

Summary
The immune and stromal contexture within the tumor microenvironment (TME) interact with cancer cells and jointly determine disease process and therapeutic response. We aimed at developing a risk scoring model based on TME-related genes of squamous cell lung cancer to predict patient prognosis and immunotherapeutic response. TME-related genes were identified through exploring genes that correlated with immune scores and stromal scores. LASSO-Cox regression model was used to establish the TME-related risk scoring (TMErisk) model. A TMErisk model containing six genes was established. High TMErisk correlated with unfavorable OS in LUSC patients and this association was validated in multiple NSCLC datasets. Genes involved in pathways associated with immunosuppressive microenvironment were enriched in the high TMErisk group. Tumors with high TMErisk showed elevated infiltration of immunosuppressive cells. High TMErisk predicted worse immunotherapeutic response and prognosis across multiple carcinomas. TMErisk model could serve as a robust biomarker for predicting OS and immunotherapeutic response.
Subject areas: Biological sciences, Immunology, Cancer, Machine learning
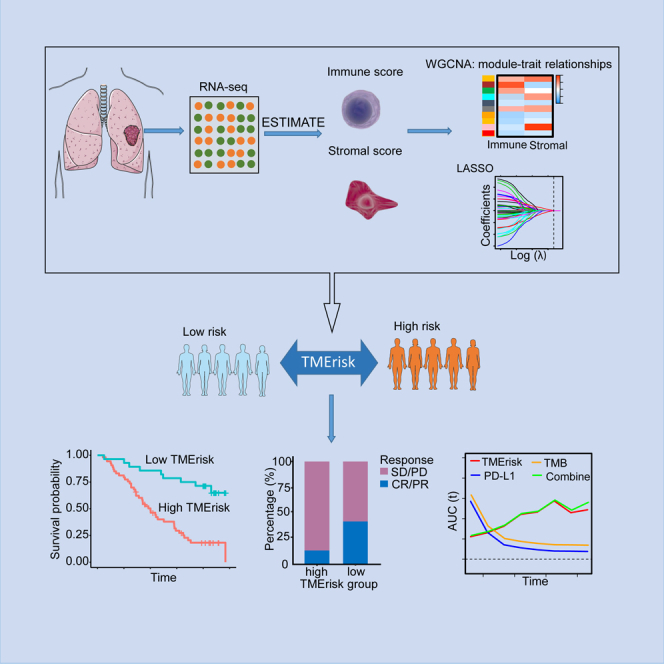
Graphical abstract
Highlights
-
•
A TMErisk model was established based on six immune/stroma-related genes
-
•
High TMErisk predicts poor prognosis and immunotherapy efficacy in multiple cancers
-
•
High TMErisk correlated with immune-suppressive tumor micro-milieu
-
•
TMErisk outperformed PD-L1 and TMB in predicting immunotherapy efficacy
Biological sciences; Immunology; Cancer; Machine learning
Introduction
Antibodies targeting programmed cell death 1 (PD-1) immune checkpoint or its ligand PD-L1 have yielded substantial overall survival (OS) and therapeutic response benefits in non-small cell lung cancer (NSCLC).1,2,3,4 Their clinical application, however, faces challenges which mainly lie in the relatively low response rate of 14%–20% for single-agent treatment in unselected patient population,4,5,6 and the lack of well-established predictive biomarkers. PD-L1 by immunohistochemistry (IHC) and tumor mutation burden (TMB) have been supported by multiple evidence to serve as predictive biomarkers for immunotherapeutic response and have gained approval for routine clinical use,7,8,9 whereas in some cases they are confronted by paradoxical prediction. It is difficult to draw a precise screening of potential beneficiaries using a single biomarker. A multidimensional indicator based on tumor cells, tumor microenvironment (TME) and host immunity holds promise to integrate complementary factors for individualized immunotherapy.
Tumor micro-milieu is an ecological niche where tumor cells exist and is also composed of other nonmalignant components including infiltrating immune cells, stromal cells, other types of cells, extracellular matrix (ECM), vasculature, and signaling molecules.10,11 Components in the tumor ecosystem interact with each other and collectively control tumor initiation and progression, patient prognosis and drug sensitivity.10,12 Current published studies mainly put emphasis on crosstalk between cancer cells and two major benign cell components (immune cells and stromal cells). The type, proportion, and spatial distribution of infiltrating immune cells in the TME determine the immune phenotype of tumor and therefore affect drug sensitivity of cancer cells and patient survival.12,13 The stromal cells bridge the crosstalk between cancer cells and other TME components, which plays a crucial regulatory role in tumor development and progression as well as anti-tumor immunity, such as the immunosuppressive function of cancer-associated fibroblasts (CAFs).14,15 A previous study published in Nature unraveled a dynamic co-evolution of pre-invasive bronchial cells and the immune response during carcinogenesis, which highlighted the feasibility of developing immune biomarkers.16 Considering the critical role of TME in carcinogenesis and tumor progression, evaluating TME status could provide robust personalized prediction of drug sensitivity and patient prognosis.
In this study, we employed the Estimation of STromal and Immune cells in MAlignant Tumors using Expression data (ESTIMATE) algorithm and the Least Absolute Shrinkage and Selection Operator (LASSO) regression analysis to screen TME-related prognostic genes in the lung squamous cell carcinoma (LUSC) patients from The Cancer Genome Atlas (TCGA) database and established a TME-related risk scoring (TMErisk) model. Prognostic efficacy of TMErisk and its ability to predict immunotherapeutic response were comprehensively assessed and validated in multiple cohorts. On development of the TMErisk model, we aimed to provide a reliable and efficient quantitative instrument to select patients likely or unlikely to be immunotherapy responders, and provide insights into development of new combined therapy strategies.
Results
Correlation of immune/stromal scores with patient prognosis
494 patients containing complete prognosis information in TCGA-LUSC cohort were enrolled in our study to develop TMErisk model (Figure 1, Table S1). Based on ESTIMATE algorithm, these patients had a median immune score of 993.75 (range, −1189.75–3436.15), and a median stromal score of −92.13 (range, −2217.10–1856.99) (Table S2). The optimal cut-points of immune score and stromal score determined by maximally selected rank statistics were 1904.86 and 191.14, respectively (Figures S1A and S1B), based on which, patients were divided into low and high immune/stromal score groups. Kaplan–Meier survival analysis showed that patients with higher immune/stromal scores had unfavorable OS compared with those with lower scores (immune score: hazard ratio [HR] 1.41, 95% confidence interval [CI] 1.01–1.97, p = 0.047; stromal score: HR 1.52, 95% CI 1.15–2.00, p = 0.003) (Figures S1C and S1D).
Figure 1.

A flowchart delineating process of model building and our data analysis protocol
TCGA, the cancer genome atlas; LUSC, lung squamous cell carcinoma; FPKM, Fragments Per Kilobase of exon model per Million mapped fragments; ESTIMATE, Estimation of STromal and Immune cells in MAlignant Tumors using Expression data; WGCNA, weighted gene co-expression network analysis; TME, tumor microenvironment; NSCLC, non-small cell lung cancer; LUAD, lung adenocarcinoma; TMB, tumor mutation burden; ssGSEA, single sample gene set enrichment analysis; TIDE, tumor immune dysfunction and exclusion; GDSC, Genomics of Drug Sensitivity in Cancer; CTRP, Cancer Therapeutics Response Portal; PD-L1, programmed cell death ligand 1; AUC, area under the receiver operating characteristic curve.
