

Abstract
Neurodegenerative diseases exhibiting the pathological accumulation of tau such as Alzheimer’s disease and related disorders still have no disease-modifying treatments and the molecular mechanisms of neurodegeneration remain unclear. To discover additional suppressor of tauopathy (sut) genes that mediate or modulate the toxicity of pathological tau, we performed a classical genetic screen using a tau transgenic Caenorhabditis elegans model. From this screen, we identified the suppressing mutation W292X in sut-6, the C. elegans homolog of human NIPP1, which truncates the C-terminal RNA-binding domain. Using CRISPR-based genome editing approaches, we generated null and additional C-terminally truncated alleles in sut-6 and found that loss of sut-6 or sut-6(W292X) suppresses tau-induced behavioral locomotor deficits, tau protein accumulation and neuron loss. The sut-6(W292X) mutation showed stronger and semi-dominant suppression of tau toxicity while sut-6 deletion acted recessively. Neuronal overexpression of SUT-6 protein did not significantly alter tau toxicity, but neuronal overexpression of SUT-6 W292X mutant protein reduced tau-mediated deficits. Epistasis studies showed tauopathy suppression by sut-6 occurs independent of other known nuclear speckle-localized suppressors of tau such as sut-2, aly-1/aly-3 and spop-1. In summary, we have shown that sut-6/NIPP1 modulates tau toxicity and found a dominant mutation in the RNA-binding domain of sut-6 which strongly suppresses tau toxicity. This suggests that altering RNA-related functions of SUT-6/NIPP1 instead of complete loss of SUT-6/NIPP1 will provide the strongest suppression of tau.
Introduction
The microtubule binding protein tau, encoded by the MAPT gene, accumulates abnormally in multiple neurodegenerative diseases termed tauopathies such as Alzheimer’s disease and frontotemporal dementias (1,2). Mutations in MAPT, first discovered roughly 25 years ago, cause familial forms of frontotemporal dementia (termed frontotemporal dementia and parkinsonism linked to chromosome 17 or FTDP-17) (3–7). Patients carrying these autosomal dominant mutations undergo significant changes in behavior, reduced executive function, and the development of parkinsonism. Pathologically, these patients accumulate tau in neurofibrillary tangles predominantly in their frontal and temporal lobes. Initial in vitro studies of tau mutations found in these families showed that many of these mutations impaired tau protein binding to microtubules (8). Further studies using multiple animal and neuronal cell models showed abnormal tau can induce behavioral deficits and cell death (reviewed in (9)). However, there are still no disease-modifying treatments for tauopathies and the molecular mechanisms underlying tauopathies are still unclear (2,10,11).
To gain additional insight into the molecular mechanisms underlying tauopathies, we employed classical genetic screening approaches. We performed forward mutagenesis screening in tau transgenic Caenorhabditis elegans, a model in which tau 1N4R protein is overexpressed in all neurons (12). This C. elegans model of tauopathy recapitulates various features of human tauopathies, such as tau-induced behavioral deficits, accumulation of hyperphosphorylated and insoluble tau protein, progressive neuron loss and shortened lifespans (12). Forward genetic screening in this model has led to the discovery of novel genetic modulators of tau such as sut-1, sut-2 and spop-1 (13–15). Further investigation of the genetic modulator of tau sut-2 demonstrated that genetic modifiers of tau identified in C. elegans model have translational relevance (16). By investigating functionally related genes, we have identified additional modifiers of tau toxicity including PARN, CCR4, TOE1 and ALYREF (16–18).
Here, we report the identification of mutations in sut-6 that strongly modulate tau-induced toxicity in tau transgenic C. elegans. sut-6 is the C. elegans homolog of mammalian NIPP1, or nuclear inhibitor of protein phosphatase 1 (PP1). NIPP1 is a nuclear speckle-localized protein that binds not only to PP1 but also to RNA, splicing factors such as CDC5L and SAP155, and transcription regulators EED and EZH2 (19–23). Functionally, NIPP1 acts as a regulator of PP1-mediated dephosphorylation of protein targets in the nucleus, forming a complex with PP1 and inhibiting PP1’s dephosphorylation activities until NIPP1 is phosphorylated at certain sites and/or binds RNA at its C terminus (19,24,25). NIPP1 can also alter splicing of pre-mRNAs (21,26–28) and alter transcription of many genes involved in development, cell growth and survival in a context-dependent manner (29–36). The C terminus of NIPP1 itself has endoribonuclease activity, although it appears to be inactive in full-length NIPP1 (20). Interestingly, conditional knockout of NIPP1 in mouse neural progenitor cells was recently shown to increase phosphorylation of tau protein (36). It is unclear how many functions of NIPP1 are shared with SUT-6, but there is strong conservation of the FHA binding domain, nuclear localization sequences, PP1-interacting sites and the C-terminal binding domain between mammalian, Drosophila and C. elegans genes (37).
In this work, we show that either partial loss of RNA-binding function or complete loss of sut-6 expression suppresses tau toxicity in tau transgenic C. elegans. Loss of sut-6 ameliorated tau-induced behavioral locomotor deficits, reduced neuron loss and reduced the accumulation of tau protein, while overexpression of SUT-6 in neurons enhanced locomotor deficits under certain conditions. When tested with other previously identified nuclear speckle-localized suppressors of tau, sut-6 appeared to act in a different pathway to sut-2, aly genes and spop-1, suggesting that nuclear speckle proteins act in multiple ways to modulate tau toxicity.
Results
Genetic sut-6 loss of function suppresses tauopathy-related phenotypes in tau transgenic C. elegans
To identify novel suppressors of tau, we employed a forward mutagenesis approach in a tau transgenic C. elegans model. In this model, multicopy arrays of cDNA encoding for the human tau isoform 1N4R under the pan-neuronal aex-3 promotor were integrated into the C. elegans genome, resulting in the production, accumulation and phosphorylation of human 1N4R tau protein in neurons (12). Accumulation of human tau in tau transgenic C. elegans neurons causes significant behavioral locomotor deficits, neuron loss and shortening of lifespan (12). After using ethyl methanesulfonate to mutate the genome of tau transgenic C. elegans, individual tau transgenic C. elegans that show suppression of locomotor deficits were backcrossed against wild-type N2 C. elegans at least six times before being whole-genome sequenced. We previously used this approach successfully to identify sut-1, sut-2 and spop-1 (13–15).
With this genetics approach, we identified a c.876G- > A change in the C. elegans gene B0511.7, which we subsequently refer to as sut-6 (Fig. 1A). This mutation results in the coding change of W292X, introducing a premature stop codon 11 amino acids before the normal C terminus of the protein. sut-6 is the C. elegans homolog of mammalian NIPP1 (nuclear inhibitor of protein phosphatase 1) (Fig. 1B). Based on the amino acid alignment of the C. elegans SUT-6 protein sequence to the human NIPP1 protein sequence (Supplementary Material, Fig. S1), the W292X mutation truncates the C-terminal half of the RNA-binding domain without altering the PP1-interacting portion (20). To explore the consequence of this mutation on SUT-6 protein function, we used CRISPR to generate alleles that deleted the entire coding sequence of the sut-6 gene (subsequently referred to as sut-6(null)). We also used CRISPR to delete only the last 30 base pairs of the coding sequence of sut-6, which should make SUT-6 protein 1–292 (we will subsequently refer to this type of mutation as sut-6(CΔ10)), as well as remake the mutation identified in forward mutagenesis in a wild-type C. elegans background (Fig. 1A, Table 1). We raised monoclonal antibodies to SUT-6 protein in order to detect and quantify protein levels and isolated multiple antibodies that detected SUT-6 protein. The hybridoma clone 1A antibody only detected SUT-6 protein in wild-type worm lysates and not lysates from sut-6(null), sut-6(CΔ10) or sut-6(W292X) worms (Supplementary Material, Fig. S2A–B). The hybridoma clones 6B and 7A antibodies detected SUT-6 protein in both wild-type and sut-6(W292X) worm lysates. The level of SUT-6(W292X) protein in mutant worms was similar to the level of SUT-6 protein in wild-type worms (Supplementary Material, Fig. S2B). However, deletion of the last 30 base pairs of sut-6’s genetic coding sequence resulted in reduced expression of SUT-6 protein without altering mRNA transcript levels (Supplementary Material, Fig. S2C), suggesting that this truncated protein may be less stable. Nevertheless, residual expression of SUT-6 protein lacking the C terminus, sut-6(CΔ10) can be interpreted as a combination of partial loss of RNA-binding function and reduction in total protein levels, yielding an intermediate phenotype between sut-6(null) and sut-6(W292X).
Figure 1.

sut-6 mutations suppress tau-induced behavioral deficits. (A) sut-6 alleles used in this study and their approximate locations in comparison to the sut-6 genomic sequence. (B) Protein domains and interaction regions of human NIPP1. FHA = forkhead-associated domain. EED and PP1-interacting regions are indicated in blue and green, respectively. NLS = nuclear localization sequence. RBD = RNA-binding domain. The # indicates the approximate location of W292 of SUT-6 mapped onto human NIPP1 (at W340). (C) Swimming assays for non-transgenic (non-Tg) and tau transgenic C. elegans expressing high levels of wild-type tau (tau Tg wt high, tau background = CK144) carrying sut-6 mutations. N = 3, n = 99–146. (D) Swimming assays for non-transgenic (non-Tg) and tau transgenic C. elegans expressing mutant V337M tau (tau Tg V337M, tau background = CK10) carrying sut-6 mutations. N = 3, n = 97–127. (E) Swimming assays for tau transgenic C. elegans carrying moderate levels of wild-type tau (tau Tg wt mod, tau background = CK1443) and sut-6 mutations as homozygous or heterozygous against the hT2 balancer. N = 3, n = 90–135. For (C–E), day 1 adult worms swimming in M9 buffer were video recorded for 1 min and analyzed for frequency of turns made. Individual values are displayed as well as boxplots. One-way ANOVA followed by Games–Howell post hoc tests were performed. n.s. = not significant. ***P < 0.001. ****P < 0.0001.
Table 1.
