


Abstract
Protein phosphorylation, catalyzed by protein kinases (PKs), is one of the most important post-translational modifications (PTMs), and involved in regulating almost all of biological processes. Here, we report an updated server, Group-based Prediction System (GPS) 6.0, for prediction of PK-specific phosphorylation sites (p-sites) in eukaryotes. First, we pre-trained a general model using penalized logistic regression (PLR), deep neural network (DNN), and Light Gradient Boosting Machine (LightGMB) on 490 762 non-redundant p-sites in 71 407 proteins. Then, transfer learning was conducted to obtain 577 PK-specific predictors at the group, family and single PK levels, using a well-curated data set of 30 043 known site-specific kinase-substrate relations in 7041 proteins. Together with the evolutionary information, GPS 6.0 could hierarchically predict PK-specific p-sites for 44046 PKs in 185 species. Besides the basic statistics, we also offered the knowledge from 22 public resources to annotate the prediction results, including the experimental evidence, physical interactions, sequence logos, and p-sites in sequences and 3D structures. The GPS 6.0 server is freely available at https://gps.biocuckoo.cn. We believe that GPS 6.0 could be a highly useful service for further analysis of phosphorylation.
Graphical Abstract
Graphical Abstract.

GPS 6.0 is an online service for prediction of PK-specific phosphorylation sites in eukaryotes.
INTRODUCTION
Protein phosphorylation is one of the most studied post-translational modifications (PTMs) that plays a role in regulating biological processes (1). It involves in reversible covalently attaching a phosphate group on specific serine, threonine and tyrosine residues of proteins, catalysed by protein kinases (2–4). Protein phosphorylation alters the conformation, activity, localization, and interactions of proteins, thereby regulating processes such as gene expression, signal transduction and cell metabolism (5–8). The aberrances in PKs or their substrates have been linked to diseases like cancer (9,10), neurodegenerative diseases (11) and diabetes (12). Therefore, identifying site-specific kinase-substrate relations (ssKSRs) is vital for understanding the protein phosphorylation regulatory roles in biological functions (13), and even more importantly finding therapeutic targets for disease diagnosis and treatment (14).
To effectively identify PK-specific phosphorylation sites (p-sites), a number of in silico prediction approaches have been developed to provide information besides experimental screening. From 2004 to 2017, we developed a series of GPS algorithms, from version 1.0 to 4.0, for predictions of p-sites and other PTM sites (15–21). In 2020, our group released GPS 5.0 using the combination of position weight determination and scoring matrix optimization modules. Within classical module, GPS 5.0 could predict p-sites of 479 human PKs (21). In addition, other researchers also constructed reliable tools for the prediction of PK-specific p-sites, such as MusiteDeep (22), KinasePhos (23), NetPhos (24), and Scansite (25) (Supplementary Table S1). MusiteDeep was the first deep-learning framework for predicting general and PK-specific p-sites in 5 kinase families, including CDK, PKA, CK, MAPK and PKC (22). KinasePhos 3.0 model integrated SVM and eXtreme Gradient Boosting (XGBoost) algorithms for 10 groups, 81 families, and 302 kinases (23). NetPhos 3.1 used artificial neural network for 17 PK-specific prediction (24). Scansite 4.0 utilized motifs for matching PK-specific substrates of 81 kinase (25). However, with the accumulating data of PK-specific p-sites in eukaryotes, challenges exist in developing user-friendly and convincing predictors for more types of kinases.
Here, we present GPS 6.0, a new webserver for PK-specific p-sites prediction of 44 046 PKs in 185 species, which includes 500 PKs in human. With 10 features from previous GPS 5.0 and iLearnPlus (26), GPS 6.0 first integrated three types of machine learning algorithms for general p-sites prediction, including penalized logistic regression (PLR), deep neural network (DNN), and Light Gradient Boosting Machine (LightGMB) (27). Then we fine-tuned the general LightGBM and DNN models to each PK-specific prediction task, so that the models could inherit the prior phosphorylation knowledge. For using GPS 6.0 web server, one or multiple protein sequences could be inputted in the FASTA format and the output will be quickly shown in a tabular list with additional annotations from 22 public resources, including experimental evidence, interactions, sequence logos, physical properties and 3D structures. We also developed five modules to meet different demands. Taken together, GPS 6.0 webserver provides user-friendly and convincing PK-specific predictions with prior knowledge. We anticipate that GPS 6.0 could be helpful for further analysis of phosphorylation.
