

Translation initiation in eukaryotic cells typically begins at an AUG start codon in a Kozak sequence (1). The consensus translation initiation sequence is GCCGCCACC|AUGG, with variations leading to differences in translation efficiency (2,3). Variable efficiency of Kozak sequences and their embedded start codons may explain congenital profound recessive non-syndromic hearing loss due to Golgi snap receptor complex member 2 (GOSR2) in an extended consanguineous Palestinian family (Fig. 1A). This result was unexpected, given that known pathogenic alleles of GOSR2 are responsible for a severe recessive neurological phenotype, but with normal hearing. The paradox may be resolved by the combination of unique features of the first codon mutation in the family, and differences in GOSR2 binding partners of soluble-NSF-attachment-protein-receptor (SNARE) complexes in brain neurons versus GOSR2 binding partners of SNARE complexes in the inner ear.
Figure 1.
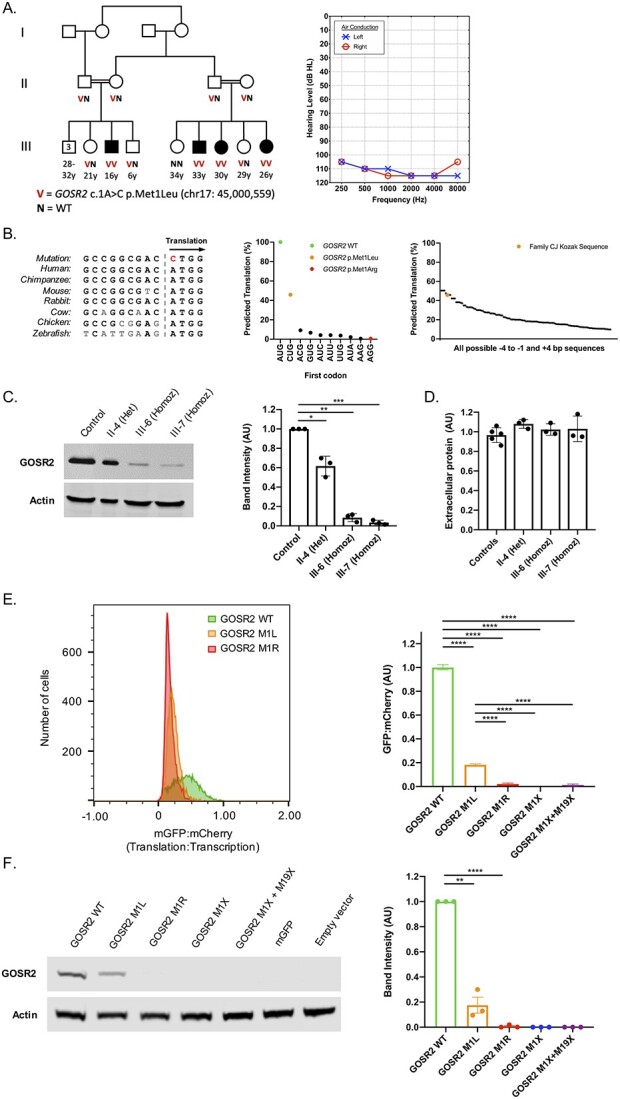
Clinical and cellular features of GOSR2 p.Met1Leu. (A) Family CJ with congenital non-syndromic sensorineural hearing loss in four children (filled symbols), with genotypes at GOSR2 c.1A>C, p.Met1Leu and an audiogram of III-3. (B) Nucleotide alignment of the GOSR2 Kozak sequence across multiple species (left), with predicted translation efficiency of human GOSR2 alternate start codons, given the GOSR2 Kozak sequence in family CJ [adapted from Ref. (3); center], and predicted translation efficiencies of CUG start codons with different surrounding Kozak sequences [adapted from Ref. (3); right]. (C) Western blot of GOSR2 in cultured lymphoblasts (LCLs) from members of family CJ and a control, with quantification of band intensities normalized to actin and to GOSR2 protein level of the control. (D) Total extracellular protein in culture media following 48 h incubation of LCLs from family members of homozygous and heterozygous GOSR2 genotypes and five controls; all P-values less than E – 10. (E) Translation:transcription ratios, indicated by monomeric green fluorescent protein:mCherry, for GOSR2 p.Met1Leu, p.Met1Arg, and reference; and cellular translation:transcription ratios, normalized to wild-type (see also Supplementary Material, Fig. S1). (F) Western blot of GOSR2 translation in transfected cells used in flow experiments, with quantification of GOSR2 band intensities, normalized to actin and to reference protein levels. Error bars indicate standard errors; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In family CJ, four children in two sibships have congenital profound hearing loss. The parents of both sibships are first cousins with normal hearing. The families reported occasional febrile seizures in infancy for each of the deaf children, but these did not persist into adolescence. As young adults, none of the deaf individuals had any signs of epilepsy, ataxia, other severe neurological abnormalities or other syndromic features (see also Supplementary Material, Supplemental Methods). Whole genome sequencing of 12 members of family CJ yielded linkage of hearing loss, under an autosomal recessive model, to a region of 4.8 MB on chromosome 17q21.3. The region harbored only one likely candidate allele, GOSR2 c.1A > C, p.Met1Leu (NM_054022) (Supplementary Material, Table S1). This variant appeared once in the gnomAD database, as a heterozygote, and not in any of ~2000 in-house controls of Palestinian ancestry. The likelihood is <0.005 that this variant co-segregates by chance with hearing loss in the family. GOSR2 is ubiquitously expressed, including in the inner ear (4).
