

Abstract
Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency is the most common inherited disorder of mitochondrial fatty acid β-oxidation (FAO) in humans. Patients exhibit clinical episodes often associated with fasting. Symptoms include hypoketotic hypoglycemia and Reye-like episodes. With limited treatment options, we explored the use of human MCAD (hMCAD) mRNA in fibroblasts from patients with MCAD deficiency to provide functional MCAD protein and reverse the metabolic block. Transfection of hMCAD mRNA into MCAD- deficient patient cells resulted in an increased MCAD protein that localized to mitochondria, concomitant with increased enzyme activity in cell extracts. The therapeutic hMCAD mRNA-lipid nanoparticle (LNP) formulation was also tested in vivo in Acadm−/− mice. Administration of multiple intravenous doses of the hMCAD mRNA-LNP complex (LNP-MCAD) into Acadm−/− mice produced a significant level of MCAD protein with increased enzyme activity in liver, heart and skeletal muscle homogenates. Treated Acadm−/− mice were more resistant to cold stress and had decreased plasma levels of medium-chain acylcarnitines compared to untreated animals. Furthermore, hepatic steatosis in the liver from treated Acadm−/− mice was reduced compared to untreated ones. Results from this study support the potential therapeutic value of hMCAD mRNA-LNP complex treatment for MCAD deficiency.
Introduction
Mitochondrial β-oxidation of fatty acids provides energy, especially during fasting conditions. Medium-chain acyl-CoA dehydrogenase (MCAD) has a wide range of substrate specificity, catalyzing the dehydrogenation of medium-chain length (carbon lengths C6–C12) fatty acid coenzyme A thioesters (1). MCAD deficiency (MCADD) is an autosomal recessive disease, considered to be the most common hereditary disease of hepatic fatty acid oxidation and one of the most common inborn errors of metabolism with an estimated incidence of 1:5000 to 1:20 000 in North America and northern Europe (2,3). In MCADD patients, the inability to catabolize medium-chain fatty acids causes the accumulation of high amounts of medium-chain alternative metabolites. In most cases, onset occurs during infancy, usually between the ages of 3–15 months. The phenotype includes hypoketotic hypoglycemia, lethargy, coma, seizures and death induced by fasting and infectious illness. MCADD patients can also present with acute liver disease and hepatomegaly. The mortality rate is as high as 20% in untreated patients but falls to near zero when identified by newborn screening. Nevertheless, significant morbidity remains. The clinical presentation of an individual with MCADD depends not only on the presence of the mutations in the ACADM gene but also on the presence of environmental or physiological stressors that require the body to depend on fatty acid oxidation for energy.
In areas with expanded newborn screening using tandem mass spectrometry (MS/MS), acylcarnitine profiles show a characteristic pattern of elevated hexanoylcarnitine (C6), octanoylcarnitine (C8), decanoylcarnitine (C10) or decenoylcarnitine (C10:1), with C8 being greater than C6 and C10. As with most other fatty acid oxidation disorders, individuals with MCADD need to avoid fasting for prolonged periods of time. Treatment of MCADD is mainly preventive by avoiding fasting and other situations where the body relies on fatty acid oxidation to supply energy. During illnesses, patients require careful management to avoid metabolic decompensation. Supplementation with simple carbohydrates or glucose during illness is key to preventing symptoms. Even with treatment, patients can experience exercise intolerance and persistent myalgias.
In vitro transcribed message RNA (IVT mRNA) is emerging as a new class of medications, enabling cells to produce replacement protein. Recent advances in mRNA technology, including modifications to the mRNA itself and improvements to delivery vehicles, have transformed the utility of mRNA as a potential therapy (4,5). This approach delivers a specially formulated in vitro generated mRNA to target organ-specific cells that is translated by the cell’s endogenous machinery under physiological conditions. To date, systemic administration of mRNA encapsulated in lipid nanoparticles (LNPs) has primarily targeted the liver (6–10), with hepatocytes as the primary target cell type (11,12). LNPs introduced via intravenous (i.v.) injection are opsonized by apolipoprotein E (ApoE) followed by receptor-mediated endocytosis (11). mRNA therapy has broad applicability across a range of therapeutic areas such as oncology/immuno-oncology (13,14), cardiovascular diseases (15) and infectious diseases (16).
Recently, preclinical proof-of-concept has been demonstrated for mRNA therapy for rare metabolic disorders including methylmalonic acidemia, acute intermittent porphyria, very long-chain acyl-CoA dehydrogenase (17) and Fabry disease (4,5). MCADD is a candidate for mRNA therapy because of limited available treatment options and its risk of serious or fatal outcome with liver being affected. The Acadm−/− mouse is an excellent model to study MCADD because it presents with many relevant features characteristic of the disease found in MCADD patients. In the present study, we investigated the ability of therapeutic human MCAD (hMCAD) mRNA and LNP-hMCAD to rescue MCAD activity in fibroblasts from MCADD patients and in Acadm−/− mice. Our data identify IVT mRNA as potentially viable therapeutic option for MCADD.
Results
Human MCAD mRNA transfection in MCADD patient–derived skin fibroblasts, mouse embryonic fibroblasts and HeLa cells
To investigate the ability of hMCAD mRNA to express MCAD protein and its localization in cells, hMCAD mRNA (Moderna, Inc., Cambridge, MA) was transfected into two MCADD patient–derived fibroblasts (MCADD 1 and MCADD 2), mouse embryonic fibroblasts (MEFs) and HeLa cells. Human enhanced green fluorescent protein (eGFP) mRNA (Moderna, Inc.) was used as control. MCAD protein expression was evaluated at 48 h post transfection. Protein signal was observed in skin fibroblasts from an MCADD patient and control cells transfected with hMCAD mRNA, as determined by western blotting (Fig. 1A). The expressed MCAD protein colocalized with mitochondrial markers (MTCO1 or Mitotracker) in patient fibroblasts (Fig. 1B), MEFs (Fig. 1C) and HeLa cells (Fig. 1D) as shown by in situ immunofluorescence.
Figure 1.

MCAD expression and localization in cell mitochondria after hMCAD mRNA transfection. (A) MCAD protein expression in normal control and two MCADD patient derived fibroblasts (MCADD 1 and MCADD 2) 48 h post transfection with hMCAD mRNA (1.0 μg). Transfection with human eGFP mRNA (1.0 μg) was used as a negative control. GAPDH was used as a loading control. (B) MCADD patient fibroblast immunofluorescence staining of MCAD (green) 24 h post transfection with hMCAD mRNA. Anti-cytochrome c oxidase subunit 1 (MTCO1) antibody was used as mitochondrial control (red). The merged image shows colocalization of the signal (yellow). Nuclei were visualized with DAPI staining (blue). Scale bars, 10 μm. MCAD expression and localization in mitochondria of MEFs and HeLa cells after hMCAD mRNA transfection. MEFs (C) and HeLa cells (D) immunofluorescence staining of MCAD (green) 24 h post transfection with hMCAD mRNA. MTCO1 antibody or Mitotracker Red was used as mitochondrial control (red). The merged image shows colocalization of the signal (yellow). Nuclei were visualized with DAPI staining (blue). Scale bars, 10 μm.
