

Summary
Systemic inflammation is established as part of late-stage severe lung disease, but molecular, functional, and phenotypic changes in peripheral immune cells in early disease stages remain ill defined. Chronic obstructive pulmonary disease (COPD) is a major respiratory disease characterized by small-airway inflammation, emphysema, and severe breathing difficulties. Using single-cell analyses we demonstrate that blood neutrophils are already increased in early-stage COPD, and changes in molecular and functional neutrophil states correlate with lung function decline. Assessing neutrophils and their bone marrow precursors in a murine cigarette smoke exposure model identified similar molecular changes in blood neutrophils and precursor populations that also occur in the blood and lung. Our study shows that systemic molecular alterations in neutrophils and their precursors are part of early-stage COPD, a finding to be further explored for potential therapeutic targets and biomarkers for early diagnosis and patient stratification.
Keywords: chronic obstructive pulmonary disease, neutrophil, blood, single-cell transcriptomics, granulopoiesis, bone marrow
Graphical abstract
Highlights
-
•
Increase in blood neutrophil molecular states in early-stage COPD
-
•
Progenitor reprogramming is linked to altered blood neutrophil states
-
•
Neutrophil state abundance correlates with lung function decline
Kapellos et al. show that systemic molecular changes in neutrophils in early-stage COPD are associated with increased granulopoiesis and demonstrate that neutrophil transcriptomic states correlate with lung function decline.
Introduction
The World Health Organization (WHO) recently elevated chronic obstructive pulmonary disease (COPD) to be the third leading cause of death worldwide, with projected substantial increases in prevalence and incidence over the next decades.1 Classified as a heterogeneous chronic respiratory disease manifesting with inflammation, progressive and persistent airflow limitation, alveolar emphysema, and acute exacerbations,1 COPD is driven by the interaction of genetics and the environment, in which smoking and air pollution are major risk factors.2
While mainly involving the lung, severe-stage COPD has systemic involvement of elevated inflammatory markers, such as C-reactive protein, TNF-α, IL-6, IL-8, and fibrinogen.3 Patients with systemic inflammation have higher exacerbation frequency and all-cause mortality.4 Furthermore, blood transcriptome studies in COPD patients support the presence of a systemic inflammatory component.5,6,7,8,9,10,11,12 Yet surprisingly, little is known of the cellular and molecular changes within the systemic immune system in early-stage COPD. In particular, neutrophils, the most abundant circulating immune cell, accumulate in the lung tissue of COPD patients and are associated with alveolar damage, lung function decline, and reduced gas exchange, as well as phagocytosis of opportunistic pathogens.13
Recently, the application of single-cell sequencing technologies has revealed an unexpected molecular and functional diversity within the neutrophil compartment,14,15,16,17,18,19,20 both under homeostatic conditions and in the context of inflammatory diseases in affected tissues and in the circulation.14 We postulated that such high-resolution technologies would enable us to characterize the molecular and phenotypic changes in this important immune cell type in COPD and that increased influx of neutrophils into the lung during early-stage disease would entail molecular changes in the bone marrow and the periphery. Such changes might serve as surrogates for disease activity and guide novel therapeutic interventions.
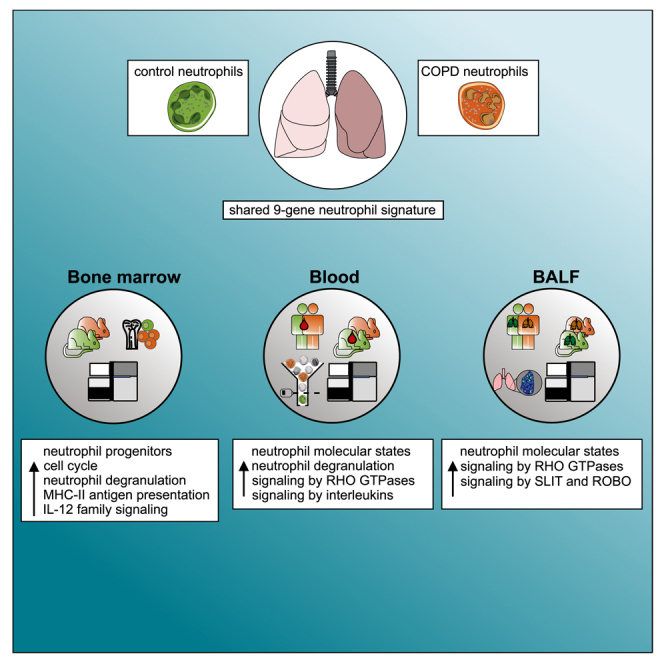
We applied several single-cell technologies to human and murine blood and human bronchoalveolar lavage fluid (BALF) samples, as well as murine bone marrow, to characterize the altered neutrophil cell states in COPD. We describe elevated and reprogrammed granulopoiesis in early-stage COPD, which is directly linked to alterations in the human blood neutrophil compartment that correlate with disease clinical manifestations.
Results
Blood neutrophils exhibit an altered molecular phenotype and are elevated in COPD
To assess systemic changes within the immune system in COPD, we collected peripheral blood from 31 individuals suffering from chronic idiopathic cough or an exquisitely sensitive cough reflex without underlying pathology (hereafter named controls), as well as 69 COPD patients (Figure 1A and Table S1), and determined the changes in the numbers of immune cell populations (Figure 1B). Whole-blood immune cell counts were significantly increased (p = 0.000016) in COPD patients, and this was attributed to the elevated numbers of neutrophils (p = 0.00012) and monocytes (p = 0.00012). Multicolor flow cytometry analysis using previously established panels for the myeloid and lymphoid compartments21 (Figure S1 and Table S1) also showed the relative elevation of neutrophil frequencies (p = 0.02), as well as decreases in natural killer (NK) cell frequencies (p = 0.048) in COPD patients (Figure 1C), consistent with previous reports.22 Furthermore, the absolute neutrophil counts correlated with disease severity, as examined by the percentage predicted forced expiratory volume in 1 s (FEV1) (r = −0.39; p = 0.0002), used as the major clinical severity score in the Global Initiative for Chronic Obstructive Lung Disease (GOLD) grade classification (Figure 1D), and the degree of emphysema (in both lungs), suggesting that they may be implicated in the loss of small airways in COPD (Figure 1E). Similarly, blood neutrophil numbers negatively correlated with the FEV1/FVC (forced vital capacity) ratio (r = −0.35; p = 0.0009) (Figure 1F). Altogether, neutrophils showed significant elevation in their numbers in peripheral blood and correlated with lung function decline and degree of emphysema in COPD.
Figure 1.

Immune-cell-specific transcriptomic signatures in the blood of control and COPD patients
(A) Sample collection and processing pipeline.
(B) Bar plot of absolute immune cell counts in the blood of 30 control and 56 COPD patients. Data are represented as the mean ± SD and statistical analysis was carried out with a Wilcoxon test, ∗∗∗p < 0.001.
(C) Bar plot of immune cell proportions in the blood of 31 control and 69 COPD patients analyzed by flow cytometry. Data are represented as the mean ± SD and analysis was carried out with a two-tailed t test (neutrophils) or a Wilcoxon test (monocytes, eosinophils, T cells, B cells, NK cells) for non-normally distributed data, ∗p < 0.05.
(D–F) Spearman correlation analysis of blood neutrophil counts with (D) percentage forced expiratory volume in 1 s (% FEV1), (E) percentage emphysema in both lungs, and (F) FEV1/FVC ratio. Color code depicts the stratification of COPD patients according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines.
All major cellular states of blood neutrophils are altered in COPD
To define the molecular alterations in different immune cell types between COPD and controls, we next analyzed blood samples from six control individuals and eight early-stage COPD (GOLD2) patients by single-cell RNA sequencing (scRNA-seq; Seq-well). Unsupervised clustering of 69,199 CD45+ cells identified 17 clusters (Figure S2A), which were annotated using published gene markers for human blood cells (Figure S2B and Table S2) and the GeneSigPro classifier labels12 (Figure S2C). The dataset consisted of major blood immune cells (monocytes, neutrophils, dendritic cells, eosinophils, and T, B, and NK cells), a minor population of erythrocytes and megakaryocytes, and lineage subsets and cellular states, for example, classical and non-classical monocytes or resting and activated CD4+ T cells (Figure 2A).
Figure 2.

Blood neutrophils are transcriptionally heterogeneous in control and COPD patients
(A) UMAP (Uniform Manifold Approximation and Projection) representation of 69,199 blood cells from six control and eight COPD patients. Cell clusters were annotated using published canonical gene markers and using label transfer from the GenSigPro classifier.12
(B) Heatmap of the union of differentially expressed (DE) genes between control and COPD patients for immune cells. Each column represents the scaled average normalized expression per cell type and disease status. Hierarchical clustering grouped the genes in 10 clusters. The bar plots indicate cluster gene cellular origin.
(C) Network of the top 5 enriched Reactome gene sets in control or COPD patients. Red arrows depict terms upregulated in COPD, blue arrows terms downregulated in COPD. In bold are gene sets mentioned in the text.
(D) UMAP representation of 20,670 neutrophils from the blood of six control and seven COPD patients.
(E) Heatmap of the top 5 markers for each neutrophil state. Each column represents the average scaled normalized expression per patient.
(F) Dot plot with markers from blood neutrophil states from Combes et al.23 Circle size represents the percentage of cells within a cluster that express a particular gene, circle color shows average scaled normalized gene expression within the cluster.
(G) Single-cell enrichment analysis of neutrophil granule proteins from Cowland and Borregaard.24 Color depicts the scaled average area under the curve score of all cells within the respective neutrophil state.
(H) Gene set enrichment analysis of blood neutrophil DE genes between control and COPD patients using the Reactome database.
(I–L) Dot plots of DE genes in (I) N1S/LCN2, (J) N2S/ISG, (K) N3S/NEAT1, and (L) N5S/S100A12 blood neutrophil states between control and COPD patients. Circle size represents the percentage of cells within a cluster that express a particular gene, circle color shows average gene expression within the cluster.
Molecular alterations in COPD were assessed by defining differentially expressed (DE) genes between control and COPD patients for all immune cell types (Figure 2B and Table S2). Groups of DE genes were unique to neutrophils, classical/intermediate monocytes, activated T cells, or dendritic cells (DCs), and there were several others that were shared by different immune cell types. Hierarchical clustering of the union of DE genes between COPD and controls resulted in 10 distinguished clusters (Figure 2B). Cluster 1 was enriched in alarmins, the neutrophil granule marker CD63,25 and the proteinase inhibitor SERPINB1,26 whereas clusters 3, 4, and 5 were enriched in interferon responses (IRF1, IFNAR1), oxidative phosphorylation (NDUFA4, ATP5L, COX5B, COX7B), and antigen presentation-related genes originating from the comparison of intermediate/non-classical monocytes. Cluster 2 included neutrophil activation markers (FCGR3B, SOD2, IFITM2, IFIT2), clusters 7 and 10 contained B cell activation genes (CD69, CXCR4) and immunoglobulins, and cluster 6 (CD8+ T cells/NK cell dominated) was enriched in cytotoxic molecules (NKG7, GZMH, GZMA, GNLY). Last, two large clusters (8, 9) characterized by mitochondrial (MT-CO2, MT-ND2, MT-CO1, MT-CO3) and protein translation machinery genes (EEF1A1, ENO1, EIF4G2), respectively, were altered in several immune cell types in COPD patients’ blood. Pathway analysis and visualization of the top 5 enriched gene sets (based on the upregulated and downregulated DE genes for each immune cell type individually) revealed two major hubs of regulated pathways for the majority of cell types (Figures 2C and S2D). Neutrophils did not downregulate common pathways in COPD, such as nonsense-mediated decay, translation, and metabolism of amino acids (Figure 2C, left) and overlapped only in some molecular pathways with monocytes (e.g., neutrophil degranulation), but they differed in their molecular deviation (e.g., TLR cascades, interferon-α signaling, interferon signaling, and interactions with lymphoid cells) (Figure 2C, right).
To further characterize the molecular phenotype of blood neutrophils in COPD, we subdivided the 20,670 neutrophils from the complete dataset. They exhibited five neutrophil cellular states that we hereafter refer to as N1S–N5S (Figures 2D and 2E). With the exception of N4S, all neutrophil states were present in both COPD patients and controls, with N3S and N5S having the highest abundance (Figure S3A). Quality control assessment showed that N1S neutrophils presented, on average, with a higher number of unique molecular identifiers (UMIs) and genes than the rest (Figures S3B and S3C). The neutrophil compartment displayed distinct functional states in terms of the expression of cluster-specific genes (Figure 2E and Table S3): N1S (LCN2, ANXA1, LYZ), N2S (IFIT1, IFIT2, IFIT3), N3S (CXCR2, CXCL8), N4S (PTPRC, RNF149, GLT1D1), and N5S (S100A12, S100P, MME). Pathway analysis revealed that the neutrophil states shared functionalities, such as degranulation and interleukin/interferon signaling, but also displayed state-specific pathways, such as translation, antigen presentation and processing, Ga12/13 signaling, and RHO GTPase signaling (Figures S3D–S3H).
The neutrophil clusters described in our study matched the population structure in a recent COVID-19 study.23 N1S corresponded to the previously described LCN2 neutrophil state, with a small contribution from SLPI and RIBO neutrophils; N2S resembled the ISG (interferon-stimulated gene) state; and N3S neutrophils corresponded to the NEAT1 state, N4S to the G0S2 state, and N5S to S100A12 neutrophils (Figure 2F). In addition, evaluation of the expression of neutrophil granule genes allowed us to classify the cell states based on their maturation status24 (Figure 2G). With the exception of N1S/LCN2 neutrophils, all other states were enriched in mature neutrophil granule genes. In fact, trajectory inference analysis connected the immature N1S/LCN2 to mature N5S/S100A12 and N3S/NEAT1 states before a bifurcation into either the N2S/ISG or the N4S/G0S2 state was identified (Figure S3I). Finally, comparison of the blood neutrophil state frequencies between control and COPD patients did not reveal any statistically significant differences (Figure S3J).
