

Abstract
Purpose
To characterize a novel neurodevelopmental syndrome due to loss-of-function (LoF) variants in Ankyrin 2 (ANK2), and to explore the effects on neuronal network dynamics and homeostatic plasticity in human-induced pluripotent stem cell-derived neurons.
Methods
We collected clinical and molecular data of 12 individuals with heterozygous de novo LoF variants in ANK2. We generated a heterozygous LoF allele of ANK2 using CRISPR/Cas9 in human-induced pluripotent stem cells (hiPSCs). HiPSCs were differentiated into excitatory neurons, and we measured their spontaneous electrophysiological responses using micro-electrode arrays (MEAs). We also characterized their somatodendritic morphology and axon initial segment (AIS) structure and plasticity.
Results
We found a broad neurodevelopmental disorder (NDD), comprising intellectual disability, autism spectrum disorders and early onset epilepsy. Using MEAs, we found that hiPSC-derived neurons with heterozygous LoF of ANK2 show a hyperactive and desynchronized neuronal network. ANK2-deficient neurons also showed increased somatodendritic structures and altered AIS structure of which its plasticity is impaired upon activity-dependent modulation.
Conclusions
Phenotypic characterization of patients with de novo ANK2 LoF variants defines a novel NDD with early onset epilepsy. Our functional in vitro data of ANK2-deficient human neurons show a specific neuronal phenotype in which reduced ANKB expression leads to hyperactive and desynchronized neuronal network activity, increased somatodendritic complexity and AIS structure and impaired activity-dependent plasticity of the AIS.
Introduction
The ankyrin 2 (ANK2) gene on chromosome 4q25-q26 encodes ankyrin-B (ANKB), an adaptor protein that links integral membrane proteins to the spectrin-actin cytoskeleton, and is important for localization and membrane stabilization of ion channels. Two major ANKB polypeptides are encoded by ANK2, of which one is 220 kDa and is expressed in multiple tissues. The other, also known as giant ANKB (gANKB), is an alternatively spliced transcript that includes a neurospecific domain (NSD) encoded by a giant exon inserted between the spectrin binding and death domain. In total, gANKB measures 440 kDa and is mainly expressed in the brain (1,2) (see Fig. 1 for gene–protein structure and distribution of variants). A set of pathogenic missense variants targeting both polypeptides was previously shown to cause a phenotype of cardiac arrhythmia and long QT syndrome [MIM:106410]. In vitro experiments showed that ANK2 missense variants lead to a disruption of several ion channels in cardiomyocytes and were therefore interpreted as representing loss-of-function (LoF) (3).
Figure 1.
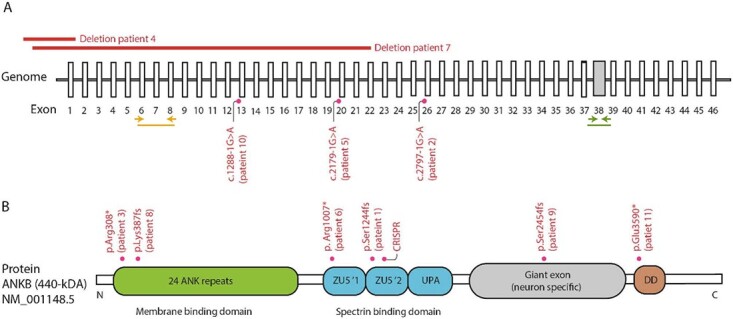
Schematic representation of ANK2 variants. (A) Schematic representation of the genomic structure of ANK2 with known intronic variants and larger deletions. Yellow arrows indicate the position of qPCR primer pair used to assess mRNA expression of the 220-kDa isoform of ANK2. Green arrows indicate the position of qPCR primer pair used to assess mRNA expression of the 440-kDa isoform of ANK2. (B) Schematic representation of the major transcript of ankyrin B, NM_001148.5 (the 440-kDA variant) and its domains. In the 440-kDA protein (gANKB), a neurospecific 6.7 kb exon is expressed and inserted between the UPA and the death domain. The PTVs are depicted as well as the variant used in our CRISPR experiment.
On the basis of a limited number of individual reports typically from genome-sequencing studies, ANK2 has been proposed as a candidate susceptibility gene for autism spectrum disorder (ASD) (4–10). In these individuals, the ASD typically was non-syndromic (noting ascertainment for most of these families was for an ASD diagnosis only), and the variants were mostly missense variants, with frameshift variants observed in some cases. De novo variants in ANK2 have been described in a small number of individuals from large cohorts with neurodevelopmental disorders (NDDs) and epilepsy as well (10–14). A recent large-scale exome-sequencing study of ASD concluded that ANK2 variants are more likely to be found in ASD compared with ID, noting there is a significant overlap between the two (15).
It has been speculated that the effect on the NSD of gANKB might underlie the phenotypic difference with the missense variants that cause cardiac arrhythmia (16,17). A frameshift variant in the giant exon of gANKB in mice causes an increased frequency of axonal branching in neurons, dependent on the interaction of gANKB with L1CAM, suggesting aberrant structural connectivity (16). Direct interaction of gANKB with microtubules causes prominent bundling of microtubules, suppressing invasion into filopodia and axon branching (17). Furthermore, there is a transient increase in excitatory synapses during postnatal development and a stochastic increase in structural connectivity in cerebral cortices of ANKB-deficient mice found on diffusion tensor imaging (16).
We considered that another possible mechanism by which variants in ANKB affect ASD risk could be through its role in the determination of length and location of the axon initial segment (AIS) (18). The AIS is a specialized membrane region in the axon, where high-density voltage-gated sodium (NaV) and potassium (KV) channels generate action potentials under the regulation of synaptic input (19). The structure of the AIS undergoes activity-dependent structural changes that can be considered as a homeostatic mechanism for modulating neuronal excitability (20,21). Such effects of ANK2 variants on neuronal excitability would be consistent with a role in autism or intellectual disability (ID) (22).
In this study, we collected detailed clinical data for 12 individuals (7 male, 5 female) with a de novo LoF variant in ANK2. In contrast to a recent large-scale compilation of ASD sequencing data (15), we find that the ANK2 LoF variants cause a wider range of neurodevelopmental phenotypes including mild to severe ID and epilepsy. We sought to determine how LoF variants in ANK2 affect neuronal network activity in human neurons derived from human-induced pluripotent stem cells (hiPSCs). To better understand how ANKB affects neuronal network activity, we analyzed the effect of heterozygous loss of ANKB on somatodendritic morphology and on the structure and location of the AIS.
Results
Patients with heterozygous LoF of ANK2 show variable severity of epilepsy
Gene discovery
We identified 12 patients in whom a LoF variant in ANK2 was identified and where phenotypic data could be obtained. These variants included four nonsense, three frameshift, three canonical splice site variants and two (partial) gene deletions (Fig. 1). Analysis of parental DNA showed that 11 of these occurred de novo. For the remaining patient, parental DNAs were not available.
Clinical phenotype
Epilepsy
We next performed in-depth phenotyping of all patients. Early onset epilepsy is reported in 7 of 12 patients (58%), with an onset of epilepsy before the age of 2 years (Table 1). Four patients had neonatal onset epilepsy (Patients 2, 3, 10 and 11). Of these four patients, three were seizure-free with medication (oxcarbazepine or phenobarbital), one of whom (Patient 3) remained seizure-free after discontinuing phenobarbital at 4 months. The fourth patient (Patient 2) developed a bilateral tonic–clinic seizure at the age of 3 with multifocal discharges on electroencephalogram (EEG), and became seizure-free on lamotrigine. Patient 5 had focal epilepsy with focal motor seizures and impaired awareness, which responded well to treatment with valproic acid. Two patients had therapy-resistant epilepsy (Patients 6 and 8), despite multiple anti-epileptic drugs and implantation of a vagal nerve stimulator (VNS). Of these two patients with therapy-resistant epilepsy, Patient 6 had West syndrome with infantile spasms, which later developed into Lennox–Gastaut syndrome. Despite treatment with valproic acid, clonazepam, clobazam, cannabidiol, rufinamide, ketogenic and VNS, Patient 6 has daily seizures. The other patient with therapy-resistant epilepsy, Patient 8, had focal seizures with clonic movements of the right upper limb, and later developed a generalized epilepsy type with absences (absences with eyelid myoclonia and myoclonic absences) as well as generalized tonic–clonic seizures (GTCS). Although valproic acid, clobazam, ethosuximide, topiramate and levetiracetam were prescribed, the seizures were not well controlled. She received VNS implantation and now uses lamotrigine, but still experiences daily seizures, including absences.
