


Abstract
Allostery refers to the biological process by which an effector modulator binds to a protein at a site distant from the active site, known as allosteric site. Identifying allosteric sites is essential for discovering allosteric process and is considered a critical factor in allosteric drug development. To facilitate related research, we developed PASSer (Protein Allosteric Sites Server) at https://passer.smu.edu, a web application for fast and accurate allosteric site prediction and visualization. The website hosts three trained and published machine learning models: (i) an ensemble learning model with extreme gradient boosting and graph convolutional neural network, (ii) an automated machine learning model with AutoGluon and (iii) a learning-to-rank model with LambdaMART. PASSer accepts protein entries directly from the Protein Data Bank (PDB) or user-uploaded PDB files, and can conduct predictions within seconds. The results are presented in an interactive window that displays protein and pockets’ structures, as well as a table that summarizes predictions of the top three pockets with the highest probabilities/scores. To date, PASSer has been visited over 49 000 times in over 70 countries and has executed over 6 200 jobs.
Graphical Abstract
Graphical Abstract.
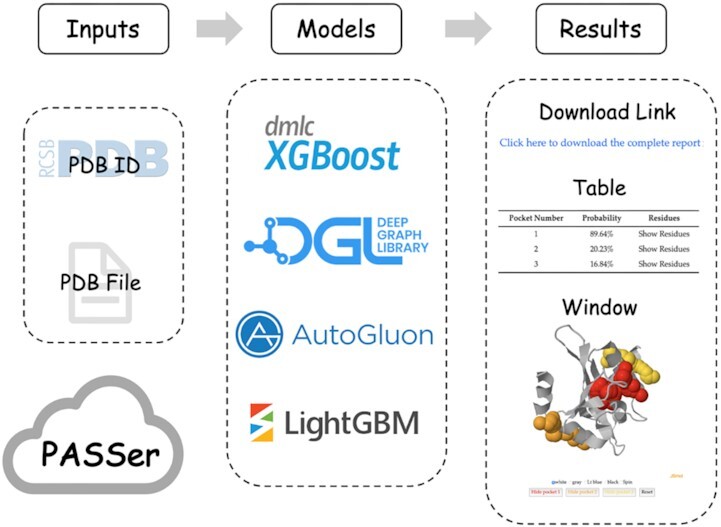
PASSer web server architecture.
INTRODUCTION
Allostery is a critical biological process in the regulation of protein activity. It involves the transmission of the effect of a small molecule binding from the allosteric site to the active site, leading to protein conformational and dynamic changes (1). There are many characteristics of allosteric processes that can be harnessed in drug design: (a) allosteric site is conserved and highly specific in the evolution of proteins (2,3), (b) allosteric drugs can activate or inhibit protein activities in a controlled manner, leading to potential therapeutic effects (4), and (c) once the allosteric site is saturated, there are no further therapeutic effects (5). For these reasons, the study of allosteric sites is vital in allosteric drug development and has gained significant attention over the last decade. In fact, it has been recognized as the ‘second secret of life’ (6).
Several computational methods have been developed for the prediction of allosteric sites based on protein dynamics including normal mode analysis (NMA) (7) and molecular dynamics (MD) simulations (8). For instance, PARS (9) employs NMA to identify protein sites that can transmit or mediate allosteric signals, while SPACER (10) combines NMA and MD simulations to evaluate allosteric sites. Recently, many machine learning-based models have been demonstrated improved prediction performance. Allosite (11) and AlloPred (12) employ support vector machines (SVMs) to learn the physical and chemical features of protein pockets. Chen et al. (13) uses random forests (RFs) to build a three-way model to predict allosteric, orthosteric, and non-functional pockets. Among the prediction models, Allosite, PARS, and AlloPred are accessible as websites.