When analyzing overall changes between COPD patients and controls for all neutrophil states together, interferon signaling was the only reduced pathway in COPD blood neutrophils, while several pathways were upregulated, including degranulation genes, RHO GTPase signaling, and ephrin signaling, which can promote inflammation (Figure 2H and Table S3). Studying individual neutrophil states (Figures 2I–2L and Table S3), alarmins (S100A8, S100A9, S100A12), which were recently linked to mucus hypersecretion, lung function decline, and alveolar destruction in COPD,27,28 were mainly upregulated in the N1S/LCN2 and N5S/S100A12 states. N1S/LCN2 neutrophils additionally had upregulated expression of resistin (encoded by RETN) (Figure 2I), which can inhibit bacterial clearance,29 and CD63. While overall interferon signaling was downregulated in COPD blood neutrophils (Figure 2H), IRF1, an important transcription factor for interferon response genes, was elevated in N2S/ISG neutrophils (Figure 2J). Notably, N2S/ISG, N3S/NEAT, and N5S/S100A12 neutrophils overexpressed genes that are involved in cell apoptosis (BNIP2, PDCD10). In contrast, the larger N3S/NEAT1 (Figure 2K) and N5S/S100A12 (Figure 2L) neutrophil states downregulated ISGs IFIT2 and IFITM2, indicating that neutrophil-state-specific regulation is apparent already in circulating blood neutrophils in COPD. Similarly surprising was the downregulation of CSF3R (Figure 2L), an important receptor for neutrophil trafficking to the lung, reactive oxygen species secretion, and degranulation,30,31,32 which may represent an attempt to control inflammation. SDCBP (coding for Syntenin-1), which has been linked to immune evasion and proangiogenic processes in cancer,33,34 was also elevated in these neutrophils. The apparent complexity of the transcriptional deviation in N5S/S100A12 neutrophils is further illustrated by the upregulation of CAP1, which on one hand is involved in apoptotic pathways as a response to cigarette smoking35; on the other hand, it may also serve as a receptor for aggravating inflammation.36
Together, while there are overall changes within the blood neutrophil compartment in COPD, in particular loss of interferon (IFN) signaling, all five neutrophil transcriptional states in the circulation show distinct deviations, which point toward a complex influence of neutrophils on disease development, progression, and dynamics of COPD, including infectious exacerbations.
Differential expression of surface molecules on blood neutrophil states in COPD
Currently, a limitation in several studies is the lack of validation of the findings of single-cell technologies in multiple cohorts to ensure reproducibility and interpretability. We therefore applied a combined single-cell transcriptome with targeted proteome analysis on an additional three control and three COPD patient cohort using the BD Rhapsody platform to define potential cell-surface markers for COPD-related neutrophil state deviation (Figures 3A and S4A–S4D and Table S4).
Figure 3.

Neutrophil transcriptional states from control and COPD patients correspond to distinct phenotypes
(A) Experimental design and analysis pipeline.
(B) UMAP representation of 4,072 neutrophils from three controls and three COPD patients.
(C) Heatmap of the top 5 marker genes for each neutrophil state. Each column represents the scaled average normalized expression per patient.
(D) Violin plots of neutrophil state-specific protein markers.
(E) Violin plots of differentially expressed protein markers between control and COPD patients for blood neutrophil states. Statistical analysis was performed with the MAST algorithm, ∗p < 0.05, ∗∗∗p < 0.001.
In total, 9,269 blood cells passed the filtering criteria and 13 clusters were annotated based on their cluster-specific gene expression (Figures S4A–S4C and Table S4). We detected 4,072 neutrophils that were further divided into five states (Figure 3B) and were detected in all studied patients (Figure S4D): N1R (LTF, LCN2, CAMP), N2R (ISG15, IFIT1, MX1), N3R (TNFAIP3, NFKBIA), N4R (S100P, S100A12, PADI4), and N5R (NEAT1, PHOSPHO1) (Figure 3C and Table S4). The N1R neutrophil state had enriched CD38 expression (p = 0.0002), consistent with a progenitor status,19 while N5R neutrophils expressed the highest levels of CD62L (p = 5.29e−93), a protein involved in neutrophil adhesion and extravasation (Figure 3D and Table S4).
The neutrophil population structure was similarly defined in both datasets (Seq-well and BD Rhapsody) when overlaying the top 20 genes for each cluster (Figures S4E–S4G). Importantly, when comparing the transcriptomic differences in the neutrophil states between control and COPD patients from the test (Seq-well) and validation (BD Rhapsody) cohorts, we observed similar patterns. These included the significant upregulation in COPD patients of cell movement-related VIM and the HLA molecule HLA-B in N3R neutrophils (Figure S4G and Table S4). Similarly, N4R neutrophils upregulated HLA-C and N5R neutrophils overexpressed alarmins (S100A12) (Figure S4G and Table S4).
The altered molecular phenotype of blood neutrophils in COPD was further supported by increase in cell-surface activation markers, such as the adhesion markers CD15 (p = 3.26e−8; N3R), CD66B (p = 0.026; N4R), and CD62L (p = 0.0007; N5R) (Figure 3E and Table S4). Collectively, molecular deviations observed for blood neutrophil states in early-stage COPD are mirrored by changes in the expression of surface proteins involved in neutrophil functions. Thus, in two independent cohorts, we identified subsets of blood neutrophils with unique gene expression patterns using two different scRNA-seq technologies.
BALF neutrophil states overlap with blood neutrophil deviations in COPD
As COPD is a pulmonary disease with systemic responses and co-morbidities, we next asked whether the changes observed in blood neutrophils are linked to changes in neutrophils from BALF, which would facilitate patient monitoring and stratification. We examined BALF from six controls and seven GOLD2 COPD patients using the Seq-well technology to define the cellular changes in early-stage COPD (Figure 4A). Dimensionality reduction and clustering revealed three BALF neutrophil clusters that were present in all donors and had comparable transcript counts (Figures 4B and S5A–S5C): N1bal (CXCL8, SOD2, TNFAIP6), N2bal (MNDA, SYNE2, SMCHD1), and N3bal neutrophils (FCGR3B, S100A8, S100A9, IFITM2) (Figure 4C and Table S5). N1bal neutrophils were enriched in interleukin signaling (Figure S5D), N2bal neutrophils featured genes related to influenza virus infection and translation (Figure S5E), whereas the N3bal neutrophil state was characterized by gene signatures of neutrophil degranulation, TLR cascades, and antigen presentation (Figure S5F). The frequencies of the three identified neutrophil states were comparable between control and COPD patients (Figure 4D). Pathway analysis on DE genes derived from all BALF neutrophils between control and COPD patients defined degranulation and TLR cascade signaling to be upregulated in COPD BALF neutrophils, while metabolism of amino acids, translation, signaling by RHO GTPases, and signaling via SLIT and Roundabout (ROBO) proteins was downregulated (Figure 4E).
Figure 4.

Bronchoalveolar neutrophils are transcriptionally heterogeneous in control and COPD patients
(A) Experimental design and analysis pipeline.
(B) UMAP representation of 1,203 neutrophils from the bronchoalveolar fluid (BALF) of six control and seven COPD patients.
(C) Heatmap of the top 5 marker genes for each BALF neutrophil state. Each column represents the scaled average normalized expression per patient.
(D) Bar plot of BALF neutrophil state frequencies in control and COPD patients.
(E) Gene set enrichment analysis of BALF neutrophil differentially expressed (DE) genes between control and COPD patients using the Reactome database.
(F–H) Dot plots of DE genes in (F) N1bal, (G) N2bal, and (H) N3bal BALF neutrophil states between control and COPD patients. Circle size represents the percentage of cells within a cluster that express a particular gene; circle color shows average gene expression within the cluster.
(I) Modified upset plot depicting the shared DE genes (COPD vs. control) between peripheral blood and BALF neutrophil states.
We next determined DE genes in BALF neutrophil states (Figures 4F–4H). N1bal had upregulated expression of the ATPase H+-transporting accessory protein 2 (ATP6AP2), which is involved in lysosomal acidification. We also identified several genes in N2bal and N3bal that were also differentially regulated in the blood of COPD patients (CSF3R, BNIP2, RETN, SERPINB1, APMAP, GYG1, ENO1, GABARAPL2, CD63, PDCD10, IRF1) (Figures 4G and 4H), demonstrating similarities in the neutrophil responses in COPD in the two compartments. This was further corroborated by the upregulation of SDCBP in all BALF neutrophil states in COPD patients and upon comparison of blood and BALF neutrophil cluster markers (Figures S5G and S5H).
When comparing DE genes in both compartments, we identified 75 genes similarly altered in blood and BALF from COPD patients (Figure 4I), although a clear one-to-one relationship between changes observed in any of the neutrophil states in blood and those seen in the three BALF neutrophil states could not be identified. To further examine the similarities in transcriptomic alterations between blood and BALF neutrophils, we next grouped genes found in a particular neutrophil state in blood to be also changed in at least one of the BALF neutrophil states in COPD. Our findings show that N1S/LCN2 blood neutrophils share DE genes between COPD and controls with BALF neutrophils that are involved in biological processes (slate blue color), such as eukaryotic translation (EIF4A2, EIF2B1, RPS29, RPL18A) and rRNA processing (DIMT1, RPS29, RPL18A). For blood N2S/ISG neutrophils (orange color), genes associated with processes such as TLR cascades (TLR2), previously associated only with COPD exacerbations37,38,39 and apoptosis (PDCD10), were DE between COPD and healthy controls, which we also identified in BALF neutrophils. The shared differences in gene expression in blood N3S/NEAT1 and BALF neutrophils (red color) were linked to RHO GTPases (ARHGAP3), lysosomal acidification (ATP6V1B2), and cytoskeleton (SDCBP). Finally, there was a larger group of DE genes in more than one blood neutrophil state which were also DE in BALF (magenta color). These genes were involved in degranulation (CD63, RAB10, DDX3X, SERPINB1), clathrin vesicle assembly and exosome formation (PICALM),40 antiviral responses (IFNAR1, IRF1), biosynthesis (APMAP), proteinase inhibition (SERPINB1), and cell apoptosis (BNIP2, CFLAR).
To link our findings to lung tissue-derived neutrophils, we defined a 21-surface-marker panel based on significant neutrophil state surface markers (Table S3), which was used for imaging mass cytometry (IMC) of lung biopsies from controls, smokers, and COPD patients (Figures S6A–S6D and Table S1). N1bal neutrophils were identified as CD44+CD74+, N2bal neutrophils as CD45+CD66A+CD16+CD44−CD74−CD62L−CD10−, and N3bal as CD62L+CD10+ cells. In line with previous findings from our laboratory,21 all neutrophil state proportions were increased in early-stage COPD patients compared with controls (p < 0.01) and smokers (p < 0.05) (Figure S6E), which was similarly true for macrophage abundance in COPD against controls (p = 0.005) and smokers (p = 0.02) (Figure S6F). Finally, CD4+ T cells were significantly higher in smokers and COPD patients compared with controls (p = 0.04; Figure S6F).
Collectively, in addition to the elevated levels of all neutrophil states in the lung of early-stage COPD patients, several of their transcriptional deviations are identical to those observed in blood neutrophils in early-stage COPD.
Murine smoke-induced COPD model reflects human neutrophil cell states in blood and BALF
As we observed molecular deviations in blood neutrophils from COPD patients, we hypothesized that neutrophil precursors might be already altered in early-stage COPD.41 Since bone marrow aspirates are not diagnostically indicated in COPD patients, we turned to an established murine model of cigarette smoke (CS) exposure that replicates the major features of human COPD.42,43,44 Blood (Figure 5), BALF (Figure S7), and bone marrow (Figures 6 and S8) were harvested from female BALB/c mice exposed to air (controls) or CS for 12 weeks (experimental COPD group) to investigate cellular composition via scRNA-seq with 10× Genomics technology.
Figure 5.

A murine model of cigarette smoke (CS)-induced COPD recapitulates the human blood neutrophil population structure
(A) Experimental design and sample processing.
(B) UMAP representation of 33,577 CD45+ cells from the blood of four air- and four CS-exposed mice.
(C) Dot plot of top 5 differentially expressed (DE) genes for each identified blood neutrophil cluster against the rest. Circle size represents the percentage of cells within a cluster that express a particular gene, circle color shows average gene expression within the cluster.
(D) UMAP representation of 10,181 blood neutrophils from four air- and four CS-exposed mice.
(E) Heatmap of the top 20 unique genes from the n1b murine blood neutrophil population from the Xie et al.15 data on the blood neutrophil states of this study.
(F) Heatmap of the top 20 unique murine blood neutrophil genes for the human neutrophil states from Figure 2. Murine genes were first converted to their human homologs.
(G) UMAP representation of 3,068 mature blood neutrophils from this study.
(H) Heatmap of the top 20 unique murine blood neutrophil state genes for the mature human neutrophil states from Figure 2. Murine genes were first converted to their human homologs.
(I) Gene set enrichment analysis of DE genes between air- and smoke-exposed mice in the blood n1b neutrophil state using the Reactome database.
Figure 6.

Smoke inhalation induces activation of neutrophil progenitors in the bone marrow
(A) Experimental design and sample processing.
(B) UMAP visualization of 57,966 bone marrow neutrophils from air- and cigarette smoke (CS)-exposed mice acquired with 38-parameter mass cytometry (CyTOF).
(C) Heatmap of selected marker expression in bone marrow neutrophil metaclusters. Each column represents the scaled average normalized expression per neutrophil metacluster.
(D) Heatmap of bone marrow neutrophil metacluster proportions in air- and CS-exposed animals.
(E) UMAP representation of 38,277 CD45+ cells from the bone marrow of four air- and four CS-exposed mice.
(F) Dot plot of top 5 differentially expressed (DE) genes for each identified bone marrow neutrophil cluster against the rest. Circle size represents the percentage of cells within a cluster that express a particular gene, and circle color shows average gene expression within the cluster.
(G) UMAP representation of 18,941 bone marrow neutrophils from four air- and four CS-exposed mice.
(H) Number of DE genes of all bone marrow neutrophil clusters between CS- and air-exposed mice.
(I) Pathway analysis of early granulocyte-monocyte progenitor (GMP) DE genes between air- and CS-exposed mice using the Reactome database.
(J) Venn diagram showing the overlap of murine bone marrow DE genes between air- and CS-exposed mice with human blood and BALF neutrophil states between control and COPD patients. Murine genes were first converted to their human homologs.
Of 33,577 CD45+ cells in murine blood (Figures 5A–5C), 10,181 were neutrophils and were separated into seven main (n1b–n7b) clusters (Figure 5D). Enrichment of murine blood neutrophil signatures from Xie et al.15 showed that n3b and n6b neutrophils contained common myeloid progenitors (CMPs) and granulocyte-monocyte progenitors (GMPs), n5b neutrophils were reminiscent of preneutrophils, n2b and n4b contained the previously described band cells, while n1b and n7b were fully mature neutrophils (Figure 5E).
