

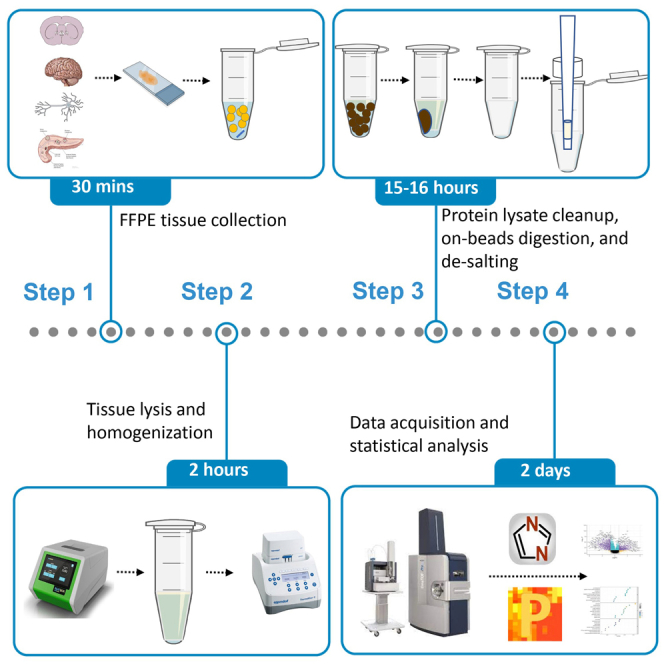
Summary
Formalin-fixed paraffin-embedded (FFPE) samples are valuable archived bio-specimens of individuals and are commonly used in biomedical research. Here, we present a protocol for deep proteomic profiling of FFPE specimens using a spectral library-free approach. We describe steps for FFPE tissue collection, tissue lysis, homogenization, protein lysate cleanup, on-beads digestion, and de-salting. We then detail data acquisition and statistical analysis. This protocol is highly sensitive, reproducible, and applicable for high-throughput proteomic profiling and can be used on various types of specimens.
Subject areas: Clinical Protocol, High-throughput Screening, Mass Spectrometry, Protein Biochemistry, Proteomics
Graphical abstract
Highlights
-
•
Protocol for rapid and reproducible proteomic profiling of FFPE specimens
-
•
Steps described for preparing samples, acquiring data, and analyzing the proteome
-
•
Perform statistical analysis to identify candidates that exhibit significant changes
-
•
Conduct gene ontology and functional analysis
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Formalin-fixed paraffin-embedded (FFPE) samples are valuable archived bio-specimens of individuals and are commonly used in biomedical research. Here, we present a protocol for deep proteomic profiling of FFPE specimens using a spectral library-free approach. We describe steps for FFPE tissue collection, tissue lysis, homogenization, protein lysate cleanup, on-beads digestion, and de-salting. We then detail data acquisition and statistical analysis. This protocol is highly sensitive, reproducible, and applicable for high-throughput proteomic profiling and can be used on various types of specimens.
