

The Authors identify an IL-12 prodrug that is selectively activated in the tumor microenvironment and generates potent antitumor immunity in mice by activating and restoring the metabolic health of tumor-infiltrating lymphocytes while preventing toxicity associated with systemic IL-12.
Abstract
IL-12 is a pleotropic inflammatory cytokine, which has broad stimulatory effects on various immune cell populations, making it an attractive target for cancer immunotherapy. However, despite generating robust antitumor activity in syngeneic murine tumor models, clinical administration of IL-12 has been limited by severe toxicity. mWTX-330 is a selectively inducible INDUKINE molecule comprised of a half-life extension domain and an inactivation domain linked to chimeric IL-12 by tumor protease–sensitive linkers. Systemic administration of mWTX-330 in mice was well tolerated, resulted in robust antitumor immunity in multiple tumor models, and preferentially activated tumor-infiltrating immune cells rather than immune cells present in peripheral tissues. Antitumor activity was dependent on in vivo processing of the protease cleavable linkers and required CD8+ T cells for full efficacy. Within the tumor, mWTX-330 increased the frequency of cross-presenting dendritic cells (DC), activated natural killer (NK) cells, skewed conventional CD4+ T cells toward a T helper 1 (TH1) phenotype, drove regulatory T cells (Treg) fragility, and increased the frequency of polyfunctional CD8+ T cells. mWTX-330 treatment also increased the clonality of tumor-infiltrating T cells by expanding underrepresented T-cell receptor (TCR) clones, drove CD8+ T and NK cells towards increased mitochondrial respiration and fitness, and decreased the frequency of TOX+ exhausted CD8+ T cells within the tumor. A fully human version of this INDUKINE molecule was stable in human serum, was reliably and selectively processed by human tumor samples, and is currently in clinical development.
Introduction
Cytokine therapy could become one of the pillars of cancer immunotherapy due to its potential to activate the immune system and promote antitumor activity (1–3). However, clinical results with cytokines have been disappointing so far. To date, only two cytokines, IL-2 and IFNα, have received approval from the FDA for specific cancer types. These approvals were based on well-documented antitumor efficacy as monotherapies, including a subset of patients with complete responses (CR; ref. 2). Unfortunately, the clinical use of cytokines has been largely impeded by their short half-life, the severe toxicity often associated with their systemic administration, and limited efficacy at their maximum tolerated dose (MTD; refs. 2, 3). However, recent advances in therapeutic bioengineering have renewed interest in cytokines as therapeutic agents (1) and have led several groups to revisit the design of IL-2 for the clinic (4–8).
IL-12 is a master regulator of innate and adaptive cell-mediated immunity with multiple mechanisms responsible for its potent antitumor effects (1, 3, 9). IL-12 is a heterodimer encoded by two genes: IL12A (p35) and IL12B (p40; ref. 9). The active heterodimer (p70) binds a cell-surface receptor formed by the subunits IL-12Rβ1 and IL-12Rβ2 (9, 10). IL-12 p40 binds to the IL-12Rβ1 receptor, and IL-12 p35 binds to the IL-12Rβ2 receptor (9–11). IL-12 has numerous immunostimulatory functions, including increasing IFNγ production by natural killer (NK) and T cells, and stimulating the growth and cytotoxicity of NK, CD8+, and CD4+ T cells. Furthermore, IL-12 promotes the differentiation of CD4+ T cells into T helper 1 (TH1) cells and enhances antibody-dependent cellular toxicity against tumor cells (1, 12, 13). In the field of cytokine therapy, IL-12 has gained particular attention due to its role as a pleotropic cytokine that induces both innate and adaptive immune responses, its well established ability to generate robust antitumor activity in preclinical studies (9, 12, 14), and the existence of some evidence of clinical activity in patients treated with the recombinant cytokine (13). Unfortunately, systemic IL-12 treatment in the clinic is associated with the rapid development of a lethal inflammatory syndrome, which has so far rendered IL-12 treatment strategies impracticable (15). While ongoing research has been aimed at finding strategies to overcome the toxicity caused by systemic delivery of IL-12 (14–20), there are currently no approved IL-12 cytokine-based cancer therapies in the clinic.
To overcome some of the difficulties associated with cytokine therapy in the clinic, we designed WTX-330, a selectively inducible polypeptide (INDUKINE molecule) that is composed of wild-type human IL-12, which is kept inactive in the periphery by tethering to a high-affinity anti–IL-12 blocking antibody domain. The INDUKINE molecule also contains a half-life extension (HLE) domain to enhance the pharmacokinetic profile of the molecule and to facilitate less frequent dosing. The inactivation and HLE domains are linked to IL-12 via tumor protease–sensitive linkers. Cleavage of the protease-sensitive linkers in the tumor microenvironment (TME) should remove the HLE and inactivation domain, potentially delivering fully active IL-12 selectively in the TME. Indeed, although in vitro–activated WTX-330 had equivalent activity to recombinant IL-12, the intact molecule was substantially less active. WTX-330 was designed to be a systemically dosed prodrug that could potentially deliver fully active IL-12 selectively into the TME via targeted intratumoral activation of the INDUKINE molecule. This selectivity was confirmed using in vitro dissociated primary human samples or primary cells derived from healthy human tissues. Although incubation of WTX-330 with primary human dissociated tumor samples resulted in activation of the molecule, the INDUKINE molecule remained inactive in the presence of primary human cells derived from healthy tissues. WTX-330 is being developed for the treatment of relapsed or refractory, advanced or metastatic solid tumors, or lymphomas, and is currently in clinical development (NCT05678998).
Herein we report the first preclinical findings for mWTX-330, a mouse surrogate for WTX-330. mWTX-330 differs from WTX-330 only in that it contains a mouse IL-12 p35 subunit instead of the human subunit, as fully human IL-12 is not active in mouse cells, whereas the chimeric molecule is (21). We demonstrate that mWTX-330 treatment was well tolerated compared with chimeric IL-12 in mice, achieved complete tumor regressions in multiple models without inducing weight loss or apparent liver toxicity, and widened the therapeutic window compared with the chimeric cytokine. Furthermore, tumor-infiltrating lymphocyte (TIL) analysis and GeoMX Digital Spatial Profiling (Nanostring) demonstrate that mWTX-330 treatment activated and reshaped various tumor-infiltrating immune cell populations, including both professional antigen-presenting and effector cells. mWTX-330 treatment resulted in the expansion of several underrepresented T-cell receptor (TCR) clones, and also reinvigorated the metabolic state of intratumoral CD8+ T cells and NK cells, which has recently been linked to the generation of robust and persistent antitumor immunity in response to PD-1 or CTLA-4 blockade (22). Furthermore, mWTX-330 treatment decreased the frequency of TOX+ exhausted CD8+ T cells within the tumor. These results demonstrate that the bioengineering of mWTX-330 enabled the practical and systemic delivery of an IL-12 prodrug that was selectively processed in the TME where IL-12 was locally released to trigger a potent antitumor effect while avoiding systemic toxicities.
Materials and Methods
INDUKINE protein production, protease activation, and SDS-PAGE analysis
mWTX-330, recombinant chimeric IL-12, WTX-330, and any noncleavable (NC) variants were produced at Werewolf Therapeutics. mWTX-330 consists of chimeric IL-12 (mouse p35 and human p40) linked to a proprietary inactivation domain and HLE domain by a proprietary linker sequence. For additional details on INDUKINE designs, please refer to patent PCT/US2021/033014 (https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021236676). The proteins were expressed using the Expi293 transient expression system from Life Technologies (A14635) according to the manufacturer's protocol. INDUKINE proteins were purified by Protein A chromatography. Briefly, transfected cultures were left to rotate overnight in the presence of Amsphere A3 resin (JSR Life Sciences, BP-AMS-A3–0500) or MabSelect protein A resin (Cytiva, 17519902). The following day, the culture/resin mixture was applied to numerous gravity columns. The resin was washed with 1× PBS, then 20 mmol/L sodium phosphate pH 7.3, 500 mmol/L NaCl; and the protein was eluted with 200 mmol/L acetic acid pH 3.5, 50 mmol/L sodium chloride into 1 M Tris pH. Recombinant chimeric IL-12 was purified by immobilized metal affinity chromatography (via a 6× histidine tag). Briefly, transfected cultures were left to rotate overnight in the presence of Ni Sepharose excel resin (GEHealthcare, 17371201). The following day, the culture/resin mix was applied to numerous gravity columns, and the resin was washed with 1× PBS, followed by 20 mmol/L Imidazole, 300 mmol/L NaCl, 50 mmol/L NaPO4 pH 7. The protein was then eluted with 250 mmol/L Imidazole, 300 mmol/L sodium chloride, 50 mmol/L sodium phosphate pH 7. Elutions were pooled, dialyzed against 1× PBS, concentrated, aliquoted, and stored for future use at −80°C. Theoretical extinction coefficients were determined for each protein and concentration was determined by A280. In experiments where protease-activated INDUKINE proteins were used, INDUKINE proteins were incubated with Cathepsin L (R&D Systems, 952-CY) at room temperature for ten minutes, then frozen for later use. Intact INDUKINE protein was also incubated at room temperature without enzyme, then frozen as a control. To determine the quality of each INDUKINE molecule, proteins were analyzed by SDS-PAGE under nonreducing conditions to characterize purity. Briefly, 5 μg of various INDUKINE molecules were loaded on a 4% to 12% Bis-Tris gel under nonreducing conditions. Gels were stained using Bio-safe Coomassie stain (Bio-Rad, 1610786), destained in water, and visualized using a MyECL Imager (Thermo Fisher Scientific).
HEK-blue IL-12 reporter assay
The HEK-Blue IL-12 reporter cell assay was performed according to the manufacturer's protocol (Invivogen, hkb-IL-12). On assay Day 1, the cells were rinsed, resuspended in RPMI-1640 media with 10% heat inactivated FBS (included in the kit) containing 1.5% human serum albumin (Sigma-Aldritch, A5843; weight/volume) and plated at a concentration of 5 × 104 cells per well in a 96-well flat bottom plate. Titrated amounts of intact and protease-activated (cleaved) INDUKINE proteins or recombinant IL-12 were added to the cells to generate a full dose–response curve. On Day 2, secreted embryonic alkaline phosphatase (SEAP) levels were measured using a Synergy HTX multi-mode plate reader (Biotek) at 620 nm according to the manufacturer's protocol.
