

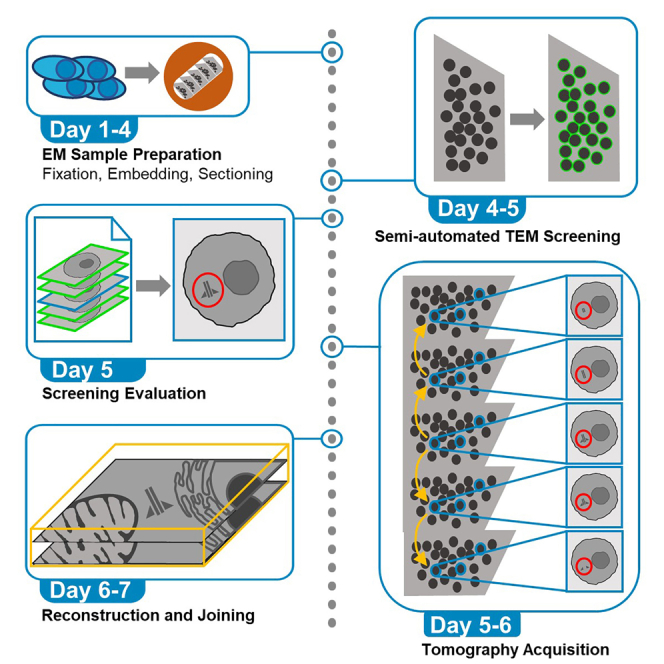
Summary
Electron microscopy is the gold standard to characterize cellular ultrastructure. However, production of significant morphometrical data is highly limited by acquisition time. Here, we describe a semi-automated high-throughput strategy using single-axis serial section electron tomography to investigate and analyze centriole ultrastructure in bone-marrow-derived, primary human CD138pos plasma cells. The protocol comprises steps for electron microscopy sample preparation, semi-automated transmission electron microscopy screening, and screening evaluation for cells of interest. Thereafter, we detail tomography acquisition, data reconstruction, and joining.
For complete details on the use and execution of this protocol, please refer to Dittrich et al.1
Subject areas: High Throughput Screening, Microscopy
Graphical abstract
Highlights
-
•
High-throughput production of volumetric EM data
-
•
From sample acquisition to tomography data in one week
-
•
Protocol can be used in both cell lines and primary patient material
-
•
Protocol is adaptable to various organelles and subcellular features
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Electron microscopy is the gold standard to characterize cellular ultrastructure. However, production of significant morphometrical data is highly limited by acquisition time. Here, we describe a semi-automated high-throughput strategy using single-axis serial section electron tomography to investigate and analyze centriole ultrastructure in bone-marrow-derived, primary human CD138pos plasma cells. The protocol comprises steps for EM sample preparation, semi-automated transmission electron microscopy screening, and screening evaluation for cells of interest. Thereafter, we detail tomography acquisition, data reconstruction, and joining.
Before you begin
The protocol below describes the specific steps for studying centriole ultrastructure using bone marrow derived, primary human CD138pos plasma cells. We used MACS-based cell sorting in order to achieve sample purities of >90%. Additionally, we have used the protocol for bone marrow-derived, primary human CD138neg mononuclear cells and cultivated U2OS cells. The workflow is applicable to cell features and/or organelles beyond centrioles if they can be visualized in electron microscopy.
Institutional permissions
Our study was performed in accordance with the principles of the Declaration of Helsinki. Both the Ethics Committee of the University of Heidelberg as well as the European Molecular Biology Laboratory (EMBL) review board approved the study (Ethics Committee of the University of Heidelberg approval reference number: S-206/2011; EMBL BIAC application number: 2019-005).
If this protocol is to be used with specimens of primary human origin, experiments must be performed according to institutional and/or national guidelines, and users should acquire all necessary permissions in advance.
Preparation of reagents for chemical fixation, agarose pre-embedding, and Epon embedding
Timing: 20 min
Prepare stock solutions and buffers if necessary. A complete list of reagents and recipes can be found in the ‘key resources table’ and ‘materials and equipment’ section.
Microscope and software set-up
We used two different electron microscopes for this workflow: A JEOL JEM 2100 Plus with a faster moving stage and a ThermoFisher / FEI Tecnai F30 for high-resolution tomography imaging at 15,500× magnification. Both microscopes were controlled using SerialEM software.2 The main advantage using two individual microscopes is that since most of the acquisition is done automatically, you can analyze multiple samples at the same time (i.e., while you screen through one sample, tomography acquisition can be set up for the other, and vice versa). Handling multiple samples simultaneously enables even faster throughput of samples than described by the timing estimations we provide in this protocol. However, this workflow can also be applied using a single electron microscope running on SerialEM. Where applicable, instructions for a single-microscope setup are provided in the respective sections in the step-by-step method details.
SerialEM supports Windows versions starting from Windows XP. As for hardware, your operating system should have sufficient resources to display stitched montage images (typically 25,000 × 20,000 pixels, 1 GB file size).
-
1.
If not available already, install SerialEM on your TEM-PC. For instructions on how to set up SerialEM with your electron microscope, refer to the link provided in the key resources table.
-
2.
To enable automated acquisition, you will need to set up a python environment (ideally Python 3.6 or higher) on your TEM-PC and install the pyEM software package. You can find instructions on how to implement all necessary files on Github (refer to key resources table).
-
3.
Additionally, KNIME (Konstanz Information Miner),3 ImageJ,4 and the IMOD5 software package should be downloaded and set up on your PC.
-
4.
Once all necessary software is installed, implement all necessary scripts and tools needed for this workflow into the SerialEM graphical user interface for easy access. Scripts and tools can be downloaded from the pyEM repository and the SerialEM Script Repository, respectively. Links to both databases are provided in the key resources table.
CRITICAL: Correct installation of and communication between software is crucial for this workflow. We have therefore created a trial navigator file to test your setup (downloadable from Mendeley Data: https://doi.org/10.17632/ct23zrnxc2.1).
-
5.Lastly, you should set up Imaging states in SerialEM and save these states for easier access throughout your sessions. For this workflow, you will need different imaging conditions:
-
a.A very low magnification to obtain overviews of the whole grid. This will help you to assess its overall quality and to navigate through long distances if needed. Use around 80× magnification.
-
b.For acquisition of section overviews as montaged image, you will need another low magnification state. We used 400× magnification and 450× magnification, respectively.
-
c.SerialEM’s ‘Realign to item’ feature is used frequently throughout this workflow. Since this feature cannot be used with low magnification images that were obtained without the objective aperture, you will need an imaging state that uses the objective aperture at a relatively low magnification. We used 1000× and 2300×, respectively.
-
d.For screening, you will need a magnification that provides you with enough details to identify your feature of interest in the cell. We used 3000× magnification to screen for centrioles in human plasma cells. Depending on cell type and feature of interest, you might need a higher or lower magnification.
-
e.Lastly, you will need a very high-magnification imaging state for tomography. We used 15,500× magnification.
-
f.Once you have prepared a set of imaging states, save them as ‘tomo_states’ to be able to quickly access them each time you use this workflow.
-
a.
Note: Imaging states can be imported into SerialEM as text files. We have provided the settings we used on both microscopes as an example for download on Mendeley Data:https://doi.org/10.17632/ct23zrnxc2.1. If you want, you can use them as a starting point for defining imaging states on your setup.
Reconstruction and joining
-
6.
Install IMOD on the hardware used for processing the tilt series.
-
7.