Before you begin
Timing: 30 min
The stock solutions and buffers should be freshly prepared as indicated in the protocol. Prepare all solutions in LC/MS grade water wherever indicated and use a glass syringe to pipette strong acidic solutions, like concentrated TFA and FA. Wear gloves and avoid inhaling any fumes from the chemicals.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Water, Optima LC/MS grade | Fisher Scientific | W6-1 |
| 3M™ Empore™ SDB-RPS Solid Phase Extraction Disks (SPE) | Fisher Scientific | 13-110-022 |
| Sodium deoxycholate (SDC) | Millipore Sigma | D6750 |
| Tris (2-carboxyethyl) phosphine hydrochloride (TCEP) | Millipore Sigma | C4706 |
| 2-Chloroacetamide (CAA) | Millipore Sigma | C0267 |
| OmniPur TRIS Solution 1.0 M pH 8.0 (Tris) | Millipore Sigma | 9290-OP |
| Sodium hydroxide (NaOH) | Sigma-Aldrich | 221465-500G |
| Sera-Mag SpeedBeads | Thermo Scientific | 4515-2105-050250 |
| Trypsin/Lys-C Mass spec grade | Promega | V5071 |
| Trifluoroacetic acid, sequencing grade (TFA) | Thermo Fisher Scientific | 28904 |
| Ethylacetate | Millipore Sigma | 270989 |
| Acetonitrile, Optima LC/MS Grade (ACN) | Fisher Scientific | A955 |
| Ammonium hydroxide solution (NH4OH) | Honeywell Fluka | 4427310X1ML |
| Formic acid, Optima LC/MS Grade (FA) | Fisher Scientific | A11710X1-AMP |
| Impulse heat sealer | Sealer Sales | KF200H |
| Eppendorf LoBind Microcentrifuge Tubes | Eppendorf | 22431081 |
| Centrifuge adapter 10 μL, 200 μL | GL Sceinces | 5010-21514 |
| Qorpak Glass Bottle with Green Thermoset F217 and PTFE Caps | Fisher Scientific | 02-992-128 |
| Savant Speed-Vac Plus concentrator | Thermo Fisher Scientific Savant | SC110A |
| Ultrasonic bath | Branson | CPX-952-218R |
| NanoDrop One Microvolume UV-Vis Spectrophotometer | Thermo Fisher Scientific | ND-ONE-W |
| Autosampler vials, 9 mm plastic screw thread | Thermo Fisher Scientific | C400011 |
| SUN-Sri StepVial II Cap | Thermo Fisher Scientific | 503790 |
| BeatBox | PreOmics | https://www.preomics.com/products/beatbox |
| Software and algorithms | ||
| R Environment | The R Project for Statistical Computing1 | https://www.r-project.org/ |
| R Studio | RStudio Team1 | https://www.rstudio.com/ |
| clusterProfiler | Wu et al.2 | https://guangchuangyu.github.io/software/clusterProfiler/ |
| EnhancedVolcano | Blighe et al.3 | https://github.com/kevinblighe/EnhancedVolcano |
| Perseus | Maxquant4 | https://maxquant.net/perseus/ |
| DIA-NN | Demichev et al.5 | https://github.com/vdemichev/DiaNN |
Materials and equipment
Tissue Lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 1M Tris-Cl (pH 8.5) | 300 mM | 100 μL |
| 10% SDC | 1% | 100 μL |
| 20% SDS | 5% | 250 μL |
| HPLC grade water | 550 μL | |
| Total volume | 1000 μL |
Note: Tissue Lysis buffer should be prepared immediately before use.
Digestion buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 1 M Tris-Cl (pH 8.5) | 100 mM | 100 μL |
| 10% SDC | 1% | 100 μL |
| HPLC grade water | 800 μL | |
| Total volume | 1000 μL |
Note: Digest buffer should be prepared immediately before use.
Reduction/Alkylation mix
| Reagent | Final concentration | Amount |
|---|---|---|
| 0.5 M CAA | 40 mM | 80 μL |
| 0.5 M TCEP | 10 mM | 20 μL |
| Total volume | 100 μL |
Note: The pH of 0.5 M TCEP should be adjusted to 8.0 using a 10 N NaOH solution. A pH strip can be used to verify the pH.
CRITICAL: The reduction/alkylation mix should be prepared immediately before use, as it is oxygen and light-sensitive.
SP3 magnetic beads mix
| Reagent | Final concentration | Amount |
|---|---|---|
| Sera-Mag Speed A beads | 50% | 50 μL |
| Sera-Mag Speed B beads | 50% | 50 μL |
| Total volume | 100 μL |
Note: Store the Sera-Mag Speed A/B beads at 4°C.
Note: Prepare the concentration of Sera-Mag Speed A/B beads as 50 μg/μL immediately before use.
Ethyl Acetate/TFA buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 100% Ethyl Acetate | 100% | 1000 μL |
| 100% TFA | 1% | 1 μL |
| Total volume | 1000 μL |
StageTip Wash buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 100% Acetonitrile | 5% | 50 μL |
| 10% TFA | 0.2% | 20 μL |
| Total volume | 930 μL |
Note: The StageTip Wash buffer can be kept in stock and used for a long time.
