


Abstract
Proksee (https://proksee.ca) provides users with a powerful, easy-to-use, and feature-rich system for assembling, annotating, analysing, and visualizing bacterial genomes. Proksee accepts Illumina sequence reads as compressed FASTQ files or pre-assembled contigs in raw, FASTA, or GenBank format. Alternatively, users can supply a GenBank accession or a previously generated Proksee map in JSON format. Proksee then performs assembly (for raw sequence data), generates a graphical map, and provides an interface for customizing the map and launching further analysis jobs. Notable features of Proksee include unique and informative assembly metrics provided via a custom reference database of assemblies; a deeply integrated high-performance genome browser for viewing and comparing analysis results at individual base resolution (developed specifically for Proksee); an ever-growing list of embedded analysis tools whose results can be seamlessly added to the map or searched and explored in other formats; and the option to export graphical maps, analysis results, and log files for data sharing and research reproducibility. All these features are provided via a carefully designed multi-server cloud-based system that can easily scale to meet user demand and that ensures the web server is robust and responsive.
Graphical Abstract
Graphical Abstract.
Proksee assembles, analyzes and visualizes prokaryotic genomes from a variety of inputs.
INTRODUCTION
High-throughput sequencing technologies have reduced the cost and difficulty in generating sequence data so that virtually any lab can routinely sequence the genomes of the organisms they study. This is especially true for bacterial genomes, which have important applications in biomedicine, agriculture, environmental sciences, public health, and industry. Bacterial genomes are used extensively to infer evolutionary relationships and to reveal the genetic underpinning of biological traits such as virulence, antimicrobial resistance, and metabolic potential.
Translating raw bacterial genomic sequence data into meaningful results typically proceeds through genome assembly, annotation, and visualization. Genome assembly is the process of computationally reconstructing a genome sequence from a collection of sequence reads. Many genome assemblers exist for this purpose; of these, the most frequently used for bacterial genome assembly include SPAdes (1), notable for its ability to generate bacterial genomes with high accuracy, and SKESA (2), which is favoured for its speed and computational efficiency. Bacterial genome annotation is the process of identifying and describing the features harboured by a bacterial genome. Typical bacterial genome annotation systems use a combination of gene-calling programs and reference databases to identify and describe protein-coding genes, rRNA, and tRNA contained in a bacterial genome. Genome annotation services can be accessed via IMG/M (3) and MicroScope (4), while BV-BRC (5) offers web-based annotation using RASTtk (6). More recently, command-line-driven programs such as Prokka (7), Bakta (8) and PGAP (9) have become popular since they allow for the annotation of large numbers of genomes in high-performance computing environments. Additional resources exist for annotating specific bacterial genetic features; for example, the CARD/RGI system for identifying determinants of antimicrobial resistance (10), IslandViewer for annotating genomic islands (11), Phigaro for annotating prophage (12), and CRISPR/Cas Finder for annotating CRISPRs (13). Assembled, annotated genomes must be visualized to aid in understanding the biological properties and evolutionary relationships they possess. Programs such as Circos (14) and the CGView family of genome viewers (15) are popular for bacterial genome visualization. Such programs generate genome maps in circular or linear layouts with the genetic features plotted in tracks along the map.
The abundance of available choices for assembling, annotating, and visualizing bacterial genomes is both a blessing and a curse. Each has different capabilities and limitations, operating environments, and complex analysis parameters, which can overwhelm researchers without specialized training. Even experienced researchers face challenges in getting these programs to work together and integrating the results. To reduce the barrier to working with bacterial genomes, we developed the Proksee web server for assembling, annotating, analysing, and visualizing bacterial genomes. We designed Proksee to be simple to use, but with powerful and rich functionality that allows even inexperienced users to generate and analyse bacterial genomes. Here, we describe the architecture and features provided by Proksee and provide several use cases to profile its capabilities.