Development and validation of TMErisk model
DESeq2 package was used to compare gene expression profile of patients with low immune/stromal scores with those with high scores. 2885 differentially expressed genes (DEGs) were identified to be related with immune score and 2007 to be related with stromal score (Figures 2A and 2B). In weighted gene co-expression network analysis (WGCNA), we identified 35 co-expressed gene modules (Figure 2C, Table S3), among which genes in the light green, brown, blue and cyan modules were robustly associated with immune score, and genes in the brown, blue, cyan and yellow modules had a strong correlation with stromal score (Figure 2D). Venn diagrams presented overlapped genes between DEGs and strong TME-related genes identified by WGCNA (Figure 2E, Table S4). These intersections were further inputted into LASSO-Cox regression analyses to achieve dimension reduction. The output genes associated with immune score and stromal score were integrated to generate 15 candidate model genes (Figure 2E, Table S5). Association of the 15 candidate genes with OS was evaluated in univariate Cox analyses (Figure S2A) and 14 genes (HLA-DMB was excluded, p > 0.15) were incorporated into a stepwise multivariate Cox model. The model containing TGM2, C11orf96, PLAAT4, PNCK, KLF5 and C4BPA had a minimum AIC value (AIC = 2225.845). Then, TMErisk could be calculated through the following formula: TMErisk = 0.272255∗C11orf96 + 0.570467∗TGM2 - 0.318056∗PNCK - 0.752920∗PLAAT4 - 0.262275∗KLF5 + 0.311932∗C4BPA.
Figure 2.

Identification of significant TME-related genes
(A and B) The volcano map shows the differentially expressed genes according to immune score (A) and stromal score strata (B).
(C) A dendrogram of clustered genes in WGCNA. Each branch in the figure represents a gene and each color lump below represents a co-expressed gene module.
(D) Correlation heatmap showing the relationship between the gene modules and immune/stromal scores. Robust TME-related gene modules are marked with black frames. The number within the color lump indicates the correlation coefficient, followed by a statistical p value in the parenthesis.
(E) Screening of candidate model genes. The Venn diagram shows the intersection genes between DEGs and robust TME-related gene modules identified by WGCNA. These overlapping genes are then inputted into a LASSO regression analysis. The vertical dashed lines in the middle panel represent the selected appropriate lambda value. Immune score-related and stromal score-related LASSO output genes are combined to generate candidate model genes. Genes in bold in the rectangular boxes are unique genes in immune-related/stromal-related genes. DEGs, differentially expressed genes; WGCNA, weighted gene co-expression network analysis; LASSO, Least Absolute Shrinkage and Selection Operator. See also Figures S1–S4.
Patients in the TCGA-LUSC cohort were scored according to this model and were stratified into low- and high-TMErisk groups based on a cut-point value of 0.27 (Figure S2B). Survival analysis revealed that high TMErisk was significantly associated with worse OS (HR 2.88, 95% CI 2.09–3.96, p < 0.001) (Figure 3A). This significant association was validated in 6 NSCLC datasets (GSE81089, GSE30219, GSE37745, GSE157011, TCGA-LUAD and GSE31210; all with a univariate Cox p-value of <0.001) (Figures 3B–3G).
Figure 3.

Prognostic value of TMErisk in multiple publicly available NSCLC datasets
(A–G) Kaplan–Meier survival analysis based on TMErisk strata in the TCGA-LUSC training cohort. (B-G) Kaplan–Meier survival analyses based on TMErisk strata in the GSE81089 (B), GSE30219 (C), GSE37745 (D), GSE157011 (E), TCGA-LUAD (F) and GSE31210 (G) cohorts.
(H) Time-dependent AUC shows accuracy of TMErisk for predicting OS in the TCGA-LUSC training cohort and 6 NSCLC validation cohorts. NSCLC, non-small cell lung cancer; TCGA, the cancer genome atlas; LUSC, lung squamous cell carcinoma; AUC, area under the receiver operating characteristic curve.
Prognostic potency of TMErisk and its correlation with clinicopathological characteristics
Predictive accuracy of TMErisk for OS was evaluated using C-index and time-dependent AUC. In the TCGA-LUSC cohort, TMErisk had a higher C-index compared with TNM stage (0.620 [95% CI 0.578–0.661] vs. 0.557 [95% CI 0.516–0.598]) and combination of TMErisk and TNM stage could yield a better C statistic (0.632, 95% CI 0.591–0.673). C-index (95% CI) for validation cohorts GSE81089, GSE30219, GSE37745, GSE157011, TCGA-LUAD and GSE31210 were 0.711 (0.607–0.815), 0.654 (0.566–0.742), 0.711 (0.649–0.787), 0.606 (0.569–0.643), 0.629 (0.582–0.676) and 0.771 (0.689–0.853), respectively. Time-dependent AUC analyses demonstrated that the training cohort and most of the validation sets possessed good accuracy for predicting OS across different time points (Figure 3H). To determine relationship between TMErisk and clinicopathological characteristics, we evaluated distribution of TMErisk across selected baseline patient characteristics in the TCGA-LUSC cohort. It was found that the elderly patients had significantly higher TMErisk (median, 0.000 versus −0.046, p = 0.027) (Figure S3A). However, we observed no significant difference between TMErisk and other clinicopathological factors including gender, TNM stage, ECOG (Eastern Cooperative Oncology Group) performance status (PS) and smoking status (all with p > 0.05) (Figures S3B–S3H). Univariate and multivariate Cox analyses were used to analyze impact of TEMrisk and other baseline parameters on patient prognosis. In univariate analysis, higher TMErisk, AJCC tumor stage of III and IV (versus stage I), and two or more ECOG PS significantly predicted poorer OS (p < 0.05) (Figure S4A). All covariates analyzed in univariate analyses were incorporated into a stepwise multivariate Cox model and the result revealed that TMErisk was an independent prognostic factors with adjustment for other baseline factors (Figure S4B).
TMErisk correlated with immunosuppressive functional features and TME landscape
To clarify the underlying cellular and molecular mechanisms for prognostic difference related to TMErisk, we sought to investigate signatures of biological function and TME landscape between the low- and high-TMErisk groups. We found that significant enriched molecular processes in the low-TMErisk group were mainly related with down-regulated KRAS signaling (Figure 4A). In contrast, gene sets associated with coagulation, TNFα signaling via NF-κB, IL6-JAK-STAT3, up-regulated KRAS signaling, complement, epithelial-mesenchymal transition (EMT), Inflammatory response, angiogenesis and IL2-STAT5 signals were significantly enriched in the high-TMErisk group (Figure 4A), which indicated chronic inflammation and formation of immunosuppressive microenvironment.
Figure 4.

Association of TMErisk with cellular and molecular signatures
(A) TME-related Functional enrichment was performed by the compareCluster function. The x-axis represents different selected genes used for conducting enrichment analysis. The left facet panel shows the enriched pathways for genes up-regulated in the high-TMErisk group, and the right facet panel shows the enriched pathways for genes up-regulated in the low-TMErisk group.
(B) The heatmap shows the normalized infiltration abundance of immune and stromal cells that estimated by CIBERSORT, EPIC and xCell algorithms. In the Wilcoxon test results, the orange asterisks indicate cells significantly enriched in the low-TMErisk group, and the lightgreen asterisks indicate cells significantly enriched in the high-TMErisk group. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ns, not significant. See also Figures S5–S8.
The abundance of infiltrating immune and stromal cells in the TME was inferred by CIBERSORT, EPIC and xCell algorithms (Tables S6, S7, and S8). We observed that tumors with high-TMErisk had higher infiltration levels of cells that could contribute to immunosuppressive microenvironment, including regulatory T cells (Tregs), endothelial cells, CAFs, macrophages M2 and neutrophil; however, abundance of CD4+ and CD8+ T cells was higher in the low-TMErisk group (Figure 4B, Table S9). Because most chemokines exert an important influence on recruitment of immune cells into the TME and the crucial role of some genes in modulating immune response, we evaluated expression levels of 41 chemokine genes and 48 immune-related genes between different TME risk stratifications. It revealed that expression levels of 34 immune-related genes (including CTLA-4, PD-1, PD-L2, TIGIT, BTLA, TNFRSF14, VISTA, etc.) and 23 chemokine genes (including CCL2, CCL5, CCL13, CCL17-24, CXCL1, CXCL2, CXCL5, etc.) were elevated in the high-TMErisk group (Figures S5A and S5B). Two co-stimulatory genes (TNFRSF18, TNFRSF25) and two chemokine gens (CCL26, XCL1) were highly expressed in the low-TMErisk group, whereas there was no significant difference in PD-L1 expression level (Figures S5A and S5B). In addition, the correlation between 6 model genes and selected immune-related genes was analyzed using Pearson method. Our results revealed that TGM2 (Figure S6A), C4BPA (Figure S6B), C11orf96 (Figure S7A) and PLAAT4 (Figure S7B) had significant positive correlation with most of the selected immune-related genes, while PNCK and KLF5 were negatively associated with most of the immune-related genes (Figures S8A and S8B).