In this study, we introduce PASSer (Protein Allosteric Sites Server, https://passer.smu.edu), a web server that provides fast and accurate allosteric site prediction. PASSer offers three trained and published machine learning-based models: (a) an ensemble learning model consisting of extreme gradient boosting (XGBoost) and graph convolutional neural network (GCNN) (14), (b) an automated machine learning model powered by AutoGluon from Amazon Web Services (AWS) (15), and (c) a learning-to-rank model with the boosted tree version of LambdaRank objective on LightGBM (16). PASSer is deployed on Southern Methodist University High-Performance Computing (HPC) clusters that can complete prediction within seconds. The website does not require any login credentials. Users can submit a Protein Data Bank (PDB) (17) ID or PDB file, and all source files are deleted after calculation is completed. PASSer displays an interactive window that showcases the protein structure with highlighted pocket structures, along with a table summarizing the top protein pockets. Users can also download a .zip file containing protein and pocket PDB files, visualization scripts for Visual Molecular Dynamics (VMD) (18) and PyMOL (19), and prediction results. Since its launch in 2020, PASSer has received over 49 000 visits and completed over 6 200 jobs.
MATERIALS AND METHODS
Website implementation
PASSer models are implemented in Python language and the web service is built using the Python Django web framework (v3.1.2). PASSer provides three methods for allosteric site prediction. Below are the dependency packages and versions for each method: (a) ensemble learning: XGBoost package v1.3.3 (20) and DGL v0.4.3 (21); (b) automated machine learning: AutoGluon v0.3.1 (22); (c) learning-to-rank: LighGBM v3.3.4 (23). On the result page, an interactive window powered by the JavaScript framework JSmol (24) is provided to visualize protein and pocket structures. The website is hosted on SMU HPC (https://www.smu.edu/Provost/Data-Science-Institute/HPC) to provide substantial computing resources.
Workflow overview
On the PASSer’s main page, users can submit jobs without login requirement. They can do so by providing an existing PDB ID from the Protein Data Bank or by uploading their own PDB files. When a PDB ID is submitted, PASSer scrapes the corresponding PDB file from the RCSB PDB website. Users can also specify the chain ID if there are multiple chains in the PDB file. FPocket, a geometry-based pocket detection package (25), is then used to detect potential protein pockets in the resulting protein structure (25). The user-selected machine learning model is applied for the prediction of detected pockets. In the ensemble learning method, XGBoost (20) learns 19 physical and chemical features calculated by FPocket. GCNN (26) builds an atomic graph for each pocket to learn the local connectivity at atomic level. The final predicted probability is the average of probabilities generated from XGBoost and GCNN. In the automated machine learning model, the pocket descriptors are fed into a AutoGluon model (22) for prediction, which consists of 14 base models, such as SVM and RF. A full list of these base models is available in the supporting information of a previous study (15). For the learning-to-rank model, all pockets in a given protein are ranked with regard to their relevance of being allosteric sites. The ensemble learning and automated machine learning models report predicted probabilities of the top 3 pockets, while the learning-to-rank model reports rank scores. A detailed description of these methods can be found in previous studies (14–16). A link is provided to download a .zip file containing protein and pocket PDB files, visualization scripts, and a list of prediction results for all detected pockets.
RESULTS AND DISCUSSION
Dataset collection
Collecting and cleaning allosteric proteins is crucial to produce high-quality datasets and well-performed models (27). Although the availability of allosteric site databases, such as AlloSteric Database (ASD) (28), ASBench (29) and CASBench (30), provides a new opportunity to design allosteric site prediction models, the lack of a standardized approach for preparing machine learning-ready datasets can hinder such development. The latest model of PASSer, i.e. the learning-to-rank model, presents a workflow to produce the training data with Python implementation. The scripts are available on GitHub at https://github.com/smu-tao-group/PASSerRank. To our knowledge, this is the first open-source repository to automate the data preparation process. This could establish a benchmark for future model training and validation. Specifically, two datasets (ASD and CASBench) were used in training and validating machine learning models.
The latest version of ASD contains 1 949 protein entries, in which each entry includes information of protein, modulator, and allosteric residues. Following a data cleaning workflow proposed by Huang et al. (11), those proteins were filtered out if they (i) have low resolution (>3 Å); (ii) have missing residues in the allosteric site; or (iii) have similar structures (sequence identity threshold ≥ 30%).
The remaining proteins were analyzed using FPocket. To automate the pocket labeling process where a pocket is labeled as allosteric (positive) or non-allosteric (negative), we define the pocket nearest to the modulator as the allosteric site and all other pockets as non-allosteric sites. In each protein, the Euclidean distances between the center of masses in its modulator and all pockets are calculated. Those proteins were removed if the closest pocket to the modulator is >10 Å. Through the data cleaning steps above, 207 proteins were included to train machine learning models.
