

Summary
Synthetic opioids are increasingly challenging to combat the opioid epidemic and act primarily at opioid receptors, chiefly the G protein-coupled receptor (GPCR) μ-opioid receptor (MOR), which signals through G protein-dependent and β-arrestin pathways. Using a bioluminescence resonance energy transfer (BRET) system, we investigate GPCR-signaling profiles by synthetic nitazenes, which are known to cause overdose and death due to respiratory depression. We show that isotonitazene and its metabolite, N-desethyl isotonitazene, are very potent MOR-selective superagonists, surpassing both DAMGO G protein and β-arrestin recruitment activity, which are properties distinct from other conventional opioids. Both isotonitazene and N-desethyl isotonitazene show high potency in mouse analgesia tail-flick assays, but N-desethyl isotonitazene shows longer-lasting respiratory depression compared to fentanyl. Overall, our results suggest that potent MOR-selective superagonists may be a pharmacological property predictive of prolonged respiratory depression resulting in fatal consequences and should be examined for future opioid analgesics.
Subject areas: Cellular neuroscience, Molecular neuroscience, Neuroscience
Graphical abstract
Highlights
-
•
Isotonitazene and N-desethyl isotonitazene display superagonism at MOR
-
•
Nitazenes are highly selective for MOR over KOR and DOR
-
•
Nitazene superagonists induce potent analgesia on par with fentanyl
-
•
Nitazene superagonists induce prolonged respiratory depression compared to fentanyl
Cellular neuroscience; Molecular neuroscience; Neuroscience
Introduction
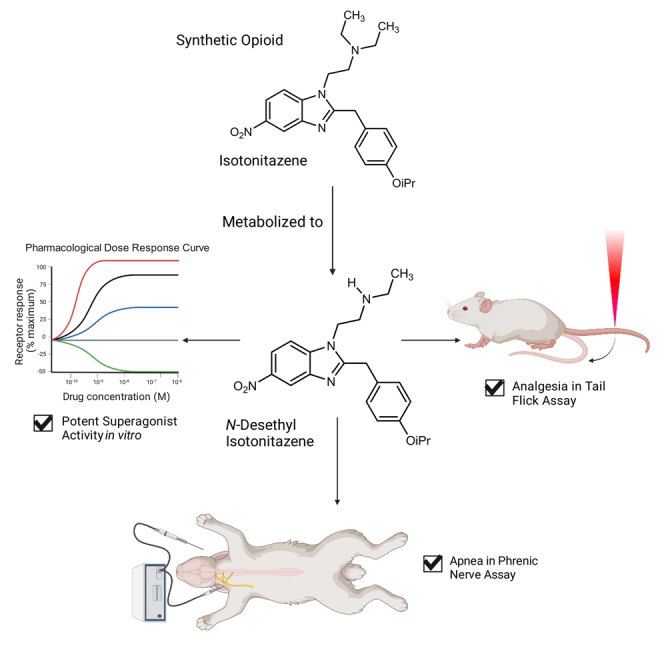
The ongoing opioid epidemic has presented new challenges in the treatment of pain, and the over-prescription of conventional opioids has been a major contributor to the problem.1 Complicating the situation is the emergence of novel synthetic or “designer” opioids. The novelty of these drugs has been used to circumvent existing drug laws and DEA scheduling.2 Synthetic opioids are responsible for an increasing number of deaths recently, and are implicated as causes of overdose occurring during the COVID-19 pandemic.3,4,5 As recreational users of synthetic opioids become aware of new compounds and because of the lack of scheduling for many of these compounds, the number of overdoses and deaths attributed to these drugs will likely increase in the future. One group of synthetic opioids that has gained attention due to its recreational use and association with several overdose deaths is the nitazene family of compounds, which includes isotonitazene.6
Nitazenes are benzimidazole derivatives originally synthesized in the 1960s to better understand the structure-activity relationships of analgesics.7 The prototypical member of this class, etonitazene, was initially developed as an analgesic medication but was subsequently used as a research compound for rodent studies of addiction and tolerance.8,9,10 In recent years, nitazenes have been used recreationally, and many members of this family have been scheduled by the DEA. Members of this family, including isotonitazene, have been found in analyses of seized illicit opioid samples and have been implicated in many overdose deaths.5,11,12 Isotonitazene has been found to undergo N- and O- dealkylation to create N-desalkyl- and O-desalkylisotonitazene species such as N-desethyl isotonitazene, and is also known to undergo reduction of the nitro group to produce 5-aminoisotonitazene; etonitazene and metonitazene have been found to produce similar metabolites.11 The most notable aspect of these compounds is that their efficacy for MOR signaling is often much greater than that of other opioids, such as morphine, hydromorphone, or even fentanyl.13 Isotonitazene has also previously been shown to selectively bind MOR over the κ- and δ-opioid receptors (KOR and DOR respectively), and potently induce analgesia in mice.14
Biased signaling is the preferential activation by ligands at a single GPCR to stabilize one signaling pathway over another. Biased agonists are a promising strategy for drug discovery to eliminate side effects mediated by the same receptor that is driving the therapeutic response.15,16 An excellent target for biased agonist drug discovery is the opioid receptors, where G protein-biased MOR-selective agonists have been developed to eliminate opioid-induced side effects, including the recently FDA-approved drug oliceridine (TRV-130).17,18 There is still controversy, however, whether activation of the β-arrestin pathway is solely responsible for many of the adverse effects of opioids, namely constipation and respiratory depression, and recent studies have challenged this idea.19,20,21,22 Previous studies have suggested that ligands that are putatively G protein biased, such as oliceridine, may have improved safety profiles and a higher therapeutic index than conventional opioids due to their behavior as partial agonists, rather than their biased signaling properties.23,24 Nevertheless, biased MOR agonists offer potential avenues for development of novel therapeutics for chronic pain and for understanding the relationship between biased agonism and in vivo effects; the development of these ligands serves as a strategy for rational opioid drug design, which seeks improved therapeutic indices and a decreased risk for respiratory depression.16,25
There is a current lack of knowledge connecting downstream MOR-signaling effects to respiratory depression, and MOR-signaling properties for the nitazene opioid class as a whole are underexplored, hampering a full understanding of the mechanisms behind the reported fatalities. In this project, we investigated the pharmacological relationships between members of the nitazene family of opioid compounds, and we compared their analgesic versus respiratory depressant properties for the purpose of expanding knowledge into determinants of opioid-induced respiratory depression.
Results
Assessment of MOR G protein and β-arrestin activity for nitazene compounds
To assess MOR-activation profiles for isotonitazene in comparison to fentanyl and DAMGO (Figure 1A), we optimized G protein dissociation and β-arrestin2 recruitment bioluminescence resonance energy transfer (BRET) platform assays, which are essential for the measurement and quantification of ligand bias (Figure 1B).26 The BRET platform provides MOR G protein dissociation and β-arrestin recruitment activity at the same time point and in the same cellular background (HEKT). BRET measurements are performed in a system that directly measures protein-protein effector activity, and unlike in second messenger assay measurement assays, BRET assays are not susceptible to receptor reserve and amplification issues.26
Figure 1.

Assessment of MOR G protein and β-arrestin activity for nitazene compounds
(A) Chemical structures of DAMGO, fentanyl, and isotonitazene.
(B) Overview of BRET assay platform measuring G protein dissociation and β-arrestin recruitment at MOR.
(C) Baseline corrected net BRET curves for DAMGO measuring G protein dissociation among the Gi/o/z subtypes (n ≥ 3 for each condition).
(D) Baseline corrected net BRET curves for the G protein (GoA) dissociation assay are shown for isotonitazene (red), DAMGO (black), and fentanyl (blue), with n ≥ 3 for each compound.
(E) Baseline corrected net BRET curves for the β-arrestin2 recruitment assay in the presence of GRK2 are shown for isotonitazene (red), DAMGO (black), and fentanyl (blue), with n ≥ 3 for each compound. Isotonitazene (F) and fentanyl (G) measuring G protein dissociation among the Gi/o/z subtypes (n ≥ 3 for each condition) and data were normalized to percent DAMGO response. Data represent mean ± S.E.M performed in duplicate.
Relates to Figures S1 and S2.
To measure G protein dissociation, we tested DAMGO in a BRET based Gα/Gγ-dissociation assay.27 Our results indicate that DAMGO induced the most robust G protein dissociation for Gi/o/z G protein subtypes (Figure 1C), as determined by change in net BRET ratio, but not for other G protein subtypes tested (Gq, G11, Gs short and long, G12, and G13 (Figure S1A). First, we interrogated G protein dissociation measuring the GoA subtype, which has been implicated in playing a role in opioid antinociception.28 Isotonitazene displays a greater maximal net BRET (0.20) compared to DAMGO (0.15), indicating that isotonitazene achieves superagonist G protein efficacy in this system (Figure 1D). Similarly, isotonitazene in MOR β-arrestin2 recruitment assays produces a net BRET signal of 0.26, whereas DAMGO only produces a maximum net BRET of 0.22, indicating isotonitazene exhibits superagonism in β-arrestin2 recruitment (Figure 1E). Maximal β-arrestin recruitment at MOR, however, requires co-expression of G protein receptor kinase 2 (GRK2).16,29 Therefore, we optimized these assays with co-expression of GRK2 in the β-arrestin2 recruitment BRET assay, which showed a significant increase in DAMGO β-arrestin2 recruitment potency (10-fold), which was blocked by the GRK2/3 inhibitor, compound 101 (Figure S1B).30 Fentanyl also showed an increase in potency (4-fold) in the β-arrestin2 recruitment BRET assay when GRK2 was co-expressed, which was blocked by compound 101 (Figure S1C). Interestingly, β-arrestin2 recruitment by isotonitazene was not GRK2 sensitive, nor was it blocked by compound 101 (Figure S1D), indicating GRK2-mediated phosphorylation may not be required for isotonitazene-induced β-arrestin2 recruitment superagonist activity. GRK2 co-expression did not show robust changes to DAMGO or isotonitazene G protein-dissociation activity (Figure S1E) indicating an exclusive effect on β-arrestin2 recruitment. Finally, we performed kinetic analysis of both GoA dissociation and β-arrestin2 recruitment BRET assays at 5, 60, and 300 min time points and found that activities at all these time points were essentially unchanged (Figures S1F–S1I). Finally, to compare isotonitazene to fentanyl and DAMGO pharmacological activities, we conducted G protein dissociation for all Gi/o/z G protein subtypes where isotonitazene demonstrates superagonism at every Gi/o/z subtype tested (Figures 1F and 1G). In summary, isotonitazene exhibits greater potency and efficacy compared to fentanyl or DAMGO in all G protein and β-arrestin2 BRET assays interrogated.
