


Abstract
Accurate and fast structure prediction of peptides of less 40 amino acids in aqueous solution has many biological applications, but their conformations are pH- and salt concentration-dependent. In this work, we present PEP-FOLD4 which goes one step beyond many machine-learning approaches, such as AlphaFold2, TrRosetta and RaptorX. Adding the Debye-Hueckel formalism for charged-charged side chain interactions to a Mie formalism for all intramolecular (backbone and side chain) interactions, PEP-FOLD4, based on a coarse-grained representation of the peptides, performs as well as machine-learning methods on well-structured peptides, but displays significant improvements for poly-charged peptides. PEP-FOLD4 is available at http://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD4. This server is free and there is no login requirement.
Graphical Abstract
Graphical Abstract.
INTRODUCTION
The FDA (Food and Drug Administration) classifies a peptide as an amino acid polymer of <40 amino acids. Peptides have diverse biological functions. Several tens of thousands of natural peptides have been identified (1,2), and full genome reanalyses have indicated that probably many more could be expressed (3). Peptides are acting as signaling entities in all domains of life, targeting receptors or interfering with molecular interactions. Families of well known peptides include hormones and their bacterial mimetics (4), antimicrobial peptides for host defence (5), or immunomodulatory peptides with a perspective of vaccine design (6). Peptides also represent a class of candidate therapeutical agents (7), particularly to target protein-protein interactions (8).
In silico assistance to the elucidation of peptide biological activity or the design of peptidic candidate therapeutics requires approaches to predict their 3D structure, in isolation, or in interaction with a receptor. However, peptide structure prediction comes with some specifics: peptides can be highly flexible (9), or can have biological activities in specific solvent conditions. Numerous protocols for peptide structure prediction have been reported so far, some of which are available as online services. Approaches available for online prediction of peptide structure in isolation include PEPStrMod (10), AlphaFold2 (11) or ColabFold (12), APPTest (13), TrRosetta (14), RaptorX (15) and PEP-FOLD (16). It has to be noted that all the servers, except PEPStrMod and PEP-FOLD, are machine-learning approaches. In particular, AlphaFold2/ColabFold, TrRosetta and RaptorX are based on protein data bank (PDB) (17) structures, multiple sequence alignments and specific algorithms to learn the backbone conformations and side chain side chain contacts. Other servers focusing on the prediction of the peptide interaction with a receptor include RosettaFlexPepDock (18), CABS-Dock (19), HPEPDOCK (20) HADDOCK (21), which perform flexible peptide docking, when approaches such as pepATTRACT (22) or ClusPro Peptidock (23) perform rigid docking. However, not all protocols are available online, particularly the latest AlphaFold2 derived ones (e.g. (24)).
All these approaches have been developed for peptides in aqueous solution, using standard conditions - neutral pH, no ionic strength, whereas it could be desirable to consider more exotic conditions, for instance to study peptides of the microbiota (25), or from extremophile organisms (26). Finally, the conformational ensemble of biologically intrinsically disordered peptides (IDPs), such as amyloid-β associated to Alzheimer’s disease and human islet amyloid polypeptide associated to type II diabetes, is difficult to assess as IDPs have flat free energy surfaces, and have different behaviors depending on solvent conditions (27).
Here, we introduce the PEP-FOLD4 server which incorporates the latest improvements to the sOPEP force field. PEP-FOLD first version (28) was based on sOPEP1, tightly derived from the coarse grained OPEP force field (29), and was applicable to linear peptides no longer than 30 amino acids. PEP-FOLD second version (30) introduced the possibility to model peptides cyclized by disulfide bonds. PEP-FOLD third version (16) proposed a faster and improved sampling to generate 3D models to up to 50 amino acids, and considered building structures of peptides in interaction with a receptor. Here, we thoroughly revisit the force field. First, PEP-FOLD4 relies on a new formalism to treat the non-bonded interactions, making it more discriminant to identify relevant conformations among non-native conformations. Second, it embeds a Debye-Hueckel formalism to treat pH conditions and salt concentration variations. To illustrate the prediction performance, we focus on the best five models obtained by PEP-FOLD4, AlphaFold2 and TrRosetta.