CASBench was used as an external test set. Proteins that did not meet the data cleaning standards mentioned above were removed, which leads to a test set consisting of 1 049 proteins. The ASD-trained machine learning models were tested on this CASBench test set.
The processed ASD and CASBench data can be downloaded from the PASSer website and users can customize the data preparation step using the Python scripts in the provided GitHub repository.
It should be noted that the previous two models (ensemble learning and automated machine learning) were trained and tested on smaller training datasets. 90 ASD proteins were used to train the ensemble learning model. A full list of these proteins is available in the supporting information of Huang et al. (11). In addition to these proteins, the core-diversity set of ASBench (138 proteins) were included to train the automated machine learning model. After removing duplicate records, this model was trained on 204 proteins.
Model training
To train the ensemble learning and automated machine learning models, the proteins were randomly split into a training set (60%), a validation set (20%) and a test set (20%). Models with different hyperparameter settings were trained on the training set with performance metrics calculated on the validation set. The hyperparameter setting leading to the highest performance was selected. The test set was used to estimate model performance in real world applications.
In the learning-to-rank model, the ASD proteins were randomly split into a training set (80%) and a test set (20%). Five-fold cross validation was performed on the training set for parameter tuning, and the best-performed parameter setting was selected and used on the test set. Due to the limited protein sample size, the n-fold cross validation was considered more effective than the previous 60/20/20 splitting to include more training data and can lead to better performance.
Data imbalance is a key issue in our model training, which the amount of data in one class is significantly smaller than other classes. In allosteric site prediction, each protein may consist of more than ten pockets while there is only one positive (allosteric) pocket. Data imbalance may cause poor performance, as a model cannot learn enough from the minority class (31). To address this issue, an undersampling strategy is applied to train the GCNN model by randomly removing negative pockets to keep a constant ratio of four between the number of positive and negative pockets. The top six pockets with the highest FPocket scores in each protein were selected to train the automated machine learning model. However, discarding pocket samples may lead to a loss of useful information for training a robust model. Oversampling is another strategy to rebalance datasets by duplicating old or generating new examples. One drawback is that the generated new pockets may not correspond to real protein pockets, thus, lack biological meaning. Moreover, it is more likely to introduce overfitting (32). Increasing the weight of the minority class is a third way. The weight of positive labels can be increased through the scale_pos_weight parameter in XGBoost and learning-to-rank models so that all data can be included in training.
Model performance
Table 1 summarizes the performance of three machine learning models. To compare the model performance, various metrics were calculated, including precision, recall, and F1 score for binary classification, and the percentage of actual allosteric sites ranked in top 1 and 3 positions.
Table 1.
Reported performance of machine learning models on PASSer
| Models | Precision | Recall | F1 score | Top 1 | Top 3 |
|---|---|---|---|---|---|
| Ensemble learning | 0.726 | 0.847 | 0.782 | 60.7% | 84.9% |
| Automated machine learning | 0.850 | 0.616 | 0.701 | 65.1% | 82.7% |
| Learning-to-rank | 0.662 | 0.662 | 0.662 | 59.5% | 83.6% |
In the learning-to-rank model, each pocket is predicted with a rank score, which reflects the relevance, i.e. how well a pocket meets the characteristics of known allosteric sites in the training set. The pockets with high rank scores suggest higher possibility of being allosteric sites and are worth further study. In our analysis, only the pocket with the highest rank score was labeled as positive in each protein, and then the metrics for binary classification can be calculated. Since we fixed the number of predicted positive labels, the false positive and false negative predictions are the same, which results the same precision and recall values. It is important to note that performance is not directly comparable across models due to differences in the training and test datasets used. Going forward, the use of standardized data preparation procedures and scripts presented in this study and the web server will enable more equitable comparison of machine learning models.
Model selection guidance
Different models require different execution time and have various output types. Table 2 summarizes the execution speed and prediction type of three machine learning models. The execution time needed in each model was extensively estimated with multiple mid-sized proteins (100–300 residues). For one prediction, the ensemble learning model requires 1–2 s on average and the learning-to-rank model is slightly faster. The automated machine learning model takes around 20 seconds due to the loading of 14 base models. For prediction types, probabilities are generated in both of the ensemble learning and automated machine learning models, and rank scores are reported in the learning-to-rank model. We recommend the users choosing ensemble learning and learning-to-rank models for time sensitive tasks, ensemble learning and automated learning models for good interpretability, and learning-to-rank model for benchmark study and performance comparison.