MATERIALS AND METHODS
Proksee workflow
Proksee accepts pre-assembled contigs or raw sequencing reads and then generates a project with a circular genome map as the focal point. Various annotation and analysis tools can then be launched, which include custom programs or published third-party software that either run in the user's browser to add results directly to the map (client-based tools) or start longer jobs that are executed on worker servers with results that can be viewed and added to the map when complete. The map as well as results from the tools can be downloaded in a variety of text and graphical formats or stored on the Proksee server (Figure 1).
Figure 1.

Proksee workflow. Proksee accepts sequencing reads, complete genomes, or map JSON as input. Genomes and reads (after being assembled) are converted into map JSON with the CGViewBuilder script. Map JSON is converted to a graphical map using CGView.js. Analyses are performed with server-based or client-based tools. Client-based tools (e.g. GC Skew) are run directly on a user's computer and the results are added to the map immediately. Server-based tool (e.g. Prokka) are run on worker servers and the results can be reviewed and added to the map when the job is complete. Server-based tools (including Assemble) produce a report with links to view and download files. Images of the map can be downloaded in SVG or PNG format. Map JSON can also be downloaded as a map archive which can be reloaded into Proksee later for further editing or to perform additional analyses.
Input and data management
Proksee accepts three types of input: pre-assembled contigs, Illumina sequencing reads, and JSON archive files (downloaded from previous Proksee sessions). The contigs can be provided as a GenBank, EMBL, or FASTA file, or as an NCBI accession. Up to 10 million bases of input in the form of one or more sequences is supported for these formats. Sequencing reads can be provided as one or two FASTQ files (the latter for paired-end sequencing) in uncompressed, zip, or gzip form. The server currently allows sequencing read files up to 1 GB in size to be uploaded. Lastly, Proksee accepts a CGView.js (https://js.cgview.ca) JSON file as input. This format contains all the details of a previously generated Proksee map, including the contig sequence(s), features, captions, and legends as well as all customizations (e.g. colours, fonts, feature widths, etc.). These JSON files can be created in Proksee, through the Download Panel, to archive a project for later viewing.
