

Abstract
Response to cardiovascular drugs can vary greatly between individuals, and the role of the microbiome in this variability is being increasingly appreciated. Recent evidence indicates that bacteria and other microbes are responsible for direct and indirect effects on drug efficacy and toxicity. Pharmacomicrobiomics aims to uncover variability in drug response due to microbes in the human body, which may alter drug disposition through microbial metabolism, interference by microbial metabolites, or modification of host enzymes. In this review, we present recent advances in our understanding of the interplay between microbes, host metabolism, and cardiovascular drugs. We report numerous cardiovascular drugs with evidence of, or potential for, gut-microbe interactions. However, the effects of gut microbiota on many cardiovascular drugs are yet uninvestigated. Finally, we consider potential clinical applications for the described findings.
Keywords: Cardiovascular agents, gastrointestinal microbiome, microbiota
Introduction
Microorganisms inhabit almost every corner of the human body, from one’s skin to their gut, and may have more biological implications than previously expected (1). These communities of microorganisms are known collectively as the human microbiota, and their genetic material, the microbiome. Due to recent breakthroughs in sequencing technologies, the human microbiome has come out from under the microscope and into a new era of scientific exploration at the molecular level (2). Culture-independent methods for identifying and characterizing the microbiota have provided a less biased method for researchers to compare microbial communities across environments based on their genetic information alone. Massive projects, such as the National Institutes of Health Human Microbiome Project, Flemish Gut Flora Project, LifeLines-DEEP, and MetaHIT (METAgenomics of the Human Intestinal Tract), have been undertaken in the last decade to understand what role these microbes play in human health (2–5). These large-scale projects have resulted in a number of discoveries, including the finding that the diversity and abundance of microbial genes far outnumbers that seen in the human genome (5). Microbial community signatures are also associated with many human disease phenotypes, such as diabetes, atherosclerosis and hypertension (4). Growing evidence supports that microbes play a role in inter-patient variability in drug response and drug toxicity (6,7). For example, metformin is shown to alter the gut microbiome to improve the therapeutic effects of the drug (8). Thus, oral drug response variability and toxicities have been linked to the variation of microbes in the gut.
While evidence-based treatments exist, cardiovascular disease remains a leading cause of death worldwide (9). This phenomenon is due in part to wide inter-individual variation in response to cardiovascular drugs. Human genetic variation has been shown to play an important role in drug response (10).Genetic variants, such as mutations in cytochrome P450 metabolic enzymes, have been shown to modify drug metabolism, drug transport or drug targets, and can be utilized to predict an individual’s treatment response (11). This heterogeneity in drug response is the primary driver of the field of pharmacogenomics, which investigates links between the human genome and drug response or adverse drug reactions. Genetic factors have been observed toaccount for between 20–95% of variability in inter-individual drug response (12) and the microbiome is being increasingly interrogated to explain this unaccounted-for variation.
Pharmacomicrobiomics, a newly emerging field, investigates the effect of population-level microbial genomic variation on drug response and disposition (13). Different microbiomes of the human body, from the gut to the vagina have been associated with variability in drug response (8,14). Pharmacomicrobiomics is concerned with this interaction between pharmacologic agents and the microbiome. Oral drugs encounter the gut microbiome during first pass metabolism and, thus have increased opportunity for microbial interactions compared to drugs administered by a different route. Further, hepatobiliary excretion of oral drugs increases interactions between the gut microbiome and drugs. In this review, we first summarize emerging evidence for microbial interaction with specific drugs, focusing on cardiovascular agents administered orally and on the bacteria of the gut microbiome. Then, we provide a new perspective on the potential translational impact of this data on clinical practice. Finally, we discuss the numerous hurdles to the clinical implementation of pharmacomicrobiomics.
Drug Disposition in the Gastrointestinal Tract and the Role of the Gut Microbiome
The gut microbiome plays a role in effective oral drug therapy. Orally ingested drugs pass through the upper GI tract and small intestine, then enter the large intestine where they encounter the largest and most diverse microbial communities in the human body (Figure 1). Most of these microbes reside in the ileum and colon (distal gut) and are part of a unique ecosystem where there is cross-talk between microbes and the host intestinal epithelium (15,16). Importantly, distinct communities of organisms reside in different parts of the gastrointestinal tract (17). Microbes provide the host with nutrients, modulate the immune system, and encode metabolic pathways missing from the human genome, such as the breakdown of polysaccharides (18). Functional capacity from microbial communities in the gut allows microbes to complement the host’s ability to extract energy from the diet, increasing the efficiency of nutrient utilization. Other known functions of the bacteria include producing vitamins B and K, protecting against pathogens, mediating innate and adaptive immune responses, and metabolizing drugs (2). The dominant bacteria in the gut belong to only a few phyla: Firmicutes, Bacteroidetes, Actinobacteria, Proteobacteria, Fusobacteria, and Verrucomicrobia (19). Together, Firmicutes and Bacteroidetes account for more than 90% of the gut microbiota (20). Although the human GI tract is mainly dominated by Firmicutes and Bacteroidetes, the entire composition of human gut microbiota is highly variable and diverse (21). Orally ingested drugs meet and compete with the microbiome in the gut.
Figure 1.
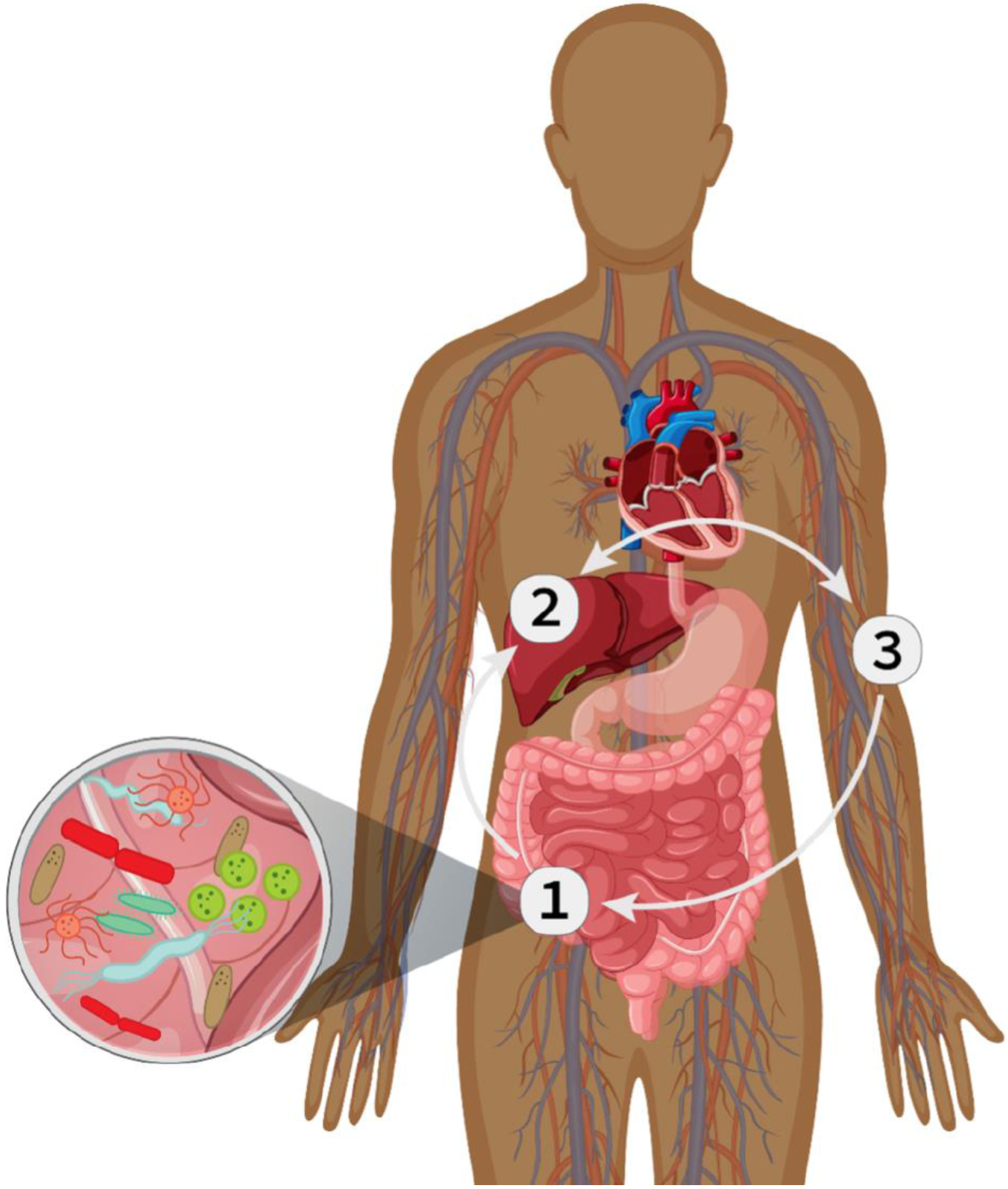
Oral Drug Therapy Meets the Gut Microbiome. 1) Oral drug compounds pass through the stomach and are absorbed in the small intestine where they first meet the gut microbiome. Gut microbes can influence the efficacy of oral drugs by participating in the metabolism of oral drugs, modulating drug transport processes, and altering components of the gastrointestinal tract.2) Compounds are transported to the liver where they may be biotransformed by Cytochrome P450 enzymes. 3) Compounds are circulated systemically and may return to the intestine where they again meet microbes in the gut.