Table 2.
Selection criteria of machine learning models on PASSer
| Models | Execution time | Type |
|---|---|---|
| Ensemble learning | Fast (1–2 s) | Probability |
| Automated machine learning | Slow (∼20 s) | Probability |
| Learning-to-rank | Fast (1–2 s) | Rank score |
Case study
We demonstrated the functionality of PASSer with a known allosteric protein, the light-oxygen-voltage domain of Phaeodactylum tricornutum Aureochrome 1a (PDB ID 5DKK) (33). On the main page (Figure 1A), we submitted a prediction job by inputting the protein identifier 5DKK and chain ID A, and choosing the ensemble prediction model. On the result page, a table (Figure 1B) displays the prediction probabilities of the top three most probable pockets as allosteric sites. Users can click on ‘Show Residues’ to view the corresponding pocket residues. Higher probability indicates higher likelihood of the pocket being an allosteric site. Figure 1C displays the interactive window showing 5DKK protein structure. The red pocket had a predicted probability of 89.65%, indicating its high potential to be an allosteric site. This result aligns well with the finding of the actual allosteric pocket based on previous research (34). Users can download the results from the provided link and interact with the protein structure and predicted pockets through this window, which supports the functions to change background colors and show or hide specific pockets.
Figure 1.

One example of allosteric site prediction results of the light-oxygen-voltage domain of Phaeodactylum tricornutum Aureochrome 1a protein. (A) The job submission form with protein identifier 5DKK, chain ID A, and ensemble learning model. (B) Predicted probabilities of the top three most probable pockets being allosteric sites. (C) An interactive window showing 5DKK protein structure with highlighted pockets.
PASSer web service has also been applied for other purposes, such as the revalidation of allosteric site prediction for other models and the screening of predicted allosteric sites. For example, PASSer was combined with Allosite Pro to validate the identified allosteric site of SARS-CoV-2 methyltransferase (MTase) (35). In another study, PASSer was employed with the Computed Atlas of Surface Topography of Proteins (CASTp) server to discover the apolipoprotein L1 (APOL1) protein (36).
CONCLUSION
PASSer is a user-friendly web application that facilitates the prediction of protein allosteric sites. It provides three pre-trained machine learning models to achieve reliable and accurate performance, along with interactive result visualization. The website is hosted on a high-performance computing platform, enabling it to complete predictions within seconds. PASSer has been widely used for the validation of known functional pockets and the discovery of new allosteric sites.
DATA AVAILABILITY
The PASSer web service is freely available at https://passer.smu.edu. The Python scripts to prepare training data is available at https://github.com/smu-tao-group/PASSerRank and https://doi.org/10.5281/zenodo.7818017.
ACKNOWLEDGEMENTS
Computational time was generously provided by the Southern Methodist University’s Center for Research Computing.
Contributor Information
Hao Tian, Department of Chemistry, Center for Research Computing, Center for Drug Discovery, Design, and Delivery (CD4), Southern Methodist University, Dallas, TX 75206, USA.
Sian Xiao, Department of Chemistry, Center for Research Computing, Center for Drug Discovery, Design, and Delivery (CD4), Southern Methodist University, Dallas, TX 75206, USA.
Xi Jiang, Department of Statistical Science, Southern Methodist University, Dallas, TX 75206, USA.
Peng Tao, Department of Chemistry, Center for Research Computing, Center for Drug Discovery, Design, and Delivery (CD4), Southern Methodist University, Dallas, TX 75206, USA.
FUNDING
National Institute of General Medical Sciences of the National Institutes of Health [R15GM122013]. Funding for open access charge: Research reported in this article was supported by the National Institute of General Medical Sciences of the National Institutes of Health [R15GM122013].
Conflict of interest statement. None declared.