To compare the cell states of murine and human blood neutrophils, we utilized the top 20 unique gene markers of the former and assessed their expression patterns on the latter (Figure 5F). We found that genes from the murine n2b–n6b clusters were similar to the N5S human neutrophil state. The n1b neutrophil signature was enriched in all mature human neutrophil states, whereas n7b neutrophils were reminiscent of the N4S/G0S2 and N5S/S100A12 states (Figure 5F). To increase the transcriptomic resolution in the mature neutrophil compartment, we further characterized the n1b murine cluster, which revealed four cellular states (S100, Il1b, Ctla2a, Isg) (Figure 5G). The top 20 unique gene markers reliably resembled the human structure; genes from the murine S100 neutrophil signature were enriched in human N5S/S100A12 neutrophils, Ccrl2 and Ctla2a-related signatures in the human N4S/G0S2 state, and the murine Isg neutrophils in the human N2S/ISG state (Figure 5H), supporting that the murine model is suitable for studying COPD-related neutrophil biology.
Similarly, we sorted live murine BALF cells to yield 18,406 cells (Figures S7A–S7C) and 7,064 neutrophils that were grouped in five clusters (Figure S7D and Table S6). The absolute BALF neutrophil counts were increased in smoke-exposed mice (Figure S7E), whereas the frequencies of BALF neutrophil states were comparable between air- and smoke-exposed animals (Figure S7F). Converting the top 20 unique murine BALF state gene markers to their human orthologs and overlaying on the respective human blood neutrophil transcriptomes showed that the n1bal and n2bal murine BALF states were enriched in the human N3bal, whereas the n4b state was enriched in N1bal (Figure S7G). Last, n5b neutrophils shared similarity with the N2bal neutrophils, while we could not clearly associate the murine n3b state, due to the low number of overlapping genes. Pathway analysis showed that the DE genes in neutrophil and BALF neutrophil states between air- and smoke-exposed animals resembled the differences observed in the human cohort, such that neutrophil degranulation, signaling by ROBO receptors, eukaryotic translation, nonsense-mediated decay, metabolism of amino acids, and signaling by RHO GTPases were similarly different in the murine model (Figures 5I and S7H).
The BALF neutrophil states were also reminiscent of the lung tissue neutrophil compartment in another murine model of smoke exposure45 that progressed from 2 to 6 months after the beginning of CS exposure. The three neutrophil clusters corresponded to the n1bal–n3bal and n4bal states of this study (Figures S7I and S7J). N4bal neutrophils have been previously defined as an aged granulocyte population in human peripheral blood and bone marrow, and they represent the terminal stage of the neutrophil development continuum.46 Comparison of the relative frequency of cluster 3 neutrophils at the 2, 4, and 6 month time points revealed a relative increase in this population over time (Figure S7K), which suggests that their accumulation might be linked to developing pathophysiology. This was additionally supported by ordering all neutrophils along pseudotime, confirming the time-dependent correlation of the aged neutrophil state (Figure S7L) and revealing a gene signature, involving tissue-degrading enzymes (Ctsb, Ctsd) and granulocyte activation markers (Ier3, Cd63, Cd24), at progressing disease stages (Figure S7M). Taken together, our results show that the CS-exposure model reliably reflected the disease pathophysiology observed in the two compartments, namely blood and BALF, of COPD patients.
Chronic CS exposure to induce experimental COPD causes bone marrow granulopoiesis and transcriptional reprogramming
Having established the molecular alterations in murine blood neutrophils, we next addressed the alterations in the neutrophil precursor compartment in the murine bone marrow (Figure 6A). In mass cytometry data (Figures 6B–6D and S8A–S8D), we identified relative increases in CD34+CD117+CD16/32+IdU+ progenitors, CD117+CD11b+ preneutrophils, and CD11b+Ly-6G+CD117−CD62L−CD172a− immature neutrophils in smoke-exposed animals, while CD11b+Ly-6G+CD117−CD62L+CD172a+ mature neutrophils were relatively decreased (Figure 6D).
To determine quantitative changes within the neutrophil precursor compartment in bone marrow, we analyzed 38,277 single-cell transcriptomes (Figures 6E and 6F). By in silico sorting of neutrophil and precursor clusters, we obtained 18,941 cells that were further clustered into eight clusters (n1bm–n8bm) (Figure 6G and Table S6). These clusters resembled previously suggested developmental stages,47 with n6bm and n8bm neutrophils expressing a mixture of multipotent progenitor (MPP) and CMP signatures, n1bm and n2bm cells equivalent to early and late GMPs, n5bm cells corresponding to promyelocytes, n4bm reminiscent of metamyelocytes, n3bm carrying signatures of band cells, and n7bm neutrophils resembling mature peripheral neutrophils (Figure S8E).
DE gene analysis between air- and CS-exposed animals revealed that the transcriptional reprogramming upon CS exposure was most pronounced in early GMPs (746 genes), followed by mature neutrophils 2 (151 genes), mature neutrophils 1 (58 genes), and promyelocytes (56 genes) (Figure 6H and Table S6), strongly supporting smoke-induced deviation of granulopoiesis. Similar to human blood neutrophils from COPD, early GMPs in CS-exposed animals had enriched expression of neutrophil degranulation, signaling by RHO GTPases, and interleukin signaling-related genes (Figure 6I), indicating that reprogramming of the neutrophil population is triggered by changes in the precursor compartment.
Interestingly, comparison of bone marrow-derived early GMP DE genes between air- and smoke-exposed mice with those of human blood (Figure 2) and BALF (Figure 4) neutrophil states between control and COPD patients identified 9 shared genes between all compartments, 43 between bone marrow and blood, and 63 between bone marrow and BALF (Figure 6J and Table S6), indicating that the neutrophil transcriptomic reprogramming observed in blood partly occurs already at immature progenitor states in the bone marrow. In fact, neutrophils from all three compartments regulate the expression of genes that are involved in cellular metabolism (GYG1, ATP6AP2, APMAP, ENO1), intracellular trafficking (GABARAPL2), apoptosis (BNIP2, PDCD10), and cell activation (CD63, SERPINB1) (Figure S8F). Taken together, these results show that bone marrow neutrophil populations in a murine experimental COPD model undergo population size and transcriptomic changes that functionally include increased myelopoiesis, degranulation, and cytokine signaling.
Blood neutrophil state signature changes associated with alveolar destruction and lung function decline
To test whether the transcriptional reprogramming observed in the murine neutrophil progenitor compartment is carried over to the blood compartment, we directly assessed the enrichment of the conserved nine-gene signature altered in murine bone marrow progenitors on human peripheral neutrophil states. We found that, in addition to the human N1S/LCN2 (p = 0.015) neutrophil state, the gene signature was also significantly enriched in the N4S/G0S2 (p = 0.028) and N5S/S100A12 (p = 0.031) states from COPD patients (Figure 7A), further supporting that neutrophil cell state analysis in human blood is indicative of changes occurring in the bone marrow.
Figure 7.

Abundance of neutrophil states associates with COPD clinical traits
(A) Enrichment analysis of the conserved nine-gene differentially regulated gene signature in murine bone marrow granulocyte-monocyte progenitors (GMPs) on human neutrophil states from control and COPD patients.
(B) Sample collection and processing pipeline.
(C) Principal-component analysis of 17,127 present genes in the dataset for blood neutrophils from 10 control and 15 COPD patients.
(D) In silico deconvolution of 10 control and 15 COPD sorted neutrophil whole-transcriptome samples with single-cell signatures from the study cohort. Data analysis was carried out with a two-tailed t test (N5S/S100A12, N3S/NEAT1) or a Wilcoxon signed rank test (N1S/LCN2, N2S/ISG, N4S/G0S2) for non-normally distributed data.
(E) Correlation analysis of neutrophil state (N1S/LCN2, N2S/ISG, N3S/NEAT1, N4S/G0S2, N5S/S100A12) abundance with emphysema, % FEV1, FEV1/FVC ratio, and number of exacerbations using mixed models adjusted for age, sex, and inhaled corticosteroid treatment. Filled boxes indicate significant (p < 0.05) correlation.
(F) In silico deconvolution of 44 granulocyte whole-transcriptome samples from COVID-19 patients from EGAS00001004503 with single-cell signatures from the study cohort.
(G) Correlation analysis of neutrophil state (N2S/ISG, N3S/NEAT1, N5S/S100A12) abundance with severity and maximum WHO score using the Spearman test.
(H) In silico deconvolution of 40 whole-transcriptome samples from sepsis patients from GSE63042 with single-cell signatures from the study cohort.
(I) Correlation analysis of neutrophil state (N1S/LCN2, N2S/ISG, N3S/NEAT1 N5S/S100A12) abundance with sepsis outcome, respiratory rate, and serum creatinine using the Spearman test. Filled boxes indicate significant (p < 0.05) correlation.
Finally, we asked whether transcriptome changes in blood neutrophils are associated with clinical parameters used to diagnose disease severity in COPD. We combined cell sorting of blood CD66b+CD16+ neutrophils from an additional cohort of 10 controls and 14 COPD patients with bulk transcriptomics and subsequently computationally deconvoluted the data using cell state information from our scRNA-seq data (Figure 7B).48 Principal-component analysis using all 17,127 present genes separated controls from COPD samples (Figure 7C). In agreement with the scRNA-seq data, the N3S/NEAT1 and N5S/S100A12 states were the most prominent, with N2S/ISG and N4S/G0S2 neutrophils following in decreasing order of abundance (Figure 7D). Notably, the N1S/LCN2 neutrophil state was negligible. There was a relative decrease in N4S/G0S2 (p < 0.01) and N5S/S100A12 (p = 0.052) neutrophil states in COPD, partly compensated for by a small increase in the abundance of N2S/ISG neutrophils (p = 0.09; Figure 7D).
We next calculated the correlation between the relative abundance of neutrophil states and the critical clinical parameters of lung function to test whether certain neutrophil states may present with a biomarker potential for early-stage COPD in the clinics (Figure 7E). The N2S/ISG state negatively correlated with % FEV1 (p = 0.002) and the FEV1/FVC ratio (p < 0.001), while the N4S/G0S2 and the N5S/S100A12 states positively correlated with FEV1 (p = 0.005, p = 0.021) and the FEV1/FVC ratio (p = 0.023, p = 0.026) as a surrogate for lung function, respectively.
To determine whether the observed deviation of neutrophil states is a general hallmark of inflammatory conditions, we performed the same analysis in cohorts of granulocyte transcriptome samples from 44 COVID-1949 and whole-blood transcriptome samples from 40 sepsis patients50 (Figures 7F–7I). In the COVID-19 cohort, computational deconvolution revealed only the N2S/ISG, N3S/NEAT1, and N5S/S100A12 neutrophil states, which we found increased in severe COVID-19, particularly at later time points (Figure 7G). In contrast to COPD, the N5S/S100A12 state positively correlated with severity (severity p = 2.721e−09, WHO max score p = 5.121e−06), while the N2S/ISG state negatively correlated (severity p = 0.000123, WHO max score p = 0.033), clearly following a different pattern of deviations in this acute systemic inflammatory condition (Figure 7G). In sepsis, computational deconvolution revealed a relative elevation of N5S/S100A12 in samples derived from patients with severe sepsis, with septic shock, and who died (Figure 7H; sepsis death). Here, the N3S/NEAT1 state correlated with respiratory rate as a surrogate for low oxygen saturation and disease severity (Figure 7I; p = 0.048). These findings further support the notion that deviations in the systemic neutrophil compartment do not merely reflect a general inflammation signature, but show disease-associated changes.
For COPD, the changes in N2S/ISG, N4S/G0S2, and N5S/S100A12 neutrophils might serve as biomarkers associated with disease progression. The anti-correlation of N2S/ISG neutrophils with lung function links them to a cardinal manifestation of COPD,1 which may be mediated via an intermediate step of T cell activation by IFNs.51 On the other hand, the more prominent N4S/G0S2 and N5S/S100A12 neutrophils in patients with better lung function suggest that neutrophil endogenous programs (alarmin production, degranulation, interleukin signaling, G-protein-coupled signaling) are activated early on in COPD as a natural response to the ongoing inflammatory triggers; however, they are proportionally decreased compared with the other neutrophil states in more severe disease.
Discussion
The present study demonstrates that a systemic inflammatory response is already occurring in early-stage COPD, which involves reprogramming of the neutrophil compartment in the bone marrow, peripheral blood, and BALF. Findings in blood were validated in independent patient groups, as well as with bulk RNA-seq, providing further evidence that systemic alterations in neutrophils are already present in early-stage COPD. While COPD-related changes in blood and BALF neutrophils, such as degranulation, RHO GTPase signaling, and translation, overlapped, we observed an unexpectedly complex relationship between cellular states in blood and BALF, even in control samples, suggesting that the biology of neutrophils in these two compartments differs significantly, as also seen by others.52 As bone marrow is not part of routine diagnostics in COPD, we turned to a model of nose-only CS-induced experimental COPD that is representative of the mode of exposure in humans and develops hallmark features of disease similar to those in COPD patients.44 We observed similar changes in neutrophils in both the blood and the BALF compartments. In addition, we further elucidated the transcriptional reprogramming within the bone marrow compartment with the majority of changes occurring in early GMPs. Indeed, we identified a gene set that is changed in all three compartments, further supporting that altered transcription of neutrophil subsets in COPD is induced at the progenitor stage. Finally, we linked changes in the neutrophil compartment to clinical severity parameters in COPD and determined that the abundance of the N2S/ISG, N4S/G0S2, and N5S/S100A12 neutrophil states correlated with lung function loss as typical clinical phenotypes linked to disease severity. This was not a general observation for inflammatory conditions, as different neutrophil states were related to clinical severity in COVID-19 and sepsis patients. We conclude from these changes that the assessment of reprogramming within the neutrophil compartment warrants further assessment as a predictor of COPD pathophysiology and progression.
We establish that the neutrophil compartment in peripheral blood is clinically relevant, with cell-type frequency, molecular, and phenotypic changes associated with clinical parameters in COPD patients. Blood neutrophil counts are elevated in a disease severity-associated manner in COPD, and certain molecular states (ISG, S100A12) within the neutrophil compartment defined by single-cell transcriptomics show unique activation patterns in this disease, including upregulation of S100 molecules and exosome formation machinery, with certain IFN-induced genes being reduced. In particular, the abundance of the N2S/ISG neutrophil state was associated with lung function loss, a major hallmark of disease severity. As demonstrated in our murine model system, changes in blood neutrophils are evoked by the major cause of COPD in humans, namely smoking, and are directly linked to altered granulopoiesis in the bone marrow that leads to changes in blood and airway neutrophils, most likely as a consequence of signals derived from continuous challenge and damage to the lung. Collectively, we propose that specific systemic cellular responses to environmental damage of the lungs in the context of COPD lead to elevated granulopoiesis in the bone marrow.