Members of the nitazene family are potent superagonists at MOR
To compare isotonitazene and other nitazenes (metonitazene, N-desethyl isotonitazene, Figure 2A) to conventional opioids such as morphine, fentanyl and DAMGO, we tested these compounds in parallel in the optimized BRET G protein (GoA) and β-arrestin2 recruitment assays (Figures 2B and 2C, Table 1). We chose to assess the metabolite of isotonitazene, N-desethyl isotonitazene, which exhibited sub-nanomolar potency and superagonism in both β-arrestin recruitment and G protein dissociation assays, similar to isotonitazene. Notably, all three nitazene compounds had greater potency than conventional opioids and DAMGO, with isotonitazene demonstrating greater than 100-fold potency compared to fentanyl (Table 1). Between the nitazene compounds, the rank order of potency in the G protein dissociation BRET assay was isotonitazene (EC50 = 107 pM) > N-desethyl isotonitazene (EC50 = 252 pM) > metonitazene (EC50 = 1.74 nM), and all three nitazenes had superagonist activities with efficacy greater than DAMGO (Emax > 100%). The evidence for greater MOR activity by nitazene compounds is further demonstrated by the fact that the DAMGO-normalized transduction coefficients (Δlog(Emax/EC50) values31 for all three nitazenes at both pathways are ≥0 (Table 1). The G protein (GoA) dissociation calculated bias factors for the nitazenes were 3.85, 1.41, and 4.66 for isotonitazene, metonitazene, and N-desethyl isotonitazene, respectively, which may indicate a slight bias for G protein signaling over β-arrestin recruitment (Table 1).
Figure 2.

Members of the nitazene family are potent superagonists at MOR
(A) Chemical structures of N-desethyl isotonitazene, metonitazene, and morphine.
(B and C) G protein (GoA) dissociation (B) and β-arrestin2 recruitment (C) dose-response curves are shown, with efficacies reported as % DAMGO response. DAMGO (black), fentanyl (blue), morphine (purple), isotonitazene (red), N-desethyl isotonitazene (brown), and metonitazene (orange) were tested in both assays following incubation with the drug for 1 h; n ≥ 3 for all compounds.
(D) Dose-response curves from the GloSensor Gi/o-mediated cAMP inhibition assay are shown for isotonitazene (red), N-desethyl isotonitazene (brown), metonitazene (orange), morphine (purple), DAMGO (black), and fentanyl (blue), with data normalized to DAMGO and displayed as a percentage of DAMGO mediated cAMP inhibition; n ≥ 3 for all compounds.
(E) Radioligand binding derived affinity (pKi) values are shown for isotonitazene (red), N-desethyl isotonitazene (brown), metonitazene (orange), morphine (purple), DAMGO (black), and fentanyl (blue); n ≥ 3 for all compounds; displayed error bars represent mean and SEM.
(F) Hierarchical clustering heatmap and dendrograms for all tested compounds at the Gi/o/z subtypes G protein-dissociation BRET and GRK2-mediated β-arrestin recruitment BRET are shown. The data displayed are transduction coefficients log(Emax/EC50) calculated using control-normalized Emax and EC50. The color scale is set such that red indicates higher values, blue indicates lower values, and white indicates intermediate values relative to the minima and maxima of the data displayed. The top dendrogram shows the clustering of tested compounds across the assays performed, and the left dendrogram shows the clustering of values observed between each assay.
(G) Circular bar charts depicting the % DAMGO Emax values calculated from the Gi/o/z subtypes and GRK2 mediated β-arrestin recruitment BRET assays are shown for each compound. The black circle in the center of each graph indicates 100% Emax relative to DAMGO and is set as the baseline for the bars; bars projecting away from the center indicate values greater than 100%, while those projecting toward the center indicate values less than 100%. The color scale is set such that the red bar represents the global maxima of Emax values observed across all drugs at all shown assays, and blue represents the respective global minima.
Relates to Figure S2.
Table 1.
MOR affinities, G protein dissociation BRET, and β-arrestin2 recruitment potency and efficacy
| Compound | [3H]-DAMGO Binding |
G protein (GoA) dissociation |
β-Arrestin2 recruitment |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ki nM (pKi ±SEM) | EC50 (nM) | pEC50 ± SEM | Emax (% DAMGO) | Log (Emax/EC50) | ΔLog (Emax/EC50) | ΔΔLog (Emax/EC50) | Bias Factor (for G protein) | EC50 (nM) | pEC50 ± SEM | Emax (% DAMGO) | Log (Emax/EC50) | ΔLog (Emax/EC50) | ΔΔLog (Emax/EC50) | Bias Factor (for β-arr) | |
| DAMGO | 4.37∗ (8.36 ± 0.07) | 12.03 | 7.92 ± 0.06 | 100.0 | 7.92 | 0 | 0 | 1 | 18.7 | 7.73 ± 0.03 | 100.0 | 7.73 | 0 | 0 | 1 |
| Isotonitazene | 0.490 (9.31 ± 0.14) | 0.107 | 9.97 ± 0.09 | 118.8 ± 2.3 ∗∗ | 10.05 | 2.13 | 0.59 | 3.85 | 0.705 | 9.15 ± 0.06 | 132.4 ± 2.5 ∗∗ | 9.27 | 1.54 | −0.60 | 0.26 |
| Fentanyl | 6.03 (8.22 ± 0.13) | 9.21 | 8.03 ± 0.09 | 112.2 ± 2.7 ∗∗ | 8.10 | 0.18 | 0.53 | 3.38 | 38.7 | 7.41 ± 0.04 | 92.0 ± 1.5 ∗∗ | 7.38 | −0.35 | −0.53 | 0.29 |
| N-desethyl isotonitazene | 0.741 (9.13 ± 0.22) | 0.252 | 9.60 ± 0.12 | 114.5 ± 3.6 ∗∗ | 9.66 | 1.74 | 0.67 | 4.66 | 2.01 | 8.70 ± 0.05 | 127.0 ± 1.9 ∗∗ | 8.80 | 1.07 | −0.67 | 0.21 |
| Metonitazene | 0.776 (9.11 ± 0.11) | 1.74 | 8.76 ± 0.10 | 107.4 ± 3.1 ∗∗ | 8.78 | 0.86 | 0.15 | 1.41 | 3.83 | 8.42 ± 0.05 | 104.4 ± 1.5 | 8.44 | 0.71 | −0.15 | 0.71 |
| Morphine | 2.63 (8.58 ± 0.17) | 50.2 | 7.30 ± 0.09 | 98.94. ± 3.9 | 7.23 | −0.68 | 0.22 | 1.65 | 74.7 | 7.13 ± 0.09 | 50.8 ± 2.0 ∗∗ | 6.83 | −0.90 | −0.22 | 0.60 |
MOR affinities for compounds in this study were determined using [3H]-DAMGO competition binding. G protein (GoA) dissociation and β-arrestin2 recruitment activities were determined by BRET in HEK293T cells. All data represent average and standard error of the mean (S.E.M) from at least three independent experiments performed in duplicate, and are represented as mean ± SEM. ∗The [3H]-DAMGO binding value shown for DAMGO is the pKd ±SEM. Emax values marked with ∗∗ are statistically significantly different from that of DAMGO as determined by the extra sum-of-squares F test, with p < 0.0001.
Next, we determined if the relationship between G protein dissociation potency and efficacy measured using BRET would be replicated in an orthogonal measure of MOR G protein activity. For example, mini-G recruitment assays have been used to examine nitazene MOR activity previously.13 Therefore, we examined the ability of nitazenes to stabilize mini-Gi recruitment to the MOR, and our results indicate that a similar rank order of potency was observed compared to BRET G protein and β-arrestin2 recruitment assays (EC50 = 110 nM for DAMGO, 0.678 nM for isotonitazene, and 61.9 nM for fentanyl) (Figures S2A–S2D). However, isotonitazene mini-Gi recruitment potencies were more similar to β-arrestin2 recruitment and not for G protein dissociation potency, or were distinct from both measures entirely as seen with DAMGO (Figures S2A–S2D). Likely, mini-G recruitment does not correlate with the functional outcome of G protein signaling due to GPCR activation requiring both association and dissociation of heterotrimeric G proteins.32 To address these assay discrepancies, we further confirmed G protein MOR activity in another orthogonal G protein activity assay using GloSensor to measure Gi/o-mediated secondary messenger cAMP inhibition. Again, we show that the rank order of potency from the BRET assays and cAMP inhibition assay are comparable: the nitazenes are more potent and efficacious agonists compared to fentanyl, morphine, or DAMGO (Figure 2D).16 Potencies and efficacies measured in the GloSensor assay (Table 2), however, were all increased relative to the G protein dissociation BRET assay, likely due to receptor reserve and amplification in a second messenger assay system.
Table 2.
MOR, KOR, and DOR Gi/o-mediated cAMP inhibition data
| Compound | MOR |
KOR |
DOR |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (nM) | pEC50 ± SEM | Emax (% DAMGO) | Log (Emax/EC50) | ΔLog (Emax/EC50) | MOR vs. KOR ΔΔLog (Emax/EC50) | MOR vs. KOR Selectivity | MOR vs. DOR ΔΔLog (Emax/EC50) | MOR vs. DOR Selectivity | EC50 (nM) | pEC50 ± SEM | Emax (% Sal A) | Log (Emax/EC50) | ΔLog (Emax/EC50) | EC50 (nM) | pEC50 ± SEM | Emax (% DADLE) | Log (Emax/EC50) | ΔLog (Emax/EC50) | |
| Isotonitazene | 0.033 | 10.48 ± 0.04 | 105.0 ± 1.1 ∗ | 10.49 | 1.85 | 4.96 | >10,000 | 4.97 | >10,000 | 335 | 6.48 ± 0.09 | 85.9 ± 4.1 | 6.41 | −3.11 | 245 | 6.61 ± 0.07 | 89.9 ± 3.4 | 6.56 | −3.13 |
| Metonitazene | 0.34 | 9.46 ± 0.06 | 104.9 ± 1.9 | 9.46 | 0.82 | 2.89 | 775 | 4.14 | >10,000 | 25.9 | 7.59 ± 0.11 | 73.1 ± 3.3 | 7.45 | −2.07 | 221 | 6.65 ± 0.16 | 51.9 ± 4.2 | 6.37 | −3.32 |
| N-desethyl isotonitazene | 0.14 | 9.85 ± 0.06 | 115.7 ± 1.2 ∗ | 9.88 | 1.24 | 5.04 | >10,000 | 4.22 | >10,000 | 1060 | 5.98 ± 0.22 | 55.8 ± 8.3 | 5.72 | −3.80 | 153 | 6.81 ± 0.18 | 77.1 ± 6.7 | 6.7 | −2.99 |
| Fentanyl | 3.28 | 8.48 ± 0.06 | 104.1 ± 1.9 | 8.49 | −0.16 | 1.74 | 55 | 3.27 | 1,850 | 19.7 | 7.70 ± 0.05 | 84.4 ± 3.6 | 7.63 | −1.89 | 466 | 6.33 ± 0.08 | 86.2 ± 3.9 | 6.27 | −3.42 |
| Morphine | 7.43 | 8.13 ± 0.06 | 86.1 ± 2.1 ∗ | 8.08 | −0.56 | 2.33 | 215 | 2.48 | 302 | 207 | 6.68 ± 0.08 | 88.3 ± 1.7 | 6.63 | −2.89 | 186 | 6.73 ± 0.06 | 82.9 ± 2.7 | 6.65 | −3.04 |
All receptors are human receptors, and expressed in HEKT cells. All data represent mean and standard error of the mean (S.E.M) from at least three independent experiments performed in triplicate, and are represented as mean ± SEM. The pEC50 values for DAMGO, salvinorin A, and DADLE at their cognate receptors are 8.65 ± 0.04, 9.52 ± 0.04, 9.69 ± 0.06, respectively. Emax values at MOR marked with ∗ are statistically significantly different from that of DAMGO as determined by the extra sum-of-squares F test, with p < 0.0001.