REFERENCES
- 1. Wodak S.J., Paci E., Dokholyan N.V., Berezovsky I.N., Horovitz A., Li J., Hilser V.J., Bahar I., Karanicolas J., Stock G.et al.. Allostery in its many disguises: from theory to applications. Structure. 2019; 27:566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Christopoulos A., May L., Avlani V.A., Sexton P.M.. G-protein-coupled receptor allosterism: the promise and the problem(s). Biochem. Soc. Trans. 2004; 32:873–877. [DOI] [PubMed] [Google Scholar]
- 3. De Smet F., Christopoulos A., Carmeliet P.. Allosteric targeting of receptor tyrosine kinases. Nat. Biotech. 2014; 32:1113–1120. [DOI] [PubMed] [Google Scholar]
- 4. Peracchi A., Mozzarelli A.. Exploring and exploiting allostery: models, evolution, and drug targeting. Biochim. Biophys. Acta (BBA) Proteins Proteomics. 2011; 1814:922–933. [DOI] [PubMed] [Google Scholar]
- 5. Wu N., Strömich L., Yaliraki S.N.. Prediction of allosteric sites and signaling: insights from benchmarking datasets. Patterns. 2022; 3:100408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu J., Nussinov R.. Allostery: an overview of its history, concepts, methods, and applications. PLoS Comput. Biol. 2016; 12:e1004966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Panjkovich A., Daura X.. Exploiting protein flexibility to predict the location of allosteric sites. BMC Bioinformatics. 2012; 13:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Laine E., Goncalves C., Karst J.C., Lesnard A., Rault S., Tang W.-J., Malliavin T.E., Ladant D., Blondel A.. Use of allostery to identify inhibitors of calmodulin-induced activation of Bacillus anthracis edema factor. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:11277–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Panjkovich A., Daura X.. PARS: a web server for the prediction of protein allosteric and regulatory sites. Bioinformatics. 2014; 30:1314–1315. [DOI] [PubMed] [Google Scholar]
- 10. Goncearenco A., Mitternacht S., Yong T., Eisenhaber B., Eisenhaber F., Berezovsky I.N.. SPACER: server for predicting allosteric communication and effects of regulation. Nucleic Acids Res. 2013; 41:W266–W272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang W., Lu S., Huang Z., Liu X., Mou L., Luo Y., Zhao Y., Liu Y., Chen Z., Hou T.et al.. Allosite: a method for predicting allosteric sites. Bioinformatics. 2013; 29:2357–2359. [DOI] [PubMed] [Google Scholar]
- 12. Greener J.G., Sternberg M.J.. AlloPred: prediction of allosteric pockets on proteins using normal mode perturbation analysis. BMC Bioinformatics. 2015; 16:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen A. S.-Y., Westwood N.J., Brear P., Rogers G.W., Mavridis L., Mitchell J.B.. A random forest model for predicting allosteric and functional sites on proteins. Mol. Inf. 2016; 35:125–135. [DOI] [PubMed] [Google Scholar]
- 14. Tian H., Jiang X., Tao P.. PASSer: prediction of allosteric sites server. Mach. Learn. Sci. Techn. 2021; 2:035015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xiao S., Tian H., Tao P.. PASSer2. 0: accurate prediction of protein allosteric sites through automated machine learning. Front. Mol. Biosci. 2022; 9:879251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tian H., Xiao S., Jiang X., Tao P.. PASSerRank: prediction of allosteric sites with learning to rank. 2023; arXiv doi:2 February 2023, preprint: not peer reviewedhttps://arxiv.org/abs/2302.01117. [DOI] [PMC free article] [PubMed]
- 17. Burley S.K., Berman H.M., Kleywegt G.J., Markley J.L., Nakamura H., Velankar S.. Protein Data Bank (PDB): the single global macromolecular structure archive. Protein Crystal. 2017; 627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Humphrey W., Dalke A., Schulten K.. VMD: visual molecular dynamics. J. Mol. Graph. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 19. Yuan S., Chan H.S., Hu Z.. Using PyMOL as a platform for computational drug design. Wiley Int. Rev.: Comput. Mol. Sci. 2017; 7:e1298. [Google Scholar]
