


Abstract
The development of AlphaFold for protein structure prediction has opened a new era in structural biology. This is even more the case for AlphaFold-Multimer for the prediction of protein complexes. The interpretation of these predictions has become more important than ever, but it is difficult for the non-specialist. While an evaluation of the prediction quality is provided for monomeric protein predictions by the AlphaFold Protein Structure Database, such a tool is missing for predicted complex structures. Here, we present the PAE Viewer webserver (http://www.subtiwiki.uni-goettingen.de/v4/paeViewerDemo), an online tool for the integrated visualization of predicted protein complexes using a 3D structure display combined with an interactive representation of the Predicted Aligned Error (PAE). This metric allows an estimation of the quality of the prediction. Importantly, our webserver also allows the integration of experimental cross-linking data which helps to interpret the reliability of the structure predictions. With the PAE Viewer, the user obtains a unique online tool which for the first time allows the intuitive evaluation of the PAE for protein complex structure predictions with integrated crosslinks.
Graphical Abstract
Graphical Abstract.
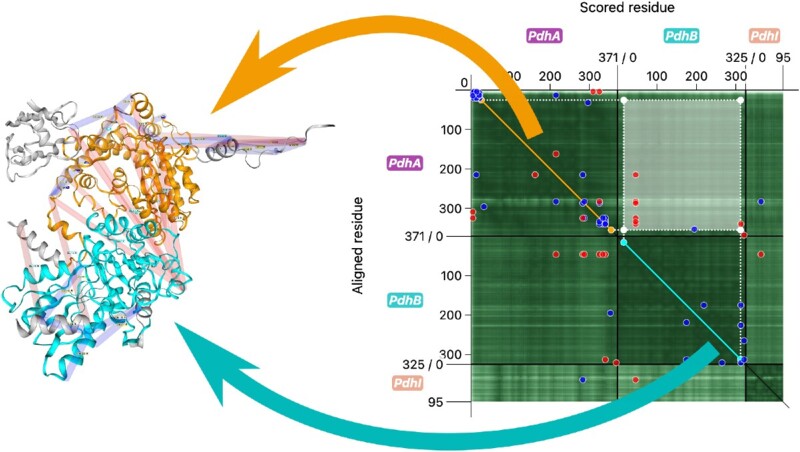
The Predicted Aligned Error (PAE) is a metric for the reliability of protein structure predictions. The PAE Viewer webserver allows the intuitive evaluation of complex structure predictions and crosslinking data.
INTRODUCTION
Many proteins act not as individual molecules but are part of larger complexes. This is particularly the case for all aspects of the replication and expression of genetic information that involve multimeric DNA polymerases, RNA polymerases and ribosomes. Also, many metabolic reactions depend on the formation of protein complexes such as ATP synthesis by a multi-subunit ATPase (1,2).
The investigation of these complexes is a major issue in current biology. Protein-protein interactions have traditionally been identified in different ways, (i) direct co-purification of unknown proteins with a specific target protein, (ii) two-hybrid assays that reconstitute enzyme activities as a result of interaction of proteins coupled to separated domains of the assay protein and (iii) direct biochemical analysis of the interaction of candidate partners. Recently, the proteome-wide identification of protein complexes by protein cross-linking or co-fractionation coupled to mass spectrometry was introduced (3,4). The further investigation of protein complexes has two major aspects: on one hand, the function of the interacting partners should be identified. In this case, interactions of unknown proteins with proteins of known functions paves the way to the development of hypotheses that can be experimentally addressed. In this way, the identification of interactions that involve unknown proteins is a major input to elucidate the functions of these often poorly studied proteins (5,6). On the other hand, the analysis of the structure of the complexes and the analysis of the molecular details of the interaction is very important for a complete understanding.
Recently, the analysis of protein structures has been revolutionized by the introduction of AlphaFold, an artificial intelligence-based tool for the computational prediction of protein structures (7). With AlphaFold, structures of all proteins that can be deduced from genome databases have been predicted and have been included in the AlphaFold Protein Structure Database (AlphaFold DB) (8). However, the prediction of protein complexes is more challenging. A novel iteration of AlphaFold, AlphaFold-Multimer, has made the accurate prediction of small protein complexes feasible (9). Since its release, the prediction of larger and larger complexes has made significant progress (10). With these advances, the interpretation of the reliability of these computational predictions has become more and more urgent. For this purpose, AlphaFold and its descendants provide several metrics which allow the quantitative assessment of the quality of structure prediction. One of these indicators is the Predicted Aligned Error (PAE), which is a measure for the confidence in the relative positions and orientations of parts of the predicted structures. A 2D plot of the PAE is prominently featured on the AlphaFold DB pages, next to the three-dimensional structure view for each of the presented predictions of the individual monomeric proteins.