Human MCAD mRNA improves activity and mitochondrial FAO in MCADD fibroblasts and wild-type MEFs
The impact of hMCAD mRNA transfection on MCAD enzyme activity was studied in MCADD fibroblasts and wild-type (WT) MEFs. The expressed MCAD 2 h after transfection in cell lysates from the two patients was active as demonstrated with the sensitive and specific anaerobic electron transfer flavoprotein (ETF) fluorescence reduction assay using octanoyl-CoA (C8-CoA) as a substrate (Fig. 2A). Both patient cell lines showed a significant increase in whole-cell FAO flux using [9, 10-3H] oleate (C18:1) as a substrate following hMCAD mRNA transfection (Fig. 2B). MEFs transfected with two hMCAD mRNA constructs (mRNA1 and mRNA2) showed a dose-dependent increase of MCAD activity (Fig. 2C).
Figure 2.

hMCAD mRNA leads to an increase of MCAD activity in skin fibroblasts from MCAD patients. Fibroblasts from normal control and two MCAD patients (MCADD 1 and MCADD 2) were seeded in 6-well plates and transfected with hMCAD mRNA or eGFP mRNA as control. (A) MCAD activity in cellular extracts was measured 48 h post transfection with octanoyl-CoA (C8-CoA) as substrate. Human eGFP mRNA transfected cells were used as a control. (B) Flux through the FAO pathway was quantified by production of 3H2O from [9,10-3H] oleate. Data are means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 compared to respective controls. (C) Test of hMCAD mRNA constructs in MEFs. Two hMCAD mRNA constructs with different doses (0, 0.3, 0.9 μg) were individually transfected into MEFs for 48 h. After transfection, MCAD activity was measured in cell lysates with octanoyl-CoA as a substrate. All values are means ± SD. **P < 0.01, ****P < 0.0001, *****P < 0.00001 compared to control (0 μg). +++P < 0.001 compared to 0.3 μg group, respectively. (D) Human MCAD mRNA reduces medium-chain acylcarnitines in tissue culture media in MCADD fibroblasts. Fibroblasts from normal control and two patients with MCADD (MCADD 1 and MCADD 2) and control normal fibroblasts were seeded in 6-well plates and incubated for 24 h in complete DMEM. The cells were then transfected with either hMCAD mRNA or eGFP mRNA as a control for 48 h. After transfection, tissue culture media was analyzed for acylcarnitines by tandem mass spectrometry. Data are means ± SD. *P < 0.05, **P < 0.01, ****P < 0.0001 compared to hMCAD mRNA group.
Treatment of MCADD patient–derived skin fibroblasts with hMCAD mRNA reduces medium-chain acylcarnitines produced by cultured cells
MCADD patients characteristically accumulate increased levels of medium-chain acylcarnitines in blood and cultured fibroblasts. To investigate the effect of hMCAD mRNA on acylcarnitines in patient cells, MCADD cells and normal control fibroblasts were transfected with hMCAD mRNA or eGFP mRNA as a control for 48 h, and acylcarnitines in tissue culture media were determined by tandem mass spectrometry. Tissue culture media from patient-derived MCADD fibroblasts showed significant elevations in medium-chain acylcarnitine species (C8 and C6) compared to normal control prior to treatment (Fig. 2D). Importantly, the accumulation of C8- and C6-acylcarnitine in tissue culture media was decreased after hMCAD mRNA treatment, indicating partial relief of the block in medium fatty acid oxidation (Fig. 2D).
A single dose of LNP-MCAD restores MCAD protein in Acadm−/− mouse liver
To investigate the ability of hMCAD mRNA to restore MCAD protein in Acadm−/− mice, a single dose (1.0 mg/kg) of therapeutic hMCAD mRNA–LNP formulation (LNP-MCAD, Moderna, Inc.) was injected i.v. into Acadm−/− mice. Liver was harvested and MCAD protein in liver homogenates was evaluated at 24, 48, 96 and 144 h post injection. There were no obvious differences in liver size or appearance postmortem. A robust MCAD protein signal was observed as early as 24 h after treatment and persisted up to 96 h (Fig. 3A). Expression levels of MCAD were quantitated by scanning and normalization to glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Fig. 3B).
Figure 3.

Longevity of MCAD protein in Acadm−/− mouse liver after LNP-MCAD injection. Acadm−/− mice were given single i.v. injection of LNP-MCAD (0.5 mg/kg). Livers were harvested at 24, 48, 96 and 144 h post injection. (A) A representative western blot for MCAD in liver homogenates. GAPDH was used as loading control. (B) Relative intensity ratio of MCAD over GAPDH as measured by scanning of triplicate western blots in multiple time points up to 144 h from three separate experiments. Liver homogenate from WT mice was used as positive control. All time point values are highly statistically different compared to WT and untransfected control (P < 0.0001).
LNP-MCAD-treated Acadm−/− mice are more cold-tolerant after treatment with LNP-MCAD
Cold exposure is a metabolic stress requiring increased energy production, and cold sensitivity is a common feature of fatty-acid oxidation deficiency in mice, including MCADD (2). To determine the effects of LNP-MCAD treatment on cold tolerance in Acadm−/− mice, an i.v. dose of either formulated LNP-MCAD (1.0 mg/kg) or eGFP mRNA (1.0 mg/kg) as control was administered on day 0, day 4 and day 8 (Fig. 4A). After the final treatment, mice were fasted for 18 h and then placed in an 8°C environment for 3 h (day 9). WT mice that received saline injections as control tolerated a 3 h cold stress with no significant change in body temperature (Fig. 4B, line a). In contrast, Acadm−/− mice treated with eGFP mRNA exhibited a significant drop in body temperature over that same period (Fig. 4B, line c). Importantly, cold tolerance was improved in Acadm−/− mice treated with LNP-MCAD (Fig. 4B, line b).
Figure 4.

Multiple injections of Acadm−/− mice with LNP-MCAD restores protein activity and improves cold tolerance. (A) Acadm−/− male mice were administered an i.v. dose of either formulated LNP-MCAD (1.0 mg/kg) or eGFP mRNA (1.0 mg/kg) for three times (day 0, day 4, and day 8) in 8 days. After the last treatment on day 8, mice were fasted for 18 h before transferred to an 8°C chamber for 3 h. WT mice received i.v.-injected saline solution as control. (B) Rectal temperature vs. time in the cold chamber was plotted. (C) MCAD activity of liver homogenates was measured using octanoyl-CoA (C8) as a substrate. (D) VLCAD activity in liver homogenates was measured using palmitoyl-CoA (C16) as a substrate. (E and F) Activity in heart and skeletal muscle homogenates, respectively, measured with octanoyl-CoA as substrate. Data are means ± SD. *P < 0.05 compared to the hMCAD mRNA group, **P < 0.01 compared to eGFP mRNA group, +P < 0.05 compared to the WT group.