Perform a manual reconstruction of one representative tilt series to determine the relevant parameters. Export these as a batch directive.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human bone marrow | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Paraformaldehyde 16%, EM grade | Electron Microscopy Sciences | Cat#15710 |
| Glutaraldehyde 25%, EM grade | Electron Microscopy Sciences | Cat#16220 |
| Sodium cacodylate trihydrate | Sigma-Aldrich | Cat#C0250 |
| PBS buffer (10× Dulbecco’s) powder | ITW Reagents | Cat#A0965 |
| Evans Blue | Sigma-Aldrich | Cat#E2129 |
| Certified low-melt agarose | Bio-Rad | Cat#161-3111 |
| Glycid ether 100 | Serva | Cat#21045 |
| Dodecenylsuccinic acid anhydride (DDSA) | Serva | Cat#20755 |
| Methylnadic anhydride (NMA) | Serva | Cat#29452 |
| 2,4,6-Tris(dimethylaminomethyl)phenol (DMP-30) | Serva | Cat#36975 |
| Osmium tetroxide in H2O | Electron Microscopy Sciences | Cat#19150 |
| Uranyl acetate in H2O | Serva | Cat#77870 |
| Deposited data | ||
| Additional supplemental data | This paper | Mendeley Data: https://doi.org/10.17632/ct23zrnxc2.1 |
| SerialEM scripts | This paper; Schorb et al.6; Dittrich et al.1 | https://serialemscripts.nexperion.net/ |
| SerialEM tools | This paper; Schorb et al.6; Dittrich et al.1 | Github: https://git.embl.de/schorb/pyem/-/tree/master/applications |
| Software and algorithms | ||
| Konstanz Information Miner (KNIME) | Berthold et al.3 | https://www.knime.com/ |
| SerialEM | Mastronarde2; Schorb et al.6 | https://bio3d.colorado.edu/SerialEM/ |
| pyEM | Schorb et al.6 | https://git.embl.de/schorb/pyem |
| IMOD – 4.10.42 | Kremer et al., 1996; Mastronarde and Held7 | https://bio3d.colorado.edu/imod/ |
| ImageJ (Fiji) – 1.53 | Schneider et al.4 | https://imagej.net/software/fiji/ |
| Other | ||
| Protein LoBind Tubes 1.5 mL | Eppendorf | Cat#0030108116 |
| Serial Sectioning Protocol | SynapseWeb, K. M. Harris, PI; University of Texas | https://synapseweb.clm.utexas.edu/serial-sectioning |
Materials and equipment
-
•Stock solutions and buffers:
-
•0.2 M cacodylate buffer: Add 10.7 g sodium cacodylate trihydrate to 200 mL dH2O. Adjust to pH 7.4 with HCl. Fill up with dH2O to a total amount of 250 mL. Store at 4°C for weeks to months.
-
•0.1 M cacodylate buffer: Mix 0.2 M cacodylate buffer (recipe above) with dH2O in a ratio of 1:1.CRITICAL: Cacodylate is toxic and should only be handled under the fume hood and disposed of according to your local health and safety requirements.
-
•1× Phosphate buffered saline (PBS): Create 10× PBS according to manufacturer’s instructions. Dilute with bidistilled water to obtain a final concentration of 1×. PBS can be stored at 2°C–8°C for 12 months.
-
•2% low melting point agarose: Under the fume hood, add 0.2 g of low melting point agarose to 10 mL of 0.1 M cacodylate buffer. Dissolve the agarose at 60°C while stirring. Cool down and store at 4°C for several weeks.
-
•Epon mixture A: Combine 62 mL (37.2 g) glycid ether 100 and 100 mL (50.1 g) DDSA and let mix well.
-
•Epon mixture B: Combine 100 mL (60 g) glycid ether 100 and 89 mL (54.25 g) MNA and let mix well.Note: Both Epon mixtures are stable separately for weeks at 4°C or for a few months if stored under argon at −20°C.
-
•
-
•
EM fixative
| Reagent | Final concentration | Amount |
|---|---|---|
| 25% glutaraldehyde (GA) | 2.5% | 1.00 mL |
| 20% paraformaldehyde (PFA) | 2% | 1.25 mL |
| 0.2 M cacodylate buffer, pH 7.4 | 0.1 M | 5 mL |
| dH2O | N/A | 2.75 mL |
| Total | N/A | 10 mL |
Fixative should always be prepared freshly on the day of the experiment.
Step-by-step method details
Day 1–2: Chemical fixation
In this first step, a standard chemical fixation is performed using glutaraldehyde and paraformaldehyde to prepare cells for electron microscopy. The protocol was optimized so that it is usable with very sparse amounts of cells.
-
1.
Prepare EM fixative according to the recipe provided above in the ‘materials and equipment’ section.
-
2.
Transfer a minimum of 100.000 live cells into a 1.5 mL Eppendorf protein LoBind tube.
Note: Usage of protein LoBind tubes is crucial especially when working with small samples to preserve as many cells as possible during preparation steps.
-
3.
Fill up the Eppendorf tube with 1× PBS.
-
4.
Spin the cells down for 10 min with 1000 g at 4°C in a swing-out centrifuge.
-
5.
Locate the newly formed cell pellet on the bottom of the tube. Gently remove and discard supernatant, then add 1 mL of 4°C cold fixative and resuspend well.
Note: Always leave the cell pellet slightly covered in liquid when discarding supernatant after centrifugation. This will prevent dehydration of the sample.
Note: If available, use a glass pipette since cells are more likely to adhere to plastic pipettes.
-
6.
Incubate at 20°C for 2 min, then centrifuge again (swing-out, 10 min, 1000 g, 4°C).
-
7.
Remove supernatant, then add 1 mL of fixative and resuspend well.
-
8.
Continue fixation in the fridge at 4°C for 12 h.
-
9.
On the next day, centrifuge again (swing-out, 10 min, 1000 g, 4°C).
-
10.
Remove supernatant and add 1 mL of 0.1 M cacodylate buffer (pH 7.4). Store sample in the fridge at 4°C.
Day 2: Agarose pre-embedding
In this step, cells are embedded into agarose in order to form a stable, dense pellet for consecutive Epon embedding and sectioning.
-
11.Preparation:
-
a.Prewarm a 37°C water bath, heat up a heating block to 37°C and prewarm a swing-out centrifuge to 37°C.
-
b.Melt low melting point agarose in a microwave (max. 10 s) and keep it in water bath at 37°C.
-
c.Cut 4 mm of pipette tips off to facilitate handling of low melting point agarose
-
d.Pre-heat pipette tips and 200 μL PCR tubes on the heating block.
-
a.
Note: Give equipment at least 30 min to reach a stable temperature. This is paramount for the formation of a proper cell pellet.
-
12.
Stain cells by adding 50 μL of 20 mg/mL Evans Blue in 0.1 M cacodylate buffer to your sample. Incubate at 20°C for 20 min.
-
13.Wash your sample 2–3 times until supernatant is almost clear and a blue pellet is visible on the bottom of the tube:
-
a.Centrifuge (swing-out, 10 min, 1000 g, 37°C).
-
b.Remove supernatant and add 1 mL of 0.1 M cacodylate buffer.
-
a.
-
14.
Centrifuge again (swing-out, 10 min, 1000 g, 37°C).
-
15.
Remove supernatant completely and immediately add 150 μL of low-melting agarose. Carefully resuspend once and put agarose and cells into 200 μL PCR tube.
-
16.
Centrifuge (swing-out, 10 min, 1000 g, 37°C).