Table 1.
Clinical features of epilepsy of seven patients
| Patient 2 | Patient 3 | Patient 5 | Patient 6 | Patient 8 | Patient 10 | Patient11 | |
|---|---|---|---|---|---|---|---|
| Age (years) | 13 | 15 | 12 | 18 | 14 | 1.75 | 1.6 |
| Sex | Female | Male | Male | Male | Female | Male | Female |
| ILAE classification | Epilepsy from unknown type with seizures unknown onset | Epilepsy from unknown type with seizures unknown onset | Focal epilepsy with focal seizures with leftward eye deviation and impaired awareness | West syndrome with infantile spasms and Lennox–Gastaut syndrome | Generalized epilepsy/epilepsy with myoclonic absences | Focal epilepsy with focal motor seizures (clonic and myoclonic) with impaired awareness | Focal epilepsy with focal motor seizures (clonic and myoclonic) with impaired awareness |
| Age of onset | 8 days | 5 days | 2 years | 4 months | 22 months | 2 weeks | 1 week |
| Description | Neonatal convulsions, at 3.5y bilateral tonic–clonic seizure | Neonatal seizures; seizure-free | Focal seizures | Refractory tonic seizures. History of infantile spasms, myoclonic seizures, absences and atonic seizures Seizures appear daily |
Complex febrile seizures. Refractory focal seizures, absences with eyelid myoclonia and myoclonic absences, GTCS Seizures appear daily |
Focal seizures, seizure-free with oxcarbazepine | Focal seizures, seizure-free with oxcarbazepine |
| EEG | Multifocal epileptic discharges | Normal | Right-sided epileptiform discharges | Abundant multifocal epileptiform activity; left>right slowing. | Generalized 3-Hz spike and wave, progressive during sleep | Multifocal, mainly central (−parietal) spikes | Focal central spikes |
| Anti-epileptic treatment | Lamotrigine (history: Valproic acid) | None | Valproic acid 500/500 mg, clonidine 0.1 mg | Ketogenic diet, clonazepam, rufinamide, clobazam, cannabidiol, valproic acid, VNS | Lamotrigine, VNS (history: valproic acid, clobazam, ethosuximide, topiramate levetiracetam) | Oxcarbazepine |
VNS: vagal nerve stimulator; GTCS: generalized tonic–clonic seizures.
EEG was performed in all patients. In five patients (71%), EEG showed focal or multifocal epileptic discharges. In the last patient (Patient 8), the EEG showed generalized 3-Hz spike and wave discharges, consistent with the dominant seizure types of absences (myoclonic absences and absences with eyelid myoclonia) and GTCS. In an eight case (Patient 9), who had no clinical seizures, EEG in 2014 showed bilateral occipital slowing without epileptic discharges, which spontaneously disappeared within a few weeks. CT scan was performed and showed no underlying abnormalities.
MRI
Six patients underwent a brain MRI scan. Of these, five had no abnormalities. One patient (Patient 8) had nonspecific hyperintensities on T2, located bilateral periventricular in the frontal and occipital regions. The ventricular horns were mildly enlarged.
NDD
Nine patients (75%) had ID or delayed motor and/or speech development (Table 2). Although most patients had mild ID, moderate or severe ID was seen in Patients 6 and 8, respectively. Both Patients 6 and 8 have an epileptic encephalopathy, which may contribute to the severity of their cognitive impairment. Patient 7 has normal cognitive abilities as judged by a score of 32 on the Raven progressive matrices. Patients 10 and 11 show no developmental delay thus far but are still being assessed because of their relatively younger age of <2 years.
Table 2.
General genetic and clinical information of 12 ANK2-related patients
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | Patient 9 | Patient 10 | Patient 11 | Patient 12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Female | Male | Female | Male | Male | Female | Female | Male | Male | Female | Male |
| cDNA location | c.3632_3633del | c.2797-1G > A | c.922C > T | c.-288710_8555700del | c.2179-1G > A | c.3019C > T | c.-65884_ 2476923del |
c.1159_1160del | c.7360_ 7361delTC insA |
c.12881G > A | c.10768G > T | c.862C > T |
| Protein change | p.Ser1244Met(fs*5) | p.(?);- | p.Arg308* | N/a | p.(?);- | p.Arg1007* | N/a | p.Lys387Glu(fs*10) | p.Ser2454 Ile(fs*7) |
p.(?);- | p.Glu3590* | p.Arg288* |
| Mutation type | Deletion leading to frameshift | Probable leading to frame shift | Nonsense | Deletion | Splice site variant | Nonsense | Deletion | Deletion leading to frameshift | Indel leading to frameshift | Splice site variant | Nonsense | Nonsense |
| Inheritance | De novo | De novo | De novo | Unknown | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo |
| ID/DD | Mild | Mild | Mild | Mild | Mild* | Severe | None | Moderate | Mild | None | None | Mild |
| ASD | Yes | Yes | Probable | Yes | Unknown | Yes | Yes | No | No | N/a | N/a | Yes |
| Other behavioral problem | Gilles de la Tourette, ADHD, DCD | Attention difficulty | ADD | Unknown | Unknown | Agitation, sleep disturbances | Unknown | ADHD, agitation and aggression | ADHD, aggression | – | – | – |
| MRI | – | Normal | Normal | – | – | Normal | – | Hyperintensities and atrophy | – | Normal | Normal | – |
| Other | NRXN1: 2p16.3(50 991 243–51 289 649)x1 mat (deletion 4–5 exons | KNCQ3 variant, paternally inherited | Severe feeding difficulties (needing tube feeding) | SCN9A SCN9a c.555G>A (p.E519K); paternally inherited TRIO c.8120G>A (p.R2707Q) ALTE 11 months |
ALG13 ALG13 (c.3406G>T, p.G1136C) – hemizygous. Inherited from mom. NM_001099922. Genomic Coordinates (GRCh37): 110924346–111003877 |
Sleep apnea | Mild increase muscle tone, mild coordination deficit |
ID: intellectual disability; NDD: neurodevelopmental delay; ASD: autism spectrum disorder; n/a: not applicable; ADHD: attention deficit hyperactivity disorder; ADD: attention deficit disorder; DCD: developmental coordination disorder; ALTE: apparent life-threatening event
Seven patients (58%) had a formal or suspected diagnosis of ASD. Further behavioral problems and psychiatric comorbidity included attention-deficit hyperactivity disorder (ADHD) in five patients (42%), and Gilles de la Tourette, agitation or aggressiveness, sleep disturbances and eating disorder in single patients (8% each). Mild and non-specific dysmorphisms were reported in five patients (42%), and congenital anomalies were uncommon (Supplementary Material, Table S1). One patient (Patient 5) showed generalized overgrowth. Mild hypotonia was reported in five patients (42%) with associated clumsiness.
Cardiac evaluation
Since (likely) pathogenic missense variants in the ANK2 gene have previously been causally related to various cardiac-related phenotypes, surveillance by a cardiologist was performed in six patients (50%) (Supplementary Material, Table S1). Cardiac abnormalities were found in two of these six patients (33%), Patients 1 and 3. Both showed intraventricular conduction delay as well as a small atrial septum defect. Patient 1 also had a patent foramen ovale.