Mutation profile for patients with low and high TMErisk in the TCGA-LUSC cohort
Genetic alteration has been recognized as a pivotal factor that can impact tumor biological behaviors and prognosis. We used “maftools” instrument to explore whether there existed genetic mechanisms that related to TMErisk. The top 15 genes mutated more frequently in the low-TMErisk group included CDK6, FAM133B, LOC101927497, MTOR, PLCH1, BCL11A, MIR4432, MIR4432HG, ATP13A2, COL6A3, IGSF3, CASZ1, KIF17, MMEL1 and PRDM2, whereas for the high-TMErisk group, the top 15 genes with higher mutation frequency compared with those in the low-TMErisk group were MYO15A, ATAD2, JMJD1C, IRF6, ITPR1, KIAA1524, VWA3A, CDKL5, DPH2, DDC, HEG1, VGLL1, SLC35F1, OR7C2 and RNF10 (Figures S9A and S9B). The top 20 mutated genes and their mutation types based on the TME risk stratification are respectively presented in Figures S9C and S9D. It has emerged that mutation heterogeneity of TP53 gene had predictive potential for immunotherapy efficacy, in which PD-L1 expression, IFN-γ signatures and TME composition were significantly distinguished between tumors with TP53 missense and nonsense mutations.17 In our work, the low-TMErisk group had higher frequency of TP53 missense mutation compared with the high-TMErisk group (53.9% versus 38.6%, Chi-square p value = 0.017), whereas frequency of nonsense mutation and truncating mutation (referring to frame_shift_del, frame_shift_ins, and nonsense mutations) was similar (Figure S9E).
TMErisk had potential in predicting drug sensitivity
Based on the Genomics of Drug Sensitivity in Cancer (GDSC) and Cancer Therapeutics Response Portal (CTRP) databases, we investigated whether TMErisk could serve as a predictive biomarker for therapeutic response to chemotherapy and targeted therapy. It turned out that patients with low TMErisk were more sensitive to most chemotherapeutic and targeted agents, including drugs that are commonly used in clinical setting, such as paclitaxel, docetaxel, cisplatin and afatinib (Figure S10A–S10H). TMB and Tumor Immune Dysfunction and Exclusion (TIDE) algorithm were utilized to infer predictive potential of TMErisk in terms of immunotherapeutic response. The results demonstrated that patients with high TMErisk had higher TIDE score (Wilcoxon p < 0.001) and lower TMB (Wilcoxon p = 0.017) than those with low TMErisk (Figures 5A and 5B), indicating a better response to immunotherapy in the low-TMErisk group.
Figure 5.

Evaluation of value of TMErisk in predicting immunotherapeutic response and validation of predictive value of TMErisk for immunotherapy in external cohorts
(A) Difference of TIDE score between the low- and high-TMErisk groups was examined by Wilcoxon test.
(B) Difference of TMB between the low- and high-TMErisk groups was examined by Wilcoxon test. Kaplan–Meier analyses for OS and PFS based on TMErisk strata in OAK cohorts (C-D, G-H), POPLAR cohort (K-L) and IMvigor210 cohort (O-P). Stacked percentage bar charts show the association of TMErisk with ORR and DCR in in OAK cohorts (E-F, I-J), POPLAR cohort (M−N) and IMvigor210 cohort (Q-R). TIDE, tumor immune dysfunction and exclusion; TMB, tumor mutation burden; HR, hazard ratio; CI, confidence interval; BOR, best overall response; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; ORR, objective response rate; DCR, disease control rate. See also Figures S10 andS11.
TMErisk predicted immunotherapeutic response in external cohorts
We first assessed the ability of TMErisk to serve as a predictive biomarker for anti-PD-1/PD-L1 therapy in two GEO transcriptomic datasets. Response and survival benefits could be observed for patients with low TMErisk in the two GEO cohorts (GSE135222: HR 5.28, 95% CI 2.06–13.51, p < 0.001; GSE78220: HR 6.49, 95% CI 1.93–21.78, p = 0.002; objective response rate [ORR] for GSE78220: 0.0% vs. 61.9%, p = 0.039) (Figures S11A–S11C). Similar results were found in another four external immunotherapy datasets from OAK, POPLAR and IMvigor210 clinical trials, in which patients with high TMErisk score had poorer OS and progression-free survival (PFS) (Figures 5C, 5D, 5G, 5H, 5K, 5L, 5O, and 5P). In the OAK cohorts, high TMErisk was significantly associated with lower ORR in LUSC patients (6.0% vs. 25.0%, p = 0.028) (Figure 5E) and reduced disease control rate (DCR) in LUAD patients (35.8% vs. 61.2%, p < 0.001) (Figure 5J). For the immunotherapy groups in the IMvigor210 and POPLAR cohorts, patients with high TMErisk had both lower ORR and DCR (Figures 5M−5N, 5Q, and 5R). TMErisk also was negatively associated with OS and PFS in the chemotherapy groups from OAK and POPLAR clinical trials (Figures 6A, 6B, 6E, 6F, 6I, and 6J); however, no significant relationship between the TMErisk and ORR as well as DCR was observed (all with a p value of >0.05) (Figures 6C, 6D, 6G, 6H, 6K, and 6L).
Figure 6.

Validation of predictive value of TMErisk for chemotherapy in external cohorts
(A-B, E-F, I-J) Kaplan–Meier analyses for OS and PFS based on TMErisk strata in OAK cohorts (A-B, E-F) and POPLAR cohort (I-J). (C-D, G-H, K-L) Stacked percentage bar charts show the association of TMErisk with ORR and DCR in in OAK cohorts (C-D, G-H) and POPLAR cohort (K-L).
(M−N) Predictive accuracy of TMErisk, TMB and PD-L1 for OS (M) and PFS (N) in the POPLAR and IMvigor210 cohorts. HR, hazard ratio; CI, confidence interval; BOR, best overall response; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; ORR, objective response rate; DCR; OS, overall survival; PFS, progression-free survival; TMB, tumor mutation burden; PD-L1, programmed cell death ligand 1. See also Figures S10 and S12.
Univariate Cox analyses of TMErisk and other clinicopathological covariates for OS and PFS in the POPLAR and IMvigor210 cohorts are shown in Figures S12A–S12D. These analyzed variables were then incorporated into multivariate Cox models with stepwise screening. TMErisk was found to be an independent predictive factor for OS and PFS in patients treated with immunotherapy or chemotherapy drugs (all with a p-value of <0.05). TMB and PD-L1 are two commonly used predictive biomarker for response to immunotherapy in clinical practice. We next compared the predictive performance of TMErisk, TMB and PD-L1 for OS and PFS in the POPLAR and IMvigor210 cohorts. Our results demonstrated that TMErisk outperformed TMB and PD-L1 in predicting OS and PFS for immunotherapy and a combination of the three indexes could promote predictive accuracy (Figures 6M−6N). Predictive advance of TMErisk for OS was also observed in the chemotherapy cohort from POPLAR, while these three biomarkers alone had similar predictive accuracy for PFS (Figures 6M−6N).
