


Abstract
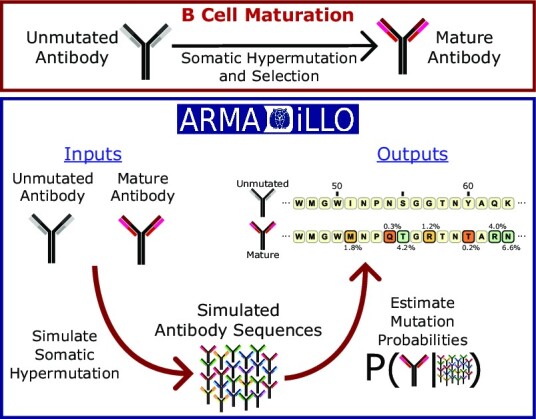
Antibodies are generated by B cells that evolve receptor specificity to pathogens through rounds of mutation and selection in a process called affinity maturation. Somatic hypermutation is mediated by an enzyme with DNA sequence context-dependent targeting and substitution resulting in variable probabilities of amino acid substitutions during affinity maturation. We have previously developed a program called Antigen Receptor Mutation Analyzer for the Detection of Low Likelihood Occurrences (ARMADiLLO) that performs simulations of the somatic hypermutation process to estimate the probabilities of observed antibody mutations. Here we describe the ARMADiLLO web server (https://armadillo.dhvi.duke.edu), an easy-to-use web interface that analyzes input antibody sequences and displays the probability estimates for all possible amino acid changes over the full length of an antibody sequence. The probability of antibody mutations can be used by immunologists studying B cell ontogenies and by vaccine designers that are pursuing strategies to elicit broadly neutralizing antibodies which are enriched with developmentally rate-limiting improbable mutations. The ARMADiLLO web server also contains precomputed results reporting the probability of amino acid substitutions in all human V gene segments and in a collection of HIV broadly neutralizing antibodies.
Graphical Abstract
Graphical Abstract.
INTRODUCTION
B cells develop receptors that can specifically recognize pathogens through an evolutionary process called affinity maturation which involves iterative rounds of somatic hypermutation (SHM) and antigenic selection within germinal centers (1). After B cells attain specificity to antigen, they can differentiate into plasma cells which secrete the soluble form of the B cell receptor as antibodies or into memory B cells which form the basis of immunological memory and guard against subsequent re-infection with the same or similar pathogen (2). Advances in antibody cloning and high throughput sequencing methods have resulted in a wealth of antibody sequencing data which has led to a greater understanding of B cell clonal evolution (3–5). Antibody sequences can be grouped into clones based on shared immunogenetics and these clonally related sequences can then be used as input to phylogenetic methods for inferring an unmutated common ancestor (UCA) sequence that represents the B cell receptor of the original B cell progenitor of the clone prior to affinity maturation (6). Mutations are then identified as differences between an observed antibody sequence and its UCA. The mutational agent that mediates somatic hypermutation in the germinal center, the anatomical microenvironment within lymphoid tissues in which affinity maturation takes place, is activation-induced cytidine deaminase (AID) (7). AID preferentially targets specific sequence motifs for mutation (‘AID hot spots’) and demonstrates bias against mutating other sequence motifs (‘AID cold spots’) (8). Due to the sequence context-dependence of AID activity, certain mutations are much more likely to occur than others (9). Amino acid changes that require multiple base substitutions to co-occur within a codon to attain a specific amino acid are also of low probability. We previously developed a computational program, Antigen Receptor Mutation Analyzer for Detection of Low Likelihood Occurrences (ARMADiLLO) to perform simulations of the somatic hypermutation process and draw upon these simulations to estimate the probabilities of amino acid substitutions in the absence of antigenic selection (9). Using ARMADiLLO, we showed that improbable mutations occur with high frequency in HIV broadly neutralizing antibodies (bnAbs) and that improbable mutations are often functionally required for these antibodies to neutralize diverse strains of HIV (9). Critical improbable mutations can act as rate-limiting steps in the development of bnAbs, and thus are high-value targets for selection by immunogens employed in vaccine strategies that aim to recapitulate the induction of similar types of antibodies in vaccinees using modern vaccine development approaches (10–12). Additionally, estimates of mutation probability can help the immunologist or vaccinologist determine whether recurrent patterns of mutations arose simply by chance in the absence of selection due to AID activity or were due to selection by the vaccine (13).