A human neuronal model of ANKB haploinsufficiency leads to hyperactive and desynchronized neuronal network
To examine the effects of heterozygous LoF variants targeting both the 220-kDa and 440-kDa isoforms of ANK2 on neuronal function, we generated a human model for heterozygous isogenic ANKB-deficient neurons. We used CRISPR/Cas9 genome editing in healthy curated control hiPSCs (WT) to induce a premature stop codon (p.Arg1225*) in exon 31 of ANK2 (hereafter referred to as ANK2+/−), which closely resembles a nonsense mutation identified in Patient 6 (Figs 1A and 2A). ANK2+/−-derived hiPSCs showed genetic integrity after genomic editing, which is tested by off-target analysis and detection of genomic abnormalities (Supplementary Material, Fig. S1A and B). We differentiated WT and ANK2+/− hiPSCs into excitatory neurons (iNeurons) by forced expression of the transcription factor transgene Ngn2 in two independent ANK2+/−-derived hiPSC clones (Fig. 2B) (23,24). ANKB localized to axons and MAP2-positive dendrites (Fig. 2C), in line with previous reports localizing the 220-kDa isoform to dendrites and the 440 kDa to axons in rodent primary cerebellar neurons (1,25). The frameshift variant in ANK2+/− iNeurons resulted in a 50% reduction on average of both isoforms in comparison with WT iNeurons (Fig. 2D–F). We also examined the somatodendritic morphology in ANK2+/− iNeurons at days in vitro (DIV) 21 (Fig. 2G). Dendritic reconstructions revealed an increase in soma size, number of dendritic nodes and total dendritic length in ANK2+/− iNeurons (Fig. 2H). No difference was observed in the number of primary dendrites compared with WT iNeurons (Fig. 2H). Sholl analysis was used to further assess the complexity of the dendritic network, which allows for a detailed quantification of dendritic branches close to and distal from the soma (Fig. 2I). The analysis confirmed an increase in dendritic length in ANK2+/− iNeurons, accompanied by an increased number of dendritic intersections and dendritic nodes (Fig. 2J). These results suggest that ANKB haploinsufficiency leads to an increased somatodendritic complexity.
Figure 2.

Heterozygous loss of 220-kDa and 440-kDa isoforms of ANK2 leads to reduced ANKB expression in iNeurons and increases somatodendritic structures. (A) Schematic diagram of CRISPR/Cas9 genetic editing in exon 31 and the DNA sequences before and after targeted gene editing. One nucleotide insertion causes a frameshift variant in WT hiPSCs. (B) Schematic presentation of the iNeuron differentiation protocol. (C) Representative images of a WT iNeuron immunostained for MAP2 (red) and ANKB (green) (scale bar 10 μm). (D) Representative images of ANK2+/− iNeurons immunostained for ANKB (green) (scale bar 5 μm), and quantification of ANKB fluorescent intensity, n = 8 for WT; n = 4 for ANK2+/− clone 1 and n = 4 for ANK2+/− for clone 2. ***P < 0.001 by two-tailed unpaired t-test. (E) Schematic presentation of protein domains expressed in 220-kDa ANKB, and qPCR analysis of 220-kDa ANK2 isoform performed in WT and ANK2+/− iNeurons at DIV 21. Values of ANK2+/− are normalized to WT (n = 8). ***P < 0.001 by two-tailed unpaired t-test. (F) Schematic presentation of protein domains expressed in 440-kDa ANKB, and qPCR analysis of 440-kDa ANK2 performed in WT and ANK2+/− iNeurons at DIV 21. Values of ANK2+/− are normalized to WT (n = 8). ***P < 0.001 by two-tailed unpaired t-test. (G) Representative somatodendritic reconstructions of WT and ANK2+/− iNeurons (scale bar 20 μm). (H) Quantification of the soma size, the number of primary dendrites, the number of dendritic nodes and the total dendritic length, n = 30 for WT; n = 30 for ANK2+/−. **P < 0.01 and ***P < 0.001 by two-tailed unpaired t-test. (I) Representative image of a WT iNeuron in sequential 10 μm rings placed from the center soma outwards for Sholl analysis. (J) Quantification per 10 μm Sholl section of the total dendritic length per ring, the number of dendritic intersections per ring and the number of dendritic nodes per ring, n = 30 for WT; n = 30 for ANK2+/−. *P < 0.05, **P < 0.001, ***P < 0.001 by two-way ANOVA with post hoc Bonferroni correction. MBD = membrane-binding domain; SBD = spectrin-binding domain; RD = regulatory domain.
To evaluate the potential effects of haploinsufficiency of both ANKB isoforms on neuronal network level, we measured spontaneous neuronal activity of WT and ANK2+/− iNeurons cultured on micro-electrode arrays (MEAs). After 28 days of neuronal differentiation, WT iNeurons showed a spontaneously active network that was organized into rhythmic, synchronous network bursts. These bursts were composed of many action potentials (spikes) occurring closely in time and across all electrodes (23) (Fig. 3A and B). This is in accordance with our previous study in which we benchmarked the use of MEAs by comparing 10 independent control lines (26). Representative Raster plots display an increased global level of activity in ANK2+/− iNeurons compared with WT iNeurons (Fig. 3B and C), which was reflected by an increase in spiking rate (Fig. 3F). This was accompanied by a significant increase in burst rate (Fig. 3G), but not with a difference in burst duration (Fig. 3H). As a consequence of the higher burst frequency, we observed a significant decrease in interburst interval (IBI) (Fig. 3I). The increased burst activity in ANK2+/− iNeurons was not accompanied by a difference in spike organization [i.e. the percentage of random spikes (Fig. 3J)] or increased network burst frequencies and duration (Fig. 3K and L), indicating that in ANK2+/− networks, an increased number of bursts occur independent of synchronous network bursts.
Figure 3.

Spontaneous electrophysiological activity of neuronal networks from WT and ANK2+/− iNeurons recorded on MEA. (A) Schematic diagram of spikes and (network) bursts shapes plotted per electrode. (B) Example of spontaneous WT neuronal network (scale bar 5 s) and burst shape (scale bar 6 s). Synchronous network bursts are indicated in gray vertical bars. (C) Example of spontaneous ANK2+/− neuronal network (scale bar 5 s) and burst shape (scale bar 6 s). Synchronous network bursts are indicated in blue vertical bars. (D) Representative image of WT-derived neuronal network on a well of a MEA plate. (E) Representative image of ANK2+/−-derived neuronal network on a well of a MEA plate. Quantification of (F) spiking rate, (G) burst rate, (H) burst duration, (I) IBI, (J) percentage of random spikes, (K) network burst rate and (L) network burst duration as recorded by MEA in WT and ANK2+/− neuronal networks, n = 5 for WT; n = 16–18 for ANK2+/−, *P < 0.05 and **P < 0.001 by two-tailed unpaired t-test.
To explore whether the increased network activity in ANK2+/−-derived cultures is caused by other neuronal characteristics, we measured the number of functional synapses. We immunostained neurons for pre- and postsynaptic markers (Synapsin and Homer1, respectively). We found no differences in the density of Synapsin/Homer1 co-localized puncta between WT and ANK2+/−-derived iNeurons (Supplementary Material, Fig. S2A and B). Combined with the observation that ANK2+/−-derived iNeurons show a longer dendritic length, these data, however, imply that the total amount of synapses per neuron is increased. This could reflect the increased neuronal network excitability.
ANKB haploinsufficiency affects AIS length and activity-dependent plasticity
Since ANKB together with the αII/βII-spectrin complex forms an intra-axonal boundary that limits ANKG clustering at the AIS (18), we wondered whether ANKB haploinsufficiency would affect AIS length. Using ANKG as a marker for the AIS, we found that the length of the AIS was significantly increased in ANK2+/− iNeurons compared with WT iNeurons at DIV 21 and DIV 28 (Fig. 4A and B), suggesting that the intra-axonal boundary is shifted toward the distal axonal ends. In particular, we found that ANKG was more diffused and interrupted at the distal part of ANK2+/− iNeurons (Fig. 4A). Because the AIS first develops in the distal parts of the axon (27), we also measured the distance from the soma to the proximal end of ANKG at DIV 14. We did not find a significant difference in the distance of ANKG to the soma in both genotypes (Supplementary Material, Fig. S3A and C). Furthermore, we did not find a positional change of the proximal end of the AIS relative to the soma between WT and ANK2+/− iNeurons at DIV 21 and DIV 28 (Fig. 4A and C). In addition, the ANKG length is already significantly longer in ANK2+/− iNeurons at DIV 14 (Supplementary Material, Fig. S3A and B). Since ANKG anchors AIS-specific membrane proteins, such as NaV channels (19), we asked whether Nav channels expression is differently expressed in the increased AIS structure of ANK2+/− iNeurons. Using an antibody that recognizes all sodium channels, as well as an antibody against Nav1.1, we found no significant differences in mean intensities for both Nav1.1 and PanNav in the AIS between WT and ANK2+/− iNeurons. However, combined with the observation that ANK2+/− iNeurons show longer AIS structure, these data suggest that the total amount of sodium channels in the AIS per neuron is increased (Supplementary Material, Fig. S3D–G).
Figure 4.