Caenorhabditis elegans mutations used in this study
| Target gene | C. elegans allele | Shorthand | Mutation details |
|---|---|---|---|
| sut-6 | bk1461 | sut-6(W292X#1) | W292X coding mutation in sut-6 |
| sut-6 | bk3067 | sut-6(null#1) | Whole-gene deletion of sut-6 |
| sut-6 | bk3068 | sut-6(null#2) | Whole-gene deletion of sut-6 |
| sut-6 | bk3083 | sut-6(CΔ10#1) | 30 base-pair deletion at C-terminal end of sut-6 |
| sut-6 | bk3084 | sut-6(CΔ10#2) | 30 base-pair deletion at C-terminal end of sut-6 |
| sut-6 | bk3104 | sut-6(W292X#2) | W292X coding mutation in sut-6 |
| sut-6 | gk325026 | sut-6(R65X) | R65X coding mutation in sut-6 |
| sut-6 | tm6820 | sut-6(tm6820) | 398 base-pair + 5 base-pair insertion in sut-6 spanning exon 4 and introns |
| sut-2 | bk3012 | sut-2 | Whole-gene deletion of sut-2 |
| aly-1;aly-3 | bk3029;bk3113 | aly-1;aly-3 | Whole-gene deletions of aly-1 + aly-3 |
| spop-1 | bk3107 | spop-1 | Whole-gene deletion of spop-1 |
We crossed all generated sut-6 alleles into two different tau transgenic C. elegans strains. One strain expressed high levels of 1N4R human tau protein in all neurons (called ‘tau Tg wt high’). The other strain expressed 1N4R human tau protein with the mutation V337M in all neurons (called ‘tau Tg V337M’). To measure the impact of sut-6 mutations on tau-related behavioral deficits, we performed swimming reflex assays to assess the effects of the different sut-6 mutations on tau-induced swimming deficits. We found that all sut-6 alleles tested significantly reduced the swimming deficits of tau transgenic C. elegans with wild-type or mutant tau protein (Fig. 1C–D). Interestingly, in the high wild-type tau expression background, tau transgenic C. elegans carrying sut-6(W292X) had significantly restored swimming compared to tau worms carrying sut-6(null) and tau worms carrying sut-6(CΔ10). We did not see as extensive a difference in the mutant tau transgenic strains, perhaps due to a ceiling effect, as deleting sut-6 already reversed swimming deficits in this model by 80%. This difference in suppression ability between alleles is not explained by non-tau effects of these mutations on swimming, as sut-6 alleles showed no major effects on swimming in non-transgenic worms (Supplementary Material, Fig. S3).
We also investigated whether heterozygous loss of sut-6 could suppress tau-induced swimming deficits. Using the chromosomal rearrangement hT2 to balance against sut-6 mutations, we examined whether tau transgenic C. elegans heterozygous for sut-6(null) or sut-6(W292X) showed improved swimming compared to control tau transgenic C. elegans. We found that heterozygous loss of sut-6 expression did not suppress tau-induced swimming deficits but heterozygous sut-6(W292X) partially suppressed tau-mediated swimming deficits (Fig. 1E). This indicates that sut-6(null) is a recessive suppressor of tau toxicity while sut-6(W292X) is semi-dominant. It also suggests that sut-6(W292X) has a different or an additional mechanism for suppressing tau that is not due to loss of SUT-6 protein function.
Loss of sut-6 reduces tau protein levels but not mRNA levels
While sut-6 function remains incompletely characterized, its homolog NIPP1 encodes for a protein that inhibits protein phosphatase 1 (PP1), a phosphatase that can dephosphorylate tau (38,39), and interacts with the transcription repressor EED (22), which might reduce tau mRNA levels. In addition, truncated forms of NIPP1 have shown endonuclease activity in vitro (20), which could possibly degrade tau mRNA and lead to reduced tau protein levels. In the context of brain-specific conditional knockout of NIPP1, an increase in phosphorylation of tau without a change in total tau levels was observed (36). Therefore, we examined the effects of the various sut-6 alleles on total tau protein levels, tau phosphorylation levels and tau mRNA levels. We found that all sut-6 alleles significantly reduced total tau protein levels in tau transgenic C. elegans with wild-type or mutant tau protein (Fig. 2A–B, Supplementary Material, Fig. S4–5). We also found reduced amounts of tau phosphorylated at S202 or T212, the latter a phosphorylation site known to be targeted by PP1 (38,39). Unlike in the behavioral studies, the different sut-6 mutations appeared to act similarly on tau protein. The proportion of tau phosphorylated at S202 or T212 was not significantly changed between tau transgenic C. elegans controls and sut-6 mutants (Fig. 2C–D), suggesting that loss of sut-6 primarily drives loss of total tau protein and not changes in tau phosphorylation. Altogether, this data suggests that (1) changes in PP1 activity may not be important in the mechanism for suppression of tau toxicity by sut-6 mutants and (2) sut-6(W292X) suppresses tau toxicity not only by reducing total tau protein but by another mechanism that does not directly affect total tau protein and phosphorylation levels.
Figure 2.

sut-6 mutations reduce total tau protein levels in tau transgenic C. elegans. (A) Representative immunoblots showing SUT-6, total tau, phosphorylated tau at pT212 and pS202, and tubulin (load control) in tau transgenic C. elegans expressing high levels of tau with sut-6 mutations. Tau background = CK144. (B) Quantification of total tau levels in tau transgenic C. elegans with sut-6 mutations. (C) Quantification of pT212 phosphorylated tau over total tau levels in tau transgenic C. elegans with sut-6 mutations. (D) Quantification of pS202 phosphorylated tau over total tau levels in tau transgenic C. elegans with sut-6 mutations. (E) Tau mRNA levels in tau transgenic C. elegans. Data is displayed as relative mRNA compared to the average control value. For (B–E), individual values are displayed as well as bars showing the means ± SEM. One-way ANOVA followed by Dunnett’s T3 post hoc tests comparing sut-6;tau transgenic to tau transgenic control were performed. ***P < 0.001.
Figure 5.

Neuronal overexpression of SUT-6 restores tau-induced deficits in whole-body sut-6 knockout tau transgenic C. elegans. (A) Swimming assays for tau transgenic C. elegans expressing high levels of wild-type tau (tau Tg wt high, tau background = CK144) with SUT-6 neuronal overexpression and/or sut-6 deletion. N = 3, n = 136–149. Individual values are displayed as well as boxplots. One-way ANOVA followed by Games–Howell post hoc tests were performed. n.s. = not significant. ****P < 0.0001. (B) Representative immunoblots showing SUT-6, total tau and tubulin (load control) in tau transgenic C. elegans expressing high levels of tau with SUT-6 neuronal overexpression and/or sut-6 deletion. Tau background = CK144. (C) Quantification of total tau levels in tau transgenic C. elegans with SUT-6 neuronal overexpression and/or sut-6 deletion. Individual values are displayed as well as bars showing the means ± SEM. One-way ANOVA followed by Dunnett’s T3 post hoc tests comparing tau transgenic mutants to tau transgenic control were performed. n.s. = not significant. *P < 0.05.
We quantified the effect of sut-6 mutation on the relative tau mRNA levels of tau transgenic C. elegans and found no significant changes to tau mRNA levels with any of the sut-6 mutations (Fig. 2E). This suggests that loss of sut-6 suppresses tau toxicity via tau protein-related changes and not transgene transcription-related changes.
Loss of sut-6 reduces neurodegeneration
The impairment in locomotion that worsens with age in tau transgenic C. elegans normally correlates with progressive loss of GABAergic motor neurons (12). To determine whether mutations in sut-6 that suppress tau-induced swimming deficits also suppressed tau-induced GABAergic motor neuron loss, we crossed tau transgenic C. elegans strains with the CZ1200 reporter strain that expresses GFP in all GABAergic motor neurons (40), and we counted how many GABAergic motor neurons were missing. We found that both sut-6(null) and sut-6(W292X) suppressed neuron loss seen in tau transgenic C. elegans (Fig. 3). The suppression was not statistically different between sut-6(W292X) and sut-6(null), consistent with the results seen with these alleles on tau protein levels.
Figure 3.
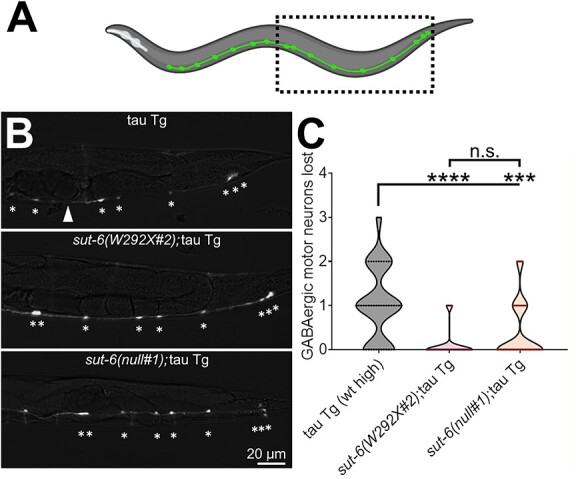
sut-6 mutations reduce loss of GABAergic motor neurons. (A) Diagram showing the GABAergic motor neurons of a C. elegans and approximate region captured by representative images in (B). (B) Representative images of GFP fluorescence in 4-day-old (approximately day 1 adult) tau transgenic C. elegans carrying unc-25::GFP and sut-6 mutations. Tau background = CK144. unc-25::GFP background = CZ1200. * indicates the location of neurons that were counted. Arrowhead indicates location where a neuron is missing. (C) Quantification of GABAergic neuron loss in tau transgenic C. elegans carrying sut-6 mutations. Individual values are displayed in violin plots. Kruskal–Wallis followed by Dunn’s post hoc tests comparing all strains to each other were performed. ***P < 0.001. ****P < 0.0001.