MATERIALS AND METHODS
Data collection and preparation
First, we downloaded 1 616 804 experimentally identified p-sites in 209 326 proteins for eukaryote from EPSD (28) and we split out the test data based on timestamp-based method (29). A widely used clustering program, CD-HIT (30), was adopted to classify this data set into different clusters with a threshold of 40% sequence similarity (Figure 1A). To avoid the homologous redundancy, only one representative sequence in each cluster was extracted into training data. From GPS 5.0 (21) and PhosphositePlus (https://www.phosphosite.org/) (31), we obtained 23195 and 22206 experimentally determined ssKSRs, respectively. Also, we used multiple keyword combinations to search the literature published from 2019 to 2022 in PubMed. The combinations included a prefix keyword of ‘phosphorylation’, ‘phosphorylate’ or ‘phosphorylated’, by adding a suffix keyword of ‘serine’, ‘threonine’, ‘tyrosine’, ‘residue’ or ‘site’. Only known ssKSRs in eukaryotes were collected, and in total, we obtained 381 additionally known ssKSRs. After de-duplication and removal of the controversial kinases, we obtained 30369 non-redundant ssKSRs (Supplementary Table S5).
Figure 1.

The experimental procedure of the study. (A) General p-site data preparation. (B) Experimentally PK-specific p-sites data collection. (C) GPS 6.0 algorithm. (D) The online service of GPS 6.0 server with annotations.
Before the PK-specific training, we classified known PK-specific p-sites into different PK clusters of group, family, subfamily, and single PK levels with the hierarchical classification information from KinBase (http://kinase.com/web/current/kinbase/genes/SpeciesID/9606/) (32). Due to the fact that multiple aliases are present for each human PK, here we only used the standard gene names taken from iEKPD (http://iekpd.biocuckoo.org). We selected 326 ssKSRs whose kinases were ‘CDK’ or ‘MAPK’ from new collected data as timestamp-based testing data (29) (Supplementary Table S4). The rest data were de-duplicates to be the benchmark data set that contained 30 043 ssKSRs for 19 846 unique p-sites (Figure 1B). Only PK clusters with more than three p-sites were kept for further training.
We defined a p-site peptide PSP(30,30) as a phosphorylation residue flanked by upstream 30 residues and downstream 30 residues, and the PSP(30,30) items around known p-sites were regarded as positive data, whereas PSP(30,30) items from other non-phosphorylation Ser/Thr or Tyr residues were taken as negative data. For Ser/Thr or Tyr residues located near to N- or C-terminus of the protein sequences, one or multiple characters ‘*’ were added to complement the PSP(30,30) items. Prior to model training, the redundant PSP(30,30) items were removed.
Feature encoding and model training
The framework of GPS 6.0 for protein p-site prediction is shown in Figure 1C. Each PSP(30,30) in the training data set were encoded by ten types of sequence features, including the peptide similarity encoded by the GPS method (21), AESNN3 learn from alignments, OPF_10bit (OPF10), OPF_7bit type 1 (OPF1), OPF_7bit type 3 (OPF3), physicochemical properties in the Amino Acid index (AAIndex) database, Z-Scale indexes including five physicochemical descriptor variables, orthogonal binary coding (OBC), Binary5bit2 and enhanced amino acid composition (EAAC). The latter nine features were chosen from iLearnPlus (26) according to the receiver operating characteristic (ROC) corresponding area under the curves (AUC) trained with LightGBM. For each feature, two models were constructed based on LightGBM and DNN algorithms. Then, 20 scores that generated by ten features with two methods were adopted by a PLR model as inputs to get the final prediction score.