Gut microbes impact the metabolism of drugs through several well-established pharmacokinetic pathways, including production of microbial enzymes that transform drug molecules, production of microbial metabolites that interfere with drug metabolism, alteration of inflammatory pathways that can alter host gene expression, and modification of drug metabolizing genes or enzymes of the host (7,8,22). The gut microbiota can produce enzymes that directly alter drug pharmacokinetics and metabolize drugs into active, inactive, and toxic metabolites (Figure 2a). Due to a large variety in enzymatic activities, the gut microbiota can metabolize compounds over and above host metabolism. This metabolism is responsible for some known toxicities, such as GI disturbances with irinotecan (23). Irinotecan is converted back to its toxic form in the intestine by microbial β-glucuronidase. This cytotoxic metabolite contributes to the dose-limiting GI side effects of irinotecan. The amount of drug that reaches the distal gut, as well as the composition of the host microbiome influences the rate and extent of the microbial metabolism (24). Therefore, drugs that are slowly or partially absorbed in the upper gut may have increased contact with gut microbiota. Moreover, diet, antibiotics, and the presence of digestive enzymes affect the composition of a microbiome (3). Given the number of factors that influence microbiota composition, and the variety of enzymatic processes in microbes, studies are now focused on quantifying the clinical importance of microbial biotransformation of pharmaceuticals (25).
Figure 2.
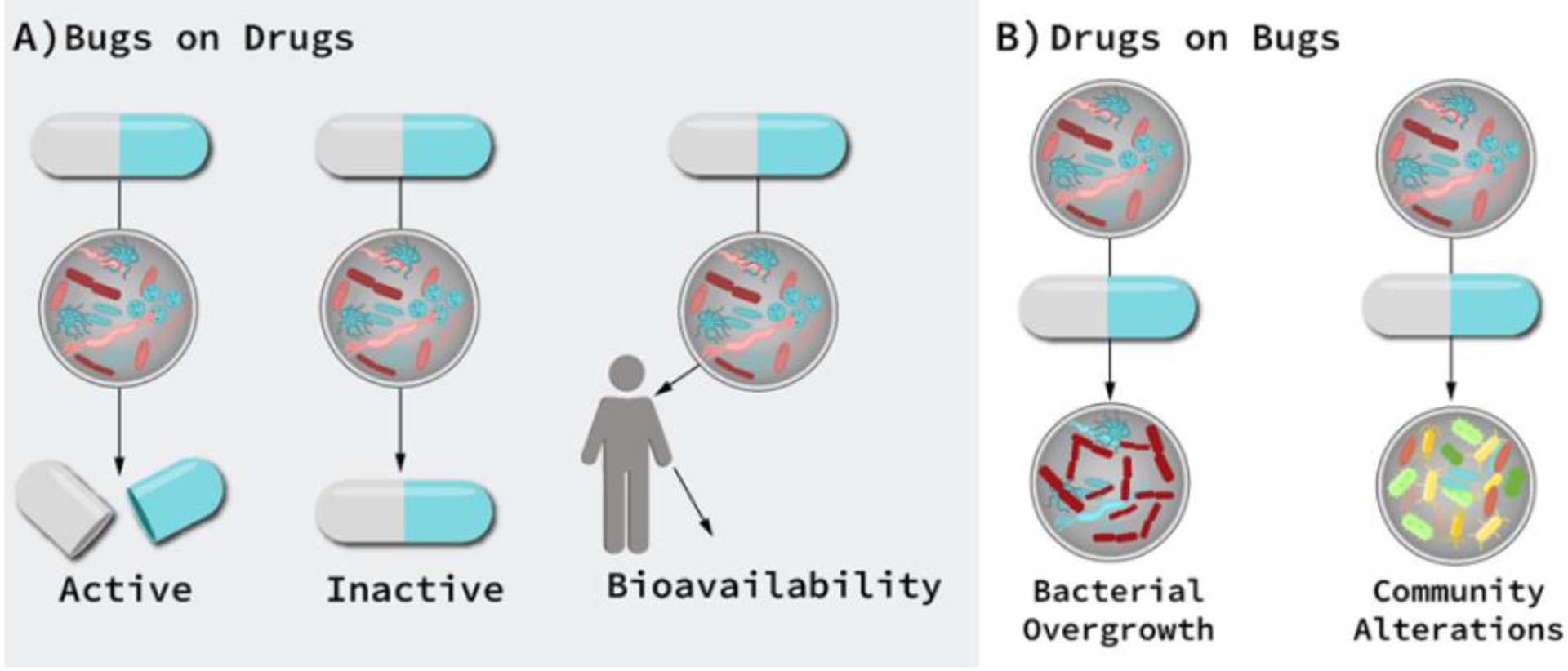
Cardiovascular Drug-microbe effects. A) Direct effect of gut microbiome on drug efficacy and toxicity: microbial transformation can produce active, inactive, or toxic metabolites. B) Indirect microbial effects: Drugs may change the intestinal microenvironment, alter microbial metabolism, and/or affect bacterial growth, thereby altering microbial community composition and function, which indirectly affects drug therapy
The field of pharmacomicrobiomics has attracted much interest in recent years with numerous reports and investigations into drug-microbiome interactions. The human microbiota has been found to activate ellagic acid, modulate the toxicity of irinotecan, and exert major impacts on other chemotherapeutic agents, such as oxaliplatin and cisplatin, through interaction with the immune system (13,26,27). A growing body of evidence has established that gut microbes have the potential to impact drug therapy and has suggested that many of these microbial interactions are clinically meaningful.
Conversely, drugs can have a surprisingly large effect on the microbiome and thus, pharmacodynamic processes. Drugs may change the intestinal microenvironment, alter microbial metabolism, and affect bacterial growth, thereby altering microbial community composition and function (Figure 2b). Antibiotics are a well-studied class of drugs with influences on the gut microbiome. Antibiotics can affect the abundance of bacteria in the gut community, causing rapid and significant drops in taxonomic richness, diversity and evenness, or microbial species abundance in an environment (28). Dysbiosis, an imbalance of microbes, is another term for a shift in the microbiome from a healthy state to one associated with disease. A common clinical example of dysbiosis is a Staphylococcus aureus infection. S. aureus is an opportunistic pathogen that grows rapidly on the skin and in the respiratory tract and may result in severe infection. Dysbiosis due to antibiotics can increase susceptibility to infections, compromise immune homeostasis and deregulate metabolism (29). Other examples of drug therapies affecting the gut microbiome is seen with proton pump inhibitors, metformin, and laxatives (30).