Each of the aforementioned inputs generates a Proksee project. There are two types of projects: Session Projects and User Projects. A Session Project is created when a user does not have a Proksee account or is not logged in. All functionality of Proksee can be securely accessed in this manner. Session Projects are deleted after one week of inactivity. User Projects (associated with a user account) have the advantage of being accessible across devices and browsers and are not automatically deleted. Users can access their Session and User Projects via the My Projects page.
Sequence assembly
Sequencing data is assembled using SPAdes (1). Assembly quality metrics are then compared to those in a custom reference database prepared from publicly available genome sequences, and the results reported to the user graphically as bar charts (Figure 2). The reference database currently contains assembly characteristics for 117 species. For assemblies that map to a species for which a reference collection does not exist in the database, the evaluation criteria fall back to NCBI reference sequence exclusion criteria according to which a prokaryotic assembly meeting any criteria of contig L50 above 500, contig N50 below 5000, or contig counts of >2000 is flagged for exclusion.
Figure 2.
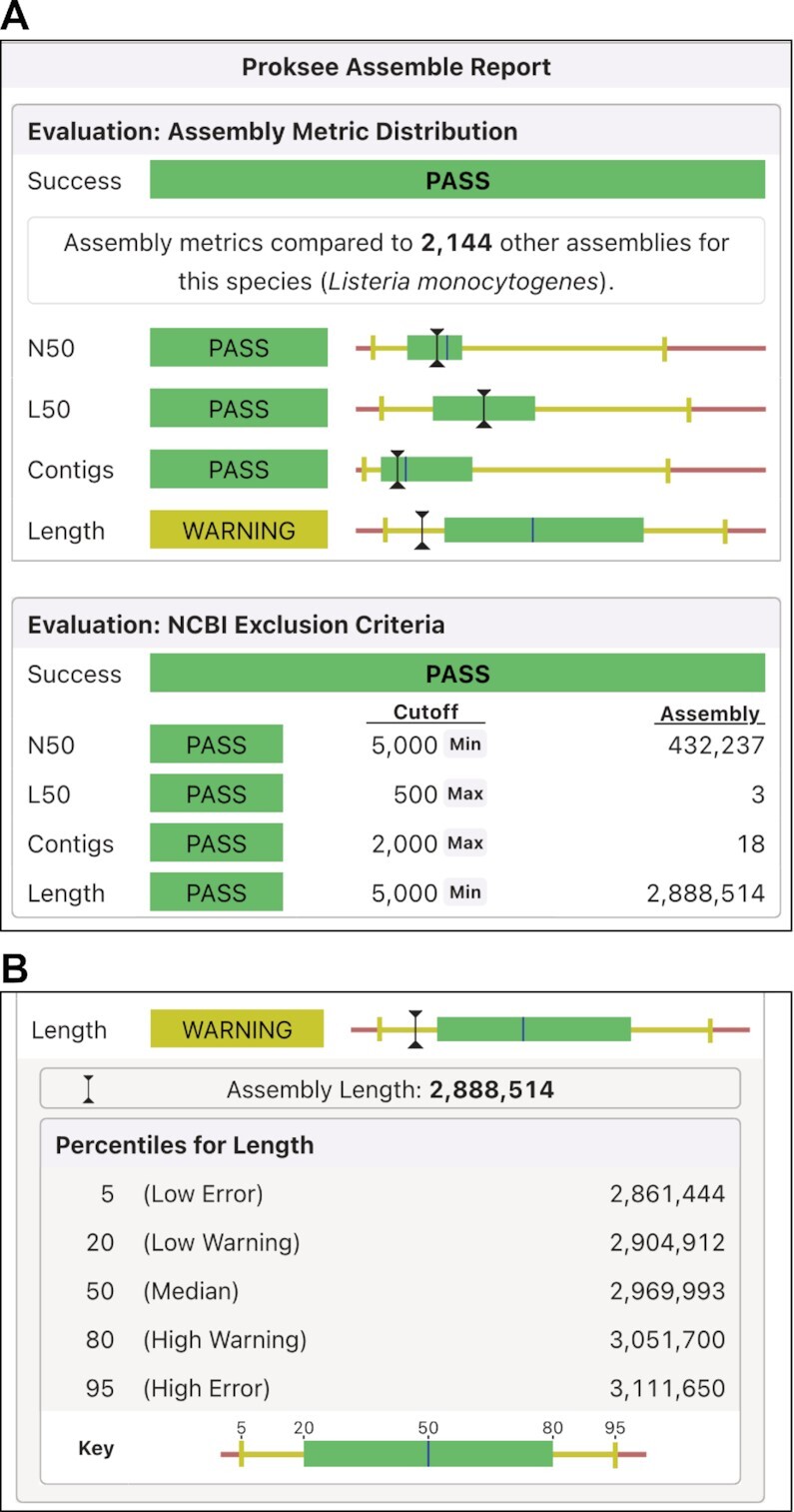
Assembly report. (A) Assembly metric distribution (top) shows the assembly values compared to Proksee's custom reference database of existing assemblies for the same species. NCBI exclusion criteria (bottom) compare the assembly to NCBI’s reference sequence exclusion criteria. (B) Metric distribution details are shown when a metric is clicked. The distribution is displayed as a bar plot with the median length shown as a blue vertical line; the 20th percentile to the 80th percentile shown as a green box; the 5th to 20th percentile and 80th to 95th percentile shown as yellow lines; and above the 95th or below the 5th percentile shown as red. A black I-beam indicates the value of each metric for the project assembly.
Project/map view
The view for a project is divided into two main sections, a set of tabs on the left for viewing the graphical map (Map Tab), map information (About Tab) and analysis outputs (job tabs), and the sidebar on the right consisting of panels for launching tools, customizing the map, monitoring jobs, and downloading results (Figure 3A).
Figure 3.

Project page and map viewer. (A) The project page has a set of tabbed windows on the left (Map Tab shown) and a sidebar with multiple panels on the right (Tool Panel shown). The Map Tab consists of the interactive map as well as the following elements: the Location Bar for viewing, editing, or bookmarking the current position on the map; the Format Bar for changing the map layout (linear or circular), inverting map colours, or changing the aspect ratio; and the Control Bar for zooming, panning, or resetting the map. (B) A zoomed in view of the map showing the map sequence in the backbone, a popup from hovering over a feature (CAS Cluster), and the colour picker.