Cell lines
All cell lines used for in vivo studies were grown and maintained by Charles River Laboratories according to ATCC guidelines and kept in culture for no longer than 2 weeks. Frozen cells were thawed and maintained for one to three passages before implantation. The MC38 cell line was obtained from the NCI Fredrick Cancer DCT Tumor Repository in 2012, the EMT-6 cell line was obtained from ATCC in 2014, the CT26 cell line was obtained from ATCC in 2017, and the B16-F10 cell line was purchased from ATCC in May of 2019. All cell lines were tested for Mycoplasma every 2 months and have not been reauthenticated within the past year. B16-F10, CT26, and EMT-6 cell lines were cultured in RPMI-1640 with L-Glutamine (Gibco, 11875–085) with 10% heat-inactivated FCS (Gibco, 35–015-CV), whereas MC38 was cultured in DMEM (Gibco, 1966–025) supplemented with 10% heat-inactivated FCS (Gibco, 16000–044). Prior to tumor implantation, cells were washed twice with PBS and counted.
Mice, tumor implantations, and in vivo dosing
All mouse in vivo work was performed in accordance with current regulations and standards of the U.S. Department of Agriculture and the NIH at Charles River Laboratories with the approval of the Charles River Laboratories (Worcester site) Institutional Animal Care and Use Committee (IACUC). Female, 6- to 8-week-old C57Bl/6 or Balb/C mice from Charles River Laboratories were shaved on their flank 1 day prior to tumor cell implantation. A total of 5 × 105 (MC38), 1 × 105 (B16-F10, EMT-6), or 3 × 105 (CT26) cells in a 50% Matrigel solution (Corning, 354234) were injected subcutaneously on the flank of the animals. Tumor volume was monitored until the group average was 100 mm3, and mice were randomized into treatment groups on Day 0. Mice receiving INDUKINE proteins were dosed twice a week, on Days 1 and 4 of the week. Mice receiving recombinant chimeric IL-12 were dosed twice a day for 5 days before receiving a 2-day break (5/2 regimen) and the cycle was repeated. The doses used in these studies are specified in the individual figure legends. In studies using FTY720 (Sigma-Aldrich, SML0700–25MG), mice were initially dosed with 25 μg on the first dose, then treated daily with 10 μg per dose. FTY720 dosing was initiated 24 hours prior to starting treatment with INDUKINE proteins (Day 0). Antibody depletions were performed with twice weekly doses of 200 μg of each respective antibody purchased from Bio X Cell (anti-CD8-Clone 2.43; anti-CD4-Clone GK1.5; and anti-NK1.1-Clone PK136). Antibodies used for depletion were administered 1 day prior to INDUKINE protein administration. All treatments were administered by intraperitoneal injection, and mice were dosed for 2 weeks unless otherwise noted. Tumors were measured in two dimensions using calipers, and volume was calculated using the formula tumor volume (mm3) = [(w2 × l) / 2], where w is the width and l is the length, in mm, of a tumor. Mice were kept on study until tumors reached 1,500 mm3 or the study reached the termination point of Day 45. In some instances, mice with complete tumor regression were saved for later immune memory experiments. For therapeutic window calculations, the MTD was defined as the highest dose where the mice maintained above 85% of their body weight throughout the course of treatment. The minimum efficacious dose (MED) was defined as the lowest dose where the treatment demonstrated clear antitumor activity (50% TGI) based on the average tumor growth of the treated animals compared with the control group. The therapeutic window was defined as the ratio of the MTD to the MED (TW = MTD/MED).
Liver toxicity assay
Liver toxicity was analyzed as previously reported (23). Briefly, naïve 6- to 8-week-old C57BL/6 female mice were dosed with either PBS (100 μL on Day 1) as a negative control or anti-CD40 (Leinco, Platinum Grade, Clone FGK45; 100 μg dose on Day 1) as a positive control. For recombinant chimeric IL-12, mice were dosed at the level and schedule required to get complete tumor rejection in the MC38 model (10 μg, twice a day for a total of five doses). Meanwhile, either 43 or 86 μg of mWTX-330 was administered once, matching or doubling the dosing level required to get complete tumor rejection in the MC38 model, respectively. Livers were collected in the afternoon of Day 3 and placed in 10% neutral buffered formalin for 48 hours before being shipped to the Harvard Rodent Histopathology core for paraffin embedding, slide preparation, and H&E staining. H&E stained sections were analyzed for cellular dysplasia and perivenular infiltration by leukocytes, as has been reported following IL-12 treatment (23, 24).
Tumor digestions and NanoString analysis
Murine tumors were chopped in HBSS containing 1.25 mg/mL Collagenase Type IV (Sigma-Aldrich, 17104–019), 0.025 mg/mL Hyaluronidase (Worthington, LS002594), and 0.01 mg/mL DNASE I (Worthington, LS002006) before being processed using a Miltenyi OctoMax Dissociator running program “37C_m_TDK_1.” Single-cell suspensions were then filtered, counted, and used for downstream applications. For TCR clonality, dead cells were depleted by negative selection (Miltenyi, 130–090–101) and CD3+ cells were enriched by positive selection (Miltenyi, 130–094–973) according to the manufacturer's protocol before being frozen and shipped to Creative Biolabs for TCR sequencing. The raw sequencing files are available in the SRA database, under accession number PRJNA952625. The clonality index specific for characterizing the situation of T-cell clonal proliferation was calculated on the basis of the following equation where pi is the proportion of sequence i relative to the total sequences and S is total quantity of clonotypes:
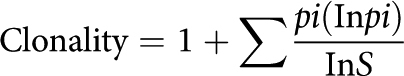 |
For NanoString analysis of bulk RNA, 5 × 105 cells were frozen in 100 μL of RLT Lysis buffer (Qiagen, 1053393). RNA samples were shipped on dry ice to LakePharma and analyzed using the nCounter Mouse PanCancer Immune Profiling Codeset with an nCounter FLEX analysis system. NanoString analysis was performed using nSolver Software with the Advanced Analysis module installed. For GeoMX Digital Spatial Profiler analysis, whole tumors were harvested at the indicated time point and placed in 10% neutral buffered formalin for at least 96 hours before being paraffin embedded by the Harvard Rodent Histopathology Core. FFPE blocks were shipped to Nanostring for GeoMX DSP analysis. Analysis was performed using the GeoMX Digital Spatial Profiler Software (v.2.5.1145), which utilizes a similar codeset-based technology as the bulk RNA analysis, just on a larger scale encompassing the whole transcriptome and selectively collecting the samples from previously identified cells (such as all CD8+ T cells in the region of interest).
Flow cytometry
All cell staining was performed in 96-well round bottom plates using FACs buffer (PBS + 0.5% BSA) or 1× permeabilization buffer (eBioscience, 00–5223–56), where appropriate. 1 to 5×106 cells were plated per well and were first treated with FC block (BioLegend, 101320) at room temperature for 15 minutes before the FC block was washed off and cells were resuspended in FACs buffer containing only the MulV p15E–specific tetramer (Thermo Fisher Scientific, 50–168–9385) for 20 minutes. After tetramer staining, cells were washed and then stained with a master mix of extracellular antibodies for 20 minutes at 4°C before being fixed/permeabilized overnight using the eBioscience Foxp3 Transcription Factor Staining Buffer Set (Thermo Fisher Scientific, 00–5521–00) according to the manufacturer's protocol. The next day, samples were washed with permeabilization buffer and stained with intracellular markers for 20 minutes at 4°C. Cells were analyzed on a Cytek Aurora system running Spectral Flow Software [2.2.0 (10212019)]. Fluorescence minus one (FMO) and single stain controls [cells or OneComp ebeads (Thermo Fisher Scientific, 01–1111–42)] were stained alongside samples. Unless otherwise noted, when flow cytometry was used to assess effector cytokine production, cells were restimulated prior to staining with PMA (50 ng/mL, Sigma-Aldrich, P1585) and Ionomycin (1μg/mL, Sigma-Aldrich, IO634–1MG) in the presence of 1× Brefeldin A (Thermo Fisher Scientific, 00–4506–51) for 4 hours at 37°C in RPMI-1640 media (Gibco, 11879–020) containing 10% heat inactivated FCS (Thermo Fisher Scientific, 10082–147) and 1× penicillin/streptomycin (Thermo Fisher Scientific, 15140–122). Cells used for 2-NDBG assay were starved in glucose-free RPMI-1640 media (Gibco, 11879–020) for 1 hour, then incubated with 150 μmol/L 2-NDBG (Cayman Chemical, No. 186689–07–6) for 1 hour at 37 °C/5% CO2 before being stained for extracellular markers. MitoTracker Deep Red FM (Thermo Fisher Scientific, No. M46753), MitoTracker Green FM (Thermo Fisher Scientific, M46750), MitoSOX Red (Thermo Fisher Scientific, M36008), and TMRM (Thermo Fisher Scientific, T668) staining were all performed according to the manufacturers protocol. Briefly, the individual dyes were diluted in RPMI-1640 media (Gibco, A10491–01) containing 10% heat inactivated FBS (Gibco, 10082–147) and penicillin/streptomycin (Gibco, 15140–122) and the cells were incubated for 1 hour at 37°C and 5% CO2. Cells were then washed with FACs buffer and stained for extracellular markers. Flow cytometry plots were generated with FlowJo Software (v10.5.30) and are representative samples.