In agreement with previous work,14,16,19,20,23,53,54,55 we identified five transcriptomic states along a developmental continuum of neutrophils, starting from LCN2 progenitors to intermediate states (N3S/NEAT1, N5S/S100A12) and two endpoints (N2S/ISG and N4S/G0S2 neutrophils). Our findings suggest that, in control individuals and in COPD patients with adequate lung function, the egress of bone marrow-derived LCN2 progenitors to the circulation is rather limited, and the trajectory of neutrophil maturation is arrested at the N5S/S100A12 state. In COPD patients with poor lung function, however, there seems to be an active mobilization of neutrophil progenitors, causing a shift from the N5S/S100A12 intermediate state to the two terminal branches. This was corroborated by in silico neutrophil population deconvolution analysis where a relative decrease in N5S/S100A12 neutrophil frequency was accompanied by a relative increase in the N2S/ISG state. Interestingly, the blood neutrophil structure is conserved between control subjects and COPD patients, suggesting that the alterations in neutrophil molecular phenotypes in COPD are mainly qualitative at the transcriptomic level. The LCN2 progenitor and the intermediate N5S/S100A12 neutrophil state were characterized by cellular activation and overexpressed anti-microbial and antigen-presentation genes (alarmins, cell polarization, HLA class I). The abundance of three of the molecular states, namely N2S/ISG, N4S/G0S2, and N5S/S100A12 neutrophils, correlated with key COPD manifestations (lung function loss), which was not the case for more acute inflammatory conditions such as sepsis,50 strongly suggesting that these COPD-related alterations are not simply a general sign of inflammation. We speculate that the increased presence of N2S/ISG neutrophils contributes to emphysema, possibly upon interactions with T cells, as has been shown in experimental lung cancer models where anti-tumor neutrophils expressed similar anti-viral gene signatures.56 On the other hand, the decreased frequency of N5S/S100A12 neutrophils may be beneficial for lung function, as this molecular state expresses inflammatory genes, including alarmins, proteases, pathogen recognition molecules, and proinflammatory cytokines.
We also linked findings in blood to tissue-related neutrophils by analyzing BALF samples, which revealed three major neutrophil states. We demonstrated the elevated cell numbers of all three molecular neutrophil states in lung samples of COPD patients via IMC. Interestingly, there was no direct link between the blood neutrophil states and those observed in BALF, indicating a complex integration of microenvironmental signals shaping cell-state structures of neutrophils in these two compartments. Our findings agree with recent work from Ballesteros et al., who showed that neutrophils display distinct tissue-specific transcriptional and chromatin profiles, whereas transition from the blood to the lung induces additional chromatin modifications.52 Despite the difficulty in directly linking neutrophil states in blood and BALF, many of the changes between COPD and controls were observed in both compartments, including genes coding for degranulation, exosome formation, infiltration in tissues, and pattern recognition receptors.
Transcriptomic changes in blood neutrophil states were most likely explained by alterations in granulopoiesis,41 which we assessed in the bone marrow of the murine model of smoke exposure. Deviations in transcription in neutrophils in human blood and in BALF neutrophils clearly showed overlaps with findings in the precursor compartment in the murine smoking model. As a sign of elevated granulopoiesis, we observed increases in neutrophil-committed populations in smoke-exposed animals. Further, these progenitors showed transcriptional alterations, including genes involved in neutrophil degranulation, interleukin signaling, and antigen presentation. Smoke-related signatures derived from these GMPs were significantly enriched in human blood neutrophil progenitors, as well as the N4S/G0S2 and N5S/S100A12 neutrophil states.
Limitations of the study
While our results from the three compartments in the murine smoking model strongly support that the changes observed in COPD are linked to chronic exposure to CS, it is important to highlight the fact that lung inflammation in COPD patients may have alternative causes, e.g., genetic predisposition, early-life events and infections, air pollution, and exposure to occupational gasses.57 As such, it cannot be ruled out that chronic lung inflammation may feedback to the bone marrow and alter granulopoiesis. The molecular changes observed in the bone marrow, blood, and BALF neutrophils upon smoke exposure point toward a complex feedback loop between chronic challenge, lung damage, and induction of granulopoiesis, for which we did not yet find a single factor or pathway responsible. Most likely, due to the complex environmental stresses involved, several mechanisms are acting simultaneously. In this respect, potential effects of active therapeutic treatments of COPD patients on the neutrophil transcriptomic changes in peripheral blood and BALF of COPD patients cannot be fully excluded. Clearly, further studies are required to determine the feedback loops between the lung and the hematopoietic system occurring in our mouse model and in COPD patients already at early stages of the disease.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD3 PE/Cy7 (UCHT1) | Biolegend | Cat# 300420; AB_439781 |
| CD3 Alexa Fluor 647 (UCHT1) | BD Biosciences | Cat# 557706; AB_396815 |
| CD4 PerCP/Cy5.5 (RPA-T4) | Biolegend | Cat# 300530; AB_893322 |
| CD8 BV650 (SK1) | Biolegend | Cat# 344730; AB_2564510 |
| CD11c FITC (3.9) | Biolegend | Cat# 301604; AB_314174 |
| CD14 APC/Cy7 (HCD14) | Biolegend | Cat# 325620; AB_830693 |
| CD14 FITC (M5E2) | BD Biosciences | Cat# 555397; AB_395798 |
| CD16 BV605 (3G8) | Biolegend | Cat# 302039; AB_2561354 |
| CD19 APC/Cy7 (HIB19) | BD Biosciences | Cat# 302218; AB_314248 |
| CD19 APC (HIB19) | Biolegend | Cat# 302212; AB_314242 |
| CD33 FITC (HIM3-4) | BD Biosciences | Cat# 555626; AB_395992 |
| CD45 BV711 (HI30) | Biolegend | Cat# 304050; AB_2563466 |
| CD45 APC/Cy7 (30-F11) | BD Biosciences | Cat#557659; AB_396774 |
| CD56 Alexa Fluor 647 (B159) | BD Biosciences | Cat# 557711; AB_396820 |
| CD56 BV510 (NCAM16.2) | BD Biosciences | Cat# 563041; AB_2732786 |
| CD66b FITC (G10F5) | Biolegend | Cat# 305104; AB_314496 |
| CD123 FITC (6H6) | Biolegend | Cat# 306006; AB_314580 |
| CD127 PE (HIL-7R-M21) | BD Biosciences | Cat# 557938; AB_2296056 |
| HLA-DR PerCP/Cy5.5 (L243) | Biolegend | Cat# 307630; AB_893567 |
| Siglec-8 PE/Cy7 (7C9) | Biolegend | Cat# 347111; AB_2629719 |
| CD1C (F10/21A3) | BD Biosciences | Cat# 940083; AB_2875974 |
| CD2 (RPA-2.10) | BD Biosciences | Cat# 940046; AB_2875937 |
| CD3 (SK7) | BD Biosciences | Cat# 940000; AB_2875891 |
| CD4 (SK3) | BD Biosciences | Cat# 940001; AB_2875892 |
| CD5 (UCHT2) | BD Biosciences | Cat# 940038; AB_2875929 |
| CD8 (RPA-T8) | BD Biosciences | Cat# 940003; AB_2875894 |
| CD10 (HI10a) | BD Biosciences | Cat# 940045; AB_2875936 |
| CD11B (M1/70) | BD Biosciences | Cat# 940008; AB_2875899 |
| CD11C (B-ly6) | BD Biosciences | Cat# 940024; AB_2875915 |
| CD14 (MфP9) | BD Biosciences | Cat# 940005; AB_2875896 |
| CD15 (W6D3) | BD Biosciences | Cat# 940274; AB_2876152 |
| CD16 (3G8) | BD Biosciences | Cat# 940006; AB_2875897 |
| CD19 (SJ25C1) | BD Biosciences | Cat# 940004; AB_2875895 |
| CD20 (2H7) | BD Biosciences | Cat# 940016; AB_2875907 |
| CD21 (B-ly4) | BD Biosciences | Cat# 940048; AB_2875939 |
| CD24 (ML5) | BD Biosciences | Cat# 940028; AB_2875919 |
| CD25 (2A3) | BD Biosciences | Cat# 940009; AB_2875900 |
| CD27 (M-T271) | BD Biosciences | Cat# 940018; AB_2875909 |
| CD32 (FLI8.26) | BD Biosciences | Cat# 940069; AB_2875960 |
| CD34 (581) | BD Biosciences | Cat# 940021; AB_2875912 |
| CD38 (HIT2) | BD Biosciences | Cat# 940013; AB_2875904 |
| CD44 (G44-26) | BD Biosciences | Cat# 940039; AB_2875930 |
| CD45 (HI30) | BD Biosciences | Cat# 940002; AB_2875893 |
| CD45RA (HI100) | BD Biosciences | Cat# 940011; AB_2875893 |
| CD49D (9F10) | BD Biosciences | Cat# 940059; AB_2875950 |
| CD56 (NCAM16.2) | BD Biosciences | Cat# 940007; AB_2875898 |
| CD62L (DREG-56) | BD Biosciences | Cat# 940041; AB_2875932 |
| CD66 (B1.1/CD66) | BD Biosciences | Cat# 940088; AB_2875979 |
| CD90 (5E10) | BD Biosciences | Cat# 940032; AB_2875923 |
| CD94 (HP-3D9) | BD Biosciences | Cat# 940081; AB_2875972 |
| CD117 (YB5.B8) | BD Biosciences | Cat# 940051; AB_2875942 |
| CD123 (7G3) | BD Biosciences | Cat# 940020; AB_2875911 |
| CD127 (HIL-7R-M21) | BD Biosciences | Cat# 940012; AB_2875903 |
| CD141 (1A4) | BD Biosciences | Cat# 940079; AB_2875970 |
| CD161 (DX12) | BD Biosciences | Cat# 940070; AB_2875961 |
| CD183 (1C6/CXCR3) | BD Biosciences | Cat# 940030; AB_2875921 |
| CD184 (12G5) | BD Biosciences | Cat# 940056; AB_2875947 |
| CD197 (3D12) | BD Biosciences | Cat# 940014; AB_2875905 |
| CD294 (BM16) | BD Biosciences | Cat# 940098; AB_2875989 |
| CD314 (1D11) | BD Biosciences | Cat# 940061; AB_2875952 |
| CD335 (9E2/NKp46) | BD Biosciences | Cat# 940064; AB_2875955 |
| CD336 (p44-8) | BD Biosciences | Cat# 940085; AB_2875976 |
| TCRαβ (IP26) | BD Biosciences | Cat# 940074; AB_2875965 |
| TCRγδ (B1) | BD Biosciences | Cat# 940057; AB_2875948 |
| IgD (IA6-2) | BD Biosciences | Cat# 940026; AB_2875917 |
| CD3 (polyclonal) | Dako | Cat# GA503 |
| CD4 (EPR19514) | Abcam | Cat# ab183685; AB_2686917 |
| CD8 (D8A8Y) | Cellsignal | Cat# 85336 |
| CD10 (HI10A) | Biolegend | Cat# 312202; AB_314913 |
| CD11b (EPR1344) | Abcam | Cat# 133357; AB_2650514 |
| CD11c (EP1374Y) | Abcam | Cat# ab52632; AB_2129793 |
| CD14 (EPR3653) | Abcam | Cat# ab133335; AB_2889158 |
| CD16a (EPR16784) | Biolegend | Cat# 302004; AB_314204 |
| CD16b (G10F5) | Biolegend | Cat# 305102; AB_314494 |
| CD20 (H1) | BD Biosciences | Cat# 347670; AB_400337 |
| CD44 (BJ18) | Biolegend | Cat# 338802; AB_1501199 |
| CD45 (D9M8I) | Cellsignal | Cat# 13917 |
| CD56 (EPR2566) | Abcam | Cat# ab133345 |
| CD62L (polyclonal) | Abcam | Cat# ab264045 |
| CD66a (B1.1/CD66) | BD Biosciences | Cat# 551354; AB_394166 |
| CD68 (KP1) | Abcam | Cat# ab955; AB_307338 |
| CD74 (PIN.1) | Abcam | Cat# ab22603; AB_447187 |
| CCR1 (polyclonal) | Abcam | Cat# ab139399 |
| EPCAM (VU-1D9) | Stemcell | Cat# 60136 |
| HLA-DR (EPR3692) | Abcam | Cat# ab92511; AB_10563656 |
| Siglec-8 (polyclonal) | Abcam | Cat# ab198690 |
| Chemicals, peptides, and recombinant proteins | ||
| AMPure beads | Beckman Coulter | Cat# A63881 |
| Dulbecco’s Phosphate Buffered Saline | Sigma-Aldrich | Cat# D8537 |
| EDTA | Merck | Cat# EX0546A |
| Fetal Bovine Serum | PAN Biotech | Cat# 3302 |
| FcR Blocking Reagent, human | Miltenyi Biotec | Cat# 130-059-901 |
| Pancoll human; density: 1.077 g/ml | PAN Biotech | Cat# P04601000 |
| SPRIselect | Beckman Coulter | Cat# B23318 |
| Stain buffer (FBS) | BD Pharmigen | Cat# 554656 |
| Critical commercial assays | ||
| BD Rhapsody cartridge kit | BD Biosciences | Cat# 633733 |
| BD Rhapsody cDNA kit | BD Biosciences | Cat# 633773 |
| BD Rhapsody WTA Amplification kit | BD Biosciences | Cat# 633801 |
| BD Rhapsody Human Single-Cell Multiplexing kit | BD Biosciences | Cat# 633781 |
| Chromium Next GEM Single Cell 3’ GEM, Library and Gel Bead Kit v3.1 | 10x Genomics | Cat# 1000121 |
| Chromium Next Gem Chip G Single Cell kit | 10x Genomics | Cat# 1000120 |
| High Sensitivity DNA assay | Agilent | Cat# 5067-4626 |
| High Sensitivity D5000 ScreenTape | Agilent | Cat# 5067-5592 |
| High Sensitivity RNA assay | Agilent | Cat# |
| High Sensitivity ScreenTape | Agilent | Cat# 5067-5579 |
| Human Single-Cell Multiplexing kit | BD Biosciences | Cat# 633781 |
| LIVE/DEAD Fixable Yellow Dead Cell Stain kit | ThermoFisher | Cat# L34967 |
| miRNeasy Micro kit | Qiagen | Cat# 217084 |
| Nextera XT DNA Library Preparation kit | Illumina | Cat# FC-131-1096 |
| NextSeq 500 High Output Kit v2.5 (150 cycles) | Illumina | Cat# 2002497 |
| NovaSeq 6000 S1 Reagent kit (100 cycles) | Illumina | Cat# 20012865 |
| NovaSeq 6000 S2 Reagent kit (100 cycles) | Illumina | Cat# 20012862 |
| Qubit dsDNA HS Assay kit | ThermoFisher | Cat# Q32854 |
| Single Index Kit T Set A | 10x Genomics | Cat# 1000213 |
| Deposited data | ||
| scRNA-seq raw data | This paper | EGAS000001006281; EGAS000001006322; EGAS000001006323; GSE205078 |
| Software and algorithms | ||
| AUCell | 58 | v1.4.1 |
| Bioconductor | https://bioconductor.org | v3.12 |
| bioMart | 59 | v2.5.1 |
| car | 60 | v3.0-3 |
| Cell profiler | https://cellprofiler.org | v4.1.3 |
| CIBERSORTx | https://cibersortx.stanford.edu | N/A |
| clusterProfiler | 61 | v3.14.3 |
| Cutadapt | 62 | v1.16 |
| Cytoscape | https://cytoscape.org | v3.7.2 |
| DESeq2 | 63 | v1.10.1 |
| DoubletFinder | 64 | v2.0.3 |
| Dropseq-tools | https://github.com/broadinstitute/Drop-seq/releases | v2.0.0 |
| fgsea | 65 | v1.16.0 |
| FlowJo | https:/flowjo.com | v10 |
| gam | https://cran.r-project.org/web/packages/gam/index.html | v1.16.0 |
| ggplot2 | 66 | v3.2.1 |
| Harmony | 67 | v1.0 |
| Histocat++ | 68 | N/A |
| limma | 69 | v3.48.1f |
| MAST | 70 | v1.20 |
| Python | https://python.org | v3.8.5 |
| pheatmap | 71 | v1.0.12 |
| R | https://r-project.org | v3.6.1; v4.0.3 |
| Rphenograph | 72 | v0.99.1 |
| Seurat | 73 | v3.2.2 |
| Slingshot | 74 | v1.4.0 |
| SoupX | 75 | v1.5.0 |
| scanpy | 76 | v1.8.0 |
| STAR | 77 | v2.6.1b |
| TXimport | 78 | v1.2.0 |
| UpsetR | 79 | v1.4.0 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Joachim L. Schultze (j.schultze@uni-bonn.de).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals
Seven-to-eight-week-old female BALB/c mice were purchased from the Australian BioResource Facility (Moss Vale, NSW, Australia). Mice were exposed to normal air or cigarette smoke through custom-designed and purpose-built nose-only exposure system (CH technologies, USA). The smoke of up to 12 3R4F reference cigarettes was administered twice per day, 5 days a week for 12 weeks as previously described.42,43,80 All experiments were approved by the institute’s animal ethics committee (protocol number: 2018–030).