Heterozygous loss of ANKB increases AIS length and impairs AIS plasticity. (A) Representative images of WT and ANK2+/− iNeurons immunostained for MAP2 (blue) and ANKG (red) at DIV 21 and 28 (scale bar 10 μm). Arrowheads indicate the start and end of the AIS. (B) Quantification of the ANKG length shown by immunostaining in WT and ANK2+/− iNeurons at DIV 21 and DIV 28, n = 19–29 cells/group. Green dots represent datapoints obtained in ANK2+/− clone 1, and brown datapoints in ANK2+/− clone 2. ****P < 0.0001 by one-way ANOVA and Tukey’s post hoc analysis. (C) Quantification of distance soma to start ANKG shown by immunostaining in WT and ANK2+/− iNeurons at DIV 21 and DIV 28, n = 19–29 cells/group. Green dots represent datapoints obtained in ANK2+/− clone 1, and brown datapoints in ANK2+/− clone 2. ****P < 0.0001 by one-way ANOVA and Tukey’s post hoc analysis. (D) Representative images show immunostainings for MAP2 (blue) and ANKG (red) in WT and ANK2+/− iNeurons under basal condition or treated with 1 μM TTX for 48 h (scale bar 10 μm). Arrowheads indicate the start and end of the AIS. (E) Quantification of the length of ANKG shown by immunostaining in WT and ANK2+/− iNeurons under basal condition or treated with 1 μM TTX for 48 h, n = 24 for WT; n = 31 for ANK2+/−. Green dots represent datapoints obtained in ANK2+/− clone 1, and blue datapoints in ANK2+/− clone 2. ****P < 0.0001 by one-way ANOVA with Tukey’s post hoc analysis. (F) Representative images show immunostainings for MAP2 (blue) and ANKG (red) in WT and ANK2+/− iNeurons under basal condition or treated with 10 mM KCl for 48 h (scale bar 10 μm). Arrowheads indicate the start and end of the AIS. (G) Quantification of the length of ANKG shown by immunostaining in WT and ANK2+/− iNeurons under basal condition or treated with 10 mM KCl for 48 h, n = 16–21 for WT; n = 20 for ANK2+/−. Green dots represent datapoints obtained in ANK2+/− clone 1, and blue datapoints in ANK2+/− clone 2. ****P < 0.0001 by one-way ANOVA with Tukey’s post hoc analysis.
Neuronal activity can modulate AIS position and length, which may act as a homeostatic mechanism for regulating neuronal excitability (20,21). If ANKB is important for AIS length, then activity-dependent AIS modulation could be dependent on the intra-axonal boundary formed by the ANKB/αII/βII-spectrin complex. To test this possibility, we chronically silenced neuronal activity with 1 μM tetrodotoxin (TTX) for 48 h at DIV 21 and measured AIS positioning and length. As previously reported in rodents (20,28), AIS length significantly increased in WT iNeurons after 48 h of TTX treatment (Fig. 4D and E). Interestingly, we did not find such an increase in AIS length in ANK2+/− iNeurons (Fig. 4D and E). In neither genotype, we observed a change in AIS position relative to the soma (Supplementary Material, Fig. S3C). One possibility is that the increase of AIS length in ANK2+/− iNeurons is occluded (i.e. AIS length cannot increase any further as opposed to impaired AIS plasticity). To further test whether ANKB is required for AIS plasticity, we induced chronic depolarization with 10 mM KCl to increase neuronal activity for 48 h as it was previously reported to reduce AIS length in human iNeurons (29). Indeed, we found that the AIS length significantly decreased in WT iNeurons after 48 h of KCl treatment (Fig. 4F and G). However, AIS length did not decrease in ANK2+/− iNeurons after KCl treatment (Fig. 4F and G), suggesting that ANKB is required for bidirectional AIS plasticity. We did not observe any repositioning relative to the soma of the AIS in both WT and ANK2+/− iNeurons (Supplementary Material, Fig. 3H and I).
Our results showed that AIS structure and plasticity are dependent on the intra-axonal boundary formed by the ANKB/αII/βII-spectrin complex. In addition, we observed that the increased AIS length leads to an increased total amount of sodium channels in the AIS of ANK2+/− iNeurons.
Discussion
Here we collected clinical information on 12 patients with predicted LoF variants in ANK2 and carefully examined their clinical- and genomic data. We observed a neurodevelopmental phenotype, with high risk of ASD and ID. We found early onset epilepsy in 7 of 12 of our patients, ranging from mild and self-limiting to a severe epileptic encephalopathy. Furthermore, we demonstrated that heterozygous loss of ANKB leads to hyperactive and desynchronized bursting activity in hiPSC-derived neuronal networks. We also showed that heterozygous loss of ANKB in human neurons increases somatodendritic complexity and AIS structure and impairs AIS plasticity.
Early onset childhood epilepsy was present in 7 of 12 patients in our cohort. The course of the epilepsy showed great variability, although onset was in the first 2 years of life. On one side, neonatal seizure had a very good treatment response and are possibly self-limiting (as was seen Patient 3). The clinical picture in four patients resembles self-limiting neonatal-infantile seizures. On the other side, the epilepsy phenotype of Patients 6 and 8 was more severe compared with other patients, and it is therapy resistant. The epileptic encephalopathy probably contributes to the more severe ID in these two patients. In Patient 6, we cannot exclude an additive effect of the maternally inherited ALG13 variant. However, we engineered a nonsense mutation (p.Arg1225*) that closely resembles the nonsense mutation identified in Patient 6 (p.Arg1007*) in ANK2. Experimental in vitro data of ANK2+/−-derived cultures on MEAs showed an increased neuronal network excitability, suggesting that the ALG13 variant would not contribute to the increased network activity.
This broad spectrum of the severity of epilepsy is seen in other genes, mainly those which affect ion channels, such as in SCN2A (30). Interestingly, variants in other genes involving the actin-spectrin-ankyrin complex have identified as major cause of severe refractory epilepsy in infancy (e.g. SPTAN1 variants affecting spectrin) (31). In Patients 2, 5, 10 and 11, a positive effect of sodium channel blockers is seen. This suggest an (secondary) effect of ANKB effect on sodium channel function in the AIS, but more research would be necessary to understand how ANKB haploinsufficiency affects ion channels in these patients.
Variants in ANK2 were initially associated with non-syndromic ASD (4–10). Two large studies with a compilation of variants found in sequencing studies of ASD and NDD concluded that LoF variants in ANK2 cause NDD, with a predominance of ASD (15,32). Satterstrom et al. have reported six protein truncating variants (PTVs), four missense variants and two synonymous (15), whereas further phenotypic data were not specified. In the preprint of Fu et al., variant counts and phenotypic data are not accessible. Therefore, it is not possible to evaluate the phenotype–genotype relationship. The current data suggest that the phenotypic spectrum for ANK2-related NDD is broad, as is seen in many other genes, especially in channelopathies (SCN1A, SCN2A) (30,33).
ANK2 can cause a cardiac disease (OMIM 600919) which is predominantly characterized by long QT syndrome. Interestingly, all cardiac phenotypes have missense mutations, whereas the patients with nonsyndromic ASD and the patients with NDD and epilepsy often have de novo LoF variants. Previous experimental work has suggested that the missense variants observed in patients with cardiac arrhythmias resulted in a LoF at the cellular level. The presumed LoF in the context of the heart could be different in the brain, yet the effect of one variant has not been studied in both the heart and the brain. Future research is necessary to distinguish the differences between nonsyndromic ASD and the wider range of NDDs.
We further explored the effects of heterozygous LoF variants targeting both 220-kDa and 440-kDa isoforms of the ANKB protein in a human model for heterozygous isogenic ANKB-deficient neurons. We presume that this variant, by affecting both isoforms, will resemble the variants of our patients. The data show that heterozygous loss of ANKB leads to changes in the somatodendritic structure. A previous report showed that ANK2 variants, that specifically targets gANKB, lead to increased ectopic axon branching without any changes in dendritic morphology (16). Therefore, our data suggest a role for 220-kDa ANKB in limiting dendritic networks through a mechanism that still needs to be identified. The 220-kDa ANKB has also been shown to localize in axons, which promotes fast axonal transport of synaptic vesicles, mitochondria, endosomes and lysosomes through interaction with Class III PI3-kinase (PIK3C3) and dynactin (34). The authors showed that loss of 220-kDa ANKB impaired retrograde organelle transport in hippocampal mouse neurons leading to shortened axon tracts. Because 220-kDa ANKB also localizes to dendrites, it is important to note that the authors found no particular role for 220-kDa ANKB in organelle transport in dendrites, further suggesting a role for 220-kDa ANKB in the development of dendrites. We did not include axonal analysis in our ANKB-deficient neurons, but it would be useful to investigate whether LoF variants targeting both 220-kDa and 440-kDa isoforms would affect axonal structures as shown in other studies (18,34).