To make antibody mutation probability analysis accessible to a wider user base, we have developed the ARMADiLLO web server (https//armadillo.dhvi.duke.edu). This web server provides a user-friendly interface for running ARMADiLLO geared towards immunologists and vaccinologists. The ARMADiLLO web server includes several features not previously available in the command-line version of ARMADiLLO including automatic inference of a UCA for the input antibody sequence using the Cloanalyst immunogenetics analysis software (14) while also providing an alternative manual input option for the user to enter a previously inferred UCA. Additionally, the web server allows users to download high-resolution images of the generated results. Finally, the ARMADiLLO web server contains precomputed results reporting the probability of amino acid substitution in all human V gene segments as well as precomputed results for a representative set of 32 HIV bnAbs. Thus, the ARMADiLLO web server is an easy-to-use tool and resource for researchers to perform antibody mutation probability analysis with applications for vaccine and immunological research.
MATERIALS AND METHODS
ARMADiLLO method
ARMADiLLO uses simulations of somatic hypermutation to estimate the probability of amino acid substitutions in an observed antibody sequence. ARMADiLLO relies on SHM simulations because the sequence context-dependent nature of SHM activity prevents direct estimation of mutation probabilities. The ARMADiLLO algorithm and its implementation as a commandline program have previously been described (9). Here, we briefly summarize the method and describe the extensions specific to the web server. Starting with the sequence of an antibody of interest and its corresponding inferred UCA, the number of nucleotide mutations N is calculated. Then, the mutability of each position in the UCA is scored for each nucleotide 5-mer in the sequence according to the S5F model of SHM targeting (8). Mutability scores are then normalized, and a nucleotide position is drawn at random from the resulting probability distribution. The base is then changed according to the S5F substitution model (8) and this process is iterated N times to generate a synthetic sequence with an identical number of nucleotide mutations as the observed antibody of interest. Using this iterative procedure, ARMADiLLO rapidly generates a set of synthetically mutated sequences (with a default of 10 000 sequences generated) which are then translated into amino acid sequences. The frequencies of each amino acid at each position in the simulated set of sequences are used as empirical probability estimates of amino acid changes. Thus, the probability estimates of amino acid changes are conditioned upon the UCA sequence and the number of mutations in the observed antibody sequence reflecting that the probability any position mutates within the antibody sequence increases with the overall number of mutations acquired (9). Importantly, ARMADiLLO’s estimates of mutation probability are made in the absence of antigenic selection, and therefore represent the probability an amino acid would change by chance in a germinal center due to SHM activity alone.
The ARMADiLLO website provides extensions upon the original ARMADiLLO command-line implementation specifically aimed at increasing ease-of-use for the researcher. The ARMADiLLO web server incorporates the Cloanalyst software package (14) for inferring a UCA of the input antibody sequence. The web server also allows the user to specify a set number of mutations to simulate to estimate the probability of amino acid changes for sequences with higher or lower numbers of nucleotide mutations than observed in the input sequence. Additionally, the web server generates high-resolution image files in both PNG and PDF formats of the results for download by the user.
ARMADiLLO web server implementation
The ARMADiLLO web server is run on a nginx (https://nginx.org/) server using the Django framework (https://www.djangoproject.com/) based on Python3 (https://www.python.org/) as Platform-as-a-Service using Dokku (https://dokku.com). Messaging and data management is handled by Redis (https://redis.io/) communicating with a PostgreSQL database (https://www.postgresql.org/). The client-side utilizes HTML5 (https://html.com/html5/) with JavaScript (https://www.javascript.com/) for interactive user-interface components. Internet protocol addresses are deidentified using an SHA-2 hash and stored for reporting usage statics only. The ARMADiLLO web server does not utilize cookies nor collect other personal information. The ARMADiLLO web server supports most web browsers, including Microsoft Edge, Google Chrome, Apple Safari, and Mozilla Firefox across the major platforms (Windows, MacOS, and Linux). The results of submitted jobs are stored for 30 days before deletion.