MATERIALS AND METHODS
PEP-FOLD principles
A flowchart of PEP-FOLD4 is presented in Figure 1. PEP-FOLD is a fragment-based approach adapted to the prediction of the structure for peptides. Unlike most fragment-based approaches for structure prediction, it does not rely on a description of fragments as combinations of ϕ/ψ angles, but on shape descriptors of fragments of 4 consecutive amino acids (see (31)). A peptide is considered as a series of 4 amino acid fragments, overlapping by 3 amino acids, a process that can be described using Hidden Markov Models (HMM)(see (31)). The best model we identified consists of 27 structural alphabet (SA) states, each associated with a specific distribution of the shape descriptors and a transition matrix of size 27x27 describing the probability that a given state is followed by another one. Given such a model, it is possible to decode a 3D structure as a series of states. To predict the structure from the sequence, PEP-FOLD relies on a two-state process: (i) the prediction of the probabilities of each state at each position in the sequence (SA profile) from an amino acid profile built from a psi-blast (32) search against a collection of sequences, and (ii) the generation of the 3D models from the SA profile. The former relies on a support vector machine (SVM) that has been described previously (33). The latter is itself decomposed in two steps. First, algorithms of the HMM paradigm such as the forward-backtrack algorithm are used to sample the conformational space described by the SA profile (see (16)), and produce series of states or trajectories likely to represent the structure, each describing one conformation in the HMM model space. Second, given a trajectory, prototype fragments associated with each of the states are then assembled rigidly to progressively build the structure of the complete peptide, starting from any amino acid in the sequence (34). Once a complete structure is generated, it is submitted to a Monte Carlo refinement in which we substitute fragments randomly. Both the initial structure generation and the Monte Carlo simulation are driven by the implicit solvent sOPEP force field, which has two specific features. (i) It is a coarse grained force field, with only one bead to represent each side chain but preserving all the backbone atoms except Hα. (ii) Backbone hydrogen bond potential is specific. It does not rely on Van der Waals and electrostatic interactions, but on the introduction of two-body and four-body potentials which may allow hydrogen bond formation and the formation of helix, parallel or anti-parallel β-sheets.
Figure 1.

Flowchart of PEP-FOLD4.
PEP-FOLD4 evolutions
In earlier versions of PEP-FOLD, the sOPEP force field was not accurate enough to correctly rank alone the best models. It was supplemented by tools such as Appolo (35) to perform additional re-ranking. PEP-FOLD4 comes with a new improved force field version which does not require Appolo any longer.
sOPEP is expressed as a sum of local, nonbonded and hydrogen-bond (H-bond) terms:
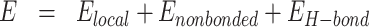 |
(1) |
In PEP-FOLD4, two major improvements are brought to the evaluation of the non-bonded interactions. The first is the replacement of the Van der Waals formalism by a Mie formalism. The second is the use of a Debye-Hueckel formalism to assess the interactions between the charges, which leads to:
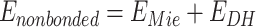 |
(2) |
Mie formulation for non-bonded interactions
The heterogeneity of the bead sizes among side chains and backbone atoms made it difficult to find a good balance between attraction and repulsion using the Van der Waals formulation. PEP-FOLD4 comes with a generalized formulation of the Van der Waals formalism to any positive values for the exponents instead of only 6-12. We now compute the non-bonded interactions between all backbone and side chain particles using the Mie formulation (36):
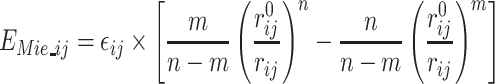 |
(3) |
where εij is the potential depth and 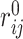 is the position of the potential minimum function of atomic types for i and j. The combination of exponents, n and m, gives the relationship between the position of the potential minimum (r0) and the position where it is zero (gR0):
is the position of the potential minimum function of atomic types for i and j. The combination of exponents, n and m, gives the relationship between the position of the potential minimum (r0) and the position where it is zero (gR0):
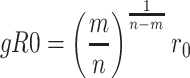 |
(4) |
The reader can refer to (37) for the parameter values.