To assess whether the potencies observed in the BRET assays are due to high affinity at MOR, we next performed radioligand binding assays in competition with [3H]-DAMGO (Figure 2E and Table 1). The binding data show that the nitazenes bind with greater affinity to MOR compared to conventional opioids; isotonitazene has the strongest binding affinity with approximately 10-fold greater MOR affinity compared to fentanyl (isotonitazene pKi = 9.31; fentanyl pKi = 8.21). Notably, the rank order of measured binding affinities mirrored the rank order of potencies seen in the BRET assay, supporting the idea that nitazene compounds exhibit sub-nanomolar potency dependent on their extremely high affinity at MOR.
To determine if the degrees of efficacy observed with nitazene superagonists were specific to the GoA subtype, we used BRET to measure the DAMGO-normalized G protein-dissociation activity for N-desethyl isotonitazene and metonitazene and compared to morphine at each member of the Gi/o/z subtypes (Figures S2E–S2H). Similar to isotonitazene, the N-desethyl isotonitazene metabolite demonstrated superagonism at every Gi/o/z subtype tested (Figure S2F).
To compare pharmacological parameters across all assay platforms (Table S1), we used hierarchical cluster analysis and generated a heatmap of each studied compound computed from the relative activity (log(Emax/EC50)) values from G protein dissociation and β-arrestin recruitment BRET assays (Figure 2F). The relative activities (log(Emax/EC50)) values were used as they allow for a comparison of pharmacological parameters across systems.33 As shown, isotonitazene and N-desethyl isotonitazene are closely related to each other amongst each pathway assessed, whereas DAMGO, fentanyl, metonitazene, and morphine are more similar to each other than either of the nitazene superagonists. Overall, isotonitazene and N-desethyl isotonitazene exhibit superagonist activity at every tested pathway, whereas all other tested compounds showed partial agonism for at least two of the tested signaling pathways (Figure 2G). This analysis reveals that isotonitazene and N-desethyl isotonitazene are unique among all tested compounds showing MOR signaling properties distinct from other conventional opioids.
Nitazene compounds are highly selective MOR agonists
Off-target activity at other opioid receptors is common for conventional opioids, and can complicate determinations of measurements in vivo.34 To determine the opioid receptor selectivity of the nitazenes, we used GloSensor and BRET assays to measure Gi/o-mediated cAMP inhibition at all 3 opioid GPCRs: MOR, κ opioid receptor (KOR), and δ opioid (DOR) receptors. Isotonitazene appears to be ≥ 15,000 times more potent at MOR than KOR or DOR in either assay (Figure 3A) demonstrating high-MOR selectivity. Similarly, N-desethyl isotonitazene is also highly selective for MOR, being more than 3,000x more potent at MOR over KOR or DOR (Figure 3B). Metonitazene, on the other hand, showed less MOR selectivity over KOR (775-fold) and DOR (>10,000) (Figures S3A and S3B), but still significantly selective for MOR compared to morphine (Figures S3C and S3D) and fentanyl (Figure 3C). Thus, the nitazene scaffold appears to be extremely selective for MOR compared to conventional opioids, fentanyl and morphine (Figure 3D).
Figure 3.

Nitazene compounds are highly selective MOR agonists
(A–C) Gi/o-mediated cAMP inhibition GloSensor (left) and G protein (GoA) dissociation BRET assays (right) showing isotonitazene (A), N-desethyl isotonitazene (B), and fentanyl (C) tested at the MOR (red), KOR (black), and DOR (blue). For each assay, data were normalized to a control compound for each receptor. DAMGO (MOR), DADLE (DOR), and salvinorin-A (KOR); Data represent mean ± SEM and n ≥ 3 for all conditions in each assay.
(D) Transduction coefficients log(Emax/EC50) derived from the cAMP inhibition assays are shown for isotonitazene, metonitazene, N-desethyl isotonitazene, fentanyl, and morphine tested at MOR (red), KOR (black), and DOR (blue). The coefficients were calculated using the control-normalized Emax and EC50 values from the curves in (A–C) and relates to Figure S3.
Nitazenes are potent analgesics but produce potent respiratory depression that is longer-lasting than fentanyl
To test the analgesic activity of isotonitazene, its metabolite N-desethyl isotonitazene, and metonitazene, we performed tail-flick assays with C57BL/6J mice. Isotonitazene (W(3,11.27) = 15.88, p = 0.0002), N-desethyl isotonitazene (W(4,16.59) = 116.5, p < 0.0001), and metonitazene (W(4,13.28) = 10.63, p = 0.0004) produced significant analgesic effects in the tail-flick experiments (Figure 4A). Based on their molar mass, isotonitazene (ED50 = 11.3 μg/kg) showed almost 4-fold higher analgesic potency than fentanyl (ED50 = 55.7 μg/kg), whereas N-desethyl isotonitazene (ED50 = 40.1 μg/kg) was equipotent with fentanyl, and metonitazene (ED50 = 321 μg/kg) had 8-fold lower potency (Table 3). While there was not a significant difference between the potency of fentanyl and N-desethyl isotonitazene (F(1,37) = 0.35, p = 0.5602), fentanyl has higher potency than metonitazene (F(1,33) = 10.66, p = 0.0026) and lower potency than isotonitazene (F(1,26) = 8.25, p = 0.008). Although isotonitazene and metonitazene did not achieve 100% MPE in the tail flick experiments (Figure 4A), higher doses of those compounds induced respiratory depression, which limited the dose range that could be tested. To confirm rodent species effects, we performed G protein dissociation BRET assays with the mouse MOR to determine if the rank order of potency was similar compared to human MOR activity. Notably, all the tested compounds had slight increases in potency at the mouse MOR (Figure S4A) compared to the human MOR (Figure S4B); however, the rank order of potency was similar between species.
Figure 4.

Nitazenes are potent analgesics but produce potent respiratory depression that is longer-lasting than fentanyl
(A) Tail-flick analgesia assay dose-response relationships, measured as the mean percentage of the maximum possible effect per dose, are shown for isotonitazene (black), N-desethyl isotonitazene (green), fentanyl (blue), and metonitazene (brown). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, significant difference vs. vehicle control. Dunnett’s T3 test was used for all compounds except fentanyl, which used Dunn’s test.
(B) The dose response relationship between drug concentration and mean respiratory rate as measured by the phrenic nerve activity assay are shown for N-desethyl isotonitazene (red, n = 6) and fentanyl (blue, n = 4). Error bars represent ±SEM for each compound.
(C) Respiratory depression measured as respiratory rate (bpm, breaths per minute) in percent of baseline respiratory rate after a single dose (1 μg/kg) of either fentanyl (blue, n = 3) or N-desethyl isotonitazene (red, n = 6). Error bars are ±SEM.
(D) The time to recovery of baseline respiratory rate after an apnea inducing dosage of either fentanyl (blue, 9.0 ± 0.5 μg/kg, n = 4) or N-desethyl isotonitazene (red, 3.5 ± 0.3 μg/kg, n = 6) is shown. Error bars are ±SEM.
(E) Example of the respiratory rate response to a 3 μg/kg bolus of N-desethyl isotonitazene. Pictured is the respiratory rate at each respiratory cycle. The “apneic dose” had been determined for the animal with titration earlier in the experiment (see B), and complete recovery of respiratory rate was accomplished (see D). After bolus injection, respiratory rate decreased over ∼3 min until complete apnea. After confirming apnea for 5 min, IV naloxone (20 μg/kg) was administered. Thirty seconds after IV injection, phrenic nerve activity restarted, and respiratory rate reached steady-state recovery after ∼3 min. Naloxone frequently increases respiratory rate above pre-opioid baseline, which likely indicates reversal of endogenous opioid activity in our preparation as previously described.35
(F) Pooled data for the recovery of respiratory rate from N-desethyl isotonitazene-induced apnea after IV naloxone injection (20 μg/kg, n = 6). Error bars are ±SEM.
Relates to Figure S4.
Table 3.
Potencies of test compounds in the tail-flick assay
| Compound | ED50 (95% CI) μg/kg | ED50 (95% CI) nmol/kg |
|---|---|---|
| Isotonitazene | 11.3 (5.7–22.3) | 27.5 (13.8–54.3) |
| N-desethyl isotonitazenea | 40.1 (28.1–64.7) | 95.7 (67.1–154) |
| Fentanylb | 55.7 (44.4–65.3) | 105 (84–124) |
| Metonitazene | 321 (179–575) | 840 (469–1504) |
Median effective doses (ED50 values) and 95% CI were calculated using nonlinear regression.
Hydrochloride salt.
Citrate salt.
To compare the respiratory depressant effects of N-desethyl isotonitazene and fentanyl, we used phrenic nerve activity using a well-established decerebrate rabbit model where pO2 and pCO2 were maintained constant throughout drug administration.32 This setup allows monitoring of more consistent drug effects compared to whole-body plethysmography where baseline ventilation as well as the magnitude of respiratory depression are more variable due to the respiratory effects of behavior and activity level. Phrenic nerve recordings were performed without background anesthetic and under physiologically relevant conditions.32 Since isotonitazene is currently a DEA-scheduled compound, we instead chose to test its metabolite, N-desethyl isotonitazene in these respiratory-depression assays, especially toward the hypothesis that nitazene active metabolites may contribute to respiratory-depressant effects. We administered each drug intravenously (IV) at 1 μg/kg to determine onset kinetics and, second, we titrated repeated small boluses (1–4 μg/kg) until apnea was achieved. When the doses were titrated to produce apnea, N-desethyl isotonitazene required less than half (3.5 ± 0.3 μg/kg, n = 6) of the dose required to cause complete apnea by fentanyl (9.0 ± 0.5 μg/kg, n = 4, p < 0.001) (Figure 4B) indicating 2-fold increased potency by IV administration. A single equal dose (1 μg/kg) of N-desethyl isotonitazene induced greater respiratory depression (59 ± 2% of baseline respiratory rate, n = 6) compared to fentanyl (75 ± 3%, n = 3, p < 0.001) (Figure 4C). The time to maximal effect for the 1 μg/kg dose was approximately 4 times longer for N-desethyl isotonitazene (10.5 ± 1 min) than for fentanyl (2.5 ± 0.5 min, p < 0.001) (Figure 4C), and the time of recovery from apnea to baseline respiratory rate was approximately 3 times longer for N-desethyl isotonitazene (208 ± 38 min) compared to fentanyl (67 ± 9 min, p = 0.018) (Figure 4D). Importantly, injection of the non-selective opioid receptor antagonist naloxone completely reversed apnea for the 3 μg/kg dose N-desethyl isotonitazene within 5.5 ± 0.6 min, n = 6 (Figures 4E and 4F). This reversal effect is similar to the time to complete reversal of fentanyl-induced respiratory depression reported previously.36 Taken together, these data show that a primary metabolite of isotonitazene, N-desethyl isotonitazene, is a more potent respiratory depressant capable of decreasing respiration to a greater extent and for a longer duration compared to the conventional opioid fentanyl.