Elution buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 100% Acetonitrile | 50% | 500 μL |
| 30% NH4OH | 1% | 33.3 μL |
| HPLC grade water | 466.7 μL | |
| Total volume | 1000 μL |
Peptide resuspension buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 100% Acetonitrile | 3% | 30 μL |
| 10% Formic acid | 0.1% | 10 μL |
| HPLC grade water | 960 μL | |
| Total volume | 1000 μL |
Note: Prepare the Peptide resuspension buffer before use.
Lys-C/Trypsin mix solution
| Reagent | Final concentration | Amount |
|---|---|---|
| 20 μg Lys-C/Trypsin Mix | 0.20 μg/ μL | 100 μL |
| HPLC grade water | 100 μL | |
| Total volume | 100 μL |
Note: Reconstitute the Lys-C and Trypsin mix in 100 μL HPLC grade water before use and store the remaining small aliquot at −80°C.
Mobile phase A
| Reagent | Final concentration | Amount |
|---|---|---|
| HPLC grade water | 1000 mL | 1000 mL |
| 100% Formic acid | 0.1% | 1000 μL |
| Total volume | 1000 mL |
Mobile phase B
| Reagent | Final concentration | Amount |
|---|---|---|
| 100% Acetonitrile | 1000 mL | 1000 mL |
| 100% Formic acid | 0.1% | 1000 μL |
| Total volume | 1000 mL |
Step-by-step method details
FFPE sample preparation for proteomics analysis
This step outlines the procedure for retrieving the FFPE tissue samples from the glass slide.
-
1.Start with FFPE tissue sections that are 10 μm thick and mounted on a glass slide.
-
a.Use a Sharpie marker to mark the tissue area on the back side of the glass slide.
-
b.Scrape only the tissue from the marked area using a scalpel blade, leaving excess wax on the slide.
-
a.
Note: In Figure 1, marking areas 1 and area 2 are approximately 8 × 5 mm on the glass slide, which provides sufficient starting material.
Figure 1.

FFPE tissue scrapping from the glass slide
(A) Mark the tissue area on the back side of the tissue on the glass slide.
(B) Scrape the tissue from the marked area and leave the excess wax.
(C) Transfer the scraped tissue to new 1.5 mL microcentrifuge tubes containing a single magnetic bead from the Beatbox kit, and briefly centrifuge.
-
2.
Transfer the scraped tissue into new 1.5 mL microcentrifuge tubes that contain a single magnetic bead from the Preomics Beatbox kit.
FFPE tissue lysis and de-crosslinking
This step describes how to lyse and de-crosslink FFPE tissue.
-
3.FFPE tissue lysis, homogenization and de-crosslinking.
-
a.Add 100 μL of tissue lysis buffer into each sample from step 2 and sonicate for 2 min in a water bath.
-
b.Place each sample into the BeatBox holder for tissue lysis and homogenization.
-
c.Select the high-power setting of BeatBox and run it for 10 min at room temperature.
-
d.Collect the solubilized protein lysate by placing the tubes on a magnetic rack and transfer it to new 1.5 mL microcentrifuge tubes.
-
e.Incubate each sample on a ThermoMixer at 95°C/1400 rpm for 90 min.
-
f.Remove the samples from the ThermoMixer and let them incubate at room temperature for 10 min.
-
a.
Reduction and alkylation
This step outlines the process for reducing and alkylating solubilized protein.
-
4.Reduction and alkylation.
-
a.Add 10 μL of Reduction/Alkylation mix to each sample and incubate for 10 min at 45°C/1400 rpm in a thermomixer.
-
b.Incubate for 5 min at room temperature and sonicate it for 2 min in water bath.
-
a.
Protein lysate cleanup with the SP3 method
These steps involve the removal of detergents, which were used to solubilize proteins during the lysis process, and paraffin, which can interfere with protein digestion.
-
5.Prepare SP3 magnetic beads and clean up the tissue lysate.
-
a.Combine Sera-Mag Speed A and B beads (supplied at 50 mg/mL) in a new 1.5 mL microcentrifuge tube to prepare 100 μL of SP3 magnetic beads mix for 10 samples.
-
b.Thoroughly mix the beads by gently vortexing with 50 μL of each bead.
-
c.Place the tube on a magnetic rack to remove the storage buffer.
-
d.Wash the bead pellet three times with 200 μL of 80% acetone without disturbing the pellet on the magnetic rack.