Structural proteomics by combining protein crosslinks and mass spectrometry provides an additional layer of information. The crosslinks indicate which residues are located in close vicinity. This can be relevant for individual proteins but also for protein complexes. Importantly, this approach even allows the detection of conformational changes of protein under different conditions. For protein complexes, the combination of AlphaFold-Multimer predictions and experimental data from structural proteomics provides an unprecedented view of the molecular organization of the complexes. Indeed, in a recent study on the Bacillus subtilis interactome, a so far unknown protein that interacts with two subunits of pyruvate dehydrogenase was identified. This protein, then renamed PdhI, was found to inhibit the enzyme activity of the pyruvate dehydrogenase. Structural modeling suggested that the protein interferes with catalysis by protruding into the active center of the enzyme. Site-directed mutagenesis based on this hypothesis finally confirmed this proposed mechanism of inhibition of the pyruvate dehydrogenase (4).
The experimental identification of crosslinks between specific regions of interacting proteins can provide independent validation of AlphaFold-Multimer complex predictions. For individual proteins, this experimental evidence can give important hints about the validity of the predicted conformation. The same is true for complexes, where crosslinks between subunits additionally provide information on the shared interface. Thus, the integration of this experimental crosslink information can help to assess the accuracy of these predictions.
Currently, several tools are available to visualize and evaluate the quality of AlphaFold predictions and the corresponding PAE. As mentioned, the pages for (monomeric) protein predictions on AlphaFold DB display multiple interactive visualizations. Users can view the 3D structure of the protein, as well as the corresponding amino acid sequence. In addition, a 2D plot of the PAE allows users to select different areas of the matrix, which in turn get highlighted in the 3D structure and in the sequence display. Other options to evaluate structure predictions include the notebook by Deepmind (https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb) and ColabFold (11). Both Google Colab notebooks allow to run AlphaFold-Multimer remotely and to download its output. They also display static PAE plots as well as several other quality metrics of the predicted structure(s). Yet another tool, the stand-alone application ChimeraX, is a popular choice for molecular visualization, and offers extensive functionality to visualize and analyze structures (12). It allows to render 3D structures interactively, display sequences, and includes an interactive display of the PAE. Using the PAE values, ChimeraX can also evaluate the contact points between the chains of a complex, and it supports clustering of domains. Additionally, it features controls to display and evaluate crosslinks.
All of these tools have certain advantages and drawbacks with respect to the evaluation of predicted structures, the PAE and crosslink data. The AlphaFold DB pages offer an integrated, easy-to-use overview over the structure and the PAE. However, as database pages, they only display precomputed monomer predictions, and are not designed for users to upload their own data. The interactivity of the PAE display is also limited. In case of the Deepmind notebook and ColabFold, complex structure predictions are possible, but the displayed PAE plots are static images. Again, the notebooks are meant to visualize the output of the performed AlphaFold run, not for uploaded structure data. Finally, ChimeraX, which requires installation as it is a stand-alone application, offers the most utility in this context. The functionality of the PAE display is more advanced than the one featured on AlphaFold DB, and the program features several tools to analyze crosslinks. However, it does not combine the crosslink information with the PAE, which could complement each other to add experimental validation to the predicted structure. And although ChimeraX features a sequence viewer, selections made in the PAE display are not reflected by it.
To help researchers with the interpretation of complex predictions and experimental crosslinking data in an integrated way, we developed the PAE Viewer webserver. With this online tool, users can view the 3D representation of a predicted protein complex, corresponding amino acid sequences as well as an interactive display of the PAE. All of these components work together, so user interactions with one of them are reflected by the others. While this resembles the structure of AlphaFold DB entry pages, particular focus was put on the representation and interactivity of the PAE display, the PAE Viewer. It is similar to the tool provided by ChimeraX, but allows for the integration of experimental data by incorporating crosslinking data into the display. With this unique combination of features, our webserver provides a comprehensive tool to interpret the quality of multimeric structure predictions. A version of the PAE Viewer is currently integrated into the database SubtiWiki on the model organism B. subtilis (13), where a predefined selection of predicted protein complex structures is presented. In contrast, the PAE viewer webserver can be used for uploading any predicted custom structure that is of interest to the user. It is designed to directly use the outputs of AlphaFold-Multimer and the mentioned online notebooks, as well as downloads from AlphaFold DB.
