


Abstract
Quantitative assessment of single cell fluxome is critical for understanding the metabolic heterogeneity in diseases. Unfortunately, laboratory-based single cell fluxomics is currently impractical, and the current computational tools for flux estimation are not designed for single cell-level prediction. Given the well-established link between transcriptomic and metabolomic profiles, leveraging single cell transcriptomics data to predict single cell fluxome is not only feasible but also an urgent task. In this study, we present FLUXestimator, an online platform for predicting metabolic fluxome and variations using single cell or general transcriptomics data of large sample-size. The FLUXestimator webserver implements a recently developed unsupervised approach called single cell flux estimation analysis (scFEA), which uses a new neural network architecture to estimate reaction rates from transcriptomics data. To the best of our knowledge, FLUXestimator is the first web-based tool dedicated to predicting cell-/sample-wise metabolic flux and metabolite variations using transcriptomics data of human, mouse and 15 other common experimental organisms. The FLUXestimator webserver is available at http://scFLUX.org/, and stand-alone tools for local use are available at https://github.com/changwn/scFEA. Our tool provides a new avenue for studying metabolic heterogeneity in diseases and has the potential to facilitate the development of new therapeutic strategies.
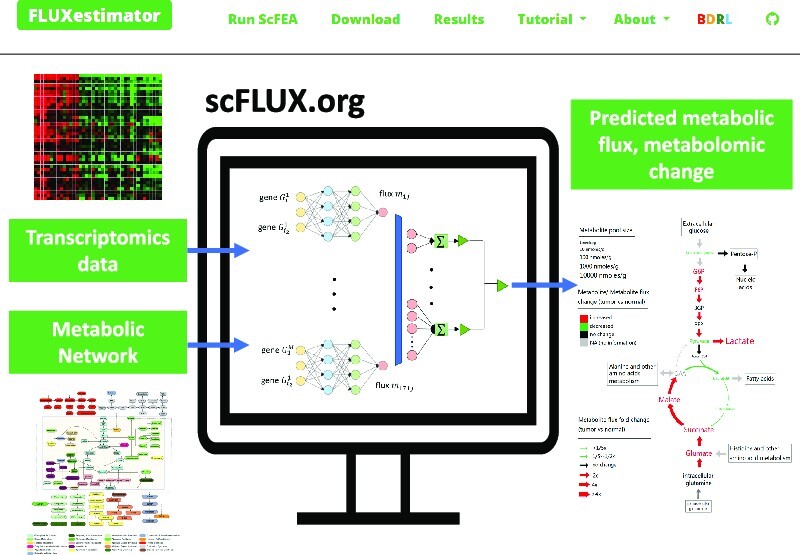
Graphical Abstract
Graphical Abstract.
INTRODUCTION
Metabolic pathways provide essential energy and building blocks for the function of all cells, and dysregulated metabolism is a hallmark of many disease types such as cancer, diabetes, cardiovascular disease, and Alzheimer's disease. Given the pervasive role of metabolism in disease pathology, an accurate and refined characterization of metabolic alterations and inference of their causes or downstream effects could have far-reaching impact on our understanding of disease biology, clinical diagnosis and prevention, and disease management. This includes the potential to: (i) significantly increase our knowledge of metabolic variation, reprogramming and heterogeneity within the disease tissue microenvironment during disease initiation and progression (1,2); (ii) identify new drug targets or novel metabolic biomarkers for early diagnosis or therapeutic optimization (3,4) and (iii) provide diet or nutrition recommendations for patients (5,6).
Numerous computational methods have been proposed to study metabolic activities in different species (7–12). However, despite substantial efforts focused on reconstructing genome-wide metabolic maps and flux analysis, a fundamental question remains un-addressed: how metabolic activities differ among cells of different morphological types, physiological states, tissues or disease backgrounds that have the same genetic constitutions? While transcriptomics data have been utilized to characterize metabolic alterations in diseases (13,14), existing analysis tends to portray the average change of intermixed and heterogeneous cell subpopulations within a given tissue (15–17). Moreover, experimental profiling of metabolomics and fluxomics at single-cell resolution is still in its infancy, particularly for large metabolic pathways. As a result, it is impossible to further study the metabolic heterogeneity and cell-wise flux changes in a complex tissue, where cells are known to rewire their metabolism and energy production in response to varied biochemical conditions (18–21). In light of this gap, we developed FLUXestimator, a webserver that enables users to predict cell-/sample-wise metabolic flux and variation using either single-cell RNA-seq (scRNA-seq) data or large-scale (general) transcriptomics data.
We have recently developed a computational method, called single-cell Flux Estimation Analysis (scFEA), which enables the estimation of cell-wise metabolic fluxome (i.e. flux distribution of the whole metabolic network) by using scRNA-seq data (22). This method incorporates an unsupervised model to estimate cell- or sample-wise flux distribution of a metabolic network, utilizing neural networks to approximate the reaction rate of each metabolic reaction based on gene expression data. In addition, scFEA employs a quadric loss to regularize the flux balance of the predicted in-flux and out-flux for each intermediate metabolite. Noted, scFEA optimizes the flux balance through all cells and enables certain levels of imbalance at individual cell level. Currently, scFEA is available as a Python package, which requires a GPU environment, pre-selection of metabolic network, species, and hyperparameters.