Transfer learning
We pre-trained two general models for phosphoserine/phosphothreonine and phosphotyrosine sites separately catalyzed by serine/threonine-specific and tyrosine-specific kinases. We used the parameters of the pre-trained models to initialize the LightGBM and DNN models for kinase-specific prediction. Then the ssKSRs in each kinase cluster were used to fine-tune the general serine/threonine (S/T) and tyrosine (Y) models through the transfer learning strategy, and the optimized models were assigned to corresponding kinase predictors. For each predictor of S/T-specific kinases, we used previous methods in GPS 5.0 (21) to determine three thresholds, including high, medium and low, based on Sp values of 98%, 94% and 90%, respectively, while 96%, 91% and 85% for predictors of T-specific kinases. In the online service of GPS 6.0, the medium threshold was chosen as the default. The eukaryotic PKs information obtained from iEKPD (33) and training data were used to develop the species-specific module. As a result, GPS 6.0 could predict the PK-specific p-sites of 44 046 PKs in 185 species.
Performance evaluation measurements
For the evaluation of GPS 6.0, four widely used measurements, including sensitivity (Sn), specificity (Sp), accuracy (Ac) and Matthew correlation coefficient (MCC), were calculated as below:
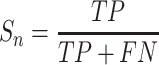 |
 |
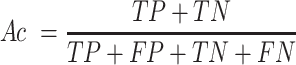 |
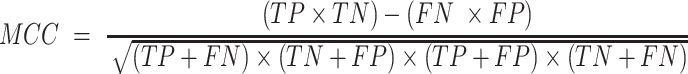 |
For general p-sites prediction, the AUC was calculated from the ROC curve illustrated based on Sn and 1-Sp scores. For kinase-specific p-sites prediction, the robustness of models with ≥30 sites were tested with 10-fold cross-validation (Supplementary Table S2A), and leave-one-out (LOO) validation was performed for other models with <30 sites (Supplementary Table S2B). The ROC curve was illustrated based on Sn and 1-Sp scores to evaluate the performance with timestamp-based testing data (29) of GPS 6.0 and other tools.
Integrated annotations
To provide an interactive user-friendly web service, we integrated other tools and resources to provide annotations. For each kinase cluster, the PSP(30,30) items in positive data were directly uploaded to the web service of WebLogo (http://weblogo.berkeley.edu/logo.cgi) (34), and then the sequence logo was generated automatically. The protein disorder propensity values were predicted by IUPred (35). The accessible surface area (ASA) of amino acids and the secondary structure were predicted by NetSurfP (36). The interaction between kinase and substrate is shown according to the BioGrid database. Also, we show 3D structures and predicted p-sites of the substrate in the PDB database by 3Dmol.js (http://3dmol.csb.pitt.edu/) (37). (Figure 1D, Supplementary Table S3).
Web server implementation
The webserver consists of front end implemented with PHP 7.0.33 and JQuery 1.4.4. The back end was written in Python 3.8 for GPS 6.0 algorithm framework. Tabular predictions are parsed and written using PHP. The visualizations of charts are generated using JavaScript libraries. We tested the online service of GPS 6.0 on a series of mainstream internet browsers, including Microsoft Edge 112.0.1722.48, Google Chrome 107.0.5304.107, Mozilla Firefox 107.0.1 and Safari 16.3. For convenience, local packages have been constructed by PyInstaller (https://pyinstaller.org/), a package in Python, to support three major operating systems, including Windows, Mac OS and Linux, and can be freely available at: https://gps.biocuckoo.cn/download.php.
RESULTS
Performance evaluation and comparison
For the general p-site prediction, we adopted 10-fold cross-validations to evaluate the performance of GPS 6.0 and each single feature on pS/pT and pY sites on training dataset. And we also evaluated the performance on a timestamp-based testing data. The AUC values on pS/pT and pY for GPS 6.0 were calculated as 0.8312 and 0.8080, increasing 2.08 and 6.42 percentage comparing to the highest AUC of single features, respectively. Meanwhile, the test AUC values on these two tasks were 0.7925 and 0.7478 (Figure 2A, B).
Figure 2.