Note: It is crucial to carry out steps 15 and 16 as quickly as possible. If the agarose cools down too much, cells cannot make it to the bottom of the tube during centrifugation and you will not obtain a usable cell pellet.
-
17.
Let the pellet solidify on ice for 10 min.
-
18.
Continue with Epon embedding or store the pre-embedded sample in the fridge at 4°C.
Day 2–4: Post-fixation, dehydration and Epon embedding
During post-fixation, heavy metals are used so that cells can be visualized in EM later with adequate contrast. Afterwards, water is replaced by acetone and cells are embedded into Epon resin as preparation for sectioning. You will end up with at least one Epon block per sample. These steps are carried out using a temperature-controlled microwave (Pelco BioWavePro) at 23°C equipped with a vacuum pump to speed up the embedding process.
Note: You can find a detailed microwave protocol on Mendeley Data: https://doi.org/10.17632/ct23zrnxc2.1
-
19.Preparation:
-
a.Place all components (Epon mixture A, Epon mixture B, DMP-30, and acetone (50%, 70%, 90%, 100%) at 20°C for 1 h.
-
b.Calculate the necessary amount of resin: You will need 15 mL Epon resin per sample.
-
c.In a 50 mL Falcon tube, mix Epon mixtures A and B according to “hard” formula:
-
i.Per 15 mL resin, you will need 4.5 mL (4.85 g) Epon mixture A and 10.5 mL (12.72 g) Epon mixture B.
-
ii.Mix them together in a 50 mL Falcon Tube.
-
iii.Let the components mix well for 30 min, then add 0.225 mL DMP-30 per 15 mL resin. Let mix well again.Note: Mix gently and do not shake to avoid the formation of small air bubbles inside the resin.
-
i.
-
d.Prepare at least 2 mL each of the following EPON concentrations (v/v%) in 100% acetone: 10%, 30%, 50%, 70%, 90%
-
a.
-
20.
On a glass dish under the fume hood, cut the PCR tube to free your cell pellet. Add a few drops of 0.1 M cacodylate buffer to prevent dehydration. If necessary, trim the pellet into appropriate pieces with a scalpel.
Note: Trimming larger pellets into small pieces (e.g., cubes of 0.5 mm side length) is necessary to ensure penetration of the post-fixation agents.
-
21.
Carefully transfer the cell pellet or its pieces into a new 1.5 mL Eppendorf tube containing 1 mL 0.1 M cacodylate buffer.
Note: In the upcoming steps, you will be required to remove the respective supernatant and add another substance. To prevent desiccation and subsequent damage to your sample pellet, it is safer to leave a few μl of supernatant in the tube. This is especially important for steps involving acetone due to its volatility.
-
22.
Remove buffer and add 400 μL of 1% OsO4 in water.
-
23.
Transfer samples into microwave and start the protocol.
-
24.
When the microwave is finished, remove OsO4 and wash with dH2O twice.
-
25.
Remove supernatant and add dH2O again. Put into the microwave, continue protocol, then repeat this step once.
-
26.
Remove supernatant and add 500 μL 1% uranyl acetate in dH2O.
-
27.
Transfer samples into microwave and continue protocol.
-
28.
Remove uranyl acetate and wash with dH2O twice.
-
29.
Remove supernatant and add dH2O again. Put into the microwave, continue protocol, then repeat this step once.
-
30.
For dehydration, remove supernatant and add 50% acetone in water.
-
31.
Transfer to the microwave and continue protocol.
-
32.
Repeat steps 30 and 31 with 70%, 90%, and 2 × 100% acetone to complete dehydration.
-
33.
Transfer cell pellets into small glass vials containing enough 100% acetone to cover the pellet(s).
-
34.Remove acetone and start Epon embedding:
-
a.Add 10% Epon in acetone, then put the sample in the microwave.
-
b.After microwave step, remove Epon.
-
c.Repeat steps 34a and 34b with the following Epon concentrations: 30%, 50%, 70%, 90%, 3 × 100%
-
a.
-
35.
Add the pellet(s) and enough Epon to an embedding mold. Incubate at 20°C for 12 h.
-
36.
Polymerize blocks in an oven at 60°C for 48 h.
Day 4: Serial sectioning and post-staining
In this step, Epon blocks are serially sectioned and then loaded onto formvar-coated grids. A post-staining is applied to enhance contrast in electron microscopy images.
Note: We used five serial sections measuring approximately 300 × 800 μm in length and width, respectively, and of 200 nm thickness each per grid, yielding a total of 1 μm in Z dimension. You can increase or decrease your final volume by either fitting more sections on one grid, increasing the section thickness, or analyzing several consecutive grids. The choice of your section thickness depends on your feature of interest and the available microscope. Thicker sections will lead to more blur in transmission electron microscopy (TEM), and since you will use 2D TEM images to screen for your target feature, still being able to identify your regions of interest limits your section thickness choice. As an example, larger features (e.g., mitochondria) might still be visible in 300 nm thick sections, whereas for small structures such as cell junctions, you might even have to use thinner sections of 100–150 nm thickness. Additionally, your choice is limited depending on your available electron microscope since for greater section thicknesses, a higher acceleration voltage (200 or even 300 kV) might be necessary to achieve an adequate electron penetration depth, especially at high tilt angles during tomography acquisition. Thus, depending on the cellular ultrastructure you want to analyze, your sample, and the available microscope, we recommend planning your sectioning conditions accordingly.
-
37.
Sectioning: Trim the block, then make serial sections of 200 nm thickness. Carefully place 5 or more consecutive sections on a formvar coated slot grid (2 × 1 mm).
Note: If you are a novice to serial sectioning of resin blocks, you should consider practicing both sectioning and picking up of the sections with a trial sample (e.g., cells from a cell line) before working with precious, scarce samples. For very detailed information regarding ultramicrotomy techniques, refer to Kolotuev et al.8 Additionally, at SynapseWeb, the University of Texas provides a well-described step-by-step protocol for serial sectioning for beginners (link in the key resources table).
-
38.
Place parafilm on a wet surface and use a lid to cover it to create a slightly moist chamber.
-
39.Uranyl acetate staining:
-
a.Per grid you want to post-stain, prepare one row of drops (content of drops see below) to the parafilm:
-
i.50% methanol.
-
ii.2% (v/v%) uranyl acetate in 70% methanol.
-
iii.4 × 50% methanol.
-
iv.3× dH2O.Note: All solutions should be filtered using a 0.22 μm filter prior to use to avoid precipitation of reagents.
-
i.
-
b.Carefully place your grid onto the methanol drop.
-
c.Proceed to uranyl acetate. Let the grid incubate for 5 min. Cover parafilm with a lid.Note: Grids should float on the surface of the drops during incubation, sections facing down.
-
d.Rinse the grid in all 4 methanol drops, then place it in the first water drop.
-
e.Let sit in the water for 1 min with the lid on, then proceed to the next drop. Repeat until last drop of water.
-
a.
-
40.Lead citrate staining:
-
a.Add potassium hydroxide (KOH) pellets to the parafilm.
-
b.Next to that, add one drop of lead citrate and cover with a lid for 2 min.
-
c.Add 4 drops of water next to lead citrate.
-
d.Place grid on lead citrate drop and incubate for 3 min with a lid on.
-
e.Place grid in first drop of water and let it sit in the water for 1 min with the lid on, then proceed to the next drop. Repeat until last drop of water is reached.
-
f.Transfer grid to a rubber plate with the sections facing upward.
-
a.