To provide a more accessible and user-friendly tool for single cell- or sample-wise metabolic flux estimation analysis using transcriptomics data, we developed FLUXestimator. This webserver offers an optimized and coding-free environment that implements the scFEA analysis pipeline, reducing the programming and computational resource burden for public users. Fluxer is an existing webserver with a similar scope to FLUXestimator (23). However, Fluxer is designed for prokaryotes genomics data, focused on visualization, and based on strict Flux Balance Analysis (FBA) model, which cannot predict sample-wise and disease context specific flux distribution. Other methods with a similar scope to FLUXestimator include COMPASS (24) and scFBA (25). However, both methods are only able to predict flux for groups of cells or samples, require high computational cost, and do not have a webserver release, which makes them significantly different from FLUXestimator. In summary, FLUXestimator offers a wider range of capabilities and a more generalized scope, making it a valuable resource for researchers in the field of metabolic flux estimation analysis. Specifically, FLUXestimator, together with the original scFEA pipeline, provides the following capabilities:
FLUXestimator is a one-stop server that allows users to upload their own data, select species and pre-constructed metabolic networks, and adjust algorithm parameters via a user-friendly interface. With the unsupervised scFEA pipeline, FLUXestimator enables sample-wise flux estimation in a single platform.
FLUXestimator takes the input of a scRNA-seq or transcriptomics data of a large sample size to predict cell-wise or sample-wise metabolic flux. FLUXestimator accepts varied input formats and gene IDs for different species, making it a versatile tool for many research applications.
FLUXestimator provides access to 15 manually curated metabolic networks for human and mouse collected by integrating RECON3D, KEGG and literature data. These include two global metabolic maps, four central metabolic networks, four neural transmitter synthesis pathways, lipids, branched chain amino acids, methionine, and iron ion metabolic pathways, and MHC class I antigen presentation pathways.
FLUXestimator provides the global metabolic map curated by using KEGG modules for human, mouse and 15 common experimental organisms.
The outputs of FLUXestimator include (i) predicted flux distribution and (ii) levels of accumulation or depletion of intermediate metabolites. To help users interpret the outputs, FLUXestimator provides a tutorial on how to analyze and interpret the results.
MATERIALS AND METHODS
Reconstruction and representation of metabolic networks for flux estimation
The whole metabolic network in human, mouse, and other common experimental organisms have been extensively studied and well annotated in databases such as the Kyoto Encyclopedia of Genes and Genomes (KEGG) (26) and Recon3D (27). However, to optimize the network representation and topological structure for fluxome estimation analysis, several issues need to be considered: (i) the constraints of flux distribution may vary depending on the optimization goal or computational assumption, such as the tolerance of the imbalance of different intermediate metabolites, (ii) the network complexity needs to be reduced to enable computational feasibility and (iii) small molecules such as H2O, CO2, and co-factors, whose concentrations are much larger than the scale of reaction rate, should be excluded from flux balance consideration.
FLUXestimator provides a set of curated metabolic pathways for human, mouse, and 15 other major experimental organisms. In FLUXestimator, a metabolic network is represented by a directed factor graph. This graph captures the topological and biochemical features of a pathway, with each reaction being represented as a variable and each metabolite as a factor (Figure 1A). Directed edges from a factor to a variable and from a variable to a factor represent the input and output of a reaction, respectively. The stoichiometric coefficients are assigned as attributes on each edge. The flux rate of each reaction can be viewed as a value or a function on each variable, and the flux balance or quasi-flux balance under steady state can be formulated as the difference between the total in-flux and the total out-flux, which should be zero or a small value close to zero.
Figure 1.

(A) Reconstructed M171 metabolic map of human and mouse. Modules belonging to different metabolic pathways (or termed super modules) are highlighted by different colors. Modules of metabolisms are presented in the central yellow block while biosynthesis modules are presented outside. Glycans are labeled as KEGG G-IDs. Intermediate metabolites in M171 typically have more than one input or output, but a few important intermediate metabolites with only one input or output were kept. (B) Chain-shaped reactions are merged into reaction modules. (C) Boundary reactions that have multiple outputs (or inputs) could be merged into one module. The metabolites of the products or inputs of boundary reactions could be merged into one-factor node that represents boundary metabolites. (D) Important intermediate metabolites were kept in the network reconstruction.
FLUXestimator provides 16 manually curated metabolic (sub)networks for human and mouse, including the reconstructed central metabolic map, namely M171 and M171_NAD, one KEGG global metabolic map, and 13 smaller and more focused metabolic sub-networks (Table 1). These networks were reconstructed using KEGG and Recon3d reactions, which encompassed 19 131 reactions and 5607 metabolites, as well as 619 transporters from Transporter Classification Database (28–30). We note that KEGG and Recon3d lack detailed annotations of biosynthesis of large molecules and metabolism of small molecules such as metal ions. To address this, we manually collected this information by literature mining and curated metabolite IDs from different databases. We also excluded 273 common molecules such as H2O, H+, and cofactors, from the intermediate metabolites, as the flux balance assumption may not be valid for these metabolite (22). Noted, network reconstruction needs to leverage the network topology, computational feasibility, and importance of metabolites and reactions. Thus, we utilized the concept of metabolic module to further reduce the complexity of the metabolic network. The concept of metabolic module has been utilized by KEGG (26). Under steady state, the metabolic flow over a reaction chain without an intermediate in-/out-branch has a unique solution. Such reaction chain can be reduced into one reaction module. In our network reduction, we (i) tightly followed the no-branch constraint (Figure 1B), (ii) generalized the definition of a metabolic module by extending the single input- (or output-) end condition to be a group of metabolites if the module is at the boundary of the system (Figure 1C), (iii) kept important intermediate metabolites (Figure 1D), and (iv) ensured a very small overlaps of enzymes shared by different modules.
Table 1.