Liver, heart and skeletal muscle were harvested after cold challenge, and homogenates were analyzed for MCAD protein by western blotting. GAPDH and very long-chain acyl-CoA dehydrogenase (VLCAD) were used as loading and enzyme activity controls, respectively. MCAD protein signal was observed in Acadm−/− liver, heart and skeletal muscle after repeated LNP-MCAD treatment (Fig. 5). In addition, the expressed MCAD protein after treatment restored MCAD activity in all three tissues as measured by the ETF fluorescence reduction assay using octanoyl-CoA (C8-CoA) as substrate (Fig. 4C, E and F). There was no significant change of VLCAD activity in liver after treatment as measured by the ETF fluorescence reduction assay using palmitoyl-CoA (C16-CoA) as a substrate (Fig. 4D). WT mouse liver was used as positive control. Digitonin-extracted mitochondrial membranes were analyzed by blue native gel electrophoresis (BN-PAGE) followed by Coomassie blue-stain to show electron transport-related bands (Fig. 6). In-gel semi-quantitative activity staining of ETC complex I, III, IV and V activity was also performed (Fig. 6B–D). No obvious enzymatic abnormalities besides MCAD were identified before or after treatment in duplicate gels.
Figure 5.

Tissue distribution of MCAD expression following LNP-MCAD. treatment. Acadm−/− male mice were treated with LNP-MCAD (1.0 mg/kg, i.v.) or eGFP mRNA with multiple i.v. injections up to 9 days. WT mice were injected on a similar schedule with saline. MCAD protein expression in tissue homogenates (heart and skeletal muscle) was determined by western blotting. Liver, skeletal muscle and heart expression of MCAD protein is shown in (A–C), respectively. Corresponding activity analysis is shown in Figure 4. GAPDH was used as loading control.
Figure 6.
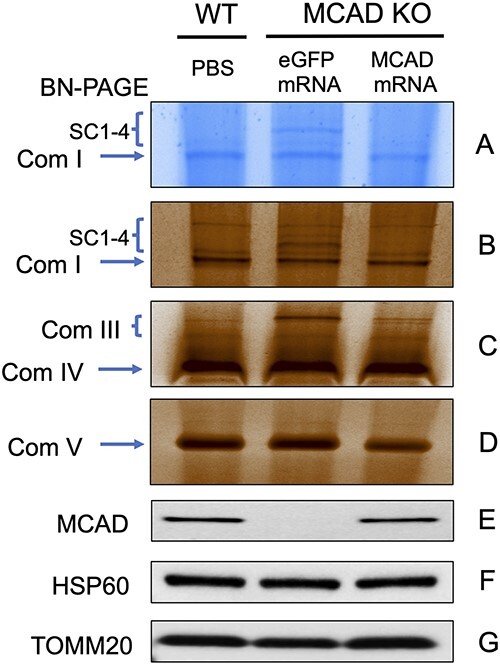
Blue native gel and complex activity in Acadm−/− mouse liver after treatment with LNP-MCAD Acadm−/− male mice were administered multiple i.v. injections of LNP-MCAD (1.0 mg/kg) or eGFP (1.0 mg/kg) for 8 days. Mitochondrial from WT control and mRNA-treated mouse liver was then isolated and treated with digitonin. Digitonin-extracted mitochondrial membranes were analyzed by Blue Native Gel Electrophoresis (BN-PAGE) followed by Coomassie Blue-stain to show electron transport-related bands (A). (B–D) In-gel staining assay of ETC complex I, III, IV and V activity. (E–G) Mitochondrial from WT control and mRNA treated mouse liver was used for measuring protein presence by western blotting with anti-MCAD, with anti-HSP60 and anti-TOMM20 as loading controls. Com I, complex I; Com III, complex IIII; Com IV, complex IV; Com V, complex V; SC, supercomplexes.
Treatment of Acadm−/− mice with therapeutic LNP-MCAD reduces medium-chain acylcarnitines in blood
MCADD patients and mice characteristically accumulate increased blood levels of medium-chain acylcarnitines. Therefore, the blood from Acadm−/− mice treated with repeated doses of LNP-MCAD and LNP-eGFR (1.0 mg/kg) was examined for acylcarnitine content. As expected, blood from control MCADD animals, treated with eGFP mRNA at 1.0 mg/kg, showed significant elevations in medium-chain acylcarnitine species (C6, C8, C10 and C10:1) compared to WT mice (Fig. 7A–D). The levels of C8- and C10:1-acylcarnitines in plasma were decreased after multiple injections with LNP-MCAD (Fig. 7B and D), indicating partial relief of the block in medium fatty acid oxidation. Acadm−/− mice showed reduced blood glucose levels after cold stress compared to WT, independent of LNP-MCAD treatment (Fig. 8), consistent with incomplete restoration of bioenergetics.
Figure 7.

Treatment of Acadm−/− mice with LNP-MCAD decreases the plasma level of medium-chain acylcarnitine Acadm−/− male mice were administered multiple i.v. injections of LNP-MCAD (1.0 mg/kg) or eGFP mRNA (1.0 mg/kg) for 8 days (Fig. 5A). WT mice received i.v.- injected saline solution as a control. After 18 h of food deprivation and 3 h of cold stress, blood from those mice was collected and plasma was obtained for acylcarnitine profile analysis by tandem mass spectrometry. The panels show the levels of (A) C6 acylcarnitine species, (B) C8 acylcarnitine species, (C) C10 acylcarnitine species and (D) C10:1 acylcarnitine species. Data are means ± SD. *P < 0.05, **P < 0.01 compared to respective controls, ns = not significant.
Figure 8.

Blood glucose and lactate levels after treatment with LNP-MCAD Acadm−/− male mice were administered multiple i.v. injections of LNP-MCAD (1.0 mg/kg) or eGFP mRNA (1.0 mg/kg) for 8 days (Fig. 5A). WT mice received i.v.-injected saline solution as a control. All mice were under 18 h of food deprivation followed by 3 h cold stress. Before and after cold stress, glucose (A) and lactate (B) levels in blood were measured using strips. Data are means ± SD. *P < 0.05, **P < 0.01 compared to respective controls.
Treatment with therapeutic LNP-MCAD reduces hepatic steatosis in Acadm−/− mice
The acute metabolic decompensation in children with inborn errors of FAO is often precipitated by fasting and is accompanied by the development of hepatic steatosis (18). Acadm−/− mice fasted for 24 h also show hepatic steatosis (2). Therefore, we treated Acadm−/− mice multiple times with i.v. injection of LNP-MCAD (1.0 mg/kg) (as shown in Fig. 4A). Following the last treatment on day 8, mice were fasted for 18 h followed by 3 h cold stress at 8°C. Livers were then harvested and stained with hematoxylin and eosin (H&E) and Oil Red O. Frozen liver sections stained with Oil Red O demonstrated abundant accumulation of fat droplets that were reduced by multiple LNP-MCAD injections (Fig. 9A). The improvement was also identified in sections stained with H&E (Fig. 8B). WT mice showed little evidence of hepatic steatosis following fast-cold stress (Fig. 9A and B).
Figure 9.