Discussion
Here we present evidence for the superagonism, MOR selectivity, and analgesic activity for three nitazene compounds along with the striking potential for potent and long-lasting respiratory depression. To our knowledge, this study is the first to characterize the degree of opioid receptor efficacy and selectivity, and in vivo characterization of the analgesic and respiratory properties possessed by isotonitazene and metabolite, N-desethyl isotonitazene. We verified that the BRET assay platforms can achieve sufficient signal to accurately differentiate between a full agonist and a superagonist, which allowed for the determination that all nitazenes tested display some degree of MOR superagonism, and that this level of G protein or β-arrestin efficacy surpasses both fentanyl and morphine. We also showed that the nitazene superagonist compounds were unique compared to the other opioids in showing a minimum of full agonism at all tested MOR secondary messenger pathways (Figure 2G). Furthermore, radioligand-binding experiments confirmed that nitazenes possess high affinity for the MOR. Experiments examining effects at KOR and DOR demonstrated a very high degree of MOR selectivity in terms of functional signaling outcomes for the nitazene class of compounds, especially when compared to morphine and fentanyl. Clustered data of all BRET-observed pharmacological parameters at MOR show that nitazenes tend to be distinct from conventional opioids, with very potent and efficacious signaling profiles more similar to each other than to morphine or fentanyl. Notably, isotonitazene and N-desethyl isotonitazene have equal or greater analgesic potency than fentanyl, but N-desethyl isotonitazene is a more potent and longer-lasting respiratory depressant than fentanyl.
Superagonism refers to the ability of a drug to achieve a greater signaling efficacy than that of endogenous ligands.37 Although some nitazenes have been identified as having robust efficacy at MOR, these studies did not compare efficacy relative toward opioid peptides, here represented by the synthetic analog DAMGO, nor across all members of the Gi/o/z subtypes, as we did in this study.13,38 Furthermore, we demonstrated nitazene pharmacological activity in several assay systems: MOR G protein-dissociation BRET assays, MOR β-arrestin2 recruitment BRET assays, MOR second messenger assays (e.g., cAMP inhibition) and affinity measurements, which all demonstrate that nitazenes are a potent and superagonist class of MOR-selective drugs.
An ongoing area of debate in the opioid field is the explanation of the improved safety profile of G protein-biased agonists. Specifically, it is unclear whether the improvement is caused by the bias itself, or because most of these compounds are only partial agonists.24,39 Questions still linger however, regarding whether the G protein pathway or the β-arrestin pathway mediates respiratory depression and long-term tolerance.19,20,21,22,39,40,41 The transduction coefficients of isotonitazene and N-desethyl isotonitazene show a slight preference for the G protein pathway over β-arrestin recruitment (Table 1). Given this finding, there is some potential of using the nitazene scaffold in the future as inspiration for the design of novel biased agonists. Considering that each of the nitazenes displays superagonism inherent in the chemical scaffold, this also suggests there is potential to design a compound that is a biased superagonist. Succeeding in this effort would assist in determining the cause of the improved safety profile of biased opioid ligands, as superagonism would exclude low efficacy as an explanation for reduced side effects.
An interesting aspect of the superagonism displayed by isotonitazene is its ability to induce β-arrestin recruitment in the absence of GRK proteins. This finding could indicate that binding of the ligand induces a novel conformation or set of conformations that is capable of recruiting β-arrestins with high affinity without the need for GRK-mediated phosphorylation of the C-terminus as the rate-limiting step. Compounds such as BU72 have been described as potential MOR superagonists because of their ability to induce a stable conformational state.42,43 This suggests the possibility that the basis of the superagonism shown by the nitazenes may come from the induction of a unique and stable conformational state able to robustly cause both G protein activation as well as β-arrestin recruitment. Given the high MOR affinity displayed in radioligand binding assays, it is also feasible that the origin of the superagonism profile of the nitazenes could arise from a slow dissociation of the compounds from the receptor, thereby maintaining the receptor in an active state for a longer period. It is clear that the structure-function link between MOR ligands, superagonism, and biased agonism is currently underexplored. Ultimately, structural and kinetic studies will need to be performed to elucidate the conformational changes and mechanisms by which these compounds achieve such high-signaling efficacy. These studies may aid in the design of novel biased nitazenes, as the role of binding affinity and kinetics in biased agonism is currently underexplored.44,45,46
The nitazenes tested here all showed a high degree of MOR selectivity. This is consistent with previous studies on isotonitazene and etonitazene which demonstrated similar selectivity for the MOR.8,14,47,48 There are very few described superagonists at the MOR which also display high MOR selectivity. The strong selectivity of the nitazene scaffold may prove useful in the design of drugs specific for the MOR, allowing for the avoidance of KOR- or DOR-associated adverse effects such as hallucinations or convulsions, respectively.49,50,51,52 Understanding the aspects of this scaffold that lead to selectivity may lead to a better understanding of the causes of MOR selectivity as a whole and could offer a way to design new MOR selective compounds.
Clearly, nitazenes are documented to produce severe respiratory depression in humans, which can be lethal.5 For example, one-fifth of the patients who received metonitazene in a clinical trial showed evidence of cyanosis and one participant lost consciousness and had to be revived using nalorphine.53 This study provides further evidence of the potential of these types of compounds to produce respiratory depression. Because N-desethyl isotonitazene showed a fast onset of respiratory depression in our study, administration would likely not allow time for a rise in pCO2 sufficient to increase respiratory chemodrive, which could alleviate the respiratory depression and reduce the risk of death from apnea. This property has been suggested as a factor in the increased rate of fatalities with illicit fentanyl use, compared to drugs with lower potency and slower onset of respiratory depression like morphine and heroin, and is likely relevant to the recreational use of nitazenes. Even if the drug dose is not sufficient to cause apnea, the long duration of action of N-desethyl isotonitazene would result in a prolonged period of hypoventilation with potential hypoxic injury. In addition, the difficulty to titrate isotonitazene to the maximal analgesic effect without eliciting apnea could indicate that the nitazene compounds may have a narrow therapeutic index, as suggested previously.54 Ultimately, the long half-life of nitazenes provides a liability for renarcotization after delivery of naloxone or another opioid antagonist, which adds to their inherent danger in society.
Isotonitazene and other nitazenes are biotransformed to active (and potentially lethal) N-desethyl metabolites. The extent to which nitazenes undergo N-deethylation after in vivo administration is not clear, but in a published analysis of samples collected during toxicology casework, N-desethyl isotonitazene and isotonitazene were detected in blood samples at similar concentrations.55 If the N-deethylation of nitazenes does occur in humans to a significant degree then that process could have serious toxicological implications for individuals who abuse these substances. Although the enzymes responsible for the N-deethylation of isotonitazene and other nitazenes have not been identified, cytochrome P450 isozymes (CYP450) are likely candidates. If nitazenes are metabolized by CYP450 then genetic polymorphisms of these isozymes, as well as the use of other drugs and medications that can influence their enzymatic activity and/or expression, could markedly alter interindividual sensitivity to these drugs and contribute to drug-drug interactions. Furthermore, re-dosing with a nitazene drug could pose a grave risk if its metabolites have relatively long half-lives and remain present in the brain when the additional dose is taken; these metabolites could potentially magnify the effects of the drug, including respiratory depression.
Overall, while the nitazenes tested here may have therapeutic indices too low for therapeutic use, the commonalities between them suggest that the nitazene scaffold may be useful for analgesic drug discovery. That said, these drugs have demonstrated their potential to produce lethal effects; their scheduling may be necessary to prevent nitazene derivatives from further contributing to the opioid epidemic. Future studies should address the pharmacological profiles of other derivatives of the nitazene scaffold including assessment of superagonist and biased agonist properties. Ultimately, the lack of extremely biased MOR-selective ligands, especially toward the β-arrestin pathway, has hindered the detailed investigation toward understanding the relationship between ligand structure, ligand bias, and clinically relevant in vivo effects. Development of this family of compounds may offer a scaffold uniquely suited to answering these questions.