RESULTS
The ARMADiLLO web server features a simple interface for performing analysis of antibody sequences and obtaining mutation probability estimates (Figure 1A). To start, a user inputs an antibody sequence of interest either by entering the sequence into a text box or by uploading a sequence in fasta format. The user then selects the chain type of the input antibody sequence (heavy, kappa or lambda) as well as the species from which the antibody sequence was recovered. Currently only human, mouse and rhesus macaque input sequences are supported because of the availability of their immunoglobulin gene segment libraries in Cloanalyst. Advanced options can be accessed if the user prefers to supply their own externally inferred UCA sequence instead of using the default UCA inference with Cloanalyst (14). An additional advanced option allows the user to specify the number of mutations to generate in the simulated sequences, instead of the default setting which simulates sequences with the same number of mutations as calculated in the input sequence. This feature can be used to explore how antibody mutation probabilities change at higher or lower levels of mutation than observed in the antibody sequence of interest. The user can also specify in the advanced options the number of sequences ARMADiLLO simulates with higher iteration numbers resulting in a reduction in the variance of the estimated mutation probabilities, however, this comes at the expense of longer wait times for results. In practice, confidence intervals are small even at the default n = 10 000 iterations (Supplementary Table S1). ARMADiLLO analysis is initiated when the user presses the start button.
Figure 1.

The ARMADiLLO web server interface and results pages. (A) The ARMADiLLO web server provides a easy-to-use interface for antibody mutation probability analysis. To estimate mutation probabilities, users input an antibody sequence, specify the heavy or light chain type and choose a host species. ARMADiLLO results are then returned in two views: (B) a simple view comparing the amino acid sequences of the UCA and antibody sequence of interest with amino acid changes color-coded according to their estimated mutation probability and (C) a detailed view comparing both the amino acid and nucleotide sequences, highlighting AID hot and cold spots, and displaying the corresponding mutation probabilities associated with amino acid changes (see main text for detailed descriptions of the outputs).
After the user initiates the analysis, a waiting page containing information about the submitted job is displayed. Typically, results are generated within 10 seconds of submission. Job results can also be recalled for 30 days by either using the link provided to the user upon job completion, or by entering the provided Job ID into the Job ID field at the upper right of the webpage.
The ARMADiLLO web server returns the mutation probability analysis results in two output views via tabs at the top of the results page. The first tab (‘Overview’) shows the UCA and input antibody sequences at the amino acid level with amino acid changes from the input sequence color coded by their estimated probability (Figure 1B). The color code is divided into seven categories ranging from over 20% probability to <0.01% probability for a given mutation. Mutations with <2% probability are defined as ‘improbable mutations’ and are colored from shades of orange to red. Improbable mutations are not expected to occur frequently in B cell clones within germinal centers in the absence of strong antigenic selection. Mutations with >2% probability are defined as ‘probable mutations’ and are colored in shades of green. Probable mutations are expected to occur frequently within B cell clones in germinal centers due to intrinsic AID activity (9,15,16). Purple lines above the sequence denote the locations of the CDR loops. The overview tab provides the user with an easy way to identify antibody mutations that are improbable and quickly detect potential evolutionary bottlenecks for antibody development.
The second tab (‘Details’) provides a more detailed view by comparing the UCA and input antibody sequence at both the nucleotide and amino acid level (Figure 1C). The first three rows of the detailed output view display the UCA amino acid sequence, the amino acid position numbering and the UCA nucleotide sequence. The mutability score (calculated with the S5F mutability model (8)) represents the propensity of a position to mutate (with scores above 1 having higher than average mutability and scores below 1 having lower than average mutability) and is listed below each base position. AID hot spots and cold spots are highlighted within the nucleotide sequences in red (mutability score > 2) and blue (mutability score < 0.3), respectively. The next three rows display the antibody input sequence using the same format. Amino acid changes resulting from nucleotide mutations in the input sequence are highlighted with a yellow background while the nucleotide mutations are shown in a red font color. The last row displays the ARMADiLLO estimated probability of each observed amino acid in the antibody of interest. This includes the probability of amino acids that have not mutated in the antibody of interest and can be interpreted as the probability that the position will not mutate in the absence of antigenic selection. Probability values for improbable mutations (<2% probability) are highlighted in purple. Additionally, any nucleotide that occurred at a cold spot is identified by a small red arrow underneath the last row. Hovering a cursor over the amino acid probability estimate brings up a list of all possible amino acids in descending order of their mutation probabilities, thus providing the user with information on the probability of alternative amino acids at specific positions of interest. Both tabbed views can be downloaded as high-resolution images in the PNG and PDF formats. ARMADiLLO results can be recalled for 30 days after submission at the ARMADiLLO website by using the provided job ID and/or link.