Treatment with therapeutic LNP-MCAD reduces hepatic steatosis in Acadm−/− mice. Acadm−/− and WT male mice were administered multiple i.v. injections of LNP-MCAD (1.0 mg/kg) or eGFP mRNA (1.0 mg/kg) for 8 days (Fig. 5A). WT male mice received i.v. injection of saline as control. Followed 18 h of food deprivation and 3 h of cold stress, livers from each group were processed and used for Oil Red O staining (A) and H&E staining (B).
Discussion
MCAD deficiency in humans can be associated with significant morbidity even when diagnosed by newborn screening, especially in the first few years of life (19) Treatment of MCAD deficiency is mainly preventive by avoiding fasting and other situations where the body relies on fatty acid oxidation to supply energy, often with a need for hospitalization and IV fluids during illness (20). As a result, infants and young children are often hospitalized for IV fluids even with otherwise minor illnesses, with increased parental anxiety regardless (21). Symptoms in older patients are less common, but sudden death even in adults has been reported (22). Thus, there is a need for additional therapeutic options, especially early in life and during acute illness. In this manuscript, we describe the use of synthetic hMCAD mRNA and mRNA-LNPs (LNP-MCAD) to improve fatty acid oxidation in MCAD-deficient cells and animals. Skin fibroblasts from MCADD patients after 48 h transfection with hMCAD mRNA showed an increase in MCAD protein and activity, while i.v. injection of LNP-MCAD into Acadm−/− mice led to expression of active MCAD in the liver, muscle and heart. Expression in the latter two tissues was unexpected, as the LNP was expected to target the liver (1 and 5). The reason for this phenomenon is unclear. Importantly, Acadm−/− mice treated with multiple injections of LNP-MCAD improved cold tolerance and decreased the plasma level of medium-chain acylcarnitines, systemic manifestations of the disease. Finally, treated Acadm−/− mice showed less micro- and macrovesicular steatosis in livers compared to untreated animals.
Impaired bioenergetics in extrahepatic disease tissues contributes significantly to symptoms seen in this disease. However, the mechanism of mitochondrial dysfunction is not yet fully understood. One hypothesis is that accumulation of toxic metabolites such as saturated and unsaturated dicarboxylic acids leads to mitochondrial toxicity (23). Uncouplers of oxidative phosphorylation and/or modulators of mitochondrial permeability transition can also lead to impairment of mitochondrial bioenergetics (24).
Mitochondria are critical for thermogenesis in mammals, caused by uncoupling of respiratory chain activity from ATP synthesis. In humans, shivering thermogenesis in response to cold exposure is a muscle-based phenomenon. In mice, it is largely performed in brown fat (25), and reduced cold tolerance is a common feature of fatty-acid oxidation deficiency in mice (2,26,27). Cold sensitivity has also been reported in UPC-1-deficient mice (28) and in SIRT3-deficient mice (29). In addition, a combination of fasting and cold exposure is fatal in Acadm−/− mice (2). Moreover, cold sensitivity persists even when animals are provided glucose by oral gavage when cold-stressed. Thus, appropriate mitochondrial bioenergetics is necessary for thermogenesis. In this study, liver-directed LNP-MCAD therapy of Acadm−/− mice improved cold tolerance, demonstrating that liver mitochondrial bioenergetics plays a critical role in response to systemic metabolic stress (30). However, thermogenesis was still not normal in treated animals, highlighting the importance of other tissues in this process. How this effect translates to humans remains to be seen.
Acylcarnitines have been hypothesized to have direct cytotoxic effects (especially in brain), but the precise nature of this effect is unknown (31,32). Additionally, long-chain acyl-CoAs are known to inhibit the mitochondrial adenine nucleotide translocator in vitro, though the physiologic ramifications of this phenomenon remain unknown (33). Regardless of their role in disease pathogenesis, markedly elevated medium-chain acylcarnitines are at least a biochemical marker of potentially life-threatening metabolic crises in severe MCADD patients. The Acadm−/− mouse is an excellent model to study MCADD because it presents with similar features that mimic the clinical, biochemical and pathologic phenotypes found in human patients (2). Of note, acylcarnitine analysis indicates that mouse MCAD is more active toward longer-chain substrates than the human MCAD enzyme (2). In this study, Acadm−/− mice indeed had significant elevations in acylcarnitine species including C6, C8, C10 and C10:1 in blood that decreased after LNP-MCAD treatment (Fig. 6).
The microvesicular and macrovesicular hepatic steatosis seen in fasted Acadm−/− mice is consistent with the primary pathological finding in human MCAD patients with fasting stress. Lipid accumulation is exacerbated by impaired mitochondrial FAO. The acute metabolic decompensation in children with inborn errors of FAO is often precipitated by fasting (18). Acadm−/− mice fasted for 24 h showed hepatic steatosis (2). Both micro- and macrovesicular steatosis were reduced in stressed deficient animals by multiple LNP-MCAD injections (Fig. 7A and B).
Clinical improvement in systemic diseases with liver-based mRNA therapy has previously been demonstrated for two inborn errors of branched-chain amino acid metabolism, propionic acidemia and methylmalonic acidemia (5,11). In both diseases, administration of mRNA to affected animals reduced the primary signals of the metabolic blocks and improved clinical outcome in animals in spite of the fact that the majority of branched-chain amino acid catabolism is based outside the liver, predominantly in muscle. Consistent with our findings with mRNA-treated MCADD mice, the methylmalonyl-CoA mutase mice did not normalize their methylmalonic acid levels in blood but nonetheless had a dramatic improvement in survival. Expressed MCAD clearance in our study was in keeping with these two models and suggests that patient dosing every 2 or 3 weeks will be sufficient to protect against symptoms. However, additional dose finding experiments likely will be necessary to move to a Federal Drug Administration new drug application.
Finally, while in vitro and in vivo models of MCAD deficiency have demonstrated the potential benefit of treatment with hMCAD mRNA, it is necessary to consider the need for such a treatment. Prior to addition of MCAD deficiency to the newborn screening panel in the United States and many other developed countries, the major risk for hypoglycemia and/or sudden death was typically in the first few years of life. However, MCAD deficiency is now managed with diet (avoidance of fasting) and hospitalization when oral intake cannot be maintained. Thus, use of a chronically injected medication is likely of limited value for a condition that is otherwise managed less invasively. Nevertheless, lessons learned in developing and accessing therapeutic mRNA for MCAD deficiency will be valuable for similar programs in other diseases.
Materials and Methods
Mice
Mice with MCAD deficiency have been previously described (2). The corresponding WT mice were bred in house under specific pathogen-free conditions by the Division of Laboratory Animal Resources at the Rangos Research Building, University of Pittsburgh. All animal experiments and procedures were performed in accordance with the institutional protocol and were approved by the institutional animal use and care committee of the University of Pittsburgh. Age- and sex-matched mice were used in studies.