Day 4–5: Transmission electron microscopy screening acquisition
In this step, images of each cell will be acquired on the central section at 1000× and 3000× magnification semi-automatically using SerialEM,2 KNIME,3 IMOD5 and pyEM.6 These should thus be installed on your TEM computer before you start. 3000× magnification images will be needed to screen through cells and identify cells with target features (in our case: centrioles), whereas 1000× images will be used to trace cells on adjacent sections on the same grid. For in-depth information, refer to Schorb et al.6
-
41.
Set up the electron microscope at 200 kV, cool it down and insert the grid.
-
42.
Start SerialEM and load your settings.
-
43.Record a low-magnification overview of the whole grid to assess its quality at 80×.
-
a.Select 80× magnification in the Imaging states window.
-
b.Using a continuous acquisition camera parameter set, draw a polygon around all sections on the grid using ‘add polygon’ in the Navigator window.
-
c.Press ‘stop adding’ in the Navigator window, then tick ‘Acquire (A)’ and ‘New File at item’.
-
d.Choose ‘Montaged images’ and ‘Fit montage to polygon’ in the upcoming dialogue, then press ‘OK’.
-
e.‘Montage Setup’ will appear. Select ‘Move stage instead of shifting image’, adjust the minimum overlap to 15%.Note: If the acquired image montage is not assembled well, increase overlap to 20% (or higher), and check that the microscope is in eucentric focus (‘Standard Focus’) when acquiring.
-
f.In the upcoming ‘File Properties’ dialog box, select ‘Unsigned integers’ and tick all information for storage in the extended header of the file (Figure 1). Press ‘OK’ and save as ‘80_ov’.
-
g.In SerialEM, run ‘Navigator’ → ‘Acquire at items’.
-
h.Select only ‘Acquire map image or montage’ and ‘Skip Z moves in initial move and Realign’, then press ‘GO’.
-
i.After acquisition, label the new map in the Navigator window.
-
a.
-
44.Record an overview of the central section for image analysis at 400×.
-
a.Select 400× magnification in the Imaging States window.
-
b.From the 80× overview map, navigate to a corner of the central section (usually section 3) by clicking on it on the map image and pressing ‘Go to marker’ in the navigator window.
-
c.In continuous mode, draw a polygon around the section and record a map/montage following the instructions from step 43.
-
d.Save as ‘400_s3’. After acquisition, label your map in the Navigator window.
-
a.
-
45.Set up a reference map (‘refmap’) at 1000×.
-
a.Use the 400× overview map and select a cell on an adjacent section close by. Move the stage to this cell by clicking ‘Go to marker’.
-
b.In the Imaging states, change to 1000× magnification. Adjust the beam and focus.
-
c.Record an image and save it as ‘refmap’.
-
d.Click ‘New map’ in the Navigator window and label the new item as ‘refmap’.
-
e.Set ‘400_s3’ to ‘Acquire’ in the Navigator window and save the navigator as ‘nav.nav’.
-
a.
-
46.Image analysis workflow using KNIME to detect all cells on the section.
-
a.Launch KNIME on your PC and open the workflow ‘cells_virtualmaps’ (Figure 2).
-
b.Double-click ‘Navigator settings’ node. Load your saved navigator file and double-check that ‘Reference map NavLabel’ is set to ‘refmap’.
-
c.Right-click on the ‘Image Viewer’ node, then click ‘Execute and View’.Note: The script will now automatically merge the 400× mosaic map from step 44 to obtain a smooth merged image using IMOD.
-
d.3dmod will launch automatically. In there, use ‘Model mode’ to create a polygon around the cells and omit section borders. When you are done, save the model by pressing ‘s’ on your keyboard and close 3dmod.
-
e.KNIME will then generate the cropped map image which will automatically open once the workflow is done. Continue with 46.f if you are content with your cropped map. If not, go to troubleshooting, problem 1.
-
f.Right-click ‘Interactive Segmentation View’ node, then click ‘Execute and View’ and wait until an image will pop up.Note: This will create a label mask of the section, marking each cell on it in a different color (Figure 3). All cells should be labelled individually.
-
g.If you are content with the results, continue with step 46.h. If not, refer to troubleshooting, problem 2.Note: This color labelling of individual cells is later used to automatically target these cells during acquisition at higher resolutions. Hence, it is important that as many cells as possible were picked up correctly.
-
h.Right-click on ‘Python Script’ node, then click execute.Note: KNIME will now create individual virtual maps at each labeled position at 1000× magnification using the reference map. This is needed because SerialEM cannot use low magnification maps for the ‘Realign to item’ command. A new Navigator file (‘nav_automaps.nav’) containing all items will be generated automatically.
-
a.
-
47.Automated acquisition of each cell on the central section at 1000× and 3000× magnification, respectively:
-
a.In SerialEM, open the new Navigator file: ‘Navigator’ → ‘Read and Open’ → select ‘nav_automaps.nav’.
-
b.Still at 1000× magnification, record an image and save it as ‘anchormaps.mrc’. Click ‘New map’ in the Navigator window and label the new item as ‘anchormap’.
-
c.For this new map, tick ‘For anchorstate’ in the Navigator window.
-
d.Double-check that ‘anchormaps.mrc’ is the only open file in SerialEM (displayed on top of the screen). If not, close all other files.
-
e.Switch to 3000× magnification in the imaging states. Check brightness and centering of the beam and apertures.
-
f.In the main window, click ‘File’ → ‘Open new’ → select ‘Series of TIFF files’. Create a new folder ‘cells’ and in there, save your file as ‘c_’. All 3000× magnification maps will be stored there individually.Optional: In the ‘Scripts’ dialog, open script ‘screen’. Adjust parameters for autofocus and eucentricity if necessary.
-
g.Click ‘Navigator’ → Acquire at items…’. Choose the following settings in the upcoming dialog:
-
i.As ‘Primary Task’, tick ‘Run script’ and select ‘screen’.
-
ii.Below, tick ‘Skip Z moves in initial move and Realign’ and ‘Turn off filament at end’.
-
iii.Click ‘Go’.Note: SerialEM will automatically create individual images at 1000× and 3000× magnification of all cells and store them in the Navigator file (‘lm_xxx’ for 1000× and ‘hm_xxx’ for 3000× magnification, respectively). Depending on your microscope, about 150 cells will be acquired per hour.
-
i.
-
h.Let the microscope run for the estimated run time. Remember to refill cooling agent (e.g., liquid nitrogen).
-
a.
Figure 1.