Performance evaluation and comparison of GPS 6.0. (A, B) The AUC of the GPS 6.0 with different features on general pS/pT and pY sites prediction on training dataset and independent test dataset. (C–F) Performance comparison with GPS 5.0, MusiteDeep, KinasePhos 3.0, Netphos 3.1 and Scansite 4.0 for MAPK, CDK, PKA and CK2 kinase families. (G) The ratio of predicted p-sites with experimental verification in CDK from GPS 6.0, MusiteDeep and GPS 5.0.
There have been many useful tools developed for prediction of kinase-specific p-sites. However, a number of tools did not provide available web service or executable files, and their online services could not be accessed any longer, due to lack of maintenance. In this regard, we selected five widely used, stable, and convenient tools for comparison, including GPS 5.0 (21), MusiteDeep (22), KinasePhos 3.0 (23), NetPhos 3.1 (24) and Scansite 4.0 (25). For performance evaluation and comparison, we selected PK families of MAPK and CDK in the time-stamped testing data, while PKA and CK2 families in training data due to the limitation of test data, (Figure 2C–F). The ROC curve was illustrated and AUC value was calculated for each tool. We found that GPS 6.0 achieved a competitive accuracy with MusiteDeep (Figure 2C). We further interpreted the experimental verification rate within PK-specific predictions of CDK on substrates of test ssKSRs from GPS 6.0, GPS 5.0 and MusiteDeep. We found that predicted results from GPS 6.0 consisted of 25.7% p-sites with CDK-specific phosphorylation verifications and 53.3% of p-sites with general phosphorylation verifications. While, the CDK-specific verified p-site ratio from MusiteDeep and GPS 5.0 were 19.9% and 15.2%, respectively. With the interpretation, we believe that GPS 6.0 could provide convincing kinase-specific prediction with prior general p-site knowledge (Figure 2D).
Usage of GPS 6.0 web server
The final model integrated with LightGBM and DNN predictions was used as the default option for the online service. For a prediction with a faster speed, users can select the lightGBM model. The input can be submitted by specifying their sequences in FASTA format (Figure 3A). The FASTA format has a description line starting with ‘>’ for each entry followed by one or more lines of protein sequence. We also provide a protein identiter module that accepts gene name, protein name or UniProt accession as input. The server of species-specific prediction provides 184 species for species-specific prediction and he comprehensive mode could predict secondary structures and surface accessibility of substrates, which consumes more time. The job was under processing after selecting the kinase(s) and clicking the ‘Submit’ button (Figure 3A).
Figure 3.

The usage of GPS 6.0 web server. (A) The example sequence input page with kinase selection. (B) The prediction results of the example. (C) The annotations of the prediction results.
After a while, the output can be visualized for each input sequence one by one, which can be retrieved by clicking the ‘Next protein’ button. The prediction result contains potential p-sites with eleven types of information, including ‘ID’, ‘Position’, ‘Code’, ‘Kinase’, ‘Peptide’, ‘Score’, ‘Cutoff’, ‘Source’, ‘Links’, ‘Interaction’ and ‘Logo’ (Figure 3B).
Experimental evidence of predicted sites could be viewed by clicking on ‘Exp’ if available in the column of ‘Source’ in the prediction page. For each kinase cluster, the sequence logo of the PSP(30,30) items in positive data could be shown in the ‘Logo’ column. The ‘Links’ column provides a link of substrate to the EPSD database. The ‘interaction’ column indicated whether there was an interaction between kinase and the substrate, and if so, the relevant literature could be viewed (Figure 3B). As the default configuration, the top 3 p-sites with the highest prediction scores were shown in a schematic diagram of the protein sequence, together with the disorder propensity score for each residue predicted by IUPred (35). On the comprehensive mode, the surface accessibility and the secondary structures including α-helix, β-strand and coil predicted could also be predicted by NetSurfP (36) according to substrate sequence, and they were shown under the schematic diagram (Figure 3C). In the same, we performed basic statistics on the distribution of selected kinase families, number in disordered region and secondary structures of predicted p-sites (Figure 3C). Also, 3D structure with predicted p-sites of substrate could be visualized by 3Dmol.js (Figure 3C). The prediction results of all inputted proteins could be downloaded in one of the four file formats, including .txt, .csv, .tsv and .xlsx. The ‘Export’ button helps to obtain the .png file for the annotation image. A help page is also provided with an example input and output.