-
e.Re-suspend the SP3 beads in 100 μL of 80% acetone for a final working concentration of 50 μg/μL.Note: SP36 method is modified.
-
f.Add 10 μL of washed SP3 beads, 400 μL of 80% acetone, and 100 mM NaCl to each sample from step 4b.
-
g.Mix the sample by gentle vortexing and incubate for 5 min on ice, followed by 1 min of incubation on a magnetic rack.
-
h.Remove the supernatant without disturbing the SP3 bead pellet and wash the pellet twice with 400 μL of 80% acetone while on the magnetic rack. Then, remove the buffer.
-
i.Resuspend the SP3 bead pellet in 50 μL of digestion buffer by gentle vortexing.
-
a.
Protein digestion and de-salting
This step includes two procedures: protein digestion and preparation of high-capacity SDB-RPS stage tips for peptide desalting.
-
6.Protein digestion.
-
a.Add 2.5 μL of a freshly prepared Lys-C/Trypsin mix (0.5 μg) to each sample from step 5i.
-
b.Digest proteins overnight at 37°C/1200 rpm on a thermomixer.
-
c.After brief centrifuging, place the tubes on a magnetic rack and transfer the supernatant to a new 1.5 mL microcentrifuge. Then, resuspend the SP3 bead pellet in 20 μL of MQ H2O and wash at 37°C/1200 rpm for 5 min on a ThermoMixer.
-
d.Place the tubes on a magnetic rack and transfer the wash to the supernatant collected in step 6c, resulting in a total volume of 72.5 μL.
-
e.Acidify each sample by adding 7.25 μL of 10% TFA solution, resulting in a final concentration of 1% TFA. The addition of TFA will cause SDC to precipitate, and the sample will become cloudy.
-
f.Centrifuge each sample for 5 min at 10,000 × g using a benchtop centrifuge to separate the precipitate.Note: Use the pH strip to verify that the pH level of the solution is between 2–3.
-
g.Prepare the StageTip and peptide desalting.
-
i.Insert one Empore SDB-RPS disk core into a P200 pipette tip by using a 14-gauge blunt-ended syringe needle. Carefully pack the disk at the broad end of the tip using a gel-loader tip. Then, position the StageTip on the tip holder and assemble it into a 1.5 mL microcentrifuge tube.
-
ii.Load the samples from step 6f onto the StageTip and centrifuge for 2 min at 2000 rpm.
-
iii.Wash with 200 μL of ethyl acetate buffer followed by 200 μL of wash buffer in a centrifuge at 3000 rpm for 2 min.
-
iv.Elute the de-salted peptides with 40 μL of elution buffer into new 1.5 mL microcentrifuge and dry them in a SpeedVac centrifuge at 45°C.Note: C18-StageTip can be used for desalting peptides.
 Pause point: The protocol can be paused here by storing the samples at −80°C for a long period of time.
Pause point: The protocol can be paused here by storing the samples at −80°C for a long period of time. -
v.Reconstitute the dried peptides in 10 μL of peptide resuspension buffer and thoroughly mix the solution. Subsequently, sonicate the sample in a water bath for 2 min, followed by centrifugation for 2 min at 10,000 × g. Measure the peptide concentration at 280 nm using a NanoDrop.
-
vi.Adjust the peptide concentration to 200 ng/μL with peptide resuspension buffer.Note: Starting with FFPE tissue sections that are 10 μm thick and marking Area 1 and Area 2 on the glass slide will yield around 1.5–2 μg of peptide.
-
vii.Transfer each sample to autosampler vials for MS analysis.
-
i.
-
a.
Mass spectrometry parameters for data acquisition
This section includes the detailed settings required for the diaPASEF7 run.
-
7.
Inject 200 ng of peptides onto a reversed-phase C18 column with an integrated CaptiveSpray Emitter (25 cm × 75 μm, 1.6 μm, IonOpticks) in the timsTOF Pro mass spectrometer.
-
8.
Elute the peptides from the column using mobile phases A and B with 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively, at a flow rate of 400 nL/min over 90 min. Gradually increase the fraction of Mobile Phase B with a linear gradient from 2% to 25% over 60 min, followed by a further increase to 35% within 10 min, and then increase to 80% before re-equilibration, while keeping the column at a constant temperature of 50°C.