Known and Reported Microbiome-Drug Interactions: High Evidence
Each major drug or drug class discussed below was classified as having “High Evidence”, “Moderate Evidence”, or “Low Evidence” for microbiome-drug interactions. These classifications are based loosely on evidence standards developed by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and were modified for the purposes of evaluation of drug-microbiome interactions (31). High evidence indicates consistent results from rigorous studies. Drugs in this section have known mechanisms, or, as a class, numerous publications observing evidence for associations with the gut microbiome, with at least one study in humans.
Aspirin
In 2016, aspirin was found to be metabolized by bacteria in the gut microbiome during an investigation of the effects of antibiotics on aspirin (32). The study showed that in rats, the antithrombotic activity of aspirin was increased after treatment with ampicillin, thus, obliteration of the gut microbiome modified the pharmacokinetics of aspirin. As a follow-up study, amoxicillin was studied in rats. Investigators noted a reduction in the species and number of gut microbes such as Helicobacter pylori and Prevotella copri in rats treated with both aspirin and amoxicillin. The reduced gut microbiome subsequently affected metabolic activity by decreasing the rate and amount of aspirin converted to the metabolite salicylic acid (33). Systemically, this would equate to increased plasma concentrations of aspirin in order to exert antithrombotic activity since less is being biotransformed in the gut. Again in 2020, aspirin was linked to the gut environment when Zhao et al. found Lysinibacillus sphaericus to influence the bioavailability in mice by degrading aspirin (34). Germ-free mice fed L. spaericus had lower plasma levels of aspirin than controls. Finally, in humans, microbial differences were so stark between aspirin users and non-users that Bacteroides (and the Ruminococcaceae family) could discriminate between users and non-users with an Area Under the Receiver Operator Curve of 0.83, suggesting a strong effect of aspirin on the gut microbiome (35).
Digoxin
The ability of the gut microbiome to interfere with the bioavailability of digoxin has been under investigation for many years after the finding that approximately 10% of patients receiving digoxin had substantial conversion to the inactive digoxin metabolite, dihydrodigoxin, and required increased doses (36,37). In the early 1980’s it was demonstrated that the inactive metabolite of digoxin was formed in the GI tract as the result of the microbe Eubacterium lentum, a member of the Actinobacteria phyla (27,37). In the late 1980’s a study found that the prevalence of E. lentum in the stool samples from New York were similar to those in Southern India, but the proportion able to inactivate digoxin was significantly less for Southern Indians. The authors speculated that the differences were environmental in nature, but the exact variables were not determined (38). In the past decade, E. letnum, now called E. lenta, was closely investigated to reveal certain strains expressed the cytochrome-encoding operon (cardiac glycoside reductase), which was upregulated due to digoxin exposure, resulting in digoxin inactivation (39). Most recently, details of the interaction between digoxin and cardiac glycoside reductase have been described (40). The inactivation of digoxin in the gut is one of the most well-established associations between a cardiovascular drug and the gut microbiome.
Statins
While the efficacy of statins has been proven through numerous randomized trials, there is evidence that statin use increases the risk of type 2 diabetes (41,42). Type 2 diabetes is multifactorial, and recent studies have highlighted the importance of gut microbiome contributing to the etiology of diabetes (43). A study investigating statin treatment and the development of type 2 diabetes in a mouse model found profound changes in the microbial composition of the gut following statin treatment. Bacteroidales spp. dominated the gut of statin-treated mice and decreased the presence of the gram-positive phylum Firmicutes. This shifts the microbiota towards a higher capacity for energy production (from butyrate towards acetate, lactate and succinate), similar to what is observed in diet-induced obesity-linked gut microbiota (44). Statin use also has proposed associations with infections. There is a growing body of literature demonstrating reduced risk of bacterial infection in patients taking statins. Retrospective studies have suggested that statins can reduce mortality among patients with bacteremia and pneumonia (45–47). Another study suggests reduced incidence of sepsis, a serious reaction to infection, among statin users (48). Epidemiological studies have reported a reduced risk of infections postoperatively among statin users. One particular study among hospitalized patients found a 0.78 risk reduction in the development of C. diff (member of the Firmicutes phylum) in statin users (45,49). Although numerous studies have reported improved survival in severe bacterial infections among statin-treated patients, a meta-analysis of these publications did not reach statistical significance (50). Below we review the statins that have individually been linked to drug-microbiome interactions using various study designs, including population analyses, animal models, and metabolomics studies.
Atorvastatin
Atorvastatin increased bacterial diversity in rats given a high fat diet towards amounts seen in the control group who were fed a normal chow diet (51). Conversely, the gut microbiome has been shown to influence the cholesterol lowering effects of atorvastatin. In mice with antibiotic induced microbiome depletion, the lipid lowering effects of atorvastatin were decreased and the expression of hepatic and intestinal cholesterol-regulating genes (Ldlr, Srebp2, and Npc1l1) were altered (52). In addition to the effects on cholesterol, mice models exhibit changes in intestinal bile acid levels, which the authors hypothesize is due to the gut microbiome. It is postulated that bacteria are needed to enzymatically catalyze the deconjugation of bile acid so that bile can be passively absorbed from the intestine to circulation (53). Changes in the microbial composition would explain why decreased bile acid in the intestine and increased fecal bile acid excretion were observed (54). In atorvastatin treated humans with hypercholesterolemia, the drug helped restore the relative abundance of several dominant and important taxa such as anti-inflammatory-associated bacteria Faecalibacterium prausnitzi and Akkermansia muciniphila, that were disrupted in hypercholesterolemia (55).
Lovastatin
Incubations of lovastatin with control and antibiotic treated human and rat fecalase preparations produced the following metabolites: demethylbutyryl metabolite, hydroxylated metabolite, active M8 hydroxy acid metabolite and hydroxylated M8 (56). Additionally, in rats concomitantly treated with antibiotics, the system concentration of active M8 metabolite was lower. These results suggest that microbes in the gut may be involved in lovastatin metabolism and antibiotic intake may reduce the intestinal bacteria’s biotransformation of oral lovastatin.
Rosuvastatin
In a mouse study, rosuvastatin treatment significantly affected bile acid metabolism (57). In rats, dysbiosis of intestinal microbes was associated with reduced efficacy of rosuvastatin on lowering blood LDL (58). These studies suggested that rosuvastatin affects gut microbial composition, prompting further studies in humans as a result. The first showed the efficacy of rosuvastatin was associated with microbial variation in community composition. Sixty-four hyperlipidemic patients were given rosuvastatin (10mg/day) and divided into rosuvastatin responders and non-responders’ groups based on the blood lipid levels being above or below normal. The gut microbiome from stool collected had major differences in bacteria taxa present. In rosuvastatin responders, there was a higher abundance of Firmicutes, butyrate-producing bacteria such as Ruminococcaceae, Lachnospiraceae, Clostridiaceae and a lower abundance of Bacteroidetes (59). The second study provides conflicting evidence as stool samples from the randomized control trial of 66 participants given either rosuvastatin (20mg/day) or placebo did not show any significant changes in microbial composition. They did find changes in microbial gene expression for those encoding proteins related to the choline/betaine-TMA-pathway, suggesting rosuvastatin affects metabolism by the microbes in the gut (60).