IMPLEMENTATION
The predicted aligned error
AlphaFold, in addition to the predicted structure, provides several metrics which allow to better assess the prediction quality. The pLDDT (predicted local-distance difference test) is a confidence measure for the per-residue accuracy of the structure (7). It predicts the Cα local-distance difference test (lDDT-Cα) accuracy of the prediction (14). The pTM (predicted template modeling score) (7) is an estimate of the TM-score (15), a metric for the similarity between protein structures (in this case, between the predicted structure and the assumed real structure). In turn, the ipTM (Interface pTM) scores interactions between residues of different chain to estimate the accuracy of interfaces (9). Another scoring function for the quality of complexes is the mpDockQ (multiple-interface predicted DockQ), which combines the interface plDDT and the number of interface contacts (10).
The PAE, another metric, is a measure for the confidence in the relative position of two residues within the predicted structure (8). The PAE for a pair of residues x and y is defined as the expected positional error in Ångströms at x if the predicted and actual structures are aligned at y. This can provide valuable information about the reliability of relative position and orientations of different domains: if the PAE between the contained residues is high, AlphaFold predicts these domains to be accurately oriented; in turn, a low value indicates limited reliability of the predicted domain orientation. Correspondingly, the same is true for AlphaFold-Multimer predictions with regard to different chains of the model. In this context, a high PAE between the residues of different chains indicates a reliable prediction of the shared interface (9).
Overview of the webserver functionality
When assessing the quality of structure predictions, it can be difficult to interpret the PAE and its relation to the predicted structure. For this reason, we developed the PAE Viewer webserver, which allows user to upload and evaluate protein multimer predictions, the corresponding PAE and crosslinking data.
The PAE Viewer webserver page presents an integrated view of an AlphaFold-Multimer structure prediction (Figure 1). In addition to the PAE Viewer itself, the page features interactive displays of the associated amino acid sequences as well as the 3D structure. The sequence viewer displays the amino acid sequences of the chains contained in the predicted multimer. By using the mouse, the user can select individual amino acids or continuous ranges, which in turn are highlighted in the 3D structure viewer. The latter is based on NGL viewer, a web-based tool for molecular visualization (16). The structure viewer shows a 3D representation of the predicted multimer, which the user can interact with by rotating, dragging or zooming with the mouse. It also features information on additional prediction quality metrics and offers options for the choice of color schemes and display of crosslinks. The PAE Viewer, the sequence viewer and the 3D structure viewer work in conjunction with each other, so user interactions with one of the viewers are reflected by the other displays.
Figure 1.

Overview of the PAE Viewer webserver page. The panel at the top of page allows to select structure data from several examples and to upload custom data. Below, the dynamic sequence viewer presents the amino acid sequences of the viewed multimer prediction. Further down on the left, a 3D structure viewer shows a molecular representation of the predicted multimer. Additionally, quality metrics and presentation options are provided. On the right, the PAE Viewer is embedded.
At the top of the page, the user can choose from a selection of example structures to test the page functionality. Furthermore, an upload form allows users to provide their own structural data. The featured example data includes structure predictions, crosslinking data and quality metrics. They are derived from a recent global interaction study on B. subtilis, where AlphaFold-Multimer was used in combination with experimental crosslinking and co-fractionation analyses (4). This approach resulted in the prediction of high-quality models of numerous potential protein-protein interactions, of which three were chosen as example data. For uploads, structure files in PDB or PDBx/mmCIF format are supported. The PAE and other quality metrics can be provided by uploading a JSON file, which is produced by the Deepmind notebook and ColabFold, and can be also downloaded from AlphaFold DB. Additionally, a Python script can be downloaded to convert the original AlphaFold-Multimer output from the ‘pickle’ format (a serialization format native to Python) to an appropriate JSON file. Crosslinking data can be provided by uploading a CSV. Alternatively, pseudobond files (.pb), as used for example by ChimeraX (12), also have (limited) support. Detailed documentation about the supported input can be found on the webpage. The functionality of the webserver page is entirely provided by the client-side web application, so no exchange of information with our server is performed when users ‘upload’ data.