Cell culture conditions and hMCAD mRNA transfection
Fibroblasts were derived from two patients on a clinical basis. A control human diploid fibroblast cell line was obtained from a healthy individual purchased from ATTC (Cat # PCS-201-012). Biopsies from patients were performed on a clinical basis with written informed consent from patients and/or parents. Mouse embryonic fibroblasts (MEFs) and HeLa cells were obtained from American Type Culture Collection (ATCC). Cells were cultured in complete Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS), 2 mM glutamine and 1% penicillin/streptomycin at 37°C in a 5% CO2 incubator. Cells were plated in 6-well plates and transfected with hMCAD mRNA for 24–48 h either using the TransIT-mRNA transfection kit (Mirus Bio LLC, Madison, WI) or the Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific Inc, Waltham, MA). Subcellular localization of MCAD to the mitochondria in cells was assessed using fluorescent microscopy.
mRNA production and formation
mRNA sequence was produced and formulated by ModernaRx, Inc. (Cambridge, MA). Complete N1-methylpseudouridine-substituted mRNA was synthesized in vitro from a linearized DNA template containing the 5′ and 3′ untranslated regions (UTRs) and a poly-A tail, as previously described (34). After purification, the mRNA was diluted in citrate buffer to the desired concentration and frozen. mRNA was encapsulated in an LNP through a modified ethanol drop nanoprecipitation process, as described previously (35). All formulations were confirmed to be between 80 and 100 nm particle size, greater than 80% of RNA encapsulation and <10 EU/ml endotoxin.
ETF fluorescence reduction assay
MCAD activity was measured using octanoyl-CoA (C8-CoA) (Sigma-Aldrich) as a substrate with the anaerobic ETF fluorescence reduction assay (36,37). Human fibroblasts (400 000 cells/well) were plated in 6-well plates and transfected with hMCAD mRNA. After 48 h transfection, the cell pellets were lysed in 50 mM Tris buffer (pH 8.0) with 1.0 mM EDTA and protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany) and disrupted by sonication at 4°C. After centrifugation at 16 000g for 20 min, the supernatants were collected and stored at −80°C. Mouse livers were resuspended in 50 mM Tris buffer (pH 8) containing 1.0 mM EDTA and protease inhibitor cocktail (Roche) and disrupted using a bullet blender at speed 6 for 2 min with zirconium beads (Next Advance, Inc., Troy, NY). Homogenates were centrifuged at 16 000 × g for 15 min at 4°C. Supernatants after the centrifugation were stored at −80°C until use. Protein concentration from the supernatants of both cells and livers was determined by the DC™ protein assay kit (Bio-Rad Laboratories, Hercules, CA). Protein was assayed for dehydrogenase activities using a Jasco FP-6300 spectrofluorometer (Easton, MD) under anaerobic conditions at 32°C, exciting ETF flavin at 342 nm and measuring emission at 496 nm as described (37).
Fatty acid oxidation flux analysis
Whole-cell fatty acid oxidation was measured by release of 3H2O from [9,10-3H] oleate (54. 5 Ci/mmol; Perkin Elmer, Waltham, MA) as previously described (38,39). Briefly, 400 000 cells were plated per well in 6-well plates and grown for 24 h in DMEM with 10% fetal bovine serum. Cells were then transfected with hMCAD or eGFP mRNA as described above. After 48 h of transfection, cells were washed once with PBS and then incubated with 0.34 μCi [9,10-3H] oleate in 0.1 mM of oleate prepared in 0.5 ml glucose-free DMEM with 0.1 mM L-carnitine and 2 mg/ml α-cyclodextrin for 2 h at 39°C. Fatty acids were solubilized with α-cyclodextrin. After incubation, 3H2O released was separated from the oleate on a column containing 1 ml of anion exchange resin (AG 1 × 8, acetate, 100–200 Mesh, Bio-Rad, Richmond, CA) prepared in water. After the incubation medium passed through the column, the plate was washed with 1 ml of water that was also transferred to the column. The resin was then washed twice with 0.5 ml of water. All eluates were collected in a scintillation vial and mixed with 10 ml of scintillation fluid (Eco-lite, MP), followed by counting in a Beckman scintillation counter in the tritium window. Assays were performed in triplicate with triplicate blanks (cell free wells). Standards contained a 10 μl ‘hot’ aliquot of the incubation mix with 2.99 ml of deionized water and 10 ml of scintillation fluid.
Electrophoresis and western blotting
Cell lysates and liver homogenates were prepared as above and used for western blotting as previously described (38,39). Briefly, 10–50 μg of protein were loaded onto SDS-PAGE gel (Bio-Rad). Following electrophoresis, separated proteins were transferred to a nitrocellulose membrane. MCAD antigen was detected using rabbit anti-MCAD polyclonal antibody (Thermo Fisher Scientific, Waltham, MA), at a 1:1000 dilution followed by a 1:3000 dilution of goat anti-rabbit IgG-HRP (Bio-Rad). Mouse anti-GAPDH monoclonal antibody (1:10 000) (Abcam, Cambridge, MA) and rabbit anti-VLCAD antibody (1:1000, Vockley laboratory) were used to verify equal loading. Blots were visualized with chemiluminescence with an ECL substrate (Bio-Rad). Additionally, samples were separated by polyacrylamine blue native gel electrophoresis and stained for protein with Coomasie blue dye or in situ enzymatic enzyme assays (40,41).
Immunofluorescence microscopy
Fibroblasts and MEFs were seeded at a density of 5 × 104 cells/ml on tissue culture–treated glass cover slips and allowed to grow overnight at 37°C in a 5% CO2, 95% humidity incubator. Cells were then fixed in 4% paraformaldehyde for 10 min followed by 0.1% Triton X100 cell permeabilization and further blocked after brief washings in 5% donkey serum (Jackson ImmunoResearch, West Grove, PA) for 1 h on ice. This was followed by double primary antibody incubation with (1) anti-MCAD antibody (Vockley laboratory, 1:1000) and (2) anti-cytochrome c oxidase subunit 1 antibody (MTCO1, Abcam, Cambridge, MA, 1:100) at 4°C overnight. After brief washing, cells were further incubated with donkey anti-rabbit secondary antibody Alexa Fluor 488 (Invitrogen, Grand Island, NY) for MCAD and donkey anti-mouse secondary antibody Alexa Fluor 555 (Invitrogen) for MTCO1. Nuclei were counterstained with DAPI (Invitrogen). Mitotracker was used for mitochondrial stain in HeLa cells. The cover slips were then mounted using mounting media before imaging. All the images were taken using a Zeiss LSM710 confocal microscopy.
Cold tolerance
Eight- to ten-week-old male mice were used in this study. Acadm−/− mice were administered an i.v. dose of either formulated LNP-MCAD (1.0 mg/kg) or eGFP mRNA (1.0 mg/kg) (Moderna, Inc.) on day 0, day 4 and day 8 for a total of three times. After final treatment, mice were fasted for 18 h and then fed orally 2,6-dimethylheptanoinc acid (dMC7) oil one time at 4.2 mg/g body weight before being placed in a cold environment (8°C). The corresponding WT mice were administered an i.v. dose of saline for three times on same days and then fasted and fed triheptanoin oil before being placed in a cold environment.
Cold tolerance was accessed by individually housing mice at 8°C. Rectal temperature was measured prior to exposure to the cold and then hourly after cold using a Thermalert TH-5 thermometer (Physitemp, Clifton, NJ). When rectal temperature dropped below 25°C or after 3 h (whichever occurred first), mice were euthanized by O2/CO2 inhalation. Mouse liver, heart and skeletal muscle tissue samples were then collected. Blood was drawn via cardiac puncture. Glucose and lactate levels in blood were also measured before and after the cold challenge.
