>90% of cases).18,19 IGH, IGK, TRB, and TRG rearrangements were all detected in the study, and a few disease clonotypes were identified in IGK, TRB, and TRG. Focusing on the IGH clonotypes, we found that the proportion of hyperexpanded IGH could better reflect the abnormal immune cell status in children with pre-B-ALL. Since the immune repertoire of patients with pre-B-ALL with multiple relapses has not been evaluated before, we focused on analyzing clonotypes among multiple relapses samples.
To further analyze the diagnostic and prognostic value of NGS-IGH in children with pre-B-ALL, we found that NGS-IGH (−) patients had a better prognosis, as seen in patients with chronic lymphocytic leukemia.20 Hyperexpanded IGH clonotypes and diversity of IGH clonotypes showed good prediction for the number of relapses and disease risk levels in children with pre-B-ALL. The ratio of IGH/TRB clonotypes also showed good prediction for disease risk level. In addition, to further elucidate the pathogenic mechanism of IGH clonotypes in the NGS-IGH (+) group, we used AlphaFold2 to predict the tertiary structures of the disease IGH clonotypes and used RMSD to analyze the similarity between NGS-IGH (+) and NGS-IGH (−) groups. To our knowledge, only one article had evaluated the CDR3 structure in B-ALL. Zha et al.21 found that the TRB CDR3 regions contained a conserved amino acid motif with different spatial conformations in B-ALL. We predicted the high accurate structure of IGH CDR3 protein based on AlphaFold2 for the first time and found that deep learning approaches are valuable for advancing MRD detection. The structure of IGH-CDR3 was more similar within the NGS-IGH (+) group (66%) than that within the NGS-IGH (−) group, which may play the same role in tumorigenesis of pre-B-ALL; in addition, the protein structures could better reflect the role of abnormal B cells in disease compared to the amino acid sequences. In 203 samples derived from other scientific papers that we cite, 119 sequences (81%) had high similarity with the consensus IGH CDR3 structures. Therefore, IGH rod-like tracer was defined as the class of disease IGH CDR3 coding domain with rod-like alpha-helices structure in IGH-NGS (+) samples. Since this marker has only been investigated in subtypes of B leukemia, we will continue to explore its predictive value in other leukemia types.
We further evaluated the predictive value of the IGH rod-like tracer in the NGS-IGH (+) group. In patients for whom clonal evolution cannot be dynamically monitored by flow cytometry, qPCR, or NGS, IGH rod-like tracer in samples collected at different time points helped to distinguish abnormal B cells from the same source and was validated in published IGH sequencing data in pre-treatment patients with pre-B-ALL. Thus, this study provided a new method to track patients with pre-B-ALL at the molecular level. IGH CDR3 structures can be predicted and identified as IGH rod tracers by visiting the IGH rod-like tracer website (https://ai-lab.bjrz.org.cn/IR); the user just needs to copy the sequence into the site and wait for the site to return the result to the page within half an hour.
Furthermore, we found that the third relapse sample had the highest relative abundance and the lowest diversity. The V-J gene usage and related motifs in the third relapse samples differed from the first and second relapse samples. The high-frequency use of IGHJ6 and IGHV4-34 genes was considered self-reactive,22,23,24,25,26,27 and the 9G4 antibody encoded by IGHV4-34 could bind to self-antigens and cause damage.26 GMDVW had similar motifs to ZNF24, TBX20, DBD1, and TBX20. YGMDV had similar motifs to ZNF410, ZNF410 DBD, and GMEB2 DBD3, indicating the motifs of IGH may promote tumorigenesis.28,29 In our pre-B-ALL samples, the biased V-J gene usage and the presence of a shared antigen-binding motif in the CDR3 region of tumor B cells demonstrated a significant antigen-driven process. Precursor B cell receptor signaling and spleen tyrosine kinase have recently been introduced as therapeutic targets for pre-B-ALL.30 These results suggested that the influence of antigen-stimulated self or foreign antigens on BCR may play a key role in disease progression and the initiation of B cell malignant transformation.