Neuronal overexpression of SUT-6 does not affect tau-induced toxicity but is sufficient to block suppression of tau-induced toxicity by sut-6(null)
Since loss of sut-6 suppresses tau-induced toxicity, we examined whether overexpression of SUT-6 in neurons might enhance tau-induced toxicity. We generated two overexpression lines that increased total SUT-6 protein levels by ~3-fold (Fig. 4A–B, Supplementary Material, Fig. S6). We crossed these lines to tau transgenic C. elegans. We examined the localization of SUT-6 and tau protein expressed in the same neuronal cell and saw SUT-6 in the nucleus, tau in the cytoplasm and no visible colocalization between these proteins (Fig. 4C). When we examined effects of SUT-6 neuronal overexpression on tau-induced toxicity, we found that neuronal overexpression of SUT-6 only enhanced tau-induced swimming deficits in low expression tau transgenic C. elegans but not high expression tau transgenic C. elegans (Fig. 4D–E). However, neuronal overexpression of SUT-6 itself significantly reduced swimming ability independent of tau expression (Fig. 4F), suggesting that the enhancement of swimming deficits seen in low expression tau transgenic C. elegans was not due to increased activity of SUT-6 protein to enhance tau toxicity but instead an independent, additive locomotor impairment. We analyzed tau protein and mRNA in tau transgenic C. elegans overexpressing neuronal SUT-6 protein. SUT-6 neuronal overexpression did not significantly alter total tau protein levels or tau phosphorylation levels in a consistent manner (Supplementary Material, Fig. S7A–D, S7F). In contrast, SUT-6 neuronal overexpression increased tau mRNA levels (Supplementary Material, Fig. S7E). Because loss of sut-6 did not change tau mRNA levels, this suggests that SUT-6 overexpression has an additional effect on tau mRNA transcription. However, since this change in mRNA levels occurs without affecting tau-induced swimming deficits or tau protein levels, the functional importance of this result is unclear.
Figure 4.

Neuronal overexpression of SUT-6 causes tau-independent locomotor deficits. (A) Diagram of the sut-6 transgene and a representative blot of SUT-6 and tubulin (load control) in the two generated SUT-6 neuronal overexpression strains (SUT-6 OE #1 and #2) compared to non-transgenic (non-Tg). (B) Quantification of SUT-6 protein levels in non-transgenic and SUT-6 neuronal overexpression strains. Individual values are displayed as well as bars showing the means ± SEM. One-way ANOVA followed by Dunnett’s T3 post hoc tests were performed. **P < 0.01. ***P < 0.001. (C) Immunofluorescence images showing DAPI (nuclear stain), SUT-6 and tau in tau transgenic;SUT-6 overexpression line #1 double transgenic worms. In the merged image, DAPI is shown in blue, SUT-6 is shown in red and tau is in green. (D) Swimming assays for tau transgenic C. elegans expressing low levels of human tau (tau Tg wt low, tau background = CK1441) with SUT-6 neuronal overexpression. N = 3, n = 101–126. (E) Swimming assays for tau transgenic C. elegans expressing high levels of wild-type tau (tau Tg wt high, tau background = CK144) with SUT-6 neuronal overexpression. N = 3, n = 107–147. (F) Swimming assays for non-transgenic (non-Tg) and SUT-6 neuronal overexpression transgenic C. elegans. N = 3, n = 120–155. For (D–F), individual values are displayed as well as boxplots. One-way ANOVA followed by Games–Howell post hoc tests were performed. n.s. = not significant. *P < 0.05. ***P < 0.001. ****P < 0.0001.
If overexpression of SUT-6 in neurons did not enhance tau toxicity, we wondered whether sut-6 might modulate tau-induced toxicity through a cell non-autonomous pathway. To determine whether SUT-6 neuronal expression affected tau-induced toxicity, we crossed one of the SUT-6 neuronal overexpression lines to sut-6(null) and determined the effect of neuron-only expression of SUT-6 on tau-induced swimming deficits. We found that restoring neuronal expression of SUT-6 protein was sufficient to block the suppression of tau-induced swimming deficits by whole worm deletion of sut-6 and reverse the reduction in tau protein levels (Fig. 5A–C, Supplementary Material, Fig. S8). This confirmed that sut-6 modulates tau through cell-autonomous pathways.
Neuronal overexpression of SUT-6(W292X) suppresses tau-induced toxicity
Because we previously observed that sut-6(W292X) heterozygotes could partially suppress tau-induced toxicity, we examined whether expression of SUT-6(W292X) in neurons might suppress tau-induced toxicity. We generated two lines that expressed SUT-6(W292X) under a neuronal promoter (Fig. 6A–B, Supplementary Material, Fig. S9A). Since we could not detect an increase in total SUT-6 protein in one of the SUT-6(W292X) transgenic lines, we crossed it to sut-6(null) and confirmed that the SUT-6 W292X transgene was producing protein (Fig. 6C, Supplementary Material, Fig. S9B). Interestingly, the strain with low-level neuronal expression of SUT-6(W292X) showed a slightly hyperactive swimming phenotype while the strain with higher level neuronal expression of SUT-6(W292X) showed swimming deficits similar to those seen in our neuronal overexpression wild-type SUT-6 transgenic C. elegans (Fig. 6D), suggesting that high level neuronal expression of SUT-6 is toxic to C. elegans regardless of W292X mutation presence. We then crossed these neuronal SUT-6(W292X) neuronal expression lines with tau transgenic C. elegans and found that neuronal expression of SUT-6(W292X) suppressed tau-induced swimming deficits and reduced tau protein levels (Fig. 6E–G, Supplementary Material, Fig. S10–S11). This confirmed that mutant SUT-6(W292X) protein acts dominantly in a cell-autonomous manner to suppress tau-induced toxicity.
Figure 6.

Neuronal overexpression of SUT-6 W292X suppresses tau. (A) Representative blot of SUT-6 total (measured by SUT-6 antibody 6B), endogenous SUT-6 as measured by SUT-6 C-terminal antibody 1A and tubulin (load control) in the two generated SUT-6 W292X neuronal expression strains (W292X neuro #1 and #2) compared to non-transgenic (non-Tg). (B) Quantification of SUT-6 total protein levels in non-transgenic and SUT-6 W292X neuronal expression strains. Individual values are displayed as well as bars showing the means ± SEM. One-way ANOVA followed by Dunnett’s T3 post hoc tests were performed. n.s. = not significant. ****P < 0.0001. (C) Representative blot showing SUT-6 protein levels (6B antibody) in non-transgenic and SUT-6 W292X neuronal expression strain #1 in a sut-6 deletion background. (D) Swimming assays for non-transgenic (non-Tg) and SUT-6 W292X neuronal expression transgenic C. elegans. N = 3, n = 138–150. (E) Swimming assays for tau transgenic C. elegans expressing mutant tau (tau Tg V337M, tau background = CK10) with SUT-6 W292X neuronal expression. N = 3, n = 95–122. For (D–E), individual values are displayed as well as boxplots. One-way ANOVA followed by Games–Howell post hoc tests were performed. n.s. = not significant. ****P < 0.0001. (F) Representative immunoblots showing SUT-6, total tau and tubulin (load control) in tau transgenic C. elegans expressing mutant V337M tau with SUT-6 W292X neuronal expression. Tau background = CK10. (G) Quantification of total tau levels in tau transgenic C. elegans expressing mutant V337M tau with SUT-6 W292X neuronal expression. Individual values are displayed as well as bars showing the means ± SEM. One-way ANOVA followed by Dunnett’s T3 post hoc tests comparing tau transgenic;SUT-6 W292X neuronal expression double mutants to tau transgenic control were performed. *P < 0.05.
Sut-6 acts independently from sut-2, aly genes and spop-1 on tau-induced toxicity
The human homolog of SUT-6, NIPP1, localizes to the nucleus and primarily in nuclear speckles (41), which are nuclear domains that mediate splicing and processing of RNAs (42). We have shown that SUT-6 protein also localizes to the nucleus (Fig. 4C). sut-6 is just one of several genes encoding nuclear speckle proteins that modulate tau-induced toxicity in tau transgenic C. elegans. This group includes sut-2, spop-1 and the aly genes (14,15,18). We explored whether these genes might interact with sut-6 to modulate tau-induced toxicity by performing epistasis analysis.
First, we investigated the relationship between sut-6 and sut-2. Since loss of sut-2 results in complete suppression of tau-induced swimming deficits, we determined the effect of neuronal overexpression of SUT-2 on suppression of tau-induced swimming deficits by loss of sut-6 and the effect of neuronal overexpression of SUT-6 on suppression of tau-induced swimming deficits by loss of sut-2. Due to the close proximity of sut-2 with the integration site of the tau transgene in our wild-type tau C. elegans model, we performed these epistasis studies with our mutant tau V337M transgenic C. elegans model. We found that overexpression of SUT-2 reduced but did not completely block the suppression of tau-induced swimming deficits seen with loss of sut-6 in tau transgenic C. elegans (Fig. 7A); when considering the effect that overexpression of SUT-2 has on tau by itself, the effects of SUT-2 overexpression and loss of sut-6 appear additive. Meanwhile, overexpression of SUT-6 partially reduced the swimming ability of tau transgenic C. elegans lacking sut-2 even though overexpression of SUT-6 alone had no effect on tau-induced swimming deficits (Fig. 7B). Closer examination of the results with sut-2 show that overexpression of SUT-6 reduced swimming turn frequency by roughly one-third; however, the turn frequency of tau transgenic C. elegans lacking sut-2 and overexpressing SUT-6 was similar to that observed for C. elegans that overexpressed SUT-6 in the absence of tau protein (see Fig. 4F). Thus, another possible interpretation of the data is that loss of sut-2 maximally reversed tau-induced swimming deficits and that overexpression of SUT-6 reduced swimming ability via an independent pathway. To confirm the independent actions of sut-6 and sut-2 on tau-induced toxicity, we investigated the effects of loss of sut-6 or sut-2 had on SUT-2 and SUT-6 protein levels, respectively. We found that mutation of sut-6 did not significantly alter SUT-2 protein levels in tau transgenic C. elegans and loss of sut-2 did not significantly alter SUT-6 protein levels in tau transgenic C. elegans (Supplementary Material, Fig. S12). Altogether, this suggests that sut-6 and sut-2 suppress tau-induced toxicity in independent, parallel pathways.
Figure 7.