We provide additional evidence that ANKB is required to restrict ANKG to the AIS in human neurons (18). During development, the AIS ‘barrier’ is formed by the ANKB/αII/βII-spectrin complex, which is important to restrict ANKG to the AIS, which in turn clusters Na+ and K+ channels at the AIS (35). The AIS therefore plays a role in fine-tuning neuronal excitability to variation in levels of input activity (28,35). We showed that the expression of Nav channels, which is amongst others responsible for both the generation and modulation of actions potentials (19), is increased in total amount in the AIS of ANK2+/− iNeurons. The data therefore imply that the increased AIS could lead to an increased neuronal excitability. We specifically addressed the question of how loss of ANKB neurons affects the neuronal network. Our in vitro data on MEAs demonstrate a hyperactive neuronal network phenotype with a higher spiking rate and an increased burst rate. We further show that these bursts occur at the single electrode level, indicative for a desynchronized neuronal network. However, the observed hyperactive neuronal network could also be explained by several other factors that co-occur with changes in excitability. These factors could be changes in input resistance (20), phosphorylation of voltage-gated K+ channels (28) or redistribution of voltage-gated K+ channels (36). Disentangling the specific contribution of AIS structure to excitability changes is therefore challenging. It is possible that the increased AIS structure in ANK2+/− iNeurons is a compensation effect to the increased neuronal excitability. In addition, a desynchronized neuronal network was previously found in neuronal models for Kleefstra syndrome patients who have ID and ASD (37), as well in five different variants causing ASD (38). In contrast to the decreased network burst frequency found in these ID and ASD syndromes (37,38), the overactive network we found is more similar to that of seizure conditions, such as in a variant in the nicotinic acetylcholine receptor (nAChR) subunit causing autosomal dominant nocturnal frontal lobe epilepsy (39).
Activity-dependent modulation of the AIS has been described as a homeostatic mechanism for modulating neuronal excitability (20,21). This mechanism compensates for chronic alterations in activity by shifting the input–output function of a neuron along its input axis by structural changes of the AIS, such as length and/or location relative to the soma. Hence, the AIS structure has a major impact on neuronal excitability. Our findings provide evidence for the role of ANKB in bidirectional AIS plasticity in humans. However, if AIS plasticity is important as a homeostatic mechanism for neuronal excitability, how do ANKB-deficient neurons fine-tune their excitability according to ongoing levels of input activity? A possible scenario could involve redistribution of voltage-gated ion channels at the AIS, which substantially augments the efficacy of regulation (21,36). Future experiments will determine if redistribution of voltage-gated ion channels on the AIS plays a role in fine-tuning neuronal excitability in ANKB-deficient neurons. Interestingly, ANKB has recently been shown to be essential for scaffolding the voltage-gated ion channel Nav1.2 to the dendritic membrane of mouse neocortical neurons (40). Haploinsufficiency of ANK2 has been shown to phenocopy the intrinsic dendritic excitability observed in SCN2A+/− conditions, indicating a convergent mechanism between two epilepsy-associated genes (40). This study suggested that the excitability is dependent on somatic sodium channel density and is insensitive to changes in dendritic channel density, and therefore demonstrated that Nav1.2 densities are at WT levels in ANK2+/− cells in the soma (40). These data may suggest that ANKB primarily localizes sodium channels to the soma to compensate for loss of dendritic sodium channels. Further studies will provide a better understanding whether this compensation mechanism applies during AIS plasticity. Unravelling the pathways linking fine-tuning mechanisms for neuronal excitability to impaired AIS plasticity in ANKB-deficient neurons could uncover potential new targets for epilepsy treatment, where fine-scale control over neuronal excitability represents a major therapeutic challenge for ANK2-related patients with ID, ASD and epilepsy.
In conclusion, de novo LoF variants in ANK2 cause early onset epilepsy, in addition to ASD and ID. We show the essential role of ANKB in neuronal network activity, which is increased by heterozygous loss of ANKB that may underlie some of the phenotypic features observed in these patients. In addition, we showed that reduced ANKB expression leads to structural changes in the somatodendritic complexity and the AIS and impairs AIS plasticity. Future studies may explore the therapeutic potential of modulating these neuronal network characteristics.
Materials and Methods
Subjects
Through (inter)national collaborations and querying databases, such as denovo-db (denovo-db.gs.washington.edu) and Genematcher (https://genematcher.org/), we aimed to identify patients with (de novo) LoF variants in ANK2. For all patients identified, we systematically collected all phenotypic information, including, but not limited to, neurodevelopmental phenotypes, epilepsy, other neurological problems or congenital anomalies, and cardiac phenotypes. Patients in literature with presumed LoF variant in ANK2 in which phenotypic information could not be obtained were excluded. The study was approved by the medical ethics committee of the Radboudumc, Nijmegen the Netherlands (2021–7530).
ANK2 variant identification processes
ANK2 variants were all identified through routine diagnostic procedures including either chromosomal microarray analysis (patient 4) or trio-base whole exome sequencing procedures (41). Of note, Patients 2, 5 and 6 were previously published as part of large-scale sequencing study of patients with autism (9,11).
hiPSC cell culture
HiPSCs used in this study were obtained from reprogrammed fibroblasts of a healthy 30-year-old male donor. To investigate the role of ANKB, CRISPR/Cas9 was used to induce a LoF mutation of ANK2 into this healthy control line. HiPSCs were cultured on a 6-well plate pre-coated with 1:15 Matrigel (Corning, #356237) that is diluted in DMEM/F12 medium. The cells were cultured in E8 Flex medium (Thermo Fisher Scientific) supplemented with primocin (0.1 mg/ml, Invivogen).
To make Neurogenin 2 (Ngn2)-stable hiPSC lines, hiPSCs were infected with Ngn2 and rtTA lentivirus using the transfer vector pLVX-(TRE-tight) -(MOUSE) Ngn2-PGK-Puromycin(R) and pLVX-EF1α-(Tet-On-Advanced)-IRES-G418(R), respectively (all the plasmids are available upon request). Medium was supplemented with puromycin (0.5 mg/ml) and G418 (50 mg/ml) at 37°C/5% CO2. Medium was refreshed every 2–3 days and cells were passaged twice per week using an enzyme-free reagent (ReLeSR, Stem Cell Technologies).
CRISPR/Cas9 editing of ANK2
CRISPR/Cas9 technology was used to create a heterozygous indel variant in exon 31 of ANK2 in a hiPSC line derived from a healthy male donor, which was obtained from the Corriel Institute (USCFi001-A, GM25256) that shows normal karyotype, expresses pluripotency markers and has the capacity to differentiate into three germ layers. In brief, two sgRNAs were designed that specifically target ANK2 (CTTCAGCCCTATAGTCACTT and CTGCATAGTAATCTTAAGGT) and cloned into pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene #62988) according to the previous studies. About 8 × 105 hiPSCs in single-cell suspension were nucleofected with 5 μg of the generated SpCas9-sgRNA plasmid using the P3 Primary Cell 4D-Nucleofector kit (Lonza, #V4XP-3024) in combination with the 4D Nucleofector Unit X (Lonza, #AAF-1002X). After nucleofection, cells were resuspended in E8 Flex supplemented with Revitacell (Thermo Fisher Scientific, #A2644501) and seeded on biolaminin 521 (Biolamina, #LN521) pre-coated wells. Twenty-four hours after nucleofection, 0.5 μg puromycin was added for 24 h. Puromycin-resistant colonies were manually picked and send for Sanger Sequence to ensure heterozygous editing of exon 31. Genomic stability was assessed by detection of recurrent abnormalities using the iCS-digital™ PSC test, provided as a service by Stem Genomics (https://www.stemgenomics.com/). Off-target analysis was performed to confirm genetic integrity after genomic editing.