GOSR2 encodes a transmembrane protein in the SNARE family of vesicle locking proteins, which mediate membrane fusion during transit of proteins to and from the endoplasmic reticulum (ER) (5). The International Mouse Phenotyping Consortium has shown that homozygous deficiency of Gosr2 is embryonic lethal (mousephenotype.org). Knockdown of GOSR2 leads to a loss in ER-Golgi transport and interferes with Golgi maintenance (6). In humans, homozygosity for a Northern European founder allele, GOSR2 p.Gly144Trp, causes a severe form of childhood-onset progressive myoclonic epilepsy with ataxia, but with apparently normal hearing (7,8), Compound heterozygosity for GOSR2 p.Gly144Trp and GOSR2 c.2T>G, p.Met1Arg, leads to the most severe GOSR2-related phenotype, including both rapidly progressive epilepsy and muscular dystrophy (8,9).
Previous analysis of start codons and Kozak sequences genome-wide (3) predicted that mutation of codon 1 from ATG to AGG (Met1Arg, the severe European allele) would nearly abolish translation, but that mutation of codon 1 from ATG to CTG (Met1Leu, the allele of family CJ) would retain partial translation (Fig. 1B). We tested this prediction by measuring GOSR2 protein in Epstein-Barr virus (EBV)-transformed human lymphoblast cell line (LCL) lysates from individuals in family CJ (Fig. 1C). Western blot revealed that GOSR2 protein was detectable in homozygous affected children III-6 and III-7, but significantly reduced, to 8.5% and 3.4% respectively, of protein levels in controls; protein of heterozygous unaffected carrier II-4 was modestly reduced compared with control. Gels were run sufficiently long to rule out translation from the downstream start codon, methionine-19, which would result in a protein of size ~23 kD.
Because GOSR2 encodes an integral component of the ER-Golgi SNARE complex, we measured Golgi function and morphology in LCLs of affected individuals (Supplementary Material, Supplementary Methods). We did not detect any significant differences among genotypes in total protein secretion (Fig. 1D). Laser scanning confocal microscopy was used to visualize gross Golgi morphology and GOSR2 subcellular localization. In cells of all genotypes, GOSR2 protein was present, located as expected exclusively outside the nucleus, and co-localized with Golgi marker GPP130 diffusely throughout the cytoplasm (Supplementary Material, Figs S2 and S3).
Finally, to test whether lower levels of GOSR2 protein were due to decreased translation efficiency, we designed a flow cytometry assay, based on previously published methods [Supplementary Material, Fig. S4; (3)], to compare GOSR2 c.1A>C, p.Met1Leu and GOSR2 c.2T>G, p.Met1Arg to wild-type GOSR2. Mutant plasmids GOSR2 p.Met1ter and GOSR2 p.Met1ter + Met19ter were also tested to assess possible translation from downstream methionine-19. Flow cytometry results indicated that the GOSR2 p.Met1Leu plasmid had 18% of the signal of wild-type plasmid (Fig. 1E); that for all mutant plasmids, translation was significantly lower than wild-type (P < E – 10); and that the GOSR2 p.Met1Leu plasmid was translated at a significantly higher level than any of the other mutants. Results from western blot experiments revealed the same profile (Fig. 1F). Transfection with GOSR2 p.Met1Leu led to 18% of wild-type protein levels. Corresponding bands were not present for either nonsense mutant, indicating that translation did not occur from Met-19.