In summary, we for the first time performed immune repertoire sequencing on bone marrow samples from children with pre-B-ALL with multiple recurrences, followed by AlphaFold2 structural similarity analyses of IGH CDR3 coding region. The IGH rod-like tracer was then extracted from mass quantitative immune repertoire sequencing data with great predictive values as a biomarker for the dynamic monitoring of MRD in children with pre-B-ALL. The high-accuracy protein structural predictions by AlphaFold2 will greatly facilitate further clinical interpretations of the mass NGS data, and AlphaFold2 itself will also be a powerful tool to identify robust biomarkers in clinical diagnosis and monitoring in future studies.
Limitations of the study
Due to the limited number of patients in our present study, we cannot provide the percentage of patients who would benefit from the study. We will further expand the number of patients and determine the proportion of patients who will benefit in subsequent studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| All the sequenced 47 samples (13 patients) were from patients who received the "CCLG-2008"31 chemotherapy regimen, including 13 pre-treatment, 13 post-treatment, and 21 samples with MRD (+) /MRD (-) change. | Peking University First hospital | NA |
| Critical commercial assays | ||
| QIAamp DNA Mini Kit | Qiagen | #51004 |
| Qubit dsDNA HS analysis kit | Thermo Fisher Scientific | Q32854 |
| EuroClonality-NGS primer sets | EuroClonality | NA |
| Deposited data | ||
| Raw sequencing data | Genome Sequence Archive | HRA003517 |
| Sequence Read Archive (SRA) | Sequence Read Archive (SRA) | SRA456729 |
| Sequence Read Archive (SRA) | Sequence Read Archive (SRA) | ERP115376 |
| Software and algorithms | ||
| FLASH | Magoč and Salzberg32 | http://www.cbcb.umd.edu/software/flash |
| Seqkit | Shen et al.33 | https://github.com/shenwei356/seqkit |
| MiXCR | Bolotin et al.34 | https://github.com/milaboratory/mixcr |
| immunarch | Nazarov | https://github.com/immunomind/immunarch |
| AlphaFold2 | Jumper et al.11 | https://alphafold.com |
| PyMOL | PyMOL open-source team | https://www.pymol.org |
| SPSS | IBM Corp, Armonk | 22.0 |
| GraphPad Prism | La Jolla | 6.0 |
| R | Vienna | 3.4.0 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Fei Xiao (xiaofei3965@bjhmoh.cn).
Materials availability
This study did not generate any new reagents.
Experimental model and study participant details
Patient characteristics, treatment, and MRD sampling
The chemotherapy process was divided into the following phases: remission induction, early intensive treatment, consolidation treatment, delayed intensive treatment I, intermediate maintenance treatment, delayed intensive treatment II, and maintenance treatment. Patients were grouped according to the different risks. Multicolor flow cytometry (FCM)-MRD samples were obtained from bone marrow at different periods, and MRD (+) /MRD (-) change in bone marrow samples was also analyzed at each time point. All the sequenced 47 samples (13 patients) were from patients who received the "CCLG-2008"31 chemotherapy regimen, including 13 pre-treatment, 13 post-treatment, and 21 samples with MRD (+) /MRD (-) change. The subjects were Han children aged 2-13 years, with a roughly equal ratio of males to females. Other IGH sequencing data from pre-B-ALL patients for validation were downloaded from the Sequence Read Archive (SRA) (SRA456729 and ERP115376), including 162 pre-treatment, 67 post-treatment, and 21 samples with MRD (+) /MRD (-) change during the chemotherapy process. The study was approved by the institutional ethics committee of our center, and written informed consent was obtained from the parents or guardians of each child (and assent from the patients as appropriate).