-
9.
The data acquisition settings are listed below:
MS/TIMS Setting
| Spray source | Positive mode |
|---|---|
| Spray voltage | 4000 kV |
| Glass capillary tube temperature | 220°C |
| MS1 resolution | 40,000 |
| MS2 resolution | 40,000 |
| Full MS Scan Cycle | 100–1,700 m/z |
| Mode | Custom |
| Mobility Range 1/KO | 0.60–1.60 |
| Accumulation Time | 100 ms |
| Ramp Time | 100 ms |
| Ramp Rate | 9.43 |
| MS Averaging | 1 |
diaPASEF Scan setting
| Mass Range | 345–1500 m/z |
|---|---|
| Collision energy mode | 1/KO- 0.60, 20 eV 1/KO-1.60, 59 ev |
| Mobility Range 1/KO | 0.66–1.43 |
| Cycle Time | 0.95s |
| Window Mass Width | 50 Da |
| Mass Steps per Cycle | 22 |
| Number of Mobility Windows | 2 |
Note: If a different MS instrument is used, the settings may need to be optimized.
Protein identification and quantification using DIA-NN
This step outlines the use of DIA-NN software to perform a proteomic search of the raw MS files. We recommend using the latest version of DIA-NN, which includes the newest features and search algorithm updates. In our analysis, we used version DIA-NN 1.8.2 beta 11, but other software can also be used with parameters adjusted accordingly.
-
10.
DIA-NN - a universal software for data-independent acquisition (DIA) proteomics data processing by Demichev, Ralser, and Lilley labs. DIA-NN5 uses deep neural networks (DNNs) and several algorithms that enable reliable, robust, and quantitatively accurate large-scale experiments using high-throughput methods. The DIA-NN pipeline is fully automated and includes both an intuitive graphical interface and a command line tool, with results being reported in a simple text format.
Note: Spectronaut,8 FragPipe,9 and MaxQuant10 are commonly used software tools for proteomics data analysis.
-
11.
Use the DIA-NN search engine with default settings of the spectral library-free search algorithm (as shown in Figure 2) to search the acquired diaPASEF raw files against the UniProt Human proteome Swiss-Prot database (UP000005640).
Note: By starting with FFPE tissue sections that are 10 μm thick and marking area 1 and area 2 on a glass slide, this workflow is able to identify and quantify over 6000 protein groups and more than 55,000 peptides in each sample with a 200 ng peptide injection (Table S2: DIA-NN search_DIA_NN.stats). This study represents the most extensive proteome profiling of FFPE tissue to date, achieving a level of depth that has not been previously attained in a single run without the use of a spectral library.
Note:Table S1: Reports.pg_matrix
Note:Table S2: DIA-NN search_DIA_NN.stats
Note: Normalization can be performed in various ways, such as global normalization and RT-dependent normalization, depending on the experiment.
Figure. 2.

DIA-NN search engine and parameters used for diaPASEF search
Differential protein expression (DEP) and gene ontology analysis
This step describes the procedures for performing a differential protein expression (DPE) analysis and a Gene Ontology (GO) analysis, which provides functional annotations for the identified proteins.
Download and install Perseus software.
Note: Perseus software can be freely downloaded at https://maxquant.net/perseus/.
Note: Perseus version 1.6.15.0 was used.
-
12.Differential protein expression (DEP)
-
a.Import Reports.pg_matrix, the output generated by DIA-NN, into the Perseus statistical package for data processing.
-
b.Perform Student’s t-test on log-transformed data using default settings to determine the statistical significance of differentially expressed proteins (DEPs).
-
c.Refer to Tyanova et al.4 for a description of the statistical analysis performed in Perseus.
-
a.
Note:Table S1: Reports.pg_matrix
Note: Perseus tutorial are available on YouTube: https://www.youtube.com/watch?v=MsSiqpMBfRc
Note: Statistical tools, such as R packages LIMMA, edgeR, and others, can be used for identifying differentially expressed proteins (DEPs).
-
13.