Human specimens
Human studies were approved by the ethics committees of the University of Bonn and University hospital Bonn (local ethics vote 076/16). All patients provided written informed consent according to the Declaration of Helsinki and were aged at least 24 years old (Table S1). They were recruited over a period of 41 months from the Department of Pneumology, were diagnosed with COPD if the FEV1/FVC ratio was less than 0.7 and were stratified in grades according to the guidelines of the global initiative for chronic obstructive lung disease (GOLD1-4).81 Current smokers had smoked in the last 3 months, ex-smokers had not smoked in the last 3 months prior to bronchoscopy and never smokers had not smoked more than 100 cigarettes in their lifetime and did not smoke at the time of recruitment. Age-matched individuals suffering from chronic idiopathic cough and an exquisitely sensitive cough reflex without underlying pathology and served as control donors.82 A diagnostic algorithm that considered medical history, including medication, physical examination, echocardiography, chest X-ray, lung function, presentation at an otolaryngologist, blood test, FeNO (excluding >50 ppb), computer tomography of the chest and bronchoscopy,83 was used for enrolment. Exclusion criteria included hypoxemia despite oxygen supplementation, hypercapnia, increased risk of bleeding, unstable cardiac disease, COPD exacerbations in the 4 weeks prior to recruitment and other pulmonary diseases, including asthma, asthma and COPD overlap, bronchiectasis, cancer, fibrosis, pneumonia and sarcoidosis. Percentage of emphysema in the upper and lower compartments of both lungs was determined radiologically. A summary of the patient clinical parameters of this study is shown in Table S1. BALF samples which satisfied the above quality criteria and their paired peripheral blood samples were processed further in single-cell analysis.
Method details
Whole blood processing
Peripheral blood was drawn in plastic tubes containing 3.2% citrate (Sarstedt) and was processed within 1 h of collection. Samples were diluted 1:1 with PBS (Sigma-Aldrich) at room temperature and carefully overlaid on 15 ml room temperature Pancoll (PAN Biotech). Gradients were separated by centrifugation at 700xg for 25 min at room temperature with no break. Subsequently, plasma was collected, and aliquots were frozen at -80oC. Peripheral blood mononuclear cells were carefully transferred to a new tube in FACS buffer (PBS/2% FBS/1 mM EDTA) and the granulocyte fraction was treated with erythrocyte lysis buffer (ELB) (15 min at 4oC, 150 mM NH4Cl, 10 mM KHCO3 and 100 uM EDTA in H2O, pH 7.4) to preferentially lyse erythrocytes. Cells were centrifuged at 300xg for 8 min at 4oC and washed twice with 20 ml FACS buffer.
Differential cell counts
Blood leukocyte counts were routinely determined in the clinic hematology devices (XN9000/XN1000; Sysmex Europe) according to the manufacturer’s instructions within 15 min of collection.
Murine sample collection
Animals were euthanized by intraperitoneal injection of 200 ul sodium pentobarbitone (65 mg/ml) and blood was collected by cardiac puncture in EDTA-treated tubes. Whole blood (400-500 ul) was diluted 1:40 with red blood cell (RBC) lysis buffer (155 mM NH4Cl, 12 mM NaHCO3, 0,1 uM EDTA) and incubated for 10 min on ice before a centrifugation (132xg, 5 min, 4oC). Cells were resuspended in FACS buffer (PBS/2%FBS/2mM EDTA) and stained with CD45 (30-F11; BD Biosciences, 30 min on ice) followed by live/dead staining with 250 ng/ml DAPI solution. Cells were filtered with 70 um filters (In Vitro Technologies, Australia) and 20,000 cells per animal were sorted in a BD Aria II instrument (Garvan Institute, NSW, Australia).
Bone marrow was collected by flushing murine femora and tibiae with RPMI media. Cells were pelleted (150xg, 5 min, 4oC) and resuspended in RBC lysis buffer (5 min on ice). After one more centrifugation, cells were resuspended again in FACS buffer and stained with CD45 (BD Biosciences, 30 min on ice) followed by live/dead staining. Cell suspensions were finally washed, filtered and 20,000 cells per animal were sorted.
BALF was obtained from the left lung lobe by two washes with 0.5 ml PBS via a cannula inserted into the trachea. BALF cells were pelleted (150xg, 5 min, 4oC) and resuspended in RBC buffer for 5 min on ice. Differential counts were carried out onto microscopy slides upon cytocentrifugation (300 x g, 7 min, 4oC). The slides were stained with May-Grunwald-Giemsa and cell counts were enumerated according to morphological criteria using a light microscope. Cells were centrifuged again and resuspended in FACS buffer followed by live/dead staining. Cell suspensions were washed, filtered and 20,000 live cells from each animal were sorted. For all compartments, 5,000 sorted cells from each mouse were pooled together for loading on a Chromium chip (10x Genomics) which was the available technology in Hansbro lab (Sydney, Australia).
Human flow cytometry/sorting
Single-cell suspensions were stained with Live/Dead yellow fluorescent dye (1:1,000, ThermoFisher) for 15 min at room temperature and washed with FACS buffer at 300xg for 5 min at 4oC. Blocking followed with 5 ul human FcR blocking reagent (Miltenyi Biotec) per 100 ul cell suspension for 15 min on ice and staining for blood immune cells was performed with the following anti-human antibodies for 30 min on ice: CD3 (1:54; UCHT1; Biolegend), CD3 (1:27; UCHT1; BD Biosciences), CD4 (1:54; RPA-T4; Biolegend), CD8 (1:54; SK1; Biolegend), CD11c (1:32; 3.9; Biolegend), CD14 (1:68; HCD14; Biolegend), CD14 (1:32; M5E2; BD Biosciences), CD16 (1:68; 3G8; Biolegend), CD19 (1:45; HIB19; BD Biosciences), CD19 (1:32; HIB19; Biolegend), CD33 (1:54; HIM3-4; BD Biosciences), CD45 (1:45; HI30; Biolegend), CD56 (1:27; B159; BD Biosciences), CD56 (1:32; NCAM16.2; BD Biosciences), CD66b (1:54; G10F5; Biolegend), CD123 (1:32; 6H6; Biolegend), CD127 (1:32; HIL-7R-M21; BD Biosciences), HLA-DR (1:68; L243; Biolegend) and Siglec-8 (1:21; 7C9; Biolegend). Cells were centrifuged at 300xg for 5 min at 4oC and resuspended in 1 ml FACS buffer. Data acquisition was performed on a 3 laser-FACS Aria III cell sorter (BD Biosciences) and were analyzed with FlowJo v10 software (BD Biosciences). Sorted neutrophils (40,000) were frozen at -80oC for further processing.
Staining for imaging on hyperion (imaging mass cytometry)
The bronchial biopsies were provided by the Tasmanian Respiratory Biobank (Ethics ID: H0013051, approved by Tasmanian Health and Medical Human Research Ethics Committee). Lung biopsies from controls, smokers (Normal Lung Function Smokers) and COPD smokers were paraffin-embedded and sectioned at 8 um. Lung sections were baked for 60 min at 60oC followed by dewaxing in xylene and descending grades of absolute ethanol before two washes in 1% TBS-Tween buffer. Heat-mediated antigen retrieval was performed with 1M Tris/0.5 M EDTA solution on sections in a microwave (Panasonic) for 3 minutes on high power and 15 minutes on low power. Sections were allowed to cool and were rinsed for 10 min in 1x TBS-Tween and 1X PBS. Blocking was performed for 45 min at 37oC with antibody diluent/block (Akoya Biosciences). Finally, sections were incubated with metal-bound antibodies: CD3-170 (polyclonal; Dako), CD4-173 (EPR19514; Abcam), CD8-146 (D8A8Y; Cellsignal), CD10-156 (HI10a; Biolegend), CD11B-163 (EPR1344; Abcam), CD11C-142 (EP1347Y; Abcam), CD14-149 (EPR3653; Abcam), CD16A-148 (EPR16784; Biolegend), CD16B-152 (G10F5; Biolegend), CD20-141 (H1; BD Biosciences), CD44-166 (BJ18; Biolegend), CD45-154 (D9M8I; Cellsignal), CD56-176 (EPR2566; Abcam), CD62L-160 (polyclonal; Abcam), CD66A-167 (B1.1/CD66; BD Biosciences), CD68-153 (KP1; Abcam), CD74-150 (PIN.1; Abcam), CCR1-169 (polyclonal; Abcam), EPCAM-159 (VU-1D9; Stemcell), HLA-DR-174 (EPR3692; Abcam), Siglec-8-165 (polyclonal; Abcam) overnight at 4oC. The following day, sections were permeabilized and washed twice with 0.1% Triton-X in PBS for 8 min. Sections were incubated with Ir-Intercalator (1:400 in PBS) for 30 min to identify nucleated cells and were rinsed with dH2O. Sections were air dried for 20 min before imaging on a Hyperion (CPS-5404).
Cytometry by time of flight
Murine bone marrow was collected from both femurs and cells were immediately incubated with 10 uM iododeoxyuridine for 1 hour at 37°C. RBCs were lysed with 1 ml RBC lysis buffer for 5 min at 4°C and single-cell suspensions were prepared for analysis by time-of-flight mass cytometry. Cell numbers and viability were assessed using trypan blue and the remaining cells were transferred to a 96-well plate for staining with 5 uM cisplatin for 5 min at room temperature. Quenching in 5% FCS/5mM EDTA quenching buffer in PBS was performed before centrifugation at 300xg for 3 min at 4°C. Fc receptor blocking with metal conjugated anti-mouse CD16/32 antibody for 30 min at 4°C followed and quenched in quenching buffer before antibody staining for 30 min at 4°C. Cells were washed three times in quenching buffer, resuspended in 4% PFA and stored at 4°C. The following day, cells were washed once in PBS, permeabilized in methanol for 10 min at 4°C and washed three times before staining with intracellular antibodies for 45 min at room temperature. Samples were washed thrice in permeabilization buffer (Thermo Fisher), resuspended in 4% PFA and stored at 4°C until analysis using a Helios instrument (Fluidigm).
BD Rhapsody library preparation
For simultaneous measurements of single transcriptomes and surface markers in neutrophils, peripheral blood from 3 control and 3 COPD patients was labeled with Sample Tags (Human Single-Cell Multiplexing Kit, BD Biosciences) and stained with Ab-seq Ab-Oligos (BD Biosciences) following the manufacturer's protocol. Briefly, a total of 1x106 cells were resuspended in 180 ul of cold Stain Buffer (BD Pharmingen) and Sample Tags were added for 20 min staining at room temperature. After incubation, samples were washed twice at 300xg for 5 min at 4°C. Subsequently cells were counted and were equally pooled to obtain a total of 1x106 cells. Pooled samples were resuspended in 100 ul blocking buffer (95 ul Stain Buffer/5 ul FcγR blocking reagent) and incubated for 10 min at room temperature. A master mix (100 ul) containing Ab-seq Ab-Oligos diluted in Stain Buffer was added to the samples for 40 min on ice. The samples were washed twice, counted and resuspended to achieve 60,000 cells in 650 ul of cold Sample Buffer (BD Biosciences). The cell suspensions were then loaded onto a primed Rhapsody cartridge (BD Biosciences). Single cells were isolated using Single Cell Capture and cDNA Synthesis with the BD Rhapsody Express Single-Cell Analysis System according to the manufacturer’s recommendations (BD Biosciences). cDNA libraries were prepared using the BD Rhapsody Whole Transcriptome Analysis Amplification Kit following the BD Rhapsody System mRNA Whole Transcriptome Analysis (WTA) and Sample Tag Library Preparation Protocol (BD Biosciences) as described previously84. Briefly, mRNA products were separated from Sample Tag and Ab-seq products by a denaturation step at 95°C for 5 min. mRNA transcripts were further amplified by PCR (11 cycles) and then purified using SPRIselect magnetic beads (Beckman-Coulter), while the Sample Tag and Ab-seq products were amplified together with the PCR master mix containing the provided specific primers. A second PCR (10 cycles) was performed for the Sample Tag product as described in the protocol. Final libraries were indexed using 2 nM of whole transcript mRNA products and 3 ng of Sample Tag and Ab-seq PCR products by PCR (6 cycles). After purification with SPRIselect magnetic beads, the index PCR products were quantified using a Qubit Fluorometer with the Qubit dsDNA HS Kit (ThermoFisher).
