

Abstract
FOXJ1 is expressed in ciliated cells of the airways, testis, oviduct, central nervous system and the embryonic left–right organizer. Ablation or targeted mutation of Foxj1 in mice, zebrafish and frogs results in loss of ciliary motility and/or reduced length and number of motile cilia, affecting the establishment of the left–right axis. In humans, heterozygous pathogenic variants in FOXJ1 cause ciliopathy leading to situs inversus, obstructive hydrocephalus and chronic airway disease. Here, we report a novel truncating FOXJ1 variant (c.784_799dup; p.Glu267Glyfs*12) identified by clinical exome sequencing from a patient with isolated congenital heart defects (CHD) which included atrial and ventricular septal defects, double outlet right ventricle (DORV) and transposition of the great arteries. Functional experiments show that FOXJ1 c.784_799dup; p.Glu267Glyfs*12, unlike FOXJ1, fails to induce ectopic cilia in frog epidermis in vivo or to activate the ADGB promoter, a downstream target of FOXJ1 in cilia, in transactivation assays in vitro. Variant analysis of patients with heterotaxy or heterotaxy-related CHD indicates that pathogenic variants in FOXJ1 are an infrequent cause of heterotaxy. Finally, we characterize embryonic-stage CHD in Foxj1 loss-of-function mice, demonstrating randomized heart looping. Abnormal heart looping includes reversed looping (dextrocardia), ventral looping and no looping/single ventricle hearts. Complex CHDs revealed by histological analysis include atrioventricular septal defects, DORV, single ventricle defects as well as abnormal position of the great arteries. These results indicate that pathogenic variants in FOXJ1 can cause isolated CHD.
Introduction
Congenital heart defects (CHD) are currently impacting nearly 11 per 1000 newborns (1). Among the genetic causes of CHD are aneuploidy, structural variation such as copy number variants and DNA sequence alterations such as single-nucleotide variants or insertions and deletions (indels) (2,3). Currently, alterations of a single gene are identified in approximately 3–10% of CHD cases (1,3). Through next-generation sequencing, novel genetic variants have been identified in isolated cases of CHD and many of these genes are involved in heart development and function (1,3). In addition, research exome sequencing of a CHD cohort of ~2900 probands uncovered that 8% of the cases are related to de novo variants whereas nearly 2% of the cases are due to rare inherited variants (4).
Mouse has frequently been used as a model organism to investigate causes and mechanisms of CHD. A global genetic analysis from ethylnitrosourea mutagenized fetal mice with various CHD phenotypes revealed that 34 of 61 CHD genes recovered with pathogenic variants were cilia-related genes, of which 22 and 12 were non-motile and motile cilia genes, respectively. Interestingly, only half of the non-motile cilia genes were related to laterality defects while all motile cilia genes were involved in laterality (5,6).
Cilia are cell-surface microtubule-formed organelles that are important for the movement of extracellular fluid (motile cilia) as well as for sensing changes in the extracellular environment (non-motile cilia) (7). Cilia are involved in the proper formation of the left–right body asymmetry during embryogenesis. Cells in the left–right organizer of mice contain motile ciliated cells in a teardrop shaped structure surrounded by immotile ciliated cells. The immotile cilia sense the leftward fluid flow produced by the clockwise rotation of the motile cilia, activating the NODAL signaling cascade and establishing and enhancing left–right asymmetry (7,8). Defective cilia lead to heterogeneous phenotypes including hydrocephalus, hearing loss, organ laterality defects and heterotaxy, infertility, chronic respiratory illnesses and retinal diseases, among others (6).
FOXJ1 (previously known as hepatocyte nuclear factor-3/forkhead homolog 4, HFH-4) is a 3-exon transcription factor expressed in motile ciliated cells of the respiratory tract, brain, reproductive tract (testis, oviduct and endocervix) and the embryonic left–right organizer and it is considered the master regulator of motile ciliogenesis (9–14). The establishment of left–right asymmetry in frogs and zebrafish is altered by the inhibition of Foxj1 expression (or the homolog foxj1a) which results in a reduced number and shorter cilia or lack of motile cilia in the gastrocoel roof plate and Kupffer’s vesicle, respectively (15,16). Foxj1 deletion in mice causes an abnormal position of the cilia in the pit cells of the left–right organizer, decreased cilia length as well as lack of motility, preventing the generation of leftward fluid flow and subsequent activation of NODAL signaling (7,8,17). Hydrocephalus, situs inversus totalis (organs, including the heart, are in a mirror-image position), situs inversus abdominalis (the abdominal viscera is reversed but the heart is in a normal position), isolated dextrocardia and transposition of the great arteries (TGA) are the most common phenotypes observed in Foxj1 null mice due to defective cilia and establishment of the left–right axis (13,14,18). Individuals carrying de novo or novel heterozygous pathogenic variants in FOXJ1 suffer from motile ciliopathies including obstructive hydrocephalus and chronic airway disease; however, among them, those carrying the FOXJ1 c.868_871dup; p.Thr291Lysfs*12, FOXJ1 c.826C>T; p.Gln276* and FOXJ1 c.939delC; p.Ile314Serfs*19 pathogenic variants also display situs inversus (19).
