

Abstract
Boroxinate complexes of VAPOL and VANOL are a chiral anionic platform that can serve as a versatile staging arena for asymmetric catalysis. The structural underpinning of the platform is a chiral polyborate core that covalently links together alcohols (or phenols) and vaulted biaryl ligands. The polyborate platform is assembled in situ by the substrate of the reaction, and thus a multiplex of chiral catalysts can be rapidly assembled from various alcohols (or phenols) and bis-phenol ligands for screening of catalyst activity. In the present study, variations in the steric and electronic properties of the phenol/alcohol component of the boroxinate catalyst are probed to reveal their effects on the asymmetric induction in the catalytic asymmetric aziridination reaction. A Hammett study is consistent with a mechanism in which the two substrates are hydrogen-bonded to the boroxinate core in the enantiogenic step. The results of the Hammett study are supported by a computational study in which it is found that the H–O distance of the protonated imine hydrogen bonded to the anionic boroxinate core decreases with an increase in the electron releasing ability of the phenol unit incorporated into the boroxinate. The results are not consistent with a mechanism in which the boroxinate catalyst functions as a Lewis acid and activates the imine by a Lewis acid/Lewis base interaction.
Graphical Abstract
INTRODUCTION
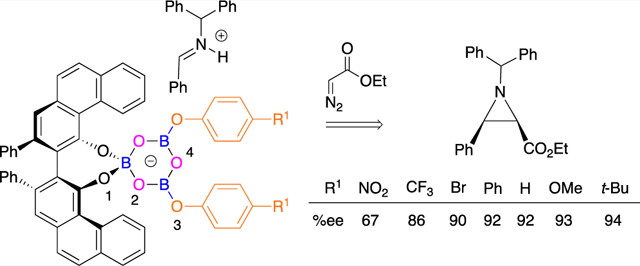
Chiral polyborate esters comprise a new class of asymmetric catalysts that provide a unique platform for templating catalytic reactions and a multifaceted forum for facilitating catalyst diversity. We have discovered1 that the catalyst system that we had originally developed2 for the asymmetric aziridination reaction is in fact a chiral polyborate. It was found that the boroxinate catalyst 2 can be prepared from the VAPOL ligand 1 and B(OPh)3 only in the presence of a base (usually the imine substrate), otherwise, no reaction occurs between VAPOL and B(OPh)3 (Scheme 1). Boroxinate formation is quite fast and is complete in the time it takes to mix everything together and introduce a sample into the NMR spectrometer. The formation of three B–O–B linkages requires 3 equiv of water but commercial B(OPh)3 is partially hydrolyzed to the point where the addition of water is typically not needed.1 The structure of the VAPOL boroxinate catalyst was determined by X-ray diffraction and found to consist of an ion pair comprised of a cation generated from a protonated molecule of the substrate and a chiral boroxinate anion.1 The boroxinate can be envisioned to be derived from a boroxine ring in which one of the borons is four coordinate as a result of being spiro-fused to a molecule of the ligand. In a similar manner, we have NMR evidence that the VANOL ligand can also form the boroxinate catalyst 4,3 and more recently this has been supported by X-ray diffraction.4 Experimentally, the simplest method for boroxinate catalyst formation involves the reaction of a molecule of the vaulted biaryl ligand VANOL or VAPOL with a triaryl or trialkyl borate ester.1
Scheme 1.

Boroxinate Catalysts from VAPOL and VANOL
These boroxinate species are the first and still only examples of chiral polyborate esters in asymmetric catalysis, although there are examples of chiral diborates that do not have a B–O–B linkage.5,6 In addition to the original method of preparation from a borate ester, the boroxinate catalysts can also be assembled in situ by a molecule of the substrate from either the VANOL or VAPOL ligand by one of several different methods (Scheme 2). The most direct method involves the reaction of a triphenoxyboroxine 6 with a molecule of the ligand.1 From the point of view of diversity-oriented catalyst generation, the method of choice is the coupling of the ligand with three molecules of water, two molecules of a phenol or alcohol, and three molecules of a boron source, either BH3·SMe22 or boric acid.7 The boroxinate catalyst can function as a chiral Brønsted acid catalyst in catalytic asymmetric aziridination reactions,2–4 aza-Cope rearrangements,8a heteroatom Diels–Alder reactions,8c reduction of quinolines,8d or as a chiral anion catalyst9 in the Ugi reaction.8b The success in identifying an effective catalyst for the Ugi reaction was made possible by the substantial diversity possible in the boroxinate catalyst as a result of the ability to introduce a variety of alcohols and phenols (HOR3 8) into the boroxinate core as indicated in Scheme 2.
Scheme 2.

Four Methods for the Preparation of Boroxinate Catalysts
A study of the mechanism of the boroxinate catalyzed asymmetric aziridination was undertaken on the reaction of the imine 10 with ethyl diazoacetate 11 which gives the cis-aziridine 12 in 94% yield and 97% ee (Scheme 3).10 Upon examination of the reaction by natural abundance 13C KIE it was found that the reaction occurs in two steps with the second ring-closing step as the rate-limiting step. Computational analysis of the boroxinate core of the boroxinate catalyst shows that the negative charge is dispersed over all of the oxygen atoms of the boroxinate core, of which there are four different types (Scheme 2).3 Computational analysis of the transition states for the first carbon–carbon bond forming step revealed that the protonated imine substrate is H-bonded to the boroxinate core at O-3 with an O–H distance of 2.17 Å (16 in Scheme 3).10 In the same transition state the ethyl diazoacetate is H-bonded via its diazo hydrogen with oxygen O-1 of the boroxinate core with an O–H distance of 1.94 Å.10 The transition state for the second step involving carbon–nitrogen bond formation still has the O–H bond (1.97 Å) between the hydrogen on the diazo carbon and O-1 of the boroxinate core (17 in Scheme 3). There is also an interaction between the diazonium ion and O-2 of the boroxinate core (N–O distance of 2.73 Å). The H-bond interaction of the protonated imine with O-3 of the boroxinate in the first transition state is replaced with a H-bonding interaction with the carbonyl oxygen from the diazo acetate (2.43 Å). Thus, it is clear that this analysis for both transition states implicates several H-bonding interactions between the chiral boroxinate anion and the substrates.
Scheme 3.

Summary of the Mechanism of Aziridine Formation
Although we have X-ray crystal structures of the catalyst/substrate complex with an imine,1,4 it does not necessarily mean that in solution the actual catalyst is a boroxinate catalyst that functions as a chiral Brønsted acid. Other catalytic species may be involved which also mediate the reaction in a two step manner with C–N bond formation in the second step as rate-limiting. It may be possible that the actual catalyst functions as a Lewis acid rather than as a Brønsted acid. For example, the complex 18 characterized by X-ray diffraction analysis as an ion pair consisting of a chiral spiro-borate anion and an iminium cation has been reported to react via the Lewis acid/Lewis base complex 19 (Scheme 4).11 One of the phenol oxygens of the two BINOL units becomes protonated, and this phenol unit is H-bonded to one of three oxygens attached to boron, thereby activating the Lewis acidity of the boron. These types of catalysts were referred to as Brønsted acid assisted Lewis acid or BLA catalysts.11
Scheme 4.

Brønsted Acid versus Lewis acid Catalyst for the Borate Ester of BINOL
Possible scenarios for a related isomerization in the boroxinate catalyst 4/2 would involve proton transfer to one of the oxygens in the boroxine ring to give either 20 or 21, each of which could be considered as a BLA catalyst where the Lewis acidity of the boron was enhanced by hydrogen bonding of an OH unit to an oxygen attached to boron. A method to differentiate between the Brønsted acid mechanism and the Lewis acid mechanism for the boroxinate catalyst would be to vary the electronic nature of the phenol substituent in the boroxinate catalyst. This should be possible from the self-assembly of the boroxinate catalyst from a VANOL or VAPOL ligand, 3 equiv of a boron source, 3 equiv of water, and 2 equiv of a substituted phenol/alcohol (OR3 in Schemes 2 and 5).8b If it functions as a Brønsted acid catalyst, the expectation is that the greater the electron density in the boroxinate catalyst as a result of a more electron-rich phenol, the more strongly bound will be the H-bonded substrates, the tighter the transition state and the higher the asymmetric inductions. On the other hand, if it functions as a Lewis acid, the greater the electron density in the boroxinate catalyst as a result of a more electron-rich phenol, the more loosely bound will be imine to the boron, and the longer B–N bond in the transition state should result in lower asymmetric inductions.