Simvastatin
In a systematic evaluation of small molecules, Kaddurah-Daouk et al. investigated the correlation between baseline metabolites and therapeutic efficacy of simvastatin in 100 individuals from a cholesterol study to predict response to statin treatment (61). Results indicate that microbial-derived secondary bile acids, including taurocholic acid and glycocholic acid, and coprostanol can predict a LDL lowering effect of simvastatin, suggesting gut microbial impacts. Coprostanol, used as a biomarker for the presence of human fecal matter, is produced by intestinal bacteria by hydroxylation of cholesterol (62). The results suggest that patients with higher pretreatment levels of coprostanol producing bacteria will respond better to statins. Interestingly, simvastatin is being investigated for roles outside of the traditional lipid lowering health effects. Many countries have concerns of antimicrobial resistance developing such that antibiotics used to treat common infection will no longer be effective. One way to combat this future is the use of non-antibiotic drugs that act as “breakers” to antimicrobial resistance through synergistic effects with antibiotics; one such drug is simvastatin. Simvastatin appears to have antibacterial effects on many gram-positive bacteria such as Staphylococcus and Streptococcus, thus making the drug a good candidate for future studies as a antimicrobial resistance breaker (63).
Likely Microbiome Drug Interactions: Moderate Evidence
Drugs in this section have likely microbial associations, however, studies have yet to be performed in humans, have limited evidence, have conflicting evidence or have not been directly investigated. The drugs are listed in order of the amount of evidence.
Angiotensin-converting-enzyme inhibitors
In rats, captopril has been shown to modulate gut microbiota. In a study investigating the involvement of the gut microbes in rat hypertension, captopril normalized blood pressure and was associated with reversal of hypertension-linked gut pathology (64). The authors conclude that dysbiosis plays a key role in hypertension and a clinical trial is underway to show this relationship in humans. Another study in rats by the same lab found increased bacterial abundance with captopril treatment, even following captopril withdrawal (65). The authors suggest these results demonstrate that long-lasting effects of captopril are associated with alterations in gut microbiota. In addition to long-lasting effects of captopril, another study in rats suggests that maternal captopril treatment attenuates hypertension in male offspring by altering the gut microbiota, specifically the Clostridiales and Erysipelotrichales orders (65,66). Thus, captopril has been shown to affect the gut microbiome with lasting and potentially transgenerational effects. Similar changes to gut microbiome were seen in a rat study with benazepril, where authors noted changes in both Firmicutes to Bacteroidetes and Coccus to Bacillus ratios (67). In a rat study using enalapril, they interestingly did not observe any changes in the species of the gut microbiome, however, they found lower plasma levels of trimethylamine N-oxide (TMAO), which is a metabolite produced bacteria that has links to increased cardiovascular risk. This suggests that enalapril could be affecting the microbiome’s ability to produce the harmful metabolite (68).
Angiotensin Receptor Blockers
As a class, angiotensin receptor blockers have evidence for perturbations in the gut microbiome. Telmisartan reduced colonic inflammation in rats with colitis (69). Inflammatory bowel diseases, including ulcerative colitis, result from inappropriate activation of the GI immune system by intestinal microbiota (70). The discovery of reduced colonic inflammation by telmisartan led to an investigation into the drug’s effect on atherosclerosis (71). In mice, a high fat diet was associated with lower colonic microbial diversity. Telmisartan therapy, however, restored an induced dysbiotic ratio of Firmicutes/Bacteroidetes, leading to positive effects downstream. In fact, telmisartan was more effective than the probiotic Lactobacillus rhamnosus in altering the gut microbiota. Candesartan and losartan therapy in hypertensive mice similarly restored the altered Firmicutes to Bacteroidetes ratio and protected against pathophysiological alterations due to hypertension in the gut (72,73). Irbesartan improves stress-induced changes of the microbiota. Stress markedly decreases percentages of fecal taxa, while treatment with irbesartan corrected these changes in community structure (74). Conversely, Olmesartan has a unique adverse drug effect called sprue like enteropathy, which is characterized by chronic diarrhea, weight loss, and biopsy revealing villous atrophy and mucosal inflammation. In patients seen at the Mayo Clinic, discontinuation of olmesartan improved symptoms and brought about histological recovery (75). It is unclear why olmesartan’s effects on the GI tract differ from other drugs of the ARB class like telmisartan, candesartan, losartan and irbesartan, which show beneficial effects to the gut microbiota.
Calcium Channel Blockers
Amlodipine is a dihydropyrimidine calcium channel blocker that is frequently prescribed for the treatment of hypertension. A study by Yoo et al. reported amlodipine metabolism in human fecalase incubations, indicating involvement of the gut microbiota in the metabolism of amlodipine (76). Unchanged amlodipine decreased in 24-hr incubations with human fecalase by 9%. Additionally, they showed antibiotic intake may increase the bioavailability of amlodipine in rats, further indicating microbial involvement. At ampicillin doses of 10mg/kg and 20mg/kg, the plasma concentration of amlodipine increased by 42 and 133% when compared to controls (76). Amlodipine alone also appears to have bactericidal properties. Of the 504 bacteria strains screened, amlodipine had varying degree of antimicrobial effects, but most notably had good sensitivity towards Staphylococcus aureus, Vibrio cholerae, Vibrio parahemolyticus, Shigella species, Salmonella species, and Bacillus species (77). Song et al. reported improvements in GI side effects of amlodipine therapy with probiotics (78). Amlodipine, as well as bacterial dysbiosis, has been associated with diarrhea. The known side effect and association between dysbiosis and diarrhea led the authors to investigate amlodipine and probiotics in a rat model. The probiotic blend alleviated intestinal complications and induced compositional changes in the gut microbiota of rats. Ahn et al. followed up by investigating what metabolite alterations occurred due to the induced microbiota compositional change in amlodipine versus amlodipine and probiotic treated mice (79). Lipid metabolites in amlodipine treated mice had increased levels of phosphatidylcholines, triglycerides with large numbers of double bonds, and cholesterols. The amlodipine treated group also had decreased levels of triglycerides with small numbers of double bonds. The authors believed this to be evident that amlodipine shifted the composition and balance of bacterial produced metabolites that ultimately contribute to the GI side effects (79).
Similarly, the calcium channel blockers nifedipine and nimodipine have been investigated for microbial attenuation. Nifedipine and nimodipine have similar mechanisms of action in the treatment of hypertension and thus evidence for one drug’s relationship with the gut microbiome is potential evidence for the other. Due to the high permeability of nimodipine through the GI tract, a study investigated its bacterial degradation in human fecal dilutions. Using simulated colonic bacteria as a model, Vertzoni et al. observed rapid degradation of nimodipine in fecal material (80). More recently, Zhang et al. demonstrated gut microbes are involved in the metabolism of nifedipine in vitro (81). After a 24-hr incubation in rat fecal suspensions, nifedipine concentrations were reduced by over 50%, proving intestinal microbes are associated with metabolism and biotransformation of nifedipine. Finally, although unclear which calcium channel blocker the study is referring to, a high-throughput drug screen found these drugs are rich with anticommensal activity, reflecting their potential to modulate the human gut microbiota (82).
Warfarin
Dietary vitamin K is known to interact with warfarin efficacy: Patients are instructed to keep vitamin K levels stable throughout warfarin therapy. Because there are known bacteria in the gut that produce vitamin K, such as Escherichia coli of the Proteobacteria phyla, it is likely that bacterially produced vitamin K plays a role in warfarin efficacy (83). Numerous antibiotics interact with warfarin therapy, causing over-anticoagulation (84). Many antibiotics disrupt gut flora, thereby reducing intestinal vitamin K synthesis. Notably, some antibiotics also inhibit CYP enzymes which metabolize warfarin. Some specific gut bacteria implicated in warfarin response were identified when stool and plasma samples of 200 patients undergoing heart valve replacement were analyzed based on their response to warfarin being low, high, or normal. The genera Escherichia and Shigella were more abundant in the low responder group and the amount of vitamin K produced was increased compared to the high responder group, which had had more Enterococcus and less vitamin K produced by their gut microbiota (85). Finally, in a large previously mentioned drug screening study, three of four vitamin K antagonists studied showed anti-commensal activity, reflecting their ability to affect microbes (82).