Method details
Detection of MRD with NGS
DNA from bone marrow samples was extracted using the QIAamp DNA Mini Kit (Qiagen) and quantified with the Qubit dsDNA HS analysis kit (Thermo Fisher Scientific) and Nanodrop 2000 (Thermo Fisher Scientific). According to the NGS of immunoglobulin (BCR) and T-cell receptor (TCR) gene recombination for MRD identification in acute lymphoblastic leukemia.18,35 The SOP (20190611-EuroClonality-NGS_SOP_for_two-step-marker-identification_version_1.0_FINAL) downloaded from the web page ( https://euroclonality.org/ngs/protocols/) provided detailed instructions on how to prepare amplicon sequencing libraries for target screening using EuroClonality-NGS primer sets for IGH (VJ + DJ), IGK (VJ-Kde + intron-Kde), TRB (VJ + DJ), TRG and TRD. The 1st step primers and 2nd step primers were synthesized according to the sequences in Table 7 and Table 8 in the SOP (20190611-EuroClonality-NGS_SOP_for_two-step-marker-identification_version_1.0_FINAL). The 1st step PCR, purification of TRB (VJ, DJ) PCR products by gel extraction, and the 2nd step PCR were performed according to this SOP. The IGH, IGK, TRB, and TRG VDJ regions were sequenced on an Illumina NovaSeq system (PE250).
Detection of MRD using multicolor flow cytometry
MRD was detected using the 8-color FCM-MRD assay. Bone marrow samples were collected at different time points during the treatment. Approximately 106 nucleated cells were collected. BD FACSDivaTM software was used for analysis, FSC-A/FSC-H was used to set gates to remove adhesion cells, FSC-A/SSC-A was used to remove debris, SSC-A/CD45 was used to find lymphocytes at different stages, and CD45/CD19 was used to gate B lymphocytes. B cell markers (CD10/CD45, CD34/CD10, CD38/CD45, CD34/CD20 and CD20/CD10) were used to observe the differentiation and developmental trajectory of B cells. CD58/CD45 and CD13-33-15-117/CD45 were used to observe whether there were abnormal B cells (according to leukemia-associated immunophenotype [LAIP] and different from normal [DFN]).
Immune repertoire analysis
Complementarity-determining region 3 (CDR3) is the most hypervariable region in BCR and TCR genes and also the most critical structure in antigen recognition. The length and sequence of this region determine antibody specificity, thereby determining the fates of developing and responding lymphocytes. There are millions of different TCR Vβ chain or BCR heavy chain CDR3 sequences in human blood. When high-throughput sequencing becomes widely used, CDR3 sequence variations are still a quicker and cheaper method of assessing repertoire diversity. Thus, the region of CDR3 was chosen to analyze the BCR and TCR.
FLASH32 was used to merge paired-end reads from NGS experiments. Seqkit33 was used to convert FASTQ to FASTA. Gene quantification and BCR/TCR clonotype assignment were performed using MiXCR.34 All sequences we analyzed have excluded sequences with untranscribed/translated stop codons. The “immunarch” R package (https://github.com/immunomind/immunarch) was used for further analyses, including computing the number of clones or distributions of lengths and counts (repExplore), computing the clonality of repertoires (repClonality), computing the repertoire overlap (repOverlap), computing the distributions of V or J genes (geneUsage), and estimating the diversity of repertoires (repDiversity). Clonally related sequences were identified by sorting based on their CDR3 nucleotide sequences. Group means were compared with the Wilcoxon rank-sum test if data were grouped. P-values were adjusted according to the Holm method.
CDR3 protein structure prediction and similarity comparison
AlphaFold211 was used to predict the protein structure of CDR3 from the amino acid sequence of CDR3. Next, PyMOL (https://www.pymol.org) was used to calculate the structural similarity in NGS-IGH (+) and NGS-IGH (-) groups. The root mean squared deviations (RMSD) parameter was used to compare the similarity of protein tertiary structures. An RMSD of 0 between two structures suggested that they were the same and could be completely overlapped, and an RMSD (0-3) considered the two structures to be similar.