An example for using GPS 6.0
We randomly selected mTOR kinase as an example. The mammalian target of rapamycin (mTOR) kinase belongs to the phosphatidylinositol (PI) kinase-related kinase (PIKK) family, atypical kinase group. The hyperactivated mTOR could enhance the progression of breast cancer through downstream substrate human estrogen receptor (ESR1) (38,39). So, we used GPS 6.0 for mTOR-specific prediction of ESR1 to find significant p-sites as potential therapeutic targets.
In the prediction results of ESR1, we got five mTOR-specific p-sites, including S104, S106, S118, S154 and S294. It was reported that mTORC1, a complex of mTOR, could phosphorylate ESR1 on S104, S106 and S118 in AF1 domain (38,40). Phosphorylation of AF1 domain on ESR1 is recognized by MACROD1, a co-activator of ESR1 (41–43), which would introduce progression of hormone-dependent cancers by feed-forward mechanism that activates ESR1 transactivation (43,44). Based on our prediction, not only the three serine sites could be catalysed by mTORC1, S154 would be another possible specific residue within AF1 domain, which was previously verified as a general p-site (Figure 4A).
Figure 4.

Convincing prediction based on general knowledge. (A) Predicted p-sites on ESR1 by MTOR kinase. (B) The annotation of ESR1 p-sites on AF1 domain.
Besides the prediction results, we presented annotations of p-sites on ESR1, including the calculated disorder scores, ASA scores, zoomed 3D structure for S154 and experimental curation information from EPSD (Figure 4B). According to disorder and ASA scores, S154 could be a more accessible p-site comparing to S104, S106 and S118, due to a more flexible substrate structure for kinase catalysis. The general p-site identification of ESR1 S154 was also conducted in breast cells and breast cancer cells, which also supported our predictions. Taken together, we indicated a new potential mTOR-specific p-site S154 on ESR1, which could help for understanding the therapeutic strategies through targeting phosphorylation signals in breast cancer.
DISCUSSION
In this work, GPS 6.0 brings convincing PK-specific p-sites prediction for 44 046 PKs in 185 species. This would help more users to predict up-stream kinases and p-sites on substrates in the research areas of interest. We also integrated much visualized annotations for interpreting the p-sites and substrates. With the appended information of these p-sites and substrates, the prediction results would be presented in more comprehensive ways. GPS 6.0 was constructed as a user-friendly webserver with highly customizable ssKSRs prediction, including selectable kinase and different modules.
For using GPS 6.0 and other similar tools, a prerequisite hypothesis is that the selected PKs can recognize specific sequence motifs around p-sites to phosphorylate the inputted protein(s). This hypothesis is correct in vitro, since purified kinases can be easily close to potential substrates in tubes. However, such a sequence specificity might not be enough for the recognition of substrates by PKs in vivo, and a number of contextual factors, such as physical interaction, co-localization, coexpression, and co-complex, contribute the additional specificity (45–47). Previously, by integrating both sequence-based prediction and protein-protein interaction (PPI) information between PKs and substrates, NetworKIN (45,46) and iGPS (47), were constructed for prediction of potentially in vivo ssKSRs. Of note, the interactions between PKs and substrates might be weak and transient, and could not be detected from standard PPI screenings. Thus, a considerable proportion of real in vivo ssKSRs might be missed by NetworKIN or iGPS. In GPS 6.0, only sequence features around p-sites were considered, while the contextual factors were not included. Thus, further experimental validations should be carried out to validate the interactions between PKs and substrates, after obtaining the prediction results.
In the future, more annotations will be collected from additional public resources and tools for interpretation of results. Also, we will integrate more features and update our dataset to improve the performance. GPS 6.0 will be continuously maintained and improved for academic research.