The sidebar consists of a Display Panel that can be used to customize map contents and appearance. The Regions Panel contains tables describing project contigs, features, plots, and bookmarks (used to navigate between genome regions of interest). This panel is commonly used to search for, select, and modify features of interest (e.g. to move certain features to a new track on the map). The Download Panel provides access to a PNG or SVG image of the current map view, a CGview.js JSON file for archiving the map for future editing and analysis, and contig and feature sequences for offline storage or analysis.
Map viewer/genome browser
The Map Tab consists of the graphical map and includes a Location Bar, Format Bar, and Control Bar (Figure 3A). The Location Bar displays the base pair position being viewed and the current zoom level. The Mark Button in the Location Bar creates a bookmark for the currently viewed location. Bookmarks permit quick navigation to regions of interest via shortcut keys or using the Regions Panel. The Format Bar has buttons to invert map colours as well as to change the view format (linear or circular) and the aspect ratio (square or full sized). The Control Bar has buttons to reset, zoom in/out and pan the map. Users can also pan and zoom (right to the level of individual base pairs) using standard mouse and touch gestures. Hovering over map elements (e.g. features, contigs, plots) displays informative popups, and map elements can be clicked to open details in the sidebar (Figure 3B).
Tools and jobs
The sidebar displays the Tool Panel when a project is first opened (Figure 3A), which contains a list of programs that can be used to annotate and analyze project contigs. There are two types of tools: client-based and server-based. Client-based tools (e.g. GC Skew) are run directly in the browser and the results are immediately added to the map. Server-based tools (e.g. Prokka, BLAST) are run as jobs on Proksee servers and can be added to a map once the job is complete. When a server-based tool is launched (Figure 4A), a job tab is displayed, initially showing the Log Card, which displays real time job progress and messages. Job tabs can be closed and accessed later from the Jobs Panel. Once a job is complete, the corresponding job tab is populated with additional information cards. The Report Card displays a summary of any features found by the tool including a button to add the features to the map (Figure 4B). When adding features, a dialog opens to provide options specifying which features to add and what track and legend to use for the added features (Figure 4C). The Map Tab then opens to show the newly added features (Figure 4D). Depending on the tool, the Report Card may contain summary plots or tables. The Report Card also includes links for downloading key output files and accessing helpful information (e.g. additional documentation), and a citation for the tool that can be included in publications. The Files Card allows input and output files to be previewed and downloaded (Figure 4E). Data cards provide customized views of key output files (e.g. tabular data with hyperlinks, or PDF, HTML or graphical output). Multiple data cards are provided for some tools.
Figure 4.

Server-based tool workflow. The mobileOG-db tool is shown as an example. (A) Starting a server-based tool will show the Start Dialog where the name for the job can be provided as well as any tool-specific options. (B) Completed jobs display a report card with a summary of features found and a button to add them to the map. The report also includes a list of featured files (i.e. key results files) with links to view or download each file. (C) Add Dialog for adding job results to the map with options for selecting which features to add and which track and legend to use for the added features. (D) Map with added features. Shown are the original features (i.e. CDS, tRNA, rRNA) extracted from a GenBank file (NZ_CP007470), the mobileOG-db features split into five categories (e.g. stability/transfer/defense, replication/recombination/repair, integration/excision, transfer and phage) and the results of the GC Content and GC Skew tool. (E) File Card showing the file tree of input and output files for this job (top) and the file viewer for one of the output files (bottom).
Device compatibility and usability
The user interface for Proksee has been refined so that it renders equally well on a wide range of screen sizes. Termed ‘responsive design’ this approach to web design allows users and Proksee administrators to access and interact with all aspects of the Proksee system from laptops, tablets and smart phones.
Proksee has light and dark themes that the user can change depending on their preference. Dark themes have become popular on web pages and in application because they can reduce eyestrain and make it easier to focus on the content rather than the control elements.
An integrated Help system, example input, and tutorials are provided to allow quick testing of Proksee and to demonstrate the many features and capabilities to new and existing users.