Reconstructed metabolic networks in FLUXestimator
| Network name | Network description | #Modules | #Enzymes | #Intermediate metabolites | #Genes (in human, mouse) |
|---|---|---|---|---|---|
| M171 | Central Metabolic Network | 171 | 451 | 70 | 663, 719 |
| M171_NAD | M171 + Redox balance of NAD+/NADH | 172 | 464 | 71 | 683, 740 |
| GlucoseGlutamine SubcellularLocalization (GGSL) | Glycolysis, TCA cycle, glutamine, and glutathione metabolic network in the resolution of subcellular localization. | 41 | 241 | 37 | 254, 237 |
| GlucoseTCAcycle | Glycolysis and TCA cycle | 15 | 45 | 12 | 65, 66 |
| GlucoseGlutaminolysis | Glycolysis, TCA cycle, and glutaminolysis pathways | 23 | 98 | 17 | 132, 134 |
| GlucoseGlutamine | General glucose and general glutamine metabolic pathways | 27 | 146 | 17 | 165, 176 |
| Branched Chain Amino Acids (BCAA) | Branched chain amino acids metabolic pathways | 14 | 52 | 6 | 60, 64 |
| Acetylcholine | Acetylcholine biosynthesis and metabolism | 15 | 30 | 6 | 80, 86 |
| Dopamine | Dopamine biosynthesis and metabolism | 9 | 30 | 5 | 23, 24 |
| Histamine | Histamine biosynthesis and metabolism | 6 | 18 | 3 | 23, 24 |
| Serotonin | Serotonin biosynthesis and metabolism | 8 | 18 | 4 | 24, 26 |
| IronIon | Sub-cellular localization specific metabolic network of iron ion | 15 | 27 | 8 | 141, 152 |
| Methionine Glutathione Folic Acid (MGF) | Methionine, DNA methylation and related metabolism | 8 | 72 | 5 | 98, 91 |
| Lipid metabolism | Metabolism and biosynthesis of lipids | 112 | 165 | 93 | 320, 334 |
| MHC Class I Antigen Presentation (MHC-I) | MHC Class I antigen presentation pathway | 9 | 9 reaction steps | 8 | 325, 302 |
Among the 16 curated networks, M171 covers most parts of the central metabolic network of human and mouse. To ensure a comprehensive coverage of the global metabolic map, we collected reactions from KEGG and Recon3d, Transporter Classification Database, and additional literature data (26,31). The final metabolic map covers the metabolism, transport, and biosynthesis of carbohydrates, amino acids, fatty acids and lipids, glycan, nucleic acids and other co-factors in human and mouse, including 663 human genes (and 719 mouse genes) of 451 enzymes, 116 transporters, 1471 reactions, and 1561 metabolites. Figure 1 illustrates the M171 network after network reduction, which includes 171 connected reaction modules of 22 super modules that have 66 intermediate substrates. Here, each super module is a manually defined group of modules with a similar function (See Supplementary Table S1). It is worth noting that the size of M171 is much smaller than the whole set of human and mouse metabolic reactions in KEGG and Recon3d since it only covers the connected reactions in the central metabolic pathways. Some pathways like fatty acids biosynthesis and metabolism contain highly connected reactions, but they connect to the central metabolic map only by a few important modules, such as 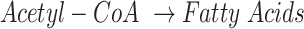 for fatty acids biosynthesis. For such pathways, we (i) reduce the whole pathway as a boundary (or intermediate) module in M171 to reduce computational cost and (ii) reconstruct them into a specific sub-network if such networks are important. FLUXestimator also provides a M171_NAD network by including the biosynthesis of
for fatty acids biosynthesis. For such pathways, we (i) reduce the whole pathway as a boundary (or intermediate) module in M171 to reduce computational cost and (ii) reconstruct them into a specific sub-network if such networks are important. FLUXestimator also provides a M171_NAD network by including the biosynthesis of 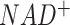 and flux balance of
and flux balance of 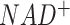 and
and 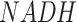 (see details in Supplementary Information).
(see details in Supplementary Information).
As KEGG directly offers module-based representation of genome scale metabolic network for different species, we adopted, curated, and annotated the network from KEGG modules for human, mouse, and 15 other experimental organisms (Table 2). Compared to the M171 network, the KEGG module network omits more branches and contains multiple disconnected subnetworks.
Table 2.
KEGG module-based network reconstruction for selected experimental organisms
| Species | KEGG organism code | #Modules | #Sub-networks | #Intermediate metabolites | #Genes |
|---|---|---|---|---|---|
| Homo sapiens | hsa | 100 | 10 | 133 | 570 |
| Mus musculus | mmu | 100 | 10 | 134 | 569 |
| Ciona robusta | cin | 59 | 6 | 82 | 222 |
| Zea mays | zma | 86 | 4 | 104 | 1093 |
| Danio rerio | dre | 95 | 10 | 126 | 577 |
| Gallus gallus | gga | 82 | 6 | 112 | 402 |
| Xenopus tropicalis | xtr | 94 | 10 | 126 | 502 |
| Rattus norvegicus | rno | 99 | 10 | 132 | 581 |
| Escherichia coli K-12 | eco | 84 | 7 | 102 | 378 |
| Bacillus subtilis subsp. subtilis str. 168 | bsu | 63 | 4 | 85 | 258 |
| Pseudomonas fluorescens SBW25 | pfs | 66 | 7 | 88 | 345 |
| Arabidopsis thaliana | ath | 86 | 9 | 105 | 581 |
| Azotobacter vinelandii DJ | avn | 65 | 8 | 86 | 347 |
| Synechocystis sp. PCC 6803 | syn | 51 | 4 | 64 | 199 |
| Methanococcus voltae A3 | mvo | 22 | 3 | 38 | 137 |
| Streptomyces coelicolor A3(2) | sco | 64 | 7 | 82 | 380 |
| Methanosarcina acetivorans C2A | mac | 42 | 2 | 51 | 216 |
In addition to these two whole metabolic maps, we also manually curated 13 subnetworks for human and mouse, which include (i) four networks of glucose and glutamine metabolism focusing on carbon source and energy metabolism at different resolutions, (ii) four networks of neuron transmitters that support the analysis of nervous systems, (iii) two amino acids centric networks focusing on branched chain amino acids and methionine metabolism, (iv) lipids metabolism and biosynthesis pathway, (v) subcellular localization specific iron ion metabolism, which is the largest metabolic network of metal ion and (vi) MHC class I antigen presentation network (Table 1). In our current network reconstruction, reaction directions are fixed, and subcellular localization of enzymes were considered in a few networks. All networks consider elemental balance of the flux of carbon-based molecules while M171_NAD also considers the balance of 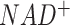 and
and 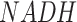 . Detailed biological characteristics and information of the M171, M171_NAD, KEGG modules, and 13 sub-networks can be found in the Supplementary Information, Supplementary Figures S1–S11, and Supplementary Tables S1 and S2. Description of network is also provided on the main page of FLUXestimator when the networks were selected for analysis.