Potential for Microbiome-Drug Interactions: Weak Evidence
Drugs in this section have the potential for microbial modulation mechanisms. For each drug listed, study results were non-specific, evidence was not translatable to humans, or design was not directly investigating the drug-microbiome relationship. For example, oral heparin was investigated for a gut microbial association, however, heparin is not delivered orally in humans. The drugs are listed in order of amount of evidence.
Amiodarone
Amiodarone induced organ toxicity is potentially severe due to the long half-life of the drug, and thus the bioavailability of amiodarone is of special importance. Amiodarone has been shown to affect and be affected by representative gut bacterial strains. Concomitant administration of the probiotic Escherichia coli Nissle 1917 demonstrated increases in bioavailability of amiodarone (43%) and its metabolite N-desethylamiodarone in rats (86). While increased activity of CYP enzymes which metabolize amiodarone is always possible, the authors suggest possible modulation of the metabolism of amiodarone by E. coli Nissle 1917 administration, leading to better drug absorption in the GI tract. Another study suggested that amiodarone has in vitro bactericidal effects on human pathogenic strains such as S. epidermidis, E. coli, and Klebsiella (87). Both studies suggest that amiodarone therapy has a microbiomic component, albiet through different mechanisms.
Clopidogrel
Clopidogrel was nominally associated with numerous gut microbiota profiles in the Twins United Kingdom cohort, suggesting a potential interaction between clopidogrel and the gut microbiome (25). In another large population-based metagenomic analysis, clopidogrel was among the class of platelet inhibitors that was associated with increased microbial diversity (4). Finally, a metabolomic analysis on plasma metabotypes (groups of metabolically similar metabolites) brought to light the potential role of dysbiotic gut microbiota inducing the adverse drug event high on treatment platelet reactivity , which hinders achieving therapeutic outcome. Plasma metabotypes contained dysbiotic gut microbiota associated metabolites such as choline, trimethylamine, and L-phenylalanine. Although high on treatment platelet reactivity is multifactorial, it is associated with a dysbiosis in the gut microbiome (88).
Heparin
Although heparin is not currently administered orally in humans due to poor bioavailability, oral administration of heparin was shown to increase bacterial taxa in mice (89). Predominant bacterial communities were different, where some species of bacteria were increased and some decreased, in heparin treated rats compared to saline treated controls. The authors conclude that orally administering heparin could act as an effective gut microbiota modulator by facilitating colonization of Lactobacillus, a member of the Firmicutes phyla. In humans, fecal samples from six volunteers were treated with heparin and two strains of heparin-degrading bacteria, Bacteroides ovatus A2 and Bacteroides cellulosilyticus B19 were identified. The degradation products from bacteria affected the heparins antithrombin binding site, leading to loss of therapeutic activity (90). Although oral delivery of heparin may affect gut microbial community structure, in humans the drug is delivered intravenously. In the 1990’s, however, heparin was serendipitously used as an ulcerative colitis therapy, showing evidence of a potential gut microbiome modulation. Heparin was being used to treat thromboembolic problems, that patients with colitis are highly susceptible to, and showed complete remission of colitis in patients treated with heparin (91,92).
Future Studies and Translational Pharmacomicrobiomics
We have provided a summary of evidence for cardiovascular drugs that have the potential to interact with microbes in and on the human body, however clinical implications for many of these microbes remain unknown. Many reports show alterations in pharmacokinetics of drugs in conditions affecting the gut microbiome, such as germ-free animals or antibiotic treatment. Although only a small number of these studies directly investigated human gut microbiota interactions with xenobiotics, the sheer number of associations with the gut microbiome suggests the need to further investigate these interactions.
Most drugs, let alone cardiovascular drugs, have yet to be investigated regarding their relationship with the human gut microbiome. The lack of investigations is potentially due to the challenges of studying the microbiome such as the need for closely matched controls, differences in the gut microbiome due to diet, diseases, or environmental factors. While gut microbiome samples such as stool samples are abundantly available non-invasively, the relevance of variation in microbial communities in such samples to the site of interaction with a drug or drug metabolite is not always clear. Marques et al. provide an in-depth guideline for reporting on gut microbiome analysis on animal models and human cohorts in experimental hypertension which can be applied to additional phenotypes (93). Using such guidelines, results from different studies are likely to be more easily comparable and conclusions more easily drawn.
Future pharmacological studies should aim to collect data on patients that will best describe variability of a patient’s microbiome, with the complex drug-host-microbe interaction in mind. For example, it will be important to assess whether microbiomic alterations explain inter or intra dose variability and responses across demographic characteristics or comorbidities. Potential confounders of the drug-microbiome interactions should also be evaluated, including whether regional microbe variation of the GI tract impacts drug response through physiologic conditions such as bowel transit time or competition of metabolites with drug transporters. For drug-microbe interactions with sufficient evidence of clinical validity, future study design should ideally be able to assess causality as well as clinical utility in real world settings. Finally, the clinical utility of the gut microbiome above and beyond standard clinical factors, such as liver or kidney function, needs to be assessed prior to clinical implementation. Additional studies with models of the GI tract, advanced sequencing approaches, and computational prediction techniques will be required to assess clinical utility by characterizing the relationship between the gut microbiome and cardiovascular drug therapy. Ex vivo colon models and model systems, such as rodent or worm models, may be an approach to replicate the complexity and dynamics of the communities of the human gut microbiome. Further, community level analysis of microbiome function through multi-omic sequencing combined with in silico assessment of relationships between the gut microbiome and drug therapy may provide insights into the effects of gut microbes ondrug kinetics and eventually help to predict clinical outcomes (94).
Involvement of gut microbiota may account for therapeutic variability in drugs that thus far has been undescribed. There are potential clinical applications for the association between the gut microbiota and cardiovascular drug efficacy. Specific microbial taxa or indices of diversity in the gut microbiome may be of use in predicting response to drug treatment, or to stratify individuals who may be best suited for a therapy. One such drug that can potentially be utilized clinically is warfarin, which has a narrow therapeutic index(95). Indices of gut microbiome taxa or diversity might improve understanding of variables affecting warfarin anticoagulation, or adverse drug events, as well as be used as a biomarker to keep patients within the safe therapeutic window or to find patients who may be better suited for alternative anticoagulation therapies. Early evidence indicates the gut microbiome of warfarin users with higher doses is enriched with menaquinone biosynthesizing genes. Microbial profiles could provide valuable information about treatment outcomes and could contribute to a more personalized approach to therapy. However, the clinical utility of the gut microbiome above and beyond standard clinical prediction needs to be assessed prior to implementation of warfarin pharmamicrobiomics in the clinic. It may eventually be feasible to alter the composition of the microbes in the GI tract in order to increase efficacy or decrease toxicity of cardiovascular therapies via probiotic treatment or fecal transplant. Additionally, investigation into the involvement of the gut microbiota on cardiovascular drug therapy might also identify novel roles for proteins that constitute druggable targets, leading to development of novel drugs or repurposing of existing drugs.
Conclusion
More than ten years after the completion of the human microbiome project, we are still likely at the tip of the iceberg with our understanding of microbial interactions with drugs. Gut microbes possess various biotransformation capabilities to metabolize drugs, and otherwise indirectly affect drug metabolism. Moreover, microbial communities can vary given host genetic variation, inflammation in the gut, and crosstalk at the interface between gut epithelial cells and microbiome (96). Thus, the gut microbiome is emerging as a key to personalized medicine. Existing evidence suggests that many drugs used for the treatment of cardiovascular disease have strong interactions with the gut microbiome. Translational pharmacomicrobiomics has the potential to combine genetic profiles with personal microbiomes to predict drug response and modulate the gut microbiome to improve drug efficacy on the individual level.