Introduction of a Debye–Hueckel formalism
To take into account charge variation and pH dependence, we consider a Debye-Hueckel contribution, defined as:
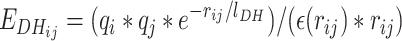 |
(5) |
q i and qj correspond to the charge of particles i and j, j > i + 1, respectively. rij is the distance between the particles, the charges being positioned at the center of the beads representing the side chains, lDH is the Debye length that depends of the ionic strength of the solvent, and ε(rij) is the dielectric constant that depends on the distance between the charges. It is evaluated as:
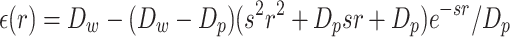 |
(6) |
where Dw is the dielectric constant of water, Dp is the dielectric constant inside a protein, and s is the slope of the sigmoidal function. Note that since charges are associated with particles of heterogeneous sizes, we have considered shifting the energy curve to have energy values compatible with those of the Mie formulation. More details about practical implementation and parameter setup can be found in (38).
WEB SERVER IMPLEMENTATION
Input
The main input consists of the sequence in FASTA format limited to 40 amino acids. Optional parameters are related to
3D model generation: parameters are related to the algorithm used to sample the SA profile, the number (100 or 200) of models generated, the Monte Carlo procedure, and the pseudo-random generator seed. The SA profile sampler can be either the forward-backtrack, or the taboo sampling (see (16) algorithms). For the taboo sampling the size of the fragments to avoid redundancy can vary from 3 to 5. Note that for sequences of <12 amino acids, the taboo sampling is not recommended. For the Monte Carlo simulation, it is possible to invalidate it setting the number of steps to 0, and the temperature can be chosen between 700 K and 370 K. The pseudo-random generator seed gives control over reproducibility: the same seed associated with the same combination of parameters will return the same results. Thus, varying the seed can result in increasing the sampling over different job submissions.
Debye-Hueckel contribution: it is possible to switch on/off its use, and specify the pH (varying from 2 to 13 by 0.5 increments) and the ionic strength in NaCl concentration from 1 to 1000 mM. It is possible to use the zwitterionic forms of the peptides or block their extremities by acetyl and N-methyl. The default pKa values of 3.6, 4.2, 6, 10.5 and 12.5 for ASP, GLU, HIS, LYS and ARG residues can be superseded in a specific manner for each residue in a sequence. This provides a mean to take into account pKa variations due to amino acid local environment and conformations, if necessary using on-line servers (see (38).)
Output
The main output consists of the models generated, clustered and ranked according to the sOPEP energy. The best five models can be explored interactively online using a wrapper around the NGL javascript viewer (39). Various molecular representations, surface representations, and color schemes are proposed, which can be mixed and matched, and the surface transparency can be adjusted. Another output consists of the SA profile that can be downloaded and re-used to generate new series of models (see the input section), skipping the SA profile generation step. Typical PEP-FOLD4 execution times are only a few minutes, depending on server load.
USE-CASES
Well-structured peptides
Three recent articles compared the performance of PEP-FOLD with respect to high performance methods. Using NMR structures and a dataset of 588 peptides between 10 and 40 amino acids, including membrane-associated peptides, soluble peptides and disulfide-rich peptides, it was found that AlphaFold2 performed at least as well as PEP-FOLD version 3 using our 2016 parameters and TrRosetta, without considering pH variations and membrane presence (40).
Next, we compared the predictions of PEP-FOLD4, TrRosetta and AlphaFold2 on (i) 15 peptides between 8 and 35 amino acids and pH from 4.3 to 7 for which a PDB structure is present and (ii) 4 peptides between 11 and 38 amino acids without nuclear magnetic resonance (NMR) structures, but with a topological description in literature. The three methods performed similarly on a total 17 peptides, but TrRosetta and AlphaFold2 failed on two peptides of 10 and 17 amino acids which are described as β-hairpins experimentally(38).
A third article revealed that the predictions of PEP-FOLD4 free of Debye-Hueckel formalism are better, in particular with respect to the prediction of small β-targets, than those of APPTest and RaptorX.(37) As an example of the quality of the PEP-FOLD4 model, we show in Figure 2 the superposition of the models predicted by PEP-FOLD4, AlphaFold2 and TrRosetta on a 14-residue peptide, which showed by NMR experiments a percentage of β-hairpin structure higher than 70% (41). Note that PEP-FOLD relying on a coarse grained model, side chain positioning is just performed to return all-atom models. Side chain conformations are not optimized. They are just set in the most frequent rotameric state observed in protein structures (42), which we found a convenient starting point for peptides. Users are free to re-optimize them using standard side-chain positioning programs.