Proksee requires a modern web browser and has been successfully tested on Chrome 108, Edge 108, Firefox 108, and Safari 16. Chrome, Edge and Firefox were tested on Windows, Linux, macOS, iPadOS and iOS. Safari was tested on macOS, iPadOS and iOS.
Implementation
The Proksee backend is written in Ruby on Rails and uses a distributed architecture with one main server responsible for the web server, file system and databases (PostgreSQL and Redis) and multiple worker servers for running jobs. Servers are hosted on Digital Research Alliance of Canada cloud resources (https://alliancecan.ca).
The frontend is written in JavaScript and uses the React framework (https://reactjs.org) for UI components. The genome viewer uses CGView.js (https://js.cgview.ca). Created for Proksee and now available for use in other projects, it is named after the popular Java-based circular genome viewer CGView (16). The advantage of using CGView.js is that it creates high-quality, interactive maps that can be easily embedded in web pages, and a comprehensive API provides actions to manipulate map components and hooks for integration with the Proksee user interface. Redux (https://redux.js.org) is used to maintain the state between CGView.js and react components. Any changes made in CGView.js are propagated throughout the interface using Redux and the CGView.js API.
Proksee uses a custom tool framework called Prokan to facilitate the incorporation of third-party software into Proksee. Prokan tools use a YAML file to describe tool attributes, inputs, outputs, and tool-specific components. Each tool runs a custom wrapper for external software, using either Conda or Docker for handling software setup and dependencies.
Job management is handled by Sidekiq (https://sidekiq.org) which uses a Redis (https://redis.io) database for queues. Sidekiq on worker servers continually checks the Redis database queue for jobs. When a new job is available, a worker will copy the input files from the Proksee server, run the job using prokan tools and then copy the results back to the Proksee server. As Proksee matures, additional worker servers can be easily added. Multiple queues are used to handle jobs with different resource requirements. The initial map for a project is created on a separate job queue from the tools to ensure there is no delay starting a project.
Assembly reference metrics are generated using a Snakemake workflow that performs API queries on the NCBI Assembly database to download respective genomic assemblies and obtain the corresponding metadata on assembly information, sequencing platforms, and inclusion/exclusion criteria. Using the assemblies, the pipeline also calculates the following assembly metrics: N50, L50, number of contigs, overall GC content and assembly length.
RESULTS
Usage
In one year since the public release of Proksee (14 February 2022 to 14 February 2023), Proksee has been used to create 55 616 projects and run 110 325 jobs. Over 33 594 map images have been downloaded. In that time, Proksee has had 39 862 visitors, 305 000 page views and an average visit duration of 8 min and 21 s. 1285 user accounts have been created, however, approximately 90% of projects are created without a user account. Users have come from over 90 countries with the top 10 countries in terms of visitors being China, India, United States, South Korea, Canada, Japan, United Kingdom, Thailand, Brazil and France.
Case studies
Assembly of Staphylococcus aureus sequence reads
Paired-end Illumina sequence reads were downloaded from NCBI using accession SRR13968505. Reads were assembled using Proksee, yielding various assembly metrics and a map which was customized to emphasize the presence of multiple contigs (Figure 5A). Through comparison with Proksee's internal database of assemblies it is apparent that the characteristics of this assembly are typical for genomes from this species, in terms of N50, L50, number of contigs and total length. In the graphical map, contigs are displayed using alternating colours (red and black in this map). An added GC Skew plot illustrates how the display of contig boundaries is helpful when interpreting other map contents/features. For example, it is apparent from the map that many of the abrupt changes in GC Skew coincide with contig boundaries. This relationship indicates that the absence of the typical GC Skew pattern seen in complete genomes (arising from the asymmetric nature of replication) is due at least in part to the draft nature of the assembly (multiple contigs ordered by size).
Figure 5.