DATA AVAILABILITY
GPS 6.0 is freely available for all users at: https://gps.biocuckoo.cn/ and the source at https://github.com/BioCUCKOO/GPS6.0 (permanent DOI: 10.5281/zenodo.7875616).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank numerous users of GPS 6.0 for their valuable comments and communications with us during the past years.
Contributor Information
Miaomiao Chen, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, 430074, China.
Weizhi Zhang, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, 430074, China.
Yujie Gou, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, 430074, China.
Danyang Xu, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China.
Yuxiang Wei, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, 430074, China.
Dan Liu, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, 430074, China.
Cheng Han, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, 430074, China.
Xinhe Huang, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, 430074, China.
Chengzhi Li, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, 430074, China.
Wanshan Ning, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, 430074, China.
Di Peng, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, 430074, China.
Yu Xue, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, 430074, China; Nanjing University Institute of Artificial Intelligence Biomedicine, Nanjing, 210031, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: National Key R&D Program of China [2021YFF0702000, 2022YFC2704300, 2021ZD0201300] Natural Science Foundation of China [31930021, 31970633, 81701567]; Hubei Innovation Group Project [2021CFA005]; Research Core Facilities for Life Science (HUST).
Conflict of interest statement. None declared.
REFERENCES
- 1. Mann M., Ong S.E., Gronborg M., Steen H., Jensen O.N., Pandey A.. Analysis of protein phosphorylation using mass spectrometry: deciphering the phosphoproteome. Trends Biotechnol. 2002; 20:261–268. [DOI] [PubMed] [Google Scholar]
- 2. Bilbrough T., Piemontese E., Seitz O.. Dissecting the role of protein phosphorylation: a chemical biology toolbox. Chem. Soc. Rev. 2022; 51:5691–5730. [DOI] [PubMed] [Google Scholar]
- 3. Johnson S.A., Hunter T.. Kinomics: methods for deciphering the kinome. Nat. Methods. 2005; 2:17–25. [DOI] [PubMed] [Google Scholar]
- 4. Sharma K., D'Souza R.C., Tyanova S., Schaab C., Wisniewski J.R., Cox J., Mann M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014; 8:1583–1594. [DOI] [PubMed] [Google Scholar]
- 5. Mateus A., Kurzawa N., Becher I., Sridharan S., Helm D., Stein F., Typas A., Savitski M.M.. Thermal proteome profiling for interrogating protein interactions. Mol. Syst. Biol. 2020; 16:e9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Potel C.M., Kurzawa N., Becher I., Typas A., Mateus A., Savitski M.M.. Impact of phosphorylation on thermal stability of proteins. Nat. Methods. 2021; 18:757–759. [DOI] [PubMed] [Google Scholar]
- 7. Needham E.J., Hingst J.R., Parker B.L., Morrison K.R., Yang G., Onslev J., Kristensen J.M., Hojlund K., Ling N.X.Y., Oakhill J.S.et al.. Personalized phosphoproteomics identifies functional signaling. Nat. Biotechnol. 2022; 40:576–584. [DOI] [PubMed] [Google Scholar]
- 8. Cantley L.C. The phosphoinositide 3-kinase pathway. Science. 2002; 296:1655–1657. [DOI] [PubMed] [Google Scholar]
- 9. Drake J.M., Paull E.O., Graham N.A., Lee J.K., Smith B.A., Titz B., Stoyanova T., Faltermeier C.M., Uzunangelov V., Carlin D.E.et al.. Phosphoproteome integration reveals patient-specific networks in prostate cancer. Cell. 2016; 166:1041–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen Y., Shao X., Cao J., Zhu H., Yang B., He Q., Ying M.. Phosphorylation regulates cullin-based ubiquitination in tumorigenesis. Acta Pharm. Sin. B. 2021; 11:309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gan Z.Y., Callegari S., Cobbold S.A., Cotton T.R., Mlodzianoski M.J., Schubert A.F., Geoghegan N.D., Rogers K.L., Leis A., Dewson G.et al.. Activation mechanism of PINK1. Nature. 2022; 602:328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang Q., Liu S., Zhai A., Zhang B., Tian G.. AMPK-mediated regulation of lipid metabolism by phosphorylation. Biol. Pharm. Bull. 2018; 41:985–993. [DOI] [PubMed] [Google Scholar]