- 20. Chen T., Guestrin C.. Xgboost: a scalable tree boosting system. Proceedings of the 22nd ACM Sigkdd International Conference on Knowledge Discovery and Data Mining. 2016; 785–794. [Google Scholar]
- 21. Wang M., Zheng D., Ye Z., Gan Q., Li M., Song X., Zhou J., Ma C., Yu L., Gai Y.et al.. Deep graph library: A graph-centric, highly-performant package for graph neural networks. 2019; arXiv doi:3 September 2019, preprint: not peer reviewed 10.48550/arXiv.1909.01315. [DOI]
- 22. Erickson N., Mueller J., Shirkov A., Zhang H., Larroy P., Li M., Smola A.. Autogluon-tabular: robust and accurate automl for structured data. 2020; arXiv doi:13 March 2020, preprint: not peer reviewed 10.48550/arXiv.2003.06505. [DOI]
- 23. Ke G., Meng Q., Finley T., Wang T., Chen W., Ma W., Ye Q., Liu T.-Y.. Lightgbm: A highly efficient gradient boosting decision tree. Adv. Neural Inf. Proc. Syst. 2017; 30:3149–3157. [Google Scholar]
- 24. Hanson R.M., Prilusky J., Renjian Z., Nakane T., Sussman J.L.. JSmol and the next-generation web-based representation of 3D molecular structure as applied to proteopedia. Israel J. Chem. 2013; 53:207–216. [Google Scholar]
- 25. Le Guilloux V., Schmidtke P., Tuffery P.. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 2009; 10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kipf T.N., Welling M.. Semi-supervised classification with graph convolutional networks. 2016; arXiv doi:9 September 2016, preprint: not peer reviewed 10.48550/arXiv.1609.02907. [DOI]
- 27. Xiao S., Verkhivker G.M., Tao P.. Machine learning and protein allostery. Trends Biochem. Sci. 2022; 48:375–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang Z., Zhu L., Cao Y., Wu G., Liu X., Chen Y., Wang Q., Shi T., Zhao Y., Wang Y.et al.. ASD: a comprehensive database of allosteric proteins and modulators. Nucleic Acids Res. 2011; 39:D663–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang W., Wang G., Shen Q., Liu X., Lu S., Geng L., Huang Z., Zhang J.. ASBench: benchmarking sets for allosteric discovery. Bioinformatics. 2015; 31:2598–2600. [DOI] [PubMed] [Google Scholar]
- 30. Zlobin A., Suplatov D., Kopylov K., Švedas V.. CASBench: a benchmarking set of proteins with annotated catalytic and allosteric sites in their structures. Acta Naturae. 2019; 11:74–80. [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao X.-M., Li X., Chen L., Aihara K.. Protein classification with imbalanced data. Proteins: Struct.Funct. Bioinformatics. 2008; 70:1125–1132. [DOI] [PubMed] [Google Scholar]
- 32. Chang C.-Y., Hsu M.-T., Esposito E.X., Tseng Y.J.. Oversampling to overcome overfitting: exploring the relationship between data set composition, molecular descriptors, and predictive modeling methods. J. Chem. Inf. Model. 2013; 53:958–971. [DOI] [PubMed] [Google Scholar]
- 33. Takahashi F., Yamagata D., Ishikawa M., Fukamatsu Y., Ogura Y., Kasahara M., Kiyosue T., Kikuyama M., Wada M., Kataoka H.. AUREOCHROME, a photoreceptor required for photomorphogenesis in stramenopiles. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:19625–19630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tian H., Trozzi F., Zoltowski B.D., Tao P.. Deciphering the allosteric process of the Phaeodactylum tricornutum Aureochrome 1a LOV domain. J. Phys. Chem. B. 2020; 124:8960–8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Faisal S., Badshah S.L., Kubra B., Sharaf M., Emwas A.-H., Jaremko M., Abdalla M.. Identification and inhibition of the druggable allosteric site of SARS-CoV-2 NSP10/NSP16 methyltransferase through computational approaches. Molecules. 2022; 27:5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Phong N.V., Min B.S., Yang S.Y., Kim J.A.. Inhibitory effect of coumarins and isocoumarins isolated from the stems and branches of acer mono maxim. against Escherichia coli β-glucuronidase. Appl. Sci. 2022; 12:10685. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The PASSer web service is freely available at https://passer.smu.edu. The Python scripts to prepare training data is available at https://github.com/smu-tao-group/PASSerRank and https://doi.org/10.5281/zenodo.7818017.