Specific antibody clones are detailed as follows. Fluorescent dye–conjugated antibodies specific for the following proteins were purchased from Biolegend: CD8α APC, clone 53–67; CD4 BV650, clone RM4–5; CD3 AF700, clone 17A2; CD45 BV605, clone 30-F11; CD49b APC/Cy7, clone DX5; CD25 BV421, clone PC61; CD25 APC/Fire 750, clone PC61; Ki67 PeCy7, clone 16A8; Ki67 AF700, clone 16A8; granzyme B FITC, clone GB11; IFNγ PE, clone XMG1.2; F4/80 Pe/Dazzle 594, clone BM8; CD3 Complex PeCy7, clone 17A2; FC Block, clone 93. Fluorescent dye–conjugated antibodies specific for the following proteins were purchased from eBioscience: CD45 BUV395, clon30-F11; CD4 BUV496, clone GK1.5; CD8 BUV563, clone 53.6–7; TNF BV750, clone MP6-XT22; CD49b Pe-Cy5, clone DX5, FoxP3 AF488, clone FJK-16s; FoxP3 eFlour450, clone FJK-16s. Fluorescent dye–conjugated antibodies specific for the following proteins were purchased from Becton Dickinson: TIM-3 BV650, clone 5D12; LAG-3 BV711, clone C9B7W; TCF-7/TCF-1 FITC, clone S33–966; FOXO1 PE, clone C29H4. The fluorescent dye–conjugated tetramer against the MulV p15E peptide KSPWFTTL (50–168–9385) and the anti-TOX mAb (eFlour660, Clone TXRX10) was purchased from Thermo Fisher Scientific. The Live/Dead Blue Dye was also purchased from Thermo Fisher Scientific (L23105).
Pharmacokinetic analysis
Plasma and tumor samples were collected at indicated time points by Charles River Laboratories and shipped on dry ice to Werewolf Therapeutics, where they were stored at –80°C. MC38 tumor lysates were generated by homogenizing each tumor with a Qiagen TissueRuptor with disposable probes (Qiagen) in ice cold Lysis Buffer [1× Tris buffered saline (Sigma-Aldrich, T5912–1L), 1 mmol/L EDTA (Sigma-Aldrich, 3690–100 mL), 1% Triton X-100 (Sigma-Aldrich, X100–1000 mL), with protease inhibitors (Sigma-Aldrich, P8340–1L) in diH2O]. Plasma and tumor samples were analyzed using a sandwich ELISA on the MSD platform, which detects both intact mWTX-330 and free/released IL-12. Briefly, MSD multiarray plates were coated with capture antibody (human IL-12/IL-23 p40 antibody, R&D Catalog No. MAB1510–500) in PBS (pH 7.4), then washed with PBS plus 0.05% Tween 20 and blocked with PBS + 3% BSA. Diluted controls, study samples, and calibration standards were then added to the wells for 1 hour before the wells were washed and Biotin-tagged detector antibody (biotinylated human IL-12 antibody, R&D Catalog No. BAF219) was added to each well. Following incubation, plates were washed, and diluted Sulfo-tagged Streptavidin was added to each well. After incubation, the plates were washed and 1× Read Buffer T surfactant (MSD Catalog No. R92TG-1) was added to each well. Plates were immediately analyzed using an MSD Imager (MSD MESOTM QuickPlex SQ 120). Free IL-12 level was quantified using an in-house developed electrochemiluminescence immunoassay (ECLIA) assay. Briefly, a human IL-12/IL-23 p40 mAb was used as the capture antibody (Biolegend, Catalog No. 508808) and biotinylated mouse IL-12 (p35) mAb was used as the detector antibody (MabTech, Catalog No. 3456–6-250). Data acquisition and analysis were performed using MSD Workbench 4.0.12, and pharmacokinetic parameters were calculated using Phoenix WinNonlin Version 8.1. The concentration of samples and controls was calculated from the standard curve.
WTX-330 stability in human serum
WTX-330 was incubated in human serum (BioIVT) from healthy donors in duplicate for each timepoint. Time zero (T0) samples were immediately frozen at −80°C. The remaining samples were incubated at 37°C for 24 (T24) or 72 (T72) hours before being stored at −80°C. Stability of WTX-330 was assessed by Western blot analysis using the JESS system (Protein Simple, SM-W004) according to the manufacturer's general protocol. Input controls (intact and protease cleaved) were also analyzed. Samples, primary antibody (anti–IL-12; R&D Systems, AB-219-NA), and secondary antibody (Jackson Labs, 805–035–180) were loaded into a 12–230 kDA Jess separation module and run using a Jess system set to the standard settings for chemiluminescence, with modifications to the standard protocol for nonreducing conditions. Analysis of the resulting Western blot analysis was performed using Compass for Simple Western Software (v4.1.0).
Human primary cell assays
Human PBMCs were isolated from whole blood (BioIVT) using Ficoll-Paque Plus (GE Healthcare, GE17–1440–03) according to the manufacturer's protocol and frozen in Recovery Cell Culture Freezing Media (Gibco, 12648010) for later use. To generate activated T cells (Tblasts), PBMCs were thawed, counted, resuspended at 1 × 106 cells per mL in Tblast media (RPMI-1640 +10% heat inactivated FCS + 1× penicillin/streptomycin + 10 mmol/L HEPES), and stimulated with 5 μg/mL of PHA (Sigma-Aldrich, L1668–5MG) for 72 hours before being frozen for later use. To measure IL-12 activity in human primary cells, previously frozen Tblasts were thawed, counted, and plated in a 96-well round bottom plate in X-Vivo 15 media (Lonza, 04418Q), and incubated with titrated amounts of intact or protease-activated (cleaved) INDUKINE proteins or chimeric IL-12. After 72 hours, IFNγ production was measured using a Human IFNγ specific AlphaLisa Kit (Perkin Elmer, AL217C) according to the manufacturer's protocol with a Perkin Elmer Enspire Αlpha Reader running Enspire Manager Software (V4.13.3005.1482).
Ex vivo INDUKINE protein processing assay
Primary human healthy cells were purchased from either ATCC (Healthy Lung Fibroblasts, Lots 70002722/70014625/801; primary bronchial/tracheal epithelial cells, Lot 70031252; primary renal mixed epithelial cells, Lot 70018214; primary small airway epithelial cells, Lot 70026720; renal cortical epithelial cells, Lot 70001233), Lonza (intestinal myofibroblasts, Lots 437306/662132), or Zen-Bio (human bladder smooth muscle cells, Lot Hu1033; human dermal fibroblasts, Lots DFM012114C/DFM020314A/DFM062217B), and cultured according to the manufacturers’ protocols. Dissociated human tumor samples were purchased from Discovery Life Sciences and included the following indications: bladder cancer, glioblastoma multiforme, gastric cancer, colorectal cancer, endometrial cancer, melanoma, kidney cancer, NSCLC-adenocarcinoma, NSCLC-squamous, cervical cancer, ovarian cancer, and non-Hodgkin lymphoma. These samples are generated from primary human tumor samples that were surgically removed and enzymatically digested on site prior to being frozen. All purchased samples were shipped to Werewolf Therapeutics on dry ice and were stored in a liquid nitrogen freezer. To examine INDUKINE protein processing, tumor samples were thawed, washed, and counted. Cells were then resuspended in X-Vivo 15 media containing intact WTX-330, a NC variant of WTX-330, or precut WTX-330 for 48 hours before cell culture supernatants were collected and frozen for later analysis. To measure the level of INDUKINE molecule processing, the cell culture supernatants from the dissociated tumor samples were thawed and added to primary human Tblasts as described above (Human primary cell assays). After 72 hours, IFNγ production was measured using a Human IFNγ specific AlphaLisa Kit (Perkin Elmer, AL217C), and was normalized using the following equation to assess the level of processing:
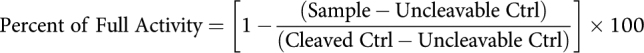 |
Data representation, bioinformatic analysis, and statistical analysis
Flow cytometry plots were generated with FlowJo Software (v10.5.30) and are representative samples. All the quantitative plots were generated using GraphPad Prism 8 Software for Windows (64-Bit). For in vitro activity assays, data were analyzed using a nonlinear sigmoidal, 4PL curve fit model without constraints. Statistical analysis was also performed using GraphPad Prism software [(v9.4.1(681)]. Two sample comparisons used a Student t test while comparisons of more than two groups used an ANOVA test with multiple comparisons. Antitumor effects over time were analyzed by using a mixed-effects model. For the NanoString dataset, statistical analysis was performed using nSolver software with the Advanced Analysis Module installed. Pathway analysis (utilizing the KEGG database for mus musculus v1) and GSEA (utilizing Gene Ontology pathways for mus musculus) was performed using Partek software (v 10.0.22.0428), based on transcripts that had significant differences in expression following mWTX-330 treatment with an FDR step-up of 0.05.
Data availability statement
All data needed to evaluate the conclusions in the manuscript are either present in the manuscript or the Supplementary Materials and Methods, available in publicly accessible databases, or are available upon reasonable request from the corresponding author with the appropriate legal protections in place. All Z scores used to generate pathway specific heat maps or analyze GSEO analysis are included in the supplemental files, as are Log2 fold change and Log10P values for both bulk RNA NanoString analysis as well as those from the Nanostring DSP system. Raw data for TCR sequencing are available in the SRA database, accession no. PRJNA952625.