Representation of data with the PAE viewer
The PAE Viewer was designed to facilitate the interpretation of the PAE for multimeric predictions. To this end, a two-dimensional, interactive plot was developed which features intuitive selection of structure parts, as well as integration of crosslinking data. Figure 2A shows the PAE Viewer for one of the example complexes, the PdhA-PdhB-PdhI trimer in B. subtilis.
Figure 2.
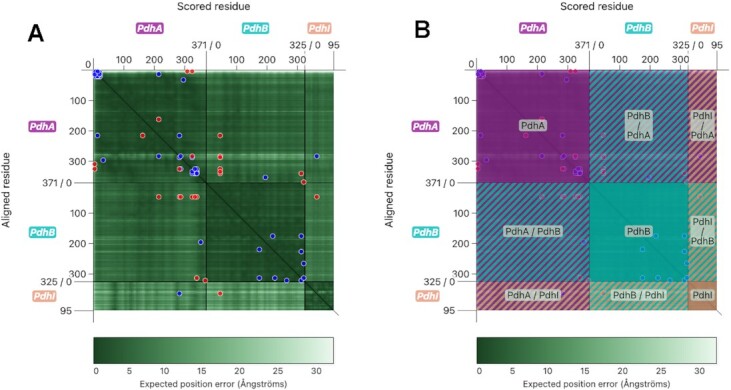
The PAE Viewer display. (A) The interactive PAE Viewer plot, which features a PAE heatmap and additional graphical elements. (B) The PAE Viewer with enabled ‘region overlay’, which allows to highlight the plot segments corresponding to different chains or interfaces.
As can be seen, the representation of the PAE is similar to the display used by the AlphaFold DB and ChimeraX. However, additional graphical elements were added to distinguish between the different chains of the multimer. Furthermore, our PAE Viewer allows the integration of crosslinking data, where circular markers indicate crosslinks between residues.
The 2D plot displays the PAE at the scored residue x with regards to the aligned residue y as a so-called heatmap, where the value of the PAE is indicated by the color. A dark green corresponds to a low PAE, which indicates a high reliability of the relative position of the residues. Conversely, a lighter color corresponds to a lower confidence. This color scheme is the same as the one used by AlphaFold DB. For scaling, the maximum PAE can be provided by the user, and the color legend is adjusted appropriately. If not provided, the maximum value of the PAE matrix is used.
The x and y axes are segmented by the multimer chains, so the position of a residue within a subunit can be easily seen. Special axis ticks also indicate the total sequence length of the individual chains. Additional color-coded labels denote the names of the chains. The color scheme is consistent for the PAE viewer, the sequence viewer and the 3D models. A colorblind-friendly scheme based on the Okabe/Ito palette was used (https://jfly.uni-koeln.de/color/).
The heatmap itself is segmented into a grid, where rectangular parts of the plot correspond to different chains, or interfaces between two chains. These can be highlighted and labeled by toggling the ‘region overlay’, which applies a color-coded mask to the plot as shown in Figure 2B.
In addition to the PAE itself, crosslinking data can be integrated. A crosslink between two residues at x and y is displayed as a pair of circular markers at (x, y) and (y, x) within the plot. Color coding is used to denote satisfaction (blue) or violation (red) of crosslink restraints. In the example (Figure 2A), a distance restraint was imposed, where a Cα–Cα distance ≥30 Å was considered a restraint violation, as such a distance was deemed physically impossible for the used crosslinker. Combined with the PAE, the existence of crosslinks with either satisfied or violated distance restraints provides additional hints about the confidence of the conformation and orientation of structure parts.
Interactivity of the PAE viewer
Further differences to the PAE plot of the AlphaFold DB arise when comparing interactivity of selections, which is more similar to the one of ChimeraX. Figure 3 shows the state of the webserver page when a part of the heatmap is selected.
Figure 3.

Selection functionality of the PAE Viewer. When a selection is performed on the PAE Viewer, the corresponding parts of the multimer are highlighted in the sequence viewer as well as the 3D structure viewer using a consistent color scheme.