Scheme 5.

Brønsted versus Lewis Acid Catalysts for the Boroxinate Catalyst System
We report here the first study that examines how the enantioselectivity of aziridines from the catalytic asymmetric aziridination of imines with diazo compounds mediated by the boroxinate catalyst can be modulated by the incorporation of different alcohols and/or phenols into the boroxinate core of the boroxinate catalysts. This study will include boroxinate catalysts generated from both the VANOL and VAPOL ligands and various sterically and electronically differentiated alcohols and phenols. A series of electronically modified para-substituted phenols were chosen to probe the Hammett relationship between the electronic nature of the para-substituent and the asymmetric induction in the aziridination reaction. This was supported by computational studies that indicate that the strength of the H-bond between the protonated imine and the anionic boroxinate core increases with the increasingly electron releasing ability of the phenol unit incorporated into the boroxinate. This study includes imines generated from cyclohexane carboxaldehyde and benzaldehyde. The findings support a mechanism involving substrates H-bonded to a chiral anionic catalyst where the boroxinate catalyst functions as a chiral Brønsted acid.
RESULTS AND DISCUSSION
Aziridination Reactions of Phenyl Imine 22a and Cyclohexyl Imine 22b with Self-Assembled Boroxinate Catalysts from Nineteen Different Alcohols and Phenols.
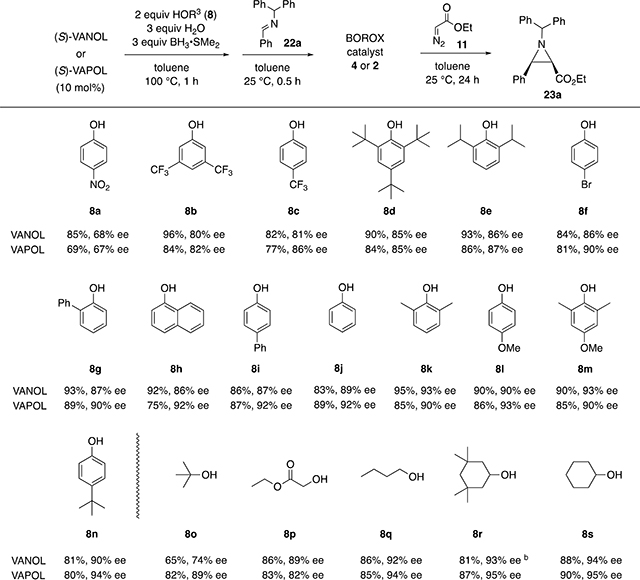
The set of boroxinate catalysts that were examined in the present work were those generated from either the VANOL or VAPOL ligand and the set of 19 different alcohols and phenols indicated in Tables 1 and 2.12 This electronic study was carried out on the benzhydryl imines 22 rather than the bis(3,5-dimethylanisyl)methyl imines of the type 10 (Scheme 3) since the asymmetric inductions in the aziridination with the latter are too high10 to give a useful measure of the relationship between and the electronic nature of the phenol/alcohol substituent in the boroxinate catalyst and the asymmetric induction over a range of electronically different alcohols and phenols. Each of the boroxinate catalysts were examined with the two major classes of imines, those generated from benzhydryl amine and aryl aldehydes and those from aliphatic aldehydes (Tables 1 and 2, respectively). The boroxinate catalyst was assembled by first generating a precatalyst by heating a mixture of 0.1 equiv of the ligand, 0.2 equiv of the alcohol or phenol, 0.3 equiv of water, and 0.3 equiv of BH3·SMe2 in toluene at 100 °C for 1 h. The resulting precatalyst solution is treated with 0.1 equiv of the imine at room temperature for 30 min. Ethyl diazo acetate 11 (1.2 equiv) is then added to the solution of the boroxinate catalyst along with the remaining 0.9 equiv of imine and allowed to react at room temperature for 24 h. This reaction is typically complete in an hour or so with the boroxinate catalyst prepared from phenol, but an extended time period was chosen to ensure that all catalysts can bring each reaction to >95% completion.
Table 1.
Catalytic Asymmetric Synthesis of Aziridine 23a via Boroxinate Catalysts Prepared from Various Alcohols and Phenolsa
|
Each reaction was performed with 10 mol % catalyst at 0.5 M of imine 22a with 1.2 equiv of EDA 11 at room temperature in toluene for 24 h and went to >95% completion. The catalyst was prepared by heating a mixture of either VANOL or VAPOL with 2 equiv of the alcohol or phenol 8, 3 equiv of H2O and 3 equiv of BH3·SMe2 in toluene at 100 °C for 1 h. The solution is cooled to 25 °C and 10 equiv of imine 22a is added to induce boroxinate formation over a period of 30 min. The reaction is initiated by the addition of 1.2 equiv of EDA 11 and allowed to proceed for 24 h at 25 °C. The yields are those of material isolated and purified by chromatography on silica gel. The optical purities were measured by HPLC.
NMR yield with internal standard
Table 2.
Catalytic Asymmetric Synthesis of Aziridine 23b via Boroxinate Catalysts Prepared from Various Alcohols and Phenolsa
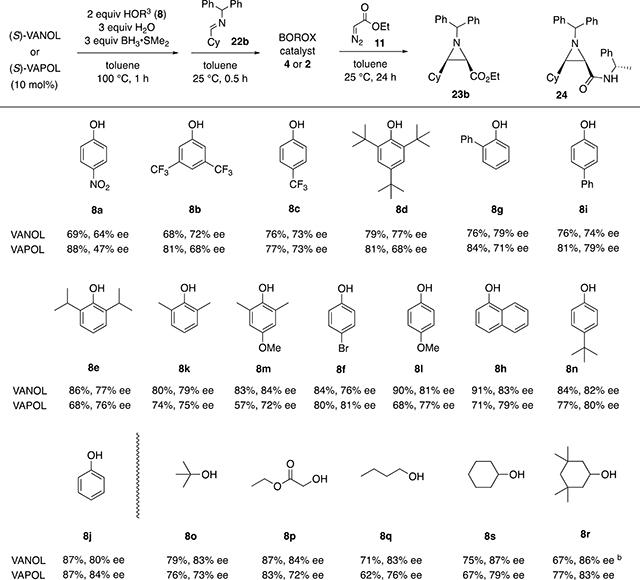
|
Each reaction was performed with 10 mol % catalyst at 0.5 M of imine 22b with 1.2 equiv of EDA 11 at room temperature in toluene for 24 h and went to >95% completion. The catalyst was prepared by heating a mixture of either VANOL or VAPOL with 2 equiv of the alcohol or phenol 8, 3 equiv of H2O, and 3 equiv of BH3–SMe2 in toluene at 100 °C for 1 h. The solution is cooled to 25 °C and 10 equiv of imine 22b is added to induce boroxinate formation over a period of 30 min. The reaction is initiated by the addition of 1.2 equiv of EDA 11 and allowed to proceed for 24 h at 25 °C. The yields are those of material isolated and purified by chromatography on silica gel. The optical purities were measured by HPLC.
NMR yield with internal standard.