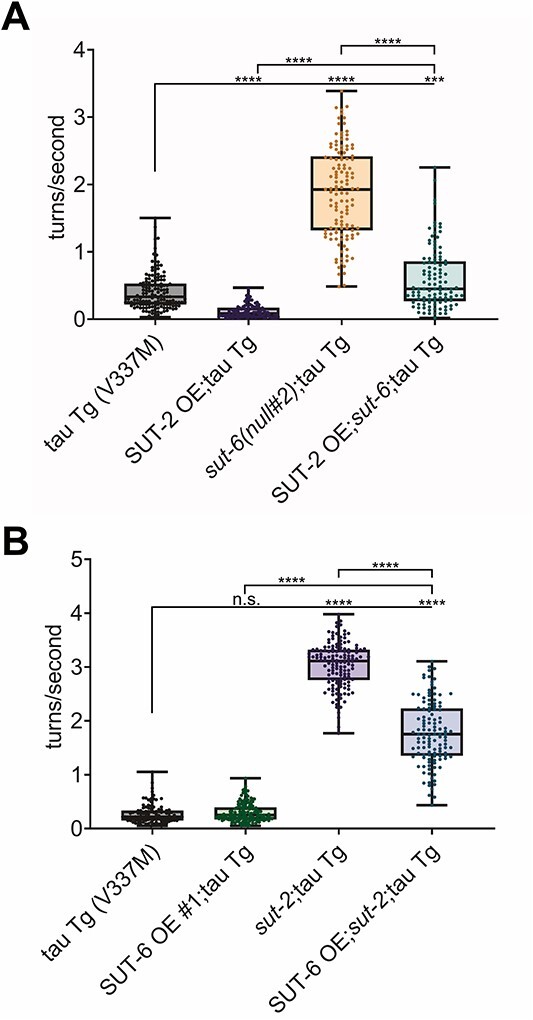
sut-2 and sut-6 act independently on tau-induced swimming behavior deficits. (A) Swimming assays for tau transgenic C. elegans expressing mutant tau (tau Tg V337M, tau background = CK10) with SUT-2 overexpression and/or sut-6 deletion. N = 3, n = 98–146. (B) Swimming assays for tau transgenic C. elegans expressing mutant tau (tau Tg V337M, tau background = CK10) with SUT-6 neuronal overexpression (line #1) and/or sut-2 deletion. N = 3, n = 102–148. For both graphs, individual values are displayed as well as boxplots. One-way ANOVA followed by Games–Howell post hoc tests were performed. n.s. = not significant. ***P < 0.001. ****P < 0.0001.
Next, we investigated the relationship between sut-6 and the aly genes. Because neither loss of sut-6 nor the aly genes show complete suppression of tau toxicity, we examined the effect of loss of sut-6 and the aly genes aly-1 and aly-3 on tau-induced swimming deficits. We chose aly-1 and aly-3 because this aly gene combination was previously shown to produce the strongest suppression of tau-induced swimming deficits by aly genes (18). We found that loss of sut-6, aly-1 and aly-3 resulted in suppression of tau-induced swimming deficits that appeared additive (Fig. 8A), suggesting that these genes act separately on tau. Experiments using ALY-1 or SUT-6 overexpression suggest that despite working separately, there may be an order to their activities on tau. We found that neuronal overexpression of ALY-1 caused a minor enhancement of tau-induced swimming deficits seen with loss of sut-6, but neuronal overexpression of SUT-6 significantly blocked the suppression of tau-induced swimming deficits by loss of aly-1 and aly-3 despite not enhancing tau-induced swimming deficits on its own (Fig. 8B–C). To better understand this latter finding, we investigated whether SUT-6 overexpression might block the reduction of total tau protein levels caused by loss of aly gene expression. Interestingly, we found that, in contrast to the swimming results, neuronal overexpression of SUT-6 did not block the decrease in total tau protein seen with loss of aly-1 and aly-3 (Fig. 8D–E, Supplementary Material, Fig. S13). This suggests that neuronal overexpression of SUT-6 alters tau-induced swimming deficits in aly-1;aly-3 mutants in a tau protein-independent manner. Taken together, these data show that sut-6 and the aly genes act independently on tau-induced toxicity.
Figure 8.

aly genes and sut-6 act independently on tau-induced swimming behavior deficits. (A) Swimming assays for tau transgenic C. elegans expressing high levels of wild-type tau (tau Tg wt high, tau background = CK144) with sut-6 deletion and/or aly-1 and aly-3 deletion. N = 3, n = 119–129. (B) Swimming assays for tau transgenic C. elegans expressing mutant tau (tau Tg V337M, tau background = CK10) with ALY-1 neuronal overexpression and/or sut-6 deletion. N = 3, n = 133–148. (C) Swimming assays for tau transgenic C. elegans expressing mutant tau (tau Tg V337M, tau background = CK10) with SUT-6 neuronal overexpression (line #1) and/or aly-1 and aly-3 deletion. N = 3, n = 141–150. For (A–C), individual values are displayed as well as boxplots. One-way ANOVA followed by Games–Howell post hoc tests were performed. n.s. = not significant. *P < 0.05. **P < 0.01. ****P < 0.0001. (D) Representative immunoblots showing SUT-6, total tau and tubulin (load control) in tau transgenic C. elegans expressing mutant V337M tau with SUT-6 neuronal overexpression (line #1) and/or aly-1 and aly-3 deletion. Tau background = CK10. (G) Quantification of total tau levels in tau transgenic C. elegans expressing mutant V337M tau with SUT-6 neuronal overexpression (line #1) and/or aly-1 and aly-3 deletion. Individual values are displayed as well as bars showing the means ± SEM. One-way ANOVA followed by Dunnett’s T3 post hoc tests were performed. **P < 0.01.
Finally, we examined the relationship between sut-6 and spop-1. Due to the close proximity of spop-1 with the integration site of the tau transgene in our mutant tau V337M C. elegans model, we performed these epistasis studies with our wild-type tau transgenic C. elegans model. We first examined the effect of sut-6 and spop-1 loss on tau-induced swimming deficits and found combined loss of sut-6 and spop-1 caused significant suppression of tau-induced deficits in a manner that could be synergistic (Fig. 9A). To determine whether this meant that sut-6 and spop-1 act in the same pathway, we investigated the effect of neuronal overexpression of SPOP-1(ΔNLS), which was previously shown to significantly enhance tau-induced toxicity (15), on suppression of tau-induced swimming deficits by loss or W292X mutation of sut-6 and the effect of neuronal overexpression of SUT-6 on suppression of tau-induced swimming deficits by loss of spop-1. We found that neuronal overexpression of SPOP-1(ΔNLS) completely blocked suppression of tau-induced swimming deficits by loss of sut-6 and partially blocked suppression of tau-induced swimming by sut-6(W292X), while overexpression of SUT-6 partially blocked suppression of tau-induced swimming deficits by loss of spop-1 (Fig. 9B–C). This suggests that sut-6 and spop-1 are acting in distinct pathways. Because neuronal overexpression of SPOP-1(ΔNLS) was shown to increase tau protein levels (15), we investigated whether SPOP-1(ΔNLS) overexpression blocked suppression of tau toxicity by loss of sut-6 by altering tau protein levels. We found that SPOP-1(ΔNLS) neuronal overexpression completely reversed the decrease in tau protein level caused by loss of sut-6 but not the decrease in tau protein level caused by sut-6(W292X), mirroring the behavior results (Fig. 9D–E, Supplementary Material, Fig. S14). Interestingly, loss of sut-6 or sut-6(W292X) reduced the total levels of SPOP-1 protein in overexpressed SPOP-1(ΔNLS) strains (Fig. 9F, Supplementary Material, Fig. S14). Neuronal overexpression of SPOP-1(ΔNLS) protein is dramatically increased in the presence of neuronal tau, a sign of tau-induced proteostasis deficits (15), suggesting that sut-6 loss-of-function mutants protect against tau-induced proteostasis deficits even in the face of SPOP-1(ΔNLS) overexpression mediated enhancement of tauopathy in worms. Next, we examined the effect of SUT-6 overexpression on tau protein levels in tau transgenic C. elegans lacking spop-1. Loss of spop-1 reduced total tau protein levels as expected (15), but overexpression of SUT-6 did not block the decrease in total tau protein (Fig. 9G–H, Supplementary Material, Fig. S15). Once again, we observed that neuronal overexpression of SUT-6 reduced swimming ability of tau transgenic C. elegans in the presence of a suppressor mutation in another gene with no effect on tau protein changes, strongly suggesting that any effects that SUT-6 overexpression has on swimming are independent of tau protein. Altogether, these data show that spop-1 and sut-6 also act independently to modulate tau-induced toxicity.
Figure 9.

spop-1 and sut-6 act independently on tau-induced swimming behavior deficits. (A) Swimming assays for tau transgenic C. elegans expressing high levels of wild-type tau (tau Tg wt high, tau background = CK144) with sut-6 deletion and/or spop-1 deletion. N = 3, n = 129–150. (B) Swimming assays for tau transgenic C. elegans expressing high levels of wild-type tau (tau Tg wt high, tau background = CK144) with SPOP-1(ΔNLS) neuronal overexpression and/or sut-6 mutations. N = 5, n = 146–198. (C) Swimming assays for tau transgenic C. elegans expressing high levels of wild-type tau (tau Tg wt high, tau background = CK144) with SUT-6 neuronal overexpression (line #1) and/or spop-1 deletion. N = 3, n = 107–150. For (A–C), individual values are displayed as well as boxplots. One-way ANOVA followed by Games–Howell post hoc tests were performed. n.s. = not significant. *P < 0.05. ****P < 0.0001. (D) Representative immunoblots showing SPOP-1, SUT-6, total tau and tubulin (load control) in tau transgenic C. elegans expressing high levels of wild-type tau with SPOP-1(ΔNLS) neuronal overexpression and/or sut-6 mutations. Tau background = CK144. (E) Quantification of total tau levels in tau transgenic C. elegans expressing high levels of wild-type tau with SPOP-1 neuronal overexpression and/or sut-6 mutations. (F) Quantification of SPOP-1 levels in in tau transgenic C. elegans expressing high levels of wild-type tau with SPOP-1(ΔNLS) neuronal overexpression and/or sut-6 mutations. (G) Representative immunoblots showing SPOP-1, SUT-6, total tau and tubulin (load control) in tau transgenic C. elegans expressing high levels of wild-type tau with SUT-6 neuronal overexpression (line #1) and/or spop-1 deletion. Tau background = CK144. (H) Quantification of total tau levels in tau transgenic C. elegans expressing high levels of wild-type tau with SUT-6 neuronal overexpression (line #1) and/or spop-1 deletion. For (E), (F) and (H), individual values are displayed as well as bars showing the means ± SEM. One-way ANOVA followed by Dunnett’s T3 post hoc tests on the displayed comparisons were performed. n.s. = not significant. *P < 0.01. ****P < 0.0001.