TMErisk predicted benefits of immunochemotherapy in the in-house cohort
For the internal ORIENT-11 cohort, a total of 113 samples from immunochemotherapy (combo) group and 58 samples from chemotherapy group were included in our study to conduct transcriptomic analysis. Detailed sample screening procedure have been delineated in our previously published study.18 Median TMErisk was 0.00 (range, −1.68 – 1.93) for the combo group and −2.00 (−3.56 – 1.16) for the chemo group. In the combo group, patients with high-TMErisk (42/113, 37.2%) had worse median OS (11.7 vs. 34.0 months; HR 3.42, 95% CI 2.10–5.56, p < 0.001) and PFS as well as lower ORR and DCR than those with low-TMErisk (Figures 7A–7C). For the chemotherapy group from ORIENT-11, high-TMErisk was significantly associated with shorter OS (10.6 versus not reached months, HR 4.04, 95% CI 1.84–8.87, p < 0.001) whereas there was no significant association between TMErisk and PFS (p = 0.423) as well as ORR (p = 0.195) (Figures 7D–7F).
Figure 7.

Validation of predictive value of TMErisk for immunotherapy and chemotherapy in the internal cohort
(A and B) Kaplan–Meier analyses for OS and PFS based on TMErisk strata in the immunochemotherapy group from ORIENT-11 cohort.
(C) Stacked percentage bar chart shows the distribution of BOR according to TMErisk (left panel) and association of TMErisk with ORR (right panel) in the immunochemotherapy group from ORIENT-11 cohort.
(D and E) Kaplan–Meier analyses for OS and PFS based on TMErisk strata in the chemotherapy group from ORIENT-11 cohort.
(F) Stacked percentage bar chart shows the distribution of BOR according to TMErisk (left panel) and association of TMErisk with ORR (right panel) in the chemotherapy group from ORIENT-11 cohort.
(G–J) Predictive accuracy of TMErisk and PD-L1 for OS and PFS in the ORIENT-11 immunochemotherapy and chemotherapy groups. Chemo, chemotherapy; IO + Chemo, immunotherapy plus chemotherapy; HR, hazard ratio; CI, confidence interval; BOR, best overall response; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; ORR, objective response rate; R, responder; NR, non-responder; AUC (t), time-dependent area under the receiver operating characteristic curve; OS, overall survival. See also Figure S12.
Association of other clinicopathological factors with OS and PFS for two treatment groups in ORIENT-11 was also evaluated using univariate Cox analyses and the results are presented in Tables S10 and S11. Multivariate analysis corroborated that TMErisk was an independent predictive factor for OS in the combo (HR 3.81, 95% CI 2.33–6.24, p < 0.001) and chemo (HR 4.04, 95% CI 1.84–8.87, p < 0.001) group, and for PFS in the combo group (HR 3.01, 95% CI 1.83–4.94, p < 0.001); whereas TMErisk failed to predict PFS in the chemotherapy group (Figures S12E and S12F). We next specifically compared the predictive efficacy of TMErisk and PD-L1 on patient survival and therapeutic response. It was found that, irrespective of treatment groups, TMErisk did better than PD-L1 in predicting OS at different time points (Figures 7G and 7H). In terms of predictive accuracy for PFS, TMErisk was significantly superior to PD-L1 in the combo group (Figure 7I), while both of the two biomarkers had low and similar predictive accuracy in the chemo group (Figure 7J). Intriguingly, we observed that predictive efficacy of PD-L1 had a reduction tendency over time; predictive accuracy of TMErisk, however, decreased first and then raised (Figures 7G–7J).
Discussion
Immune cells and stromal cells, as two major components in the tumor micro-milieu, both can exert an important influence on biology and prognosis of tumors and their response to treatment through direct interaction with cancer cells or other indirect mechanisms.10,12,15 In this study, we developed a risk scoring (TMErisk) model with 6 gene signatures that related to immune and stromal scores, and dissected the association of TMErisk with TME as well as cellular and molecular signatures. Performance of TMErisk for predicting OS and immunotherapeutic efficacy were evaluated and validated in an internal cohort and multiple external cohorts. Our results demonstrated that TMErisk could mirror the composition and features of TME, and showed significant association with unfavorable OS and poorer response (PFS, ORR and DCR) to checkpoint blockade immunotherapy.
Accumulating studies have employed the ESTIMATE algorithm to infer proportion of infiltrating immune and stromal cells in the tumor samples, which highlighted reliability and effectiveness of prognosis prediction according to TME signatures and the predictive advantage of TME scores than other single biomarkers.19,20 Based on methodology of WGCNA and a machine-learning method LASSO-Cox, we conducted a dimension-reduced screening for gene expression matrix from TCGA-LUSC samples and a simplified model containing 6 gene signatures (TGM2, C11orf96, PLAAT4, PNCK, KLF5 and C4BPA) was generated.
Prognostic performance of TMErisk was comprehensively evaluated via several methodologies in the present study. Compared with TNM stage, TMErisk had a higher C statistic for predicting OS and combination of the two variables could enhance the predictive efficacy. Then, TMErisk was corroborated to be an independent prognosticator in adjusted multivariate Cox regression analysis. Ability of TMErisk for OS prediction was validated in 6 NSCLC datasets, in which C-index ranged from 0.606 to 0.711 and favorable predictive accuracy at different time points was exhibited in time-dependent AUC. These assessments indicated that TMErisk could serve as a quantitative and promising risk stratification tool to guide personalized treatment. Furthermore, analysis of drug sensitivity revealed a better sensitivity to most of the tested chemotherapeutic and targeted agents in the low-TMErisk group than that in the high-TMErisk group, which provided a preliminary hint of therapeutic prediction. However, drug sensitivity was estimated based on gene expression matrix and drug response data of tumor cell lines and therefore, should be interpreted with this caveat in mind. It appeared that TMErisk was unable to significantly stratify PFS and was marginally associated with therapeutic response in the chemotherapy groups from OAK, POPLAR and ORIENT-11 cohorts. The chemotherapeutic sensitivity of patients screened using TMErisk remains to be evaluated in studies with larger sample size and different cancer types.
Antibodies blocking PD-1 or PD-L1 checkpoint have revolutionized the treatment paradigm of various cancer types, whereas only a fraction of patients benefit from this treatment, and a reliable and accurate predictive biomarker has not been established. To investigate the potential role of TMErisk in predicting benefits of immunotherapy, we analyzed the relationship of TMErisk with response and survival benefits in multiple external cohorts and an in-house cohort. We identified robust value of TMErisk for predicting therapeutic response and patient prognosis in all these immunotherapy cohorts, though only a marginal statistical significance was observed for LUSC patients from OAK trial. We thought the small sample size and the retrospective nature might be, in part, responsible for this marginal association. In the atezolizumab group from POPLAR and IMvigor210 clinical trials, high TMErisk was demonstrated to be an independent factor both for OS and PFS in the multivariate analyses. In addition, predictive accuracy (as measured using C-index) of TMErisk for OS and PFS was significantly superior to that of PD-L1 and TMB. A strategy assessing combined TMErisk, PD-L1 and TMB could promote predictive performance, and therefore, might provide additional information for screening of potential beneficiaries. In the internal cohort ORIENT-11, time-dependent AUC also uncovered a remarkable predictive advantage of TMErisk than PD-L1 for OS and PFS across different time points. More interestingly, predictive accuracy of TMErisk first attenuated, and then escalated, whereas there was a diminishing tendency for PD-L1, which, to some extent, revealed inadequacy of PD-L1 as a predictive biomarker.