Neuronal differentiation
HiPSCs were differentiated into iNeurons using the Ngn2-protocol as previously described (23). Briefly, hiPSCs were directly differentiated into excitatory cortical layer 2/3 neurons by overexpressing Ngn2 upon doxycycline treatment. Neuronal maturation was supported by rat astrocytes, which were added to the culture in a 1:1 ratio 2 days after hiPSC plating. At DIV 3, the medium was changed to Neurobasal medium (Thermo Fisher Scientific, #21103049) supplemented with B-27 (Thermo Fisher Scientific, #17504001), glutaMAX (Thermo Fisher Scientific, #35050061), primocin (0.1 μg/ml), NT3 (10 ng/ml) (Thermo Fisher Scientific, #PHC7036), BDNF (10 ng/ml) and doxycycline (4 μg/ml). Cytosine β-D-arabinofuranoside (2 μM) (Sigma, #C1768) was added to remove proliferating cell from the culture at DIV 3. From DIV 6 onward, half of the medium was refreshed every other day. From DIV 10 onward, the medium was additionally supplemented with 2.5% FBS to support astrocyte viability. Neuronal cultures were kept through the whole differentiation process at 37°C/5%CO2.
Immunocytochemistry
Cells were treated with either 1 μM TTX or 10 mM KCl for 48 h and fixed with 4% paraformaldehyde supplemented with 4% sucrose for 15 min and permeabilized with 0.2% triton in 1× PBS for 10 min at RT. Nonspecific binding sites were blocked by incubation in blocking buffer (5% normal goat serum diluted in 1× PBS) for 1 h at RT. Primary antibodies were diluted in blocking buffer and incubated overnight at 4°C. Secondary antibodies, conjugated to Alexa-fluorochromes, were also diluted in blocking buffer and added for 1 h at RT. Hoechst 33342 was used to stain the nucleus before cells were mounted using DAKO fluorescent mounting medium. The following primary antibodies were used: rabbit anti-MAP2 (1:1000; Abcam, #ab32454); mouse anti-AnkG (1:250; Thermo Fisher Scientific, #33–8800); mouse anti-AnkB (1:1000, Santa Cruz Biotechnology, #sc-365 757); mouse anti-Homer1 (1:500, Synaptic Systems, #160011); rabbit anti-Synapsin (1:500, Merck Millipore, #AB1543P); rabbit anti-Nav1.1 (1:500, Alomone Labs, #ASC-001); rabbit anti-PanNav (1:500, Alomone Labs, #ASC-003). Secondary antibodies that were used are: goat-anti rabbit Alexa 568 (1:1000, Invitrogen, #A11036); goat-anti-mouse Alexa 488 (1:1000, Invitrogen, #A11029). Cells were imaged at a ×63 magnification using the Zeiss Axio Imager Z1 equipped with apotome. The ANKG length was determined per individual cell using 13.8889 pixels/μm. Synapse puncta were counted manually and normalized per 10 μm dendritic length where they reside. PanNav and Nav1.1 expressions were measured on the basis of mean intensity on ANKG length where they reside. PanNav and Nav1.1 mean intensities were measured on non-enhanced images. For visualization purposes (Supplementary Material, Fig. S3D and E), the PanNav and Nav1.1 fluorescent signals were enhanced. Fluorescent signals were quantified using ImageJ software.
Quantitative polymerase chain reaction
RNA samples were isolated from mature iNeurons (DIV 21) using the Nucleospin RNA isolation kit (Machery Nagel, 740955) according to the manufactures’ instructions. cDNAs were synthesized by iScript cDNA synthesis kit (Bio-Rad, 1708890). GoTaq master mix 2x with SYBR Green (Promega, A6002) was used for the PCR reactions according to the manufacturer’s protocol. PCR reactions were performed on the QuantStudio™ 3 Real-Time PCR System, using a 96-well, 0.2 ml format (Thermo Fisher Scientific). All samples were analyzed in duplicate in the same run. As a negative control, we used reverse transcriptase-negative conditions and no template-controls in all runs. We used the arithmetic mean of the Ct values of the technical replicates to calculate the relative mRNA expression levels of either 220 kDa or 440 kDa of ANK2. The mRNA expression level was calculated using the 2−ΔΔCt method with standardization to PPIA (Peptidylprolyl Isomerase A), GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and TBP (TATA-Box Binding protein). Primers used to assess mRNA expression of the 220 kDa of ANK2 are Fw (5′-3′) GACGTACAATCCAAGAGTGGT and Rv (5′-3′) TGTGAAGTCCACAGCAGCTC. To detect mRNA expression of the 440 kDa of ANK2, we used Fw (5′-3′) TTCCTGTCCTAGCAAGTCC and Rv (5′–3′) CCCATTCCTCTTCAACTAATCC.
Somatodendritic reconstruction
Neuron reconstruction was performed using Neurolucida 360 (Version 2017.01.4, Microbrightfield Bioscience). Neurons were fixed (4%PFA/sucrose in 1× PBS) and labeled with MAP2 antibody (1:1000, Synaptic Systems, #188004). Fluorescent images of MAP2-labelled neurons were taken at ×20 magnification using Zeiss Axio Imager Z1 equipped with ApoTome.
FiJi 2018 software was used to stitch the images with the stitching plugin. The stitched images were used for reconstruction using Neurolucida, which allows three-dimensional reconstructions and quantitative morphometrical analysis of the somatodendritic organization of the neurons. We selected neurons that had at least two primary dendrites for reconstruction and further analysis. For morphometrical analysis, we determined soma size, number of primary dendrites, dendritic nodes and ends and total or mean dendritic length as well as covered surface by dendritic trees. In addition, we also investigated dendritic complexity by performing Sholl analysis. Sholl profile was obtained by applying a series of concentric circles at 20 μm intervals from the soma center. Subsequently, dendritic length, number of intersections and number of nodes of the neurons were measured for each distance interval.
MEA recordings and analysis
All recordings were performed using the 24-wells MEA system (Multichannel Systems, MCS GmbH, Reutlingen, Germany). Recordings and analysis were performed according to previously published methods (20,23). In brief, MEA devices were composed of 24 independent wells with embedded microelectrodes (i.e. 12 electrodes/well, 80 μm in diameter and spaced 300 μm apart). Spontaneous electrophysiological activity of hiPSC-derived neuronal networks cultured on MEAs was recorded for 10 min in a recording chamber that was continuously maintained 37°C/95% O2/5% CO2. Before recording, MEAs were allowed to acclimate for 10 min in the recording chamber. The recording was sampled at 10 kHz and filtered with a high-pass filter with a 100 Hz cut-off frequency and a low-pass filter with a 3500 Hz cut-off frequency. The spike detection threshold was set at ±4.5 standard deviations. Spike detection, burst detection and network burst detection were performed by a built-in algorithm in Mulitwell Analyzer software (Multichannel Systems), and a custom-made MATLAB (The Mathworks, Natrick, MA, USA) code to extract parameters describing network activity. Mean firing rate was calculated as the average of the spike frequency of all channels across one MEA well. The algorithm for burst detection was set to define a burst when the interspike interval starts and ends with a maximum of 50 ms, with a minimum of 100 ms IBI and 4 spikes per burst. Network burst was detected when at least 50% of the channels in one well displayed a synchronous burst.
Conflict of Interest statement. S.W.S is GlaxoSmithKline-CIHR Endowed Chair in Genome Science of University of Toronto McLaughlin Centre, SickKids Foundation.
Funding
This work was supported by grants from the Netherlands Organization for Health Research and Development ZonMw Grant 91217055 (to N.N.K.); SFARI Grant 890042 (to N.N.K.).
Data availability
All genotypic and phenotypic data supporting the findings of this study are available within the paper and supplementary files.
Supplementary Material
Contributor Information
Maria W A Teunissen, Department of Neurology, Maastricht University Medical Center, Maastricht, HX 6229, The Netherlands; Academic Center for Epileptology Kempenhaeghe/Maastricht University Medical Center, Heeze 5591 VE, The Netherlands.
Elly Lewerissa, Department of Human Genetics, Radboudumc, Donders Institute for Brain, Cognition, and Behaviour, Nijmegen, HB 6500, the Netherlands.
Eline J H van Hugte, Department of Human Genetics, Radboudumc, Donders Institute for Brain, Cognition, and Behaviour, Nijmegen, HB 6500, the Netherlands.
Shan Wang, Department of Human Genetics, Radboudumc, Donders Institute for Brain, Cognition, and Behaviour, Nijmegen, HB 6500, the Netherlands.