Here, we show that clinical exome sequencing uncovered a novel FOXJ1 indel (c.784_799dup; p.Glu267Glyfs*12) in exon 3 from a patient with isolated and complex CHD. This indel results in a frameshift and incorporation of an early stop codon and was interpreted as a variant of uncertain significance (VUS) by the reference laboratory on clinical exome. To investigate the functional impact of this variant, we analyzed cilia in the deceased patient’s lung tissue via immunofluorescence and performed in vivo and in vitro functional testing of the novel FOXJ1 variant revealing that it failed to induce ectopic cilia formation in the epidermis of Xenopus at developmental stage 26 or to activate the ADGB promoter, a downstream target of FOXJ1 also expressed in cilia. In addition, we investigate rare, predicted damaging variants in FOXJ1 in patients with heterotaxy, a laterality disorder where abnormal position of multiple organs including the heart is a rare cause of CHD, as determined by variant analysis in two separate cohorts of CHD patients and patients with heterotaxy and related CHD. Finally, we analyze embryonic hearts from Foxj1 loss-of-function mice to identify the diverse phenotypes of cardiac looping and heart defects.
Results
Identification of a novel indel in FOXJ1 in a patient with isolated CHD
Clinical exome sequencing identified a novel heterozygous duplication resulting in a frameshift in FOXJ1 (NM_001454.4) (c.784_799dup; p.Glu267Glyfs*12) from a 1-month-old male proband with complex CHD. Mixed peaks caused by the duplication can be observed in the Sanger sequencing electropherogram used to confirm the exome findings (Fig. 1A). The FOXJ1 variant was not present in paternal DNA; however, the maternal DNA was not available to determine de novo status of the variant. The novel FOXJ1 c.784_799dup; p.Glu267Glyfs*12 variant localizes in exon 3, relatively close to the forkhead DNA transcription binding domain (Fig. 1B), and it is predicted to result in a premature termination codon. The clinical laboratory interpreted the finding as a VUS. The patient subsequently died as a result of complex CHD and multiorgan failure. According to the autopsy report, the proband’s body and facial features developed normally. Upon internal examination of the organs, the lungs showed the usual shape and lobation. The heart, however, displayed several defects including an atrial septal defect (ASD), dilated posterior left ventricle and a smaller anterior right ventricle with trabecular hypertrophy, double outlet right ventricle (DORV), dextro-transposition of the great arteries (D-TGA) with a large malalignment ventricular septal defect (VSD) and mild left ventricular outflow obstruction. This FOXJ1 c.784_799dup; p.Glu267Glyfs*12 variant is associated with isolated CHD as no left–right asymmetry anomaly, hydrocephalus or heterotaxy was identified, a novel finding indicating the need for additional analysis of the functional impact of the variant.
Figure 1.

Localization of the FOXJ1 novel pathogenic variant (c.784_799dup) from an affected individual with severe congenital heart defects. (A) Sanger sequencing electropherogram showing the FOXJ1 exon-3 region where part of the c.784_799dup sequence is located. Below the electropherogram is the sequence from the wild-type and mutated alleles showing the duplicated sequence in red. (B) Schematic overview of the FOXJ1 gene consisting of two coding exons (gray boxes) in which the forkhead DNA-binding domain is located (black dotted area). The FOXJ1 c.784_799dup variant is localized downstream of the forkhead DNA-binding domain. (C) Representative images of lung sections from an unaffected pediatric control (upper panels) and from the proband affected by the FOXJ1 c.784_799dup; p.Glu267Glyfs*12 variant (mid and lower panels) immunostained against acetylated α-TUBULIN (green). Nuclei were stained with DAPI (blue). Center and right panels are higher magnifications of the white boxed areas. Scale bars from left to right are 500, 50 and 20 μm.