- 13. Hodgson D.R., Schroder M.. Chemical approaches towards unravelling kinase-mediated signalling pathways. Chem. Soc. Rev. 2011; 40:1211–1223. [DOI] [PubMed] [Google Scholar]
- 14. Tong M., Yu C., Zhan D., Zhang M., Zhen B., Zhu W., Wang Y., Wu C., He F., Qin J.et al.. Molecular subtyping of cancer and nomination of kinase candidates for inhibition with phosphoproteomics: reanalysis of CPTAC ovarian cancer. EBioMedicine. 2019; 40:305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou F.F., Xue Y., Chen G.L., Yao X.. GPS: a novel group-based phosphorylation predicting and scoring method. Biochem. Biophys. Res. Commun. 2004; 325:1443–1448. [DOI] [PubMed] [Google Scholar]
- 16. Xue Y., Zhou F., Zhu M., Ahmed K., Chen G., Yao X.. GPS: a comprehensive www server for phosphorylation sites prediction. Nucleic Acids Res. 2005; 33:W184–W187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xue Y., Ren J., Gao X., Jin C., Wen L., Yao X.. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics. 2008; 7:1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xue Y., Liu Z., Cao J., Ma Q., Gao X., Wang Q., Jin C., Zhou Y., Wen L., Ren J.. GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng. Des. Sel. 2011; 24:255–260. [DOI] [PubMed] [Google Scholar]
- 19. Liu Z., Yuan F., Ren J., Cao J., Zhou Y., Yang Q., Xue Y.. GPS-ARM: computational analysis of the APC/C recognition motif by predicting D-boxes and KEN-boxes. PLoS One. 2012; 7:e34370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao Q., Xie Y., Zheng Y., Jiang S., Liu W., Mu W., Liu Z., Zhao Y., Xue Y., Ren J.. GPS-SUMO: a tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic Acids Res. 2014; 42:W325–W330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang C., Xu H., Lin S., Deng W., Zhou J., Zhang Y., Shi Y., Peng D., Xue Y.. GPS 5.0: an update on the prediction of kinase-specific phosphorylation sites in proteins. Genomics Proteomics Bioinformatics. 2020; 18:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang D., Zeng S., Xu C., Qiu W., Liang Y., Joshi T., Xu D. MusiteDeep: a deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics. 2017; 33:3909–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma R., Li S., Li W., Yao L., Huang H.D., Lee T.Y.. KinasePhos 3.0: redesign and expansion of the prediction on kinase-specific phosphorylation sites. Genomics Proteomics Bioinformatics. 2022; 10.1016/j.gpb.2022.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blom N., Sicheritz-Ponten T., Gupta R., Gammeltoft S., Brunak S.. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004; 4:1633–1649. [DOI] [PubMed] [Google Scholar]
- 25. Obenauer J.C., Cantley L.C., Yaffe M.B.. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003; 31:3635–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Z., Zhao P., Li C., Li F., Xiang D., Chen Y.Z., Akutsu T., Daly R.J., Webb G.I., Zhao Q.et al.. iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 2021; 49:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ke G.L., Meng Q., Finley T., Wang T.F., Chen W., Ma W.D., Ye Q.W., Liu T.Y.. LightGBM: a highly efficient gradient boosting decision tree. Adv Neur In. 2017; 30:3149–3157. [Google Scholar]
- 28. Lin S., Wang C., Zhou J., Shi Y., Ruan C., Tu Y., Yao L., Peng D., Xue Y.. EPSD: a well-annotated data resource of protein phosphorylation sites in eukaryotes. Brief Bioinform. 2021; 22:298–307. [DOI] [PubMed] [Google Scholar]