Acknowledgements:
We thank reviewers whose comments helped improve and clarify this manuscript. This work is supported by an institutional career development award from the University of Arizona Health Science Center (JHK) and a Seed Grant to Promote Translational Research in Precision Medicine from the Flinn Foundation (JHK). JHK is supported by the National Heart, Lung, and Blood Institute (NHLBI, K01HL143137, R01 HL158686), JBG is supported by the National Institute of Environmental Health Sciences (T32 ES007091).
Abbreviations:
- GI
indicates gastrointestinal
- CYP
indicates cytochrome P450
- spp
indicates multiple species
- OTU
operational taxonomic unit
- AUC
area under the curve
- ROC
receiver operating curve
- LC-MS
Liquid Chromatography– Mass Spectrometry
- GC-MS
Gas chromotragraphy – mass spectrometry
- LDL
low density lipoprotein
- TMAO
trimethylamine N-oxide
- HPLC
high performance liquid chromatography
- ULPC-MS
Ultra-performance liquid chromoatography-Mass Spectrometry
Footnotes
Conflict of Interest Statement: The authors declare no conflicts of interest.
References
- 1.Gilbert J, Blaser MJ, Caporaso JG, Jansson J, Lynch SV, Knight R. Current understanding of the human microbiome. Nat Med. 2018 Apr 10;24(4):392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007 Oct 18;449(7164):804–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, et al. Population-level analysis of gut microbiome variation. Science. 2016 Apr 29;352(6285):560–4. [DOI] [PubMed] [Google Scholar]
- 4.Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T, et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science. 2016 Apr 29;352(6285):565–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010 Mar 4;464(7285):59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015 Dec 10;528(7581):262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018 Jan 5;359(6371):91–7. [DOI] [PubMed] [Google Scholar]
- 8.Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Mannerås-Holm L, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med. 2017. Jul;23(7):850–8. [DOI] [PubMed] [Google Scholar]
- 9.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet Lond Engl. 2012 Dec 15;380(9859):2095–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roden DM, Wilke RA, Kroemer HK, Stein CM. Pharmacogenomics: The genetics of variable drug responses. Circulation. 2011 Apr 19;123(15):1661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang K-L, Weitzel K, Schmidt S. Pharmacogenetics: Using Genetic Information to Guide Drug Therapy. Am Fam Physician. 2015 Oct 1;92(7):588–94. [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, McLeod HL, Weinshilboum RM. Genomics and Drug Response. N Engl J Med. 2011 Mar 24;364(12):1144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doestzada M, Vila AV, Zhernakova A, Koonen DPY, Weersma RK, Touw DJ, et al. Pharmacomicrobiomics: a novel route towards personalized medicine? Protein Cell. 2018. May;9(5):432–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klatt NR, Cheu R, Birse K, Zevin AS, Perner M, Noël-Romas L, et al. Vaginal bacteria modify HIV tenofovir microbicide efficacy in African women. Science. 2017 Jun 2;356(6341):938–45. [DOI] [PubMed] [Google Scholar]
- 15.Wells JM, Rossi O, Meijerink M, van Baarlen P. Epithelial crosstalk at the microbiota-mucosal interface. Proc Natl Acad Sci U S A. 2011 Mar 15;108 Suppl 1:4607–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe. 2015 May 13;17(5):662–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crespo-Piazuelo D, Estellé J, Revilla M, Criado-Mesas L, Ramayo-Caldas Y, Óvilo C, et al. Characterization of bacterial microbiota compositions along the intestinal tract in pigs and their interactions and functions. Sci Rep. 2018 Aug 24;8(1):12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rowland I, Gibson G, Heinken A, Scott K, Swann J, Thiele I, et al. Gut microbiota functions: metabolism of nutrients and other food components. Eur J Nutr. 2018;57(1):1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Argenio V, Salvatore F. The role of the gut microbiome in the healthy adult status. Clin Chim Acta Int J Clin Chem. 2015 Dec 7;451(Pt A):97–102. [DOI] [PubMed] [Google Scholar]
- 20.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. 2011 May 12;473(7346):174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajilić-Stojanović M, Smidt H, de Vos WM. Diversity of the human gastrointestinal tract microbiota revisited. Environ Microbiol. 2007. Sep;9(9):2125–36. [DOI] [PubMed] [Google Scholar]
- 22.Sultana J, Cutroneo P, Trifirò G. Clinical and economic burden of adverse drug reactions. J Pharmacol Pharmacother. 2013. Dec;4(Suppl 1):S73–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wardill HR, Gibson RJ, Van Sebille YZA, Secombe KR, Coller JK, White IA, et al. Irinotecan-Induced Gastrointestinal Dysfunction and Pain Are Mediated by Common TLR4-Dependent Mechanisms. Mol Cancer Ther. 2016. Jun;15(6):1376–86. [DOI] [PubMed] [Google Scholar]
- 24.Hall SD, Thummel KE, Watkins PB, Lown KS, Benet LZ, Paine MF, et al. Molecular and physical mechanisms of first-pass extraction. Drug Metab Dispos Biol Fate Chem. 1999. Feb;27(2):161–6. [PubMed] [Google Scholar]
- 25.Jackson MA, Verdi S, Maxan M-E, Shin CM, Zierer J, Bowyer RCE, et al. Gut microbiota associations with common diseases and prescription medications in a population-based cohort. Nat Commun. 2018 Jul 9;9(1):2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El-Kattan A, Varma M. Oral Absorption, Intestinal Metabolism and Human Oral Bioavailability [Internet]. Topics on Drug Metabolism. IntechOpen; 2012. [cited 2021 Jun 9]. Available from: https://www.intechopen.com/books/topics-on-drug-metabolism/oral-absorption-intestinal-metabolism-and-human-oral-bioavailability- [Google Scholar]
- 27.Saha P, Yeoh BS, Singh R, Chandrasekar B, Vemula PK, Haribabu B, et al. Gut Microbiota Conversion of Dietary Ellagic Acid into Bioactive Phytoceutical Urolithin A Inhibits Heme Peroxidases. PloS One. 2016;11(6):e0156811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011 Mar 15;108 Suppl 1:4554–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Francino MP. Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front Microbiol. 2015;6:1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weersma RK, Zhernakova A, Fu J. Interaction between drugs and the gut microbiome. Gut. 2020 Aug 1;69(8):1510–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Relling MV, Klein TE. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin Pharmacol Ther. 2011. Mar;89(3):464–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim IS, Yoo D-H, Jung I-H, Lim S, Jeong J-J, Kim K-A, et al. Reduced metabolic activity of gut microbiota by antibiotics can potentiate the antithrombotic effect of aspirin. Biochem Pharmacol. 2016 Dec 15;122:72–9. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Sun Y, Wang R, Zhang J. Gut Microbiota-Mediated Drug-Drug Interaction between Amoxicillin and Aspirin. Sci Rep. 2019 Nov 7;9(1):16194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao R, Coker OO, Wu J, Zhou Y, Zhao L, Nakatsu G, et al. Aspirin Reduces Colorectal Tumor Development in Mice and Gut Microbes Reduce its Bioavailability and Chemopreventive Effects. Gastroenterology. 2020. Sep;159(3):969–983.e4. [DOI] [PubMed] [Google Scholar]