. Detailed biological characteristics and information of the M171, M171_NAD, KEGG modules, and 13 sub-networks can be found in the Supplementary Information, Supplementary Figures S1–S11, and Supplementary Tables S1 and S2. Description of network is also provided on the main page of FLUXestimator when the networks were selected for analysis.
On the FLUXestimator website, users can choose to analyze specific metabolic networks or download the network annotations. As each module contains highly dependent metabolic reactions, they could also serve as analysis units for pathway or gene set enrichment analysis. To facilitate this, we provide the gene information of each reconstructed module in .gmt format that can be directly implemented with gene set or single-sample gene set enrichment analysis (GSEA or ssGSEA) (32,33).
Method overview of the scFEA flux estimation pipeline
FLUXestimator is webserver version of scFEA (22), a method we recently developed to estimate cell-/sample- wise metabolic fluxome of a predefined metabolic network using scRNA-seq or tissue transcriptomics data (Figure 2A). scFEA utilizes a graph neural network architecture to approximate the non-linear dependency between the metabolic flux of each module and the expression of genes involved in the module. Specifically, two computational assumptions were utilized by scFEA (Figure 2B): (1) the flux rate of each metabolic module can be modelled as a neural network of the genes involved in the module, such as enzymes and transporters, and (2) the imbalance between the predicted in-flux and out-flux for intermediate metabolites should be minimized, i.e. steady state flux balance assumption. The primary output of scFEA is the predicted cell- or sample-wise metabolic flux (Figure 2C), and the metabolomic changes of each intermediate metabolite in each sample could be estimated as the difference between the predicted influx and outflux of the metabolite. Additionally, we have developed downstream functions to compute (i) the subset of cells or samples having distinct variations of certain metabolic modules and (ii) the impact of each gene on metabolic fluxome in each sample (22,34), which are detailed on the FLUXestimator tutorial page. For a more in-depth understanding of the model and computational considerations of scFEA, please refer to Supplementary Information, Supplementary Figure S12, and the original scFEA paper (22).
Figure 2.

The computational pipeline of scFEA. (A) The input of the scFEA pipeline includes a predefined metabolic network reconstructed as a directed factor graph and transcriptomics data with a substantial number of samples. (B) The computational model of scFEA assumes that the non-linear dependency between the reaction rate of each reaction module and gene expression can be modeled by a neural network and that the difference between the influx and outflux of each intermediate metabolite should be minimized. (C) The outputs of scFEA include predicted sample-wise metabolic flux and metabolomic changes of each intermediate metabolite. The metabolite pool size shown in the figure was exacted from HMDB (37).
Compared to existing methods like flux balance analysis or enrichment analysis-based approaches, scFEA has distinct advantages. Firstly, it models the non-linear dependency between gene expression and metabolic flux using neural network-based non-linear functions. Secondly, it assesses flux and metabolomic changes in each individual cell or sample. Thirdly, scFEA accounts for the biochemical conditions that can vary across samples or cells and allows a certain level of flux imbalance for intermediate metabolites. As a result, scFEA is more suitable for characterizing metabolic heterogeneity and variations among a group of cells or samples. Like FBA-based methods, scFEA does not consider kinetics but follows the mass balance constraint (35). For a more detailed discussion of the comparison between scFEA and other methods, please refer to Supplementary Information and the original scFEA paper (22).
Training information: scFEA is an unsupervised method based on flux balance assumptions, which does not require training data. Method validations, quantitative evaluations, and tuning of hyperparameters are detailed in Supplementary Information and the original scFEA paper (22).
FLUXestimator web server
FLUXestimator is a user-friendly web server that allows users to conduct cell-/sample-wise flux estimation analysis using scFEA in a coding free environment. As illustrated in Figure 3, FLUXestimator takes in single cell or tissue transcriptomics data, along with a user selected metabolic network, and carries out data preprocessing and flux estimation analysis. It then generates outputs including predicted cell-/sample-wise metabolic flux and metabolomic changes, as well as result annotation files. The web server implementation is described below, and further details on its implementation can be found in the Supplementary Information.
Figure 3.

Flowchart of the FLUXestimator webserver. The inputs, analysis pipeline and outputs are depicted in green, blue and yellow blocks, respectively.
Front-end
The FLUXestimator web-based server is implemented in Python using the Django framework. FLUXestimator utilizes the SQLite database for storage and retrieval of requested input or output data. The Nginx HTTP server is utilized as a secure application gateway, which accelerates data uploading, serves as a reverse proxy, and provides a caching mechanism. The python program of FLUXestimator is deployed via the uWSGI server and the program communicates with uWSGI by the WSGI spec. Here the uWSGI server enables efficient management and resource allocation for multiple processes. The website interface was built using the Bootstrap framework, the jQuery JavaScript library, and extension packages.
Back-end
Data input and processing are conducted by using Pandas, NumPy and Pyreadr. Data preprocessing consists of three major steps: (i) an evaluation step that checks if the input file is in a correct format. Warnings will be returned if the data format does not meet the requirement of FLUXestimator; (ii) data normalization and imputation step by using MAGIC (36) and (iii) passing of the processed expression data and user selected metabolic network to the flux estimator.