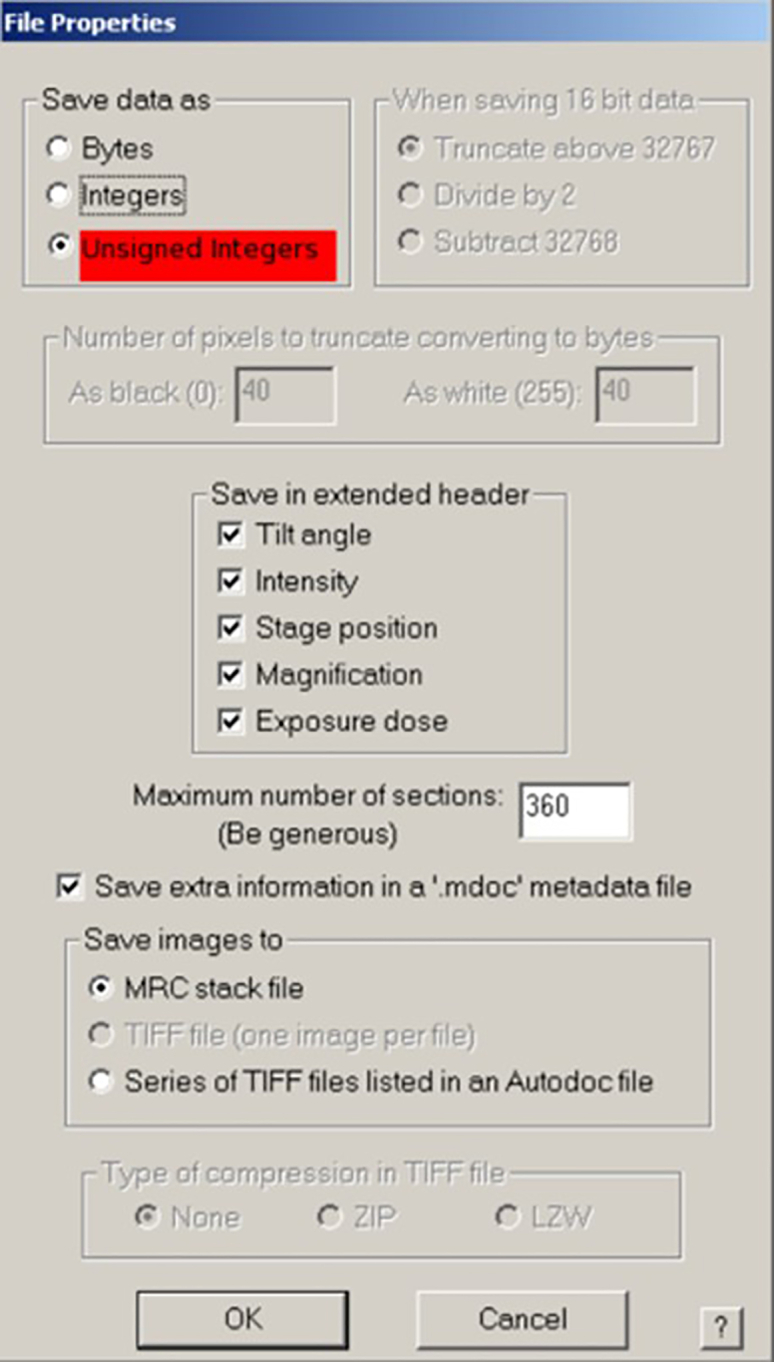
File Properties dialog
Choose ‘Unsigned integers’ (highlighted in red) on top and tick all information to store it in the extended header.
Figure 2.

‘cells_virtualmaps’ workflow in KNIME
You can edit node settings by double clicking on them. Top: Overview of the whole workflow. Bottom: Detailed view of ‘Image analysis’ metanode.
Figure 3.

Label mask of the section displayed in KNIME
The algorithm marks each cell as an individual object (displayed as different colors).
Day 5: Screening evaluation
In this step, 3000× magnification images from the previous step will be used to identify cells of interest (i.e., cells containing target features, in our case centrioles) using ImageJ software.4 Images/maps of these cells will be kept for further targeting, whereas all other items, such as images of cells without interesting features, will be discarded after this step.
-
48.
Launch ImageJ on your PC and drag and drop your folder ‘cells’ there. Load it as a stack.
-
49.
Check your images for features of interest and note the image number of your target cell. You can use your own sheet or the excel template provided on Mendeley Data: https://doi.org/10.17632/ct23zrnxc2.1
-
50.In SerialEM, prepare your Navigator file:
-
a.Set the following items to ‘Acquire’ in the Navigator window:
-
i.All polygons.
-
ii.All virtual maps.
-
iii.The reference map (‘refmap’).
-
iv.All maps (both lower and higher magnification) of cells without target features.
-
v.All 3000× magnification maps (‘hm_xxx’) of cells containing target features.
-
vi.The ‘anchormap’.
-
i.
-
b.The following items need to be kept and therefore must not be set to ‘Acquire’:
-
i.1000× magnification maps (‘lm_xxx’) of cells containing target features.
-
ii.The 400× magnification overview of the central section (‘400_s3’).
-
i.
-
c.Save the Navigator file, then save as ‘nav_prepared’.
-
d.In the main window, select ‘Tools’, run ‘Delete Navigator’s Acquire Items’. Once the script is finished, re-open ‘nav_prepared’.
-
e.Check that all relevant data is still stored in the Navigator file. If not, re-open your old navigator (‘nav_automaps.nav’) and check that all items are set to ‘Acquire’ correctly. Repeat from step 50.a.
-
a.
Day 5–6: Targeting and tomography acquisition
In this step, maps of cells containing centrioles and points defining the exact location of the centrioles will be created to serve as target coordinates for tomography on the central section. Afterwards, these will be duplicated and then shifted to all adjacent sections of the grid using registration/transformation and SerialEM’s RealignToItem feature. Thereafter, an automated tomography acquisition is initiated at the points of interest.
Note: Steps 51 to 56 only need to be performed if you removed the grid or if you use two different electron microscopes for the workflow. If in your setup, everything is performed on a single microscope, you will only have to acquire the section overviews as described in step 54 and then continue with step 57.
-
51.
Set up your microscope: Cool it down and load the grid. Launch SerialEM and load your settings.
-
52.
Open the Navigator file ‘nav_prepared’. In the Navigator window, change ‘Registration’ to 2 using the arrow buttons.
-
53.
Switch to 110× in the Imaging states. Add a polygon around the whole grid in Preview mode and acquire a montaged image using the instructions from step 43. Name it ‘ov_new’ and label the item in the Navigator window as well.
-
54.Acquisition of section overviews at low magnification:Note: These section overviews will be used for transformation of duplicate items in step 59.
-
a.Switch to 450× magnification in the Imaging states.
-
b.Open ‘ov_new’ and click on a corner of one of the outmost sections. Navigate to this position by clicking ‘Go to marker’.
-
c.Draw a polygon around each section. When you are finished, set all polygons to ‘Acquire’ in the Navigator window.
-
d.Mark the first polygon in the Navigator window, then click ‘New file at item’ and ‘Montaged images’ with 15% overlap. Choose MRC format and name the file ‘s1’.Note: SerialEM will now automatically name the subsequent files (‘s2’, ‘s3’, and so on)
-
e.In the main window, go to ‘Navigator’ → Acquire at items. Select ‘Acquire map image or montage’ as Primary Task, and tick ‘Skip Z moves’, then click ‘GO’.
-
f.Once the acquisition is finished, relabel the new maps in the Navigator file and save the Navigator.
-
a.
-
55.Registration:
-
a.Display your new image of the central section (in our case ‘s3’) by double-clicking on it in the Navigator window.
-
b.Click ‘New window’ in the buffer control menu of the main window.
-
c.Now double-click on ‘400_s3’. It will appear in your new window. Arrange both windows so that they are displayed side by side.
-
d.Set registration points to shift the coordinates from the old items to the newly acquired ones: Set corresponding points on correlating structures on both images by clicking ‘Add points’ in the Navigator window. Evenly distribute 6–8 pairs of these points on the whole section.
-
e.Make them ‘registration points’ in the Navigator window.
-
f.Save the Navigator file, then save the Navigator as ‘s3_pretransform’.
-
g.In the main window, click ‘Navigator’ → ‘Transform items’.Note: This will transform the coordinates of all images acquired during the screening to the new, correct coordinates.
-
h.If the mean deviation is higher than 10 μm between the old and the new coordinates, an error box will show up. In that case, refer to troubleshooting, problem 3, potential solution 1.
-
a.