The data in Table 1 were generated from the reactions of the imine 22a, prepared from benzaldehyde and benzhydryl amine, with ethyl diazoacetate to give the aziridine 23a. Nineteen different boroxinate catalysts were prepared from the 14 different phenols and 5 different alcohols shown in the Table. The 14 phenols are ordered in the table according to the degree of asymmetric induction observed for the aziridine 23a ranging from 4-nitrophenol 8a (67–68% ee) to 4-tert-butylphenol 8n (90–94% ee). There is a general correlation between the asymmetric induction observed for aziridine 23a and the electronic nature of the phenol with increased electron density in the phenol leading to higher asymmetric induction. With nitro, bromo, and trifluoromethyl groups in the para-position, lower asymmetric inductions are observed and with methoxyl and tert-butyl groups in the para-position, higher asymmetric inductions are observed.
Small steric effects are observed as well. While the 2,6-dimethylphenol 8k generates a slightly more selective catalyst than phenol 8j, perhaps due to the electron releasing nature of the methyl groups, the 2,6-di-isopropylphenol 8e shows a drop in induction and this is presumably related to the increased size of the substituents in the 2- and 6-positions. A slight drop is seen for the 2,4,6-tri-tert-butylphenol 8d although this drop probably would have been much larger in the absence of the tert-butyl group in the 4-position considering the increased induction observed for the phenol 8n. The five alcohols shown in Table 1 are also ordered according to induction from lowest to highest. The sterically encumbered tert-butyl alcohol 8o gives the lowest induction (74–89% ee), but there is no correlation between the induction and the degree of α-branching in the alcohol. Cyclohexanol gives a higher induction than n-butanol, and this may be due to competing steric and electronic effects with the latter providing cyclohexanol with the capacity to give the highest induction out of all 19 alcohols and phenols tested.
The data in Table 2 were generated from the reaction of the benzhydryl imine 22b, prepared from cyclohexane carbox-aldehyde and benzhydryl amine, with ethyl diazoacetate which gives the aziridine 23b. All entries in Table 2 went to >95% completion. It was deemed important to include a benzhydryl imine derived from an aliphatic aldehyde in the present study since aldehydes in this class tend to give lower asymmetric inductions (78–87% ee) in the aziridination reaction with boroxinate catalysts than do benzhydryl imines derived from aromatic aldehydes (90–95% ee).2,4
The same set of 19 alcohols and phenols were screened in boroxinate catalysts for the aziridination of the cyclohexyl imine 22b, and the results can be found in Table 2. One interesting feature is that of all 38 of the boroxinate catalysts, those generated from VANOL are more effective in the asymmetric induction for the cyclohexyl imine 22b with 16 out of the 19 different alcohols and phenols. The exceptions are phenols 8i, 8f, and 8j. The average difference between the VANOL and VAPOL catalyst for all 19 alcohols and phenols is 4.8% ee. It is interesting that the reverse is true for the imine 22a derived from benzaldehyde. For this imine, the boroxinate catalyst derived from VAPOL gives higher inductions than the VANOL catalyst, albeit by only 2.2% ee averaged over all 19 alcohols and phenols (Table 1). Although the absolute configuration of the phenyl aziridine 23a produced with a boroxinate catalyst has been previously established,2 the cyclohexyl aziridine 23b was just assumed to be homochiral. Thus, we confirmed this assumption by converting aziridine 23b to the amide 24 with (S)-α-methylbenzylamine and the relative stereochemistry was determined by X-ray diffraction (see Supporting Information).
The alcohols and phenols in Table 2 are also ordered according to the degree to which their boroxinate catalysts provide asymmetric induction to the aziridine 23b. The ordering is similar to that observed for the aziridine 23a (Table 1) especially for phenols with electron-withdrawing groups. As with the phenyl imine 22a, there is a trend of higher asymmetric inductions from the reactions of the cyclohexyl imine 22b with catalysts prepared from phenols having electron-donating para-substituents and lower inductions from catalysts prepared from phenols having electron-withdrawing para-substituents. Essentially the same trend is observed for the two imines 22a and 22b with catalysts prepared from aliphatic alcohols with the exception that tetramethylcyclohexanol 8r gives a slightly more effective catalyst than does cyclohexanol 8s for the imine 23b.
Hammett Analysis.
The set of phenols and alcohols shown in Tables 1 and 2 that were chosen include several para-substituted phenols such that a Hammett analysis could be preformed on the nature of these substituents and the asymmetric induction in the formation of aziridines 23a and 23b. The original Hammett relationship was set up for the ionization of para-and meta-benzoic acids. However, the resulting constants and do not work well for the ionization of phenols and for this reason the constant was developed which is based on the ionization of phenols and anilines and which are used in the present analysis.13 All of the data points in Figures 1 to 4 represent reactions that went to >95% completion.12 While some data points represent the average for reactions run more than once, most of the data points are from reactions with a single run.
Figure 1.

Correlation between enantiomer ratio in the formation of aziridine 22a and for substituents in VANOL boroxinate catalyst 4.
Figure 4.

Correlation between enantiomer ratio in the formation of aziridine 23b and for substituents in VAPOL boroxinate catalyst 2.
As can be seen from the plots in Figures 1 and 2 there is a strong correlation between the enantiomeric ratio of the phenyl substituted aziridine 23a and the constants over seven different phenols for both the VANOL and VAPOL ligands. For the VANOL ligand the range is from an enantiomeric ratio of 84:16 for the 4-nitrophenol 8a to 95:5 for the 4-tert-butylphenol 8n (R2 = 0.9829). For the VAPOL ligand the range is from an enantiomeric ratio of 83.5:16.5 for the 4-nitrophenol 8a to 97:3 for the 4-tert-butylphenol 8n (R2 = 0.9656). All of the numerical data can be found in the Supporting Information. It is to be noted that the slopes of the plots for both the VAPOL (−0.50) and VANOL ligands (−0.37) are negative. This is consistent with a situation in which a negatively charged catalyst is mediating a reaction with the substrates H-bonded to the catalyst as in species 4 or 2 (Scheme 1) rather than a catalyst that is acting as a Lewis acid by interacting with Lewis basic substrates as in species 20 or 21 (Scheme 5). The fact that the slope for the VAPOL catalyst is more negative than that for the VANOL catalyst may indicate that the hydrogen bonding of the substrates to the VAPOL boroxinate anionic catalyst are a little more important than those in the VANOL boroxinate anionic catalyst, and this would be consistent with the fact that the VAPOL ligand is more selective than VANOL by 3.3% ee averaged over the seven boroxinate catalysts represented in the plots in Figures 1 and 2. The structures presented in Figures 1 and 2 are intended to be indicative of the catalyst itself where the H-bonding of the iminium substrate to O2 of the boroxinate core was observed by X-ray crystallography.1 The transition state for the aziridination reaction undoubtedly involves the binding of both the imine and diazo substrates and possible transition states have been previously supported by computationally studies.3,10
Figure 2.

Correlation between enantiomer ratio in the formation of aziridine 23a and for substituents in VAPOL boroxinate catalyst 4.
There is also a correlation between the asymmetric induction observed in the formation of the cyclohexyl substituted aziridine 23b and the nature of the para-substituents in the phenoxy group in the boroxinate core, but as can be seen in Figures 3 and 4, the correlation is not as strong as in the formation of the phenyl substituted aziridine 23a (Figures 1 and 2). For the VANOL ligand the range is from an enantiomeric ratio of 82:18 for the 4-nitrophenol 8a to 91:9 for the 4-tert-butylphenol 8n (R2 = 0.8506). For the VAPOL ligand the range is from an enantiomeric ratio of 73.5:26.5 for the 4-nitrophenol 8a to 90:10 for the 4-tert-butylphenol 8n (R2 = 0.7514). The relative ordering of the slopes for the catalysts from the VANOL and VAPOL ligands is the same for the aziridine 23b (−0.21 and −0.33, respectively) as it is for the aziridine 23a (−0.37 and −0.50, respectively). The slopes for the plots with the VANOL and VAPOL catalysts with the cyclohexyl imine 22b (−0.21 and −0.33, respectively) are of a smaller magnitude than those for the VANOL and VAPOL catalysts with the phenyl imine 22a (−0.37 and −0.50, respectively). This is consistent with the higher asymmetric inductions observed for the phenyl imine 22a averaged over the reactions indicated in Figures 1 and 2 (86.1% ee) versus the inductions observed for the cyclohexyl imine 22b averaged over all the reactions indicated in Figures 3 and 4 (75.1% ee).