Charlotte W Ockeloen, Department of Human Genetics, Radboud University Medical Center, Nijmegen, GA 6525, the Netherlands.
David A Koolen, Department of Human Genetics, Radboudumc, Donders Institute for Brain, Cognition, and Behaviour, Nijmegen, HB 6500, the Netherlands.
Rolph Pfundt, Department of Human Genetics, Radboud University Medical Center, Nijmegen, GA 6525, the Netherlands.
Carlo L M Marcelis, Department of Human Genetics, Radboud University Medical Center, Nijmegen, GA 6525, the Netherlands.
Eva Brilstra, Department of Human Genetics, University Medical Center Utrecht, Utrecht, CX 3584, The Netherlands.
Jennifer L Howe, The Centre for Applied Genomics and Genetics and Genome Biology, The Hospital for Sick Children, Toronto, ON M5G 1X8, Canada.
Stephen W Scherer, The Centre for Applied Genomics and Genetics and Genome Biology, The Hospital for Sick Children, Toronto, ON M5G 1X8, Canada; McLaughlin Centre and Department of Molecular Genetics, University of Toronto, Toronto, ON M5S 3H7, Canada.
Xavier Le Guillou, Department of Medical Genetics, Centre Hospitalier Universitaire de Poitiers, Poitiers 86000, France.
Frédéric Bilan, Department of Medical Genetics, Centre Hospitalier Universitaire de Poitiers, Poitiers 86000, France; Laboratory of Experimental and Clinical Neurosciences University of Poitiers, INSERM U1084, Poitiers 86000, France.
Michelle Primiano, Department of Clinical Genetics, Morgan Stanley Children’s Hospital of New York-Presbytarian, New York, NY, 10032, USA.
Jasmin Roohi, Department of Clinical Genetics, Morgan Stanley Children’s Hospital of New York-Presbytarian, New York, NY, 10032, USA; Clinical Genetics, Kaiser Permanente Mid-Atlantic Permanente Medical Group, Rockville, MD 20852, USA.
Amelie Piton, Laboratoire de Diagnostic Génétique, Institut de Génétique Médicale d’Alsace (IGMA), Hôspitaux Universitaire de Strasbourg, Strasbourg, BP 426 67091, France; Institut de Génétique et de Biologie Moléculaire et Cellulaire, Illkirch 67400, France.
Anne de Saint Martin, Institut de Génétique et de Biologie Moléculaire et Cellulaire, Illkirch 67400, France; Department of Pediatric Neurology, Strasbourg University Hospital, Hôspital de Hautepierre, Strasbourg, BP 426 67091, France.
Sarah Baer, Institut de Génétique et de Biologie Moléculaire et Cellulaire, Illkirch 67400, France; Department of Pediatric Neurology, Strasbourg University Hospital, Hôspital de Hautepierre, Strasbourg, BP 426 67091, France.
Simone Seiffert, Department of Neurology and Epileptology, Hertie Institute for Clinical Brain Research, Tuebingen, 72076, Germany.
Konrad Platzer, Institute of Human Genetics, University Medical Center Leipzig, Leipzig 04103, Germany.
Rami Abou Jamra, Institute of Human Genetics, University Medical Center Leipzig, Leipzig 04103, Germany.
Steffen Syrbe, Division of Paediatric Epileptology, Centre for Paediatric and Adolescent Medicine, University Hospital Heidelberg, Heidelberg 69120, Germany.
Jan H Doering, Division of Paediatric Epileptology, Centre for Paediatric and Adolescent Medicine, University Hospital Heidelberg, Heidelberg 69120, Germany.
Shenela Lakhani, Department of neurogenetics, Weill Cornell Medicine, Brain and Mind Research Institute, New York, NY, 10065, USA.
Srishti Nangia, Department of Pediatrics, Division of Child Neurology, New York Presbyterian Hospital-Weill Cornell Medical Center, New York, NY, 10032, USA.
Christian Gilissen, Department of Human Genetics, Radboud University Medical Center, Nijmegen, GA 6525, the Netherlands.
R Jeroen Vermeulen, Department of Neurology, Maastricht University Medical Center, Maastricht, HX 6229, The Netherlands.
Rob P W Rouhl, Department of Neurology, Maastricht University Medical Center, Maastricht, HX 6229, The Netherlands; Academic Center for Epileptology Kempenhaeghe/Maastricht University Medical Center, Heeze 5591 VE, The Netherlands; School for Mental Health and Neuroscience, Maastricht University, Maastricht, MD 6200, the Netherlands.
Han G Brunner, Academic Center for Epileptology Kempenhaeghe/Maastricht University Medical Center, Heeze 5591 VE, The Netherlands; Department of Human Genetics, Radboudumc, Donders Institute for Brain, Cognition, and Behaviour, Nijmegen, HB 6500, the Netherlands; School for Mental Health and Neuroscience, Maastricht University, Maastricht, MD 6200, the Netherlands; Department of Clinical Genetics and GROW School for Oncology and Developmental Biology, Maastricht University Medical Centre+, Maastricht, MD 6299, the Netherlands.
Marjolein H Willemsen, Department of Human Genetics, Radboud University Medical Center, Nijmegen, GA 6525, the Netherlands.
Nael Nadif Kasri, Department of Human Genetics, Radboudumc, Donders Institute for Brain, Cognition, and Behaviour, Nijmegen, HB 6500, the Netherlands.
References
- 1. Kunimoto, M., Otto, E. and Bennett, V. (1991) A new 440-kD isoform is the major ankyrin in neonatal rat brain. J. Cell Biol., 115, 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Otto, E., Kunimoto, M., McLaughlin, T. and Bennett, V. (1991) Isolation and characterization of cDNAs encoding human brain ankyrins reveal a family of alternatively spliced genes. J. Cell Biol., 114, 241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mohler, P.J., Le Scouarnec, S., Denjoy, I., Lowe, J.S., Guicheney, P., Caron, L., Driskell, I.M., Schott, J., Norris, K., Leenhardt, A. et al. (2007) Defining the cellular phenotype of "ankyrin-B syndrome" variants: human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation, 115, 432–441. [DOI] [PubMed] [Google Scholar]
- 4. Willsey, A.J., Sanders, S.J., Li, M., Dong, S., Tebbenkamp, A.T., Muhle, R.A., Reilly, S.K., Fertuzinhos, S., Miller, J.A., Murtha, M.T. et al. (2013) Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell, 21(155), 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Iossifov, I., O'Roak, B.J., Sanders, S.J., Ronemus, M., Krumm, N., Levy, D., Stessman, H.A., Witherspoon, K.T., Vives, L., Patterson, K.E. et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature, 13(515), 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Rubeis, S., He, X., Goldberg, A.P., Poultney, C.S., Samocha, K., Ercument Cicek, A., Kou, Y., Liu, L., Fromer, M., Walker, S. et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 13(515), 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krumm, N., Turner, T.N., Baker, C., Vives, L., Mohajeri, K., Whitherspoon, K., Raja, A., Coe, B.P., Stessman, H.A., He, Z. et al. (2015) Excess of rare, inherited truncating mutations in autism. Nat. Genet., 47, 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yuen, R.K., Thiruvahindrapuram, B., Merico, D., Walker, S., Tammimies, K., Hoang, N., Chrysler, C., Nalpathamkalam, T., Pellecchia, G., Liu, Y. et al. (2015) Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med., 21, 185–191. [DOI] [PubMed] [Google Scholar]
- 9. Farwell, K.D., Shahmirzadi, L., El-Khechen, D., Powis, Z., Chao, E.C., Davis, B.T., Baxter, R.M., Zeng, W., Mroske, C., Parra, M.C. et al. (2015) Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet. Med., 17, 578–586. [DOI] [PubMed] [Google Scholar]
- 10. Stessman, H.A., Xiong, B., Coe, B.P., Wang, T., Hoekzema, K., Fenckova, M., Kvarnung, M., Gerdts, J., Trinh, S., Cosemans, N. et al. (2017) Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet., 49, 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lelieveld, S.H., Reijnders, M.R., Pfundt, R., Yntema, H.G., Kamsteeg, E., de Vries, P., de Vries, B.B.A., Willemsen, M.H., Kleefstra, T., Lohner, K. et al. (2016) Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci., 19, 1194–1196. [DOI] [PubMed] [Google Scholar]