-
56.Update microscope states for the old 1000× magnification maps:Note: Moving a grid from one microscope to another usually requires applying a large scaling factor and rotation to maps. This script therefore creates virtual maps with a modified header from the old 1000× modification maps which can then be utilized for the ‘RealignToItem’ procedure on the second microscope. These virtual maps are eventually replaced by newly acquired maps on the second microscope (step 57).
-
a.Select 2300× in the Imaging states and adjust the beam.
-
b.Record an image, save the image as ‘refmap_new’, make it a map in the Navigator window and label the new item as ‘refmap’.Note: Even a blank image with a blocked beam is fine.
-
c.Set all 1000× magnification maps (‘lmxxx’) to ‘Acquire’ and save the Navigator file.
-
d.In the main window, click ‘Tools’ → Update Microscope States for Selected Maps’ to generate a new Navigator file called ‘s3_pretransform_updated’.
-
e.Click ‘Navigator’ → ‘Read and Open’ and open the newly generated Navigator file. All maps acquired at 1000× earlier will now be converted into virtual maps at 2300× magnification.
-
f.Check whether your transformation was successful: Choose a virtual map in the Navigator window and click ‘Realign to item’. Continue with the next step if you are content with your results. If not, refer to troubleshooting, problem 3.
-
a.
-
57.Re-Acquisition of target cells and defining target points.Note: In this step, you will reacquire the maps of cells containing centrioles to replace the virtual maps from step 56. On these new maps, you will define your points of interest (where tomography will be performed eventually) by setting points to the centrioles. These points and maps on the central section of the grid will be used in step 59 to semi-automatically target the same cells on adjacent sections.
-
a.Set all created virtual maps to ‘Acquire’.
-
b.Move the stage to a point on the central section. In the main window, click ‘Task’ → ‘Eucentric Rough’.
-
c.Click ‘Navigator’ → ‘Acquire at items’. In the upcoming dialog, choose ‘Run script’ as a Primary Task and select the script ‘serial section_initial map’ from the drop-down menu. Then click ‘GO’.
-
d.After the first map, an error message will pop up asking for the saving directory of the new maps. Simply click ‘yes’.
-
e.When the script has finished, control whether all cells were identified correctly comparing the newly acquired maps with the virtual maps. If an incorrect cell was targeted, refer to troubleshooting, problem 4.
-
f.While controlling all maps, set points to your feature of interest (in our case, centrioles) by clicking ‘Add points’ in the Navigator window.
-
a.
-
58.Rename points and add a section index.
-
a.Set all points to ‘Acquire’.
-
b.In the main window, click ‘Navigator’ → ‘Acquire at items’. As Primary Task, run the script ‘Name_pts’, and tick ‘Skip initial stage move’ and ‘Skip initial Z move and realign’. Click ‘GO’ and confirm the upcoming error message.Note: This will rename all points according to the map they were added on (e.g., a point on the map ‘lm001’ will be named ‘c001’)
-
c.Set all 2300× maps and all corresponding points to ‘Acquire’.
-
d.In the main window, click ‘Navigator’ → ‘Acquire at items’. Run the script ‘Add Section Index’.Note: This will add the index ‘_s03’ to the name of your maps and points. If you did not screen on section 3, edit the script accordingly in the ‘Scripts’ window.
-
e.Save the Navigator file.
-
a.
-
59.Transfer acquired maps and coordinates to adjacent sections to locate target cells:Note: In this step, three major steps are involved: First, the maps and points on the central section are duplicated. Second, these duplicates are shifted from the central section to another using transformation on low-magnification overviews of the respective sections. Third, a SerialEM script utilizes the ‘RealignToItem’ feature to locate the exact cell and centriole point on the adjacent section and labels the points automatically.
-
a.Mark the map ‘s3’ in the Navigator window and change its Registration to 3. To do that, in the main window, click ‘Navigator’ → ‘Change registration’.
-
b.Display ‘s3’ side by side with the overview map of an adjacent section by clicking ‘New window’ in the Buffer controls menu.
-
c.Add 6–8 pairs of registration points (as described in step 55).
-
d.Change the registration of s3 back to ‘2’ using ‘Navigator’ → ‘Change registration’ in the main window.
-
e.Set the target points (the ones named ‘cxxx_s03’) to ‘Acquire’ and save the Navigator file.
-
f.In the main window, click ‘Tools’ → Duplicate Navigator’s Acquire Points and associated maps’ to create a new Navigator file with the suffix ‘_duplicated’.Note: This tool duplicates the selected points and maps. Subsequently, the duplicates will be shifted to an adjacent section using a transformation, while the original items on the central section remain unmoved.
-
g.Read and open the new Navigator file.
-
h.Find the points from the central section and ‘De-Acquire’ them. Instead, find the duplicated points at the bottom of the Navigator file and set them to ‘Acquire’.
-
i.The registration was automatically set to ‘3’ in the process. Set it back to ‘2’ using the arrow keys in the Navigator window.
-
j.Save the Navigator file.
-
k.In the main window, click ‘Navigator’ → ‘Transform items’. If an error message pops up (Figure 4), refer to troubleshooting, problem 3, potential solution 1.Note: The duplicated points and maps should now be moved to the adjacent section you chose before.
-
l.Check for 3 to 4 cells if they have been identified correctly: Select the map or point in the Navigator window and click ‘Realign to Item’. If you are content with your results, continue with the next step. If not, refer to troubleshooting, problem 3, potential solution 2.
-
m.In the ‘Scripts’ dialog, find the script ‘serialsections_pts_map’. Change the section number to the one you have just transferred your points to (Figure 5).
-
n.Move the stage to a point on the adjacent section. In the main window, click ‘Task’ → ‘Eucentric Rough’.
-
o.Click ‘Navigator’ → ‘Acquire at items’. Run the script ‘serialsections_pts_map’ as a Primary Task. Below, only ‘Skip initial Z moves and realign’ should be ticked.Note: This script uses the shifted maps from the central section to identify the same cells on the adjacent section. It will automatically realign to the points, create new map images of each cell, mark the target features with points and then rename the maps and points accordingly.
-
p.Set the old virtual maps and the registration points to ‘Acquire’ in the Navigator window.
-
q.Save the Navigator file, then save as a new file (e.g., ‘s1_pretransform’).
-
r.In the main window, click ‘Tools’ → ‘Delete Navigator’s acquire items’.
-
a.
-
60.
Read and open the new Navigator file and repeat step 59 for all sections on the grid.
-
61.Prepare the Navigator file for tomogram acquisition:
-
a.Sort the Navigator file by clicking ‘Tools’ → ‘Sort Navigator’ to generate a new Navigator file with the suffix ‘_sorted’.
-
b.Browse through the Navigator file and check that all points were set correctly on all sections. Use ‘Edit mode’ in the Navigator window if a point was not set accurately and correct its position or, if a cell was not imaged correctly, refer to troubleshooting, problem 4.
-
c.Set all points to ‘Acquire’ and save the Navigator file.
-
d.Click ‘Tools’ → ‘Sort by Section’.
-
a.
-
62.Start the automated tomogram acquisition:
-
a.Switch to your desired magnification for tomography in the imaging states and adjust the beam.
-
b.Generate a new folder as a directory for the tomograms. Name it ‘tomo’ and copy its file path.
-
c.Edit the script ‘quicktomo_master’ in the Scripts’ ‘dialog: paste the file path to the directory you have just created and set ‘realign to item’ (=1) as strategy.
-
d.Check the available space on your hard drive: approximately 1 GB will be used per tomogram.