Precomputed results
In addition to performing analysis on user-provided antibody sequences, the ARMADiLLO web server provides results from previously run ARMADiLLO analysis for a collection of representative bnAbs for which heavy and light chain nucleotide sequences are publicly available, as well as pre-simulated data for all human V gene segments. The precomputed results are accessible via tabs at the top of the page. The ‘Precomputed BNAb Results’ tab displays a table of HIV bnAbs that have over 50% neutralization breadth on a large multi-clade virus panel (Figure 2A) (17). This table includes neutralization breadth and potency for each bnAb along with its immunogenetic information and the number of improbable mutations in the heavy and light chain. The table is grouped according to the HIV Env epitope class and bnAbs within each epitope class are sorted by their neutralization breadth. Each of the bnAbs can be explored in more detail by clicking on the bnAb name or pressing the explore button. This takes the user to a more detailed view that shows the overview and detailed tabs for the bnAb as well as a tab displaying a color-coded table of the positional amino acid probabilities. The ‘Human V Alleles’ tab is intended to be a quick reference for finding mutation probabilities for a given V gene at a given mutation frequency (Figure 2B). This page shows ARMADiLLO results for each human V heavy, kappa and lambda gene allele with mutation probabilities for each position in the V gene computed from pre-simulated data over a range of mutation frequencies (1%, 5%, 10%, 15% and 20%). For each combination of V gene and mutation frequency level, the user can view amino acid probabilities in a similar format to the Details tab returned by the ARMADiLLO web server when a user inputs a sequence. Additionally, the user can view a positional amino acid probability table which is color-coded by probability. This resource allows the user to quickly look up the probability of mutating to any specific amino acid in a V gene of interest over a range of low, medium or high mutation frequencies.
Figure 2.

Precomputed Results for HIV-1 BnAbs and Human V Alleles. The ARMADiLLO web server provides results from precomputed ARMADiLLO simulations for users to quickly get mutation probability estimates within (A) a representative set of HIV-1 bnAbs and (B) human V gene alleles for heavy, kappa and lambda chains where mutation probabilities are estimated over a range of mutation frequencies. This feature is intended to be used as a reference for looking up mutation probabilities for any amino acid position within any V gene at a given mutation frequency.
Case study
The HIV bnAb DH270.6 can neutralize 51% of HIV-1 strains (18) and is the type of antibody that a protective HIV vaccine would need to elicit. DH270.6 arose in an HIV infected subject after a period of 3 years and we have shown through antibody-HIV co-evolution studies that a critical rate-limiting step in its development was the acquisition of the G57R mutation in the heavy chain CDR2 loop (9,18). As an example of how the ARMADiLLO web server can help users identify improbable mutations like G57R, we have included the DH270.6 heavy chain sequence as example data that can be used to populate the input sequence text box on the ARMADiLLO web server launch page. Upon running ARMADiLLO analysis for DH270.6, the user can quickly see in the ‘Overview’ tab that G57R is shaded orange and thus is an improbable mutation between 1–2% probability (Figure 3, bottom panel) and the purple line above it demarcates that G57R mutation occurs in the CDRH2. Shifting to the ‘Details’ tab allows the user to get more information on why the mutation is improbable. In the case of G57R, this view reveals that the arginine mutation was the result of a base change that occurred in a cold spot (highlighted in blue) in the first position of the glycine codon at position 57. Mutations can also be improbable due to requiring two or even three base changes to transition to a specific amino acid. One example of this type of mutation in DH270.6 is the Y60T improbable mutation (Figure 3, top panel) which requires the first two base positions of the tyrosine codon to mutate in order to encode for a threonine. In DH270.6, Y60T has an estimated probability of 0.22%. The user may be interested in whether functionally similar amino acids have higher probabilities at a position of interest. By hovering the cursor over a position of interest in the ‘Details’ view, the user can view the mutation probabilities of alternative amino acids. In DH270.6, hovering the cursor over the Y60T mutation reveals that the functionally similar amino acid serine, which is estimated at 10% mutation probability, is substantially more likely to occur than threonine at this position. In addition, probable mutations like S55T in DH270.6, which is estimated to occur with 4.4% probability (Figure 3), are not likely to require strong antigenic selection to be elicited during affinity maturation. The quick identification of G57R as an improbable mutation allows the immunologist to then study the function of this mutation. We have shown that selection of functionally-relevant improbable mutations are critical to the successful elicitation of bnAbs with a vaccine (13). Overall, the DH270.6 case study serves as an excellent example of how the ARMADiLLO web server can quickly and easily provide the immunologist with useful information about the maturation of an antibody of interest and how this information can guide vaccine design strategies.