Aberrant cilia detected in the conducting zone of the respiratory tract of the affected proband
In the conducting zone of the respiratory tract, ciliated cells are localized in the trachea, bronchi, bronchioles and terminal bronchioles, and their main function is to drive the movement of mucous across the airway tract (20). We performed immunostaining on lung sections from an unaffected control as well as from the affected proband to determine any deleterious effect that the novel FOXJ1 variant might have on motile cilia. In the unaffected control, α-TUBULIN immunostaining demonstrated consistent dense cilia in the respiratory epithelium, displaying the typical hair-like structure projecting from the cell surface (Fig. 1C, upper panels). In contrast, cilia were notably sparse in areas of the respiratory epithelium of the affected proband (Fig. 1C, mid and lower panels). Thus, the FOXJ1 variant c.784_799dup; p.Glu267Glyfs*12 was associated with disrupted cilia formation on the respiratory epithelium.
Ectopic cilia development is not induced in Xenopus by the novel FOXJ1 variant c.784_799dup; p.Glu267Glyfs*12
Previous experiments in Xenopus and zebrafish demonstrated that misexpression of the master ciliogenesis transcription factor Foxj1 or the homolog foxj1a promotes ectopic cilia formation (15,16). To test the functionality of the FOXJ1 variant, we used the same experimental approach in which 2-cell stage Xenopus laevis embryos were injected with either FOXJ1 or the mutant FOXJ1 c.784_799dup mRNA, and ectopic cilia formation examined in stage 26 embryos. As expected, in non-injected controls, single inner epidermal cells containing motile cilia clusters were uniformly distributed among intercalating non-ciliated cells and epidermal outer cells (Fig. 2A) (15,21). Upon injection of FOXJ1 mRNA, single cilia developed ectopically on the skin of frog embryos (Fig. 2B). Unlike the FOXJ1 mRNA, no ectopic single cilia were observed on the skin of embryos injected with the FOXJ1 mutant mRNA (Fig. 2C), suggesting that FOXJ1 c.784_799dup is unable to activate the ciliogenic program. Real-time PCR experiments confirmed the presence of FOXJ1 mRNA as well as the mutant FOXJ1 c.784_799dup mRNA in stage-26 frog embryos injected with either RNA at the 2-cell stage (Fig. 2D), indicating the stability of the transcripts during these experiments.
Figure 2.
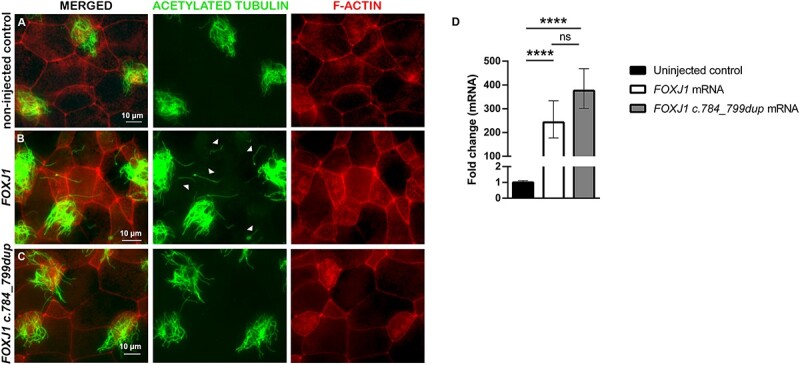
The FOXJ1 c.784_799dup mRNA failed to generate ectopic single cilia on Xenopus skin cells. Representative images of Xenopus skin sections from (A) non-injected control embryos, and embryos either injected at the 2-cell stage with (B) FOXJ1 mRNA or with (C) the novel FOXJ1 variant c.784_799dup mRNA. At developmental stage 26, embryos (n = 16 non-injected controls, n = 15 injected with FOXJ1 mRNA and n = 6 receiving the FOXJ1 mRNA variant) were fixed and stained with antibodies against acetylated tubulin (green) and F-ACTIN labeled with phalloidin (red). Arrowheads in (B) point to ectopic single cilia. Scale bars represent 10 μm. (D) Analysis of FOXJ1 and FOXJ1 c.784_799dup mRNA expression of pooled Xenopus embryos at stage 26 via qPCR. The results are presented as fold change values relative to the non-injected control and statistical analysis was performed on average ΔCt values. P-values were derived from a one-way ANOVA followed by the Tukey’s test for multiple comparisons from n = 8 non-injected controls (black bar); n = 4 FOXJ1 mRNA (white bar) and n = 4 FOXJ1 c.784_799dup mRNA (gray bar) treated embryos. ****P < 0.001; ns, no significant.