Quantification and statistical analysis
Progression-free survival (PFS) was defined as the time from the start of treatment until disease progression or death from any cause. Survival curves were calculated using Kaplan-Meier analysis. Receiver operating characteristic (ROC) curves were used to evaluate the performance of diagnostic tests and the accuracy of logistic regression that divided subjects into two categories. Continuous variables were described by the mean ± standard deviation (±s) (for normal distribution), or the median and interquartile range M (P25, P75) (for non-normal distribution). Moreover, the t-test or Mann-Whitney U test was used to identify statistically significant differences between two groups. The proportion of patients in each category was assessed using the chi-square test or Fisher’s exact test for categorical variables. All P values were 2-sided, with a significance level of 0.05. Statistical analyses were performed using SPSS version 22 (IBM Corp, Armonk, NY), GraphPad Prism 6 (La Jolla, CA), and R version 3.4.0 (Vienna, Austria).
Acknowledgments
This work was supported by National High Level Hospital Clinical Research Funding (BJ-2022-169), CAMS Innovation Fund for Medical Sciences (2021-I2M-1-050), National Key Research and Development Program of China (Grant 2022YFC2705000).
Author contributions
Z.Z.L. and W.Q.C. analyzed the data and wrote the main text of the manuscript. L.C., Z.L.L., Z.L.X., Y.R., G.Y., and H.Y. analyzed the data. M.L.Z., B.J.F., Z.C.L., F.R., C.M., and S.F. prepared figures. Q.C.X. and F.X. supervised this work. All authors reviewed and approved the submitted manuscript.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Published: June 12, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107107.
Contributor Information
Chenxue Qu, Email: qucx2012@163.com.
Fei Xiao, Email: xiaofei3965@bjhmoh.cn.
Supplemental information
Data and code availability
-
•
The sequencing data has been submitted to the Genome Sequence Archive and is available under the accession number HRA003517.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Pui C.H., Evans W.E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 2006;354:166–178. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 2.Hunger S.P., Mullighan C.G. Acute lymphoblastic leukemia in children. N. Engl. J. Med. 2015;373:1541–1552. doi: 10.1056/NEJMra1400972. [DOI] [PubMed] [Google Scholar]
- 3.Conter V., Bartram C.R., Valsecchi M.G., Schrauder A., Panzer-Grümayer R., Möricke A., Aricò M., Zimmermann M., Mann G., De Rossi G., et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. 2010;115:3206–3214. doi: 10.1182/blood-2009-10-248146. [DOI] [PubMed] [Google Scholar]
- 4.Campana D., Pui C.H. Minimal residual disease-guided therapy in childhood acute lymphoblastic leukemia. Blood. 2017;129:1913–1918. doi: 10.1182/blood-2016-12-725804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berry D.A., Zhou S., Higley H., Mukundan L., Fu S., Reaman G.H., Wood B.L., Kelloff G.J., Jessup J.M., Radich J.P. Association of minimal residual disease with clinical outcome in pediatric and adult acute lymphoblastic leukemia: a meta-analysis. JAMA Oncol. 2017;3:e170580. doi: 10.1001/jamaoncol.2017.0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faham M., Zheng J., Moorhead M., Carlton V.E.H., Stow P., Coustan-Smith E., Pui C.H., Campana D. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120:5173–5180. doi: 10.1182/blood-2012-07-444042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu D., Emerson R.O., Sherwood A., Loh M.L., Angiolillo A., Howie B., Vogt J., Rieder M., Kirsch I., Carlson C., et al. Detection of minimal residual disease in B lymphoblastic leukemia by high-throughput sequencing of IGH. Clin. Cancer Res. 2014;20:4540–4548. doi: 10.1158/1078-0432.CCR-13-3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gawad C., Pepin F., Carlton V.E.H., Klinger M., Logan A.C., Miklos D.B., Faham M., Dahl G., Lacayo N. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood. 2012;120:4407–4417. doi: 10.1182/blood-2012-05-429811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bashford-Rogers R.J.M., Nicolaou K.A., Bartram J., Goulden N.J., Loizou L., Koumas L., Chi J., Hubank M., Kellam P., Costeas P.A., Vassiliou G.S. Eye on the B-ALL: B-cell receptor repertoires reveal persistence of numerous B-lymphoblastic leukemia subclones from diagnosis to relapse. Leukemia. 2016;30:2312–2321. doi: 10.1038/leu.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tunyasuvunakool K., Adler J., Wu Z., Green T., Zielinski M., Žídek A., Bridgland A., Cowie A., Meyer C., Laydon A., et al. Highly accurate protein structure prediction for the human proteome. Nature. 2021;596:590–596. doi: 10.1038/s41586-021-03828-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brüggemann M., Raff T., Flohr T., Gökbuget N., Nakao M., Droese J., Lüschen S., Pott C., Ritgen M., Scheuring U., et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood. 2006;107:1116–1123. doi: 10.1182/blood-2005-07-2708. [DOI] [PubMed] [Google Scholar]
- 13.Coustan-Smith E., Gajjar A., Hijiya N., Razzouk B.I., Ribeiro R.C., Rivera G.K., Rubnitz J.E., Sandlund J.T., Andreansky M., Hancock M.L., et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia after first relapse. Leukemia. 2004;18:499–504. doi: 10.1038/sj.leu.2403283. [DOI] [PubMed] [Google Scholar]
- 14.Paganin M., Zecca M., Fabbri G., Polato K., Biondi A., Rizzari C., Locatelli F., Basso G. Minimal residual disease is an important predictive factor of outcome in children with relapsed 'high-risk' acute lymphoblastic leukemia. Leukemia. 2008;22:2193–2200. doi: 10.1038/leu.2008.227. [DOI] [PubMed] [Google Scholar]
- 15.Raetz E.A., Borowitz M.J., Devidas M., Linda S.B., Hunger S.P., Winick N.J., Camitta B.M., Gaynon P.S., Carroll W.L. Reinduction platform for children with first marrow relapse of acute lymphoblastic Leukemia: a Children's Oncology Group Study[corrected] J. Clin. Oncol. 2008;26:3971–3978. doi: 10.1200/JCO.2008.16.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leung W., Campana D., Yang J., Pei D., Coustan-Smith E., Gan K., Rubnitz J.E., Sandlund J.T., Ribeiro R.C., Srinivasan A., et al. High success rate of hematopoietic cell transplantation regardless of donor source in children with very high-risk leukemia. Blood. 2011;118:223–230. doi: 10.1182/blood-2011-01-333070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bader P., Kreyenberg H., Henze G.H.R., Eckert C., Reising M., Willasch A., Barth A., Borkhardt A., Peters C., Handgretinger R., et al. Prognostic value of minimal residual disease quantification before allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia: the ALL-REZ BFM Study Group. J. Clin. Oncol. 2009;27:377–384. doi: 10.1200/JCO.2008.17.6065. [DOI] [PubMed] [Google Scholar]
- 18.van Dongen J.J.M., Langerak A.W., Brüggemann M., Evans P.A.S., Hummel M., Lavender F.L., Delabesse E., Davi F., Schuuring E., García-Sanz R., et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17:2257–2317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 19.Szczepański T., Beishuizen A., Pongers-Willemse M.J., Hählen K., Van Wering E.R., Wijkhuijs A.J., Tibbe G.J., De Bruijn M.A., Van Dongen J.J. Cross-lineage T cell receptor gene rearrangements occur in more than ninety percent of childhood precursor-B acute lymphoblastic leukemias: alternative PCR targets for detection of minimal residual disease. Leukemia. 1999;13:196–205. doi: 10.1038/sj.leu.2401277. [DOI] [PubMed] [Google Scholar]