Figure 3.

Correlation between enantiomer ratio in the formation of aziridine 23b and for substituents in VANOL boroxinate catalyst 4.
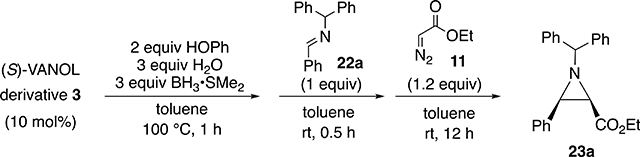
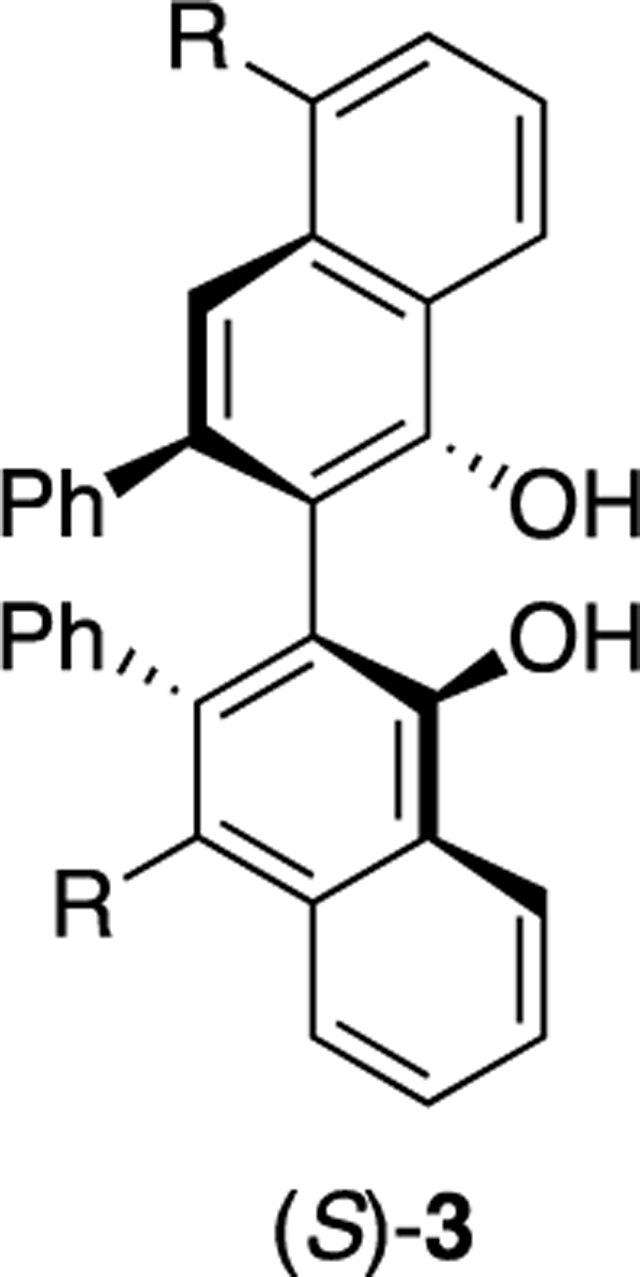
The effect of electronic modulation in the VANOL ligand was also probed on the aziridination of imine 22a with ethyl diazoacetate 11 with the boroxinate catalyst generated from the series of 5,5-disubstituted VANOL ligands shown in Table 3. The 5,5′-disubstituted VANOL ligands 3a to 3f14 were chosen since substituents in the 5- and 5′-positions would be in conjugation with the phenol groups of the VANOL ligands and hence with the boroxinate core of the VANOL boroxinated catalysts of the type 4 generated therefrom (Scheme 1). In addition, substituents in the 5- and 5′-positions would be expected to exert the least steric influence upon the reactions occurring in the active site of the VANOL boroxinate catalysts. However, upon generation and screening of all six of these catalysts, a correlation between the asymmetric induction in the aziridines and the electronic nature of the substitutent in the 5- and 5′-positions of the ligand was not found. Instead, there was no change in the asymmetric induction for any of the substituents except trifluoromethyl where the induction dropped from 89% to 84%. These results suggest that variations of the electronic density in the core of the boroxinate catalyst are much more sensitive to variations in the electron density on the oxygen O-3 in the boroxinate catalyst 4 (or 2) in Scheme 1 than to variations in the VANOL ligand itself. As indicated in Scheme 3, previous mechanistic studies suggested that in the transition state for C–C bond formation the protonated iminium cation is H-bonded (2.17 Å) to O-3 of the boroxinate core (Structure 16).
Table 3.
Aziridination with VANOL Derivatives 3a to 3fa
| ||||
|---|---|---|---|---|
| entry | VANOL ligand 3 | R | % yield 23b | % ee 23a |
| 1 | 3b | OMe | 81 | 89 |
| 2 | 3c | Me | 65 | 89 |
| 3 | 3a | H | 83 | 89 |
| 4 | 3d | Br | 78 | 89 |
| 5 | 3e | Cl | 83 | 89 |
| 6 | 3f | CF3 | 82 | 84 |
The yield and % ee in each entry is the average of two runs.
Isolated yield.
Computational Analysis.
The Hammett analysis is consistent with a mechanism in which the protonated imine is H-bonded to a boroxinate anionic core (Figure 5). Also, the increasing asymmetric induction resulting from addition to the protonated imine with increasing electron releasing ability of the phenol, further supporting the H-bonded model. We previously found by both computation3,10 and X-ray analysis1 that, of the four oxygen atoms in the boroxinate core, the Boroxinate-H-Imine complex 4 places the protonated imine preferentially H-bonded to O2. To gain further support for this mechanistic picture, we performed DFT analysis on the Boroxinate-H-Imine complexes 4 (Figure 5) formed from cyclohexanol, 4-methoxyphenol, phenol, and 4-nitrophenol and from both imines 22a and 22b. The different catalyst-protonated imine complexes were optimized using M06–2X/6–31G* as implemented by Gaussian 16.15 In all of the computed structures, the imine preferentially binds to O2 of the boroxinate core of the catalyst. We analyzed the structural features of the complex and found two significant interactions that correlate with enantioselectivity. A strong H-bonding distance between O2 and the iminium proton in the catalyst-protonated imine complexes (Imine–H–O2) and a weak nonconventional H-bonding interaction between the ortho hydrogen on one of the phenyl rings of the benzhydryl group (benzhydryl–CH–O3). The distances of these interactions along with the corresponding experimental enantioselectivity for the aziridination reaction of these complexes are listed in Table 4.
Figure 5.

Boroxinate H-imine complex with H-bonding of the iminium to O-2.
Table 4.
Calculated H-Bonding Distance between VANOL-Boroxinate Catalyst and Protonated Imines 22a and 22ba
| Boroxinate-H-Imine Complex |
H-Bond Distance (A) |
||||||
|---|---|---|---|---|---|---|---|
| Entry | Imine | R3 | R | %ee | Imine—H—O2 | benzhydryl—CH—O3 | |
| 1 | 22a | 4a | C6H11 | C6H5 | 94 | 1.70 | 2.45 |
| 2 | 22a | 4b | 4-MeOC6H4 | C6H5 | 90 | 1.86 | 2.71 |
| 3 | 22a | 4c | C6H5 | C6H5 | 89 | 1.87 | 2.71 |
| 4 | 22a | 4d | 4-NO2C6H4 | C6H5 | 68 | 1.93 | 2.83 |
| 5 | 22b | 4e | C6H11 | C6H11 | 87 | 1.79 | 2.53 |
| 6 | 22b | 4f | 4-MeOC6H4 | C6H11 | 81 | 1.82 | 2.68 |
| 7 | 22b | 4g | C6H5 | C6H11 | 80 | 1.83 | 2.73 |
| 8 | 22b | 4h | 4-NO2C6H4 | C6H11 | 64 | 1.87 | 2.82 |
The catalyst—iminium complexes were calculated using the M06–2X/6–31G* method.