Figure 2.

Structure predictions of the 14-residue peptide RGKWTYNGITYEGR (PDB: 1j4m). Gray: NMR, green: PEP-FOLD4, orange: TrRosetta, blue: AlphaFold2. Side chains at two positions are depicted to show the structural agreement of the models.
Poly-charged peptides
We recently compared the performances of PEP-FOLD4, TrRosetta and AlphaFold2 on six poly-charged peptides, (EK)15, (EK)5, (H)30, (K)15, (E)15 and (R)25 at pH varying between 3 and 13 (38), and at a salt concentration of 1 mM. The presence of α-helix in these peptides is pH-dependent, which cannot be captured by pH-independent methods such as TrRosetta and AlphaFold2. Circular dichroism (CD) experiments revealed that at pH 7.4, the secondary structure contents of these peptides are dominated by β-turns and random coil, which was faithfully reproduced by PEP-FOLD4. In contrast, both TrRosetta and AlphaFold2 predicted a very high helical content with high confidence (LDDT (local distance difference test) metric >80% for all amino acids). A particularly striking result is that the CD α-helix content of (K)15 and (E)15 change inversely with the pH. This feature was fully reproduced by PEP-FOLD4, and Figure 3 shows the predicted conformations of(E)15 at pH 2.0 and 7.4, and (K)15 at pH 3.6 and 13.
Figure 3.
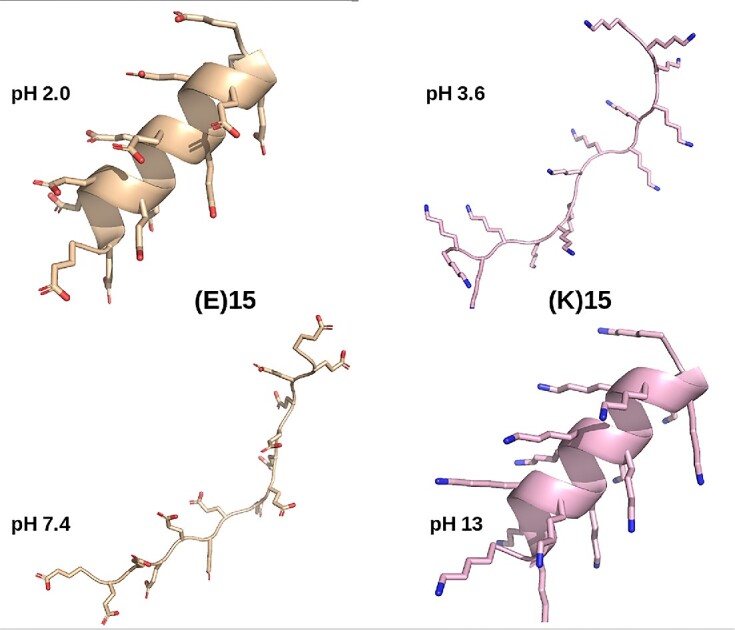
pH-dependent conformations of (E)15 and (K)15 peptides by PEP-FOLD4.
CONCLUSIONS AND PERSPECTIVES
PEP-FOLD4 comes with innovative features to identify relevant conformations among non-native conformations, and the treatment of pH and ionic strength variations. The server interface is, we hope, simple to use and allows online exploration of the generated models. A limitation is the length of the sequences. Indeed, with the recent progress of deep learning for well-structured peptides of more than 40 amino acids, PEP-FOLD4 despite being able to generate native or near-native models by using a coarse grained model and a de novo approach cannot presently generate models as accurate as AlphaFold2 and TrRosetta results. Another limitation is the possibility to grow peptides on the vicinity of a protein receptor, as in PEP-FOLD3. The impact of both Mie and Debye–Hueckel formalisms on such a facility is presently unknown and this aspect is under investigation. Many servers including AlphaFold2 offer this possibility for standard pH and ionic strength. However, PEP-FOLD4 server strong point is undoubtedly its Debye-Hueckel formalism, to the best of our knowledge, a unique feature today.