- 20.Thompson P.A., Srivastava J., Peterson C., Strati P., Jorgensen J.L., Hether T., Keating M.J., O'Brien S.M., Ferrajoli A., Burger J.A., et al. Minimal residual disease undetectable by next-generation sequencing predicts improved outcome in CLL after chemoimmunotherapy. Blood. 2019;134:1951–1959. doi: 10.1182/blood.2019001077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zha X., Chen S., Yang L., Li B., Chen Y., Yan X., Li Y. Characterization of the CDR3 structure of the Vbeta21 T cell clone in patients with P210(BCR-ABL)-positive chronic myeloid leukemia and B-cell acute lymphoblastic leukemia. Hum. Immunol. 2011;72:798–804. doi: 10.1016/j.humimm.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 22.Meffre E., Davis E., Schiff C., Cunningham-Rundles C., Ivashkiv L.B., Staudt L.M., Young J.W., Nussenzweig M.C. Circulating human B cells that express surrogate light chains and edited receptors. Nat. Immunol. 2000;1:207–213. doi: 10.1038/79739. [DOI] [PubMed] [Google Scholar]
- 23.Larimore K., McCormick M.W., Robins H.S., Greenberg P.D. Shaping of human germline IgH repertoires revealed by deep sequencing. J. Immunol. 2012;189:3221–3230. doi: 10.4049/jimmunol.1201303. [DOI] [PubMed] [Google Scholar]
- 24.Aguilera I., Melero J., Nuñez-Roldan A., Sanchez B. Molecular structure of eight human autoreactive monoclonal antibodies. Immunology. 2001;102:273–280. doi: 10.1046/j.1365-2567.2001.01159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaplinsky J., Li A., Sun A., Coffre M., Koralov S.B., Arnaout R. Antibody repertoire deep sequencing reveals antigen-independent selection in maturing B cells. Proc. Natl. Acad. Sci. USA. 2014;111:E2622–E2629. doi: 10.1073/pnas.1403278111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhat N.M., Lee L.M., van Vollenhoven R.F., Teng N.N.H., Bieber M.M. VH4-34 encoded antibody in systemic lupus erythematosus: effect of isotype. J. Rheumatol. 2002;29:2114–2121. [PubMed] [Google Scholar]
- 27.Vencovský J., Zd'árský E., Moyes S.P., Hajeer A., Ruzicková S., Cimburek Z., Ollier W.E., Maini R.N., Mageed R.A. Polymorphism in the immunoglobulin VH gene V1-69 affects susceptibility to rheumatoid arthritis in subjects lacking the HLA-DRB1 shared epitope. Rheumatology. 2002;41:401–410. doi: 10.1093/rheumatology/41.4.401. [DOI] [PubMed] [Google Scholar]
- 28.Huang X., Liu N., Xiong X. ZNF24 is upregulated in prostate cancer and facilitates the epithelial-to-mesenchymal transition through the regulation of Twist1. Oncol. Lett. 2020;19:3593–3601. doi: 10.3892/ol.2020.11456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X., Ge X., Zhang Z., Zhang X., Chang J., Wu Z., Tang W., Gan L., Sun M., Li J. MicroRNA-940 promotes tumor cell invasion and metastasis by downregulating ZNF24 in gastric cancer. Oncotarget. 2015;6:25418–25428. doi: 10.18632/oncotarget.4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Köhrer S., Havranek O., Seyfried F., Hurtz C., Coffey G.P., Kim E., Ten Hacken E., Jäger U., Vanura K., O'Brien S., et al. Pre-BCR signaling in precursor B-cell acute lymphoblastic leukemia regulates PI3K/AKT, FOXO1 and MYC, and can be targeted by SYK inhibition. Leukemia. 2016;30:1246–1254. doi: 10.1038/leu.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu J., Lu A.D., Zhang L.P., Zuo Y.X., Jia Y.P. [Study of clinical outcome and prognosis in pediatric core binding factor-acute myeloid leukemia] Zhonghua Xue Ye Xue Za Zhi. 2019;40:52–57. doi: 10.3760/cma.j.issn.0253-2727.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magoč T., Salzberg S.L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen W., Le S., Li Y., Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 2016;11:e0163962. doi: 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolotin D.A., Poslavsky S., Mitrophanov I., Shugay M., Mamedov I.Z., Putintseva E.V., Chudakov D.M. MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods. 2015;12:380–381. doi: 10.1038/nmeth.3364. [DOI] [PubMed] [Google Scholar]
- 35.Brüggemann M., Kotrová M., Knecht H., Bartram J., Boudjogrha M., Bystry V., Fazio G., Froňková E., Giraud M., Grioni A., et al. Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia. 2019;33:2241–2253. doi: 10.1038/s41375-019-0496-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The sequencing data has been submitted to the Genome Sequence Archive and is available under the accession number HRA003517.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.