Analysis of the catalyst-protonated imine complexes revealed a correlation between the asymmetric induction and the H-bonding distance of the protonated imine and O2 of the boroxinate core. The decreasing enantioselectivity upon going from electron-rich cyclohexanol (4a/4e) to the electron-poor p-nitrophenol (4d/4h) as the alcohol used for boroxinate formation is perfectly correlated with the increasing imine H–O2 and benzhydryl-CH–O3 distances in the corresponding catalyst-protonated imine complexes. For imine 22a, the imine H–O2 distance varies from 1.70 Å for cyclohexanol (94% ee) to 1.93 Å for p-nitrophenol (68% ee) while the benzhydryl–CH–O3 varies from 2.45 Å for cyclohexanol to 2.83 Å for p-nitrophenol. For imine 22b, the imine H–O2 distance varies from 1.79 Å for cyclohexanol (87% ee) to 1.87 Å for p-nitrophenol (64% ee) while the benzhydryl–CH–O3 varies from 2.53 Å for cyclohexanol to 2.82 Å for p-nitrophenol. Thus, the trend is that the highest asymmetric induction is observed for those boroxinate complexes that have the shortest H-bond between the protonated imine and O2 of the boroxinate anion. The simplest explanation for this observation is that closer proximity of the protonated imine to the chiral environment of the catalyst anion allows for better energetic discrimination between the transition states leading to the major and minor enantiomer of the product aziridine. This is consistent with the observations made in the Hammett analysis where the most electron-rich phenol in the boroxinate core leads to the highest asymmetric induction in the aziridination reaction.
An electrostatic potential (ESP) slice map in the plane of the NH···O interaction for the strongest (4a) and weakest (4d) H-bonded complexes of imine 22a is shown in Figure 6.16 Here, O2 corresponds to the oxygen to which the dotted line is drawn. When R3 = Cy, O2 is encapsulated by a blue area, indicating a region that is stabilizing for negative charges, and the imine-bound hydrogen is surrounded by a large red area, which is stabilizing for positive charges. When R3 = 4-NO2Ph, the red area is significantly diminished, indicating an area less stabilizing for positive charges, such as the electron-deficient imine-bound hydrogen. Quantitative ESP data calculated with this map indicate that the H-bonding interaction (NH···O) for the R3 = Cy structure is stabilized by 0.8 kcal/mol with respect to the R3 = 4-NO2Ph structure (for details see SI pp 50–53). This is consistent with the trend in the enantiomeric excess for each catalyst derivative (ee = 96% versus ee = 68%, respectively). Finally, the preference for O2 over O4 as the site for imine binding likely results from the fact that O4 is too far away from the vaulted biaryl groups to allow for favorable and stacking interactions between the imine protecting group and the vaulted biaryl catalyst backbone.
Figure 6.

Electrostatic potential slice maps of the Boroxinate-H-Imine complexes with the highest (R3 = Cy, ee = 96%) and lowest (R3 = 4-NO2Ph, ee = 68%) enantioselectivities for catalyst derivatives bearing phenyl substituents on the iminium complex in the plane of the NH···O interaction; red areas are stabilizing for positive charges, blue areas are stabilizing for negative charges. Bond distances are shown in angstroms, and the atoms from the catalyst backbone (this is the blue area in the bottom right-hand quadrant of both structures) have been removed for clarity.
CONCLUSION
A mixture of a vaulted biaryl ligand (either VANOL or VAPOL), 3 equiv of borane–dimethyl sulfide, 3 equiv of water, and 2 equiv of an alcohol or phenol can be used to generate boroxinate catalysts in situ upon addition of an imine. These species are known to mediate the asymmetric catalytic synthesis of aziridines from an imine and ethyl diazoacetate. In this work 38 different boroxinate catalysts were generated in situ from either VANOL and VAPOL and a set of 19 different alcohols and phenols and screened in the aziridination reaction with two different imines. The general observation made is that the more electron-donating the alcohol or phenol, the greater the asymmetric induction in the aziridine product. A set of 7 para-substituted phenols were included in the set to be part of a Hammett study which revealed a strong correlation between the constant and the enantiomeric ratio of the product. The slopes for the catalysts generated from both the VANOL and VAPOL ligands were negative indicating the reaction is favored for catalysts that have increased electron density in the boroxinate core of the boroxinate catalysts. This supports a mechanism in which the boroxinate catalyst functions by activating the imine by proton donation and the resulting chiral anion organizes the iminium as well as the diazo compound by H-bonding interactions. These results are not consistent with an alternative mechanism in which the boroxinate catalyst functions as a Lewis acid and activates the imine by a Lewis acid/Lewis base interaction.
EXPERIMENTAL SECTION
General Experimental.
Methylene chloride and acetonitrile were distilled from calcium hydride under nitrogen. Toluene, tetrahydrofuran, and benzene were distilled from sodium under nitrogen. Hexanes and ethyl acetate were ACS grade and used as purchased. All borane reagents and ethyl diazoacetate were purchased from Sigma-Aldrich and used as is. Benzhydrylamine, benzaldehyde, and cycylohexanecarboxaldehyde were purchased from Sigma-Aldrich. All of the alcohols and phenols 8a to 8s were purchased from Sigma-Aldrich except 8r which was prepared as described below. Liquid alcohols were purified by distillation under reduced pressure while solid alcohols and/or phenols were purified via sublimation. VANOL and VAPOL were prepared according to a literature procedure17 and were determined to be at least 99% optically pure.
Melting points were determined on a Thomas-Hoover capillary melting point apparatus and were uncorrected. IR spectra were taken on a Mattson Galaxy series FTIR-3000 spectrometer. 1H NMR, 13C NMR, and 11B NMR were recorded on a Varian Inova-300 MHz, Varian UnityPlus-500 MHz, or Varian Inova-600 MHz instrument in CDCl3, unless otherwise noted. CDCl3 was used as an internal standard for both 1H NMR (δ = 7.24) and 13C NMR (δ = 77.0), unless otherwise noted. BF3·OEt was used as an internal standard for 11B NMR (δ = 0.0). HRMS was performed in the Department of Biochemistry at Michigan State University using either a Waters Xevo G2-S QT of or a Waters QT of Ultima API mass spectrometer. Analytical thin-layer chromatography (TLC) was performed on silica gel IB2-F plates. Visualization was by short wave (254 nm) and long wave (365 nm) ultraviolet light or by staining with phosphomolybdic acid (PMA) in ethanol or potassium permanganate. Column chromatography was performed with silica gel 60 (230–450 mesh). HPLC analyses were carried out using a Varian Prostar 210 Solvent Delivery Module with a Prostar 330 PDA Detector and a Prostar Workstation. Optical rotations were obtained on a PerkinElmer 341 polarimeter at a wavelength of 589 nm (sodium D line) using a 1.0-decimeter cell with a total volume of 1.0 mL. Specific rotations are reported in degrees per decimeter at 20 °C.
General Procedure for the Preparation of Aldimines.