Case studies performed using Proksee. (A) Genome map showing contig boundaries and GC Skew following assembly of Staphylococcus aureus reads (left), and Assembly Report showing assembly metrics (right). (B) Zoomed in view of region of Haemophilus influenzae genome containing mobile genetic elements as identified using a variety of Proksee tools. (C) Prophage ϕLMC1 found in Listeria monocytogenes strain 08-5578 but not strain 08-5923 based on BLAST (top) and FastANI (bottom) comparisons. (D) Identification of methicillin-resistance gene (mecA) in Staphylococcus aureus genome and assessment of presence in 36 related genomes using BLAST.
Candidate mobile genetic elements in haemophilus influenzae
An annotated genome sequence was retrieved from NCBI in GenBank format using accession NZ_CP007470 and submitted to Proksee. A variety of tools / databases for identifying mobile genetic elements were then used within Proksee: VirSorter (17), Alien Hunter (https://www.sanger.ac.uk/tool/alien-hunter), mobileOG-db (18) and Phigaro (12). When the output from these analyses is added to the map a region supported by all tools is apparent (Figure 5B). Although all results need to be interpreted with the limitations and methodologies of the underlying tools in mind, the ability to quickly compare results from multiple programs can be helpful when prioritizing regions for further investigation bioinformatically or in the lab.
Prophage content difference between two listeria monocytogenes clinical isolates
Two complete Listeria monocytogenes genome sequences were downloaded in FASTA format from NCBI using accessions CP001602 and CP001604, corresponding to strains 08-5578 and 08-5923 (19). Proksee was used to generate a map for 08-5578 and to generate and display gene information from Prokka (7), prophage locations from Phigaro (12) and to compare 08-5578 and 08-5923 via BLAST (20) and FastANI (21). Five prophage were identified, one of which is located in a region of the genome that is missing from 08-5923 according to the BLAST and FastANI results (Figure 5C). Previously identified as ϕLMC1 (19), this prophage is labelled on the map using Proksee's built-in caption system.
Methicillin-resistance in Staphylococcus aureus
A Staphylococcus aureus genome was downloaded from NCBI in GenBank format using accession NC_007793 and submitted to Proksee. The CARD Resistance Gene Identifier (10) was then used to identify bacterial antimicrobial resistance (AMR) genes. Multiple additional S. aureus genomes were compared to the map genome using BLAST. The CARD Resistance Gene Identifier results highlight a mecA gene, which confers methicillin-resistance (22). Based on the BLAST comparisons, particularly when a zoomed in view is examined, it is apparent which of the comparison genomes exhibit similarity to this region of the NC_007793 (strain USA300) (Figure 5D).
DISCUSSION
Proksee is an easy-to-use web server that assembles and annotates bacterial genomes and that allows the predictions from various third-party tools to be visualized on a single graphical map. It is this integration of tools and visualization that permits the discovery of genome elements of interest, related to, for example, adaptations or divergence that contribute to important properties. Unlike the static maps produced by predecessors of Proksee, like CGView Server (23) and BRIG (which provides a GUI wrapper to CGView) (24), the maps generated by Proksee support rapid zooming to the DNA level, allowing quick identification and assessment of regions of interest and precise determination of feature boundaries.
Maps can be extensively customized with Proksee, permitting the creation of impressive and informative maps that can be downloaded in SVG format and used in publications. The source code of this browser (CGView.js) is provided so that it can be incorporated into other web-based tools. Complementing the maps are other forms of output that can be visualized and downloaded, for example tables of results, log files tracking program parameters, additional graphical output and a JSON representation of maps that can be updated with additional results from Proksee.
Other programs with overlapping functionality to Proksee include BRIG (24), GView (25) and the BV-BRC (5). BRIG is a cross-platform desktop software package capable of making static circular maps with multiple BLAST comparisons as rings; GView is a web server with similar functionality as BRIG with added support for pangenome and core genome analyses; and BV-BRC provides access to separate web-based services for genome assembly, annotation, and comparison. The distinguishing features of Proksee are: its breadth of supported analysis types (BLAST but also genome annotation and a variety of other specialized tools); its visualization capabilities (supporting rapid zooming to the DNA sequence level); its map-centric interface that allows the sequential addition and evaluation of analysis results in the context of the interactive map; and it's job management capabilities that keep the input parameters, log files, and output files from any analyses that are performed associated with the map. This latter feature is particularly helpful when publishing figures as it allows the underlying process used to add map contents to be reviewed and described.