We conducted in-depth bioinformatics analyses to elucidate mechanisms that contributed to differential prognosis and response status between the low- and high-TMErisk groups. CIBERSORT, EPIC and xCell algorithms were applied to assess infiltration levels of immune and stromal cells for each sample in the TCGA-LUSC cohort. It was found that high-TMErisk group had higher infiltration abundance of most immune and stromal cells than the low-TMErisk group. Specifically, cells that can catalyze formation of immune-suppressive microenvironment, such as Tregs, endothelial cell, CAFs, macrophages M2 and neutrophil, were enriched in the high-TMErisk group. Communication between these cells and tumor cells provides compensatory inhibitory mechanisms that contribute to tumorigenesis and development as well as immune evasion, and thus may affect the response to immunotherapy.21 By contrast, CD4+ and CD8+ T cells, as the prime effectors of anti-tumor immunity, were enriched in the low-TMErisk group. Accordingly, we found that chemokines that guide Tregs (CCL17 and CCL22), microphages (CCL2 and CCL5), and neutrophils (CXCL1, CXCL2 and CXCL5) into solid tumors upregulated in the high-TMErisk group. Most of the selected immune-related genes were highly expressed in the high-TMErisk group, including negative checkpoints CTLA-4, PD-1, PD-L2, TIGIT, BTLA, TNFRSF14 and VISTA, which might demonstrate a series of immune evasion events in tumors with high TMErisk.
In functional enrichment analysis, we found that gene sets involved in immunosuppression-related biological procedures including IL6-JAK-STAT3, complement, EMT, inflammatory response, angiogenesis and IL2-STAT5 signaling, were enriched in the high-TMErisk group. EMT is a procedure characterized as phenotype transition from epithelial cells to mesenchymal cells, in which series of biological events can occur, such as induction of tumor stem cells, angiogenesis and immune evasion.22 A recent study uncovered that there existed complex and dynamic immunomodulatory crosstalk between EMT and immune evasion, which was relevant to immunotherapeutic response and patient survival.23
Taken together, we identified that tumors in the high-TMErisk group was characterized by immunosuppressive micro-milieu, low abundance of infiltrating CD4+ and CD8+ T cells, and low TMB. These phenotypes might contribute to inferior therapeutic response to anti-PD1/PD-L1 antibody and OS in patients with high TMErisk. In regard to the deserted infiltration of T cells and immune-suppressive microenvironment, we speculated that differential intrinsic mechanisms might be responsible for the two microenvironment phenotypes, and it is worthwhile to explore the underlying mechanisms.
Our work had advantage in developing a simplified and clinically friendly risk scoring model based on the 6 TME-related genes. We identified that high TMErisk significantly predicted unfavorable prognosis and immunotherapeutic response in multiple validation cohorts. High TMErisk was associated with immune-suppressive micro-milieu, deserted infiltration of CD4+ and CD8+ T cells, and low TMB. This model can assist clinicians in screening patients who are likely or unlikely to be immunotherapy responders and the relevant findings provide a theoretical basis for developing new combined treatment strategies.
Limitations of the study
The retrospective nature in the training set made our study subject to potential biases and affected the statistical power. The association of TMErisk with immunotherapeutic efficacy and prognosis remained to be validated in multicentric prospective studies with larger sample size. Furthermore, we established the TMErisk model using RNA-seq data, and therefore it remained to be determined whether it could employ more economic detection technology, such as IHC or a PCR assay panel, to generate a risk score. Whereas, currently we can detect gene expression levels based on a panel containing 6 model genes, which is more cost-effective compared with the whole transcriptomic sequencing. In addition, the bulk RNA-seq data limited us identifying immune or stromal components that were directly relevant to TMErisk, and leading to inadequate mechanism exploration. Our ongoing effort is to identify key regulatory mechanisms of these model genes through mechanistic and clinical validations.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Bulk tissues from NSCLC patients in ORIENT-11 clinical trial | Yang et al.18 | N/A |
| Critical commercial assays | ||
| PD-L1 IHC 22C3 pharmDx | Agilent Technologies | Cat#SK006; RRID: AB_2889976 |
| Deposited data | ||
| TCGA-LUSC RNA-seq data | The Cancer Genome Atlas | N/A |
| TCGA-LUAD RNA-seq data | The Cancer Genome Atlas | N/A |
| Bulk tissue RNA-seq from LUSC patients | Mezheyeuski et al.24 | GSE81089 |
| Microarray data from LUSC patients | Rousseaux et al.25 | GSE30219 |
| Microarray data from LUSC patients | Botling et al.26 | GSE37745 |
| Microarray data from LUSC patients | Bueno et al.27 | GSE157011 |
| Microarray data from LUAD patients | Okayama et al.28 | GSE31210 |
| Bulk RNA-seq data from NSCLC patients who received immunotherapy | Jung et al.29 | GSE135222 |
| Bulk RNA-seq data from melanoma patients who received immunotherapy | Hugo et al.30 | GSE78220 |
| Bulk RNA-seq data from POPLAR clinical trial | Patil et al.31 | EGAS00001005013 |
| Bulk RNA-seq data from OAK clinical trial | Patil et al.31 | EGAS00001005013 |
| Bulk RNA-seq data from IMvigor210 clinical trial | Banchereau et al.32 | EGAS00001004343 |
| Software and algorithms | ||
| R 4.1.0 | R Development Core Team | https://cran.r-project.org/ |
| estimate | Yoshihara et al.19 | https://sourceforge.net/projects/estimateproject/ |
| maxstat | Hothorn et al.33 | https://cran.r-project.org/web/packages/maxstat/index.html |
| WCGNA | Langfelder et al.34 | https://cran.r-project.org/web/packages/WGCNA/index.html |
| immunedeconv | Sturm et al.35 | https://github.com/omnideconv/immunedeconv |
| clusterProfiler | Wu et al.36 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| oncoPredict | Maeser et al.37 | https://cran.r-project.org/web/packages/oncoPredict/index.html |
| TIDE | Jiang et al.38 | http://tide.dfci.harvard.edu |
| X-tile | Camp et al.39 | https://medicine.yale.edu/lab/rimm/research/software/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Shaodong Hong (hongshd@sysucc.org.cn).
Materials availability
This study did not generate new unique reagents.
Gene expression data and clinical data for the TCGA-LUSC, TCGA-LUAD and validation cohorts from Gene Expression Omnibus (GEO) (GEO: GSE30219, GSE37745, GSE157011, GSE31210, GSE30219, GSE37745, GSE157011, GSE31210) can be retrieved from public repositories. RNA-seq data and corresponding clinical data for OAK, POPLAR and IMvigor210 are available in the European Genome-phenome Archive (EGA) (https://ega-archive.org/) with restricted access (EGA: EGAS00001004343 and EGAS00001005013). The remaining data analyzed during this study are included within the published article and its supplementary information files. Other materials relevant to the study and data for ORIENT-11 are available from the corresponding authors upon reasonable request.
Experimental model and study participant details
Public and in-house datasets
For model development, we downloaded TCGA-LUSC RNA-seq data (FPKM normalized) and corresponding clinical information via R package “TCGAbiolinks”. Publicly available gene expression datasets for NSCLCs in Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) and TCGA were systematically searched to retrieve external validation cohorts. Four normalized microarray datasets, GSE30219 (LUSC),25 GSE37745 (LUSC),26 GSE157011 (LUSC),27 GSE31210 (lung adenocarcinoma, LUAD)28 and two RNA-sequencing datasets, GSE81089 (LUSC)24 and TCGA-LUAD, were identified. Samples lacking complete prognosis information were excluded from further evaluation. The TCGA-LUSC somatic mutation data (data category: Simple Nucleotide Variation, workflow type: MuTect2 Variant Aggregation and Masking) were obtained through R package “TCGAbiolinks”. Mutation status was analyzed and visualized via R package “maftools”.