Figure 3.

Case Study. Example application of the ARMADiLLO web server using the HIV-1 bnAb DH270.6 as input. Mutations are color-coded by their probability (bottom panel, tiled amino acid sequence view). The improbable G57R mutation in the CDRH2 (purple line) occurs due to a nucleotide mutation in an AID cold spot (top panel, detailed view). The nearby Y60T mutation is improbable because two base changes in the codon are required to encode a threonine.
CONCLUSIONS
The ARMADiLLO web server provides a simple interface for estimating the probabilities of amino acid substitutions in antibody sequences. It gives researchers a tool to rapidly identify mutations that occur infrequently during antibody development in the absence of antigenic selection. In the context of HIV bnAbs, these improbable mutations can be used as high-value targets in vaccine design as their selection could remove critical developmental bottlenecks during affinity maturation potentially accelerating the elicitation of bnAbs with a vaccine. In general, mutation probability analysis with the ARMADiLLO web server adds an additional level of information about how B cells evolve to attain specific recognition of antigens. The maturational pathways that B cell clones take in developing from unmutated precursor B cells to fully matured B cells that produce antibodies with high affinity for a pathogen can be thought of as evolutionary roadmaps. Analyzing the probability of mutations allows the researcher to see the topographical features on these evolutionary roadmaps, with improbable mutations representing uphill climbs that likely require strong antigenic selection and probable mutations representing downhill stretches in which intrinsic AID activity may be sufficient for their acquisition. Overall, the ARMADiLLO web server is a useful tool for researchers to study antibody maturation and to inform modern vaccine design strategies in which antibodies are used as templates for vaccine design.
DATA AVAILABILITY
The ARMADiLLO web server is freely available to all users at https://armadillo.dhvi.duke.edu. The source code for a standalone version of ARMADiLLO can be downloaded from https://github.com/WieheLab/ARMADiLLO. An ARMADiLLO commandline version has also been implemented in an Apptainer container, bundling Cloanalyst and ARMADiLLO into a single executable package. Due to the large size of the container, it is only available upon request. Detailed documentation for running ARMADiLLO, the containerized version and the ARMADiLLO web server can be found at https://armadillo-docs.readthedocs.io.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Madison Berry, Max He and Helene Kirshner for thoughtful discussions, design feedback and testing the web server. We are especially thankful to Elizabeth Van Itallie for her detailed feedback on the web server and the manuscript. We would also like to thank SciMed Solutions, Inc. for prototyping the original version of the ARMADiLLO website, and the DHVI IT department and Duke Office of Information Technology for their technical support.
Contributor Information
Joshua S Martin Beem, Duke Human Vaccine Institute, Duke University Medical Center; Durham, NC, 27710, USA.
Sravani Venkatayogi, Duke Human Vaccine Institute, Duke University Medical Center; Durham, NC, 27710, USA.
Barton F Haynes, Duke Human Vaccine Institute, Duke University Medical Center; Durham, NC, 27710, USA; Department of Immunology, Duke University Medical Center; Durham, NC, 27710, USA.
Kevin Wiehe, Duke Human Vaccine Institute, Duke University Medical Center; Durham, NC, 27710, USA; Department of Medicine, Duke University Medical Center; Durham, NC, 27710, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health Division of AIDS [UM1AI144371] for the Duke Consortia for HIV/AIDS Vaccine Development (BFH). Funding for open access charge: Helmholtz Centre for Infection Research.
Conflict of interest statement. K.W. and B.F.H. hold a patent on utilizing mutation probability analysis for developing vaccine immunogens.