Limitations of the study
Although we have assessed the function and selectivity of the three tested nitazenes at other opioid receptors, we cannot exclude the potential for other off-target targets. The use of mice for the analgesia assays and rabbits for the respiratory depression assays prevents direct comparison of the relationship between analgesic and respiratory depressant doses. Since fentanyl readily achieves the maximal physiologic response (i.e., apnea), the MOR superagonist property of nitazenes that can be demonstrated in the human cell assay cannot be completely differentiated from the in vivo assays. Another limitation of the study is that we did not examine isotonitazene in respiratory depression studies due to its DEA scheduling status. Therefore, further investigations are needed to determine if the properties and relationships shown in this study hold true across the nitazene family of compounds and other conventional opioids, especially as they pertain to their superagonist efficacy properties.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| DAMGO | Cayman Chemical | 21553 |
| Isotonitazene | Cayman Chemical | 30880 |
| N-desethyl isotonitazene hydrochloride | Cayman Chemical | 30216 |
| Metonitazene | Cayman Chemical | 26398 |
| Morphine | Cayman Chemical | ISO60147 |
| Fentanyl | Cayman Chemical | ISO60197 |
| Salvinorin-A | Cayman Chemical | 11487 |
| DADLE | Cayman Chemical | 28928 |
| [3H]-DAMGO | PerkinElmer | NET902250UC |
| Forskolin | Sigma | F6886-25MG |
| Compound 101 | Fisher | 564210 |
| Fentanyl citrate | USP | 1269902 |
| Tween-80 | Sigma | P1754 |
| sevoflurane | Akorn | NDC59399-107-01 |
| isoflurane | Priamal | NDC66794-013-25 |
| vecuronium | Sun Phamaceutical Ind. Ltd | NDC 47335-931-40 |
| lidocaine | Fresenius Kabi | NDC 63323-486-05 |
| aminocaproic acid | Sigma | A2504 |
| dexamethasone | Bimeda | 1DX023 |
| epinephrine | Sparhawk Laboratories, Inc | E-4000-02 |
| lactated ringer's | Baxter | NDC 0338-0117-04 |
| KCl | Fresenius Kabi | NDC 63323-965-02 |
| Mannitol | Neogen.Vet | NDC59051-8061-5 |
| Heparin | Meitheal | NDC71288-402-30 |
| Coelenterazine H | Prolume | 301-1 hCTZ |
| Coelenterazine 400a (Deep Blue C) | Prolume | 340-1 CTZ 400a |
| D-luciferin, sodium salt | GoldBio | LUCNA-1G |
| Poly-L-lysine | Sigma | P2636 |
| Penicillin Streptomycin Solution | VWR | 45000-652 |
| Carbenicillin (disodium) | GoldBio | C-103-5 |
| DMEM | VWR | 45000-306 |
| FBS | VWR | 97068-085 |
| Dialyzed FBS | Omega Scientific | FB-03 |
| 10xHBSS | Invitrogen | 14065-056 |
| BSA-fatty acid free | Akron | AK8909-0100 |
| OptiMEM Reduced Serum Medium | LifeTech | 31985070 |
| Expi293 Expression Medium | ThermoFisher Supply Center | A1435102 |
| L-Ascorbic acid | Sigma Aldrich | A5960-25G |
| Critical commercial assays | ||
| ExpiFectamine™ 293 Transfection Kit | ThermoFisher Supply Center | A14525 |
| TransIT-2020 Transfection Reagent | VWR | 10766-852 |
| pGloSensor-22F | Promega | E2301 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-3216 |
| Expi293F Cells | ThermoFisher Supply Center | A14527 |
| Experimental models: Organisms/strains | ||
| C57BL/6J mice (∼0.03-0.04kg, males) | The Jackson Laboratory | N/A |
| New Zealand White rabbits (3-4kg, both sexes) | Kuiper Rabbit Farm, Gary, IN | N/A |
| Recombinant DNA | ||
| pcDNA3.1-human MOR | Kroeze et al.56 | Addgene Cat#1000000068 |
| pcDNA3.1-human KOR | Kroeze et al.56 | Addgene Cat#1000000068 |
| pcDNA3.1-human DOR | Kroeze et al.56 | Addgene Cat#1000000068 |
| pcDNA3.1-mouse MOR | Gift from Tao Che | N/A |
| pcDNA3.1-MOR-Rluc8 | This paper | N/A |
| pcDNA3.1-Venus-β-arr2 | This paper | N/A |
| pcDNA3.1-human-GRK2 | This paper | N/A |
| pcDNA3.1-Gβ1 | cDNA Resource Center | Cat#GNB0100000 |
| pcDNA3.1-Gβ3 | cDNA Resource Center | Cat#GNB0300000 |
| pcDNA5/FRT/TO-GoA-Rluc8 | Olsen et al.27 | Addgene Cat#140976 |
| pcDNA5/FRT/TO-GoB-Rluc8 | Olsen et al.27 | Addgene Cat#140977 |
| pcDNA5/FRT/TO-Gi1-Rluc8 | Olsen et al.27 | Addgene Cat#140973 |
| pcDNA5/FRT/TO-Gi2-Rluc8 | Olsen et al.27 | Addgene Cat#140974 |
| pcDNA5/FRT/TO-Gi3-Rluc8 | Olsen et al.27 | Addgene Cat#140975 |
| pcDNA5/FRT/TO-Gz-Rluc8 | Olsen et al.27 | Addgene Cat#140978 |
| pcDNA5/FRT/TO-Gq-Rluc8 | Olsen et al.27 | Addgene Cat#140982 |
| pcDNA5/FRT/TO-G11-Rluc8 | Olsen et al.27 | Addgene Cat#140983 |
| pcDNA5/FRT/TO-G12-Rluc8 | Olsen et al.27 | Addgene Cat#140985 |
| pcDNA5/FRT/TO-G13-Rluc8 | Olsen et al.27 | Addgene Cat#140986 |
| pcDNA5/FRT/TO-Gs(short)-Rluc8 | Olsen et al.27 | Addgene Cat#140980 |
| pcDNA5/FRT/TO-Gs(long)-Rluc8 | Olsen et al.27 | Addgene Cat#140981 |
| pcDNA3.1-GFP2-γ2 | Olsen et al.27 | Addgene Cat#166774 |
| pcDNA3.1-GFP2-γ1 | Olsen et al.27 | Addgene Cat#140989 |
| pcDNA3.1-GFP2-γ9 | Olsen et al.27 | Addgene Cat#140991 |
| Software and algorithms | ||
| Prism 9 | GraphPad Software | Ver. 9.0.2 |
| R | R Core Team | 4.2.1 |
| ChemDraw Professional 16.0 | PerkinElmer | N/A |
| Powerlab 16SP | AD Instruments, Australia | LabChart 7pro v 7.8 |
| SigmaPlot | Systat Software USA | Version 11 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, John D. McCorvy (jmccorvy@mcw.edu).
Materials availability
All plasmids and cells generated from this study could be obtained directly from lead contact with a completed Materials Transfer Agreement if there is potential for commercial application.
Experimental model and study participant details
GPCR signaling studies utilized human embryonic kidney (HEK) derived-cell lines, HEK293T (ATCC) and Expi293F cells (Invitrogen). HEK293T cells were cultured in a humidified incubator at 37°C at 5% CO2 in high-glucose DMEM (VWR) supplemented with 10% FBS (Life Technologies), and authenticated and tested to be mycoplasma-free. Expi293F cells were cultured in a humidified incubator at 37°C at 8% CO2 in Expi293 expression medium, and authenticated and tested to be mycoplasma-free. Sex of HEK-derived cell lines are female. Male C57BL/6J mice (6–8 weeks old) were obtained from Jackson Labs (Bar Harbor, ME). The mice were housed on a reversed light–dark cycle (lights on at 1900 h, off at 0700 h) in an AALAC-approved vivarium at the University of California San Diego. Mice were housed up to four per cage in a climate-controlled room and with food and water provided ad libitum except during behavioral testing. Testing was performed between 1000 h and 1800 h (during the dark phase of the light-dark cycle). This study was conducted in accordance with National Institutes Health (NIH) guidelines and was approved by the University of California San Diego Institutional Animal Care and Use Committee. Respiratory depression studies were approved by the Institutional Animal Care and Use Committee, Medical College of Wisconsin, in accordance with provisions of the Animal Welfare Act, and the Public Health Service Guide for the Care and Use of Laboratory Animals. Experiments were carried out on adult (3–4 kg) New Zealand White rabbits of either sex.
Method details
Bioluminescence resonance energy transfer (BRET) assays
Bioluminescence resonance energy transfer (BRET) was used to measure β-arrestin2 recruitment and G protein heterotrimer dissociation after receptor activation as previously described.27 MOR, κ-opioid receptor (KOR), and δ-opioid receptor (DOR) constructs were derived from the Tango library with IDTG linker/V2 tail/TEV/tTA sequences removed to yield the following encoded sequences accession numbers (MOR NCBI: NP_001138751.1; KOR NCBI: NP_000903.2; DOR NCBI: NP_000902.3). TRUPATH library cDNA library was obtained from Addgene.27,56 Mouse MOR construct was a gift from Tao Che (Washington University). To measure β-arrestin2 recruitment to the MOR (NCBI: NP_001138751.1) with or without the co-expression of GPCR kinase 2 (GRK2), HEK293T cells were co-transfected with human MOR containing C-terminal fused Renilla luciferase (RLuc8), GRK2, and N-terminal Venus-tagged β-arrestin2 at a 1:1:5 ratio respectively using TransiT-2020 (Mirus). At least 24 hours after transfection, cells were plated in poly-lysine coated 96-well clear-bottom white cell culture plates in a 1% FBS in DMEM plating media at a density of 30,000-50,000 cells 200 μl per well, and then incubated overnight. The next day, the media was decanted before washing the cells with 60 μl of drug buffer (1×HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4) per well, 60 μl of drug buffer was then added to each well, and plates were incubated at 37°C for at least 10 min before drug stimulation. For experiments involving GRK2 inhibition, compound 101 was added to a concentration of 100 μM in each well of the plate at least 1 hour prior to plate reading.30 For the drug stimulation, 30 μl of drug was added to each well, and then the plate was incubated at 37°C in the dark for 1 hour; for the time-point experiments the incubation times were 5 min, 60 min, and 300 min. Coelenterazine h (Promega, 5 μM final concentration) was added, 10 μl per well, 15 minutes before each plate was read. Plates were read for both luminescence at 485 nm and fluorescent eYFP at 530 nm for 1 s per well using a Mithras LB940 microplate reader. The ratio of eYFP/RLuc was calculated per well and plotted as a function of drug concentration using GraphPad Prism 9.0 (GraphPad Software Inc., San Diego, CA).
To measure MOR mediated G protein heterotrimer dissociation, HEK293T cells were co-transfected in a 1:1:1:1 ratio of GoA-RLuc8, Gβ1, GFP2-Gγ2, and human MOR (NCBI: NP_001138751.1), respectively. Experiments co-expressing GRK2 were transfected in a 1:1 ratio with the receptor. The cells were plated, and the assay was performed almost identically to above, except coelenterazine 400a (NanoLight, 5 μM final concentration) was used as the RLuc substrate. As in the β-arrestin2 recruitment assay, 10 μl of the substrate were added to each well 15 minutes prior to the plate being read. The incubation times between drug additions and plate reading were as above: 5 min, 60 min and 300 min with the 60 min incubation time being used for all experiments other than the timepoint experiments. Plates were read using a Mithras LB940 measuring luminescence at 400 nm and fluorescent GFP2 emission at 515 nm for 1 s per well. The ratio of GFP2/RLuc8 emissions was calculated per well and plotted as a function of drug concentration using GraphPad Prism 9.0.27
A similar procedure was used to compare MOR-ligand binding induced G protein dissociation across a variety of G protein subtypes. The procedure was performed as above with the substitution of each of the following G proteins for GoA in a 1:1:1:1 transfection: GoA, GoB, Gi1, Gi2, Gi3, GZ, G11, G12, G13, Gq, GS-Short, and GS-Long. Prior to the transfection, cells meant for transfection with non-Gi/o subtype proteins were incubated with 100 ng/mL pertussis toxin (PTX; Cayman Chemical) to prevent MOR coupling to endogenous Gi/o proteins.
To measure DOR and KOR mediated GoA heterotrimer dissociation, the above procedure DOR and KOR experiments were normalized to DADLE (Cayman Chemical) and salvinorin A (Cayman Chemical), respectively and analyzed using the nonlinear regression sigmoidal dose–response function built into GraphPad Prism 9.0.