The Proksee user-interface and underlying codebase are designed to accommodate the ongoing addition of new functionality, primarily in the form of new tools. Changes to Proksee are announced in the About section of the web site, and new tools will automatically appear in the Tools Panel. Specific examples of new tools planned for addition over the coming months include those for mitochondrial genome annotation and analysis, SNP identification, and COG classification. Other planned improvements include the creation of a long-read assembly pipeline, changes to the user interface to make it easier to manage map colours and legends, and changes to the label layout engine of CGView.js to allow feature labels to be packed more densely.
In summary, by virtue of its analytical and visualization capabilities, as well as carefully designed user interface, we expect Proksee to be a useful addition to the collection of software tools available to biologists seeking to learn more about the characteristics of bacterial genomes.
DATA AVAILABILITY
Proksee is freely accessible at https://proksee.ca. The CGView.js genome browser source code and instructions on how to embed Proksee maps into web pages can be accessed at https://js.cgview.ca. The Proksee assembly pipeline code can be accessed at https://github.com/proksee-project/proksee-cmd and https://doi.org/10.5281/zenodo.7825816.
ACKNOWLEDGEMENTS
This research was enabled in part by support provided by the Digital Research Alliance of Canada (alliancecan.ca).
Contributor Information
Jason R Grant, Agriculture, Food & Nutritional Science, University of Alberta, Edmonton, Alberta T6G 2P5, Canada.
Eric Enns, National Microbiology Laboratory, Public Health Agency of Canada, Winnipeg, Manitoba R3E 3R2, Canada.
Eric Marinier, National Microbiology Laboratory, Public Health Agency of Canada, Winnipeg, Manitoba R3E 3R2, Canada.
Arnab Mandal, Medical Microbiology & Infectious Diseases, University of Manitoba, Winnipeg, Manitoba R3E 0J9, Canada.
Emily K Herman, Agriculture, Food & Nutritional Science, University of Alberta, Edmonton, Alberta T6G 2P5, Canada.
Chih-yu Chen, National Microbiology Laboratory, Public Health Agency of Canada, Winnipeg, Manitoba R3E 3R2, Canada; Biochemistry & Medical Genetics, University of Manitoba, Winnipeg, Manitoba R3E 0J9, Canada.
Morag Graham, National Microbiology Laboratory, Public Health Agency of Canada, Winnipeg, Manitoba R3E 3R2, Canada; Medical Microbiology & Infectious Diseases, University of Manitoba, Winnipeg, Manitoba R3E 0J9, Canada.
Gary Van Domselaar, National Microbiology Laboratory, Public Health Agency of Canada, Winnipeg, Manitoba R3E 3R2, Canada; Medical Microbiology & Infectious Diseases, University of Manitoba, Winnipeg, Manitoba R3E 0J9, Canada.
Paul Stothard, Agriculture, Food & Nutritional Science, University of Alberta, Edmonton, Alberta T6G 2P5, Canada.