The flux estimation procedure is conducted by using PyTorch, Pandas and NumPy. First, a factor graph model of each metabolic network is created, where each module is a variable, and each intermediate metabolite is a factor. Parallel three-layer neural networks were constructed to model the non-linear dependency between gene expression involved in the modules and their flux rates. Note that a separate neural network is constructed for each module. The loss function is created based on the in-/out-flux of each intermediate metabolite and minimized by using the Adam optimizer, which is the most efficient stochastic optimization approach in Pytorch. The loss curves are drawn by using Matplotlib and shown on the Results page (Figure 4).
Figure 4.

A screenshot of the results page of FLUXestimator. At the top of the page, the task ID, running time, and a URL of the result download page are displayed. Users could copy or email the result page using this information. The figure in the middle of the page shows the convergence of loss terms used in scFEA (see details in Supplementary Information). The x-axis and y-axis represented the number of epochs and the level of loss. The ‘total’, ‘balance’, ‘negative’, ‘cellVar’ and ‘moduleVar’ represent the total loss, and individual loss in terms of flux balance, non-negative, coherence between predicted flux and total gene expression level of each super module, and the relative scale of flux, respectively. The table at the bottom includes the input data, two output files, and two annotation files that could be directly downloaded by clicking the file name.
Job management
FLUXestimator manages asynchronous tasks by using Celery via a distributed task queue. Users can run multiple analysis tasks simultaneously. A unique task ID and link will be generated when a task is submitted, by which the user can track the progress of the analysis task. When the task is completed, the user can use the link to access, download or share the analysis results. Anyone with the task ID (or link) can access the results. Analysis results will be stored on the server for at least one year. The list of user's tasks is implemented by using a cookie mechanism with a consent form provided on the web page. If the cookie is enabled by a user, the Results page will list previous tasks submitted by this user on the same browser.
Browser compatibility
FLUXestimator has been tested on major modern browsers including Google Chrome, Mozilla Firefox, Safari and Microsoft Edge.
Running time
For a data of 1000 samples, the running time for a subnetwork is about 5–10 min and the running time for the M171 or KEGG network is about 20–30 min. Longer analysis time may be needed if multiple tasks have been submitted.
RESULTS
Server input
The FLUXestimator web server requires two inputs: (i) a transcriptomics data set of at least 25 samples for subnetwork analysis or at least 100 samples for global network analysis (see details in Supplementary Information) and (ii) a species-specific metabolic network and analysis parameters selected by users. The webserver currently hosts the central metabolic maps (M171, M171_NAD and KEGG) and 13 specific metabolic sub-networks of human and mouse, and KEGG central metabolic map for 15 other species. Users could select among the species and the metabolic networks from the boxes on the left-hand side of the home page. The input is a scRNA-seq or general RNA-Seq dataset that should have genes on its rows, and samples on its columns. We recommend using TPM (or CPM/FPKM) normalized data. FLUXestimator accepts comma-(.csv), space-(.txt), or tab-(.txt) delimited input files in a matrix format that contain row/column names. Row names can be gene symbol or Ensembl gene ID for human and mouse, and KEGG ID or Gene ID for other species. For a large data set, we recommend users upload only the gene expression data of the metabolic genes provided on the download page of FLUXestimator, as other genes will not be used in the flux estimation. Notably, the neural network-based formulation of FLUXestimator allows for the flexibility that expression values of some metabolic genes may be missing from the input data.
FLUXestimator provides two pre-processing options for the input transcriptomics dataset: (i) Imputation. If the input transcriptomics data, such as an scRNA-seq data, is highly sparse, an imputation procedure is recommended. The default imputation method is MAGIC (36). (ii) Normalization. Four options are provided by FLUXestimator: (i) no normalization, (ii) log transformation, namely log(x + 1), where x represents the original input expression matrix, (iii) CPM normalization, and (iv) log transformation of CPM normalized values, namely, log(CPM + 1). Users need to decide whether and how these two procedures will be performed by specifying two relevant hyperparameters on the running page of FLUXestimator.
The input transcriptomics data will be checked by a format validator, and a job will be submitted only when the input file meets the requirements of FLUXestimator. To help users submit their tasks correctly, FLUXestimator provides sample input files on the home page.
Server output
Once an analysis task is completed, users can obtain the output files of the job on its Results page (Figure 4). FLUXestimator provides four downloadable results for each task:
A .csv file containing the predicted cell- or sample-wise metabolic flux. The file has modules on its rows, and single cells or samples on its columns, and each entry is predicted flux rate of the metabolic module in the corresponding cell or sample.
A .csv file containing the imputed cell- or sample-wise metabolomics change. The file has metabolites on its rows, and cells or samples on its columns, and each entry is an imputed metabolomics change of an intermediate metabolite in the selected network in the corresponding cell or sample. Because FLUXestimator does not require stringent flux balance condition, sample-wise metabolomics change of each intermediate metabolite could be predicted by the difference between the predicted influx and outflux of the metabolite in each sample. A consistent negative and positive value of the predicted metabolomics change in a group of cells or samples suggests that the metabolite tends to be depleted or accumulated in the sample group.
A .csv file containing module information, where each row contains the detailed information of the reactions and metabolic compounds involved in each module. These modules are the ones involved in the user defined metabolic network.
A .gmt file containing the module-gene information, where each row contains the gene symbols of the genes in each module of the analysed metabolic network. These genes are the ones used in the flux estimation process.
Users can also view or download the convergence curves of the optimization process, for both the total loss, as well as the four individual loss terms in the optimization function. The predicted cell- or sample-wise metabolic flux can be used for downstream analyses, for which a number of analysis tools are provided by the FLUXestimator website under Tutorial/Package tutorial section. The estimated flux matrix could be integrated into the standard Seurat analysis pipeline for comparative analysis, dimensional reduction, clustering, and visualization (37). In addition, FLUXestimator provides functions to assess levels of accumulation or depletion of metabolites and detect subsets of cells or samples that have distinct variation of certain metabolic modules.