To a flame-dried round-bottom flask equipped with a stir bar was added magnesium sulfate (1.50–4.00 equiv). Cotton was placed in the neck of the vacuum adapter to prevent the MgSO4 from entering the vacuum line followed by placing the MgSO4 under vacuum and flame-drying the flask until the MgSO4 stopped bouncing (indication of removal of water). Once cooled, CH2Cl2 (2–4 mL/mmol) was added followed by amine (1.00 equiv) and finally aldehyde (1.05 equiv). The reaction flask was sealed with a rubber septum and nitrogen balloon and stirred at room temperature for the specified time. The slurry was then filtered over a pad of Celite 545 on a sintered glass funnel, and the filtrate was concentrated by rotary evaporation to give the crude product. Liquid aldimines were then subjected to the AZ reaction without further purification while solid aldimines were recrystallized prior to the AZ reaction.
Preparation of N-Benzylidene-1,1-diphenylmethanamine (22a).
The general procedure was followed with benzhydryl amine (0.92 g, 5.00 mmol, 1.00 equiv), benzaldehyde (0.56 g, 5.25 mmol, 1.05 equiv), dry CH2Cl2 (15 mL), MgSO4 (1.00 g, 8.40 mmol, 1.70 equiv, freshly dried), and a reaction time of 24 h. The crude product was placed under high vacuum (0.1 mmHg) for 4–6 h to give the imine 22a as a white solid. The imine was purified by crystallization using ethyl acetate and hexanes to give the pure product as white solid crystals (mp = 102–104 °C) in 82% yield (1.11 g, 4.1 mmol). Spectral data for 22a: 1H NMR (CDCl3, 500 MHz) δ 8.45 (s, 1H, CHN), 7.86–7.88 (m, 2H, Ar–H), 7.43–7.44 (m, 7H, Ar–H), 7.35 (t, 4H, J = 7.5 Hz, Ar–H), 7.26 (t, 2H, J = 7.0 Hz, Ar–H), 5.63 (s, 1H, CH) ppm; 13C{1H} NMR (CDCl3, 125 MHz) δ 160.8, 143.9, 136.3, 130.7, 128.5, 128.5, 128.4, 127.7, 127.0, 77.9 ppm. These spectra match those previously reported for this compound.2
Preparation of N-(Cyclohexylmethylene)-1,1-diphenyl-methanamine (22b).
The general procedure was followed with benzhydryl amine (9.15 g, 8.6 mL, 50.0 mmol, 1.00 equiv), cyclohexylcarbaldehyde (5.83 g, 6.3 mL, 52.5 mmol, 1.05 equiv), dry CH2Cl2 (150 mL), MgSO4 (12.0 g, 100 mmol, 2.00 equiv), and a reaction time of 16 h. The crude product was placed under high vacuum (0.1 mmHg) for 4–6 h to give the imine 22b as a white solid. The imine was purified by crystallization using ethyl acetate and hexanes to give the pure product as solid white crystals (mp = 52–54 C°) in 72% yield (9.99 g, 36.0 mmol). Spectral data for 22b: 1H NMR (CDCl3, 500 MHz) δ 7.70 (d, 1H, J = 5.0 Hz), 7.28–7.32 (m, 8H), 7.20–7.24 (m, 2H), 5.32 (s, 1H), 2.29–2.33 (m, 1H), 1.66–1.88 (m, 5H), 1.20–1.37 (m, 5H) ppm; 13C{1H} NMR (CDCl3, 125 MHz) δ 169.0, 144.0, 128.3, 127.6, 126.8, 78.0, 43.5, 29.7, 26.0, 25.4, ppm. These spectra match those previously reported for this compound.2
General Procedure for the Catalytic Asymmetric Aziridination Reaction Given in Tables 1 and 2.
A 50 mL Schlenk flask equipped with a stir bar and Teflon cap was placed under reduced pressure (0.1 mmHg) and flame-dried. The flask was allowed to cool under vacuum followed by purging the flask with nitrogen. Under a nitrogen atmosphere, 10 mol % (S)-ligand (22.0 mg, 0.05 mmol, 0.10 equiv) and a phenol or alcohol (9.4 mg, 0.10 mmol, 0.20 equiv) were added. Toluene (0.5 mL) was added, and the mixture was stirred to dissolve all solids. Borane dimethyl sulfide (2 M in toluene, 75 μL, 0.15 mmol, 0.30 equiv) was added followed by water (2.7 μL, 0.15 mmol, 0.30 equiv). The Teflon valve was sealed under a nitrogen atmosphere, and the reaction was stirred and heated in an oil bath at 100 °C for 1 h. The reaction was then cooled to room temperature and imine (0.50 mmol, 1.00 equiv) was added followed by toluene (0.5 mL). The reaction was stirred at room temperature for 0.5 h followed by the addition of ethyl diazoacetate 11 (62 μL, 0.60 mmol, 1.20 equiv). The Teflon valve was sealed under a nitrogen atmosphere, and the reaction mixture stirred at room temperature for 24 h. Once completed, hexanes (3.0 mL) were added to the Schlenk flask and a precipitate formed. The heterogeneous solution was then transferred to a preweighed 25 mL round-bottom flask. The Schlenk flask was rinsed with methylene chloride (3 mL × 2) that was transferred to the round-bottom flask. The volatiles were removed by rotary evaporation followed by placing the round-bottom flask under reduced pressure (0.1 mmHg) for 4–6 h. The cis/trans ratio, percent conversion, and enamine side products were determined by 1H NMR on the crude reaction mixture. The cis/trans ratios were determined on the crude mixture by assigning the integration of the cis-aziridine ring protons to 1.00 H each. The cis (J = 7–8 Hz) and trans (J = 2–3 Hz) coupling constants were used to identify the two diastereomers. The enamines (8.8 and 9.4 ppm) were calculated based upon the isolated yield of pure aziridine and integration of enamines in the crude 1H NMR. Conversion was 100% unless otherwise stated. The aziridine was purified by column chromatography (silica gel).
Preparation of (2S,3S)-Ethyl 1-Benzhydryl-3-phenylaziridine-2-carboxylate (23a) Using Catalyst Generated from Phenol.
Aziridine 23a was generated from imine 22a (136 mg, 0.50 mmol, 1.00 equiv) according to the Method described above using 10 mol % (R)-VANOL (22.0 mg, 0.05 mmol, 0.10 equiv) to generate the catalyst. Upon workup, the crude 1H NMR spectrum indicated a 100:1 cis/trans ratio (cis J = 7–8 Hz and trans J = 2–3 Hz). The product was purified by column chromatography (silica gel, 3 cm × 30 cm, hexanes/EtOAc 19:1), and 23a was obtained as a white solid (mp = 127–128 °C on 89% ee material) in 83% isolated yield (150.1 mg, 0.42 mmol). The optical purity of 23a was determined to be 89% ee by HPLC analysis (Chiralcel-OD-H, 95:5 hexane/2-propanol = 95/5, at 222 nm, flow rate = 1.0 mL/min, I = 222 nm, tR = 3.40 min (major), 8.36 min (minor)). Spectral data for 23a: Rf = 0.3 (1:9 EtOAc/hexanes); 1H NMR (CDCl3, 500 MHz) δ 7.63 (d, 2H, J = 7.0 Hz), 7.52 (d, 2H, J = 7.0 Hz), 7.43 (d, 2H, J = 7.0 Hz), 7.37 (t, 2H, J = 7.5 Hz), 7.21–7.29 (m, 7H), 3.95–3.99 (m, 3H), 3.24 (d, 1H, 6.5 Hz), 2.70 (d, 1H, J = 6.5 Hz), 1.01 (t, 3H, J = 7.0 Hz) ppm; 13C{1H} NMR (CDCl3, 125 MHz) δ 167.8, 142.6, 142.4, 135.1, 128.5, 127.8, 127.8, 127.6, 127.5, 127.4, 127.2, 77.7, 60.6, 48.01 46.4, 14.0 ppm. These spectra match those previously reported for this compound.2
Preparation of (2S,3S)-Ethyl 1-Benzhydryl-3-cyclohexyl-laziridine-2-carboxylate (23b) Using Catalyst Generated from Phenol.