Results
mWTX-330 generates a robust, cleavage-dependent antitumor immune response in multiple models
To address the clinical shortcomings of free cytokine therapeutics, a selectively inducible cytokine prodrug (INDUKINE polypeptide) was developed. mWTX-330 is a surrogate INDUKINE polypeptide that contains chimeric IL-12 (human p40, murine p35), a HLE anti-human serum albumin single-domain antibody, and an inactivation domain consisting of an scFv antibody fragment that prevents the binding of IL-12 to its receptor (Fig. 1A). The use of chimeric IL-12 allows for this molecule to be active in murine syngeneic tumor models (21); the fully human molecule (WTX-330) is discussed later. The domains of mWTX-330 are linked by a proprietary protease-cleavable linker sequence, such that when the molecule is processed in the TME, IL-12 is locally released. To measure the difference in activity between intact and protease-activated (cleaved) mWTX-330, HEK-Blue IL-12 reporter cells were incubated with either intact or protease-activated mWTX-330, and IL-12 signaling was assessed. In vitro protease activation resulted in the release of the inactivation and HLE domains, as expected (Supplementary Fig. S1A). In this assay, intact mWTX-330 had 175-fold less activity than either cleaved mWTX-330 or chimeric IL-12 based on the EC50 value (Fig. 1B). To test whether mWTX-330 could generate antitumor activity in vivo, MC38 tumor–bearing animals were treated with titrated amounts of mWTX-330, and tumor growth was monitored over time. A variant of mWTX-330 with a NC linker (Supplementary Fig. S1B) was also included at the highest dose as a control. In this model, even the lowest tested dose of mWTX-330 (7 μg/dose) generated statistically significant tumor growth inhibition, with 43 μg/dose being sufficient to generate complete tumor rejections (Fig. 1C). In contrast, the NC variant of mWTX-330 had less activity than even the lowest dose of mWTX-330, demonstrating that the full potency of mWTX-330 is dependent on in vivo processing of the molecule. mWTX-330 treatment also generated robust antitumor activity in less immune cell infiltrated (“colder") syngeneic tumor models, including CT26 (Fig. 1D), B16-F10 (Fig. 1E), and EMT-6 (Fig. 1F), demonstrating the broad activity of this molecule in vivo. Similar to the MC38 model, in vivo cleavage was also required for the full activity of mWTX-330 in the EMT-6 tumor model (Supplementary Fig. S1C). Although the use of a NC control demonstrated the necessity of processing for full activity, these data did not directly indicate that processing was occurring in the TME. FTY720 is a small molecule inhibitor of Sphingosine-1-phosphate receptor-1, which prevents lymphocyte egress from secondary lymphoid tissues (25) and effectively isolates TILs from the normal recirculating population of immune cells in vivo. mWTX-330/FTY720 cotreated animals retained the potent early antitumor activity associated with mWTX-330 treatment, although tumor control was less complete after dosing with the INDUKINE molecule had stopped (Fig. 1G). These data demonstrate that systemically administered mWTX-330 is processed in the TME, and that this local release of IL-12 is sufficient to generate early tumor growth inhibition.
Figure 1.

mWTX-330 generates robust antitumor immunity in a cleavage-dependent manner. A, The components of mWTX-330, including chimeric IL-12 (yellow), a half-life extending HSA-specific single-domain antibody (blue), the activity blocking Ab domain (teal), and the protease-cleavable linkers (red). B,In vitro activity of mWTX-330 in the HEK-Blue IL-12 reporter assay comparing intact (blue) and protease-activated (cleaved) mWTX-330 (red) to chimeric IL-12 (black), where error bars represent the mean (± SD) for duplicate wells. Tumor growth curves of MC38 (C), CT26 (D), B16-F10 (E), or EMT-6 (F) tumors. Mice were dosed twice a week for 2 weeks, with the specific doses reported in the figure, using between 8 and 12 mice per group. In some experiments, mice were treated with a NC variant of mWTX-330 (C; NC, blue). G, MC38 tumor bearing mice were treated with mWTX-330 ± daily FTY720 treatment, and tumor growth was monitored over time. H, MC38 tumor bearing mice were dosed twice a week with depleting antibodies in conjunction with mWTX-330, and tumor growth was monitored over time. Mice that completely rejected either MC38 tumors (I) or EMT-6 tumors (J) were rechallenged with the same tumor cell line at least 60 days after complete rejection of the primary tumor. As a control, age-matched, tumor naïve animals were also challenged, and tumor growth was monitored over time. In all tumor plots, average tumor volume (± SEM) over time is reported.
To identify the effector cell populations responsible for the mWTX-330–induced antitumor immunity, MC38 tumor–bearing mice were treated with mWTX-330 in conjunction with antibody-based depletion of individual effector cell populations. Although depletion of the CD8+ T-cell population did not inhibit early tumor control, mWTX-330 treated mice without CD8+ T cells were ultimately unable to control tumor growth (Fig. 1H). In contrast, depletion of either NK cells or total CD4+ T cells alone did not inhibit mWTX-330–induced antitumor activity (Fig. 1H). However, in mice where all three populations were depleted, mWTX-330 treatment had little antitumor activity, suggesting that these cell types work jointly to reject MC38 tumors in response to treatment. Furthermore, when mWTX-330 treated mice that previously rejected either MC38 tumors (Fig. 1I) or EMT-6 tumors (Fig. 1J) were rechallenged, 100% of the animals were protected against tumor growth. Together, these data suggest that mWTX-330 treatment generates a robust and durable antitumor immune response that is dependent on in vivo cleavage/activation of the molecule.
The INDUKINE design of mWTX-330 expands the therapeutic window of chimeric IL-12
In the clinic, free cytokines have poor pharmacokinetic profiles with short half-lives, resulting in rapid clearance and poor exposure in patients, which in turn leads to unreasonable dosing schedules. To examine whether the INDUKINE design of mWTX-330 enhanced the half-life and exposure of the molecule, MC38 tumor–bearing mice were dosed with a single dose of either chimeric IL-12 or mWTX-330, and peripheral blood and tumor samples were collected over time. Using a unique set of detection reagents, it was possible to separately measure the amount of total IL-12 (blocked + unblocked) or to selectively measure the amount of unblocked IL-12 present. In the plasma of tumor-bearing mice, chimeric IL-12 had a half-life of only 4 hours (Supplementary Fig. S2A), whereas mWTX-330 had a half-life of nearly 16 hours (Fig. 2A). Furthermore, only about 2% of the mWTX-330 found in the plasma was in the form of the cleaved molecule. In contrast, when the same analysis was performed in tumor samples (Fig. 2B), nearly 45% of the molecule was unmasked IL-12 and intratumoral exposure was maintained far beyond what was achieved by treatment with chimeric IL-12, which is consistent with ongoing intratumoral processing of mWTX-330 (Supplementary Fig. S2B). To further corroborate the selective processing of mWTX-330 in the tumor, the activation status of tumor-infiltrating CD8+ T cells, CD4+ T conventional cells (FoxP3−), and NK cells was compared with those same populations in the tumor-draining and nondraining lymph nodes, as well as the peripheral blood following mWTX-330 treatment. Although mWTX-330 treatment resulted in a significant increase in the frequency of polyfunctional CD8+ T cells within the MC38 tumors, no such increase was observed in the lymph nodes or in the peripheral blood (Fig. 2C). Likewise, among conventional CD4+ T cells (Supplementary Fig. S2C) and NK cells (Supplementary Fig. S2D), mWTX-330 preferentially increased the frequency of cells producing effector cytokines in the tumor compared with the peripheral tissues, consistent with selective intratumoral processing of the INDUKINE molecule.
Figure 2.

mWTX-330 is preferentially activated in the TME and has a greater therapeutic window than chimeric IL-12. A and B, Plasma (A) and tumor (B) samples from MC38 tumor-bearing mice treated with mWTX-330 were analyzed at various timepoints for either the presence of the total INDUKINE protein (tan) or free chimeric IL-12 (red). The AUC was calculated, and the ratio of total INDUKINE molecule to free chimeric IL-12 was calculated. C, MC38 tumor-bearing mice were dosed twice with mWTX-330, and the frequency of polyfunctional CD8+ T cells in the tumor, peripheral blood, tumor draining or nontumor draining lymph nodes was measured by examining coexpression of IFNγ, TNF, and Granzyme B after PMA/ionomycin restimulation. D, Representation of the therapeutic window of recombinant chimeric IL-12 or mWTX-330 in MC38 tumor-bearing mice based on multiple experiments identifying the MED and the MTD of each molecule (Supplementary Fig. S3). Where applicable, error bars represent the mean (± SD).
Systemic administration of IL-12 and the subsequent system-wide activation of the immune system results in serious, and sometimes fatal, toxicity in patients (26). Therefore, it was important to compare the tolerability and efficacy of mWTX-330 directly to that of recombinant IL-12. Titrated amounts of mWTX-330 and recombinant chimeric IL-12 were given to MC38 tumor–bearing mice, and the antitumor activity of the two treatments were compared on the basis of the moles of chimeric IL-12 administered in these two formats. In the case of chimeric IL-12, treatment induced antitumor activity at a dose as low as 0.684 nanomoles, making this the minimum efficacious dose (MED), whereas 3.42 nanomoles was required to generate complete tumor rejection (Supplementary Fig S3A). Meanwhile, 0.283 nanomoles of mWTX-330 also induced potent tumor growth inhibition, with a dose of only 1.73 nmol required to generate complete tumor rejection (Supplementary Fig. S3B), suggesting that less of the INDUKINE molecule is required to achieve similar activity. In terms of toxicity, tumor-bearing mice treated with enough chimeric IL-12 to generate complete tumor rejections (3.42 nmol) had a statistically significant loss of body weight during the dosing period (Supplementary Fig. S3C and S3E). In contrast, no statistically significant body weight loss was observed when mice were treated with up to four times the amount of mWTX-330 required for complete tumor rejection (6.885 nmol; Supplementary Fig. S3D and S3E), demonstrating that the INDUKINE molecule is better tolerated than the recombinant cytokine.
One of the adverse side effects of systemic IL-12 treatment in mice is the development of liver infiltration by immune cells (24). Indeed, one report has demonstrated that the rapid liver infiltration associated with anti-CD40 therapy is due to IL-12 production (23). Therefore, we treated naïve mice with the doses of IL-12 required to generate complete tumor rejection as identified previously (Supplementary Fig. S3) and examined their livers 2 days after the initiation of dosing. Treatment of mice with either anti-CD40 or chimeric IL-12 resulted in perivenular infiltration of the liver by leukocytes, as well as some liver dysplasia (Supplementary Fig. S4). In contrast, administration of mWTX-330 at up to twice the dose required for complete tumor rejection did not drive liver infiltration by immune cells or cause cellular dysplasia.