Acylcarnitine profile assays in fibroblasts and mouse blood
Fibroblasts were grown with unlabeled palmitic acid for the acylcarnitine assay as described with minor modifications (42). Cells were seeded at 400 000 per well in duplicate in six-well plates and incubated for 24 h in complete DMEM supplemented with 10% fetal calf serum (FCS), 2 mM glutamine and 1% penicillin/streptomycin at 37°C in 5% CO2. The cells were transfected with hMCAD mRNA or eGFP mRNA as a control. Twenty-four hours post transfection, the media was removed, and the cells were washed twice with phosphate-buffered saline (PBS). An aliquot (1 ml) of MEM with 200 μM palmitic acid, 400 μM L-carnitine and 0.4% fatty acid free BSA was added to each well and the fibroblasts incubated for 96 h. After incubation, the media was collected and stored at −80°C. Blood was collected from each mouse after completion of the cold tolerance study, and plasma was obtained by centrifugation at 1500 × g for 15 min at 4°C; the plasma was transferred to an Eppendorf tube, stored at −80°C. Acylcarnitine profiling was adapted from published methods (42–45) with minor modifications. Briefly, aliquots (75 μl of medium or 20 μl of plasma) were mixed with methanol (20 μl) containing isotope labeled carnitine standards and the protein precipitated by addition of absolute ethanol (905 or 960 μl, respectively) and centrifugation (13 000 rpm, 10 min). A portion of the supernatant (50 μl) was dried under a stream of nitrogen gas and the acylcarnitine butyl esters generated by reaction (60°C for 15 min) in 100 μl of 3 N HCl in butanol. Dried residues were reconstituted in acetonitrile–water (80:20) for flow-injection ESI-MS–MS analysis. Analysis was performed on a triple quadrupole API4000 mass spectrometer (AB Sciex™, Framingham, MA) equipped with an ExionLC™ 100 HPLC system (Shimadzu Scientific Instruments™, Columbia, MD). Acylcarnitine standards were purchased from Amsterdam UMC—VUmc (MB Amsterdam, NL) and Cambridge Isotope Laboratories, Inc. (Andover, MA). Acylcarnitines were measured using multiple reaction monitoring (MRM) for free carnitine (C0, m/z 218 ⇒ m/z 103) and acetylcarnitine (C2, m/z 260 ⇒ m/z 85) and precursor scan for precursor ions (Q1) of acylcarnitines (C3 to C18, scan range m/z 270 to 502) that generated a product ion (Q3) at m/z 85.
Histopathology
Acadm −/− mice were given multiple i.v. injections of LNP-MCAD (1.0 mg/kg) followed by fasting and cold stress. Mice were then examined for liver histological abnormalities. Liver was fixed by immersion in buffered 10% formalin, processed routinely for paraffin sectioning, sectioned at 5 μm and stained with H&E using standard methodology. Another portion of liver was embedded in the O.C.T. compound, frozen and sectioned at 5 μm thick for Oil Red O staining.
Statistical analysis
All assays were performed at least in triplicate. Data are shown as mean ± SD. Statistical analysis was performed with GraphPad software. Analysis of variance was used for comparison of more than two conditions. Statistical comparisons between groups were done with Student’s t-test, with P < 0.05 considered to be statistically significant.
Acknowledgements
We thank Lorna Cropcho, Biochemical Genetics, UPMC Children’s Hospital of Pittsburgh, for both acylcarnitine standards solutions and technical advice and methods transfer to the API4000 mass spectrometer. LC/MS/MS service was performed in collaboration with the Rangos Metabolic Core Facility, University of Pittsburgh.
Conflict of Interest statement. JV received research support from Moderna Therapeutics. K.A.C, S.S., L.M.R., S.H., E.G., C.D., A.H.G. and P.G.V.M. are employees of Moderna Therapeutics and may have received stock in part as compensation.
Contributor Information
Xue-Jun Zhao, Division of Genetic and Genomic Medicine, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, 15224, USA.
Al-Walid Mohsen, Division of Genetic and Genomic Medicine, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, 15224, USA; Department of Human Genetics, University of Pittsburgh, Pittsburgh, PA, 15261, USA.
Stephanie Mihalik, Division of Genetic and Genomic Medicine, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, 15224, USA.
Keaton Solo, Division of Genetic and Genomic Medicine, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, 15224, USA.
Shakuntala Basu, Division of Genetic and Genomic Medicine, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, 15224, USA.
Ermal Aliu, Division of Genetic and Genomic Medicine, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, 15224, USA.
Huifang Shi, Division of Genetic and Genomic Medicine, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, 15224, USA.
Catherine Kochersberger, Division of Genetic and Genomic Medicine, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, 15224, USA.
Anuradha Karunanidhi, Division of Genetic and Genomic Medicine, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, 15224, USA.
Clinton Van’t Land, Division of Genetic and Genomic Medicine, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, 15224, USA.
Kimberly A Coughlan, Moderna Therapeutics, Rare Diseases, Cambridge, MA, 02139, USA.
Summar Siddiqui, Moderna Therapeutics, Rare Diseases, Cambridge, MA, 02139, USA.
Lisa M Rice, Moderna Therapeutics, Rare Diseases, Cambridge, MA, 02139, USA.
Shawn Hillier, Moderna Therapeutics, Rare Diseases, Cambridge, MA, 02139, USA.
Eleonora Guadagnin, Moderna Therapeutics, Rare Diseases, Cambridge, MA, 02139, USA.
Christine DeAntonis, Moderna Therapeutics, Rare Diseases, Cambridge, MA, 02139, USA.
Paloma H Giangrande, Moderna Therapeutics, Rare Diseases, Cambridge, MA, 02139, USA.
Paolo G V Martini, Moderna Therapeutics, Rare Diseases, Cambridge, MA, 02139, USA.
Jerry Vockley, Division of Genetic and Genomic Medicine, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA, 15224, USA; Department of Human Genetics, University of Pittsburgh, Pittsburgh, PA, 15261, USA.
Funding
This work was supported by ModernaRx, Inc. J.V. and A.-W.M. were supported in part by NIH grant R01 DK78755.
Author Contributions
X.J.Z., manuscript drafting, experimental design and execution, mouse experiments, figure preparation; A.-W.M., project direction, personnel assignments, experimental design and manuscript preparation; S.M., experimental design and execution, mice experiments, figures preparation; K.S., experimental and mice experiments; S.B., mice husbandry and experiments; E.A., experimental and mice experiments; H.S., mice husbandry and testing; C.K., experimental and mice experiments; A.K., experimental and manuscript preparation; C.V.L., acylcarnitine MS analyses and manuscript preparation; K.C., experimental design and manuscript editing; S.S., experimental design, execution of preliminary experiments; L.R., experimental design and manuscript editing; S.H., experimental design, execution of preliminary experiments; E.G., manuscript editing; C.D., manuscript editing; P.G., manuscript editing; P.M., program oversight and manuscript editing; J.V., overall project direction, experimental design and manuscript preparation.