- 35.Rogers M a. M, Aronoff DM The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin Microbiol Infect Off Publ Eur Soc Clin Microbiol Infect Dis. 2016. Feb;22(2):178.e1–178.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peters U, Falk LC, Kalman SM. Digoxin metabolism in patients. Arch Intern Med. 1978. Jul;138(7):1074–6. [PubMed] [Google Scholar]
- 37.Lindenbaum J, Rund DG, Butler VP, Tse-Eng D, Saha JR. Inactivation of digoxin by the gut flora: reversal by antibiotic therapy. N Engl J Med. 1981 Oct 1;305(14):789–94. [DOI] [PubMed] [Google Scholar]
- 38.Mathan VI, Wiederman J, Dobkin JF, Lindenbaum J. Geographic differences in digoxin inactivation, a metabolic activity of the human anaerobic gut flora. Gut. 1989. Jul;30(7):971–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science. 2013 Jul 19;341(6143):295–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar K, Jaiswal SK, Dhoke GV, Srivastava GN, Sharma AK, Sharma VK. Mechanistic and structural insight into promiscuity based metabolism of cardiac drug digoxin by gut microbial enzyme. J Cell Biochem. 2018. Jul;119(7):5287–96. [DOI] [PubMed] [Google Scholar]
- 41.Mansi I, Frei CR, Wang C-P, Mortensen EM. Statins and New-Onset Diabetes Mellitus and Diabetic Complications: A Retrospective Cohort Study of US Healthy Adults. J Gen Intern Med. 2015. Nov;30(11):1599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karnes JH, Cooper-DeHoff RM. Antihypertensive medications: benefits of blood pressure lowering and hazards of metabolic effects. Expert Rev Cardiovasc Ther. 2009. Jun;7(6):689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012 Oct 4;490(7418):55–60. [DOI] [PubMed] [Google Scholar]
- 44.Caparrós-Martín JA, Lareu RR, Ramsay JP, Peplies J, Reen FJ, Headlam HA, et al. Statin therapy causes gut dysbiosis in mice through a PXR-dependent mechanism. Microbiome. 2017 Aug 9;5(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coleman CI, Lucek DM, Hammond J, White CM. Preoperative statins and infectious complications following cardiac surgery. Curr Med Res Opin. 2007. Aug;23(8):1783–90. [DOI] [PubMed] [Google Scholar]
- 46.Liappis AP, Kan VL, Rochester CG, Simon GL. The effect of statins on mortality in patients with bacteremia. Clin Infect Dis Off Publ Infect Dis Soc Am. 2001 Oct 15;33(8):1352–7. [DOI] [PubMed] [Google Scholar]
- 47.Mortensen EM, Restrepo MI, Anzueto A, Pugh J. The effect of prior statin use on 30-day mortality for patients hospitalized with community-acquired pneumonia. Respir Res. 2005 Jul 25;6:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hackam DG, Mamdani M, Li P, Redelmeier DA. Statins and sepsis in patients with cardiovascular disease: a population-based cohort analysis. Lancet Lond Engl. 2006 Feb 4;367(9508):413–8. [DOI] [PubMed] [Google Scholar]
- 49.Motzkus-Feagans CA, Pakyz A, Polk R, Gambassi G, Lapane KL. Statin use and the risk of Clostridium difficile in academic medical centres. Gut. 2012. Nov;61(11):1538–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Björkhem-Bergman L, Bergman P, Andersson J, Lindh JD. Statin treatment and mortality in bacterial infections--a systematic review and meta-analysis. PloS One. 2010 May 19;5(5):e10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khan TJ, Ahmed YM, Zamzami MA, Mohamed SA, Khan I, Baothman OAS, et al. Effect of atorvastatin on the gut microbiota of high fat diet-induced hypercholesterolemic rats. Sci Rep. 2018 Jan 12;8(1):662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zimmermann F, Roessler J, Schmidt D, Jasina A, Schumann P, Gast M, et al. Impact of the Gut Microbiota on Atorvastatin Mediated Effects on Blood Lipids. J Clin Med. 2020 May 25;9(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dawson PA. Role of the Intestinal Bile Acid Transporters in Bile Acid and Drug Disposition. Handb Exp Pharmacol. 2011;(201):169–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fu ZD, Cui JY, Klaassen CD. Atorvastatin induces bile acid-synthetic enzyme Cyp7a1 by suppressing FXR signaling in both liver and intestine in mice. J Lipid Res. 2014. Dec;55(12):2576–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khan TJ, Ahmed YM, Zamzami MA, Siddiqui AM, Khan I, Baothman OAS, et al. Atorvastatin Treatment Modulates the Gut Microbiota of the Hypercholesterolemic Patients. Omics J Integr Biol. 2018. Feb;22(2):154–63. [DOI] [PubMed] [Google Scholar]
- 56.Yoo D-H, Kim IS, Van Le TK, Jung I-H, Yoo HH, Kim D-H. Gut microbiota-mediated drug interactions between lovastatin and antibiotics. Drug Metab Dispos Biol Fate Chem. 2014. Sep;42(9):1508–13. [DOI] [PubMed] [Google Scholar]
- 57.Nolan JA, Skuse P, Govindarajan K, Patterson E, Konstantinidou N, Casey PG, et al. The influence of rosuvastatin on the gastrointestinal microbiota and host gene expression profiles. Am J Physiol Gastrointest Liver Physiol. 2017 May 1;312(5):G488–97. [DOI] [PubMed] [Google Scholar]
- 58.Wang L, Wang Y, Wang H, Zhou X, Wei X, Xie Z, et al. The influence of the intestinal microflora to the efficacy of Rosuvastatin. Lipids Health Dis. 2018 Jun 30;17(1):151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y, Song X, Zhou H, Zhou X, Xia Y, Dong X, et al. Gut Microbiome Associates With Lipid-Lowering Effect of Rosuvastatin in Vivo. Front Microbiol. 2018;9:530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kummen M, Solberg OG, Storm-Larsen C, Holm K, Ragnarsson A, Trøseid M, et al. Rosuvastatin alters the genetic composition of the human gut microbiome. Sci Rep. 2020 Mar 25;10(1):5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaddurah-Daouk R, Baillie RA, Zhu H, Zeng Z-B, Wiest MM, Nguyen UT, et al. Enteric microbiome metabolites correlate with response to simvastatin treatment. PloS One. 2011;6(10):e25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bethell PH, Goad L, Evershed R, Ottaway J. The Study of Molecular Markers of Human Activity: The Use of Coprostanol in the Soil as an Indicator of Human Faecal Material. 1994;
- 63.Ko HHT, Lareu RR, Dix BR, Hughes JD. Statins: antimicrobial resistance breakers or makers? PeerJ. 2017;5:e3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Santisteban MM, Qi Y, Zubcevic J, Kim S, Yang T, Shenoy V, et al. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ Res. 2017 Jan 20;120(2):312–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H-B, Yang T, Richards EM, Pepine CJ, Raizada MK. Maternal Treatment With Captopril Persistently Alters Gut-Brain Communication and Attenuates Hypertension of Male Offspring. Hypertens Dallas Tex 1979. 2020 May;75(5):1315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang T, Aquino V, Lobaton GO, Li H, Colon-Perez L, Goel R, et al. Sustained Captopril-Induced Reduction in Blood Pressure Is Associated With Alterations in Gut-Brain Axis in the Spontaneously Hypertensive Rat. J Am Heart Assoc. 2019 Feb 19;8(4):e010721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu X, Zhang X, Jin H, Wu Z, Yan C, Liu Z, et al. Zhengganxifeng Decoction Affects Gut Microbiota and Reduces Blood Pressure via Renin-Angiotensin System. Biol Pharm Bull. 2019;42(9):1482–90. [DOI] [PubMed] [Google Scholar]