Total RNA extraction
Total RNA was isolated with the miRNeasy Micro kit (Qiagen) according to the manufacturer’s protocol and RNA concentration and integrity were determined with the High Sensitivity RNA assay on a TapeStation 4200 system (Agilent).
Bulk RNA-seq library preparation and sequencing
cDNA libraries were prepared from at least 400 pg total RNA with the SMART-seq2 protocol85 and 200 pmol cDNA were tagmented with the Nextera XT DNA Library Preparation kit (Illumina). Library size selection was carried out with AMPure beads (Beckman Coulter) and the distribution was measured with the High Sensitivity D5000 assay on a TapeStation 4200 system (Agilent). Molar RNA sequencing library concentration was determined by combining size distribution information with a Qubit High Sensitivity dsDNA assay (Invitrogen) for concentration. Libraries were clustered at 1.4 pM and sequenced SR 75 cycles on a NextSeq500 system (Illumina) using High Output v2 chemistry. Upon sequencing, base call files were converted to fastq format and demultiplexed using bcl2fastq2 v2.20. Raw reads were pseudoaligned to the human transcriptome (GRCh38, Gencode v27 primary assembly) using Kallisto with default settings (v0.44.2).86
BD Rhapsody library sequencing
The Rhapsody experiment libraries were generated using the BD Rhapsody™ Whole Transcriptome Analysis (WTA) Amplification Kit according to the manufacturer’s protocol. Molar library concentration was determined using a combination of High Sensitivity D5000 assays on a Tapestation 4200 system (Agilent) for average size distribution and a Qubit High Sensitivity dsDNA assay (Invitrogen) for concentration. Libraries were pooled and clustered at 250 pM final concentration and sequenced PE (2 x 75 cycles) on a NovaSeq 6000 with NovaSeq 6000 S2 Reagent Kit (200 cycles) chemistry. Upon sequencing, base call files were converted to fastq format and demultiplexed using bcl2fastq2 v2.20.
Murine 10x genomics library generation and sequencing
The 10x Chromium libraries were prepared using the 10x Genomics Chromium Next GEM single cell 3’ kit v3.1 at the Garvan-Weizmann Centre for Cellular Genomics according to the manufacturer’s instructions. The libraries were sequenced on an Illumina NovaSeq 6000 instrument using a NovaSeq S4 kit (200 cycles) v1 chemistry at an aimed depth of 30,000 paired-end reads per cell. The sequencer generated raw data files in binary base call (BCL) format. The BCL files were demultiplexed and converted to the FASTQ file formats using Illumina Conversion Software (bcl2fastq v2.19.0.316). The 10x Genomics cellranger -v (3.1.0) count pipeline was used for alignment, filtering, barcode counting, and UMI counting from FASTQ files and was executed on a high-performance cluster with 2.6.32-754.17.1.el6.x86_64 operating system.
Quantification and statistical analysis
To facilitate reproducibility of the results, R-based analyses were performed in a dedicated docker environment based on R 4.0.3 and Bioconductor 3.12 (lorenzobonaguro/flowtools:v3).
Pre-processing of BD rhapsody scRNA-seq data
A barcode whitelist provided by BD Biosciences was used to filter the paired-end scRNA-seq reads for valid cell barcodes. Cutadapt (v1.16) R package62 was used to trim adaptor sequences and to filter reads for a PHRED quality score of 20 or above. Next, STAR (v2.6.1b)77 was used for alignment against the Gencode v27 (GRCh38p13) human reference genome and Dropseq-tools (v2.0.0) were used to quantify gene expression and collapse to UMI count data (https://github.com/broadinstitute/Drop-seq/). For SampleTag oligo-based demultiplexing of single-cell transcriptomes and subsequent assignment of cell barcodes to their sample of origin, the respective multiplexing tag sequences and AB-seq sequences were added to the reference genome and quantified, as well.84
Analysis of human Seq-well data
The single-cell datasets were analyzed with the Seurat (v3.2.2) R package.73 FindVariableFeatures calculated the 2,000 most variable genes, ScaleData scaled the expression of all present genes, RunPCA reduced the dimensionality to 50 principal components, FindNeighbors found local neighorhoods for 20 principal components and FindClusters with resolution equal to 0.6 and RunUMAP were run to cluster and project the cells.
Neutrophils were “in silico sorted” at the cluster level based on the expression of mature (S100A8, S100A9, CXCR2, CSF3R, IFITM2, FCGR3B) and immature (DEFA3, DEFA4, MMP8, MMP9, CAMP) neutrophil markers. Further subsetting of cells labeled as “Mature Neutrophil”, “Immature Neutrophil” and “Inflammatory Neutrophil” by the GenSigPro classifier from12 followed. The 2,000 most variable genes were identified, the dataset was scaled as above, dimensionality was reduced to 8 principal components and data were clustered with a resolution of 0.2 for the blood dataset and 30 principal components for the BALF dataset. Differentially expressed (DE) gene analysis for blood cluster comparisons was carried out using the FindMarkers and FindAllMarkers functions and the MAST algorithm for min.pct=0.25 and were plotted with the pheatmap (v1.0.12) R package.
Classification and integration of human single-cell RNA-seq data
Seq-well datasets from patients (“Pat 182”, “Pat 190”, “Pat 192”, “Pat 233”) were sequenced and pre-processed,12 and were added to the blood dataset. Cells from each dataset were normalized with LogNormalize by a scale factor of 10,000 to account for variations in sequencing depth and the 2,000 most variable genes were calculated with the FindVariableFeatures function in Seurat and the vst method. Next, classification followed the GenSigPro annotation with the anchoring method using FindTransferAnchors and the labels were transferred with TransferData. Finally, the new datasets were integrated to12 with FindIntegrationAnchors for 30 principal components and IntegrateData on 91,870 identified anchors. Standard integration methods assume that there are shared or similar variations between the integrated parts and attempt to understand very specific cell type batch effects. Here we describe neutrophil states from two different human compartments (BALF, peripheral blood) which, apart from anticipated donor and cell state differences, include additional biological and technical variations. For example, BALF neutrophils are not functionally identical to peripheral blood neutrophils due to the influence of the alveolar space microenvironment, the differences of which between control and COPD patients may not be comparable with those of peripheral blood in the same donors. Moreover, technical differences originating from cell isolation protocols will lead to loss of biological interpretation when integrating data from different tissue compartments. Indeed, cell dissociation protocols often result in different sampling effects, as well as different cellular ratios which makes learning a challenging task and impedes the identification and differentiation of distinct effects across organs. Forcing the integration of the two compartments could result in loss of information and the reduction in cell type resolution which would be counterintuitive to providing higher cell type resolution. Since there are currently no fully developed methods for cross-organ integration, we chose a conservative, but at the same time more robust way to investigate the similarities between neutrophil states in the two anatomical spaces.
Analysis of human BD Rhapsody data
The single-cell data generated with the BD Rhapsody platform were pre-processed to remove cells with less than 150 genes and more than 20% mitochondrial genes. Two new assays for the Sample Tag and Ab-seq data were added to the generated Seurat object and were normalized with the centered log-ratio (CLR) method. The HTODemux function with positive.quantile=0.99 was used to demultiplex the samples and doublets were discarded. The 2,000 most variable genes were calculated with FindVariableFeatures and the expression of all present genes were scaled with ScaleData. Dimensionality reduction to 20 principal components was performed with RunPCA and local neighborhoods were found with FindNeighbors and FindClusters at resolution equal to 0.4. Neutrophils clusters were subsequently subsetted and the aforementioned pipeline was followed. Fifteen principal components were used and the dataset was clustered with resolution of 0.3. DE gene analysis for all comparisons was carried out using the FindMarkers and FindAllMarkers functions and the MAST algorithm for min.pct=0.15 and logfcthreshold=0.25 for genes and logfcthreshold=0.2 for protein markers.
Analysis of murine 10x genomics data
The murine single-cell datasets were analyzed with Seurat. Cells with at least 100 genes and less than 10% mitochondrial genes were kept. Genes expressed in less than 5 cells were discarded. The 2,000 most variable genes were used in PCA calculations. BALF cells were analyzed with 30 principal components (PCs) and clustered with 0.3 resolution, blood cells were analyzed with 25 PCs and clustered with 0.3 resolution and bone marrow cells were analyzed with 30 PCs and clustered with 0.4 resolution. Doublets were identified in each sample with the DoubletFinder (v2.0.3) R package.64 An estimate of 8% doublets was assumed and the doublet number was adjusted for homotypic doublets. DE genes between clusters for each compartment were calculated with FindMarkers with min.pct=0.25 and logfc.threshold=0.4 using the MAST algorithm.
Neutrophils were identified based on the expression of canonical markers (Camp, Ngp, Ltf, Pglyrp1, Mmp8, Cd177, Ly6g, Chil3, Elane, Mpo) and singlets were in silico sorted for further analysis in each compartment. The BALF neutrophil dataset was reduced to 20 PCs and clustered with 0.2 resolution, blood neutrophils were analyzed with 15 PCs and clustered with 0.2 resolution and bone marrow neutrophils were analyzed with 20 PCs and clustered with 0.2 resolution. DE genes between clusters for each compartment were calculated with FindMarkers with min.pct=0.25 and logfc.threshold=0.25 using the MAST (v1.20) algorithm.70 The parameters for the DE gene calculation between neutrophils from air and smoke-exposed mice were min.pct=0.20 and logfc.threshold=0.25. BALF neutrophils were additionally integrated with the Harmony (v1.0) R package67 to remove minor batch effects.
In addition to the biological differences of the microenvironments in the three studied anatomical sites (BALF, blood, bone marrow), there are technical differences which are related to: 1) the collection of cells following specialized protocols; BALF: collection via a cannula in PBS and subsequent red blood cell lysis, blood: red blood cell lysis, bone marrow: femora/tibiae flushing of cells and red blood cell lysis and 2) cell isolation methods to optimize information gain; BALF: sorting of all live cells, blood and bone marrow: sorting of live CD45+ cells. Since there are currently no fully developed methods for cross-organ integration, we opted for the separate analysis of neutrophil state-specific gene signatures in the three anatomical spaces.
Analysis of publicly available murine scRNA-seq datasets
The single-cell multiple time point smoke exposure murine dataset45 was retrieved from GSE151674 and GSE185006. Cells with more than 20% of mitochondrial genes or less than 200 detected genes were excluded for further analysis and cell barcodes with 400 to 6,000 counts per cell and genes detected in at least 3 cells were retained. For this study, cells from mice treated with LTβR-Ig were further discarded. Downstream analysis was performed using the scanpy (v1.8.0) python package.76 The SoupX (v1.5.0) R package75 was used to mitigate background mRNA contamination and the contamination fraction was set to 0.3 for count matrix correction with adjustCounts(). Data were normalized with the scran size factor based approach and log transformed via scanpy’s pp.log1p() function. The top 4,000 variable genes in at least 5 samples excluding known cell cycle genes were established with the function pp.highly_variable_genes(flavor=”cellranger”) and used as input for PCA (8,696 genes). Neutrophils were in silico sorted based on known cell type markers (Camp, Ngp, S100a8, Il1b) and visualization was achieved with the UMAP embedding specifying the input parameters as 20 PCs and 20 nearest neighbors. Clustering on this subset was performed via scanpy’s leiden method at resolution 0.3.
Distribution-free DE gene analysis across patient groups
DE gene analysis was performed with a distribution-free method which was developed specifically for single-cell data of multiple patients belonging to different groups.12 This method is independent of any distribution assumptions and takes all normalized single-cell data points into account, thus it is not based on mini-bulk or pooling cells from individuals.
DE gene analysis was performed to investigate two fundamental aspects: (1) differences between the control and patient cohorts for a particular neutrophil cluster, and (2) differences between neutrophil clusters. As input, cluster-annotated cells were used. Before each DE analysis, individuals not possessing cells in a subcluster and genes with less than 10% of expressed cells were disregarded. For each gene, the differences between all possible pairs of samples between the two groups were quantified using the non-parametric Wilcoxon rank sum score. For the overall difference between the two groups, the median Wilcoxon scores of the pairwise tests were considered as the test statistic. The significance of the test statistic was assessed with the permutation test, with the null hypothesis of no differences between the two groups. For all possible permutations, the test statistic was evaluated, providing the exact null distribution. P-values were determined by the fraction of permutations that led to a higher or equal test statistic value than the value of the test statistic of the observed patient arrangement.
Effect size
Data were sorted according to the effect size being the median of the Wilcoxon–Mann–Whitney odds (WMW odds) (39). The WMW odds for each sample combination were calculated by dividing the Whitney U-score of one sample by the U-score of another sample;
with the U-scores defined as
with R1 being the sum of the ranks belonging to sample 1, and n1 and n2 being the sample sizes of sample 1 and sample 2, respectively.
The WMW odds consider all possible pairs of data points between the two considered samples and summarize how many pairs of data points are actually larger in sample 1 compared to sample 2, in relation to how many pairs of data points are larger in sample 2 compared to sample 1.
For the comparison of the neutrophil clusters the same DE analysis method was performed with an adjustment of the permutation test to ensure independence of the samples within one group. For each individual, the data were divided into two groups of cluster comparisons (e.g., Patient 1 - cluster 0 vs Patient 1 - rest of clusters). The individuals with differing cluster annotations were subsequently compared. For this setting, the permutation test needed to be adjusted, such that one individual with different cluster annotations belonged to separate groups.
Pathway analysis
Gene set enrichment analysis of cluster-specific markers was performed with the clusterProfiler (v3.14.3) R package.61 Reactome or Gene Ontology gene sets were downloaded from the MSigDB and obtained from “c2.cp.reactome.all.v7.2.symbols.gmt” and “c5.go.bp.v7.4.symbols.gmt”, respectively. Enrichment was computed with the enricher function using a minimum and maximum gene set sizes of 10 and 500, respectively and all expressed genes in the cluster as the background. Enriched gene sets with less than 3 genes were discarded and the top 5 enriched gene sets for blood immune cells were extracted and loaded in Cytoscape (v3.7.2) (https://cytoscape.org/) for visualization with the Prefuse Force Directed layout. If a gene set appeared as both upregulated and downregulated, the direction with the highest number of genes only is shown. An Upset plot was used to graphically visualize the overlap of enriched gene sets using the UpsetR (v1.4.0) R package.79 To connect gene ontology terms, the pairwise similarity matrix was calculated with pairwise_termsim.
Correlation between blood immune cell proportion and clinical parameters
The correlation between blood neutrophil counts and clinical parameters was calculated with the cor and cor.test base R functions and the Spearman method and the data were visualized with the ggplot2 (v3.2.1) R package.66
Gene set enrichment analysis
The AUCell (v1.4.1) R package58 was used for human neutrophil granule content gene signature enrichment from.24 Cells were first filtered for genes with more than 3 total counts. Genes were expression-ranked within every cell with AUCell_buildRankings and the enrichment of gene signatures was computed with AUCell_calAUC in the 5% highest genes of each cell. Data were visualized in violin plots with the ggplot2 R package with the fill depicting the mean AUC score of all cells within each group.