While the structures of many peptides are resolved by NMR in aqueous solution, other conditions such as 30% trifluoroethanol, sodium dodecyl sulfate micelles, and phosphate buffer are often encountered. We expect to include these conditions in future versions. Also, it has to be noted that treating properly poly-charged peptides is a first step toward predicting the conformational ensemble of IDPs which often contain a high percentage of titratable amino acids. For example, the 16-residue N-terminus of amyloid-β peptide consists of DAEFRHDSGYEVHHQK,(27) the 24-residue histatin 5 peptide consists of DSHAKRHHGYKRKFHEKHHSHRGY (43), and the 24-residue RS peptide consists of residues (RS)8 in its C-terminal (44). As an example, Figure 4 shows that the predicted (RS)8 conformations are diverse and random coil with PEP-FOLD4 (Figure 4A), and lack diversity with TrRosetta (single helix, Figure 4B) and AlphaFold2 (fully extended, Figure 4C). More work is, however, needed to make PEP-FOLD4 suitable for IDPs (44).
Figure 4.

Best five models of the 16-residue (RS)8 peptide at pH 7. (A) PEP-FOLD4, (B) TrRosetta, (C) AlphaFold2.
Overall, PEP-FOLD4 is a first step toward a unified peptide structure prediction approach in varied conditions.
DATA AVAILABILITY
PEP-FOLD4 is available at http://bioserv.rpbs.univparis-diderot.fr/services/PEP-FOLD4. This server is free and there is no login requirement.
ACKNOWLEDGEMENTS
The authors acknowledge financial support from the ‘Initiative d’Excellence’ programs from the French State (Grants ‘IdEx Université de Paris’, ANR-18-IDEX-0001; ‘DYNAMO’, ANR-11-LABX-0011-01, and ANR-11-INBS-0013 (French Institute of Bioinformatics - IFB).
Contributor Information
Julien Rey, Université Paris Cité, CNRS UMR 8251, INSERM U1133, RPBS, Paris, France.
Samuel Murail, Université Paris Cité, CNRS UMR 8251, INSERM U1133, RPBS, Paris, France.
Sjoerd de Vries, Université de Lorraine, CNRS, Inria, LORIA, F-54000, Nancy, France.
Philippe Derreumaux, CNRS, Université Paris Cité, UPR 9080, Laboratoire de Biochimie Théorique, Institut de Biologie Physico-Chimique, Fondation Edmond de Rothschild, 13 rue Pierre et Marie Curie, 75005 Paris, France; Institut Universitaire de France (IUF), 75005 Paris, France.
Pierre Tuffery, Université Paris Cité, CNRS UMR 8251, INSERM U1133, RPBS, Paris, France.
FUNDING
Agence Nationale de la Recherche [ANR-11-INBS- 0013, ANR-11-LABX-0011-01, ANR-18-IDEX-0001]. Funding for open access charge: INSERM [U1133].
Conflict of interest statement. None declared.