- 68.Konop M, Radkowski M, Grochowska M, Perlejewski K, Samborowska E, Ufnal M. Enalapril decreases rat plasma concentration of TMAO, a gut bacteria-derived cardiovascular marker. Biomark Biochem Indic Expo Response Susceptibility Chem. 2018. Jun;23(4):380–5. [DOI] [PubMed] [Google Scholar]
- 69.Arab HH, Al-Shorbagy MY, Abdallah DM, Nassar NN. Telmisartan attenuates colon inflammation, oxidative perturbations and apoptosis in a rat model of experimental inflammatory bowel disease. PloS One. 2014;9(5):e97193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008. Feb;134(2):577–94. [DOI] [PubMed] [Google Scholar]
- 71.Chan YK, Brar MS, Kirjavainen PV, Chen Y, Peng J, Li D, et al. High fat diet induced atherosclerosis is accompanied with low colonic bacterial diversity and altered abundances that correlates with plaque size, plasma A-FABP and cholesterol: a pilot study of high fat diet and its intervention with Lactobacillus rhamnosus GG (LGG) or telmisartan in ApoE−/− mice. BMC Microbiol. 2016 Nov 8;16(1):264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Robles-Vera I, Toral M, de la Visitación N, Sánchez M, Gómez-Guzmán M, Muñoz R, et al. Changes to the gut microbiota induced by losartan contributes to its antihypertensive effects. Br J Pharmacol. 2020. May;177(9):2006–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu D, Tang X, Ding L, Cui J, Wang P, Du X, et al. Candesartan attenuates hypertension-associated pathophysiological alterations in the gut. Biomed Pharmacother Biomedecine Pharmacother. 2019. Aug;116:109040. [DOI] [PubMed] [Google Scholar]
- 74.Yisireyili M, Uchida Y, Yamamoto K, Nakayama T, Cheng XW, Matsushita T, et al. Angiotensin receptor blocker irbesartan reduces stress-induced intestinal inflammation via AT1a signaling and ACE2-dependent mechanism in mice. Brain Behav Immun. 2018. Mar;69:167–79. [DOI] [PubMed] [Google Scholar]
- 75.Rubio-Tapia A, Herman ML, Ludvigsson JF, Kelly DG, Mangan TF, Wu T-T, et al. Severe spruelike enteropathy associated with olmesartan. Mayo Clin Proc. 2012. Aug;87(8):732–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yoo HH, Kim IS, Yoo D-H, Kim D-H. Effects of orally administered antibiotics on the bioavailability of amlodipine: gut microbiota-mediated drug interaction. J Hypertens. 2016. Jan;34(1):156–62. [DOI] [PubMed] [Google Scholar]
- 77.Kumar KA, Ganguly K, Mazumdar K, Dutta NK, Dastidar SG, Chakrabarty AN. Amlodipine: a cardiovascular drug with powerful antimicrobial property. Acta Microbiol Pol. 2003;52(3):285–92. [PubMed] [Google Scholar]
- 78.Song S-C, An Y-M, Shin J-H, Chung M-J, Seo J-G, Kim E Beneficial effects of a probiotic blend on gastrointestinal side effects induced by leflunomide and amlodipine in a rat model. Benef Microbes. 2017 Oct 13;8(5):801–8. [DOI] [PubMed] [Google Scholar]
- 79.Ahn Y, Nam MH, Kim E. Relationship Between the Gastrointestinal Side Effects of an Anti-Hypertensive Medication and Changes in the Serum Lipid Metabolome. Nutrients. 2020 Jan 13;12(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vertzoni M, Kersten E, van der Mey D, Muenster U, Reppas C. Evaluating the clinical importance of bacterial degradation of therapeutic agents in the lower intestine of adults using adult fecal material. Eur J Pharm Sci Off J Eur Fed Pharm Sci. 2018 Dec 1;125:142–50. [DOI] [PubMed] [Google Scholar]
- 81.Zhang J, Chen Y, Sun Y, Wang R, Zhang J, Jia Z. Plateau hypoxia attenuates the metabolic activity of intestinal flora to enhance the bioavailability of nifedipine. Drug Deliv. 2018. Nov;25(1):1175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. 2018 Mar 29;555(7698):623–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cooke G, Behan J, Costello M. Newly identified vitamin K-producing bacteria isolated from the neonatal faecal flora. Microb Ecol Health Dis. 2006 Jan 1;18(3–4):133–8. [Google Scholar]
- 84.Wells PS, Holbrook AM, Crowther NR, Hirsh J. Interactions of warfarin with drugs and food. Ann Intern Med. 1994 Nov 1;121(9):676–83. [DOI] [PubMed] [Google Scholar]
- 85.Wang L, Liu L, Liu X, Xiang M, Zhou L, Huang C, et al. The gut microbes, Enterococcus and Escherichia-Shigella, affect the responses of heart valve replacement patients to the anticoagulant warfarin. Pharmacol Res. 2020. Sep;159:104979. [DOI] [PubMed] [Google Scholar]
- 86.Matuskova Z, Anzenbacherova E, Vecera R, Tlaskalova-Hogenova H, Kolar M, Anzenbacher P. Administration of a probiotic can change drug pharmacokinetics: effect of E. coli Nissle 1917 on amidarone absorption in rats. PloS One. 2014;9(2):e87150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ittzes B, Szentkiralyi E, Szabo Z, Batai IZ, Gyorffy O, Kovacs T, et al. Amiodarone that has antibacterial effect against human pathogens may represent a novel catheter lock. Acta Microbiol Immunol Hung. 2020 Jul 6;67(2):133–7. [DOI] [PubMed] [Google Scholar]
- 88.Amin AM, Sheau Chin L, Teh C-H, Mostafa H, Mohamed Noor DA, Abdul Kader MASK, et al. Pharmacometabolomics analysis of plasma to phenotype clopidogrel high on treatment platelets reactivity in coronary artery disease patients. Eur J Pharm Sci Off J Eur Fed Pharm Sci. 2018 May 30;117:351–61. [DOI] [PubMed] [Google Scholar]
- 89.Duan R, Chen X, Wang F, Zhang T, Ling P. Oral administration of heparin or heparosan increases the Lactobacillus population in gut microbiota of rats. Carbohydr Polym. 2013 Apr 15;94(1):100–5. [DOI] [PubMed] [Google Scholar]
- 90.Pan L, Sun W, Shang Q, Niu Q, Liu C, Li G, et al. In vitro fermentation and isolation of heparin-degrading bacteria from human gut microbiota. Anaerobe. 2021. Apr;68:102289. [DOI] [PubMed] [Google Scholar]
- 91.Folwaczny C, Wiebecke B, Loeschke K. Unfractioned heparin in the therapy of patients with highly active inflammatory bowel disease. Am J Gastroenterol. 1999. Jun;94(6):1551–5. [DOI] [PubMed] [Google Scholar]
- 92.Evans RC, Wong VS, Morris AI, Rhodes JM. Treatment of corticosteroid-resistant ulcerative colitis with heparin--a report of 16 cases. Aliment Pharmacol Ther. 1997. Dec;11(6):1037–40. [DOI] [PubMed] [Google Scholar]
- 93.Marques FZ, Jama HA, Tsyganov K, Gill PA, Rhys-Jones D, Muralitharan RR, et al. Guidelines for Transparency on Gut Microbiome Studies in Essential and Experimental Hypertension. Hypertension. 2019 Dec 1;74(6):1279–93. [DOI] [PubMed] [Google Scholar]
- 94.Guthrie L, Kelly L. Bringing microbiome-drug interaction research into the clinic. EBioMedicine. 2019 Jun 1;44:708–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Johnson JA. Warfarin Pharmacogenetics: A Rising Tide for its Clinical Value. Circulation. 2012 Apr 24;125(16):1964–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Daniel SG, Ball CL, Besselsen DG, Doetschman T, Hurwitz BL. Functional Changes in the Gut Microbiome Contribute to Transforming Growth Factor β-Deficient Colon Cancer. mSystems. 2017. Oct;2(5). [DOI] [PMC free article] [PubMed] [Google Scholar]