For the enrichment of the conserved murine bone marrow neutrophil progenitor 9-gene signature on human blood neutrophil states, the fgsea (v1.16.0) R package65 was used. As input, the expressed genes for each human blood neutrophil state in both the control and COPD groups was used and the analysis was performed upon log2 fold change (COPD/control)-ranking with the fgsea function. Results were visualized in tables with the plotGseaTable command (gseaParam=0.5). Murine gene names were converted to their human orthologs with convertMouseGeneList() and the bioMart (v2.50.1) R package.59 Where stated, human and murine neutrophil cluster-specific markers were filtered for unique genes and the top 20 were selected as signatures for comparisons.
Trajectory inference
Developmental relationships for blood neutrophils were inferred with the Slingshot (v1.4.0) R package.74 The function slingshot was run on the first 50 principal components of the converted 20,670 blood neutrophil SingleCellExperiment with N1S as the starting cluster. The curves were then embedded onto the UMAP space with embedCurves. Cells belonging to one lineage were selected and a generalized additive model was fit to the 3,000 most variable genes with the gam R package (v1.20). The 30 most correlating genes were plotted in a heatmap along pseudotime with the pheatmap (v1.0.12) R package.71
Whole transcriptome analysis
Processed data were imported into the DESeq2 (v1.10.1) R package63 using the TXimport (v1.2.0) R package.78 DESeq2 was used for the calculation of normalized counts for each transcript using default parameters. All transcripts with a maximum over all group means lower than 10 were excluded resulting in 17,189 genes. Unwanted sources of variation, such as sequencing and smoking were removed using the function removeBatchEffect from the limma (v3.48.1f) R package69 and used for all visualizations. The same factors were also modeled within DESeq2.
In silico cytometry
CIBERSORTx (https://cibersortx.stanford.edu/) was employed to computationally deconvolute the identified blood neutrophil states in sorted blood neutrophil whole transcriptome samples. The normalized count table of 5,000 downsampled blood neutrophils was provided as the input to generate a single-cell signature matrix by setting the minimum expression to 0, number of replicates to 5 and sampling fraction to 0.5. The algorithm was subsequently run with 1,000 permutations with the normalized count table of the sorted neutrophil samples as the mixture sample. Correlation of neutrophil state abundance was calculated using mixed models adjusted for age, sex and inhaled corticosteroid treatment.
Analysis of imaging mass cytometry data
Ablated regions of interest (ROIs) from lung tissue were viewed using Histocat++68 and the images of the desired ROIs were exported as one TIFF file for every channel. All TIFF files were opened in cell profiler (v4.1.3) to perform cell segmentation and generate nucleus, cellular and cytoplasmic masks. R was used to convert masks and TIFF files into FCS files for use in Flowjo for spatial analysis.
Pseudotime analysis
For the diffusion pseudotime (dpt) analysis, the root to the cell with the highest UMAP1 value was set and the scanpy tl.dpt() function was employed in Python (v3.8.5). The dpt values were used to order the visualization. The cells were grouped into 500 equally sized bins based on their dpt value and the bin-wise average expression of the top 20 marker genes for each neutrophil cluster were used to produce a row-scaled heatmap.
Analysis of mass cytometry data
The expression levels of 38 markers measured by mass cytometry (IdU, CD34, Ki67, CD64, SCA_1, CD117, CD150, CD8a, NK1.1, ABCA1, Cyclin_B1, CD138, CD48, CD135, CD62L, CD115, CCR2, CD11c, FceR1a, CD16_32, CD4, Siglec-F, CD11b, pRb, CD45, Ly6G, CD103, CD19, CD3e, F4_80, B220, CD172a, CD24, CD80, CD86, CD127, MHCII, Ly6C) in immune cells was used as input for downstream analysis including dimensionality reduction and Phenograph clustering. Ten thousand cells for each sample were randomly selected and protein expression levels were transformed with a logicle transformation (width=0.25, top=16,409, full width=4.5). Transformed values were then used for dimensionality reduction. UMAP was calculated using the umap R package (v0.2.7) with number of neighbors=15, number of components=2, distance metric=Euclidean and minimum distance=0.2. Cell clustering was calculated with the Rphenograph (v0.99.1) R package72 with default settings and number of nearest neighbors (k)=60.
Clusters 6, 11, 18 and 20 were used for downstream analysis of the neutrophil compartment. All transformed channels were used for UMAP dimensionality reduction (umap 0.2.7) and Phenograph clustering (v0.99.1) (k=60, Rphenograph, Github JinmiaoChenLab). Clustering analysis resulted in 11 clusters of coherent marker expression which were merged in 4 metaclusters according to the expression of hallmark markers. A confusion matrix was calculated normalizing first the number of cells of each experimental group to 1,000 and later calculating the contribution of each condition in each cluster. This normalization was performed to avoid bias deriving from a different number of cells in the three experimental groups. Finally, marker expression heatmap was calculated as the mean marker expression in each cluster. The values for each marker were scaled for visualization. All data were visualized with R using the ggplot2 package for the UMAPs and pheatmap for confusion matrix and heatmap of marker expression.
Statistics
Two group comparisons were performed with two-tailed t-test (equal variances) or Welch’s test (unequal variances) for parametrical data. A Wilcoxon signed rank test was used for non-parametric data. For multiple group comparisons, one-way ANOVA with Tukey post-hoc corrections were employed. Normality was tested with the shapiro.test base R function and homogeneity of variance with the leveneTest function from the car (v3.0-3) R package.60 Data are represented as mean ± standard deviation or mean ± standard error of the mean and statistical comparisons were considered significant when p<0.05.
Acknowledgments
The authors would like to thank Anne O’Garra for critical evaluation and feedback on the manuscript. W.F. was supported by a fellowship of the Alexander von Humboldt Foundation (JPN-1186019-HFST-P). J.L.S. received funds from the BMBF-funded excellence project Diet-Body-Brain (DietBB) under grant number 01EA1809A, the EU Horizon 2020 project SYSCID under grant 733100, the EU Horizon 2020 Research and Innovation Program under grant agreement no. 874656 (discovAIR), the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation, projects numbers 347286815, 329123747, and 423957469), Germany’s Excellence Strategy (EXC2151-390873048, ImmunoSensation2), the Helmholtz-funded consortium Sparse2Big, and the Joint Programming Initiative, A Healthy Diet for a Healthy Life (JPI HDHL; project 529051018). M.C.N. acknowledges funding from the EU Horizon 2020 Research and Innovation Program under grant agreement no. 874656 (discovAIR). S.S.S. is funded by grants from Clifford Craig Foundation Launceston General Hospital. M.C. is funded by Helmholtz Zukunftsthema Immunology and Inflammation (ZT-0027). P.M.H. is funded by a fellowship and grants from the National Health and Medical Research Council (NHMRC) of Australia (1175134) and by UTS.
Author contributions
Conceptualization, D.S. and J.L.S.; formal analysis, T.S.K., K.B., W.F., C.N., A.C.S., L.B., T.P., S.A., A.S., E.D., A. Frishberg, D.G.-O., T.E.G., M.A., N.O., I.A., B.S., K.H., M. Becker, and T.U.; investigation, T.S.K., K.B., W.F., C.N., I.G., E.d.D., A.H., C.D., R.Y.K., D.G.-O., J.S.-S., N.O., I.A., C.O.-S., J.K., T.S., A. Feißt, K.H., C.P., and D.S.; writing – original draft, T.S.K. and J.L.S.; writing – review & editing, K.B., W.F., L.B., T.P., A. Frishberg, J.S.-S., M.v.d.B., H.C.D., H.J.M.G., A.Ö.Y., M.C.N., M.C., T.U., P.M.H., and D.S.; resources, W.L., M.S.E., H.C.D., M.v.d.B., H.J.M.G., S.S.S., M.C.N., A.Ö.Y., and H.B.S.; data curation, H.C.D.; visualization, M.v.U.; supervision, M.C.N., M. Beyer, M.C., J.H., F.J.T., P.M.H., D.S., and J.L.S.; funding acquisition, J.L.S.
Declaration of interests
The authors have no competing interests.
Published: May 26, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112525.
Supplemental information
Data and code availability
-
•
The human raw bulk and single-cell RNA sequencing (scRNA-seq) datasets are available through the European Genome-phenome Archive (EGAS00001006281; EGAS00001006322; EGAS00001006323). The murine raw scRNA-seq dataset is available through the Gene Expression Omnibus (GSE205078).
-
•
All original code has been deposited at FASTGenomics (www.beta.fastgenomics.org) and is publicly available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Venkatesan P. GOLD COPD report: 2023 update. Lancet Respir. Med. 2023;11:18. doi: 10.1016/S2213-2600(22)00494-5. [DOI] [PubMed] [Google Scholar]
- 2.Agustí A., Hogg J.C. Update on the pathogenesis of chronic obstructive pulmonary disease. N. Engl. J. Med. 2019;381:1248–1256. doi: 10.1056/NEJMra1900475. [DOI] [PubMed] [Google Scholar]
- 3.Gan W.Q., Man S.F.P., Senthilselvan A., Sin D.D. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004;59:574–580. doi: 10.1136/thx.2003.019588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agustí A., Edwards L.D., Rennard S.I., MacNee W., Tal-Singer R., Miller B.E., Vestbo J., Lomas D.A., Calverley P.M.A., Wouters E., et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One. 2012;7 doi: 10.1371/journal.pone.0037483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fabbri L.M., Rabe K.F. From COPD to chronic systemic inflammatory syndrome? Lancet. 2007;370:797–799. doi: 10.1016/S0140-6736(07)61383-X. [DOI] [PubMed] [Google Scholar]
- 6.Bahr T.M., Hughes G.J., Armstrong M., Reisdorph R., Coldren C.D., Edwards M.G., Schnell C., Kedl R., LaFlamme D.J., Reisdorph N., et al. Peripheral blood mononuclear cell gene expression in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2013;49:316–323. doi: 10.1165/rcmb.2012-0230OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reinhold D., Morrow J.D., Jacobson S., Hu J., Ringel B., Seibold M.A., Hersh C.P., Kechris K.J., Bowler R.P. Meta-analysis of peripheral blood gene expression modules for COPD phenotypes. PLoS One. 2017;12 doi: 10.1371/journal.pone.0185682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morrow J.D., Chase R.P., Parker M.M., Glass K., Seo M., Divo M., Owen C.A., Castaldi P., DeMeo D.L., Silverman E.K., Hersh C.P. RNA-sequencing across three matched tissues reveals shared and tissue-specific gene expression and pathway signatures of COPD. Respir. Res. 2019;20:65. doi: 10.1186/s12931-019-1032-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrow J.D., Qiu W., Chhabra D., Rennard S.I., Belloni P., Belousov A., Pillai S.G., Hersh C.P. Identifying a gene expression signature of frequent COPD exacerbations in peripheral blood using network methods. BMC Med. Genom. 2015;8:1. doi: 10.1186/s12920-014-0072-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obeidat M., Nie Y., Chen V., Shannon C.P., Andiappan A.K., Lee B., Rotzschke O., Castaldi P.J., Hersh C.P., Fishbane N., et al. Network-based analysis reveals novel gene signatures in peripheral blood of patients with chronic obstructive pulmonary disease. Respir. Res. 2017;18:72. doi: 10.1186/s12931-017-0558-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang Y., Glass K., Liu Y.-Y., Silverman E.K., Crapo J.D., Tal-Singer R., Bowler R., Dy J., Cho M., Castaldi P. COPD subtypes identified by network-based clustering of blood gene expression. Genomics. 2016;107:51–58. doi: 10.1016/j.ygeno.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baßler K., Fujii W., Kapellos T.S., Dudkin E., Reusch N., Horne A., Reiz B., Luecken M.D., Osei-Sarpong C., Warnat-Herresthal S., et al. Alveolar macrophages in early stage COPD show functional deviations with properties of impaired immune activation. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.917232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jasper A.E., McIver W.J., Sapey E., Walton G.M. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Res. 2019;8 doi: 10.12688/f1000research.18411.1. [version 1; peer review: 2 approved. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie X., Shi Q., Wu P., Zhang X., Kambara H., Su J., Yu H., Park S.-Y., Guo R., Ren Q., et al. Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat. Immunol. 2020;21:1119–1133. doi: 10.1038/s41590-020-0736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evrard M., Kwok I.W.H., Chong S.Z., Teng K.W.W., Becht E., Chen J., Sieow J.L., Penny H.L., Ching G.C., Devi S., et al. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions. Immunity. 2018;48:364–379.e8. doi: 10.1016/j.immuni.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Grieshaber-Bouyer R., Radtke F.A., Cunin P., Stifano G., Levescot A., Vijaykumar B., Nelson-Maney N., Blaustein R.B., Monach P.A., Nigrovic P.A., ImmGen Consortium The neutrotime transcriptional signature defines a single continuum of neutrophils across biological compartments. Nat. Commun. 2021;12:2856. doi: 10.1038/s41467-021-22973-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Y.P., Padgett L., Dinh H.Q., Marcovecchio P., Blatchley A., Wu R., Ehinger E., Kim C., Mikulski Z., Seumois G., et al. Identification of an early unipotent neutrophil progenitor with pro-tumoral activity in mouse and human bone marrow. Cell Rep. 2018;24:2329–2341.e8. doi: 10.1016/j.celrep.2018.07.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwok I., Becht E., Xia Y., Ng M., Teh Y.C., Tan L., Evrard M., Li J.L.Y., Tran H.T.N., Tan Y., et al. Combinatorial single-cell analyses of granulocyte-monocyte progenitor heterogeneity reveals an early uni-potent neutrophil progenitor. Immunity. 2020;53:303–318.e5. doi: 10.1016/j.immuni.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Dinh H.Q., Eggert T., Meyer M.A., Zhu Y.P., Olingy C.E., Llewellyn R., Wu R., Hedrick C.C. Coexpression of CD71 and CD117 identifies an early unipotent neutrophil progenitor population in human bone marrow. Immunity. 2020;53:319–334.e6. doi: 10.1016/j.immuni.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujii W., Kapellos T.S., Baßler K., Händler K., Holsten L., Knoll R., Warnat-Herresthal S., Oestreich M., Hinkley E.R., Hasenauer J., et al. Alveolar macrophage transcriptomic profiling in COPD shows major lipid metabolism changes. ERJ Open Res. 2021;7 doi: 10.1183/23120541.00915-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halper-Stromberg E., Yun J.H., Parker M.M., Singer R.T., Gaggar A., Silverman E.K., Leach S., Bowler R.P., Castaldi P.J. Systemic markers of adaptive and innate immunity are associated with chronic obstructive pulmonary disease severity and spirometric disease progression. Am. J. Respir. Cell Mol. Biol. 2018;58:500–509. doi: 10.1165/rcmb.2017-0373OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Combes A.J., Courau T., Kuhn N.F., Hu K.H., Ray A., Chen W.S., Chew N.W., Cleary S.J., Kushnoor D., Reeder G.C., et al. Global absence and targeting of protective immune states in severe COVID-19. Nature. 2021;591:124–130. doi: 10.1038/s41586-021-03234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cowland J.B., Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol. Rev. 2016;273:11–28. doi: 10.1111/imr.12440. [DOI] [PubMed] [Google Scholar]