To investigate value of established model for predicting treatment response and prognosis in patients who received immunotherapy or chemotherapy, we collected gene expression data and corresponding clinical information from several cohorts, including 5 external datasets (GSE135222 [NSCLC], GSE78220 [melanoma], POPLAR, OAK and IMvigor210) and an internal cohort (ORIENT-11). POPLAR (phase 2) and OAK (phase 3) are multicenter, open-label, randomized controlled trials, in which previously treated NSCLC patients were administrated with atezolizumab or docetaxel.4,40 IMvigor210 is a multicenter, single-arm phase 2 trial, evaluating efficacy of atezolizumab in patients with metastatic urothelial carcinoma who have progressed on platinum-based chemotherapy.41 Gene expression data and clinical information for these three cohorts have been stored in European Genome-phenome Archive (EGA) (https://ega-archive.org/) with controlled access (EGA: EGAS00001004343 and EGAS00001005013).31,32 We submitted data access applications to the corresponding Data Access Committee (DAC) and downloaded relevant data using Python after approval. ORIENT-11 is a multi-center, randomized, double-blind, phase 3 study that compared sintilimab or placebo, in combination with pemetrexed and platinum, for locally advanced or metastatic nonsquamous NSCLC in China.42 We have prospectively collected the baseline demographical and clinicopathological data as well as follow-up and survival information for patients enrolled in the ORIENT-11 study. Formalin-fixed, paraffin-embedded baseline tumor samples have also been prospectively collected to perform PD-L1 IHC analysis (evaluated using 22C3 pharmDx, Agilent Technologies) and RNA sequencing on the NovaSeq 6000 system (Illumina), as previously described.18 Baseline clinicopathological characteristics of patients in the POPLAR, IMvigor210 and ORIENT-11 cohorts are presented in Tables S12 and S13.
Study protocol of ORIENT-11 was approved by the respective institutional review boards and ethics committees and all participants provided written informed consent. Ethic approval and patient informed consents for TCGA, GEO and EGA were waived due to their public availability. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013). A flowchart delineating process of model building and our data analysis protocol is presented in Figure 1.
Method details
DEGs associated with TME scores and weighted gene co-expression network analysis
TCGA-LUSC was utilized as a training set to evaluate the prognostic TME-related genes from expression matrix. Immune scores and stromal scores were calculated based on ESTIMATE algorithm via the R package “estimate”.19 Tumor purity was inferred by ESTIMATE scores that generated from the combination of immune and stromal scores.19 Based on the immune and stromal scores calculated for each sample, patients were stratified into two groups (high vs. low immune scores or high vs. low stromal scores) through methodology of maximally selected rank statistics which can be employed using R package “maxstat”33 to select optimum prognosis-based cut-off. Differentially expressed genes (DEGs) between the low and high immune/stromal scores were identified using R package “DESeq2”.43 An adjusted p-value of <0.05 and simultaneously an absolute value of log2(fold change) of >1 were set as the significant criteria to filter DEGs. Weighted gene co-expression network analysis (WCGNA)34 was performed to investigate co-expressed gene modules correlated with immune/stromal scores and modules with |correlation coefficient| > 0.45 were considered as strong TME-related gene modules. we started with constructing a co-expression network based on interaction patterns among genes in the TCGA-LUSC cohort and genes with similar interaction patterns were allocated into a module. We then calculated correlation coefficients between gene modules and immune scores as well as stromal scores (inputted as continuous variables), through which co-expressed modules were connected to phenotypic variables.
Development and validation of TME-related risk scoring model
The intersections of DEGs and robust TME-related gene modules generated by WGCNA were input into the LASSO regression analysis which was performed through R package “glmnet” to identify candidate model genes. The LASSO technique is able to modulate complexity and reduce redundant variables when constructing a general linear model. A method combined with LASSO and Cox regression model can be used to screen prognostic biomarkers.44 The optimal penalty parameter λ was identified through running 10-fold cross-validations. Generated candidate genes were dichotomized (0 for low expression and 1 for high expression) using X-tile software39 and univariate Cox analyses were performed to evaluate association of these candidate genes with OS. Gene variables with a univariate p-value of <0.15 were integrated into an Akaike information criterion (AIC)-based multivariate stepwise Cox regression model to establish the final TMErisk model. The TMErisk for each sample was calculated by the formula: TMErisk = , where indicates gene expression level of a given patient and is the corresponding model coefficient for this gene. Patients were then divided into two groups, low- and high-TMErisk groups, based on maximally selected rank statistics method. Value of TMErisk for predicting OS was then validated in four LUSC cohorts (GEO: GSE81089, GSE30219, GSE37745 and GSE157011) and two LUAD cohorts (TCGA-LUAD and GSE31210), during which cut-points were derived in corresponding datasets using maximally selected rank statistics method (Table S14).
TME landscape and pathway enrichment analysis
CIBERSORT,45 EPIC,46 and xCell47 were utilized to extrapolate infiltrating abundance of immune and stromal cells. The R package “immunedeconv” provides a unified access to these algorithms.35 Expression levels of chemokine genes and immune-related genes48 were also compared to evaluate TME signature between the low- and high-TMErisk groups. In addition, we analyzed association of gene signatures in the TMErisk model with immune-related genes using Pearson method and results were visualized by R package “ggpubr”. DEGs between the low- and high-TMErisk groups were identified using R package “DESeq2”, and an adjusted p-value of <0.05 and simultaneously an absolute value of log2(fold change) of >0 were set as the significant criteria to filter DEGs. Gene set enrichment analysis of these DEGs were conducted via “compareCluster” function in R package “clusterProfiler” to analyze biological functions associated with TMErisk.36 The GMT file named “h.all.v2022.1.Hs.symbols.gmt”, containing 50 hallmark gene sets that represent specific biological states or processes, was downloaded from the Molecular Signatures Database (MSigDB) (MSigDB: http://www.gsea-msigdb.org/gsea/msigdb/collections.jsp#H).49
Prediction of drug response in the low- and high-TMErisk groups
The Genomics of Drug Sensitivity in Cancer (GDSC)50 and Cancer Therapeutics Response Portal (CTRP)51,52 databases are two important resources for predicting drug response and therapeutic biomarkers based on high-throughput cancer cell line screening data. Data from the two databases have been processed and included in the R package “oncoPredict”.37 The function calcPhenotype in “oncoPredict” package utilized a pipeline to predict therapeutic response based on baseline tumor expression data. Briefly, gene expression and drug screening data from these databases are used as training sets to build ridge regression model that can be applied to a new gene expression matrix to generate drug sensitivity prediction.37 We used the R package “oncoPredict” to infer the half maximal inhibitory concentration (IC50) value of various drugs for TCGA-LUSC patients. Predictive potential of TMErisk for immunotherapy was inferred by the tumor immune dysfunction and exclusion (TIDE) algorithm. TIDE is a computational approach to reflect immune evasion capacity of tumor and a lower TIDE score indicates a favorable response to immunotherapy.38
Evaluation of TMErisk in immunotherapy/chemotherapy cohorts
We validated predictive value of TMErisk for predicting immunotherapy or chemotherapy response and patient survival in five external public datasets (GSE135222, GSE78220, OAK, POPLAR and IMvigor210) and an in-house cohort (ORIENT-11). Since GSE135222 lacks response data, we failed to investigate association of TMErisk with immunotherapeutic response in this cohort. As per the established TMErisk model, patients were scored and then stratified into two groups using maximally selected rank statistics method in corresponding datasets (Table S14).
Quantification and statistical analysis
Continuous variables between two groups were compared using Wilcoxon rank-sum test or unpaired t-test depending on the normality of distribution. Fisher’s exact test or Chi-square test, where appropriate, was used to examine contingency tables. Correlation between two continuous variables was examined by Pearson method for normally distributed variables, and Spearman method for non-normally distributed variables. The Kaplan–Meier method was conducted to generate survival curves between different TMErisk groups in each cohort and the statistical difference was compared using log rank test. Association of TMErisk and other baseline factors with OS or progression-free survival (PFS) was investigated by univariate Cox regression models and multivariate Cox analysis was used to determine independent factors. Aforementioned survival analyses were conducted via R package “survival” and “survminer”. We utilized concordance index (C-index) and time-dependent area under the receiver operating characteristic curve (time-dependent AUC) to evaluate predictive accuracy of TMErisk, PD-L1 and TMB for OS and PFS. The distribution of response categories between the low- and high-TMErisk groups were visualized through percentage stacked bar charts generated by R package “ggplot2”. All statistical analyses and visualization were conducted using R software (http://www.R-project.org, version 4.1.0) and two-sided p-value of less than 0.05 was considered statistically significant.