FUNDING
Genome Alberta and Genome Canada. Funding for open access charge: Genome Canada.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D.et al.. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2012; 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Souvorov A., Agarwala R., Lipman D.J.. SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018; 19:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen I.-M.A., Chu K., Palaniappan K., Ratner A., Huang J., Huntemann M., Hajek P., Ritter S.J., Webb C., Wu D.et al.. The IMG/M data management and analysis system v.7: content updates and new features. Nucleic Acids Res. 2023; 51:D723–D732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vallenet D., Calteau A., Dubois M., Amours P., Bazin A., Beuvin M., Burlot L., Bussell X., Fouteau S., Gautreau G.et al.. MicroScope: an integrated platform for the annotation and exploration of microbial gene functions through genomic, pangenomic and metabolic comparative analysis. Nucleic Acids Res. 2020; 48:D579–D589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Olson R.D., Assaf R., Brettin T., Conrad N., Cucinell C., Davis J.J., Dempsey D.M., Dickerman A., Dietrich E.M., Kenyon R.W.et al.. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023; 51:D678–D689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brettin T., Davis J.J., Disz T., Edwards R.A., Gerdes S., Olsen G.J., Olson R., Overbeek R., Parrello B., Pusch G.D.et al.. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015; 5:8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinforma. Oxf. Engl. 2014; 30:2068–2069. [DOI] [PubMed] [Google Scholar]
- 8. Schwengers O., Jelonek L., Dieckmann M.A., Beyvers S., Blom J., Goesmann A.. Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb. Genomics. 2021; 7:000685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tatusova T., DiCuccio M., Badretdin A., Chetvernin V., Nawrocki E.P., Zaslavsky L., Lomsadze A., Pruitt K.D., Borodovsky M., Ostell J.. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016; 44:6614–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alcock B.P., Huynh W., Chalil R., Smith K.W., Raphenya A.R., Wlodarski M.A., Edalatmand A., Petkau A., Syed S.A., Tsang K.K.et al.. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023; 51:D690–D699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bertelli C., Laird M.R., Williams K.P.Simon Fraser University Research Computing Group Simon Fraser University Research Computing Group Lau B.Y., Hoad G., Winsor G.L., Brinkman F.S.L.. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017; 45:W30–W35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Starikova E.V., Tikhonova P.O., Prianichnikov N.A., Rands C.M., Zdobnov E.M., Ilina E.N., Govorun V.M.. Phigaro: high-throughput prophage sequence annotation. Bioinforma. Oxf. Engl. 2020; 36:3882–3884. [DOI] [PubMed] [Google Scholar]
- 13. Couvin D., Bernheim A., Toffano-Nioche C., Touchon M., Michalik J., Néron B., Rocha E.P.C., Vergnaud G., Gautheret D., Pourcel C.. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018; 46:W246–W251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A.. Circos: an information aesthetic for comparative genomics. Genome Res. 2009; 19:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stothard P., Grant J.R., Van Domselaar G.. Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinform. 2019; 20:1576–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stothard P., Wishart D.S.. Circular genome visualization and exploration using CGView. Bioinformatics. 2005; 21:537–539. [DOI] [PubMed] [Google Scholar]
- 17. Guo J., Bolduc B., Zayed A.A., Varsani A., Dominguez-Huerta G., Delmont T.O., Pratama A.A., Gazitúa M.C., Vik D., Sullivan M.B.et al.. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome. 2021; 9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brown C.L., Mullet J., Hindi F., Stoll J.E., Gupta S., Choi M., Keenum I., Vikesland P., Pruden A., Zhang L.. mobileOG-db: a Manually Curated Database of Protein Families Mediating the Life Cycle of Bacterial Mobile Genetic Elements. Appl. Environ. Microbiol. 2022; 88:e0099122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gilmour M.W., Graham M., Van Domselaar G., Tyler S., Kent H., Trout-Yakel K.M., Larios O., Allen V., Lee B., Nadon C.. High-throughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak. BMC Genomics [Electronic Resource]. 2010; 11:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 21. Jain C., Rodriguez-R L.M., Phillippy A.M., Konstantinidis K.T., Aluru S.. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018; 9:5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Diep B.A., Gill S.R., Chang R.F., Phan T.H., Chen J.H., Davidson M.G., Lin F., Lin J., Carleton H.A., Mongodin E.F.et al.. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006; 367:731–739. [DOI] [PubMed] [Google Scholar]
- 23. Grant J.R., Stothard P.. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008; 36:W181–W184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alikhan N.-F., Petty N.K., Ben Zakour N.L., Beatson S.A.. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. Bmc Genomics [Electronic Resource]. 2011; 12:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Petkau A., Stuart-Edwards M., Stothard P., Van Domselaar G.. Interactive microbial genome visualization with GView. Bioinformatics. 2010; 26:3125–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Proksee is freely accessible at https://proksee.ca. The CGView.js genome browser source code and instructions on how to embed Proksee maps into web pages can be accessed at https://js.cgview.ca. The Proksee assembly pipeline code can be accessed at https://github.com/proksee-project/proksee-cmd and https://doi.org/10.5281/zenodo.7825816.