- 29. Wang D., Liu D., Yuchi J., He F., Jiang Y., Cai S., Li J., Xu D. MusiteDeep: a deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res. 2020; 48:W140–W146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fu L., Niu B., Zhu Z., Wu S., Li W.. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012; 28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E.. PhosphoSitePlus, 2014: mutations, ptms and recalibrations. Nucleic Acids Res. 2015; 43:D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Manning G., Whyte D.B., Martinez R., Hunter T., Sudarsanam S.. The protein kinase complement of the human genome. Science. 2002; 298:1912–1934. [DOI] [PubMed] [Google Scholar]
- 33. Guo Y., Peng D., Zhou J., Lin S., Wang C., Ning W., Xu H., Deng W., Xue Y.. iEKPD 2.0: an update with rich annotations for eukaryotic protein kinases, protein phosphatases and proteins containing phosphoprotein-binding domains. Nucleic Acids Res. 2019; 47:D344–D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dosztanyi Z., Csizmok V., Tompa P., Simon I.. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005; 21:3433–3434. [DOI] [PubMed] [Google Scholar]
- 36. Petersen B., Petersen T.N., Andersen P., Nielsen M., Lundegaard C.. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009; 9:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rego N., Koes D. 3Dmol.js: molecular visualization with WebGL. Bioinformatics. 2015; 31:1322–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alayev A., Salamon R.S., Berger S.M., Schwartz N.S., Cuesta R., Snyder R.B., Holz M.K.. mTORC1 directly phosphorylates and activates eralpha upon estrogen stimulation. Oncogene. 2016; 35:3535–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yamnik R.L., Holz M.K.. mTOR/S6K1 and MAPK/RSK signaling pathways coordinately regulate estrogen receptor alpha serine 167 phosphorylation. FEBS Lett. 2010; 584:124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Masaki T., Habara M., Sato Y., Goshima T., Maeda K., Hanaki S., Shimada M.. Calcineurin regulates the stability and activity of estrogen receptor alpha. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2114258118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin L.A., Ribas R., Simigdala N., Schuster E., Pancholi S., Tenev T., Gellert P., Buluwela L., Harrod A., Thornhill A.et al.. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat Commun. 2017; 8:1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kumagai K., Takanashi M., Ohno S.I., Harada Y., Fujita K., Oikawa K., Sudo K., Ikeda S.I., Nishi H., Oikawa K.et al.. WAPL induces cervical intraepithelial neoplasia modulated with estrogen signaling without HPV E6/E7. Oncogene. 2021; 40:3695–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Han W.D., Zhao Y.L., Meng Y.G., Zang L., Wu Z.Q., Li Q., Si Y.L., Huang K., Ba J.M., Morinaga H.et al.. Estrogenically regulated LRP16 interacts with estrogen receptor alpha and enhances the receptor's transcriptional activity. Endocr. Relat. Cancer. 2007; 14:741–753. [DOI] [PubMed] [Google Scholar]
- 44. Meng Y.G., Han W.D., Zhao Y.L., Huang K., Si Y.L., Wu Z.Q., Mu Y.M.. Induction of the LRP16 gene by estrogen promotes the invasive growth of Ishikawa human endometrial cancer cells through the downregulation of E-cadherin. Cell Res. 2007; 17:869–880. [DOI] [PubMed] [Google Scholar]
- 45. Linding R., Jensen L.J., Ostheimer G.J., van Vugt M.A., Jorgensen C., Miron I.M., Diella F., Colwill K., Taylor L., Elder K.et al.. Systematic discovery of in vivo phosphorylation networks. Cell. 2007; 129:1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Linding R., Jensen L.J., Pasculescu A., Olhovsky M., Colwill K., Bork P., Yaffe M.B., Pawson T.. NetworKIN: a resource for exploring cellular phosphorylation networks. Nucleic Acids Res. 2008; 36:D695–D699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Song C., Ye M., Liu Z., Cheng H., Jiang X., Han G., Songyang Z., Tan Y., Wang H., Ren J.et al.. Systematic analysis of protein phosphorylation networks from phosphoproteomic data. Mol. Cell Proteomics. 2012; 11:1070–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GPS 6.0 is freely available for all users at: https://gps.biocuckoo.cn/ and the source at https://github.com/BioCUCKOO/GPS6.0 (permanent DOI: 10.5281/zenodo.7875616).