The therapeutic window (TW) of a treatment is defined as the ratio of the MTD and the MED, thereby measuring the distance between clinical activity and serious adverse events. In our model, the MTD was defined as the highest dose where the mice maintained above 85% of their body weight throughout the course of treatment (Supplementary Fig. S3E). On the basis of the MED (0.684 nanomoles) and MTD (3.42 nanomoles) in the MC38 model (Supplementary Fig. S3), chimeric IL-12 had a therapeutic window of approximately 5. In contrast, mWTX-330 had a slightly lower MED (0.283 nanomoles) compared with chimeric IL-12, and the INDUKINE molecule was tolerated up to a much higher dose level (MTD: 13.77 nanomoles; Supplementary Fig. S3). This resulted in the TW of mWTX-330 being nearly 50, representing a 10-fold improvement as a result of the INDUKINE design (Fig. 2D). Together, these data demonstrate that systemic treatment with mWTX-330 does not result in widespread and nonspecific activation of effector cells but instead selectively activates tumor-infiltrating lymphocytes, thus improving the safety and tolerability of IL-12.
mWTX-330 treatment activates various TIL populations in the MC38 model
To better understand the mechanism by which mWTX-330 treatment generates antitumor immunity, MC38 tumor–bearing mice were randomized into treatment groups on Day 0 and treated with either vehicle or mWTX-330 on Day 1 and Day 4. Tumors were harvested 24 hours after the second dose and analyzed by flow cytometry (Supplementary Fig. S5) or NanoString analysis using the PanCancer Mouse Immune Profiling Panel. Systemic treatment with mWTX-330 had a clear effect on the transcriptional profile of the TME, with 364 of the 770 investigated transcripts having statistically significant differences in expression after treatment (Fig. 3A). mWTX-330 treatment resulted in significant enrichment of several immune-related signaling pathways, including “PD-L1 Expression and PD-1 Checkpoint in Cancer,” “NK Cell Cytotoxicity,” and “TH1 and TH2 Differentiation” (Fig. 3B). In agreement with this analysis, mWTX-330 treatment resulted in a significant increase in the frequency of tumor-infiltrating NK cells producing IFNγ, TNF, and Granzyme B (Fig. 3C). Moreover, mWTX-330 treatment resulted in NK, NKT, conventional CD4+ T cells, and CD8+ T cells producing such elevated levels of IFNγ that it was measurable by intracellular cytokine staining without ex vivo restimulation (Supplementary Fig. S6). However, the signaling pathway with the highest enrichment score following treatment with mWTX-330 was “Antigen Processing and Presentation.” Gene set enrichment analysis revealed that mWTX-330 treatment significantly enriched several gene sets associated with antigen presentation of exogenous peptides and/or antigens in either MHC class I or MHC class II proteins (Supplementary Table S1). Presentation of exogenously derived antigens in MHC class I proteins is a phenomenon known as cross presentation, and it is exclusively mediated by a unique population of dendritic cells (DC), identified by the expression of CD103 (27). In agreement with the bioinformatic analysis, flow cytometric analysis demonstrated that mWTX-330 treatment significantly increased the frequency of the cross presenting CD103+ DC population among total DCs within the tumor (Fig. 3D and E).
Figure 3.

mWTX-330 activates various tumor-infiltrating immune-cell populations in the MC38 syngeneic tumor model. MC38 tumor-bearing mice were dosed twice a week with mWTX-330 or vehicle. Tumors (n = 5–6 mice per condition) were collected 24 hours after the second dose and dissociated into single cell suspensions for further analysis. RNA from each tumor was isolated and subjected to immune profiling with the NanoString PanCancer Mouse Immune Profiling Panel. A, Heat map of transcripts with statistically significant differences in expression between the two treatments (Supplementary Table S2). Transcripts were excluded from the heat map if they had average normalized counts below 50. Each lane represents an individual animal. B, Pathway enrichment analysis was performed using Partek software. C, The frequency of tumor-infiltrating NK cells producing IFNγ, TNF, or Granzyme B. Representative flow plots of CD11b+ and CD103+ tumor-infiltrating DCs (D) and the resulting ratio (E). F, Volcano plot of transcripts differentially expressed between mWTX-330 and vehicle-treated mice (Supplementary Table S2). G, The frequency of polyfunctional, tetramer positive CD8+ T cells was measured by examining coexpression of IFNγ, TNF, and Granzyme B after PMA/ionomycin restimulation. H, The frequency of conventional CD4+ T cells with a TH1 phenotype (Tbet+IFNγ+TNF+). I, The frequency of tumor-infiltrating FoxP3+ Tregs producing IFNγ and TNF (I–J) or expressing Tbet (K). Unless otherwise stated, data are presented a box and whiskers plot, where the box represents the 25th to 75th percentile, and the middle line represents the mean, whereas P values are derived from t tests. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Given the role of CD8+ T cells in mWTX-330–mediated tumor rejection (Fig. 1H) and the finding that mWTX-330 increased tumor infiltration by cross-presenting DCs, it seemed likely that mWTX-330 treatment also enhanced CD8+ T-cell activation. Indeed, differential expression analysis of the RNA isolated from bulk tumor samples (including both immune cells and tumor cells) demonstrated that mWTX-330 treatment significantly increased the expression of many transcripts associated with the activation of cytotoxic CD8+ T cells, including Ifng (Interferon gamma), Gzmb (Granzyme B), Prf1 (Perforin), and Tnf (Tumor necrosis factor), in addition to several chemoattractant molecules (Fig. 3F; Supplementary Table S2). Although mWTX-330 did not increase the density of various T-cell populations (Supplementary Fig. S7A) or the frequency of tumor-specific CD8+ T cells among total CD8+ T cells within the tumor at this early time point (Supplementary Fig. S7B), treatment did result in robust activation of the tumor-specific CD8+ T-cell population, as demonstrated by an increase in the frequency of tetramer+ polyfunctional CD8+ T cells (Fig. 3G), with nearly 100% of the tumor-specific T cells producing IFNγ (Supplementary Fig. S7C). Comparable results were seen when considering the entire tumor-infiltrating CD8+ T-cell population, and not just the tetramer+ ones (Supplementary Fig. S7D–S7E). In addition, among CD4+ T cells, mWTX-330 treatment significantly increased the frequency of conventional CD4+ T cells with a TH1 phenotype (Tbet+ IFNγ+ TNF+; Fig. 3H; Supplementary Fig. S7F). Finally, recent publications have highlighted the role of IFNγ in driving regulatory T cells (Treg) away from regulatory activity and toward an effector phenotype, in a phenomenon known as Treg fragility (28). mWTX-330 treatment resulted in a significant subset of the FoxP3+ Treg population co-producing the effector cytokines TNF and IFNγ (Fig. 3I and J) and expressing Tbet (Fig. 3K), demonstrating that systemic treatment with mWTX-330 can induce Treg fragility in the TME. Altogether, these data demonstrate that systemic administration of mWTX-330 results in the transcriptional reprogramming of the TME and the subsequent activation of various tumor-infiltrating effector-cell populations.
mWTX-330 treatment expands unique TCR clones and increases TCR clonality in the TME
In the MC38 tumor model, mWTX-330 treatment resulted in rapid tumor rejection (Fig. 1C), making it technically challenging to fully investigate the kinetics of immune activation. In contrast, mWTX-330 treatment of the EMT-6 tumor model generated complete rejections over a longer period, which is favorable to a more thorough analysis of an ongoing CD8+ T-cell response (Fig. 1F). Therefore, mice bearing established EMT-6 tumors were randomized into treatment groups and dosed twice a week for 2 weeks with either vehicle or mWTX-330. Tumors and plasma were then harvested at various time points. As in the MC38 model, treatment with mWTX-330 did not affect the density of tumor-infiltrating T cells at any of the examined time points as measured by flow cytometry (Supplementary Fig. S8A and S8C). In the vehicle-treated animals, the frequency of polyfunctional CD8+ T cells did expand over the course of the experiment, but eventually retracted in line with subsequent tumor growth. In contrast, mWTX-330 treatment increased the frequency of polyfunctional CD8+ T cells over that of the vehicle treated animals as early as Day 5 after the start of treatment (Fig. 4A), and this frequency continued to expand even after exposure to mWTX-330 was undetectable (Fig. 4A; Supplementary Fig. S8D).
Figure 4.

Systemic treatment with mWTX-330 expands novel TCR clones and increases overall clonality of the TCR repertoire. EMT-6 tumor-bearing mice (n = 30 per group) were dosed twice a week for 2 weeks with mWTX-330 or vehicle. Tumors were collected at various time points for analysis (n = 6 per condition and time point). A, The frequency of polyfunctional CD8+ T cells was measured by examining coexpression of IFNγ, TNF, and Granzyme B after PMA/ionomycin restimulation. Tumors from vehicle mice at day 21 and 25 exceeded tumor burden and were not included in analysis. B, FFPE EMT-6 tumors from Day 11 were analyzed using the nanostring DSP system, and immunofluorescence staining of DAPI (blue), PanCK (pink), and CD8 (green) was performed. C, Volcano plot of transcripts differentially expressed by tumor-infiltrating CD8+ T cells between mWTX-330 and vehicle-treated mice. Pathway analysis was performed using the Nanostring DSP software and heat maps of genes associated with IL-12 signaling (D), IFNγ signaling (E), or TCR signaling (F) are reported (Supplementary Table S4). G–J, Live T cells were isolated from EMT-6 tumors on Day 11 and TCR sequencing was performed. G, The frequency of individual VDJ recombinations on the TCRβ chain. H, The frequency of the top 10 clones for each animal as a fraction of the total TCR repertoire. Each column represents the TCR repertoire of a single animal. I, The overall clonality index was calculated for these samples by treatment group. J, The top 100 TCR clones based on frequency were identified on the basis of CDR3 sequencing for each individual animal, and any clones that were present in the tumors of at least two of the vehicle-treated or two of the mWTX-330–treated animals were compared.