- 25.Mincham K.T., Bruno N., Singanayagam A., Snelgrove R.J. Our evolving view of neutrophils in defining the pathology of chronic lung disease. Immunology. 2021;164:701–721. doi: 10.1111/imm.13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basilico P., Cremona T.P., Oevermann A., Piersigilli A., Benarafa C. Increased myeloid cell production and lung bacterial clearance in mice exposed to cigarette smoke. Am. J. Respir. Cell Mol. Biol. 2016;54:424–435. doi: 10.1165/rcmb.2015-0017OC. [DOI] [PubMed] [Google Scholar]
- 27.Railwah C., Lora A., Zahid K., Goldenberg H., Campos M., Wyman A., Jundi B., Ploszaj M., Rivas M., Dabo A., et al. Cigarette smoke induction of S100A9 contributes to chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2020;319:L1021–L1035. doi: 10.1152/ajplung.00207.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang J.H., Hwang S.M., Chung I.Y. S100A8, S100A9 and S100A12 activate airway epithelial cells to produce MUC5AC via extracellular signal-regulated kinase and nuclear factor-κB pathways. Immunology. 2015;144:79–90. doi: 10.1111/imm.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller L., Singbartl K., Chroneos Z.C., Ruiz-Velasco V., Lang C.H., Bonavia A. Resistin directly inhibits bacterial killing in neutrophils. ICMx. 2019;7:30. doi: 10.1186/s40635-019-0257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castellani S., D’Oria S., Diana A., Polizzi A.M., Di Gioia S., Mariggiò M.A., Guerra L., Favia M., Vinella A., Leonetti G., et al. G-CSF and GM-CSF modify neutrophil functions at concentrations found in cystic fibrosis. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-49419-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang D., Wang X., Luo M.T., Wang H., Li Y.H., Satzke C., Dunne E.M., Licciardi P.V., Seow H.J., Nichol K., et al. CSF3R/CD114 mediates infection-dependent transition to severe asthma. Front. Neurosci. 2019;13:785–788. doi: 10.1016/j.jaci.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 32.Wang H., Aloe C., Wilson N., Bozinovski S. G-CSFR antagonism reduces neutrophilic inflammation during pneumococcal and influenza respiratory infections without compromising clearance. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-54053-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsoyi K., Osorio J.C., Chu S.G., Fernandez I.E., De Frias S.P., Sholl L., Cui Y., Tellez C.S., Siegfried J.M., Belinsky S.A., et al. Lung adenocarcinoma syndecan-2 potentiates cell invasiveness. Am. J. Respir. Cell Mol. Biol. 2019;60:659–666. doi: 10.1165/rcmb.2018-0118OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menezes M.E., Shen X.-N., Das S.K., Emdad L., Sarkar D., Fisher P.B. MDA-9/Syntenin (SDCBP) modulates small GTPases RhoA and Cdc42 via transforming growth factor β1 to enhance epithelial-mesenchymal transition in breast cancer. Oncotarget. 2016;7:80175–80189. doi: 10.18632/oncotarget.13373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pouwels S.D., Faiz A., den Boef L.E., Gras R., van den Berge M., Boezen H.M., Korstanje R., Ten Hacken N.H.T., van Oosterhout A.J.M., Heijink I.H., Nawijn M.C. Genetic variance is associated with susceptibility for cigarette smoke-induced DAMP release in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2017;313:L559–L580. doi: 10.1152/ajplung.00466.2016. [DOI] [PubMed] [Google Scholar]
- 36.Lee S., Lee H.-C., Kwon Y.-W., Lee S.E., Cho Y., Kim J., Lee S., Kim J.-Y., Lee J., Yang H.-M., et al. Adenylyl cyclase-associated protein 1 is a receptor for human resistin and mediates inflammatory actions of human monocytes. Cell Metab. 2014;19:484–497. doi: 10.1016/j.cmet.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Girkin J., Loo S.-L., Esneau C., Maltby S., Mercuri F., Chua B., Reid A.T., Veerati P.C., Grainge C.L., Wark P.A.B., et al. TLR2-mediated innate immune priming boosts lung anti-viral immunity. Eur. Respir. J. 2021;58 doi: 10.1183/13993003.01584-2020. [DOI] [PubMed] [Google Scholar]
- 38.Pouwels S.D., van Geffen W.H., Jonker M.R., Kerstjens H.A.M., Nawijn M.C., Heijink I.H. Increased neutrophil expression of pattern recognition receptors during COPD exacerbations. Respirology. 2017;22:401–404. doi: 10.1111/resp.12912. [DOI] [PubMed] [Google Scholar]
- 39.Simpson J.L., McDonald V.M., Baines K.J., Oreo K.M., Wang F., Hansbro P.M., Gibson P.G. Influence of age, past smoking, and disease severity on TLR2, neutrophilic inflammation, and MMP-9 levels in COPD. Mediators Inflamm. 2013;2013 doi: 10.1155/2013/462934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gregson A.L., Hoji A., Injean P., Poynter S.T., Briones C., Palchevskiy V., Weigt S.S., Shino M.Y., Derhovanessian A., Sayah D., et al. Altered exosomal RNA profiles in bronchoalveolar lavage from lung transplants with acute rejection. Am. J. Respir. Crit. Care Med. 2015;192:1490–1503. doi: 10.1164/rccm.201503-0558OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schultze J.L., Mass E., Schlitzer A. Emerging principles in myelopoiesis at homeostasis and during infection and inflammation. Immunity. 2019;50:288–301. doi: 10.1016/j.immuni.2019.01.019. [DOI] [PubMed] [Google Scholar]
- 42.Beckett E.L., Stevens R.L., Jarnicki A.G., Kim R.Y., Hanish I., Hansbro N.G., Deane A., Keely S., Horvat J.C., Yang M., et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J. Allergy Clin. Immunol. 2013;131:752–762. doi: 10.1016/j.jaci.2012.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim R.Y., Sunkara K.P., Bracke K.R., Jarnicki A.G., Donovan C., Hsu A.C., Ieni A., Beckett E.L., Galvão I., Wijnant S., et al. A microRNA-21-mediated SATB1/S100A9/NF-κB axis promotes chronic obstructive pulmonary disease pathogenesis. Sci. Transl. Med. 2021;13:eaav7223. doi: 10.1126/scitranslmed.aav7223. [DOI] [PubMed] [Google Scholar]
- 44.Jones B., Donovan C., Liu G., Gomez H.M., Chimankar V., Harrison C.L., Wiegman C.H., Adcock I.M., Knight D.A., Hirota J.A., Hansbro P.M. Animal models of COPD: what do they tell us? Respirology. 2017;22:21–32. doi: 10.1111/resp.12908. [DOI] [PubMed] [Google Scholar]
- 45.Conlon T.M., John-Schuster G., Heide D., Pfister D., Lehmann M., Hu Y., Ertüz Z., Lopez M.A., Ansari M., Strunz M., et al. Inhibition of LTβR signalling activates WNT-induced regeneration in lung. Nature. 2020;588:151–156. doi: 10.1038/s41586-020-2882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qi J., D’Souza D., Dawson T., Geanon D., Stefanos H., Marvin R., Walker L., Rahman A.H. Multimodal single-cell characterization of the human granulocyte lineage. bioRxiv. 2021 doi: 10.1101/2021.06.12.448210. Preprint at. [DOI] [Google Scholar]
- 47.Calzetti F., Finotti G., Tamassia N., Bianchetto-Aguilera F., Castellucci M., Canè S., Lonardi S., Cavallini C., Matte A., Gasperini S., et al. CD66b-CD64dimCD115- cells in the human bone marrow represent neutrophil-committed progenitors. Nat. Immunol. 2022;23:679–691. doi: 10.1038/s41590-022-01189-z. [DOI] [PubMed] [Google Scholar]
- 48.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D., et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aschenbrenner A.C., Mouktaroudi M., Krämer B., Oestreich M., Antonakos N., Nuesch-Germano M., Gkizeli K., Bonaguro L., Reusch N., Baßler K., et al. Disease severity-specific neutrophil signatures in blood transcriptomes stratify COVID-19 patients. Genome Med. 2021;13:7. doi: 10.1186/s13073-020-00823-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsalik E.L., Langley R.J., Dinwiddie D.L., Miller N.A., Yoo B., van Velkinburgh J.C., Smith L.D., Thiffault I., Jaehne A.K., Valente A.M., et al. An integrated transcriptome and expressed variant analysis of sepsis survival and death. Genome Med. 2014;6:111. doi: 10.1186/s13073-014-0111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Majo J., Ghezzo H., Cosio M.G. Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur. Respir. J. 2001;17:946–953. doi: 10.1183/09031936.01.17509460. [DOI] [PubMed] [Google Scholar]
- 52.Ballesteros I., Rubio-Ponce A., Genua M., Lusito E., Kwok I., Fernández-Calvo G., Khoyratty T.E., van Grinsven E., González-Hernández S., Nicolás-Ávila J.Á., et al. Co-Option of neutrophil fates by tissue environments. Cell. 2020;183:1282–1297.e18. doi: 10.1016/j.cell.2020.10.003. [DOI] [PubMed] [Google Scholar]
- 53.Deniset J.F., Kubes P. Neutrophil heterogeneity: bona fide subsets or polarization states? J. Leukoc. Biol. 2018;103:829–838. doi: 10.1002/JLB.3RI0917-361R. [DOI] [PubMed] [Google Scholar]
- 54.Ng L.G., Ostuni R., Hidalgo A. Heterogeneity of neutrophils. Nat. Rev. Immunol. 2019;19:255–265. doi: 10.1038/s41577-019-0141-8. [DOI] [PubMed] [Google Scholar]
- 55.Silvestre-Roig C., Hidalgo A., Soehnlein O. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood. 2016;127:2173–2181. doi: 10.1182/blood-2016-01-688887. [DOI] [PubMed] [Google Scholar]
- 56.Gungabeesoon J., Gort-Freitas N.A., Kiss M., Bolli E., Messemaker M., Siwicki M., Hicham M., Bill R., Koch P., Cianciaruso C., et al. A neutrophil response linked to tumor control in immunotherapy. Cell. 2023;186:1448–1464.e20. doi: 10.1016/j.cell.2023.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stolz D., Mkorombindo T., Schumann D.M., Agusti A., Ash S.Y., Bafadhel M., Bai C., Chalmers J.D., Criner G.J., Dharmage S.C., et al. Towards the elimination of chronic obstructive pulmonary disease: a Lancet Commission. Lancet. 2022;400:921–972. doi: 10.1016/S0140-6736(22)01273-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Durinck S., Moreau Y., Kasprzyk A., Davis S., De Moor B., Brazma A., Huber W. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- 60.Fox J., Weisberg S. 2019. An R Companion to Applied Regression Third. (Sage) [Google Scholar]
- 61.Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. j. 2011;17:10. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 63.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2016 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- 66.Wickham H. 2nd ed. Springer International Publishing; 2016. ggplot2 - Elegant Graphics for Data Analysis. [DOI] [Google Scholar]
- 67.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.-R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schapiro D., Jackson H.W., Raghuraman S., Fischer J.R., Zanotelli V.R.T., Schulz D., Giesen C., Catena R., Varga Z., Bodenmiller B. histoCAT: analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat. Methods. 2017;14:873–876. doi: 10.1038/nmeth.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M., et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kolde R. 2019. Pheatmap: Pretty Heatmaps. [Google Scholar]
- 72.Chen H. 2015. Rphenograph: R Implementation of the Phenograph Algorithm. [Google Scholar]
- 73.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Street K., Risso D., Fletcher R.B., Das D., Ngai J., Yosef N., Purdom E., Dudoit S. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018;19:477. doi: 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Young M.D., Behjati S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience. 2020;9 doi: 10.1093/gigascience/giaa151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Soneson C., Love M.I., Robinson M.D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.2. [version 2; peer review: 2 approved. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Conway J.R., Lex A., Gehlenborg N. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics. 2017;33:2938–2940. doi: 10.1093/bioinformatics/btx364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu G., Cooley M.A., Jarnicki A.G., Hsu A.C.-Y., Nair P.M., Haw T.J., Fricker M., Gellatly S.L., Kim R.Y., Inman M.D., et al. Fibulin-1 regulates the pathogenesis of tissue remodeling in respiratory diseases. JCI Insight. 2016;1 doi: 10.1172/jci.insight.86380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.GOLD . 2021. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease.https://goldcopd.org/wp-content/uploads/2020/11/GOLD-REPORT-2021-v1.1-25Nov20_WMV.pdf [Google Scholar]
- 82.Haque R.A., Usmani O.S., Barnes P.J. Chronic idiopathic cough: a discrete clinical entity? Chest. 2005;127:1710–1713. doi: 10.1378/chest.127.5.1710. [DOI] [PubMed] [Google Scholar]
- 83.Kardos P., Dinh Q., Fuchs K.H., Gillissen A., Klimek L., Koehler M., Sitter H., Worth H. [Guidelines of the German respiratory society for diagnosis and treatment of adults suffering from acute, subacute and chronic cough] Pneumologie. 2019;73:143–180. doi: 10.1055/a-0808-7409. [DOI] [PubMed] [Google Scholar]
- 84.De Domenico E., Bonaguro L., Schulte-Schrepping J., Becker M., Händler K., Schultze J.L. Optimized workflow for single-cell transcriptomics on infectious diseases including COVID-19. STAR Protoc. 2020;1 doi: 10.1016/j.xpro.2020.100233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Picelli S., Björklund Å.K., Faridani O.R., Sagasser S., Winberg G., Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 86.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The human raw bulk and single-cell RNA sequencing (scRNA-seq) datasets are available through the European Genome-phenome Archive (EGAS00001006281; EGAS00001006322; EGAS00001006323). The murine raw scRNA-seq dataset is available through the Gene Expression Omnibus (GSE205078).
-
•
All original code has been deposited at FASTGenomics (www.beta.fastgenomics.org) and is publicly available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.