Discussion
In this work, we have identified a novel suppressor mutation of tau toxicity in the gene sut-6 which is the C. elegans homolog of human NIPP1. While functions of sut-6 are unknown, the human homolog NIPP1 encodes a protein that localizes to nuclear speckles where it can bind and regulate the activity of protein phosphatase 1 (PP1), splicing factors and transcription regulators (19,21–23,25,28,43). We have also identified a mutation, sut-6(W292X), which not only shows stronger suppression of tau toxicity than a complete deletion but can act dominantly to suppress tau toxicity when expressed in neurons. By comparing the effects of sut-6(W292X) to sut-6(null) and sut-6(CΔ10), we found that the reduction in tau toxicity by sut-6 mutations is partially reflected by a change in total tau protein levels rather than tau phosphorylation state or tau mRNA levels. However, despite causing a similar decrease in tau protein, sut-6(W292X) showed stronger suppression of tau toxicity in locomotor assays. In addition, neuronal overexpression of SUT-6 protein could enhance locomotor deficits in low toxicity tau models without altering tau protein levels. These two findings suggest that there are other mechanisms besides regulation of tau protein levels in which sut-6 modulates tau-related toxicity.
The fact that expressing SUT-6(W292X) protein in neurons suppresses tau toxicity suggests a few possible mechanisms in which sut-6 regulates tau toxicity. One possibility is that SUT-6(W292X) is a dominant negative mutation, inducing stronger inhibition of certain activities of its interactors compared to loss of SUT-6 protein expression. The C terminus of the sut-6 homolog NIPP1 has been shown to interact with the NIPP1:PP1 holoenzyme, with binding of RNA to the C terminus releasing the inhibitory activity of NIPP1 on PP1 and allowing PP1 to dephosphorylate substrates that are binding to NIPP1 (25). In that sense, NIPP1 acts more like an adaptor/regulator for PP1 than strictly as an inhibitor, allowing RNA-regulated control of PP1 activity. In fact, overexpression of NIPP1 has been shown to increase dephosphorylation of Sap155 by PP1 (28). With loss of the C terminus of NIPP1 in sut-6(W292X) but not loss of SUT-6 protein expression levels like in sut-6(CΔ10), this C-terminally truncated SUT-6/NIPP1 could bind PP1 as a dominant negative inhibitor, bringing PP1 to its target but not allowing PP1 to dephosphorylate it due to the fact that RNA can no longer release the inhibition of PP1 by SUT-6/NIPP1.
Another possibility is that SUT-6(W292X) alters the balance of activities of SUT-6/NIPP1 interactors to favor those that suppress of tau toxicity. It has been shown in human cell culture studies that phosphorylation of Y335 of the C terminus of NIPP1, an event dependent on NIPP1 association with RNA, can block the ability of PP1 to dephosphorylate myelin basic protein but not phosphorylase (25). In cell culture studies, overexpression of NIPP1 lacking the C-terminal 22 amino acids exclusively associated with unspliced pre-mRNA and inhibited pre-mRNA splicing while wild-type NIPP1 bound to both spliced and unspliced pre-mRNA (28). These are just two examples of activities altered by C-terminal truncation of NIPP1. Whether these are important in the mechanism in which SUT-6(W292X) modulates tau remains to be examined.
Moreover, it remains possible that loss of the RNA-binding domain of SUT-6 seen with SUT-6(W292X) strongly suppresses tau toxicity, but loss of other SUT-6 domains such as the FHA domain or the central core domain enhances tau toxicity or blocks the effects of losing the RNA-binding domain. This type of result has been seen with splicing, where mutation of the FHA domain or the PP1 interaction residues removed the block C-terminal truncated NIPP1 had on splicing (28). Future studies examining the consequences of sut-6 mutations will allow us to clarify which activities of sut-6 regulate tau toxicity.
We do not fully understand the molecular mechanism underlying sut-6 suppression of tau toxicity, but sut-6 is one of several recently identified tau suppressors that localize to nuclear speckles. Data from Alzheimer’s disease and tauopathy models show that nuclear speckle scaffold protein SRRM2 mislocalizes to the cytoplasm under pathological tau conditions, demonstrating a relationship between nuclear speckles and tau toxicity (44,45). Through epistasis studies, we showed that none of the previously identified suppressors of tau toxicity known to localize to nuclear speckles (sut-2, aly genes, spop-1) regulate tau toxicity in the same pathway as sut-6 (Fig. 10). This indicates that while multiple suppressors of tau toxicity localize to nuclear speckles, there are multiple parallel pathways in which these nuclear speckle resident proteins can regulate tau toxicity. Perhaps nuclear speckles serve as a hub of activity where multiple pathways that can regulate tau can intersect. In this case, disrupting the nuclear speckle resident proteins SRRM2 or SON may influence the ability of all these previously identified nuclear speckle proteins to suppress tau toxicity. Such studies are currently being pursued but are beyond the scope of this paper.
Figure 10.
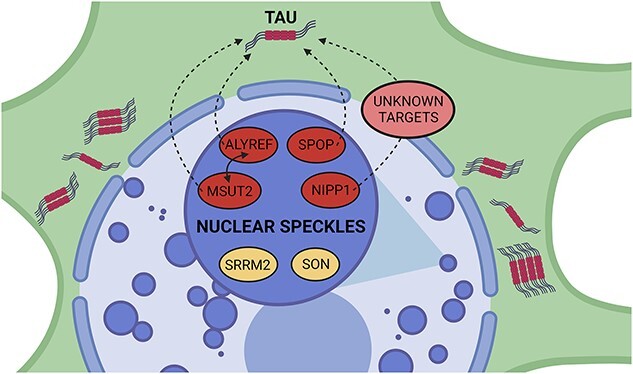
Summary of nuclear speckle suppressors of tau toxicity. sut-2/MSUT2 (14), aly/ALYREF (18), spop-1/SPOP (15) and sut-6/NIPP1 (this manuscript) are all demonstrated suppressors of tau-induced toxicity when deleted in C. elegans tauopathy models. For simplicity, mammalian protein names are used exclusively in the figure which was created with BioRender (BioRender.com). They are all known to localize to nuclear speckles (46–48). SRRM2 and SON are nuclear speckle resident proteins (49). The mechanisms in which these nuclear speckle suppressors suppress tau are still unknown. spop-1 and sut-2 are known to act in parallel to modulate tauopathy (15). MSUT2 and ALYREF can be found in the same protein complex in a RNA-dependent interaction (50). sut-6/NIPP1 probably modulates tau indirectly via unknown targets. These unknown targets may be proteins and/or RNAs.
In our results and discussion so far, we have proposed that sut-6 and its mammalian homolog, NIPP1, have similar functions due to the high protein sequence homology. However, some evidence suggests differences in the functional consequences of altering NIPP1 and SUT-6. In mammals, whole animal knockout of NIPP1 is embryonic lethal (51), while whole animal knockout of sut-6 is not. Cell-specific knockout of NIPP1 in adult mammalian brain impairs development, causes deficits in myelination, and induces death at P30 or earlier (36). The significant neurodevelopmental defects caused by loss of NIPP1 in the mouse brain might explain why McKee et al. found increased phosphorylation but no change in total levels of tau with NIPP1 knockout whereas we found decreased total tau with sut-6 knockout. The expression levels of NIPP1 in mammals versus SUT-6 in C. elegans may explain the differential requirements for NIPP1 versus SUT-6 in cell viability. According to the human protein atlas (www.proteinatlas.org, see (52)), NIPP1 (PPP1R8) RNA and protein is expressed throughout the entire brain with no regional specificity. In contrast, sut-6 (B0511.7) gene expression levels are very low in C. elegans, with the C. elegans Neuronal Gene Expression Map and Network (www.cengen.org, see (53)) reporting less than 100 transcripts per million in all neuronal types with some having no expression. In addition, we were unable to visualize any endogenous SUT-6 protein by immunostaining. When compared to PP1 (PPP1CA) RNA expression, NIPP1 levels are ~ 2–4-fold less in human brains. Meanwhile sut-6 transcripts levels are ~ 10–100-fold less than PP1 homologs gsp-1 and gsp-2 transcript levels in neurons. This suggests that NIPP1, due to its relative abundance, could influence PP1 activity more strongly in mammals than SUT-6 does in C. elegans. This may also be the case for the effects of NIPP1 on its other interactors, although whether SUT-6 binds to the C. elegans homologs of known NIPP1 interactors remains to be seen. While beyond the scope of this paper, examining whether neuronal expression of NIPP1 W340X protein, which would be the equivalent to SUT-6 W292X, also suppresses tau toxicity in a mammalian model of tauopathy would help determine whether this is a potential therapeutic means of intervention. Alternatively, if direct manipulation of NIPP1 activity is too toxic in mammals, discerning which function(s) of SUT-6 are important in modulating tau toxicity may allow us to target the analogous tau toxicity mechanisms downstream of SUT-6/NIPP1’s activity allowing for safer and more effective therapeutic strategies.
In summary, we have discovered a novel, dominant mutation in sut-6/NIPP1 that suppresses tau toxicity. We found that loss of sut-6 also suppressed tau but to a lesser extent, yet the effects on tau protein were similar between sut-6 alleles. Epistatic investigations between sut-6 and other previously identified nuclear speckle-localized suppressors of tau showed that sut-6 acts independently from all 3 (sut-2, aly-1;aly-3, spop-1) tested. Further investigation of sut-6 functions and interacting proteins in relation to tau will help elucidate the mechanisms behind this novel pathway of tau regulation.
Materials and Methods
Caenorhabditis elegans strains, genome editing and transgenics
N2 (Bristol) was used as wild-type C. elegans (54). Caenorhabditis elegans mutants were generated by CRISPR-Cas9 (described below) and crossed into the following tau transgenic (tau Tg) C. elegans strains: CK10 (bkIs10[aex-3p::tau-V337M+ myo-2p::GFP) expressing human 1N4R tau with a V337M mutation (12); CK144 (bkIs144[aex-3p::tau-1N4R+ myo-2p::GFP) expressing human 1N4R tau at high levels; CK1441 (bkIs1441[aex-3p::tau-V337M+ myo-2p::dsRed) expressing human 1N4R tau at low levels; CK1443 (bkIs1443[aex-3p::tau-1N4R + myo-2p::dsRed) expressing human 1N4R tau at moderate levels similar to that in CK10. The presence of alleles of interest was verified with PCR genotyping. All non-transgenic and tau transgenic strains were maintained at 20°C on standard nematode growth media (NGM) plates containing OP50 Escherichia coli as previously described (55). For protein studies, worms were grown on NGM plates containing 5× more peptone (5XPEP) at 20°C for at least 2 generations (1 week) prior to collection.