The selection functionality of the PAE viewer was designed to explore the PAE and the corresponding parts of the multimer prediction intuitively. When clicking and holding the left mouse button while dragging the cursor across the heatmap, a rectangular area of the plot can be selected as demonstrated in Figure 3. Such a selection corresponds to the PAE of one range of residues with regards to another. Both ranges are projected onto the diagonal of the plot to illustrate the relation of the selected ranges within the sequence. The x range, which corresponds to the selection of scored residues, is marked cyan, while the y range of aligned residues is marked orange. The same color scheme is applied to the sequence viewer and the 3D structure viewer, which makes it easy for the user to identify the different parts of the selection. By looking at the color-coded PAE values in the heatmap, the user can get an idea about the reliability of the orientation of the cyan-colored part of the model with regards to the orange-colored one. Additionally, the user can see the corresponding amino acid sequences at one glance.
On AlphaFold DB, selections with the featured PAE plot also allow highlighting of different sequence and 3D structure parts. However, the AlphaFold DB display highlights the ranges of scored and aligned residues the same way, and also includes residues between the two selected ranges. This makes the identification of the corresponding parts in the 3D structure viewer impossible. In contrast, the PAE Viewer makes a distinction between the ranges which are highlighted in different colors. This makes it easier to identify the relation of the highlighted structure parts and sequences with the selected PAE. ChimeraX also uses two colors to highlight the selected parts of the region. However, the PAE Viewer visually supports the relation of the selection to the sequence by projecting the corresponding ranges onto the diagonal and by highlighting them in the sequence viewer. Additionally, parts of the rectangular selection overlapping with the diagonal, which are difficult to interpret, are assigned a special color (magenta).
Aside from the selection of rectangular areas of the PAE heatmap, the PAE Viewer allows different selections to be made. Clicking on the PAE heatmap selects a single pair of residues, for which the distance is shown in the structure viewer, and which are highlighted in the sequence viewer. Crosslinks, which are denoted by circular markers, can be clicked to highlight corresponding representations in the 3D structure viewer and crosslinked residues in the sequence viewer. The togglable region overlay (a feature absent from AlphaFold DB and ChimeraX) also features interactivity. When clicking an area of the overlay, the corresponding chain or interface is highlighted in the 3D structure viewer as well as the sequence viewer, using the multimer color scheme. For all of these selections, corresponding (mean) PAE values are displayed numerically under the heatmap display.
CONCLUSION
Protein complexes play an essential role in cellular life, and the investigation of their functions is a major objective in biology. Structural proteomics can provide crucial insights into the workings of these molecular machineries, but experimental approaches are challenging. However, with the rise of powerful machine-learning algorithms such as AlphaFold and its offspring tools, the accurate structure prediction of larger and larger protein complexes has become a reality. In turn, the interpretation of these computational predictions and the evaluation of their quality has become more important than ever. While programs such as AlphaFold can provide quantitative measures for the confidence of their predictions, the interpretation can be difficult for the non-specialist. One of these metrics, the Predicted Aligned Error (PAE), is an important indicator for the accuracy of orientation of parts of the structures to one another. In addition to quality estimates generated by AlphaFold, experimental validation is a vital step to verify the reliability of complex structure predictions. Approaches such as crosslinking analysis can deliver important hints about the molecular organization of protein complexes.
The PAE Viewer webserver provides an intuitive tool to interactively explore the quality of AlphaFold-Multimer predictions by integrating the PAE with sequence, structure, and crosslink information. In this context, the unique combination of features of the PAE Viewer offers advantages over already established tools, such as the presentation used by AlphaFold DB, the output of the Deepmind notebook and ColabFold, and the implementation of ChimeraX. Although the focus of the PAE Viewer webserver is specialized, the introduced interactivity could be implemented as a stand-alone library. This way, it could be used more flexibly, for example by integrating it into existing notebooks or into plug-ins for programs such as ChimeraX. We hope that the PAE Viewer presents a helpful tool for the investigation of complex structure predictions and crosslinking data.
DATA AVAILABILITY
The source code for the main components of the webpage is available at https://gitlab.gwdg.de/general-microbiology/pae-viewer.
ACKNOWLEDGEMENTS
We are grateful to Andrea Graziadei, Francis O’Reilly and Juri Rappsilber for helpful discussion. In addition, we would like to thank Thornton Fokkens for providing sample data to test the implementation.