To better understand the transcriptional effects of mWTX-330 treatment specifically on the tumor-infiltrating CD8+ T-cell population, Geospatial NanoString analysis was performed on EMT-6 tumor samples from animals treated either with vehicle or mWTX-330. This technology merges immunofluorescence with whole transcriptome analysis of specific cells, allowing for transcriptional analysis of a specific cell population while maintaining spatial information that would otherwise be lost during tissue dissociation. As noted earlier, mWTX-330 treatment did not affect the overall infiltration of EMT-6 tumors by T cells as determined by flow cytometry (Supplementary Fig. S8A–S8C). However, using Geospatial NanoString analysis, CD8+ T cells were found to be largely confined to the outer margins of the tumor in the control group (Fig. 4B). In contrast, mWTX-330 treatment induced significant infiltration of the EMT-6 tumors, with CD8+ T cells penetrating deeply into the tumor tissue (Fig. 4B). Whole transcriptome analysis of the tumor-infiltrating CD8+ T cells demonstrated that mWTX-330 treatment resulted in substantial transcriptional reprogramming of these cells, including upregulation of many genes associated with T-cell activation such as Tbet, Ifng, Cd25, and chemoattractants known to be responsible for the recruitment of additional immune cells (Fig. 4C; Supplementary Table S3).
Among the pathways significantly upregulated in tumor-infiltrating CD8+ T cells by mWTX-330 treatment were IL-12 (Fig. 4D; Supplementary Table S4) and IFNγ signaling (Fig. 4E; Supplementary Table S4), confirming the local release of unmasked IL-12 and subsequent production of IFNγ within the TME. mWTX-330 treatment also increased expression of transcripts downstream of TCR signaling (Fig. 4F; Supplementary Table S4). Given the effects on cross-presenting DCs (Fig. 3), we hypothesized that mWTX-330 treatment may result in the activation of new or previously underrepresented T-cell clones to generate antitumor immunity. To test this, T cells were isolated from EMT-6 tumors following mWTX-330 or vehicle treatment and sent for TCR sequencing. Although the tumor-infiltrating TCR repertoire of control animals was dominated mainly by low frequency combinations of VDJ regions, mWTX-330 treatment drove the robust expansion of several specific combinations (Fig. 4G). The top 10 tumor-infiltrating TCR clones (as defined by the amino acid sequence of the CDR3 region) accounted for only about 13% of the total TCR repertoire in vehicle-treated animals on average, compared with over half of the TCR repertoire in mWTX-330–treated animals (Fig. 4H), resulting in a significant increase in the overall clonality index of the tumor-infiltrating TCR repertoire (Fig. 4I). To better understand the effects of mWTX-330 on the tumor-infiltrating T-cell repertoire, the 100 TCR clones with the highest frequency from each animal were compared to identify TCR clones common to at least two animals within a treatment group (Supplementary Fig. S9A). It was found that only about 30% of the clones (5/18) common to the mWTX-330–treated animals were also found in the vehicle-treated animals (Fig. 4J), and the raw frequency of those clones was not impacted by mWTX-330 treatment (Supplementary Fig. S9B). Instead, the majority of the TCR clones common to mWTX-330–treated animals were poorly represented in the tumors of vehicle-treated animals and expanded ∼1,600 -fold on average with mWTX-330 treatment (Supplementary Fig. S9C). Overall, these data indicate that mWTX-330 treatment increases the clonality of tumor infiltrating T-cell populations primarily by expanding clones that were previously underrepresented, rather than increasing the frequency of dominant clones already present.
mWTX-330 substantially increases mitochondrial activity in tumor-infiltrating CD8+ T cells and NK cells
Recently activated CD8+ T cells have substantial energy requirements and rely heavily on glucose uptake and glycolysis to quickly generate the energy necessary to perform their effector functions, before transitioning towards mitochondria-dependent oxidative phosphorylation as they develop into long-lived memory cells (29). However, recent publications have demonstrated that tumor-infiltrating CD8+ T cells often fail to generate significant mitochondrial respiration compared with those activated in the spleen or lymph nodes (30–32), suggesting that the TME negatively impacts the metabolic health of effector cells and thereby prevents the generation of effective antitumor immunity. Among tumor-infiltrating CD8+ T cells, mWTX-330 treatment resulted in the significant enrichment of transcripts associated with glycolysis (Supplementary Fig. S10A; Supplementary Table S4). Therefore, we hypothesized mWTX-330 treatment may increase glucose uptake by tumor-infiltrating CD8+ T cells, and thereby lead to increased glycolysis. However, when these cells were incubated with a nonmetabolizable fluorescent glucose analog (2-NDBG), tumor-infiltrating CD8+ T cells from mWTX-330–treated animals had slightly less glucose uptake than those from vehicle-treated animals (Fig. 5A and B). Therefore, rather than simply increasing glucose uptake by tumor-infiltrating CD8+ T cells, mWTX-330 treatment was instead reprogramming those cells to utilize glucose more efficiently than those from vehicle-treated animals.
Figure 5.

mWTX-330 treatment drives increased mitochondrial respiration and fitness. EMT-6 tumor-bearing mice were dosed twice a week with mWTX-330 or vehicle with n = 5–10 animals per group, and tumors were collected on day 11 for analysis. A and B, EMT-6 TILs from either vehicle-treated or mWTX-330–treated animals were incubated with 2-NDBG, and uptake was measured by flow cytometry. Pathway analysis was performed on tumor-infiltrating CD8+ T cells and heat maps of genes associated with the TCA cycle (C) was generated (Supplementary Table S4). EMT-6 infiltrating CD8+ T cells (D, E, H, I, L, M) or NK cells (F, G, J, K, N, O) from either vehicle-treated or mWTX-330–treated animals were stained with Mitotracker Red (D–G), TMRM (H–K), or MitoSOX (L–O). EMT-6–infiltrating CD8+ T cells were assessed on day 11 for expression of TOX (P), coexpression of PD-1 and TIM-3 (Q), or coexpression of PD-1, TIM-3, and LAG-3 (R). Data are presented using a box and whiskers plot, where the box represents the 25th to 75th percentile, and the middle line represents the mean. P values are derived from t tests. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
In addition to driving increased glycolysis, mWTX-330 treatment also enriched for transcripts associated with the TCA cycle (Fig. 5C; Supplementary Table S4), mitochondrial biogenesis (Supplementary Fig. S10B; Supplementary Table S4), and mitochondrial translation (Supplementary Fig. S10C; Supplementary Table S4), suggesting that mWTX-330 treatment may enhance the mitochondrial activity and health of tumor-infiltrating effector cells. To test this, TILs were isolated from vehicle or mWTX-330-treated animals and mitochondrial phenotyping was performed by flow cytometry (Supplementary Fig. S10D). Mitotracker Red is a dye that specifically stains actively respirating mitochondria due to its pH sensitivity. Although tumor-infiltrating CD8+ T cells from vehicle-treated animals had limited evidence of ongoing active mitochondrial respiration, those from mWTX-330–treated animals had significantly increased levels of active respiration (Fig. 5D and E). This finding extended to NK cells (Fig. 5F and G) and total CD4+ T cells (Supplementary Fig. S10E). This increase was primarily due to increased mitochondrial activity, rather than simply an increase in total mitochondrial mass, as mWTX-330 treatment only slightly increased the total mitochondrial mass of tumor-infiltrating NK cells, CD8+ T cells, and total CD4+ T cells (Supplementary Fig. S10F). Furthermore, TMRM staining also revealed that mWTX-330 treatment significantly increased the mitochondrial membrane potential in both CD8+ T cells (Fig. 5H and I) as well as NK cells (Fig. 5J and K). Mitochondrial reactive oxygen species (ROS) have previously been linked both to NFAT signaling and subsequent production of IL-2 (33), as well as IFNγ production by memory CD4+ T cells (34), and can be detected using the dye MitoSOX Red. mWTX-330 treatment increased the production of mitochondrial ROS species in both CD8+ T cells (Fig. 5L and M) and NK cells (Fig. 5N and O).
Oxidative phosphorylation is the primary energy source for memory T cells, and increased dependence on this pathway has been associated with superior antitumor immunity and a shift away from exhaustion toward a “stem-cell like” phenotype (35). Tumor-infiltrating CD8+ T cells from mWTX-330–treated mice showed significantly upregulated expression of genes associated with T-cell stemness, including Tcf1, Foxo1, Cxcr3, and Il2rg (Supplementary Table S3) while significantly downregulating expression of several genes associated with CD8+ T-cell exhaustion, including Pdcd1, Havcr2, and Lag3 (Supplementary Table S4). However, flow cytometry analysis revealed that tumor-infiltrating CD8+ T cells from mWTX-330–treated animals had no difference in expression of either TCF7 (Supplementary Fig. S10G), or FOXO1 (Supplementary Fig. S10H), suggesting that a change in the stemness phenotype was not contributing to the activity of the INDUKINE molecule. Akin to polyfunctionality, coexpression of multiple checkpoint proteins has been associated with T-cell exhaustion in viral and tumor models (36, 37). In agreement with the transcriptional data, CD8+ T cells from mWTX-330–treated animals had significantly less expression of the transcription factor TOX (Fig. 5P), and significantly less coexpression of PD-1 and TIM-3 (Fig. 5Q). When examining the coexpression of PD-1, TIM-3, and LAG-3, mWTX-330 treatment significantly decreased the frequency of tumor-infiltrating CD8+ T cells coexpressing multiple checkpoint proteins, while significantly increasing the frequency of CD8+ T cells that expressed none of the three (Fig. 5R). Furthermore, mWTX-330 treatment also drove the majority of the tumor-infiltrating CD8+ T cells to express the activation marker CD25, demonstrating that the checkpoint negative cells were still activated, and not simply naive (Supplementary Fig. S10I). Together, these data demonstrate that systemic administration of mWTX-330 is sufficient to restore mitochondrial respiration in tumor-infiltrating CD8+ T and NK cells and to protect the CD8+ T-cell population from exhaustion, resulting in a robustly activated CD8+ T-cell effector population, which translates into robust antitumor immunity.