Caenorhabditis elegans alleles generated in this study are listed in Table 1. Alleles were generated for sut-6 as indicated in the table using CRISPR-Cas9 genome editing technology by introducing purified active recombinant Cas9 protein and synthetic CRISPR guide RNAs (gRNAs) as previously described (55). The aly-1;aly-3 strain was generated using the same guides as previously described (18).
| S.p. crRNA name | Protospacer |
|---|---|
| sut-6 null 5′ | ATTAGGTTTATTTCAGAAAA |
| sut-6 null 3′ | CAACTCAACAATTAAATTAG |
| sut-6 CΔ10 5′ | GTAACATATTGATGATGTCC |
| sut-6 CΔ10 3′ | CAACTCAACAATTAAATTAG |
| sut-6 W292X | CAAAGGAAGCGTGGCCAGGA |
| Synthetic oligonucleotide repair template |
Sequence |
| sut-6 CΔ10 RT | GATCTCGAATTATATGGGAAATTGCTTCCCGAGCCAGGACACCACCAGTATGTTACATCGACTGCAACTGAAGATGAAGCTGATGATCATCACAAGAAAAGATATGCAAAGTAAACTTTAATATTTACATTTTATAAGCTTAATAAG |
| sut-6 W292X | GAAAAGATATGCAAAGGAAGCGTGACCAGGAAGAAAGCCTGGAGCTGGCATTTTTTAAAC |
Cas9 gRNAs were prepared by mixing equimolar amounts (0.5 μg/μl) of Alt-R crRNA and Alt-R tracrRNA (Integrated DNA Technologies) and then the gRNAs were complexed to equimolar quantities of Cas9 protein (Alt-R S.p. Cas9 Nuclease V3, Integrated DNA Technologies as described). Dpy-10 or rol-6 were utilized as a gene co-conversion or co-injection marker as previously described (56) and removed by outcrossing. To generate the null alleles, two gRNAs were introduced targeting the promoter/start codon and 3′UTR/stop codon sequences of the sut-6 genomic loci, respectively. To generate the sut-6 CΔ10 allele, two guide RNAs were introduced flanking the C-terminal end of SUT-6 coding sequences and a repair template as indicated in the table above was also introduced. To generate the sut-6 W292X mutation, we used a single guide and repair template-based strategy.
SUT-6 wild-type and W292X neuronal overexpression transgenic lines were constructed by introducing sut-6 genomic sequence under the pan-neuronal rgef-1 promotor (Prgef-1::sut-6 or Prgef-1::sut-6(W292X)) into the C. elegans genome. A mix containing Prgef-1::sut-6 or Prgef-1::sut-6(W292X) at 10 ng/μl and marker elt-2::mCherry (30 ng/μl) was microinjected into N2 strain C. elegans. Progeny were screened for mCherry in the gut as sign of transmission of extrachromosomal arrays. Extrachromosomal arrays were integrated by sub-lethal doses of 312 nm UV irradiation. Integrated lines were then backcrossed with N2 twice before use in experiments.
Mutagenesis
We used ethyl nitrosurea (ENU) as described (57). Briefly, tau transgenic worms were exposed to 1 mm ENU in M9 for 4 h with gentle rocking. F2 progeny that had the desired mutant phenotype of restored motility in the presence of the tau transgene were selected as previously described (13). Mutants that bred true were subjected to a secondary screen which consisted of testing each mutation in an independent tau transgenic background. Suppressing mutants were backcrossed repeatedly to remove non-causal mutations prior to whole-genome sequencing (WGS) to identify tau suppressing mutations. Genome sequence analysis was conducted using the Lasergene genomics SeqMan NGen software package (DNASTAR).
Behavior in C. elegans
Swimming assays were performed as follows. NGM plates containing tau transgenic C. elegans at roughly day 1 of adulthood or non-transgenic C. elegans at day 1 or day 2 of adulthood were flooded with 1 ml of M9 buffer. Swimming worms were then pipetted with a P1000 pipette onto a 35 mm unseeded NGM plate. Videos of swimming worms were recorded for 1 min at 14 frames per second approximately 1 min following the addition of M9 buffer. Recorded videos were analyzed using WormLab 2020 (MBF Bioscience). The frequency of body bends, defined as ‘turns’ by the software, was quantified as follows. A turn was defined as a change in the body angle defined by the quarter points and midpoint of the worm that was at least 20 degrees positive or negative from a straight line. Worms that were tracked for less than 30 s were omitted from analysis. Roughly 15–60 worms from at least three independent replicates were counted per strain.
Immunization of mice with recombinant SUT-6 protein
Recombinant GST-SUT-6 fusion protein was produced in E. coli as previously described (14). Recombinant GST-SUT-6 protein was employed as the immunogen in mice to produce monoclonal antibodies in the Washington State University (WSU) Monoclonal Antibody Center as a fee-for-service project using established standard approaches (58). Monoclonal antibodies produced at WSU are covered by mAb Center IACUC protocol #6317 using standard operating procedures for mAb production. Briefly, GST-SUT-6 protein was mixed in Sigma Adjuvant System at 50% (w/v) as an emulsion prior to immunization as suggested by the manufacturer’s protocol (Sigma Chemical, catalog S6322). Mice were immunized with an initial dose of 200 μg of GST-SUT-6 protein and subsequently received an identical boost immunization 3 weeks later. Harvest of spleens occurred 3 days after boost immunization and were used for hybridoma production. Clonal hybridoma lines were screened by enzyme-linked immunosorbent assay based on SUT-6 binding activity and the absence of GST binding activity using standard protocols (58). The SUT-6 reactive mAb expressing hybridoma monoclonal cell lines were isolated and cloned (designated SUT-6 mAb #1A, 6B and 7A, respectively).
Immunoblotting
Staged young adult C. elegans were grown from eggs at 20°C for 3–4 days on 5XPEP plates until the majority of the population was day 1 adults, washed off plates in M9 buffer and collected by centrifugation. Worm pellets were snap frozen in liquid nitrogen and stored at −80°C. Worm pellets were weighed and diluted with 1× sample buffer (0.009 m Tris, 0. 001 m EDTA, 0.04 m dithiothreitol, 10% sucrose, 1% sodium dodecyl sulfate, 0.01% bromophenol blue), at 4 μl of sample buffer added to 1 μg of worm pellet, sonicated three times, boiled for 10 min and centrifuged at 13 000×g for 2 min prior to being loaded onto 18-well or 26-well 4–15% pre-cast Criterion sodium dodecyl sulfate polyacrylamide gel electrophoresis gradient gels and transferred to PVDF membranes as recommended by the manufacturer (Bio-Rad). Then, 5 μl or 10 μl of prepared lysed worm sample was loaded onto 26-well or 18-well gels, respectively, for each analysis. Primary antibodies used were rabbit polyclonal anti-tau antibody (DAKO) at 1:500 000, rabbit polyclonal anti-pT212 tau antibody 44-740G (Invitrogen) at 1:1000, mouse monoclonal anti-pS202 tau antibody CP13 (Peter Davies) at 1:1000, mouse monoclonal anti-SUT-6 antibodies at 1:50 000, mouse monoclonal anti-SPOP-1 antibody clone 8B at 1:100, rabbit polyclonal anti-SUT-2 antibody RB841 at 1:5000 and mouse anti-tubulin antibody E7 (Developmental Studies Hybridoma Bank) at 1:5000. Secondary antibodies used were anti-rabbit HRP (Jackson Immuno Research) and anti-mouse HRP (Jackson Immuno Research) at 1:5000. ECL substrate (Bio-Rad) was added to the membrane and chemiluminescence signals were detected with a LICOR Odyssey Fc. Quantification of band signal intensity was performed on raw images, prior to adjusting images for brightness, contrast and sharpness for publication.
SUT-6 immunofluorescence in C. elegans
For each strain, age synchronized D1A worms were picked and transferred to three successive droplets of M9 then to a drop of 1% PFA on poly-L-lysine-coated slides. The worms were permeabilized by freeze-cracking and fixed by methanol/acetone fixation. Slides with fixed and permeabilized animals were moved to a humidifying chamber, washed once with PBS, then placed in blocking solution (10% goat serum in antibody buffer) for 1 h at room temperature. After blocking, the worms were incubated with SUT-6 6B primary antibody at a dilution of 1:100 and tau monoclonal antibody SP70 (Invitrogen) at 1:1000 in blocking solution at 4°C overnight. Slides were then washed 3× with PBS, 2× with blocking solution, then incubated in AlexaFluor 647F(ab′)2 fragment of goat anti-mouse IgG (H + L) (Jackson Immunological) and AlexaFluor 488F(ab′)2 fragment of goat anti-rabbit IgG (H + L) (Jackson Immunological) both at 1:1000 in blocking solution at room temperature for 1 h. Slides were then washed 3× with PBS, incubated with 300 nm DAPI for 5 min at room temperature, washed 2× more with PBS, then mounted with ProLong Gold (Thermo Fisher Scientific). Animals were imaged with a DeltaVision Elite fluorescent microscope (Applied Precision). Images shown in Figure 4C were threshold-adjusted consistently across the entire image in Photoshop to reduce background signal and allow for better visualization of nuclei in print.
GABAergic neuron loss assay
Tau transgenic worms carrying sut-6(bk3104) or sut-6(bk3067) were crossed into CZ1200 (juIs76 [unc-25p::GFP + lin-15(+)]) to generate tau Tg;unc-25p::GFP, tau Tg;unc-25p::GFP;sut-6(bk3104) and tau Tg;unc-25p::GFP;sut-6(bk3067) worms. Worms were synchronized by timed egg lays and mounted at 4 days post-hatch (roughly day 1 of adulthood) on 2% agarose pads and paralyzed with 500 mm sodium azide. The number of visible GABAergic neurons (up to 16 due to the 3 nearest the head being obscured) in the ventral nerve cord were counted with a DeltaVision Elite microscope (Applied Precision) using 40× magnification. All replicates were blinded to genotype. At least 15 animals per replicate for a total of 45+ animals were counted per strain.