Aziridine 23b was generated from imine 22b (139 mg, 0.50 mmol, 1.00 equiv) according to the Method described above using 10 mol % (R)-VANOL (22.0 mg, 0.05 mmol, 0.10 equiv) to generate the catalyst. Upon workup, the crude 1H NMR spectrum indicated a 50:1 cis/trans ratio. The product was purified by column chromatography (silica gel, 3 cm × 30 cm, hexanes/EtOAc 19:1), and 23b was obtained as a white solid (mp = 163–165 °C on 79% ee material) in 78% isolated yield (142 mg, 0.39 mmol). The optical purity of 23b was determined to be 79% ee by HPLC analysis (Chiralcel-OD-H, hexane/2-propanol = 99/1, flow rate = 1.0 mL/min, I = 222 nm, tR = 3.26 min (major), 6.66 min (minor)). Spectral data for 23b: Rf = 0.26 (1:9 EtOAc/hexanes); 1H NMR (CDCl3, 500 MHz) δ 7.53 (d, 2H, J = 7.0 Hz), 7.24–7.38 (m, 8H), 4.20–4.28 (m, 2H), 3.64 (s, 1H), 2.29 (d, 1H, J = 7.0 Hz), 1.83 (t, 1H, J = 8.0 Hz), 0.97–1.67 (m, 12H), 0.53–0.55 (m, 1H) ppm; 13C{1H} NMR (CDCl3, 125 MHz) δ 169.7, 142.8, 142.4, 128.4, 128.4, 128.3, 127.5, 127.5, 127.1, 126.9, 78.2, 60.7, 52.2, 43.4, 36.3, 30.8, 30.2, 25.6, 25.4, 14.3 ppm. These spectra match those previously reported for this compound.2
Preparation of the Alcohol 8r.
The reduction of 3,3,5,5-tetramethylcyclohexanone was best performed with sodium borohydride in the presence of silica gel according to the method of Hirano.18 To a flame-dried round-bottom flask equipped with a stir bar was added 3,3,5,5-tetramethylcyclohexanone (0.35 mL, 2.00 mmol, 1.00 equiv), hexanes (20 mL), SiO2 (3.0 g; dried under reduced vacuum (0.1 mmHg) prior to use), and NaBH4 (114 mg, 3.00 mmol, 1.50 equiv). A condenser was placed on the flask with a rubber septum and nitrogen balloon sealing the top of the condenser. The reaction mixture was stirred and heated in an oil bath at 40 °C for 10 h under reflux conditions. The flask was cooled to room temperature, and the mixture was poured over a pad of Celite. The flask was rinsed with ether, and the Celite was flushed with ether. The solution was then concentrated via rotary evaporation to give the crude alcohol 8r (mp = 82–85 °C) in 98% conversion as a white solid (5 h refluxing gave 50% conversion). The alcohol was purified via sublimation (5 mmHg at 35 °C) to give pure 8r (mp = 84–86 °C) in 88% isolated yield (274 mg, 1.80 mmol). Spectral data for alcohol 8r: 1H NMR (CDCl3, 300 MHz) δ 3.88 (dt, 1H, J = 22.4 Hz, 3.3 Hz), 1.70 (d of mult, 2H, J = 12.3 Hz), 1.42 (s, 1H), 1.18 (td, 1H, J = 13.5 Hz, 2.1 Hz), 0.93–1.04 (m, 9H), 0.90 (s, 6H) ppm; 13C{1H} NMR (CDCl3, 125 MHz) δ 66.1, 51.5, 48.8, 35.2, 32.6, 27.7 ppm. These data match those previously reported for this compound.19
Preparation of Aziridine 24 To Verify Absolute Configuration of Aziridine 23b.
Preparation of (2R,3R)-1-Benzhydryl-3-cyclohexylaziridine-2-carboxylic Acid (41).
To a flame-dried 25 mL round-bottom flask equipped with a stir bar was added aziridine 23b (210 mg, 0.57 mmol, 1.00 equiv) and ethanol (2.0 mL). To this suspension was added aqueous KOH (164 mg, 2.80 mmol, 4.90 equiv dissolved in 2.0 mL of water). A condenser was added to the round-bottom flask and sealed with a rubber septum and nitrogen balloon. The mixture was refluxed by heating in an oil bath for 1 h (all solid dissolved). The reaction mixture was cooled, and aqueous citric acid (2 N, 3.0 mL) was added to generate a precipitate. The precipitate was filtered and rinsed with water. The solid was then dissolved in methylene chloride and dried over MgSO4. The drying agent was filtered off, and the solution was concentrated via rotary evaporation. Aziridine carboxylic acid 41 was then directly subjected to the following reaction without further purification. Spectral data for acid 41: 1H NMR (CDCl3, 500 MHz) δ 8.80 (br s, 1H), 7.40 (t, 4H, J = 8.5 Hz), 7.32 (t, 4H, J = 4.0 Hz), 7.26 (q, 2H, J = 7.5 Hz), 3.76 (s, 1H), 2.47 (d, 1H, J = 7.0 Hz), 1.99 (t, 1H, J = 8.5 Hz), 1.49–1.66 (m, 4H), 0.96–1.30 (m, 6H), 0.59–0.64 (m, 1H) ppm; 13C{1H} NMR (CDCl3, 125 MHz) δ 171.2, 141.8, 141.3, 128.7, 128.6, 127.9, 127.8, 127.5, 126.8, 77.5, 53.1, 43.1, 37.1, 30.8, 29.9, 25.9, 25.4, 25.3 ppm. Acid 41 has been previously prepared by this same reaction.20
Preparation of (2R,3R)-1-Benzhydryl-3-cyclohexyl-N-((S)-1-phenylethyl)aziridine-2-carboxamide 24.
To a flame-dried 25 mL round-bottom flask equipped with a stir bar was added aziridine 41 (35.5 mg, 0.10 mmol, 1.00 equiv), DMAP (18.3 mg, 0.15 mmol, 1.50 equiv), and methylene chloride (1.0 mL). The reaction was cooled to 0 °C followed by the addition of DCC (31.0 mg, 0.15 mmol, 1.50 equiv) and (S)-methylbenzyl amine (36.3 mg, 0.30 mmol, 3.00 equiv). The solution solidified directly. However, the reaction was brought to room temperature and stirred overnight as a slurry. The urea was filtered, and the filtrate was washed with HCl (1 M) followed by NaOH (1 M). The organic phase was dried over MgSO4. The drying agent was filtered, and the volatiles were removed via rotary evaporation to give the crude aziridine 24. After purification by column chromatography (silica gel, 2.5 cm × 25 cm, hexanes/EtOAc 4:1), the product 24 was obtained as a white solid (mp = 145.5–148 °C) in 29% yield (13.5 mg, 0.03 mmol), which was recrystallized from hexanes/EtOAc. The solvent evaporated overnight leaving one large crystal, which was submitted to X-ray analysis, and the result has been deposited with the Cambridge Crystallographic Data Centre: CCDC 1863369. Spectral data for aziridine 24: Rf = 0.25 (4:1 hexanes/EtOAc); 1H NMR (CDCl3, 300 MHz) δ 7.06–7.19 (m, 15H), 5.05–5.14 (m, 1H), 3.57 (s, 1H), 2.24 (d, 1H, J = 7.2 Hz), 0.79–1.70 (m, 15H), 0.45–0.59 (m, 1H) ppm; 13C{1H} NMR (CDCl3, 125 MHz) δ 128.6, 128.5, 128.4, 127.8, 127.5, 127.4, 127.3, 127.1, 126.2, 78.1, 52.7, 47.7, 45.1, 37.1, 31.1, 29.8, 26.0, 25.1, 21.4 (one extra sp3 carbon peak than expected) ppm; IR (thin film, cm−1) 3389 (m), 2924 (s), 1655 (s), 1510 (s), 700 (s); HRMS (ESI) m/z: [M + H]+ Calcd for C30H35N2O 439.2749; Found 439.2739; [α]20D +31.6° (c = 1.0, CDCl3).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences (GM 094478).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c01769.