Data Availability Statement
Primary data for all experiments described in this manuscript are represented in the figures. Raw data are available for review on request.
References
- 1. Rinaldo, P., Matern, D. and Bennett, M.J. (2002) Fatty acid oxidation disorders. Annu. Rev. Physiol., 64, 477–502. [DOI] [PubMed] [Google Scholar]
- 2. Tolwani, R.J., Hamm, D.A., Tian, L., Sharer, J.D., Vockley, J., Rinaldo, P., Matern, D., Schoeb, T.R. and Wood, P.A. (2005) Medium-chain acy-CoA dehydrogenase deficiency in gene-targeted mice. PLoS Genet., 1, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karaceper, M.D., Khangura, S.D., Wilson, K., Coyle, D., Brownell, M., Davies, C., Dodds, L., Feigenbaum, A., Fell, D.B., Grosse, S.D. et al. (2019) Health services use among children diagnosed with medium-chain acyl-CoA dehydrogenase deficiency through newborn screening: a cohort study in Ontario, Canada. Orphanet J. Rare Dis., 14, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berraondo, P., Martini, P.G.V., Avila, M.A. and Fontanellas, A. (2019) Messenger RNA therapy for rare genetic metabolic diseases. Gut, 68, 1323–1330. [DOI] [PubMed] [Google Scholar]
- 5. Martini, P.G.V. and Guey, L.T. (2019) A new era for rare genetic diseases: messenger RNA therapy. Hum. Gene Ther., 30, 1180–1189. [DOI] [PubMed] [Google Scholar]
- 6. Pardi, N., Tuyishime, S., Muramatsu, H., Kariko, K., Mui, B.L., Tam, Y.K., Madden, T.D., Hope, M.J. and Weissman, D. (2015) Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. JOCR., 217, 345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pardi, N., Secreto, A.J., Shan, X., Debonera, F., Glover, J., Yi, Y., Muramatsu, H., Ni, H., Mui, B.L., Tam, Y.K. et al. (2017) Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun., 8, 14630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kauffman, K.J., Dorkin, J.R., Yang, J.H., Heartlein, M.W., DeRosa, F., Mir, F.F., Fenton, O.S. and Anderson, D.G. (2015) Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano Lett., 15, 7300–7306. [DOI] [PubMed] [Google Scholar]
- 9. Kauffman, K.J., Mir, F.F., Jhunjhunwala, S., Kaczmarek, J.C., Hurtado, J.E., Yang, J.H., Webber, M.J., Kowalski, P.S., Heartlein, M.W., DeRosa, F. et al. (2016) Efficacy and immunogenicity of unmodified and pseudouridine-modified mRNA delivered systemically with lipid nanoparticles in vivo. Biomaterials, 109, 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramaswamy, S., Tonnu, N., Tachikawa, K., Limphong, P., Vega, J.B., Karmali, P.P., Chivukula, P. and Verma, I.M. (2017) Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. U. S. A., 114, E1941–E1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. An, D., Frassetto, A., Jacquinet, E., Eybye, M., Milano, J., DeAntonis, C., Nguyen, V., Laureano, R., Milton, J., Sabnis, S., Lukacs, C.M. and Guey, L.T. (2019) Long-term efficacy and safety of mRNA therapy in two murine models of methylmalonic acidemia. E. Bio. Medicine., 45, 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang, L., Berraondo, P., Jericó, D., Guey, L.T., Sampedro, A., Frassetto, A., Benenato, K.E., Burke, K., Santamaría, E., Alegre, M. et al. (2018) Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat. Med., 24, 1899–1909. [DOI] [PubMed] [Google Scholar]
- 13. Pastor, F., Berraondo, P., Etxeberria, I., Frederick, J., Sahin, U., Gilboa, E. and Melero, I. (2018) An RNA toolbox for cancer immunotherapy. Nat. Rev. Drug Discov., 17, 751–767. [DOI] [PubMed] [Google Scholar]
- 14. Hewitt, S.L., Bai, A., Bailey, D., Ichikawa, K., Zielinski, J., Karp, R., Apte, A., Arnold, K., Zacharek, S.J., Iliou, M.S. et al. (2019) Durable anticancer immunity from intratumoral administration of IL-23, IL-36 gamma, and OX40L mRNAs. Sci. Transl. Med., 11, 477. [DOI] [PubMed] [Google Scholar]
- 15. Gan, L.M., Lagerström-Fermér, M., Carlsson, L.G., Arfvidsson, C., Egnell, A.C., Rudvik, A., Kjaer, M., Collén, A., Thompson, J.D., Joyal, J. et al. (2019) Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat. Commun., 10, 871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sahin, U., Karikó, K. and Türeci, Ö. (2014) mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov., 13, 759–780. [DOI] [PubMed] [Google Scholar]
- 17. Zhao, X.J., Mohsen, A.W., Mihalik, S., Solo, K., Aliu, E., Shi, H., Basu, S., Kochersperger, C., Van’t Land, C., Karunanidhi, A. et al. (2023) Synthetic mRNA rescues very long-chain acyl-CoA dehydrogenase deficiency in patient fibroblasts and a murine model. Mol. Genet. Metab., 138, 106982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cox, K.B., Hamm, D.A., Milington, D.D., Matern, D., Vockley, J., Rinaldo, P., Pinkert, C.A., Rhead, W.J., Lindsey, J.R. and Wood, P.A. (2001) Gestational, pathologic and biochemical differences between very long-chain acyl-CoA dehydrogenase deficiency and long-chain acyl-CoA dehydrogenase deficiency in the mouse. Hum. Mol. Genet., 19, 2069–2077. [DOI] [PubMed] [Google Scholar]
- 19. Yusupov, R., Finegold, D.N., Naylor, E.W., Sahai, I., Waisbren, S. and Levy, H.L. (2010) Sudden death in medium chain acyl-coenzyme a dehydrogenase deficiency (MCADD) despite newborn screening. Mol. Genet. Metab., 101, 33–39 PMC 20580581. [DOI] [PubMed] [Google Scholar]
- 20. Wilson, C.J., Champion, M.P., Collins, J.E., Clayton, P.T. and Leonard, J.V. (1999) Outcome of medium chain acyl-CoA dehydrogenase deficiency after diagnosis. Arch. Dis. Child., 80, 459–462 PMC 10208954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vockley, J., Singh, R.H. and Whiteman, D.A. (2002) Diagnosis and management of defects of mitochondrial beta-oxidation. Curr. Opin. Clin. Nutr. Metab., 5, 601–609 PMC 12394635. [DOI] [PubMed] [Google Scholar]
- 22. Randall, M., Rolf, C., Gibson, S.M., Hall, P.L., Rinaldo, P. and Davis, G.J. (2015) Medium-chain acyl-CoA dehydrogenase deficiency in adulthood: a potential diagnosis in a patient with mental status changes suspected of drug toxicity. J. Forensic Sci., 60, 1101–1103 PMC 26223762. [DOI] [PubMed] [Google Scholar]