WTX-330, a human IL-12 INDUKINE molecule, is preferentially activated by primary human tumors
For preclinical murine studies, it was important to use a surrogate molecule that was active in mice. However, for clinical development, a fully human IL-12 payload will be used. WTX-330 is identical to mWTX-330 except that it contains fully human IL-12 as the payload (Fig. 6A). As with the murine surrogate molecule, intact WTX-330 had substantially less activity than either cleaved WTX-330 or recombinant human IL-12 in a HEK-Blue IL-12 reporter assay (Fig. 6B). Likewise, when exposed to stimulated primary human Tblasts from multiple donors, intact WTX-330 was 61-fold less active on average than the cleaved molecule based on EC50 values (Fig. 6C; Supplementary Table S5). In both in vitro assays, cleaved WTX-330 had activity similar to recombinant human IL-12. Furthermore, when WTX-330 was incubated in serum from healthy human donors (n = 6), no free IL-12 was detected after 72 hours at 37°C (Fig. 6D), confirming the stability of the molecule.
Figure 6.

WTX-330 is inducible, stable in human serum, and selectively processed by dissociated primary human tumor samples. A, The components of WTX-330, including fully human IL-12 (yellow), a half-life extending HSA-specific single-domain antibody (blue), the activity blocking Ab domain (teal), and the protease-cleavable linkers (red). B,In vitro activity of WTX-330 in the HEK-Blue IL-12 reporter assay comparing intact (blue), and protease-activated (cleaved) WTX-330 (red) to rhIL-12 (black). C,In vitro activity of intact (blue) and cleaved (red) WTX-330 in primary human Tblasts compared with rhIL-12 (black). D, WTX-330 was diluted into healthy human serum from n = 6 donors and incubated at 37°C for 24 or 72 hours before WTX-330 processing was measured by capillary Western blot analysis. E, WTX-330 was exposed to primary human tumor samples (n = 88) or primary human healthy cells (n = 13) for 48 hours before INDUKINE protein cleavage was measured. For B and C, error bars represent the mean (± SD) of duplicate wells. For E, box plots represent the 25th and 75th percentile, whereas the line represents the median value for each indication.
To examine whether WTX-330 would be selectively activated by primary human tumor samples, an in vitro cleavage assay was developed (4). Briefly, primary human dissociated tumor samples from various indications were incubated with either WTX-330, pre-cut WTX-330, or a NC variant of WTX-330 for 48 hours. After 48 hours, the cell culture supernatants (now containing the processed INDUKINE molecule) were collected and used to stimulate primary human Tblasts. Because human Tblasts responded much more strongly to cleaved WTX-330 compared with the same amount of intact WTX-330 (Fig. 6C), the production of IFNγ by the Tblasts could be used as a surrogate marker for the amount of WTX-330 processing that was performed by the dissociated tumor samples. The amount of IFNγ produced was then normalized to the NC negative control (0% processed) and the precleaved positive control (100% processed). Among the n = 88 primary human tumor samples evaluated, WTX-330 was efficiently processed across all tested indications (Fig. 6E). In contrast, incubation of WTX-330 with primary human cells from various healthy tissues (n = 13) resulted in no evidence of processing. These data suggest that WTX-330 is efficiently and selectively processed by primary human tumor samples and support the continued clinical development of this molecule.
Discussion
IL-12 has long been a cytokine of great interest for oncology due to its potential to induce innate and adaptive immune responses (9, 12) and its promising antitumor preclinical data (13, 18, 20, 38, 39). Nevertheless, despite this interest, the poor pharmacokinetic properties of this cytokine and the unacceptable levels of toxicity associated with its systemic administration have prevented its use in clinical settings (9, 14, 26, 40). To address these concerns, we developed an IL-12 containing INDUKINE molecule, WTX-330. WTX-330 is a prodrug molecule, designed to be an infrequently administered, systemically delivered therapy with targeted intratumoral activation that releases native IL-12 into the TME. Our data with mWTX-330 (a chimeric IL-12 INDUKINE surrogate molecule) demonstrated antitumor activity in the MC38 tumor model that was dependent on in vivo cleavage of the INDUKINE molecule by the tumor. Furthermore, mWTX-330 was a very potent monotherapy in several mouse tumor models with varying levels of baseline infiltration, including complete responses in a model refractory to anti–PD-1 treatment (EMT-6). These complete responses translated into robust immune memory against subsequent rechallenge with the same tumor cell line, highlighting the role of the immune system in tumor rejection. In the MC38 model, depletion of various immune cell populations demonstrated that long-term efficacy was dependent on the presence of CD8+ T cells, but overall tumor growth inhibition was driven by contributions from three main effector cell types, CD8+ T cells, conventional CD4+ T cells, and NK cells. The INDUKINE design also resulted in increased exposure and a favorable ratio of active IL-12 versus the blocked prodrug molecule in the tumor tissue compared to plasma, which correlated with selectively localized pharmacodynamic changes (effector cell polyfunctionality) observed in the tumor versus peripheral tissues. Importantly, mWTX-330 also proved to be well-tolerated in mice compared with recombinant chimeric IL-12 treatment, while maintaining the potential to induce complete tumor regressions, resulting in an almost 10-fold improvement of the therapeutic window compared with the unblocked cytokine. Improvement of the therapeutic window is a key feature of INDUKINE molecules and is necessary to facilitate clinical development of potent cytokines for oncology treatment.
mWTX-330 treatment robustly activated various tumor-infiltrating innate and adaptive effector cell populations, supporting a mechanism of action where infiltration and activation of multiple effector cells plays a fundamental role in initial tumor control. The tumor-specific delivery of active IL-12 and subsequent induction of intratumoral IFNγ also induced Treg fragility, which likely contributes to the potent efficacy delivered by mWTX-330 treatment. However, of equal importance, is the effect that mWTX-330 treatment had on antigen processing and presentation and the observed increase in tumor infiltration by cross-presenting DCs. These cells are responsible for the de novo generation of new T-cell responses to novel tumor antigens, and several publications have identified their importance in the generation of preclinical antitumor immunity (41, 42). The dual role of IL-12 as a direct activator of effector-cell populations and as a potent activator of cross-presenting DCs will likely set apart therapeutics based on this cytokine compared with other treatments when it comes to triggering efficacy in “cold” tumors. Indeed, we observed this effect using a model of a “cold” tumor, the poorly infiltrated EMT-6 model. NanoString Digital Spatial Profiling demonstrated that systemic treatment with mWTX-330 enhanced deep infiltration of EMT-6 tumors by CD8+ T cells and confirmed the intratumoral increase in IL-12 and IFNγ signaling, as well as the significant upregulation of transcripts associated with robust CD8+ T-cell activation. mWTX-330 treatment also significantly increased the clonality of the TCR repertoire among tumor-infiltrating T cells by driving the expansion of underrepresented T-cell clones, suggesting that systemic mWTX-330 treatment resulted in the activation of a de novo T-cell response to unique tumor antigens, which may be key to the CD8+ T cell–dependent tumor rejection mentioned earlier.
Furthermore, systemic mWTX-330 treatment had a substantial effect on the metabolism of the tumor-infiltrating effector cells, transforming the metabolic status of not just the activated, tumor-infiltrating CD8+ T cells but also that of the intratumoral NK cells. The TME is known to have several distinctive characteristics when compared with a typical cellular environment, including a lower pH, hypoxic conditions, and substantial competition for extracellular glucose, all of which may impair effector-cell activity. Despite the evidence for increased glycolysis, we observed a slight reduction in extracellular glucose uptake following mWTX-330 treatment, suggesting that mWTX-330 treatment is not simply driving greater glucose uptake by tumor-infiltrating cells, but is instead facilitating an increase in metabolic efficiency and increasing oxidative phosphorylation. Recent studies have demonstrated that tumor-infiltrating T cells often fail to robustly activate mitochondrial respiration compared with those that have been activated in the spleen or lymph nodes, suggesting that the TME impairs the metabolic health of effector cells (29, 30, 32). In agreement with these studies, EMT-6 tumor-infiltrating CD8+ T cells and NK cells from vehicle-treated animals had very little evidence of ongoing mitochondrial respiration, despite efficiently taking up 2-NDBG in vitro. In contrast, effector cells from WTX-330–treated animals robustly upregulated active mitochondrial respiration, mitochondrial membrane potential, and mitochondrial ROS. The transition of these cells strongly towards oxidative phosphorylation could be important in the context of the highly dysregulated metabolic environment within the tumor, where there is stiff competition for glucose.
In conjunction with the metabolic effects of the TME, effector cells also have to contend with another mechanism of immune regulation, exhaustion. This state is characterized by high coexpression of checkpoint proteins (such as PD-1, LAG-3, TIGIT, and TIM-3; ref. 37), the loss of effector cytokine production, and the failure to proliferate following restimulation. However, when EMT-6 tumor-bearing mice were treated with mWTX-330, the tumor-infiltrating CD8+ T-cell population had lower expression of TOX, a transcription factor known to drive T-cell exhaustion (43, 44), and a much smaller frequency of CD8+ T cells coexpressed multiple checkpoint proteins. Together, these data suggest a model where local IL-12 activity resulting from systemic mWTX-330 treatment can attract and activate tumor infiltrating antigen presenting cells and local effector cells already present in the tumor, such as CD8+ T cells and NK cells, thereby both achieving early tumor control while driving the activation of previously under-represented T-cell clones over the longer term. The presence of local IL-12 within the TME then prevents the metabolic dysfunction usually imposed by the TME on tumor-infiltrating T cells and protects these cells from exhaustion, resulting in a potent and fully activated effector-cell population, without the toxicity typically associated with systemic administration of IL-12.
Additional studies with fully human WTX-330 showed that the INDUKINE molecule was highly inducible and was activated by a majority of human tumor samples in vitro while demonstrating stability when incubated with normal primary cells and serum. However, it remains to be seen whether cleavage of mWTX-330 by murine tumors in vivo and by human tumor samples in vitro will predict in vivo processing in the tumors of treated patients. Given the history of IL-12 toxicity in the clinic and the novel nature of WTX-330 as a therapeutic compound, clinical studies evaluating the safety and antitumor efficacy are required and have recently been initiated (NCT05678998). These studies will test the hypothesis that the design of WTX-330 results in preferential activation of the molecule in the TME to enhance antitumor immunity with minimal systemic toxicity.