R software and RStudio1: https://www.rproject.org/ https://rstudio.com/products/rstudio/download/,
Note: The R software version 4.2.3 and RStudio version 2023.03.0 + 386 were used for this data analysis.
-
14.
GO enrichment2: https://github.com/YuLab-SMU/clusterProfiler
Note: GSEA, ShinyGO, Enrichr, and DAVID are commonly used tools for functional enrichment analysis.
-
15.
Volcano plot3: https://github.com/kevinblighe/EnhancedVolcano
Note: VolcanoPlotR, Perseus, and GraphPad are commonly used tools for visualizing and analyzing proteomics data.
-
16.
Installs and loads packages into R Studio.
> install.packages(“EnhancedVolcano”)
> install.packages(“ClusterProfiler”)
> install.packages(“dplyr”)
> install.packages(“ggplot2”)
> install.packages(“xlsx”)
> library(EnhancedVolcano)
> library(ClusterProfiler)
> library(ggplot2)
> library(dplyr)
> library(xlsx)
-
17.Visualize the differentially expressed proteins (DEPs) by using the EnhancedVolcano package in R Studio with the following code.Note: Load the output generated by Perseus for differentially expressed proteins (DEPs), specifically Table S3 VolcanoPlot.Note: Convert the Excel file “Table S3. VolcanoPlot” to CSV format.
-
a.Read data from CSV file.>DEP_FFPE_Tissues<-read.csv("Table S3. VolcanoPlot.csv",header=T)
-
b.Simply run the following to generate the volcano plot.
-
a.
>EnhancedVolcano(DEP_FFPE_Tissues,lab=DEP_FFPE_Tissues$Genes,x = 'Log2FC',y = 'Pvalue',pCutoff = 0.05,FCcutoff = 1,pointSize = 2.0,labSize =3.0,boxedLabels = FALSE,xlim = c(-8, 8),ylim = c(0,8),legendPosition = 'top',colAlpha = 1, col = c("black", "#f7ca64", "#46bac2","#7e62a3"), legendLabSize = 14,legendIconSize = 4.0,drawConnectors = TRUE, widthConnectors = 1,colConnectors = 'black',xlab = "Log2FC",titleLabSize= 24)
Note: In Figure 3A, a Volcano Plot was generated, and the data used for this plot can be found in Table S3, specifically labeled as VolcanoPlot.
Figure 3.

The results of the differential protein expression and Gene Ontology analysis are presented
The data analysis settings are described above and provided in the R codes.
(A) shows the VolcanoPlot created using the EnhancedVolcano package, with significantly changed protein abundance determined by a threshold for significance of p < 0.05 (permutation-based FDR correction) and a 1.0 log2FC.
(B and C) (B) and (C) display the results of GO and functional analysis for brain-specific and breast-specific significantly enriched proteins, respectively, using the ClusterProfiler package.
-
18.Perform GO and functional analysis of differentially expressed proteins (DEPs) by executing the following code in R Studio using the ClusterProfiler package.Note: DEPs table from Perseus should be separated into two tables: Brain-Specific DEPs with log2FC > 1, and Breast-Specific DEPs with Log2FC < −1.Note: Convert the Excel file “Table S4 Brain-Specific DEPs” to CSV format.
-
a.Read data from CSV file.>DEP_FFPE_Tissue_BrainSpecific<-read.csv("Table S4Brain-Specific DEPs.csv",header=T)
-
b.Simply run the following to filter the genes and convert them into Entrez gene ID.>all_gene_id_BrainSpecific<-DEP_FFPE_Tissue_BrainSpecific$Genes>all_gene_id_BrainSpecific_to_ENTREZID = bitr(all_gene_id_BrainSpecific, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
-
c.Perform the gene ontology over-representation test using the enrichGO() function implemented in the clusterProfiler package.>EnrichGO_all_BrainSpecific<-enrichGO(gene = all_gene_id_BrainSpecific_to_ENTREZID$ENTREZID,OrgDb = org.Hs.eg.db,ont= "all", pAdjustMethod = "BH",pvalueCutoff = 0.05,qvalueCutoff = 0.2,readable = TRUE)
-
d.Simply run the following to visualize the dot plot of enriched terms.>p1 <- dotplot(EnrichGO_all_BrainSpecific, split="ONTOLOGY") + facet_grid(ONTOLOGY∼., scale="free")>p2 <- p1 + scale_y_discrete(labels = function(y) str_wrap(y, width = 50))>p3 <- p2 + scale_color_gradientn(colours=c("#f7ca64", "#46bac2","#7e62a3"),trans = "log10",guide=guide_colorbar(reverse=TRUE,order=1))>p3Note: Convert the Excel file “Table S5. Breast-Specific DEPs” to CSV format.