Case study
We have previously validated scFEA using our in-house and public matched scRNA-seq and metabolomics data and demonstrated its accuracy in predicting cell- or sample-wise flux and metabolomics changes. Our findings showed that scFEA outperforms pathway-enrichment based methods for metabolic pathway activity prediction (22) (see Supplementary Information). We first validated the robustness of FLUXestimator by evaluating (i) the predicted flux of overlapped reaction modules from different networks and (ii) the predicted flux with respect to different normalization methods of input data on selected data. Our analyses demonstrated that FLUXestimator achieved high correlations of the predicted flux of overlapped reaction modules from different networks and robust predictions with respect to different normalization approaches of the input data (see details in Supplementary Information andSupplementary Figure S13 and S14).
We further demonstrated the utility of FLUXestimator on (i) scRNA-seq datasets collected from human and mouse cancer microenvironment and (ii) ROSMAP single nucleus RNA-seq (snRNA-seq) data collected from brain tissues of Alzheimer's disease (AD) patients and healthy donors (38).
Cancer data
We applied FLUXestimator to scRNA-seq data collected from cancer microenvironments that contains cancer, myeloid and T cells, including eight human and two orthotopic mouse data sets, for flux estimation of the Glucose-Glutamine network (see details in Supplementary Information). Our analysis identified that: (i) cancer cells consistently have the highest glucose metabolic rate, including lactate production, TCA cycle, and nucleotide and serine biosynthesis, followed by myeloid cell and then T cells, in most human cancer and injected mouse tumor tissues analyzed (Supplementary Figure S15A,B); (ii) the rates of the total glucose consumption strongly correlate with the rates of proliferation (Supplementary Figure S15C) and (iii) the glycolytic flux distributions in human cancer and injected mouse tumor cells are considerably different, particularly in terms of the fractions into nucleotide and serine biosynthesis, which matches existing knowledge in (i) the roles played by serine in transplant rejection [9] and (ii) the prevalently increased nucleotide biosynthesis in proliferating cancer cells [10]. Differential test of predicted flux is conducted by using non-parametric Mann–Whitney test and P < 0.01 was utilized as a significant cutoff.
ROSMAP AD data
We also applied FLUXestimator on ROSMAP snRNA-seq data to predict AD-specific metabolic variations by using the M171 network. We have identified that the metabolic flux in neuron cells of reaction modules in glucose and amino acid metabolism is consistently higher than other cell types in the central nervous system, suggesting that metabolic activity is higher in neuron cells than in other brain cell types. We further focused on the metabolomic changes predicted by FLUXestimator. Supplementary Table S3 lists 14 metabolites whose concentrations have the largest distinctions for neuron cells from AD and healthy control brain. Among them, increased glycolytic substrates and GABA, and decreased glucose, nucleic acids and branched chain amino acids (valine, leucine and isoleucine) have been reported (39,40), while aspartate, serine and methionine may serve as new biomarkers (22).
DISCUSSION
In this paper, we present FLUXestimator, the first web server for cell- or sample-wise metabolic flux estimation using single cell or general transcriptomics data. The key methodology behind this server is based on our previous works on factor graph representation of a complex metabolic network, as well as a graph neural network-based solver for fluxome estimation (22). FLUXestimator provides an end-to-end prediction and enables the interrogation of the flux rate of metabolic modules and concentration changes of metabolites, which can be directly utilized to understand possible metabolic reprogramming events and guide targeted metabolomics experiments. We anticipate the applications of FLUXestimator could increase our understanding in (i) key metabolic reprogramming events and causes and (ii) the impact of metabolic abnormalities to other biological characteristics, which together will contribute to precision medicine such as biomarker screening and drug target prediction.
However, in terms of characterizing the context specific metabolic activities, there are still a few unsolved challenges. A complex tissue microenvironment may be constituted by cells of different metabolic abnormalities, heterogeneous metabolic networks, varied preferences, and dependencies (41–45). In future work, we will enable the reconstruction of context specific metabolic networks and modules, especially disease, tissue, and cell type specific ones, to maximize the discoveries of hidden and dynamic relationships among the metabolic units under different biological conditions. In addition, recent evidence suggests that the direction of certain reversible reactions may not be constant for cells within one disease microenvironment, which represents one way for the cells to increase their fitness level by reprogramming the metabolic exchange mechanisms under a highly perturbed environment (46). Hence, a second future direction is to enable the assessment of sample-wise directions of reversible reactions and inter-cell metabolic exchange or competition by using single cell data. We will also extend flux estimation capability for other omics data types, such as proteomics or metabolomics data. The M171_NAD network considers the balance of redox molecule 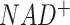 and
and 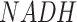 . Another future direction is to extend the flux estimation method by allowing the implementation of more biochemical constraints, such as redox balance, pH balance or energy balance, or imbalance of certain metabolites. As scFEA utilizes a quadratic loss function for flux balance, other balance or imbalance condition can be easily implemented as additional quadratic functions. A few newly developed analysis features, including a perturbation analysis to determine the contribution of each gene to each flux, and newly curated metabolic modules including methionine and copper ion metabolic pathways, are currently available in the stand-alone version of FLUXestimator. Such features will be updated to FLUXestimator web server after thorough validations have been conducted.
. Another future direction is to extend the flux estimation method by allowing the implementation of more biochemical constraints, such as redox balance, pH balance or energy balance, or imbalance of certain metabolites. As scFEA utilizes a quadratic loss function for flux balance, other balance or imbalance condition can be easily implemented as additional quadratic functions. A few newly developed analysis features, including a perturbation analysis to determine the contribution of each gene to each flux, and newly curated metabolic modules including methionine and copper ion metabolic pathways, are currently available in the stand-alone version of FLUXestimator. Such features will be updated to FLUXestimator web server after thorough validations have been conducted.