Supplementary Material
Gene Ontology Analysis of RNA from MC38 Tumors Following mWTX-330 Treatment
Differential Gene Expression Analysis of Bulk RNA from MC38 Tumors After mWTX-330 Treatment
Differential Gene Expression Analysis of EMT-6 Tumor Infiltrating CD8+ T cells
Z Scores from EMT-6 Tumor Infiltrating CD8+ T cells from Select Pathways
EC50 Values and Fold Change in Activity of WTX-330 in Primary Human Tblasts
Supplemental Figures 1-10
Acknowledgments
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
Authors' Disclosures
C.J. Nirschl reports personal fees from Werewolf Therapeutics during the conduct of the study. H.R. Brodkin reports other support from Werewolf Therapeutics during the conduct of the study; also has a patent for PCT/US2021/033014 pending. C. Domonkos reports personal fees from Werewolf Therapeutics during the conduct of the study. C.J. Dwyer reports personal fees from Werewolf Therapeutics during the conduct of the study. D.J. Hicklin reports personal fees from Werewolf Therapeutics during the conduct of the study; personal fees from MPM Capital outside the submitted work; also has a patent for PCT/US2021/033014 pending. N. Ismail reports personal fees from Werewolf Therapeutics during the conduct of the study. C. Seidel-Dugan reports personal fees from Werewolf Therapeutics during the conduct of the study; also has a patent for PCT/US2021/033014 pending. P. Steiner reports personal fees from Werewolf Therapeutics during the conduct of the study; also has a patent for PCT number not known pending. Z. Steuert reports personal fees from Werewolf Therapeutics during the conduct of the study. J.M. Sullivan reports other support from Werewolf Therapeutics during the conduct of the study. W.M. Winston reports personal fees from Werewolf Therapeutics during the conduct of the study; also has a patent for PCT/US2021/033014 pending. A. Salmeron reports personal fees from Werewolf Therapeutics during the conduct of the study; also has a patent for PCT/US2021/033014 pending.
Authors' Contributions
C.J. Nirschl: Conceptualization, resources, formal analysis, supervision, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. H.R. Brodkin: Resources, formal analysis, investigation, visualization, methodology, writing–review and editing. C. Domonkos: Resources, formal analysis, investigation, visualization, methodology, writing–review and editing. C.J. Dwyer: Resources, formal analysis, methodology, writing–review and editing. D.J. Hicklin: Formal analysis, funding acquisition, writing–review and editing. N. Ismail: Resources, formal analysis, investigation, visualization, methodology. C. Seidel-Dugan: Formal analysis, funding acquisition, writing–review and editing. P. Steiner: Formal analysis, funding acquisition, writing–review and editing. Z. Steuert: Resources, formal analysis, investigation, visualization, methodology, writing–review and editing. J.M. Sullivan: Resources, formal analysis, investigation, writing–review and editing. W.M. Winston: Formal analysis, funding acquisition, writing–review and editing. A. Salmeron: Conceptualization, formal analysis, supervision, funding acquisition, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Propper DJ, Balkwill FR. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol 2022;19:237–53. [DOI] [PubMed] [Google Scholar]
- 2. Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Pérez-Gracia JL, et al. Cytokines in clinical cancer immunotherapy. Br J Cancer 2019;120:6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Conlon KC, Miljkovic MD, Waldmann TA. Cytokines in the treatment of cancer. J Interf Cytokine Res 2019;39:6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nirschl CJ, Brodkin HR, Hicklin DJ, Ismail N, Morris K, Seidel-Dugan C, et al. Discovery of a conditionally activated IL-2 that promotes antitumor immunity and induces tumor regression. Cancer Immunol Res 2022;10:581–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, et al. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin Cancer Res 2016;22:680–90. [DOI] [PubMed] [Google Scholar]
- 6. Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology 2017;6:e1277306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lopes JE, Fisher JL, Flick HL, Wang C, Sun L, Ernstoff MS, et al. ALKS 4230: a novel engineered IL-2 fusion protein with an improved cellular selectivity profile for cancer immunotherapy. J Immunother Cancer 2020;8:673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sharma M, Khong H, Fa'ak F, Bentebibel SE, Janssen LME, Chesson BC, et al. Bempegaldesleukin selectively depletes intratumoral tregs and potentiates T cell-mediated cancer therapy. Nat Commun 2020;11:661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Waldmann TA. Cytokines in cancer immunotherapy. Cold Spring Harb Perspect Biol 2018;10:a028472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tait Wojno ED, Hunter CA, Stumhofer JS. The immunobiology of the interleukin-12 family: room for discovery. Immunity 2019;50:851–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Glassman CR, Mathiharan YK, Jude KM, Su L, Panova O, Lupardus PJ, et al. Structural basis for IL-12 and IL-23 receptor sharing reveals a gateway for shaping actions on T versus NK cells. Cell 2021;184:983–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Del Vecchio M, Bajetta E, Canova S, Lotze MT, Wesa A, Parmiani G, et al. Interleukin-12: biological properties and clinical application. Clin Cancer Res 2007;13:4677–85. [DOI] [PubMed] [Google Scholar]
- 13. Lasek W, Zagożdżon R, JM. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother 2014;63:419–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nguyen KG, Vrabel MR, Mantooth SM, Hopkins JJ, Wagner ES, Gabaldon TA, et al. Localized interleukin-12 for cancer immunotherapy. Front Immunol 2020;11:575597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang P, Li X, Wang J, Gao D, Li Y, Li H, et al. Re-designing interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat Commun 2017;8:1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wittrup KD, Kaufman HL, Schmidt MM, Irvine DJ. Intratumorally anchored cytokine therapy. Expert Opin Drug Deliv; 2022;19:725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mirlekar B, Pylayeva-Gupta Y. IL-12 family cytokines in cancer and immunotherapy. Cancers (Basel) 2021;13:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hewitt SL, Bailey D, Zielinski J, Apte A, Musenge F, Karp R, et al. Intratumoral IL12 mRNA therapy promotes TH1 transformation of the tumor microenvironment. Clin Cancer Res 2020;26:6284–98. [DOI] [PubMed] [Google Scholar]
- 19. Strauss J, Heery CR, Kim JW, Jochems C, Donahue RN, Montgomery AS, et al. First-in-human phase I trial of a tumor-targeted cytokine (NHS-IL12) in subjects with metastatic solid tumors. Clin Cancer Res 2019;25:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T cells expressing IL-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Mol Ther Oncolytics 2017;8:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schoenhaut DS, Chua AO, Wolitzky AG, Quinn PM, Dwyer CM, McComas W, et al. Cloning and expression of murine IL-12. J Immunol 1992;148:3433–40. [PubMed] [Google Scholar]
- 22. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015;162:1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Siwicki M, Gort-Freitas NA, Messemaker M, Bill R, Gungabeesoon J, Engblom C, et al. Resident kupffer cells and neutrophils drive liver toxicity in cancer immunotherapy. Sci Immunol 2021;6:eabi7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reed SD, Li S. Pre-clinical toxicity assessment of tumor-targeted interleukin-12 low-intensity electrogenetherapy. Cancer Gene Ther 2011;18:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004;427:355–60. [DOI] [PubMed] [Google Scholar]
- 26. Cohen J. IL-12 deaths: explanation and a puzzle. Science 1995;270:908. [DOI] [PubMed] [Google Scholar]
- 27. Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol 2009;10:488–95. [DOI] [PubMed] [Google Scholar]
- 28. Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, et al. Interferon-γ drives T reg fragility to promote anti-tumor immunity. Cell 2017;169:1130–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science 2013;342:1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 2016;45:374–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol 2019;37:457–95. [DOI] [PubMed] [Google Scholar]
- 32. Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2017;2:e93411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013;38:225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rackov G, Tavakoli Zaniani P, Colomo del Pino S, Shokri R, Monserrat J, Alvarez-Mon M, et al. Mitochondrial reactive oxygen is critical for IL-12/IL-18-induced IFN-γ production by CD4+ T cells and is regulated by Fas/FasL signaling. Cell Death Dis 2022;13:531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mo F, Yu Z, Li P, Oh J, Spolski R, Zhao L, et al. An engineered IL-2 partial agonist promotes CD8+ T cell stemness. Nature 2021;597:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 2009;10:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res 2013;19:4917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hong Y, Robbins Y, Yang X, Mydlarz WK, Sowers A, Mitchell JB, et al. Cure of syngeneic carcinomas with targeted IL-12 through obligate reprogramming of lymphoid and myeloid immunity. JCI Insight 2022;7:e157448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xue D, Moon B, Liao J, Guo J, Zou Z, Han Y, et al. A tumor-specific pro-IL-12 activates preexisting cytotoxic T cells to control established tumors. Sci Immunol 2022;7:eabi6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of single-dose interleukin-12 exposure on interleukin-12 associated toxicity and interferon-γ production. Blood 1997;90:2541–8. [PubMed] [Google Scholar]
- 41. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008;322:1097–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014;26:638–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019;571:211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019;571:270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene Ontology Analysis of RNA from MC38 Tumors Following mWTX-330 Treatment
Differential Gene Expression Analysis of Bulk RNA from MC38 Tumors After mWTX-330 Treatment
Differential Gene Expression Analysis of EMT-6 Tumor Infiltrating CD8+ T cells
Z Scores from EMT-6 Tumor Infiltrating CD8+ T cells from Select Pathways
EC50 Values and Fold Change in Activity of WTX-330 in Primary Human Tblasts
Supplemental Figures 1-10
Data Availability Statement
All data needed to evaluate the conclusions in the manuscript are either present in the manuscript or the Supplementary Materials and Methods, available in publicly accessible databases, or are available upon reasonable request from the corresponding author with the appropriate legal protections in place. All Z scores used to generate pathway specific heat maps or analyze GSEO analysis are included in the supplemental files, as are Log2 fold change and Log10P values for both bulk RNA NanoString analysis as well as those from the Nanostring DSP system. Raw data for TCR sequencing are available in the SRA database, accession no. PRJNA952625.