- 12. Deciphering Developmental Disorders Study (2017) Prevalence and architecture of de novo mutations in developmental disorders. Nature, 542, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hata, Y., Yoshida, K., Kinoshita, K. and Nishida, N. (2017) Epilepsy-related sudden unexpected death: targeted molecular analysis of inherited heart disease genes using next-generation DNA sequencing. Brain Pathol., 27, 292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ji, J., Shen, L., Bootwalla, M., Quindipan, C., Tatarinova, T., Maglinte, D.T., Buckley, J., Raca, G., Saitta, S.C., Biegel, J.A. et al. (2019) A semiautomated whole-exome sequencing workflow leads to increased diagnostic yield and identification of novel candidate variants. Cold Spring Harb. Mol. Case Stud., 5, a003756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Satterstrom, F.K., Kosmicki, J.A., Wang, J., Breen, M.S., De Rubeis, S., An, J., Peng, M., Collins, R., Grove, J., Klei, L. et al. (2020) Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell, 180, 568–584.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang, R., Walder-Christensen, K.K., Kim, N., Wu, D., Lorenzo, D.N., Badea, A., Jiang, Y.H., Yin, H.H., Wetsel, W.C. and Bennett, V. (2019) ANK2 autism mutation targeting giant ankyrin-B promotes axon branching and ectopic connectivity. Proc. Natl. Acad. Sci. U. S. A., 116, 15262–15271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen, K., Yang, R., Li, Y., Zhou, J.C. and Zhang, M. (2020) Giant ankyrin-B suppresses stochastic collateral axon branching through direct interaction with microtubules. J. Cell Biol., 219, e201910053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Galiano, M.R., Jha, S., Ho, T.S., Zhang, C., Ogawa, Y., Chang, K., Stankewich, M.C., Mohler, P.J. and Rasband, M.N. (2012) A distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell, 25(149), 1125–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kole, M.H. and Stuart, G.J. (2012) Signal processing in the axon initial segment. Neuron, 26(73), 235–247. [DOI] [PubMed] [Google Scholar]
- 20. Grubb, M.S. and Burrone, J. (2010) Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature, 465, 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuba, H., Oichi, Y. and Ohmori, H. (2010) Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature, 465, 1075–1078. [DOI] [PubMed] [Google Scholar]
- 22. Frega, M., Selten, M., Mossink, B., Keller, J.M., Linda, K., Moerschen, R., Qu, J., Koerner, P., Jansen, S., Oudakker, A. et al. (2020) Distinct pathogenic genes causing intellectual disability and autism exhibit a common neuronal network hyperactivity phenotype. Cell Rep., 30, 173–186.e6. [DOI] [PubMed] [Google Scholar]
- 23. Frega, M., van Gestel, S.H., Linda, K., van der Raadt, J., Keller, J., van Rhijn, J., Schubert, D., Albers, C.A. and Nadif Kasri, N. (2017) Rapid neuronal differentiation of induced pluripotent stem cells for measuring network activity on micro-electrode arrays. J. Vis. Exp., 8, 54900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang, Y., Pak, C., Han, Y., Ahlenius, H., Zhang, Z., Chanda, S., Marro, S., Patzke, C., Acuna, C., Covy, J. et al. (2013) Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron, 78, 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kunimoto, M. (1995) A neuron-specific isoform of brain ankyrin, 440-kD ankyrinB, is targeted to the axons of rat cerebellar neurons. J. Cell Biol., 131, 1821–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mossink, B., Verboven, A.H.A., van Hugte, E.J.H., Klein Gunnewiek, T.M., Parodi, G., Linda, K., Schoenmaker, C., Kleefstra, T., Kozicz, T., van Bokhoven, H. et al. (2021) Human neuronal networks on micro-electrode arrays are a highly robust tool to study disease-specific genotype-phenotype correlations in vitro. Stem Cell Reports., 14(16), 2182–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lindhout, F.W., Kooistra, R., Portegies, S., Herstel, L.J., Stucchi, R., Snoek, B.L., Altelaar, A.M., MacGillavry, H.D., Wierenga, C.J. and Hoogenraad, C.C. (2020) Quantitative mapping of transcriptome and proteome dynamics during polarization of human iPSC-derived neurons. elife, 9, e58124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Evans, M.D., Dumitrescu, A.S., Kruijssen, D.L.H., Taylor, S.E. and Grubb, M.S. (2015) Rapid modulation of axon initial segment length influences repetitive spike firing. Cell Rep., 10(13), 1233–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sohn, P.D., Huang, C.T., Yan, R., Fan, L., Tracy, T.E., Camargo, C.M., Montgomery, K.M., Arhar, T., Mok, S., Freilich, R. et al. (2019) Pathogenic tau impairs axon initial segment plasticity and excitability homeostasis. Neuron, 104, 458–470 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wolff, M., Brunklaus, A. and Zuberi, S.M. (2019) Phenotypic spectrum and genetics of SCN2A-related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia, 60, S59–S67. [DOI] [PubMed] [Google Scholar]
- 31. Syrbe, S., Harms, F.L., Parrini, E., Montomoli, M., Mutze, U., Helbig, K.L., Polster, T., Albrecht, B., Bernbeck, U., van Binsbergen, E. et al. (2017) Delineating SPTAN1 associated phenotypes: from isolated epilepsy to encephalopathy with progressive brain atrophy. Brain, 1(140), 2322–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fu, J.M., Satterstrom, F.K., Peng, M., Brand, H., Ryan, L., Collins, R.L. and Dong, S. (2022) Rare coding variation illuminates the allelic architecture, risk genes, cellular expression patterns, and phenotypic context of autism. Nat. Genet., 54, 1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scheffer, I.E. and Nabbout, R. (2019) SCN1A-related phenotypes: epilepsy and beyond. Epilepsia, 60, S17–S24. [DOI] [PubMed] [Google Scholar]
- 34. Lorenzo, D.N., Badea, A., Davis, J., Hostettler, J., He, J., Zhong, G., Zhuang, X. and Bennett, V. (2014) A PIK3C3-ankyrin-B-dynactin pathway promotes axonal growth and multiorganelle transport. J. Cell Biol., 22(207), 735–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rasband, M.N. (2010) The axon initial segment and the maintenance of neuronal polarity. Nat. Rev. Neurosci., 11, 552–562. [DOI] [PubMed] [Google Scholar]
- 36. Kuba, H., Yamada, R., Ishiguro, G. and Adachi, R. (2015) Redistribution of Kv1 and Kv7 enhances neuronal excitability during structural axon initial segment plasticity. Nat. Commun., 6, 8815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Frega, M., Linda, K., Keller, J.M., Gumus-Akay, G., Mossink, B., van Rhijn, J., Negwer, M., Klein Gunnewiek, T., Foreman, K., Kompier, N. et al. (2019) Neuronal network dysfunction in a model for Kleefstra syndrome mediated by enhanced NMDAR signaling. Nat. Commun., 10, 4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deneault, E., White, S.H., Rodrigues, D.C., Ross, P.J., Faheem, M., Zaslavsky, K., Wang, Z., Alexandrova, R., Pellecchia, G., Wei, W. et al. (2018) Complete disruption of autism-susceptibility genes by gene editing predominantly reduces functional connectivity of isogenic human neurons. Stem Cell Reports., 13(11), 1211–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gullo, F., Manfredi, I., Lecchi, M., Casari, G., Wanke, E. and Becchetti, A. (2014) Multi-electrode array study of neuronal cultures expressing nicotinic beta2-V287L subunits, linked to autosomal dominant nocturnal frontal lobe epilepsy. An in vitro model of spontaneous epilepsy. Front. Neural Circuits., 8, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nelsons, A.D., Catalfio, A.M., Gupta, J.M., Min, L., Caballero-Floran, R.N., Dean, K.P., Elvira, C.C., Derderian, K.D., Kyoung, H., Sahagun, A. et al. (2022) Physical and functional convergence of the autism risk genes Scn2a and Ank2 in neocortical pyramidal cell dendrites. bioRxiv, 494205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. de Ligt, J., Willemsen, M.H., van Bon, B.W., Kleefstra, T., Yntema, H.G., Kroes, T., Vulto-van Silfhout, A.T., Koolen, D.A., de Vries, P., Gilissen, C. et al. (2012) Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med., 15(367), 1921–1929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genotypic and phenotypic data supporting the findings of this study are available within the paper and supplementary files.