REFERENCES
- 1. Victora G.D., Nussenzweig M.C.. Germinal centers. Annu. Rev. Immunol. 2022; 40:413–442. [DOI] [PubMed] [Google Scholar]
- 2. Mesin L., Ersching J., Victora G.D.. Germinal center B cell dynamics. Immunity. 2016; 45:471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liao H.X., Lynch R., Zhou T., Gao F., Alam S.M., Boyd S.D., Fire A.Z., Roskin K.M., Schramm C.A., Zhang Z.et al.. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature. 2013; 496:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Krebs S.J., Kwon Y.D., Schramm C.A., Law W.H., Donofrio G., Zhou K.H., Gift S., Dussupt V., Georgiev I.S., Schätzle S.et al.. Longitudinal analysis reveals early development of three MPER-directed neutralizing antibody lineages from an HIV-1-infected individual. Immunity. 2019; 50:677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doria-Rose N.A., Schramm C.A., Gorman J., Moore P.L., Bhiman J.N., DeKosky B.J., Ernandes M.J., Georgiev I.S., Kim H.J., Pancera M.et al.. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature. 2014; 509:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Olson B.J., Matsen F.A.t.. The Bayesian optimist's guide to adaptive immune receptor repertoire analysis. Immunol. Rev. 2018; 284:148–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Di Noia J.M., Neuberger M.S.. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 2007; 76:1–22. [DOI] [PubMed] [Google Scholar]
- 8. Yaari G., Vander Heiden J.A., Uduman M., Gadala-Maria D., Gupta N., Stern J.N., O’Connor K.C., Hafler D.A., Laserson U., Vigneault F.et al.. Models of somatic hypermutation targeting and substitution based on synonymous mutations from high-throughput immunoglobulin sequencing data. Front. Immunol. 2013; 4:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wiehe K., Bradley T., Meyerhoff R.R., Hart C., Williams W.B., Easterhoff D., Faison W.J., Kepler T.B., Saunders K.O., Alam S.M.et al.. Functional relevance of improbable antibody mutations for HIV broadly neutralizing antibody development. Cell Host Microbe. 2018; 23:759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haynes B.F., Wiehe K., Borrrow P., Saunders K.O., Korber B., Wagh K., McMichael A.J., Kelsoe G., Hahn B.H., Alt F.et al.. Strategies for HIV-1 vaccines that induce broadly neutralizing antibodies. Nat. Rev. Immunol. 2022; 23:142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burton D.R. What are the most powerful immunogen design vaccine strategies? Reverse vaccinology 2.0 shows great promise. Cold Spring Harb. Perspect. Biol. 2017; 9:a030262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kwong P.D., DeKosky B.J., Ulmer J.B.. Antibody-guided structure-based vaccines. Semin. Immunol. 2020; 50:101428. [DOI] [PubMed] [Google Scholar]
- 13. Wiehe K., Saunders K.O., Stalls V., Cain D.W., Venkatayogi S., Martin Beem J.S., Berry M., Evangelous T., Henderson R., Hora B.et al.. Mutation-guided vaccine design: a strategy for developing boosting immunogens for HIV broadly neutralizing antibody induction. 2022; bioRxiv doi:13 November 2022, preprint: not peer reviewed 10.1101/2022.11.11.516143. [DOI] [PubMed]
- 14. Kepler T.B. Reconstructing a B-cell clonal lineage. I. Statistical inference of unobserved ancestors. F1000Res. 2013; 2:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bonsignori M., Zhou T., Sheng Z., Chen L., Gao F., Joyce M.G., Ozorowski G., Chuang G.Y., Schramm C.A., Wiehe K.et al.. Maturation pathway from germline to broad HIV-1 neutralizer of a CD4-mimic antibody. Cell. 2016; 165:449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schramm C.A., Douek D.C.. Beyond hot spots: biases in antibody somatic hypermutation and implications for vaccine design. Front. Immunol. 2018; 9:1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoon H., Macke J., West A.P. Jr, Foley B., Bjorkman P.J., Korber B., Yusim K.. CATNAP: a tool to compile, analyze and tally neutralizing antibody panels. Nucleic Acids Res. 2015; 43:W213–W219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bonsignori M., Kreider E.F., Fera D., Meyerhoff R.R., Bradley T., Wiehe K., Alam S.M., Aussedat B., Walkowicz W.E., Hwang K.K.et al.. Staged induction of HIV-1 glycan-dependent broadly neutralizing antibodies. Sci. Transl. Med. 2017; 9:eaai7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ARMADiLLO web server is freely available to all users at https://armadillo.dhvi.duke.edu. The source code for a standalone version of ARMADiLLO can be downloaded from https://github.com/WieheLab/ARMADiLLO. An ARMADiLLO commandline version has also been implemented in an Apptainer container, bundling Cloanalyst and ARMADiLLO into a single executable package. Due to the large size of the container, it is only available upon request. Detailed documentation for running ARMADiLLO, the containerized version and the ARMADiLLO web server can be found at https://armadillo-docs.readthedocs.io.