The discovery of GOSR2 c.1A>C, p.Met1Leu as the best candidate allele for hearing loss in family CJ suggests a paradox. Why do homozygotes for this allele have only hearing loss, given that known pathogenic variants of GOSR2 are associated with severe neurological features (7,8,9)? Conversely, why do homozygotes and compound heterozygotes for the known GOSR2 pathogenic variants have apparently normal hearing? More generally, why do phenotypically severe alleles not necessarily impact organ systems affected by milder alleles? In this case, the underlying biology of vesicular transport by the ER-Golgi SNARE system and the role in this system of GOSR2 may be responsible. Vesicular transport is highly regulated, with each protein having a required topological expression and tissue-specific binding partners (10). In mammals, the synaptic SNARE complex of brain neuronal cells differs from the analogous SNARE complex of inner ear hair cells both in structural components and in protein regulators (11,12). In yeast, GOSR2 localizes to the cis-Golgi and binds the SNARE-Qa molecule syntaxin-5/Sed5p (13). The yeast ortholog of mammalian GOSR2 p.Gly144Trp changes the primary structure of the GOSR2 SNARE-binding motif, leading to deficits in SNARE-mediated ER-to-Golgi membrane fusion and hence to failure of the SNARE complex to bind properly to the Golgi (14). We speculate that this process models the binding of GOSR2 p.Gly144Trp to the ER-Golgi SNARE complex in mammalian brain neuronal cells, but does not model GOSR2 binding to the SNARE complex in hair cells of the mammalian inner ear. Given the differences in mammals between synaptic SNARE complexes of brain neurons and hair cells, GOSR2 may bind to an ER-Golgi SNARE molecule in the inner ear that does not rely on the binding motif including Gly-144. GOSR2 p.Gly144Trp correctly localizes and does not affect Golgi morphology (7), so in the absence of an effect of this missense on SNARE binding, the hair cells of the inner ear could remain unaffected. In contrast, GOSR2 p.Met1Leu has normal function in all cell types, with reduced protein levels that are apparently adequate for function of brain neuronal cells but insufficient for highly sensitive inner ear hair cells.
In proposing this model, we recognize that the biology of non-synaptic SNARE complexes in the inner ear is not fully understood (11), so the mechanism by which the inner ear SNARE complexes might evade the effects of GOSR2 p.Gly144Trp remains speculative. The paradox of GOSR2 recalls the observations that mutations in the inner-ear-specific synaptic SNARE-associated protein otoferlin (OTOF) lead to hearing loss without syndromic neurological features, and that mutations in genes of the brain-neuronal-specific SNARE complex (STX1B, VAMP2, SNAP25, STXBP1, UNC13A) lead to epileptic phenotypes that do not include hearing loss (15). The GOSR2 paradox is particularly intriguing, however, because GOSR2 is active in both brain neuronal and inner ear hair cells.
Finally, the story of family CJ is a reminder that the impact of mutations involving translation initiation depends on the strength of the underlying Kozak translation initiation sequence. The relatively mild effect of GOSR2 c.1A>C, p.Met1Leu depended not only on the specific mutation in codon 1, but also on the close-to-ideal (non-mutant) Kozak sequence at base pairs −1 to −4. This observation suggests that for gene and variant discovery projects, the entire Kozak sequence, at least positions −4 to +4, be evaluated.
Supplementary Material
Acknowledgements
The project was approved by the human subjects’ committees of Bethlehem University, the University of Washington, Tel Aviv University and the Palestinian and Israeli Ministries of Health. We thank family CJ for their continued engagement and support, and the teachers of Qalqilya School for Children with Hearing Loss for referring the family to our research study. We also thank the laboratory of Ephrat Levy-Lahad at the Share Zedek Medical Center, Jerusalem, for generating LCLs from participants. This work was performed in partial fulfillment of the requirements for a Ph.D. degree by Amal AbuRayyan at the School of Medicine, Tel Aviv University.
Contributor Information
Amal Aburayyan, Department of Genome Sciences and Department of Medicine, University of Washington, Seattle, WA, USA; Department of Human Molecular Genetics and Biochemistry, Faculty of Medicine and Sagol School of Neuroscience, Tel Aviv University, Tel Aviv, Israel; Hereditary Research Laboratory, Department of Biology, Bethlehem University, Bethlehem, Palestine.
Ryan J Carlson, Department of Genome Sciences and Department of Medicine, University of Washington, Seattle, WA, USA.
Grace N Rabie, Hereditary Research Laboratory, Department of Biology, Bethlehem University, Bethlehem, Palestine.
Ming K Lee, Department of Genome Sciences and Department of Medicine, University of Washington, Seattle, WA, USA.
Suleyman Gulsuner, Department of Genome Sciences and Department of Medicine, University of Washington, Seattle, WA, USA.
Tom Walsh, Department of Genome Sciences and Department of Medicine, University of Washington, Seattle, WA, USA.
Karen B Avraham, Department of Human Molecular Genetics and Biochemistry, Faculty of Medicine and Sagol School of Neuroscience, Tel Aviv University, Tel Aviv, Israel.
Moien N Kanaan, Hereditary Research Laboratory, Department of Biology, Bethlehem University, Bethlehem, Palestine.
Mary-Claire King, Department of Genome Sciences and Department of Medicine, University of Washington, Seattle, WA, USA.