Quantification of mRNA levels of target genes in C. elegans
Staged young adult C. elegans were grown from eggs at 20°C for 3 days on 5XPEP plates, washed off plates in M9 buffer and collected by centrifugation. Worms were snap frozen in liquid nitrogen and stored at −80°C. RNA was isolated from C. elegans pellets of ~ 50–100 mg using Tri-Reagent (Molecular Research Center) following the manufacturer’s instructions and resuspended in RNAse-free water. After Tri-Reagent isolation, 10 μg of RNA was treated with DNase using DNA-free DNase Treatment and Removal Kit (Invitrogen) following the manufacturer’s instructions. Then, 2.5 μl of the DNase-treated RNA sample was transcribed into cDNA using iScript Reverse Transcription Supermix (Bio-Rad) following the manufacturer’s instructions. Quantitative PCR was performed using iTaq Universal SYBR Green Supermix (Bio-Rad) in a CFX Connect Real-Time PCR System (Bio-Rad) with primers against rpl-32 (control), sut-6 or tau. Each genotype was tested in four biological replicates and three technical replicates. Sut-6 or tau mRNA levels were normalized to rpl-32 following the deltadeltaCT method (59).
| Target gene | Forward primer | Reverse primer |
|---|---|---|
| rpl-32 | GGTCGTCAAGAAGAAGCTCACCAA | TCTGCGGACACGGTTATCAATTCC |
| sut-6 | CGAGCAATTAGATCACCAAACTTC | TCGTTTCTTTCGTCGAGCTT |
| tau | GTGTGGCTCATTAGGCAACATCC | CGTTCTCGCGGAAGGTCAG |
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8. Grubbs’ tests for extreme outliers were performed and values that fell outside the critical Z value, calculated with an alpha level of 0.05, were removed prior to statistical analysis. Behavior and immunoblot data with just two groups was analyzed by Student’s t-test. Behavior, immunoblot and mRNA data with three or more groups was analyzed by one-way ANOVA followed by post hoc multiple comparison testing (Games–Howell for behavior studies and Dunnett’s T3 for immunoblots or mRNA). Kruskal–Wallis followed by post hoc Dunn’s tests was used for neuron loss data. To reduce type 2 errors from multiple comparison testing, only the comparisons indicated on the graphs and figure legends were performed.
Supplementary Material
Acknowledgements
We thank the reviewers for useful suggestions and comments. We thank Aleen Saxton, Jade Stair, Isaac Miller and Rikki Ulrich for exceptional technical assistance. We thank Victoria Hulubei and William Davis at the Washington State University Monoclonal Antibodies Center for outstanding technical assistance in producing SUT-6 reactive mAbs. We thank Randall Eck for scientific insight into spop-1 function. We thank Dr Yishi Jin for the CZ1200 C. elegans strain. We thank the Developmental Studies Hybridoma Bank (NICHD) for the β-tubulin antibody E7. We thank Peter Davies for the CP13 tau antibody. We thank WormBase (WS251) for C. elegans genetic information. We thank the CGC (funded by the NIH Office of Research Infrastructure Programs (P40 OD010440)) and the National Bioresource Project (Japan) for provision of some C. elegans strains used in this study.
Conflict of Interest statement. The authors have no financial conflicts of interest.
Contributor Information
R L Kow, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA; Division of Gerontology and Geriatric Medicine, Department of Medicine, University of Washington, Seattle, WA 98104, USA.
A H Black, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA.
B P Henderson, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA.
B C Kraemer, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA; Division of Gerontology and Geriatric Medicine, Department of Medicine, University of Washington, Seattle, WA 98104, USA; Department of Psychiatry and Behavioral Sciences; Department of Laboratory Medicine and Pathology, University of Washington, Seattle, WA 98195, USA.
Funding
Grants from the Department of Veterans Affairs [Merit Review Grant #I01BX005742 and Career Development Award Level-2 #BX004341] and National Institutes of Health [R01NS064131].
Data availability
Additional data is available to be shared upon request.
References
- 1. Chung, D., Eun, C., Roemer, S., Petrucelli, L. and Dickson, D.W. (2021) Cellular and pathological heterogeneity of primary tauopathies. Mol. Neurodegener., 16, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Limorenko, G. and Lashuel, H.A. (2021) To target tau pathologies, we must embrace and reconstruct their complexities. Neurobiol. Dis., 161, 105536. [DOI] [PubMed] [Google Scholar]
- 3. Spillantini, M.G., Murrell, J.R., Goedert, M., Farlow, M.R., Klug, A. and Ghetti, B. (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. U. S. A., 95, 7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hutton, M., Lendon, C.L., Rizzu, P., Baker, M., Froelich, S., Houlden, H.H., Pickering-Brown, S., Chakraverty, S., Isaacs, A., Grover, A. et al. (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature, 393, 702–704. [DOI] [PubMed] [Google Scholar]
- 5. Clark, L.N., Poorkaj, P., Wszolek, Z., Geschwind, D.H., Nasreddine, Z.S., Miller, B., Li, D., Payami, H., Awert, F., Markopoulou, K. et al. (1998) Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc. Natl. Acad. Sci. U. S. A., 95, 13103–13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Poorkaj, P., Bird, T.D., Wijsman, E., Nemens, E., Garruto, R.M., Anderson, L., Andreadis, A., Wiederholt, W.C., Raskind, M. and Schellenberg, G.D. (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol., 43, 815–825. [DOI] [PubMed] [Google Scholar]
- 7. Bird, T.D., Nochlin, D., Poorkaj, P., Cherrier, M., Kaye, J., Payami, H., Peskind, E., Lampe, T.H., Nemens, E., Boyer, P.J. et al. (1999) A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (P301L). Brain, 122, 741–756. [DOI] [PubMed] [Google Scholar]
- 8. Hong, M., Zhukareva, V., Vogelsberg-Ragaglia, V., Wszolek, Z., Reed, L., Miller, B.I., Geschwind, D.H., Bird, T.D., McKeel, D., Goate, A. et al. (1998) Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science, 282, 1914–1917. [DOI] [PubMed] [Google Scholar]
- 9. Dujardin, S., Colin, M. and Buée, L. (2015) Invited review: Animal models of tauopathies and their implications for research/translation into the clinic. Neuropathol. Appl. Neurobiol., 41, 59–80. [DOI] [PubMed] [Google Scholar]
- 10. Chang, C.W., Shao, E. and Mucke, L. (2021) Tau: enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science, 371, eabb8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soeda, Y. and Takashima, A. (2020) New insights into drug discovery targeting tau protein. Front. Mol. Neurosci., 13, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kraemer, B.C., Zhang, B., Leverenz, J.B., Thomas, J.H., Trojanowski, J.Q. and Schellenberg, G.D. (2003) Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. U. S. A., 100, 9980–9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kraemer, B.C. and Schellenberg, G.D. (2007) SUT-1 enables tau-induced neurotoxicity in C. elegans. Hum. Mol. Genet., 16, 1959–1971. [DOI] [PubMed] [Google Scholar]
- 14. Guthrie, C.R., Schellenberg, G.D. and Kraemer, B.C. (2009) SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum. Mol. Genet., 18, 1825–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eck, R.J., Kow, R.L., Black, A.H., Liachko, N.F. and Kraemer, B.C. (2022) SPOP loss of function protects against tauopathy. Proc. Natl. Acad. Sci. U. S. A., 120, e2207250120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wheeler, J.M., McMillan, P., Strovas, T.J., Liachko, N.F., Amlie-Wolf, A., Kow, R.L., Klein, R.L., Szot, P., Robinson, L., Guthrie, C. et al. (2019) Activity of the poly(a) binding protein MSUT2 determines susceptibility to pathological tau in the mammalian brain. Sci. Transl. Med., 11, eaao6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kow, R.L., Strovas, T.J., McMillan, P.J., Jacobi, A.M., Behlke, M.A., Saxton, A.D., Latimer, C.S., Keene, C.D. and Kraemer, B.C. (2021) Distinct poly(a) nucleases have differential impact on sut-2 dependent tauopathy phenotypes. Neurobiol. Dis., 147, 105148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kow, R.L., Black, A.H., Saxton, A.D., Liachko, N.F. and Kraemer, B.C. (2022) Loss of aly/ALYREF suppresses toxicity in both tau and TDP-43 models of neurodegeneration. Geroscience, 44, 747–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beullens, M., van Eynde, A., Vulsteke, V., Connor, J., Shenolikar, S., Stalmans, W. and Bollen, M. (1999) Molecular determinants of nuclear protein phosphatase-1 regulation by NIPP-1. J. Biol. Chem., 274, 14053–14061. [DOI] [PubMed] [Google Scholar]
- 20. Jin, Q., Beullens, M., Jagiello, I., van Eynde, A., Vulsteke, V., Stalmans, W. and Bollen, M. (1999) Mapping of the RNA-binding and endoribonuclease domains of NIPP1, a nuclear targeting subunit of protein phosphatase 1. Biochem. J., 342, 13–19. [PMC free article] [PubMed] [Google Scholar]
- 21. Boudrez, A., Beullens, M., Groenen, P., van Eynde, A., Vulsteke, V., Jagiello, I., Murray, M., Krainer, A.R., Stalmans, W. and Bollen, M. (2000) NIPP1-mediated interaction of protein phosphatase-1 with CDC5L, a regulator of pre-mRNA splicing and mitotic entry. J. Biol. Chem., 275, 25411–25417. [DOI] [PubMed] [Google Scholar]
- 22. Jin, Q., van Eynde, A., Beullens, M., Roy, N., Thiel, G., Stalmans, W. and Bollen, M. (2003) The protein phosphatase-1 (PP1) regulator, nuclear inhibitor of PP1 (NIPP1), interacts with the polycomb group protein, embryonic ectoderm development (EED), and functions as a transcriptional repressor. J. Biol. Chem., 278, 30677–30685. [DOI] [PubMed] [Google Scholar]
- 23. Roy, N., van Eynde, A., Beke, L., Nuytten, M. and Bollen, M. (2007) The transcriptional repression by NIPP1 is mediated by Polycomb group proteins. Biochim. Biophys. Acta, 1769, 541–545. [DOI] [PubMed] [Google Scholar]