Crystallographic data for compound 24, data for the Hammett analysis and the computational studies (PDF)
Accession Codes
CCDC 1863369 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Contributor Information
Wynter E. G. Osminski, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States
Zhenjie Lu, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
Wenjun Zhao, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
Aliakbar Mohammadlou, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
Xiaopeng Yin, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
Emily C. Matthews, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States
Virginia M. Canestraight, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States
Richard J. Staples, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States
Connor J. Allen, Department of Chemistry, Binghamton University, Binghamton, New York 13902, United States
Jennifer S. Hirschi, Department of Chemistry, Binghamton University, Binghamton, New York 13902, United States
William D. Wulff, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States
REFERENCES
- (1).Hu G; Gupta AK; Huang RH; Mukherjee M; Wulff WD Substrate-Induced Covalent Assembly of a Chemzyme and Crystallographic Characterization of a Chemzyme-substrate Complex. J. Am. Chem. Soc. 2010, 132, 14669–14675. [DOI] [PubMed] [Google Scholar]
- (2).Zhang Y; Desai A; Lu Z; Hu G; Ding Z; Wulff WD Asymmetric Catalytic Aziridination with Borate Catalysts Derived from the VANOL and VAPOL Ligands – Scope and Mechanistic Studies. Chem. - Eur. J. 2008, 14, 3785–3803. [DOI] [PubMed] [Google Scholar]
- (3).Vetticatt MJ; Desai AA; Wulff WD How the Binding of Substrates to a Chiral Polyborate Counterion Governs Diastereoselection in an Aziridination Reaction: H-Bonds in Equipoise. J. Am. Chem. Soc. 2010, 132, 13104–13107. [DOI] [PubMed] [Google Scholar]
- (4).Guan Y; Lu Z; Yin X; Mohammadlou A; Staples RJ; Wulff WD Catalytic Asymmetric Aziridination of Benzhydryl Imines and Diazoacetate Esters with BOROX Catalysts from 3,3′-Disubstituted VANOL Ligands. Synthesis 2020, 52, 2073–2091. [Google Scholar]
- (5).(a) Kaufmann D; Boese R A Borate Propeller Compound as Chiral Catalyst for an Asymmetrically Induced Diels-Alder Reaction. Angew. Chem., Int. Ed. Engl. 1990, 29, 545–546. [Google Scholar]; (b) Thormeier S; Carboni B; Kaufmann DE Chiral boronates–versatile reagents in asymmetric synthesis. J. Organomet. Chem. 2002, 657, 136–145. For a review, see: [Google Scholar]; (c) Dimitrijevic A; Taylor MS Organoboron Acids and Their Derivatives as Catalysts for Organic Synthesis. ACS Catal. 2013, 3, 945. [Google Scholar]
- (6).Loewer Y; Weiss C; Biju AT; Fröhlich R; Glorius F Synthesis and Application of a Chiral Diborate. J. Org. Chem. 2011, 76, 2324–2327. [DOI] [PubMed] [Google Scholar]
- (7).Wang Z; Li F; Zhao L; He Q; Chen F; Zheng C An efficient enantioselective synthesis of florfenicol via asymmetric aziridination. Tetrahedron 2011, 67, 9199–9203. [Google Scholar]
- (8).(a) Ren R; Wulff WD Direct Catalytic Asymmetric Aminoallylation of Aldehydes – Synergism of Chiral- and Non-Chiral Brønsted Acids. J. Am. Chem. Soc. 2011, 133, 5656–5659. [DOI] [PubMed] [Google Scholar]; (b) Zhao W; Huang L; Guan Y; Wulff WD Three Component Asymmetric Catalytic Ugi Reaction – Concinnity from Diversity via Substrate Mediated Catalyst Assembly. Angew. Chem., Int. Ed. 2014, 53, 3436–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Newman CA; Antilla JC; Chen P; Wulff WD Regulation of Orthogonal Functions in a Dual Catalyst System. Subservient Role of a Non-chiral Lewis Acid in an Asymmetric Catalytic Heteroatom Diels-Alder Reaction. J. Am. Chem. Soc. 2007, 129, 7216–7217. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Desai AA; Guan Y; Odom AL; Majumder S; Wulff WD; Vetticatt MJ Self-Assembly of a Library of Polyborate Chiral Anions for Asymmetric Catalysis. Tetrahedron Lett. 2015, 56, 3481–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Briere J-F; Oudeyer S; Dalla V; Levacher V Recent advances in cooperative ion pairing in asymmetric organocatalysis. Chem. Soc. Rev. 2012, 41, 1696–1707. [DOI] [PubMed] [Google Scholar]; (b) Phipps RJ; Hamilton GL; Toste FD The progression of chiral anions from concepts to applications in asymmetric catalysis. Nat. Chem. 2012, 4, 603–614. [DOI] [PubMed] [Google Scholar]
- (10).Vetticatt MJ; Desai AA; Wulff WD Isotope Effects and Mechanism of the Asymmetric BOROX Brønsted Acid Catalyzed Aziridination Reaction. J. Org. Chem. 2013, 78, 5142–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ishihara K; Miyata M; Hattori K; Tada T; Yamamoto H A new Chiral BLA Promoter for Asymmetric Aza Diels-Alder and Aldol-Type Reactions of Imines. J. Am. Chem. Soc. 1994, 116, 10520. [Google Scholar]
- (12).Much of the data from Tables 1 and 2 and from Figures 1 to 4 are from contributions by: Osminski, W. E. G. A Multiplex of Polyborate Brønsted Acids Employed in the Catalytic Asymmetric Aziridination Reaction, Ph. D. Thesis, 2013, Michigan State University.
- (13).Hansch C; Leo A; Taft RW A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar]
- (14).Yin X; Zheng L; Mohammadlou A; Cagnon BR; Wulff WD Resolution of Vaulted Biaryl Ligands via Borates Esters of Quinine and Quinidine. J. Org. Chem. 2020, 85, 10432–10450. [DOI] [PubMed] [Google Scholar]
- (15).Zhao Y; Truhlar DG The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, non-covalent interactions, excited states, and transition elements; two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- (16).(a) Maji R; Mallojjala SC; Wheeler SE Chiral Phosphoric Acid Catalysis: From Numbers to Insights. Chem. Soc. Rev. 2018, 47, 1142–1158. [DOI] [PubMed] [Google Scholar]; (b) Maji R; Wheeler SE Importance of Electrostatic Effects in the Stereoselectivity of NHC-Catalyzed Kinetic Resolutions. J. Am. Chem. Soc. 2017, 139, 12441–12449. [DOI] [PubMed] [Google Scholar]; (c) Seguin TJ; Wheeler SE Stacking and Electrostatic Interactions Drive the Stereoselectivity of Silylium Ion Asymmetric Counteranion Directed Catalysis. Angew. Chem., Int. Ed. 2016, 55, 15889. [DOI] [PubMed] [Google Scholar]
- (17).Ding Z; Osminski WEG; Ren H; Wulff WD Scalable Syntheses of the Vaulted Biaryl Ligands VAPOL and VANOL via the Cycloaddition/Electrocyclization Cascade. Org. Process Res. Dev. 2011, 15, 1089. [Google Scholar]
- (18).Yakabe S; Hirano M; Morimoto T Practical Reduction of Carbonyl Compounds with NaBH4 and Silica Gel in an Aprotic Solvent. Synth. Commun. 1999, 29, 295. [Google Scholar]
- (19).Dean C; Whittaker D Reactions of Cyclohexane Derivatives in Superacids. J. Chem. Soc., Perkin Trans. 2 1991, 1541–1543. [Google Scholar]
- (20).Huang L; Zhao W; Staples RJ; Wulff WD Multifaceted interception of 2-chloro-2-oxoacetic anhydrides: a catalytic asymmetric synthesis of β-lactams. Chem. Sci. 2013, 4, 622. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.