Funding
This work was supported by National Institutes of Health grants R01DC011835 to K.B.A, M.K. and M.C.K; F30DC018702, T32DC005361 and T32GM007266 to R.J.C; and by fellowship support to A.A. from the Mauerberger Foundation and to R.J.C. from the Achievement Rewards for College Scientists (ARCS) Foundation.
Conflict of Interest statement. The authors have no conflicts of interests to disclose.
Data Availability
Data has been submitted to ClinVar. Additional information available from the authors on request.
References
- 1. Kozak, M. (1978) How do eucaryotic ribosomes select initiation regions in messenger RNA? Cell, 15, 1109–1123. [DOI] [PubMed] [Google Scholar]
- 2. Kozak, M. (1987) An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res., 15, 8125–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Diaz de Arce, A.J., Noderer, W.L. and Wang, C.L. (2018) Complete motif analysis of sequence requirements for translation initiation at non-AUG start codons. Nucleic Acids Res., 46, 985–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kolla, L., Kelly, M.C., Mann, Z.F., Anaya-Rocha, A., Ellis, K., Lemons, A., Palermo, A.T., So, K.S., Mays, J.C., Orvis, J. et al. (2020) Characterization of the development of the mouse cochlear epithelium at the single cell level. Nat. Commun., 11, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hay, J.C., Chao, D.S., Kuo, C.S. and Scheller, R.H. (1997) Protein interactions regulating vesicle transport between the endoplasmic reticulum and Golgi apparatus in mammalian cells. Cell, 89, 149–158. [DOI] [PubMed] [Google Scholar]
- 6. Gordon, D.E., Bond, L.M., Sahlender, D.A. and Peden, A.A. (2010) A targeted siRNA screen to identify SNAREs required for constitutive secretion in mammalian cells. Traffic, 11, 1191–1204. [DOI] [PubMed] [Google Scholar]
- 7. Corbett, M.A., Schwake, M., Bahlo, M., Dibbens, L.M., Lin, M., Gandolfo, L.C., Vears, D.F., O’Sullivan, J.D., Robertson, T., Bayly, M.A. et al. (2011) A mutation in the Golgi Qb-SNARE gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am. J. Hum. Genet., 88, 657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Larson, A.A., Baker, P.R., Milev, M.P., Press, C.A., Sokol, R.J., Cox, M.O., Lekostaj, J.K., Stence, A.A., Bossler, A.D., Mueller, J.M. et al. (2018) TRAPPC11 and GOSR2 mutations associate with hypoglycosylation of α-dystroglycan and muscular dystrophy. Skelet. Muscle, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Polet, S.S., Anderson, D.G., Koens, L.H., van Egmond, M.E., Drost, G., Brusse, E., Willemsen, M.A., Sival, D.A., Brouwer, O.F., Kremer, H.P. et al. (2020) A detailed description of the phenotypic spectrum of North Sea progressive myoclonus epilepsy in a large cohort of seventeen patients. Park. Relat. Disord., 72, 44–48. [DOI] [PubMed] [Google Scholar]
- 10. Praschberger, R., Balint, B., Mencacci, N.E., Hersheson, J., Rubio-Agusti, I., Kullmann, D.M., Bettencourt, C., Bhatia, K. and Houlden, H. (2015) Expanding the phenotype and genetic defects associated with the GOSR2 gene. Mov. Disord. Clin. Pract., 2, 271–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ramakrishnan, N.A., Drescher, M.J. and Drescher, D.G. (2012) The SNARE complex in neuronal and sensory cells. Mol. Cell. Neurosci., 50, 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nouvian, R., Neef, J., Bulankina, A.V., Reisinger, E., Pangršié, T., Frank, T., Sikorra, S., Brose, N., Binz, T. and Moser, T. (2011) Exocytosis at the hair cell ribbon synapse apparently operates without neuronal SNARE proteins. Nat. Neurosci., 14, 411–413. [DOI] [PubMed] [Google Scholar]
- 13. Malsam, J. and Söllner, T.H. (2011) Organization of SNAREs within the Golgi stack. Cold Spring Harb. Perspect. Biol., 3, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Praschberger, R., Lowe, S.A., Malintan, N.T., Giachello, C.N.G., Patel, N., Houlden, H., Kullmann, D.M., Baines, R.A., Usowicz, M.M., Krishnakumar, S.S. et al. (2017) Mutations in Membrin/GOSR2 reveal stringent secretory pathway demands of dendritic growth and synaptic integrity. Cell Rep., 21, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cali, E., Rocca, C., Salpietro, V. and Houlden, H. (2022) Epileptic phenotypes associated with SNAREs and related synaptic vesicle exocytosis machinery. Front. Neurol., 12, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data has been submitted to ClinVar. Additional information available from the authors on request.