DATA AVAILABILITY
FLUXestimator is available as a web server at http://scFLUX.org/. The stand-alone tools to run the functions of FLUXestimator on a local machine are available at the GitHub repository (https://github.com/changwn/scFEA).
Supplementary Material
ACKNOWLEDGEMENTS
C.Z and S.C want to thank Dr Ying Xu and Dr Xiongbin Lu for their constructive suggestions and advice on this work.
Contributor Information
Zixuan Zhang, College of Software, College of Computer Science and Technology, Jilin University, Changchun 130012, China; Department of Medical and Molecular Genetics and Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Haiqi Zhu, Department of Medical and Molecular Genetics and Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA; Department of Computer Sciences, Indiana University, Bloomington, IN 47405, USA.
Pengtao Dang, Department of Medical and Molecular Genetics and Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA; Department of Electric Computer Engineering, Purdue University, Indianapolis, IN 46202, USA.
Jia Wang, Department of Medical and Molecular Genetics and Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA; Department of Computer Sciences, Indiana University, Bloomington, IN 47405, USA.
Wennan Chang, Department of Medical and Molecular Genetics and Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Xiao Wang, Department of Medical and Molecular Genetics and Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA; Department of Computer Sciences, Indiana University, Bloomington, IN 47405, USA.
Norah Alghamdi, Department of Medical and Molecular Genetics and Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Alex Lu, Department of Medical and Molecular Genetics and Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Yong Zang, Department of Medical and Molecular Genetics and Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA; Department of Biostatistics and Health Data Science, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Wenzhuo Wu, Department of Industrial Engineering, Purdue University, West Lafayette, IN 47907, USA.
Yijie Wang, Department of Computer Sciences, Indiana University, Bloomington, IN 47405, USA.
Yu Zhang, College of Software, College of Computer Science and Technology, Jilin University, Changchun 130012, China; Department of Medical and Molecular Genetics and Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Sha Cao, Department of Medical and Molecular Genetics and Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA; Department of Biostatistics and Health Data Science, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Chi Zhang, Department of Medical and Molecular Genetics and Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NSF [IIBR 2047631, IIS 2145314]; American Cancer Society [RSG-22-062-01-MM]. Funding for open access charge: American Cancer Society [RSG-22-062-01-MM].
Conflict of interest statement. None declared.
REFERENCES
- 1. Pavlova N.N., Thompson C.B.. The emerging hallmarks of cancer metabolism. Cell Metab. 2016; 23:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mattson M.P., Arumugam T.V.. Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell Metab. 2018; 27:1176–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jensen M.K., Bertoia M.L., Cahill L.E., Agarwal I., Rimm E.B., Mukamal K.J.. Novel metabolic biomarkers of cardiovascular disease. Nat. Rev. Endocrinol. 2014; 10:659–672. [DOI] [PubMed] [Google Scholar]
- 4. Chen J., Liu X., Shen L., Lin Y., Shen B.. CMBD: a manually curated cancer metabolic biomarker knowledge database. Database (Oxford). 2021; 2021:baaa094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen Y., Michalak M., Agellon L.B.. Importance of nutrients and nutrient metabolism on human health. Yale. J. Biol. Med. 2018; 91:95–103. [PMC free article] [PubMed] [Google Scholar]
- 6. Fortmann S.P., Burda B.U., Senger C.A., Lin J.S., Whitlock E.P.. Vitamin and mineral supplements in the primary prevention of cardiovascular disease and cancer: an updated systematic evidence review for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2013; 159:824–834. [DOI] [PubMed] [Google Scholar]
- 7. Coquin L., Feala J.D., McCulloch A.D., Paternostro G.. Metabolomic and flux-balance analysis of age-related decline of hypoxia tolerance in Drosophila muscle tissue. Mol. Syst. Biol. 2008; 4:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mintz-Oron S., Meir S., Malitsky S., Ruppin E., Aharoni A., Shlomi T.. Reconstruction of Arabidopsis metabolic network models accounting for subcellular compartmentalization and tissue-specificity. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nookaew I., Bordel S., Nielsen J.. Mapping condition-dependent regulation of metabolism in yeast through genome-scale modeling. BMC Syst. Biol. 2013; 7:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sigurdsson M.I., Jamshidi N., Steingrimsson E., Thiele I., Palsson B.Ø.. A detailed genome-wide reconstruction of mouse metabolism based on human Recon 1. BMC Syst. Biol. 2010; 4:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weaver D.S., Keseler I.M., Mackie A., Paulsen I.T., Karp P.D.. A genome-scale metabolic flux model of Escherichia coli K–12 derived from the EcoCyc database. BMC Syst. Biol. 2014; 8:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yilmaz L.S., Walhout A.J.. A Caenorhabditis elegans genome-scale metabolic network model. Cell Syst. 2016; 2:297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hirayama A., Kami K., Sugimoto M., Sugawara M., Toki N., Onozuka H., Kinoshita T., Saito N., Ochiai A., Tomita M.et al.. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009; 69:4918. [DOI] [PubMed] [Google Scholar]
- 14. Ortmayr K., Dubuis S., Zampieri M.. Metabolic profiling of cancer cells reveals genome-wide crosstalk between transcriptional regulators and metabolism. Nat. Commun. 2019; 10:1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Damiani C., Maspero D., Di Filippo M., Colombo R., Pescini D., Graudenzi A., Westerhoff H.V., Alberghina L., Vanoni M., Mauri G.. Integration of single-cell RNA-seq data into population models to characterize cancer metabolism. PLoS Comput. Biol. 2019; 15:e1006733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Evers T.M.J., Hochane M., Tans S.J., Heeren R.M.A., Semrau S., Nemes P., Mashaghi A.. Deciphering metabolic heterogeneity by single-cell analysis. Anal. Chem. 2019; 91:13314–13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wagner A., Wang C., Fessler J., DeTomaso D., Avila-Pacheco J., Kaminski J., Zaghouani S., Christian E., Thakore P., Schellhaass Bet al.. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell. 2021; 184:4168–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thompson C., Bauer D., Lum J., Hatzivassiliou G., Zong W.-.X., Zhao F., Ditsworth D., Buzzai M., Lindsten T.. How do cancer cells acquire the fuel needed to support cell growth? Cold Spring Harbor symposia on quantitative biology. 2005; Cold Spring Harbor Laboratory Press. [DOI] [PubMed] [Google Scholar]