-
e.Read data from CSV file.>DEP_FFPE_Tissue_BreastSpecific<-read.csv("Table S5. Breast-Specific DEPs.csv",header=T)
-
f.Simply run the following to filter the genes and convert them into Entrez gene ID.>all_gene_id_BreastSpecific<-DEP_FFPE_Tissue_BreastSpecific$Genes>all_gene_id_BreastSpecific_to_ENTREZID = bitr(all_gene_id_BreastSpecific, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
-
g.Perform the gene ontology over-representation test using the enrichGO() function implemented in the clusterProfiler package.>EnrichGO_all_BreastSpecific<-enrichGO(gene = all_gene_id_BreastSpecific_to_ENTREZID$ENTREZID,OrgDb = org.Hs.e.g.,.db, ont = "all",pAdjustMethod = "BH", pvalueCutoff = 0.05,qvalueCutoff = 0.2,readable = TRUE)
-
h.Simply run the following to visualize the dot plot of enriched terms.
-
a.
p1 <- dotplot(EnrichGO_all_BreastSpecific, split="ONTOLOGY") + facet_grid(ONTOLOGY∼., scale="free")
>p2 <- p1 + scale_y_discrete(labels = function(y) str_wrap(y, width = 50))
>p3 <- p2 + scale_color_gradientn(colours=c("#f7ca64","#46bac2","#7e62a3"),trans = "log10",guide=guide_colorbar(reverse=TRUE,order=1))
>p3
Note: In Figures 3B and 3C, a GO (Gene Ontology) and functional analysis dot plot were generated, and the data used for these plots can be found in Table S4 and S5, respectively. In Table S4, the data is labeled as "Brain-Specific DEPs," while in Table S5, it is labeled as "Breast-Specific DEPs.
Expected outcomes
Our protocol allowed us to consistently identify and quantify more than 6,000 protein groups in FFPE brain and breast tissues in triplicate. The depth of proteome coverage achieved using this protocol can be influenced by several factors, such as the amount of sample loaded onto the column, the length of the LC gradient, the type of column used, and the performance of the mass spectrometer.
Limitations
Formalin-fixed paraffin-embedded (FFPE) tissue samples are a valuable source of clinical research material. However, the extraction of soluble proteins from FFPE tissues is challenging because the fixation process can result in the cross-linking of proteins, leading to low yields, protein degradation, reduced accessibility of membrane-bound and low-abundant proteins. These factors can make it difficult to achieve deep proteome profiling of FFPE tissues.
Troubleshooting
Problem 1
In step 1, it is important to check the glass slide to identify any poor-quality mounted tissue samples. When collecting the samples in the 1.5 mL microcentrifuge tube, one should also check for any excess wax that may have accumulated in the tube, as this can interfere with downstream sample processing steps.
Potential solution
In order to improve the quality of the samples for analysis, it may be necessary to consider using another slide. When collecting tissue samples, it is important to be precise and only scrape the marked area, as collecting additional tissue can result in excess wax and poor sample quality.
Problem 2
The protein lysates appear cloudy after completing step 3e.
Potential solution
It is possible for protein lysates to appear cloudy after completing step 3e due to the presence of excess wax in FFPE tissues. To improve protein extraction efficiency by dissolving the wax, it is recommended to increase the volume of tissue extraction buffer and the incubation time. For instance, one can increase the buffer volume to 50 μL and extend the incubation time from 90 to 120 min at 95°C to ensure proper wax dissolution.
Problem 3
The insufficient yield of digested peptides.