Additional resources
Clinical trial registry number of ORIENT-11: NCT03607539, https://clinicaltrials.gov/ct2/show/NCT03607539.
Acknowledgments
This study was supported by National Natural Science Foundation of China (grants numbers 81972898, 82172713, and 82072558), the Natural Science Foundation of Guangdong Province (2023B1515020008) and the Fundamental Research Funds for the Central Universities, Sun Yat-sen University (22ykqb15).
Study protocol for the ORIENT-11 clinical trial was approved by the respective institutional review boards and ethics committees and all participants provided written informed consent. Ethics approval and patient informed consents for TCGA, GEO and EGA were waived due to their public availability.
Author contributions
S.H., L.Z. and Y.Y.: Conceptualization, Study design, Methodology, Project administration, Data Curation, Funding acquisition, Supervision. L.-N.H., H.L., S.F. and W.D.: Resources, Investigation, Data Curation, Formal analysis. T.C., H.Y., Y.J., H.Z. and Y.H.: Data Curation, Visualization. W.F., Y.Z. and Z.L.: Software, Validation. L.-N.H. and H.L.: Writing - original draft. S.H., Y.H.W. and Y.X.W.: Writing - Reviewing & Editing. All authors: Final approval of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: June 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107058.
Contributor Information
Yunpeng Yang, Email: yangyp@sysucc.org.cn.
Li Zhang, Email: zhangli6@mail.sysu.edu.cn.
Shaodong Hong, Email: hongshd@sysucc.org.cn.
Supplemental information
Data and code availability
-
•
Gene expression data and clinical data for the TCGA-LUSC, TCGA-LUAD and validation cohorts from GEO (GEO: GSE30219, GSE37745, GSE157011, GSE31210, GSE30219, GSE37745, GSE157011, GSE31210) are publicly available. RNA-seq data and corresponding clinical data for OAK, POPLAR and IMvigor210 are available in the European Genome-phenome Archive (EGA) (https://ega-archive.org/) with restricted access (EGA: EGAS00001004343 and EGAS00001005013). These accession numbers for the datasets are listed in the key resources table. RNA-seq data for ORIENT-11 are available from the lead contact upon reasonable request.
-
•
This paper dose not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Blumenthal G.M., Zhang L., Zhang H., Kazandjian D., Khozin S., Tang S., Goldberg K., Sridhara R., Keegan P., Pazdur R. Milestone analyses of immune checkpoint inhibitors, targeted therapy, and conventional therapy in metastatic non-small cell lung cancer trials: a meta-analysis. JAMA Oncol. 2017;3:e171029. doi: 10.1001/jamaoncol.2017.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mok T.S.K., Wu Y.-L., Kudaba I., Kowalski D.M., Cho B.C., Turna H.Z., Castro G., Srimuninnimit V., Laktionov K.K., Bondarenko I., et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. 2019;393:1819–1830. doi: 10.1016/s0140-6736(18)32409-7. [DOI] [PubMed] [Google Scholar]
- 3.Gettinger S., Horn L., Jackman D., Spigel D., Antonia S., Hellmann M., Powderly J., Heist R., Sequist L.V., Smith D.C., et al. Five-year follow-up of nivolumab in previously treated advanced non-small-cell lung cancer: results from the ca209-003 study. J. Clin. Oncol. 2018;36:1675–1684. doi: 10.1200/jco.2017.77.0412. [DOI] [PubMed] [Google Scholar]
- 4.Rittmeyer A., Barlesi F., Waterkamp D., Park K., Ciardiello F., von Pawel J., Gadgeel S.M., Hida T., Kowalski D.M., Dols M.C., et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet (London, England) 2017;389:255–265. doi: 10.1016/s0140-6736(16)32517-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brahmer J., Reckamp K.L., Baas P., Crinò L., Eberhardt W.E.E., Poddubskaya E., Antonia S., Pluzanski A., Vokes E.E., Holgado E., et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Y.L., Lu S., Cheng Y., Zhou C., Wang J., Mok T., Zhang L., Tu H.Y., Wu L., Feng J., et al. Nivolumab versus docetaxel in a predominantly Chinese patient population with previously treated advanced NSCLC: CheckMate 078 randomized phase III clinical trial. J. Thorac. Oncol. 2019;14:867–875. doi: 10.1016/j.jtho.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Pai-Scherf L., Blumenthal G.M., Li H., Subramaniam S., Mishra-Kalyani P.S., He K., Zhao H., Yu J., Paciga M., Goldberg K.B., et al. FDA approval summary: pembrolizumab for treatment of metastatic non-small cell lung cancer: first-line therapy and beyond. Oncol. 2017;22:1392–1399. doi: 10.1634/theoncologist.2017-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yarchoan M., Hopkins A., Jaffee E.M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 2017;377:2500–2501. doi: 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hellmann M.D., Ciuleanu T.E., Pluzanski A., Lee J.S., Otterson G.A., Audigier-Valette C., Minenza E., Linardou H., Burgers S., Salman P., et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018;378:2093–2104. doi: 10.1056/NEJMoa1801946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 11.Quail D.F., Joyce J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Binnewies M., Roberts E.W., Kersten K., Chan V., Fearon D.F., Merad M., Coussens L.M., Gabrilovich D.I., Ostrand-Rosenberg S., Hedrick C.C., et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018;24:541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi H., Na K.J. Integrative analysis of imaging and transcriptomic data of the immune landscape associated with tumor metabolism in lung adenocarcinoma: clinical and prognostic implications. Theranostics. 2018;8:1956–1965. doi: 10.7150/thno.23767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salmon H., Remark R., Gnjatic S., Merad M. Host tissue determinants of tumour immunity. Nat. Rev. Cancer. 2019;19:215–227. doi: 10.1038/s41568-019-0125-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denton A.E., Roberts E.W., Fearon D.T. Stromal cells in the tumor microenvironment. Adv. Exp. Med. Biol. 2018;1060:99–114. doi: 10.1007/978-3-319-78127-3_6. [DOI] [PubMed] [Google Scholar]
- 16.Mascaux C., Angelova M., Vasaturo A., Beane J., Hijazi K., Anthoine G., Buttard B., Rothe F., Willard-Gallo K., Haller A., et al. Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature. 2019;571:570–575. doi: 10.1038/s41586-019-1330-0. [DOI] [PubMed] [Google Scholar]
- 17.Sun H., Liu S.Y., Zhou J.Y., Xu J.T., Zhang H.K., Yan H.H., Huan J.J., Dai P.P., Xu C.R., Su J., et al. Specific TP53 subtype as biomarker for immune checkpoint inhibitors in lung adenocarcinoma. EBioMedicine. 2020;60:102990. doi: 10.1016/j.ebiom.2020.102990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y., Sun J., Wang Z., Fang J., Yu Q., Han B., Cang S., Chen G., Mei X., Yang Z., et al. Updated overall survival data and predictive biomarkers of sintilimab plus pemetrexed and platinum as first-line treatment for locally advanced or metastatic nonsquamous NSCLC in the phase 3 ORIENT-11 study. J. Thorac. Oncol. 2021;16:2109–2120. doi: 10.1016/j.jtho.2021.07.015. [DOI] [PubMed] [Google Scholar]
- 19.Yoshihara K., Shahmoradgoli M., Martínez E., Vegesna R., Kim H., Torres-Garcia W., Treviño V., Shen H., Laird P.W., Levine D.A., et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S., Yu G., Liu L., Zou X., Zhou L., Hu E., Song Y. Identification of prognostic stromal-immune score-based genes in hepatocellular carcinoma microenvironment. Front. Genet. 2021;12:625236. doi: 10.3389/fgene.2021.625236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saleh R., Elkord E. Acquired resistance to cancer immunotherapy: role of tumor-mediated immunosuppression. Semin. Cancer Biol. 2020;65:13–27. doi: 10.1016/j.semcancer.2019.07.017. [DOI] [PubMed] [Google Scholar]