-
e.Start the acquisition by clicking ‘Navigator’ → ‘Acquire at items’. Run the script ‘quicktomo_master’ as Primary Task and tick ‘Skip initial Z move and realign’ and ‘Close column valves at the end’ / ‘Turn off filament at end’.
-
a.
Note: Depending on your microscope and your settings, around 7 tomograms will be acquired per hour automatically. Remember to refill your cooling agent and let the microscope run for the projected time given in SerialEM.
Figure 4.
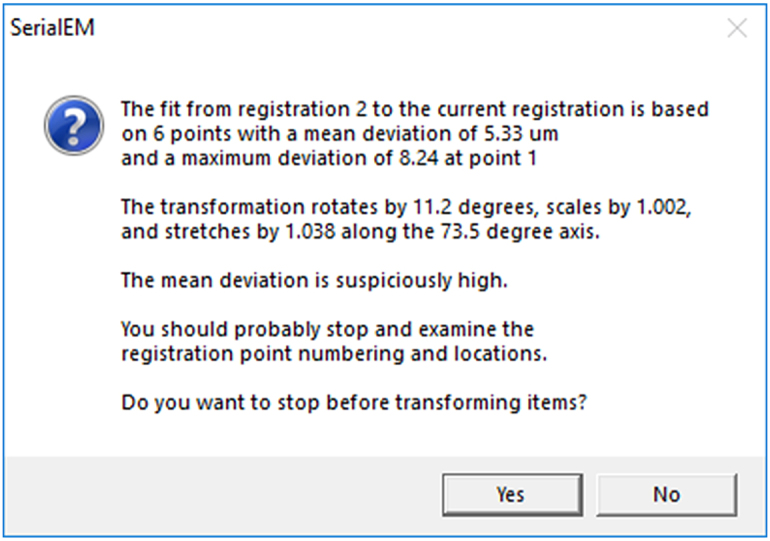
Error box showing up if the registration points were not set correctly
This comes up if the coordinates between the new and old maps deviate too much from each other. In that case, refer to troubleshooting, problem 3.
Figure 5.

Script dialog for ‘serialsections_pts_map’ script
Each time you use the script on a new section, the section index should be edited accordingly (highlighted in blue).
Day 6–7: Data reconstruction and joining
After acquisition, you will end up with single axis tomograms of cells on individual sections. In this step, raw tomography data are first converted into actual volume data (reconstruction), and then tomograms of the same cell on different sections are joined to form the final volume.
Note: We used a set of Python commands to launch IMOD’s automated pipeline on a high-performance computer cluster as reported elsewhere.7 Instructions for implementation and usage of the batchruntomo feature can be found in the IMOD user manual (link provided in the key resources table).
-
63.
Reconstruction: launch IMOD’s automated reconstruction either through scripts on dedicated HPC hardware or use the etomo graphical user interface to manage the procedure. Provide the location to the acquired tilt series and the batch directive with the reconstruction parameters.
Note: The parameters used in our study are provided in Mendeley Data: https://doi.org/10.17632/ct23zrnxc2.1
-
64.
During reconstruction, new ‘.rec’ files will be generated in the folder where the original tilt series are stored. Copy these files to a new folder called ‘reconstructed’.
-
65.Join single tomograms to obtain final volume:
-
a.Launch etomo and create a new join by clicking ‘File’ → ‘New’ → ‘Join Serial Tomograms’.
-
b.Set your working directory and name your output file.
-
c.Add all individual section tomograms of one cell: E.g., if you have acquired cell 001 on 5 sections, add ‘c001_s01’ to ‘c001_s05’.
-
d.Display each tomogram in 3dmod using the etomo graphical user interface and check the lower and higher frames. Trim away frames without information by adding the respective frame numbers in the ‘Start’ and ‘End’ panel.
-
e.Create a stack by clicking ‘Make samples’ and wait until the program has finished.
-
f.In the upcoming ‘Align’ window, you can calculate a coarse or fine alignment or manually align your stacks.
-
g.Once you are content with your alignment, switch to the ‘Join’ window and finish the serial tomogram. You can display your result directly from etomo or open it with IMOD.
-
a.
Expected outcomes
In an average experiment with a pellet size of 300,000 to 500,000 plasma cells, about 350 to 700 cells will be featured on one section and thus be screened on day 4. In our case, centrioles could be detected in 5% or more of the cells, leading to a yield of 20 or more joined tomograms in excellent imaging quality featuring at least 30 measurable, full-length centrioles per sample. This was in line with our goal of measuring at least 30 centrioles per patient to reach significance. When we applied the workflow to smaller sample sizes (e.g., 70,000 cells), we achieved the same imaging quality. However, as fewer cells will end up on one section, it was necessary to screen a second grid for some samples. For this, sections were taken at a distance (at least 5 μm deep) from the first set of serial sections.
Limitations
The method we present is applicable to various kinds of biological source materials. We have used both cultured as well as primary human cells, but applying the method to more adherent cells or even tissues should be feasible. Before starting the protocol, a pioneer study optimizing the preparation of the material for conventional transmission electron microscopy is highly recommended. As for sample size, a sample containing as little as 70,000 cells was adequate to be handled for electron tomography. Adaption of sample preparation to other types of specimens could allow for even smaller sample sizes.
Our method is applicable for targeting various subcellular structures and enables high-throughput acquisition of target features in multiple cells. Importantly, however, this workflow will always visualize only a fraction of all target features (in our case: centrioles) present in a given cell population and also potentially not all centrioles within single cells. For this reason, our methodology is not applicable for quantification of numeric organelle aberrations at the cell population or single-cell level. For analyses that focus on numeric aberrations or characterization of one organelle in singular cells that require lower throughput, such as characterization of a mitochondrial network in a single cell, other electron microscopy methods producing isotropic volume data, e.g., focused ion beam scanning electron microscopy (FIB-SEM), might be more suitable. These methods, however, are much more limited in their acquisition capacity.
Troubleshooting
Problem 1
The cropped map in KNIME is too dim/bright and cells are hardly visible (step 46.e).
Potential solution
Try with another polygon again, carefully omitting section borders.
-
•
If still open in IMOD, close the polygon and the new map.
-
•
The files were stored in the same folder that your Navigator file was saved to. Find the newly created files and delete them.
-
•
In KNIME, reset the first node (‘Set parameters’) and start the workflow from scratch.
-
•
When you are asked to draw the polygon again, make sure you omit all section borders. This will enhance the contrast of your final image.
Problem 2
Automated cell labelling in KNIME is not picking up cells correctly and/or not at all (Step 46.f).
Potential solution
Adjust the image analysis parameters.
-
•
As all samples are different depending on cell type, experimental, and imaging conditions, you might have to experiment with the image analysis settings the KNIME workflow uses. You can edit them in KNIME on two different levels:
-
•First (and usually sufficient), you can edit the general settings. For that, double-click the ‘Parameters’ node in the ‘Set parameters’ box on the left. Adjust the settings according to:
-
○Your sample and the featured cell size: Edit the minimal cell diameter according to the expected size of your cells.
-
○Your image conditions: Try with a different rolling ball radius or increase/decrease binning size. Note that a low binning might increase the runtime significantly.
-
○
-
•
Second, you can customize more settings in the ‘image analysis’ node. While holding CTRL on your keyboard, double-click the grey node. A new layer of nodes will show up (Figure 2, bottom) where all different steps of the image analysis pipeline can be edited. Try with different settings for ‘Global Thresholder’ and ‘Waehlby Cell Clump Splitter’ nodes first as this will usually suffice.