Potential solution
This is likely due to several factors, including poor-quality tissue samples collected in Step 1, poor binding of the extracted protein lysate on SP3 beads, and insufficient tryptic digestion. To improve the yield of digested peptides, it is recommended to use high-quality preserved tissue samples and ensure that the tissue extraction buffer and SP3 beads are of good quality. Additionally, ensure that the Lys-C/trypsin mix is prepared fresh before use and not subjected to freeze-thaw cycles.
Problem 4
The depth of proteome coverage is low, and the data is inconsistent.
Potential solution
This issue may be attributed to several factors, including inadequate sample preparation, imprecise peptide quantification, and suboptimal performance of the LC-MS/MS instrument. To address this, it is recommended to implement the solutions mentioned previously and explore alternative quantitative measurement techniques, such as colorimetry and fluorimetry. To ensure consistency and accuracy, always include quality control (QC) samples in the analysis and carefully monitor chromatography peak patterns, MS1 and MS2 quality, and quantitative precision.
Problem 5
Software versions and operating system specific requirements.
Potential solution
Ensure that the versions of timsControl, DIA-NN, Perseus, R, and R Studio are up-to-date and compatible with the operating system being used.
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Rajesh Kumar Soni (rs3869@cumc.columbia.edu).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This research was funded in part through the NCI Cancer Center Support Grant P30CA013696.
Author contributions
Conceptualization, methodology, analysis, writing, review, and editing, R.K.S.
Declaration of interests
The author declares no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2023.102381.
Supplemental information
Data and code availability
Processed DIA-NN search data and statistical analysis are uploaded as Tables S1, S2, S3, S4 and S5 . The raw data (diaPASEF) files have been uploaded on MassIVE and are available in MassIVE (accession number MassIVE MSV000091446). All additional information needed to reanalyze data reported in this publication is available from the lead contact, Rajesh Kumar Soni (rs3869@cumc.columbia.edu).
References
- 1.The R Project for Statistical Computing. https://www.r-project.org/about.html.
- 2.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blighe K., Rana S., Lewis M. EnhancedVolcano: publication-ready volcano plots with enhanced colouring and labeling. 2018. https://github.com/kevinblighe/EnhancedVolcano
- 4.Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 5.Demichev V., Messner C.B., Vernardis S.I., Lilley K.S., Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods. 2020;17:41–44. doi: 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hughes C.S., Moggridge S., Müller T., Sorensen P.H., Morin G.B., Krijgsveld J. Share Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019;14:68–85. doi: 10.1038/s41596-018-0082-x. [DOI] [PubMed] [Google Scholar]
- 7.Meier F., Brunner A.D., Frank M., Ha A., Bludau I., Voytik E., Kaspar-Schoenefeld S., Lubeck M., Raether O., Bache N., et al. diaPASEF: parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods. 2020;17:1229–1236. doi: 10.1038/s41592-020-00998-0. [DOI] [PubMed] [Google Scholar]
- 8.Bruderer R., Bernhardt O.M., Gandhi T., Reiter L. High-precision iRT prediction in the targeted analysis of data-independent acquisition and its impact on identification and quantitation. Proteomics. 2016;16:2246–2256. doi: 10.1002/pmic.201500488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Demichev V., Szyrwiel L., Yu F., Teo G.C., Rosenberger G., Niewienda A., Ludwig D., Decker J., Kaspar-Schoenefeld S., Lilley K.S., et al. dia-PASEF data analysis using FragPipe and DIA-NN for deep proteomics of low sample amounts. Nat. Commun. 2022;13:3944. doi: 10.1038/s41467-022-31492-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sinitcyn P., Hamzeiy H., Salinas Soto F., Itzhak D., McCarthy F., Wichmann C., Steger M., Ohmayer U., Distler U., Kaspar-Schoenefeld S., et al. MaxDIA enables library-based and library-free data-independent acquisition proteomics. Nat. Biotechnol. 2021;39:1563–1573. doi: 10.1038/s41587-021-00968-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed DIA-NN search data and statistical analysis are uploaded as Tables S1, S2, S3, S4 and S5 . The raw data (diaPASEF) files have been uploaded on MassIVE and are available in MassIVE (accession number MassIVE MSV000091446). All additional information needed to reanalyze data reported in this publication is available from the lead contact, Rajesh Kumar Soni (rs3869@cumc.columbia.edu).