Author contributions: Christoph Elfmann: Conceptualization, Implementation, Writing—original draft. Jörg Stülke: Funding acquisition, Writing—review & editing.
Contributor Information
Christoph Elfmann, Department of General Microbiology, Georg-August-University Göttingen, GZMB, 37077 Göttingen, Germany.
Jörg Stülke, Department of General Microbiology, Georg-August-University Göttingen, GZMB, 37077 Göttingen, Germany.
FUNDING
Deutsche Forschungsgemeinschaft (DFG) via SFB 1565 [469281184 (P11 to J.S.)]. Funding for open access charge: Deutsche Forschungsgemeinschaft, SFB1565.
Conflict of interest statement. None declared.
REFERENCES
- 1. Keskin O., Gursoy A., Ma B., Nussinov R.. Principles of protein−protein interactions: what are the preferred ways for proteins to interact?. Chem. Rev. 2008; 108:1225–1244. [DOI] [PubMed] [Google Scholar]
- 2. Liddington R.C. Fu H. Structural basis of protein-protein interactions. Protein-Protein Interactions. 2004; 261.Methods in Molecular Biology. [DOI] [PubMed] [Google Scholar]
- 3. O’Reilly F.J., Xue L., Graziadei A., Sinn L., Lenz S., Tegunov D., Blötz C., Singh N., Hagen W.J.H., Cramer P.et al.. In-cell architecture of an actively transcribing-translating expressome. Science. 2020; 369:554–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O’Reilly F.J., Graziadei A., Forbrig C., Bremenkamp R., Charles C., Lenz S., Elfmann C., Fischer L., Stülke J., Rappsilber J.. Protein complexes in cells by AI-assisted structural proteomics. Mol. Syst. Biol. 2023; 19:e11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kustatscher G., Collins T., Gingras A.C., Guo T., Hermjakob H., Ideker T., Lilley K.S., Lundberg E., Marcotte E.M., Ralser M.et al.. Understudied proteins: opportunities and challenges for functional proteomics. Nat. Methods. 2022; 19:774–779. [DOI] [PubMed] [Google Scholar]
- 6. Wicke D., Meißner J., Warneke R., Elfmann C., Stülke J.. Understudied proteins and understudied functions in the model bacterium Bacillus subtilis – a major challenge in current research. Mol. Microbiol. 2023; 10.1111/mmi.15053. [DOI] [PubMed] [Google Scholar]
- 7. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Varadi M., Anyago S., Deshpande M., Nair S., Natassia C., Yordanova G., Yuan D., Stroe O., Wood G., Laydon A.et al.. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022; 50:D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Evans R., O’Neill M., Pritzel A., Antropova N., Senior A., Green T., Žídek A., Bates R., Blackwell S., Yim J.et al.. Protein complex prediction with AlphaFold-multimer. 2021; bioRxiv doi:10 March 2022, preprint: not peer reviewed 10.1101/2021.10.04.463034. [DOI]
- 10. Bryant P., Pozzati G., Zhu W., Shenoy A., Kundrotas P., Elofsson A.. Predicting the structure of large protein complexes using AlphaFold and Monte Carlo tree search. Nat. Commun. 2022; 13:6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mirdita M., Schütze K., Moriwaki Y., Heo L., Ovchinnikov S., Steinegger M.. ColabFold: making protein folding accessible to all. Nat. Methods. 2022; 19:679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pettersen E.F., Goddard T.D., Huang C.C., Meng E.C., Couch G.S., Croll T.I., Morris J.H., Ferrin T.E.. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021; 30:70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pedreira T., Elfmann C., Stülke J.. The current state of SubtiWiki, the database for the model organism Bacillus subtilis. Nucleic Acids Res. 2022; 50:D875–D882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mariani V., Biasini M., Barbato A., Schwede T.. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics. 2013; 29:2722–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y., Skolnick J.. Scoring function for automated assessment of protein structure template quality. Proteins. 2004; 57:702–710. [DOI] [PubMed] [Google Scholar]
- 16. Rose A.S., Bradley A.R., Valasatava Y., Duarte J.M., Prlic A., Rose P.W.. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018; 34:3755–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The source code for the main components of the webpage is available at https://gitlab.gwdg.de/general-microbiology/pae-viewer.