-
•
Third, more advanced users might also edit the whole workflow be adding and/or removing nodes from the image analysis metanode. KNIME offers several solutions for image analysis, including neural network-based approaches.
Problem 3
Registration does not work, and new coordinates generally do not resemble the correct cells (steps 55, 56, and 59).
Potential solution 1
One or multiple registration points were not set to the correct location.
-
•
If this is the case, an error box will show up telling you that the mean deviation between images is too high (i.e., higher than 10 μm between correlating registration points) (Figure 4).
-
•
In that case, check if all registration points were set to the correct location. First, you will have to undo the transformation. To do that, you can either click ‘Navigator’ → ‘Undo transformation’ in the main window if the transformation was the last step you just completed. If not, read and open the Navigator file that you saved just before transforming items by clicking ‘Navigator’ → ‘Read and open’ in the main window.
-
•
Now display both low magnification maps next to each other in the main window.
-
•
Check that all registration points are visible. If not, change the registration of the low magnification map you want to transform items from to ‘3’. Left-click on the image once to make the points visible.
-
•
Now you can double-check the positions of all points. If you find a pair that is not set correctly, delete it from the Navigator file, then set a new pair. You want to end up with 6–8 pairs of corresponding points evenly spread over the whole section image.
-
•
If you have changed the registration of a low magnification map previously, set it back to ‘2’ and check that the general registration you are working in (displayed in the Navigator window) is set to ‘2’.
-
•
Lastly, save the Navigator file and try transforming items again.
Potential solution 2
If there is no error box or alignment still does not work after correction of registration points, try with ‘shift to marker’.
-
•
Even if all registration points were set correctly and there is no error message, the coordinates might be generally inaccurate by a few micrometers due to a shift in the stage coordinates. You can correct for that by using the ‘Shift to Marker’ function:
-
•
Display one of the section overviews (‘s1’, ‘s2’, etc.) by double clicking on it in the Navigator window.
-
•
Set a point to a well distinguishable feature of the section (e.g., a uniquely shaped part of a cell membrane) by clicking ‘Add points’ in the Navigator window.
-
•
Mark this point in the Navigator window (i.e., check that it is highlighted in blue) and click ‘Go to XY’.
-
•
Switch to 2300× magnification in the Imaging states window. Locate the feature in Preview mode, then record an image.
-
•
On this image, left click on the exact position where you set your point. This will set a marker (a small crosshair) to it. Double check that the point is still highlighted in the Navigator window.
-
•
In the main window, click ‘Navigator’ → ‘Shift to marker’. Confirm all messages that will show up and try realigning to items again.
Problem 4
One cell was not picked up correctly even though registration was successful (steps 57 and 61).
Potential solution
-
•
Even if all registration steps were done correctly, some cells might not be able to realign to due to various reasons (e.g., artifacts on the section, stretches of the grid etc.). If this occurs, it does not affect more than 1 or 2 cells. You can correct these cells manually:
-
•First, you will have to locate the correct cell on your section. You can do that in two different ways:
-
○Click on the map in the Navigator window to highlight it, then click ‘Go to XY’. Select 2300× magnification in the Imaging states window, then click ‘Preview’. Move the stage and try to identify the correct cell which should be close by if the transformation was done, or
-
○Click on the respective section overview you acquired at 450× magnification and display it by double-clicking on it. Locate the cell on there. Left-click on the cell once to set a marker to it, then click ‘Go to XY’ in the Navigator window. Switch to 2300× magnification in the Imaging states window and start Preview mode. Identify your cell of interest.
-
○
-
•
When you have identified the correct cell, center the cell and record an image. Save it and make it a new map in the Navigator window. Name the new map according to the cell it resembles (i.e., if lm001_s01 was not obtained correctly, name the new item accordingly).
-
•
Set a point to your feature of interest and rename it.
-
•
Delete both the map and the point that were not obtained correctly.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Alwin Krämer (a.kraemer@dkfz-heidelberg.de).
Materials availability
This study did not generate any new unique reagents.
Data and code availability
Original data have been deposited to Mendeley Data: https://doi.org/10.17632/ct23zrnxc2.1
All original code has been deposited at Github and Mendeley and is publicly available as of the date of publication. SerialEM scripts can be downloaded from The SerialEM Scripts Repository. DOIs and links to all files and scripts are provided in the key resources table. Any additional information is available from the lead contact upon request.
Acknowledgments
We thank A. Baumann for sample preparation and the Electron Microscopy Core Facility at EMBL Heidelberg, especially M. Börmel, R. Mellwig, and V. Oorschot, for their advice and help with sectioning and post-staining. This work was supported by funding from the Deutsche Forschungsgemeinschaft (DFG) to A.K. (KR 1981/4-1). T.D. received funding from the Medical Faculty of Heidelberg within the Physician Scientist Program.
Author contributions
A.K. conceived the project. A.K., Y.S., I.H., T.D., and S.K. designed the experiments. I.H., T.D., S.K., and M.S. performed the experiments. A.K., Y.S., M.S., and S.K. wrote the manuscript. All co-authors approved the final manuscript.
Declaration of interests
A.K. received honoraria from Hoffmann-La Roche, Bayer, Daiichi Sankyo, and AbbVie and research funding from Bayer and Merck and is a paid consultant for Hoffmann-La Roche, Daiichi Sankyo, Bristol Myers Squibb, and AbbVie.
Contributor Information
Sebastian Köhrer, Email: sebastian.koehrer@embl.de.
Martin Schorb, Email: martin.schorb@embl.de.
Alwin Krämer, Email: a.kraemer@dkfz-heidelberg.de.
References
- 1.Dittrich T., Köhrer S., Schorb M., Haberbosch I., Börmel M., Goldschmidt H., Pajor G., Müller-Tidow C., Raab M.S., Hegenbart U., et al. A high-throughput electron tomography workflow reveals over-elongated centrioles in relapsed/refractory multiple myeloma. Cell Rep. Methods. 2022;2:100322. doi: 10.1016/j.crmeth.2022.100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mastronarde D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 3.Berthold M.R., Cebron N., Dill F., Gabriel T.R., Kötter T., Meinl T., Ohl P., Sieb C., Thiel K., Wiswedel B. In: KNIME: The Konstanz Information Miner. Held in Berlin, Heidelberg, 2008//. C. Preisach. Burkhardt H., Schmidt-Thieme L., Decker R., editors. Springer Berlin Heidelberg; 2008. pp. 319–326. [Google Scholar]
- 4.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kremer J.R., Mastronarde D.N., McIntosh J.R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 6.Schorb M., Haberbosch I., Hagen W.J.H., Schwab Y., Mastronarde D.N. Software tools for automated transmission electron microscopy. Nat. Methods. 2019;16:471–477. doi: 10.1038/s41592-019-0396-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mastronarde D.N., Held S.R. Automated tilt series alignment and tomographic reconstruction in IMOD. J. Struct. Biol. 2017;197:102–113. doi: 10.1016/j.jsb.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolotuev I., Bumbarger D.J., Labouesse M., Schwab Y. Targeted ultramicrotomy: a valuable tool for correlated light and electron microscopy of small model organisms. Methods Cell Biol. 2012;111:203–222. doi: 10.1016/B978-0-12-416026-2.00011-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Original data have been deposited to Mendeley Data: https://doi.org/10.17632/ct23zrnxc2.1
All original code has been deposited at Github and Mendeley and is publicly available as of the date of publication. SerialEM scripts can be downloaded from The SerialEM Scripts Repository. DOIs and links to all files and scripts are provided in the key resources table. Any additional information is available from the lead contact upon request.