- 22.Dongre A., Weinberg R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019;20:69–84. doi: 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- 23.Wang G., Xu D., Zhang Z., Li X., Shi J., Sun J., Liu H.Z., Li X., Zhou M., Zheng T. The pan-cancer landscape of crosstalk between epithelial-mesenchymal transition and immune evasion relevant to prognosis and immunotherapy response. NPJ Precis. Oncol. 2021;5:56. doi: 10.1038/s41698-021-00200-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mezheyeuski A., Bergsland C.H., Backman M., Djureinovic D., Sjöblom T., Bruun J., Micke P. Multispectral imaging for quantitative and compartment-specific immune infiltrates reveals distinct immune profiles that classify lung cancer patients. J. Pathol. 2018;244:421–431. doi: 10.1002/path.5026. [DOI] [PubMed] [Google Scholar]
- 25.Rousseaux S., Debernardi A., Jacquiau B., Vitte A.L., Vesin A., Nagy-Mignotte H., Moro-Sibilot D., Brichon P.Y., Lantuejoul S., Hainaut P., et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci. Transl. Med. 2013;5:186ra66. doi: 10.1126/scitranslmed.3005723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Botling J., Edlund K., Lohr M., Hellwig B., Holmberg L., Lambe M., Berglund A., Ekman S., Bergqvist M., Pontén F., et al. Biomarker discovery in non-small cell lung cancer: integrating gene expression profiling, meta-analysis, and tissue microarray validation. Clin. Cancer Res. 2013;19:194–204. doi: 10.1158/1078-0432.Ccr-12-1139. [DOI] [PubMed] [Google Scholar]
- 27.Bueno R., Richards W.G., Harpole D.H., Ballman K.V., Tsao M.S., Chen Z., Wang X., Chen G., Chirieac L.R., Chui M.H., et al. Multi-institutional prospective validation of prognostic mRNA signatures in early stage squamous lung cancer (alliance) J. Thorac. Oncol. 2020;15:1748–1757. doi: 10.1016/j.jtho.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okayama H., Kohno T., Ishii Y., Shimada Y., Shiraishi K., Iwakawa R., Furuta K., Tsuta K., Shibata T., Yamamoto S., et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72:100–111. doi: 10.1158/0008-5472.Can-11-1403. [DOI] [PubMed] [Google Scholar]
- 29.Jung H., Kim H.S., Kim J.Y., Sun J.M., Ahn J.S., Ahn M.J., Park K., Esteller M., Lee S.H., Choi J.K. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 2019;10:4278. doi: 10.1038/s41467-019-12159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G., et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patil N.S., Nabet B.Y., Müller S., Koeppen H., Zou W., Giltnane J., Au-Yeung A., Srivats S., Cheng J.H., Takahashi C., et al. Intratumoral plasma cells predict outcomes to PD-L1 blockade in non-small cell lung cancer. Cancer Cell. 2022;40:289–300.e4. doi: 10.1016/j.ccell.2022.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Banchereau R., Leng N., Zill O., Sokol E., Liu G., Pavlick D., Maund S., Liu L.F., Kadel E., 3rd, Baldwin N., et al. Molecular determinants of response to PD-L1 blockade across tumor types. Nat. Commun. 2021;12:3969. doi: 10.1038/s41467-021-24112-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hothorn T., Lausen B.J.R.N. Maximally selected rank statistics in R. 2002;2:3–5. [Google Scholar]
- 34.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sturm G., Finotello F., List M. Immunedeconv: an R package for unified access to computational methods for estimating immune cell fractions from bulk RNA-sequencing data. Methods Mol. Biol. 2020;2120:223–232. doi: 10.1007/978-1-0716-0327-7_16. [DOI] [PubMed] [Google Scholar]
- 36.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maeser D., Gruener R.F., Huang R.S. oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Briefings Bioinf. 2021;22:bbab260. doi: 10.1093/bib/bbab260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang P., Gu S., Pan D., Fu J., Sahu A., Hu X., Li Z., Traugh N., Bu X., Li B., et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018;24:1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Camp R.L., Dolled-Filhart M., Rimm D.L. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin. Cancer Res. 2004;10:7252–7259. doi: 10.1158/1078-0432.Ccr-04-0713. [DOI] [PubMed] [Google Scholar]
- 40.Fehrenbacher L., Spira A., Ballinger M., Kowanetz M., Vansteenkiste J., Mazieres J., Park K., Smith D., Artal-Cortes A., Lewanski C., et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet (London, England) 2016;387:1837–1846. doi: 10.1016/s0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 41.Rosenberg J.E., Hoffman-Censits J., Powles T., van der Heijden M.S., Balar A.V., Necchi A., Dawson N., O'Donnell P.H., Balmanoukian A., Loriot Y., et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet (London, England) 2016;387:1909–1920. doi: 10.1016/s0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Y., Wang Z., Fang J., Yu Q., Han B., Cang S., Chen G., Mei X., Yang Z., Ma R., et al. Efficacy and safety of sintilimab plus pemetrexed and platinum as first-line treatment for locally advanced or metastatic nonsquamous NSCLC: a randomized, double-blind, phase 3 study (Oncology pRogram by InnovENT anti-PD-1-11) J. Thorac. Oncol. 2020;15:1636–1646. doi: 10.1016/j.jtho.2020.07.014. [DOI] [PubMed] [Google Scholar]
- 43.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tibshirani R. The lasso method for variable selection in the Cox model. Stat. Med. 1997;16:385–395. doi: 10.1002/(sici)1097-0258(19970228)16:4<385::aid-sim380>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 45.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Racle J., de Jonge K., Baumgaertner P., Speiser D.E., Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife. 2017;6:e26476. doi: 10.7554/eLife.26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aran D., Hu Z., Butte A.J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu J., Li L., Zhang H., Zhao Y., Zhang H., Wu S., Xu B. A risk model developed based on tumor microenvironment predicts overall survival and associates with tumor immunity of patients with lung adenocarcinoma. Oncogene. 2021;40:4413–4424. doi: 10.1038/s41388-021-01853-y. [DOI] [PubMed] [Google Scholar]
- 49.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang W., Soares J., Greninger P., Edelman E.J., Lightfoot H., Forbes S., Bindal N., Beare D., Smith J.A., Thompson I.R., et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41:D955–D961. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seashore-Ludlow B., Rees M.G., Cheah J.H., Cokol M., Price E.V., Coletti M.E., Jones V., Bodycombe N.E., Soule C.K., Gould J., et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 2015;5:1210–1223. doi: 10.1158/2159-8290.Cd-15-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Basu A., Bodycombe N.E., Cheah J.H., Price E.V., Liu K., Schaefer G.I., Ebright R.Y., Stewart M.L., Ito D., Wang S., et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;154:1151–1161. doi: 10.1016/j.cell.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Gene expression data and clinical data for the TCGA-LUSC, TCGA-LUAD and validation cohorts from GEO (GEO: GSE30219, GSE37745, GSE157011, GSE31210, GSE30219, GSE37745, GSE157011, GSE31210) are publicly available. RNA-seq data and corresponding clinical data for OAK, POPLAR and IMvigor210 are available in the European Genome-phenome Archive (EGA) (https://ega-archive.org/) with restricted access (EGA: EGAS00001004343 and EGAS00001005013). These accession numbers for the datasets are listed in the key resources table. RNA-seq data for ORIENT-11 are available from the lead contact upon reasonable request.
-
•
This paper dose not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.