To measure recruitment of mini-Gi protein to the MOR, we co-transfected Venus-tagged mini-Gi with Rluc8-tagged MOR at a 15:1 ratio using the procedure described above. The BRET assay was conducted as explained above for β-arrestin recruitment using a 60 min drug incubation at 37°C with a 15 min pre-incubation of 10 μL coelenterazine h, and reading plates for luminescence at 485 nm and fluorescent eYFP at 530 nm for 1 s per well using a Mithras LB940 microplate reader.
Functional Gi/o-mediated cAMP inhibition assays
To measure MOR, DOR, and KOR Gi/o-mediated cAMP inhibition, HEK293T cells were co-transfected using TransiT-2020 (Mirus) in a 1:1 ratio with human MOR, DOR, and KOR and a split-luciferase based cAMP biosensor (pGloSensor-22F; Promega). After at least 24 h, transfected cells were washed with phosphate buffered saline (PBS) and trypsin was used to dissociate the cells. Cells were centrifuged, resuspended in plating media (1% dialyzed FBS in DMEM), plated at a density of 15,000–20,000 cells/mL distributed into 40 μl per well in poly-lysine coated 384-well white clear bottom cell culture plates, and incubated at 37°C with 5% CO2 overnight. The next day, drug dilutions were prepared in fresh assay buffer (20 mM HEPES, 1× HBSS, 0.1% bovine serum album (BSA), and 0.01% ascorbic acid, pH 7.4) at 5× drug concentration. Plates were decanted and 20 μl per well of GloSensor assay substrate diluted in assay buffer was added to each well. Drug addition to 384-well plates was performed by FLIPR adding 5 μl of 5x drug per well for a total volume of 25 μl. Plates were incubated for exactly 15 min in the dark at room temperature. To stimulate endogenous cAMP via adenylyl-cyclase activation, 5 μl of 6× forskolin (FSK) (1 μM final concentration) diluted in drug buffer (20 mM HEPES, 1×HBSS, pH 7.4) was added per well. Cells were again incubated in the dark at room temperature for 15 min, and luminescence intensity was quantified using a Wallac TriLux Microbeta (Perkin Elmer) luminescence counter. Data were normalized to DAMGO, DADLE, or salvinorin-A induced cAMP inhibition for MOR, DOR, and KOR, respectively.
Radioligand binding assays
To prepare membranes for radioligand binding, the Expi293 expression system (ThermoFisher) was used. Expi293F cells in Expi293 expression medium were transfected with plasmid DNA using ExpiFectamine 293 reagent. Approximately 20 hours after transfection, Transfection reagents 1 and 2 were added to the cells, and 48 h after the transfection, a 10 min hypotonic lysis buffer (1 mM HEPES, 2 mM EDTA, pH 7.4) incubation was used to lyse the cells. The lysate was resuspended and centrifuged at 30,000×g. The supernatant was decanted, and membranes were resuspended in binding buffer (50 mM Tris, 10 mM MgCl2, 0.1 mM EDTA, pH 7.4) prior to centrifugation at 13,000×g in pre-chilled 1.7 mL microcentrifuge tubes. The supernatant was decanted and membrane pellets were stored at -80°C until use.
Radioligand binding assays utilized [3H]-DAMGO (Perkin Elmer; Specific Activity = 84.7 Ci/mmol). The [3H]-DAMGO dissociation constant (Kd) was calculated using homologous competition assays with cold, unlabeled DAMGO (pKd = 8.36 ± 0.07, Table 1). Competition assays were conducted with a constant concentration of [3H]-DAMGO ranging from 2-4 nM, unlabeled ligand competitors at concentrations ranging from 100 μM to 1 pM, and membranes were resuspended in binding buffer (50 mM Tris, 10 mM MgCl2, 0.1 mM EDTA, 0.1% BSA, 0.01% ascorbic acid, pH 7.4). Binding assays were incubated at room temperature for 2 h and assays were terminated by vacuum filtration using a 96-well Filtermate harvester onto 96-well filter mats A (Perkin Elmer) pre-soaked with 0.3% polyethyleneimine. Filters were washed three times using cold wash buffer (50 mM Tris, pH 7.4) and scintillation cocktail (Meltilex) was melted onto dried filters. Radioactivity displacement was measured using a Microbeta Trilux counter (Perkin Elmer). Counts per minute (CPM) were plotted as a function of unlabeled ligand concentration and the Ki was calculated using the One-site-Fit Ki using Graphpad Prism 9.0.
Tail-flick assay
The tail-flick test was used to assess thermal antinociceptive effects.57 Tail-flick testing was performed using an IITC Tail Flick Analgesia Meter Model 336 (IITC Life Science Inc, Woodland Hills, CA), which heats the dorsal surface of the tail with a focused beam of light and automatically measures the latency for tail withdrawal. The stimulus intensity was set so that baseline latencies ranged from 2-4 sec. A 10-sec cutoff time was used. Trials were conducted while mice were restrained in a plastic tube (Braintree Scientific Inc, Braintree, MA). On Day 1, baseline withdrawal latencies were assessed as the mean of three consecutive trials. On Day 2, mice were treated with vehicle or drug and then withdrawal latencies were assessed in a single trial conducted 15 min post-injection. Compounds were dissolved in water (N-desethyl isotonitazene) or water containing 5% Tween-80 (isotonitazene and metonitazene) and injected IP (5 mL/kg). Isotonitazene was tested at 0 (n=6), 0.003 (n=8), 0.01 (n=7), and 0.03 mg/kg (n=7); N-desethyl isotonitazene was tested at 0 (n=8), 0.01 (n=8), 0.03 (n=8), 0.1 (n=8), and 0.3 mg/kg (n=8); metonitazene was tested at 0 (n=7), 0.12 (n=7), 0.24 (n=7), 0.48 (n=7), and 0.96 mg/kg (n=7).
Phrenic nerve activity respiratory depression assay
The research was approved by the Institutional Animal Care and Use Committee, Medical College of Wisconsin, in accordance with provisions of the Animal Welfare Act, and the Public Health Service Guide for the Care and Use of Laboratory Animals. Experiments were carried out on adult (3–4 kg) New Zealand White rabbits of either sex. The preparation has been previously described in detail.58 In short, animals were anesthetized and ventilated via tracheotomy with an anesthesia machine (Ohmeda CD, GE, Datex Ohmeda, Madison, WI). Inspiratory oxygen fraction, expiratory carbon dioxide concentration and expiratory isoflurane concentration were continuously displayed with an infrared analyzer (POET II, Criticare Systems, Waukesha, WI). Femoral arterial and venous lines were used for blood pressure monitoring, infusion of solutions, and bolus drug application, respectively. Lactated Ringer’s solution with 3 μg/ml epinephrine was continuously infused at 1 ml/h, and rate was increased as needed to counteract or prevent hypotension in response to drug injections and/or from blood loss. The animal was maintained at 37.0 ± 0.5°C with a warming blanket. The animal was placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). After blunt precollicular decerebration, isoflurane was continued at subanesthetic levels (0.3–0.4 vol%) for blood pressure control, and animals were paralyzed with rocuronium (15 mg/kg subcutaneous bolus), followed by vecuronium 2 mg/h infusion to avoid motion artifacts. Bilateral vagotomy was performed to achieve peripheral deafferentation. The phrenic nerve was recorded with fine bipolar electrodes through a posterior neck incision.58,59,60 Throughout the experiment animals were ventilated with hyperoxia (FiO2 0.6) and maintained at mild hypercapnia (expiratory carbon dioxide: 45-55 mmHg). Blood pressure was maintained stable throughout the protocols by adjusting the intravenous infusion rate. The phrenic neurogram, respiratory rate, arterial blood pressure, and airway carbon dioxide concentration were continuously displayed and recorded using a digital acquisition system (Powerlab/16SP; ADInstruments, Castle Hill, Australia). Before and after intravenous drug injection, steady-state conditions were obtained for respiratory parameters. At the end of the experiment, animals were euthanized with intravenous potassium chloride.
The MOR agonists fentanyl or N-desethyl isotonitazene were administered intravenously at a specified dose diluted in lactated Ringer’s solution. Full recovery of respiratory rate to baseline was awaited to determine drug kinetics. This was followed by titration of each drug until apnea. To determine the minimum required apneic dose, each additional drug dose was administered once the maximal effect of the previous dose had been reached, i.e., after 2-3 min for fentanyl and ∼10 min for N-desethyl isotonitazene. Again, we awaited full recovery of respiratory rate to baseline. This was followed by bolus administration of the previously determined apneic dose of each drug. Once apnea was achieved, we administered intravenous naloxone (20 μg/kg) and measured time to full reversal of respiratory rate.
Quantification and statistical analysis
BRET and radioligand binding data aAnalysis
Data from BRET experiments were normalized to the positive control compound, [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO), analyzed using the nonlinear regression sigmoidal dose–response function built into GraphPad Prism 9.0, and the built-in extra sum-of-squares F test was used to determine if the calculated Emax values were statistically significantly different from that of DAMGO (α=0.005). Data from radioligand binding used counts per minute (CPM) and were plotted as a function of unlabeled ligand concentration and the Ki was calculated using the One-site-Fit Ki using Graphpad Prism 9.0.
Bias and selectivity calculations
To calculate bias and selectivity factors from the BRET and cAMP inhibition data, transduction ratios ( were calculated for each drug respective to each assay (where RA is relative activity).61,62,63 The control-normalized factor was calculated as . The logarithm of the bias or selectivity factor was calculated as , and the bias or selectivity factor was calculated as .64 In the case of bias factors, pathway 1 corresponds to G protein dissociation, and pathway 2 corresponds to β-arrestin recruitment. In the case of selectivity factors, pathway 1 was the MOR cAMP inhibition activity, and pathway 2 was the KOR or DOR cAMP inhibition activity.
Data clustering
Hierarchical clustering was performed using the “hclust” function in R 4.2.1. The Euclidean metric () was used to calculate distances between data points, and the complete-linkage agglomerative hierarchical clustering algorithm was used to determine distances between clusters such that for clusters and .
Tail-flick assay data analysis
Latency in the tail-flick assay is expressed as a percentage of the maximal possible effect (%MPE), where %MPE = ((test latency – average baseline latency) ÷ (10 s – average baseline latency)) × 100. Data were analyzed using one-way Welch ANOVAs (factor: drug dose) followed by Dunnett’s T3 multiple comparisons post-hoc test. Significance was assessed using an α level of 0.05. Median effective doses (ED50 values) and 95% CI were calculated using nonlinear regression. Fentanyl was tested as a reference compound; mice were injected subcutaneously (SC) with saline vehicle (n=8) or fentanyl citrate at 0.03 (n=6), 0.06 (n=9), 0.12 (n=8), or 0.24 mg/kg (n=7). For the experiment with fentanyl, all of the mice treated with the highest dose showed identical responses, so the data were analyzed using a Kruskal-Wallis test followed by Dunn’s multiple comparisons post-hoc test. An extra sum of squares F-test was used to compare the potencies of test compounds with the ED50 for fentanyl (in nmol/kg).