REFERENCES
- 1. Fosgerau K., Hoffmann T.. Peptide therapeutics: current status and future directions. Drug Discov. Today. 2015; 20:122–128. [DOI] [PubMed] [Google Scholar]
- 2. Quiroz C., Saavedra Y.B., Armijo-Galdames B., Amado-Hinojosa J., Olivera-Nappa Á., Sanchez-Daza A., Medina-Ortiz D.. Peptipedia: a user-friendly web application and a comprehensive database for peptide research supported by machine learning approach. Database. 2021; 2021:baab055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rey J., Deschavanne P., Tuffery P.. BactPepDB: a database of predicted peptides from a exhaustive survey of complete prokaryote genomes. Database (Oxford). 2014; 2014:bau106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fetissov S.O., Legrand R., Lucas N.. Bacterial protein mimetic of peptide hormone as a new class of protein- based Drugs. Curr. Med. Chem. 2019; 26:546–553. [DOI] [PubMed] [Google Scholar]
- 5. Mookherjee N., Anderson M.A., Haagsman H.P., Davidson D.J.. Antimicrobial host defence peptides: functions and clinical potential. Nat. Rev. Drug Discov. 2020; 19:311–332. [DOI] [PubMed] [Google Scholar]
- 6. Pavlicevic M., Marmiroli N., Maestri E.. Immunomodulatory peptides—A promising source for novel functional food production and drug discovery. Peptides. 2022; 148:170696. [DOI] [PubMed] [Google Scholar]
- 7. Muttenthaler M., King G.F., Adams D.J., Alewood P.F.. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021; 20:309–325. [DOI] [PubMed] [Google Scholar]
- 8. Cabri W., Cantelmi P., Corbisiero D., Fantoni T., Ferrazzano L., Martelli G., Mattellone A., Tolomelli A.. Therapeutic peptides targeting PPI in clinical development: overview, mechanism of action and perspectives. Front. Mol. Biosci. 2021; 8:697586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Apostolopoulos V., Bojarska J., Chai T.-T., Elnagdy S., Kaczmarek K., Matsoukas J., New R., Parang K., Lopez O.P., Parhiz H.et al.. A global review on short peptides: frontiers and perspectives. Molecules. 2021; 26:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh S., Singh H., Tuknait A., Chaudhary K., Singh B., Kumaran S., Raghava G.P.. PEPstrMOD: structure prediction of peptides containing natural, non-natural and modified residues. Biol. Direct. 2015; 10:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mirdita M., Schütze K., Moriwaki Y., Heo L., Ovchinnikov S., Steinegger M.. ColabFold: making protein folding accessible to all. Nat. Methods. 2022; 19:679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Timmons P.B., Hewage C.M.. APPTEST is a novel protocol for the automatic prediction of peptide tertiary structures. Brief. Bioinform. 2021; 22:bbab308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Du Z., Su H., Wang W., Ye L., Wei H., Peng Z., Anishchenko I., Baker D., Yang J.. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021; 16:5634–5651. [DOI] [PubMed] [Google Scholar]
- 15. Xu J., Mcpartlon M., Li J.. Improved protein structure prediction by deep learning irrespective of co-evolution information. Nat. Mach. Intell. 2021; 3:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lamiable A., Thévenet P., Rey J., Vavrusa M., Derreumaux P., Tufféry P.. PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016; 44:W449–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rose P.W., Bi C., Bluhm W.F., Christie C.H., Dimitropoulos D., Dutta S., Green R.K., Goodsell D.S., Prlić A., Quesada M.et al.. The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res. 2012; 41:D475–D482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Raveh B., London N., Schueler-Furman O.. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins: Struct. Funct. Bioinformatics. 2010; 78:2029–2040. [DOI] [PubMed] [Google Scholar]
- 19. Kurcinski M., Badaczewska-Dawid A., Kolinski M., Kolinski A., Kmiecik S.. Flexible docking of peptides to proteins using CABS-dock. Protein Sci. 2020; 29:211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou P., Jin B., Li H., Huang S.-Y.. HPEPDOCK: a web server for blind peptide–protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018; 46:W443–W450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Charitou V., Van Keulen S.C., Bonvin A.M.J.J.. Cyclization and docking protocol for cyclic peptide–protein modeling using HADDOCK2. 4. J. Chem. Theor. Comput. 2022; 18:4027–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Vries S.J., Rey J., Schindler C.E.M., Zacharias M., Tuffery P.. The pepATTRACT web server for blind, large-scale peptide–protein docking. Nucleic Acids Res. 2017; 45:W361–W364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Porter K.A., Xia B., Beglov D., Bohnuud T., Alam N., Schueler-Furman O., Kozakov D.. ClusPro PeptiDock: efficient global docking of peptide recognition motifs using FFT. Bioinformatics. 2017; 33:3299–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johansson-Åkhe I., Wallner B.. Improving peptide-protein docking with AlphaFold-Multimer using forced sampling. Front. Bioinform. 2022; 2:959160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu S., Bekhit A. E.-D.A., Wu Q., Chen M., Liao X., Wang J., Ding Y.. Bioactive peptides and gut microbiota: Candidates for a novel strategy for reduction and control of neurodegenerative diseases. Trends Food Sci. Techno. 2021; 108:164–176. [Google Scholar]