- 19. DeBerardinis R.J., Lum J.J., Hatzivassiliou G., Thompson C.B.. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008; 7:11–20. [DOI] [PubMed] [Google Scholar]
- 20. Hanahan D., Weinberg R.A.. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674. [DOI] [PubMed] [Google Scholar]
- 21. Ward P.S., Thompson C.B.. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012; 21:297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alghamdi N., Chang W., Dang P., Lu X., Wan C., Gampala S., Huang Z., Wang J., Ma Q., Zang Y.et al.. A graph neural network model to estimate cell-wise metabolic flux using single-cell RNA-seq data. Genome. Res. 2021; 31:1867–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hari A., Lobo D.. Fluxer: a web application to compute, analyze and visualize genome-scale metabolic flux networks. Nucleic Acids Res. 2020; 48:W427–W35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wagner A., Wang C., Fessler J., DeTomaso D., Avila-Pacheco J., Kaminski J., Zaghouani S., Christian E., Thakore P., Schellhaass B.et al.. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell. 2021; 184:4168–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Damiani C., Maspero D., Di Filippo M., Colombo R., Pescini D., Graudenzi A., Westerhoff H.V., Alberghina L., Vanoni M., Mauri G.. Integration of single-cell RNA-seq data into population models to characterize cancer metabolism. PLoS Comput. Biol. 2019; 15:e1006733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kanehisa M., Goto S.. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brunk E., Sahoo S., Zielinski D.C., Altunkaya A., Dräger A., Mih N., Gatto F., Nilsson A., Preciat Gonzalez G.A., Aurich M.K.et al.. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018; 36:272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brunk E., Sahoo S., Zielinski D.C., Altunkaya A., Drager A., Mih N., Gatto F., Nilsson A., Preciat Gonzalez G.A., Aurich M.K.et al.. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018; 36:272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ogata H., Goto S., Fujibuchi W., Kanehisa M.. Computation with the KEGG pathway database. Biosystems. 1998; 47:119–128. [DOI] [PubMed] [Google Scholar]
- 30. Saier M.H. Jr, Reddy V.S., Tamang D.G., Vastermark A.. The transporter classification database. Nucleic Acids Res. 2014; 42:D251–D258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saier M.H. Jr, Tran C.V., Barabote R.D.. TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006; 34:D181–D186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barbie D.A., Tamayo P., Boehm J.S., Kim S.Y., Moody S.E., Dunn I.F., Schinzel A.C., Sandy P., Meylan E., Scholl C.et al.. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009; 462:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Z., Chang W., Alghamdi N., Cao S., Zhang Y., Zhang C.. scFLUX: a webserver to estimate cell-/sample-wise metabolic fluxome by using scRNA-seq or general transcriptomics data. 2022; bioRxiv doi:20 June 2022, preprint: not peer reviewed 10.1101/2022.06.18.496660. [DOI]
- 35. Hrovatin K., Fischer D.S., Theis F.J.. Toward modeling metabolic state from single-cell transcriptomics. Mol. Metab. 2022; 57:101396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Dijk D., Sharma R., Nainys J., Yim K., Kathail P., Carr A.J., Burdziak C., Moon K.R., Chaffer C.L., Pattabiraman D.et al.. Recovering gene interactions from single-cell data using data diffusion. Cell. 2018; 174:716–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M.et al.. Integrated analysis of multimodal single-cell data. Cell. 2021; 184:3573–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mathys H., Davila-Velderrain J., Peng Z., Gao F., Mohammadi S., Young J.Z., Menon M., He L., Abdurrob F., Jiang X.. Single-cell transcriptomic analysis of Alzheimer's disease. Nature. 2019; 570:332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yurov Y.B., Vorsanova S.G., Iourov I.Y.. The DNA replication stress hypothesis of Alzheimer's disease. Sci. World J. 2011; 11:2602–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Polis B., Samson A.O.. Role of the metabolism of branched-chain amino acids in the development of Alzheimer's disease and other metabolic disorders. Neur. Reg. Res. 2020; 15:1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim J., DeBerardinis R.J.. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019; 30:434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Caetano R., Ispolatov Y., Doebeli M.. Evolution of diversity in metabolic strategies. Elife. 2021; 10:e67764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Savage V.M., Allen A.P., Brown J.H., Gillooly J.F., Herman A.B., Woodruff W.H., West G.B.. Scaling of number, size, and metabolic rate of cells with body size in mammals. Proc. Natl. Acad. Sci. 2007; 104:4718–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oren A. Metabolic diversity in prokaryotes and eukaryotes. Biolog. Sci. Fund. Syst.-Volume II. 2009; 40:http://www.eolss.net/sample-chapters/c03/e6-71-04-02.pdf. [Google Scholar]
- 45. Zenobi R. Single-cell metabolomics: analytical and biological perspectives. Science. 2013; 342:1243259. [DOI] [PubMed] [Google Scholar]
- 46. Geeraerts X., Fernández-Garcia J., Hartmann F.J., de Goede K.E., Martens L., Elkrim Y., Debraekeleer A., Stijlemans B., Vandekeere A., Rinaldi G.et al.. Macrophages are metabolically heterogeneous within the tumor microenvironment. Cell Rep. 2021; 37:110171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
FLUXestimator is available as a web server at http://scFLUX.org/. The stand-alone tools to run the functions of FLUXestimator on a local machine are available at the GitHub repository (https://github.com/changwn/scFEA).