Respiratory rate data analysis
Post experiment, values for the phrenic neurogram and a comparator to mark beginning and end of inspiratory phase were exported to SigmaPlot 11 (Systat Software, San Jose, CA) for further data reduction and plotting.65 Respiratory rate was plotted for each breath versus time, signal artefacts were removed, and best fit curves plotted for each dose-response. Values for respiratory rate were calculated at 1-minute intervals from the best-fit curves and pooled for all animals. Average values were compared between fentanyl and N-desethyl isotonitazene using t-statistics (2-tailed, p<0.05). Values are reported as mean ± S.E.M.
Acknowledgments
This work was supported by the National Institutes of Health (R35 GM133421 to J.D.M and R01 HL159546 to A.S.). N.J.M. was supported by T32 GM080202 (Medical Scientist Training Program). DJS acknowledges support from the NIH (T32 HL134643) and the Medical College of Wisconsin Cardiovascular Center’s A.O. Smith Fellowship Scholars Program.
Author contributions
N.J.M. and J.D.M. conceived of the project. N.J.M., M.M.C., and J.K.L. performed the BRET assays, N.J.M. performed the radioligand-binding and cAMP-inhibition assays, all under the supervision of J.D.M. A.L.H. performed the tail-flick analgesia assays. B.P. performed the respiratory depression studies under the supervision of A.G.S. N.J.M. and J.D.M. analyzed the data and N.J.M wrote the initial draft of manuscript with input from D.J.S and J.D.M. All authors discussed, revised, and approved the manuscript.
Declaration of interests
The authors declare no competing financial interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as living with a disability.
Published: June 13, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107121.
Supplemental information
Data and code availability
-
•
All data generated in this study are included in this article and the supplemental information. All data reported in this paper will be shared by the lead contact upon request.
-
•
No custom code was used in this study.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Meisenberg B.R., Grover J., Campbell C., Korpon D. Assessment of Opioid Prescribing Practices Before and After Implementation of a Health System Intervention to Reduce Opioid Overprescribing. JAMA Netw. Open. 2018;1:e182908. doi: 10.1001/jamanetworkopen.2018.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Congress House of Representatives. (2020). House Hearing, 116th Congress - FENTANYL ANALOGUES: PRESPECTIVES ON CLASSWIDE SCHEDULING.
- 3.Jones C.M., Einstein E.B., Compton W.M. Changes in Synthetic Opioid Involvement in Drug Overdose Deaths in the United States, 2010-2016. JAMA. 2018;319:1819–1821. doi: 10.1001/jama.2018.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman J., Beletsky L., Schriger D.L. Overdose-Related Cardiac Arrests Observed by Emergency Medical Services During the US COVID-19 Epidemic. JAMA Psychiatr. 2021;78:562–564. doi: 10.1001/jamapsychiatry.2020.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shover C.L., Falasinnu T.O., Freedman R.B., Humphreys K. Emerging Characteristics of Isotonitazene-Involved Overdose Deaths: A Case-Control Study. J. Addict. Med. 2021;15:429–431. doi: 10.1097/ADM.0000000000000775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calello D.P., Aldy K., Jefri M., Nguyen T.-A.T., Krotulski A., Logan B., Brent J., Wax P., Walton S., Manini A.F., ToxIC Fentalog Study Group Identification of a novel opioid, N -piperidinyl etonitazene (etonitazepipne), in patients with suspected opioid overdose. Clin. Toxicol. 2022;60:1067–1069. doi: 10.1080/15563650.2022.2084406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casy A.F., Wright J. Some 2-benzyl-5-nitrobenzimidazoles. J. Chem. Soc. Perkin 1. 1966;17:1511–1513. doi: 10.1039/j39660001511. [DOI] [PubMed] [Google Scholar]
- 8.Ujváry I., Christie R., Evans-Brown M., Gallegos A., Jorge R., de Morais J., Sedefov R. DARK Classics in Chemical Neuroscience: Etonitazene and Related Benzimidazoles. ACS Chem. Neurosci. 2021;12:1072–1092. doi: 10.1021/acschemneuro.1c00037. [DOI] [PubMed] [Google Scholar]
- 9.Hyyatiä P., Sinclair J.D. Oral etonitazene and cocaine consumption by AA, ANA and Wistar rats. Psychopharmacology (Berl) 1993;111:409–414. doi: 10.1007/BF02253529. [DOI] [PubMed] [Google Scholar]
- 10.Walker E.A., Young A.M. Differential tolerance to antinociceptive effects of μ opioids during repeated treatment with etonitazene, morphine, or buprenorphine in rats. Psychopharmacology (Berl) 2001;154:131–142. doi: 10.1007/s002130000620. [DOI] [PubMed] [Google Scholar]
- 11.Krotulski A.J., Papsun D.M., Kacinko S.L., Logan B.K. Isotonitazene Quantitation and Metabolite Discovery in Authentic Forensic Casework. J. Anal. Toxicol. 2020;44:521–530. doi: 10.1093/jat/bkaa016. [DOI] [PubMed] [Google Scholar]
- 12.Zagorski C.M., Myslinski J.M., Hill L.G. Isotonitazene as a contaminant of concern in the illegal opioid supply: A practical synthesis and cost perspective. Int. J. Drug Policy. 2020;86:102939. doi: 10.1016/j.drugpo.2020.102939. [DOI] [PubMed] [Google Scholar]
- 13.Vandeputte M.M., van Uytfanghe K., Layle N.K., st Germaine D.M., Iula D.M., Stove C.P. Synthesis, Chemical Characterization, and μ-Opioid Receptor Activity Assessment of the Emerging Group of “Nitazene” 2-Benzylbenzimidazole Synthetic Opioids. ACS Chem. Neurosci. 2021;12:1241–1251. doi: 10.1021/acschemneuro.1c00064. [DOI] [PubMed] [Google Scholar]
- 14.Walton S.E., Krotulski A.J., Glatfelter G.C., Walther D., Logan B.K., Baumann M.H. Plasma pharmacokinetics and pharmacodynamic effects of the 2-benzylbenzimidazole synthetic opioid, isotonitazene, in male rats. Psychopharmacology (Berl) 2023;240:185–198. doi: 10.1007/s00213-022-06292-5. [DOI] [PubMed] [Google Scholar]
- 15.Tan L., Yan W., McCorvy J.D., Cheng J. Biased Ligands of G Protein-Coupled Receptors (GPCRs): Structure–Functional Selectivity Relationships (SFSRs) and Therapeutic Potential. J. Med. Chem. 2018;61:9841–9878. doi: 10.1021/acs.jmedchem.8b00435. [DOI] [PubMed] [Google Scholar]
- 16.Manglik A., Lin H., Aryal D.K., McCorvy J.D., Dengler D., Corder G., Levit A., Kling R.C., Bernat V., Hübner H., et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeWire S.M., Yamashita D.S., Rominger D.H., Liu G., Cowan C.L., Graczyk T.M., Chen X.-T., Pitis P.M., Gotchev D., Yuan C., et al. A G Protein-Biased Ligand at the μ-Opioid Receptor Is Potently Analgesic with Reduced Gastrointestinal and Respiratory Dysfunction Compared with Morphine. J. Pharmacol. Exp. Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 18.Singla N., Minkowitz H.S., Soergel D.G., Burt D.A., Subach R.A., Salamea M.Y., Fossler M.J., Skobieranda F. A randomized, Phase IIb study investigating oliceridine (TRV130), a novel μ-receptor G-protein pathway selective (μ-GPS) modulator, for the management of moderate to severe acute pain following abdominoplasty. J. Pain Res. 2017;10:2413–2424. doi: 10.2147/JPR.S137952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raehal K.M., Walker J.K.L., Bohn L.M. Morphine Side Effects in β-Arrestin 2 Knockout Mice. J. Pharmacol. Exp. Ther. 2005;314:1195–1201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- 20.Kliewer A., Gillis A., Hill R., Schmiedel F., Bailey C., Kelly E., Henderson G., Christie M.J., Schulz S. Morphine-induced respiratory depression is independent of β-arrestin2 signalling. Br. J. Pharmacol. 2020;177:2923–2931. doi: 10.1111/bph.15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gillis A., Kliewer A., Kelly E., Henderson G., Christie M.J., Schulz S., Canals M. Critical Assessment of G Protein-Biased Agonism at the μ-Opioid Receptor. Trends Pharmacol. Sci. 2020;41:947–959. doi: 10.1016/j.tips.2020.09.009. [DOI] [PubMed] [Google Scholar]
- 22.He L., Gooding S.W., Lewis E., Felth L.C., Gaur A., Whistler J.L. Pharmacological and genetic manipulations at the μ-opioid receptor reveal arrestin-3 engagement limits analgesic tolerance and does not exacerbate respiratory depression in mice. Neuropsychopharmacology. 2021;46:2241–2249. doi: 10.1038/s41386-021-01054-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gillis A., Gondin A.B., Kliewer A., Sanchez J., Lim H.D., Alamein C., Manandhar P., Santiago M., Fritzwanker S., Schmiedel F., et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal. 2020;13:eaaz3140. doi: 10.1126/scisignal.aaz3140. [DOI] [PubMed] [Google Scholar]
- 24.Azevedo Neto J., Costanzini A., de Giorgio R., Lambert D.G., Ruzza C., Calò G. Biased versus Partial Agonism in the Search for Safer Opioid Analgesics. Molecules. 2020;25:3870. doi: 10.3390/molecules25173870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grim T.W., Acevedo-Canabal A., Bohn L.M. Toward Directing Opioid Receptor Signaling to Refine Opioid Therapeutics. Biol. Psychiatry. 2020;87:15–21. doi: 10.1016/j.biopsych.2019.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolb P., Kenakin T., Alexander S.P.H., Bermudez M., Bohn L.M., Breinholt C.S., Bouvier M., Hill S.J., Kostenis E., Martemyanov K.A., et al. Community guidelines for GPCR ligand bias: IUPHAR review 32. Br. J. Pharmacol. 2022;179:3651–3674. doi: 10.1111/bph.15811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olsen R.H.J., DiBerto J.F., English J.G., Glaudin A.M., Krumm B.E., Slocum S.T., Che T., Gavin A.C., McCorvy J.D., Roth B.L., Strachan R.T. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 2020;16:841–849. doi: 10.1038/s41589-020-0535-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamberts J.T., Jutkiewicz E.M., Mortensen R.M., Traynor J.R. Mu-opioid receptor coupling to gα o plays an important role in opioid antinociception. Neuropsychopharmacology. 2011;36:2041–2053. doi: 10.1038/npp.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miess E., Gondin A.B., Yousuf A., Steinborn R., Mösslein N., Yang Y., Göldner M., Ruland J.G., Bünemann M., Krasel C., et al. Multisite phosphorylation is required for sustained interaction with GRKs and arrestins during rapid μ-opioid receptor desensitization. Sci. Signal. 2018;11:eaas9609. doi: 10.1126/scisignal.aas9609. [DOI] [PubMed] [Google Scholar]