- 26. Shang Z., Winter J.M., Kauffman C.A., Yang I., Fenical W.. Salinipeptins: integrated genomic and chemical approaches reveal unusual D-amino acid-containing ribosomally synthesized and post-translationally modified peptides (RiPPs) from a Great Salt Lake Streptomyces sp. ACS Chem. Biol. 2019; 14:415–425. [DOI] [PubMed] [Google Scholar]
- 27. Nguyen P.H., Ramamoorthy A., Sahoo B.R., Zheng J., Faller P., Straub J.E., Dominguez L., Shea J.-E., Dokholyan N.V., De Simone A.et al.. Amyloid oligomers: A joint experimental/computational perspective on Alzheimer’s disease, Parkinson’s disease, type II diabetes, and amyotrophic lateral sclerosis. Chem. Rev. 2021; 121:2545–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maupetit J., Derreumaux P., Tuffery P.. PEP-FOLD: an online resource for de novo peptide structure prediction. Nucleic Acids Res. 2009; 37:W498–W503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maupetit J., Tuffery P., Derreumaux P.. A coarse-grained protein force field for folding and structure prediction. Proteins: Struct. Funct. Bioinformatics. 2007; 69:394–408. [DOI] [PubMed] [Google Scholar]
- 30. Thevenet P., Shen Y., Maupetit J., Guyon F., Derreumaux P., Tuffery P.. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012; 40:W288–W293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Camproux A.-C., Gautier R., Tuffery P.. A hidden markov model derived structural alphabet for proteins. J. Mol. Biol. 2004; 339:591–605. [DOI] [PubMed] [Google Scholar]
- 32. Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shen Y., Maupetit J., Derreumaux P., Tufféry P.. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J. Chem. Theor. Comput. 2014; 10:4745–4758. [DOI] [PubMed] [Google Scholar]
- 34. Maupetit J., Derreumaux P., Tufféry P.. A fast method for large-scale De Novo peptide and miniprotein structure prediction. J. Comput. Chem. 2010; 31:726–738. [DOI] [PubMed] [Google Scholar]
- 35. Wang Z., Eickholt J., Cheng J.. APOLLO: a quality assessment service for single and multiple protein models. Bioinformatics. 2011; 27:1715–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mie G. Zur kinetischen Theorie der einatomigen Körper. Annalen der Physik. 1903; 316:657–697. [Google Scholar]
- 37. Binette V., Mousseau N., Tuffery P.. A generalized attraction–repulsion potential and revisited fragment library improves PEP-FOLD peptide structure prediction. J. Chem. Theor. Comput. 2022; 18:2720–2736. [DOI] [PubMed] [Google Scholar]
- 38. Tufféry P., Derreumaux P.. A refined pH-dependent coarse-grained model for peptide structure prediction in aqueous solution. Front. Bioinform. 2023; 3: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rose A.S., Hildebrand P.W.. NGL Viewer: a web application for molecular visualization. Nucleic Acids Res. 2015; 43:W576–W579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McDonald E.F., Jones T., Plate L., Meiler J., Gulsevin A.. Benchmarking AlphaFold2 on peptide structure prediction. Structure. 2023; 31:111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pastor M.T., López de la Paz M., Lacroix E., Serrano L., Pérez-Payá E.. Combinatorial approaches: a new tool to search for highly structured β-hairpin peptides. Proc. Natl. Acad. Sci. 2002; 99:614–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tuffery P., Etchebest C., Hazout S., Lavery R.. A new approach to the rapid determination of protein side chain conformations. J. Biomol. struct. dynam. 1991; 8:1267–1289. [DOI] [PubMed] [Google Scholar]
- 43. Brewer D., Hunter H., Lajoie G.. NMR studies of the antimicrobial salivary peptides histatin 3 and histatin 5 in aqueous and nonaqueous solutions. Biochem. Cell Biol. 1998; 76:247–256. [DOI] [PubMed] [Google Scholar]
- 44. Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., De Groot B.L., Grubmüller H., MacKerell A.D. Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2017; 14:71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
PEP-FOLD4 is available at http://bioserv.rpbs.univparis-diderot.fr/services/PEP-FOLD4. This server is free and there is no login requirement.