- 24. Vulsteke, V., Beullens, M., Waelkens, E., Stalmans, W. and Bollen, M. (1997) Properties and phosphorylation sites of baculovirus-expressed nuclear inhibitor of protein phosphatase-1 (NIPP-1). J. Biol. Chem., 272, 32972–32978. [DOI] [PubMed] [Google Scholar]
- 25. Beullens, M., Vulsteke, V., van Eynde, A., Jagiello, I., Stalmans, W. and Bollen, M. (2000) The C-terminus of NIPP1 (nuclear inhibitor of protein phosphatase-1) contains a novel binding site for protein phosphatase-1 that is controlled by tyrosine phosphorylation and RNA binding. Biochem. J., 352, 651–658. [PMC free article] [PubMed] [Google Scholar]
- 26. Trinkle-Mulcahy, L., Ajuh, P., Prescott, A., Claverie-Martin, F., Cohen, S., Lamond, A.I. and Cohen, P. (1999) Nuclear organisation of NIPP1, a regulatory subunit of protein phosphatase 1 that associates with pre-mRNA splicing factors. J. Cell Sci., 112, 157–168. [DOI] [PubMed] [Google Scholar]
- 27. Beullens, M. and Bollen, M. (2002) The protein phosphatase-1 regulator NIPP1 is also a splicing factor involved in a late step of spliceosome assembly. J. Biol. Chem., 277, 19855–19860. [DOI] [PubMed] [Google Scholar]
- 28. Tanuma, N., Kim, S.E., Beullens, M., Tsubaki, Y., Mitsuhashi, S., Nomura, M., Kawamura, T., Isono, K., Koseki, H., Sato, M. et al. (2008) Nuclear inhibitor of protein phosphatase-1 (NIPP1) directs protein phosphatase-1 (PP1) to dephosphorylate the U2 small nuclear ribonucleoprotein particle (snRNP) component, spliceosome-associated protein 155 (Sap155). J. Biol. Chem., 283, 35805–35814. [DOI] [PubMed] [Google Scholar]
- 29. Nuytten, M., Beke, L., van Eynde, A., Ceulemans, H., Beullens, M., van Hummelen, P., Fuks, F. and Bollen, M. (2008) The transcriptional repressor NIPP1 is an essential player in EZH2-mediated gene silencing. Oncogene, 27, 1449–1460. [DOI] [PubMed] [Google Scholar]
- 30. Minnebo, N., Görnemann, J., O’Connell, N., van Dessel, N., Derua, R., Vermunt, M.W., Page, R., Beullens, M., Peti, W., van Eynde, A. et al. (2013) NIPP1 maintains EZH2 phosphorylation and promoter occupancy at proliferation-related target genes. Nucleic Acids Res., 41, 842–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Dessel, N., Boens, S., Lesage, B., Winkler, C., Görnemann, J., van Eynde, A. and Bollen, M. (2015) Protein phosphatase PP1-NIPP1 activates mesenchymal genes in HeLa cells. FEBS Lett., 589, 1314–1321. [DOI] [PubMed] [Google Scholar]
- 32. Boens, S., Verbinnen, I., Verhulst, S., Szekér, K., Ferreira, M., Gevaert, T., Baes, M., Roskams, T., van Grunsven, L.A., van Eynde, A. et al. (2016) Brief report: The deletion of the phosphatase regulator NIPP1 causes progenitor cell expansion in the adult liver. Stem Cells, 34, 2256–2262. [DOI] [PubMed] [Google Scholar]
- 33. Winkler, C., Rouget, R., Wu, D., Beullens, M., van Eynde, A. and Bollen, M. (2018) Overexpression of PP1-NIPP1 limits the capacity of cells to repair DNA double-strand breaks. J. Cell Sci., 131, jcs214932. [DOI] [PubMed] [Google Scholar]
- 34. Verbinnen, I., Jonkhout, M., Liakath-Ali, K., Szekér, K., Ferreira, M., Boens, S., Rouget, R., Nikolic, M., Schlenner, S., van Eynde, A. et al. (2020) Phosphatase regulator NIPP1 restrains chemokine-driven skin inflammation. J. Invest. Dermatol., 140, 1576–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hanaki, S., Habara, M., Masaki, T., Maeda, K., Sato, Y., Nakanishi, M. and Shimada, M. (2021) PP1 regulatory subunit NIPP1 regulates transcription of E2F1 target genes following DNA damage. Cancer Sci., 112, 2739–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McKee, C., Shrager, P., Mazumder, A.G., Ganguly, A., Mayer, A., Foley, K., Ward, N., Youngman, M., Hou, H. and Xia, H. (2022) Nuclear inhibitor of protein phosphatase 1 (NIPP1) regulates CNS tau phosphorylation and myelination during development. Mol. Neurobiol., 59, 7486–7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parker, L., Gross, S., Beullens, M., Bollen, M., Bennett, D. and Alphey, L. (2002) Functional interaction between nuclear inhibitor of protein phosphatase type 1 (NIPP1) and protein phosphatase type 1 (PP1)in Drosophila: consequences of over-expression of NIPP1 in flies and suppression by co-expression of PP1. Biochem. J., 368, 789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu, F., Grundke-Iqbal, I., Iqbal, K. and Gong, C.X. (2005) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci., 22, 1942–1950. [DOI] [PubMed] [Google Scholar]
- 39. Rahman, A., Grundke-Iqbal, I. and Iqbal, K. (2005) Phosphothreonine-212 of Alzheimer abnormally hyperphosphorylated tau is a preferred substrate of protein phosphatase-1. Neurochem. Res., 30, 277–287. [DOI] [PubMed] [Google Scholar]
- 40. Eastman, C., Horvitz, H.R. and Jin, Y. (1999) Coordinated transcriptional regulation of the UNC-25 glutamic acid decarboxylase and the UNC-47 GABA vesicular transporter by the Caenorhabditis elegans UNC-30 homeodomain protein. J. Neurosci., 19, 6225–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jagiello, I., van Eynde, A., Vulsteke, V., Beullens, M., Boudrez, A., Keppens, S., Stalmans, W. and Bollen, M. (2000) Nuclear and subnuclear targeting sequences of the protein phosphatase-1 regulator NIPP1. J. Cell Sci., 113, 3761–3768. [DOI] [PubMed] [Google Scholar]
- 42. Galganski, L., Urbanek, M.O. and Krzyzosiak, W.J. (2017) Nuclear speckles: molecular organization, biological function and role in disease. Nucleic Acids Res., 45, 10350–10368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Dessel, N., Beke, L., Görnemann, J., Minnebo, N., Beullens, M., Tanuma, N., Shima, H., van Eynde, A. and Bollen, M. (2010) The phosphatase interactor NIPP1 regulates the occupancy of the histone methyltransferase EZH2 at Polycomb targets. Nucleic Acids Res., 38, 7500–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lester, E., Ooi, F.K., Bakkar, N., Ayers, J., Woerman, A.L., Wheeler, J., Bowser, R., Carlson, G.A., Prusiner, S.B. and Parker, R. (2021) Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron, 109, 1675–1691.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McMillan, P.J., Strovas, T.J., Baum, M., Mitchell, B.K., Eck, R.J., Hendricks, N., Wheeler, J.M., Latimer, C.S., Keene, C.D. and Kraemer, B.C. (2021) Pathological tau drives ectopic nuclear speckle scaffold protein SRRM2 accumulation in neuron cytoplasm in Alzheimer’s disease. Acta. Neuropathol. Commun., 9, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leung, S.W., Apponi, L.H., Cornejo, O.E., Kitchen, C.M., Valentini, S.R., Pavlath, G.K., Dunham, C.M. and Corbett, A.H. (2009) Splice variants of the human ZC3H14 gene generate multiple isoforms of a zinc finger polyadenosine RNA binding protein. Gene, 439, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhou, Z., Luo, M.J., Straesser, K., Katahira, J., Hurt, E. and Reed, R. (2000) The protein Aly links pre-messenger-RNA splicing to nuclear export in metazoans. Nature, 407, 401–405. [DOI] [PubMed] [Google Scholar]
- 48. Marzahn, M.R., Marada, S., Lee, J., Nourse, A., Kenrick, S., Zhao, H., Ben-Nissan, G., Kolaitis, R., Peters, J.L., Pounds, S. et al. (2016) Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles. EMBO J., 35, 1254–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ilık, İ.A., Malszycki, M., Lübke, A.K., Schade, C., Meierhofer, D. and Aktaş, T. (2020) Son and srrm2 are essential for nuclear speckle formation. elife, 9, 1–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Morris, K.J. and Corbett, A.H. (2018) The polyadenosine RNA-binding protein ZC3H14 interacts with the THO complex and coordinately regulates the processing of neuronal transcripts. Nucleic Acids Res., 46, 6561–6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vulsteke, V., Beullens, M., Boudrez, A., Keppens, S., van Eynde, A., Rider, M.H., Stalmans, W. and Bollen, M. (2004) Inhibition of spliceosome assembly by the cell cycle-regulated protein kinase MELK and involvement of splicing factor NIPP1. J. Biol. Chem., 279, 8642–8647. [DOI] [PubMed] [Google Scholar]
- 52. Sjöstedt, E., Zhong, W., Fagerberg, L., Karlsson, M., Mitsios, N., Adori, C., Oksvold, P., Edfors, F., Limiszewska, A., Hikmet, F. et al. (2020) An atlas of the protein-coding genes in the human, pig, and mouse brain. Science, 367, eaay5947. [DOI] [PubMed] [Google Scholar]
- 53. Hammarlund, M., Hobert, O., Miller, D.M. and Sestan, N. (2018) The CeNGEN project: the complete gene expression map of an entire nervous system. Neuron, 99, 430–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brenner, S. (1974) The genetics of Caenorhabditis elegans. Genetics, 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Paix, A., Folkmann, A., Rasoloson, D. and Seydoux, G. (2015) High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics, 201, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Arribere, J.A., Bell, R.T., Fu, B.X.H., Artiles, K.L., Hartman, P.S. and Fire, A.Z. (2014) Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics, 198, 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. De Stasio, E.A. and Dorman, S. (2001) Optimization of ENU mutagenesis of Caenorhabditis elegans. Mutat. Res., 495, 81–88. [DOI] [PubMed] [Google Scholar]
- 58. Elnaggar, M.M., Abdellrazeq, G.S., Venn-Watson, S.K., Jensen, E.D., Hulubei, V., Fry, L.M., Sacco, R.E. and Davis, W.C. (2017) Identification of monoclonal antibodies cross-reactive with bottlenose dolphin orthologues of the major histocompatibility complex and leukocyte differentiation molecules. Vet. Immunol. Immunopathol., 192, 54–59. [DOI] [PubMed] [Google Scholar]
- 59. Livak, K.J. and Schmittgen, T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Additional data is available to be shared upon request.