- 30.Lowe J.D., Sanderson H.S., Cooke A.E., Ostovar M., Tsisanova E., Withey S.L., Chavkin C., Husbands S.M., Kelly E., Henderson G., Bailey C.P. Role of G Protein–Coupled Receptor Kinases 2 and 3 in μ-Opioid Receptor Desensitization and Internalization. Mol. Pharmacol. 2015;88:347–356. doi: 10.1124/mol.115.098293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kenakin T., Watson C., Muniz-Medina V., Christopoulos A., Novick S. A Simple Method for Quantifying Functional Selectivity and Agonist Bias. ACS Chem. Neurosci. 2012;3:193–203. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jang W., Adams C.E., Liu H., Zhang C., Levy F.O., Andressen K.W., Lambert N.A. An inactive receptor-G protein complex maintains the dynamic range of agonist-induced signaling. Proc. Natl. Acad. Sci. USA. 2020;117:30755–30762. doi: 10.1073/pnas.2010801117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kenakin T. The Effective Application of Biased Signaling to New Drug Discovery. Mol. Pharmacol. 2015;88:1055–1061. doi: 10.1124/mol.115.099770. [DOI] [PubMed] [Google Scholar]
- 34.Goldstein A., Naidu A. Multiple opioid receptors: ligand selectivity profiles and binding site signatures. Mol. Pharmacol. 1989;36:265–272. [PubMed] [Google Scholar]
- 35.Palkovic B., Cook-Snyder D., Callison J.J., Langer T.M., Nugent R., Stuth E.A.E., Zuperku E.J., Stucke A.G. Contribution of the caudal medullary raphe to opioid induced respiratory depression. Respir. Physiol. Neurobiol. 2022;299:103855. doi: 10.1016/j.resp.2022.103855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flecknell P.A., Liles J.H., Wootton R. Reversal of fentanyl/fluanisone neuroleptanalgesia in the rabbit using mixed agonist/antagonist opioids. Lab. Anim. 1989;23:147–155. doi: 10.1258/002367789780863655. [DOI] [PubMed] [Google Scholar]
- 37.Schrage R., de Min A., Hochheiser K., Kostenis E., Mohr K. Superagonism at G protein-coupled receptors and beyond. Br. J. Pharmacol. 2016;173:3018–3027. doi: 10.1111/bph.13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vandeputte M.M., Cannaert A., Stove C.P. In vitro functional characterization of a panel of non-fentanyl opioid new psychoactive substances. Arch. Toxicol. 2020;94:3819–3830. doi: 10.1007/s00204-020-02855-7. [DOI] [PubMed] [Google Scholar]
- 39.Yudin Y., Rohacs T. The G-protein-biased agents PZM21 and TRV130 are partial agonists of μ-opioid receptor-mediated signalling to ion channels. Br. J. Pharmacol. 2019;176:3110–3125. doi: 10.1111/bph.14702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grim T.W., Schmid C.L., Stahl E.L., Pantouli F., Ho J.-H., Acevedo-Canabal A., Kennedy N.M., Cameron M.D., Bannister T.D., Bohn L.M. A G protein signaling-biased agonist at the μ-opioid receptor reverses morphine tolerance while preventing morphine withdrawal. Neuropsychopharmacology. 2020;45:416–425. doi: 10.1038/s41386-019-0491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bachmutsky I., Wei X.P., Durand A., Yackle K. B-arrestin 2 germline knockout does not attenuate opioid respiratory depression. Elife. 2021;10:e62552. doi: 10.7554/eLife.62552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Livingston K.E., Traynor J.R. Allostery at opioid receptors: modulation with small molecule ligands. Br. J. Pharmacol. 2018;175:2846–2856. doi: 10.1111/bph.13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Livingston K.E., Mahoney J.P., Manglik A., Sunahara R.K., Traynor J.R. Measuring ligand efficacy at the mu-opioid receptor using a conformational biosensor. Elife. 2018;7:e32499. doi: 10.7554/eLife.32499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pedersen M.F., Wróbel T.M., Märcher-Rørsted E., Pedersen D.S., Møller T.C., Gabriele F., Pedersen H., Matosiuk D., Foster S.R., Bouvier M., Bräuner-Osborne H. Biased agonism of clinically approved μ-opioid receptor agonists and TRV130 is not controlled by binding and signaling kinetics. Neuropharmacology. 2020;166:107718. doi: 10.1016/j.neuropharm.2019.107718. [DOI] [PubMed] [Google Scholar]
- 45.Kapoor A., Martinez-Rosell G., Provasi D., de Fabritiis G., Filizola M. Dynamic and Kinetic Elements of μ-Opioid Receptor Functional Selectivity. Sci. Rep. 2017;7:11255. doi: 10.1038/s41598-017-11483-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gillis A., Sreenivasan V., Christie M.J. Intrinsic Efficacy of Opioid Ligands and Its Importance for Apparent Bias, Operational Analysis, and Therapeutic Window. Mol. Pharmacol. 2020;98:410–424. doi: 10.1124/mol.119.119214. [DOI] [PubMed] [Google Scholar]
- 47.Emmerson P.J., Liu M.R., Woods J.H., Medzihradsky F. Binding affinity and selectivity of opioids at mu, delta and kappa receptors in monkey brain membranes. J. Pharmacol. Exp. Ther. 1994;271:1630–1637. [PubMed] [Google Scholar]
- 48.Moolten M.S., Fishman J.B., Chen J.-C., Carlson K.R. Etonitazene: An opioid selective for the mu receptor types. Life Sci. 1993;52:PL199–PL203. doi: 10.1016/0024-3205(93)90118-M. [DOI] [PubMed] [Google Scholar]
- 49.Chung P.C.S., Boehrer A., Stephan A., Matifas A., Scherrer G., Darcq E., Befort K., Kieffer B.L. Delta opioid receptors expressed in forebrain GABAergic neurons are responsible for SNC80-induced seizures. Behav. Brain Res. 2015;278:429–434. doi: 10.1016/j.bbr.2014.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haffmans J., Dzoljic M.R. Effects of delta opioid antagonists on enkephalin-induced seizures. Pharmacology. 1987;34:61–65. doi: 10.1159/000138253. [DOI] [PubMed] [Google Scholar]
- 51.Pfeiffer A., Brantl V., Herz A., Emrich H.M. Psychotomimesis Mediated by κ Opiate Receptors. Science. 1986;233:774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- 52.Butelman E.R., Kreek M.J. Salvinorin A, a kappa-opioid receptor agonist hallucinogen: pharmacology and potential template for novel pharmacotherapeutic agents in neuropsychiatric disorders. Front. Pharmacol. 2015;6:190. doi: 10.3389/fphar.2015.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bromig G. Uber neue starkwirkende Analgetika und ihre klinische Erprobung. Klin. Wochenschr. 1958;36:960–963. doi: 10.1007/BF01486702. [DOI] [PubMed] [Google Scholar]
- 54.DeStevens G. In: Analgetics. DeStevens G., editor. Academic Press; 1965. General Synthetics; pp. 405–439. [Google Scholar]
- 55.Walton S.E., Krotulski A.J., Logan B.K. A forward-thinking approach to addressing the new synthetic opioid 2-benzylbenzimidazole nitazene analogs by liquid chromatography-tandem quadrupole mass spectrometry (LC-QQQ-MS) J. Anal. Toxicol. 2022;46:221–231. doi: 10.1093/jat/bkab117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kroeze W.K., Sassano M.F., Huang X.-P., Lansu K., McCorvy J.D., Giguère P.M., Sciaky N., Roth B.L. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 2015;22:362–369. doi: 10.1038/nsmb.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.D’Amour F.E., Smith D.L. A method for determining loss of pain sensation. J. Pharmacol. Exp. Therapeut. 1941;72 [Google Scholar]
- 58.Palkovic B., Callison J.J., Marchenko V., Stuth E.A.E., Zuperku E.J., Stucke A.G. Dose-dependent Respiratory Depression by Remifentanil in the Rabbit Parabrachial Nucleus/Kölliker–Fuse Complex and Pre-Bötzinger Complex. Anesthesiology. 2021;135:649–672. doi: 10.1097/ALN.0000000000003886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dogas Z., Krolo M., Stuth E.A., Tonkovic-Capin M., Hopp F.A., McCrimmon D.R., Zuperku E.J. Differential effects of GABA A receptor antagonists in the control of respiratory neuronal discharge patterns. J. Neurophysiol. 1998;80:2368–2377. doi: 10.1152/jn.1998.80.5.2368. [DOI] [PubMed] [Google Scholar]
- 60.Cook-Snyder D.R., Miller J.R., Navarrete-Opazo A.A., Callison J.J., Peterson R.C., Hopp F.A., Stuth E.A.E., Zuperku E.J., Stucke A.G. The contribution of endogenous glutamatergic input in the ventral respiratory column to respiratory rhythm. Respir. Physiol. Neurobiol. 2019;260:37–52. doi: 10.1016/j.resp.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ehlert F.J. Analysis of allosterism in functional assays. J. Pharmacol. Exp. Ther. 2005;315:740–754. doi: 10.1124/jpet.105.090886. [DOI] [PubMed] [Google Scholar]
- 62.Tran J.A., Chang A., Matsui M., Ehlert F.J. Estimation of relative microscopic affinity constants of agonists for the active state of the receptor in functional studies on M2 and M3 muscarinic receptors. Mol. Pharmacol. 2009;75:381–396. doi: 10.1124/mol.108.051276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Griffin M.T., Figueroa K.W., Liller S., Ehlert F.J. Estimation of agonist activity at G protein-coupled receptors: Analysis of M2 muscarinic receptor signaling through Gi/o ,Gs, and G15. J. Pharmacol. Exp. Ther. 2007;321:1193–1207. doi: 10.1124/jpet.107.120857. [DOI] [PubMed] [Google Scholar]
- 64.Kenakin T. A scale of agonism and allosteric modulation for assessment of selectivity, bias, and receptor mutation. Mol. Pharmacol. 2017;92:414–424. doi: 10.1124/mol.117.108787. [DOI] [PubMed] [Google Scholar]
- 65.Eldridge F.L. Expiratory effects of brief carotid sinus nerve and carotid body stimulations. Respir. Physiol. 1976;26:395–410. doi: 10.1016/0034-5687(76)90009-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data generated in this study are included in this article and the supplemental information. All data reported in this paper will be shared by the lead contact upon request.
-
•
No custom code was used in this study.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.