- 23. Wajner, M. and Amaral, A.U. (2016) Mitochondrial dysfunction in fatty acid oxidation disorders: insights from human and animal studies. Biosci. Rep., 36, e00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cecatto, C., Amaral, A.U., Roginski, A.C., Castilho, R.F. and Wajner, M. (2020) Impairment of mitochondrial bioenergetics and permeability transition induction caused by major long-chain fatty acids accumulating in VLCAD deficiency in skeletal muscle as potential pathomechanisms of myopathy. Toxicol. in Vitro, 62, 104665. [DOI] [PubMed] [Google Scholar]
- 25. Boss, O., Muzzin, P. and Giacobino, J.P. (1998) The uncoupling proteins, a review. Eur. J. Endocrinol., 139, 1–9. [DOI] [PubMed] [Google Scholar]
- 26. Schuler, A.M. and Wood, P.A. (2002) Mouse models for disorders of mitochondrial fatty acid beta-oxidation. ILAR J., 43, 57–65. [DOI] [PubMed] [Google Scholar]
- 27. Guerra, C., Koza, R.A., Walsh, K., Kurtz, D.M., Wood, P.A. and Kozak, L.P. (1998) Abnormal nonshivering thermogenesis in mice with inherited defects of fatty acid oxidation. J. Clin. Invest., 102, 1724–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Enerback, S., Jacobsson, A., Simpson, E.M., Guerra, C., Yamashita, H., Harper, M.E. and Kozak, L.P. (1997) Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature, 387, 90–94. [DOI] [PubMed] [Google Scholar]
- 29. Hirschey, M.D., Shimazu, T., Goetzman, E., Jing, E., Schwer, B., Lombard, D.B., Grueter, C.A., Harris, C., Biddinger, S., IIkayeva, O.R. et al. (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature, 464, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cole, L.K., Mejia, E.M., Vandel, M., Sparagna, G.C., Claypool, S.M., Dyck-Chan, L., Klein, J. and Hatch, G.M. (2016) Impaired cardiolipin biosynthesis prevents hepatic steatosis and diet-induced obesity. Diabetes, 65, 3289–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Primassin, S., ter Veld, F., Mayatepek, E. and Spiekerkoetter, U. (2008) Carnitine supplementation induces acylcarnitine production in tissues of very long-chain acyl-CoA dehydrogenase-deficient mice, without replenishing low free carnitine. Pediatr. Res., 63, 632–637. [DOI] [PubMed] [Google Scholar]
- 32. Bonnet, D., Martin, D., de Lonlay, P., Villain, E., Jouvet, P., Rabier, D., Brivet, M. and Saudubray, J.M. (1999) Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation, 100, 2248–2253. [DOI] [PubMed] [Google Scholar]
- 33. Ciapaite, J., Bakker, S.J., Diamant, M., van Eikenforst, G., Heine, R.J., Westerhoff, H.V. and Karp, K. (2006) Metabolic control of mitochondrial properties by adenine nucleotide translocator determines palmitoyl-CoA effects. Implications for a mechanism linking obesity and type 2 diabetes. FEBS J., 273, 5288–5302. [DOI] [PubMed] [Google Scholar]
- 34. Nelson, J. et al. (2020) Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci. Adv., 6, eaaz6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sabnis, S., Kumarasinghe, E.S., Salerno, T., Mihai, C., Ketova, T., Senn, J.J., Lynn, A., Bulychev, A., McFadyen, I., Chan, J. et al. (2018) A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther., 26, 1509–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hale, D.E., Batshaw, M.L., Coates, P.M., Frerman, F.E., Goodman, S.I., Singh, I. and Stanley, C.A. (1985) Long-chain acyl coenzyme a dehydrogenase deficiency: an inherited cause of nonketotic hypoglycemia. Pediatr. Res., 19, 666–671. [DOI] [PubMed] [Google Scholar]
- 37. Vockley, J., Mohsen, A.W., Binzak, B., Willard, J. and Fauq, A. (2000) Mammalian branched-chain acyl-CoA dehydrogenases: molecular cloning and characterization of recombinant enzymes. Methods Enzymol., 324, 241–258. [DOI] [PubMed] [Google Scholar]
- 38. Leipnitz, G., Mohsen, A.-W., Karunanidhi, A., Seminotti, B., Roginskaya, V.Y., Markantone, D.M., Grings, M., Mihalik, S.J., Wipf, P., Van Houten, B. et al. (2018) Evaluation of mitochondrial bioenergetics, dynamics, endoplasmic reticulum-mitochondria crosstalk, and reactive oxygen species in fibroblasts from patients with complex I deficiency. Sci. Rep., 8, 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seminotti, B., Leipnitz, G., Karunanidhi, A., Kochersperger, C., Roginskaya, V.Y., Basu, S., Wang, Y., Wipf, P., Houten, B.V., Mohsen, A.-W. et al. (2019) Mitochondrial energetics is impaired in very long-chain acyl-CoA dehydrogenase deficiency and can be rescued by treatment with mitochondria-targeted electron scavengers. Hum. Mol. Genet., 28, 928–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang, Y., Mohsen, A.W., Mihalik, S.J., Goetzman, E.S. and Vockley, J. (2010) Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J. of Biol Chem., 285, 29834–29841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang, Y., Palmfeldt, J., Gregersen, N., Makhov, A.M., Conway, J.F., Wang, M., McCalley, S.P., Basu, S., Alharbi, H., St Croix, C. et al. (2019) Mitochondrial fatty acid oxidation and the electron transport chain comprise a multifunctional mitochondrial protein complex. J. of Biol. Chem., 294, 12380–12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Okun, J.G., Kölker, S., Schulze, A., Kohlmüller, D., Olgemöller, K., Lindner, M., Hoffmann, G.F., Wanders, R.J. and Mayatepek, E. (2002) A method for quantitative acylcarnitine profiling in human skin fibroblasts using unlabelled palmitic acid: diagnosis of fatty acid oxidation disorders and differentiation between biochemical phenotypes of MCAD deficiency. Biochim. Biophys. Acta, 1584, 91–98. [DOI] [PubMed] [Google Scholar]
- 43. Smith, E.H. and Matern, D. (2010) Acylcarnitine analysis by tandem mass spectrometry. Curr. Protoc. Hum. Genet. Chapter 17, Unit 17.8.1-17.8.20, 64(1), 17.8.1–17.8.20. [DOI] [PubMed] [Google Scholar]
- 44. Shen, J.J., Matern, D., Millington, D.S., Hillman, S., Feezor, M.D., Bennett, M.J. and Van Hove, J.L.K. (2000) Acylcarnitines in fibroblasts of patients with long chain 3-hydroxyacyl-CoA dehydrogenase deficiency and other fatty acid oxidation disorders. J. Inherit. Metab. Dis., 23, 27–44. [DOI] [PubMed] [Google Scholar]
- 45. Rinaldo, P., Cowan, T.M. and Matern, D. (2008) Acylcarnitine profile analysis. Genet. Med., 10, 151–156. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Primary data for all experiments described in this manuscript are represented in the figures. Raw data are available for review on request.