FOXJ1 but not FOXJ1 c.784_799dup; p.Glu267Glyfs*12 activates the proximal ADGB promoter
ADGB belongs to the globin superfamily, and it is also expressed in tissues with motile cilia (10,22). In vitro experiments have shown that overexpression of ADGB promotes cilia formation. Moreover, binding of FOXJ1 to the endogenous ADGB promoter as well as an increase of the ADGB promoter-driven luciferase activity by FOXJ1 were recently demonstrated by chromatin immunoprecipitation and reporter gene assays, respectively (10). To investigate whether the novel FOXJ1 pathogenic variant can activate the ADGB promoter, we performed a luciferase assay using the closest segment to the ADGB transcriptional start site (−1 to −140 bp), which is the highest responsive region of the promoter (10). The reporter gene assay revealed that while the ABGD promoter-driven luciferase activity was significantly stimulated by FOXJ1 (P ≤ 0.0001), stimulation of the ADGB promoter by the novel FOXJ1 variant was negligible (Fig. 3A). Western blot analysis showed the expression of FOXJ1 and the novel FOXJ1 truncated variant (Supplementary Material, Fig. S1). Since RFX2 is another transcription factor important for ciliogenesis and its association with FOXJ1 enhances the ADGB promoter activity (10,23,24), we then tested the additive effect of RFX2 and the novel FOXJ1 variant on the ABGD promoter-driven luciferase activity in the presence of 3′-AE1 (ADGB Enhancer1), one of the two critical enhancers localized right after ADGB exon 36 (10). Our results showed a significant combined effect of RFX2 and FOXJ1 on the ABGD promoter-driven luciferase activity (P ≤ 0.0001), significantly greater than the transactivation produced by FOXJ1 alone (P = 0.0070), as expected. In contrast, and despite the presence of RFX2, there was not a significant synergistic effect in the activation of the ADGB promoter by the FOXJ1 variant form (Fig. 3B). Also, it is noticeable the significant difference (P ≤ 0.0001) between FOXJ1 and the novel FOXJ1 damaging variant in the increase of the ABGD promoter-driven luciferase activity in the presence of RFX2. Taken together, the c.784_799dup; p.Glu267Glyfs*12 variant alters the ability of FOXJ1 to activate the ADGB promoter regardless of the presence of the enhancer/stabilizer RFX2.
Figure 3.

Negligible activation of the ADGB promoter by the FOXJ1 c.784_799dup; p.Glu267Glyfs*12 novel pathogenic variant. Activity of the pGL3B-AP140-1 (A) and pGL3B-AP-3′-AE1 (B) luciferase reporter assays in HEK293 cells. (A) Cells were transfected in the absence (empty; black bar) or presence of FOXJ1 (white bar) or with FOXJ1 c.784_799dup (gray bar) expression plasmids. (B) Cells were transfected with FOXJ1 or with FOXJ1 c.784_799dup expression plasmids in the absence (empty) or presence of HA-RFX2 expression plasmid. Empty: transfection with pHM6. Results are presented as the mean of relative luminescence units (RLU; firefly/Renilla) ± SEM from at least n = 3 independent experiments. The data were analyzed using one-way ANOVA followed by Tukey’s test for multiple comparisons. SEM, standard error of the mean; ns, no significant; **P = 0.007; ***P = 0.001; ****P ≤ 0.001.
FOXJ1 variants are an uncommon cause of heterotaxy spectrum CHD
Given the novel association of the FOXJ1 c.784_799dup allele with isolated CHD, we interrogated two different cohorts to identify additional potentially damaging FOXJ1 variants in patients with CHD-related heterotaxy or complex CHD. The results from the first cohort of 279 participants with CHD phenotypes associated with left–right defects and heterotaxy whereas the second cohort (n = 711) included individuals with a broader range of CHD (Table 1). After filtering rare variants (minor allele frequency < 0.001), missense FOXJ1 variants were detected in four individuals with isolated dextrocardia or CHD-related heterotaxy; these variants were categorized as benign or likely benign by ACMG interpretation. Similar results were obtained from the second cohort of 711 CHD cases where benign or likely benign FOXJ1 missense variants were detected in five individuals; however, a novel likely pathogenic FOXJ1 variant (c.107T>C; p.Leu36Pro) was identified in one participant (Table 1). Interestingly, the FOXJ1 (c.107T>C; p.Leu36Pro) variant carrier was diagnosed with heterotaxy syndrome (situs inversus totalis with a complex CHD including DORV, VSD, ASD and D-loop TGA among other defects). Overall, mutations in FOXJ1 are an infrequent cause of isolated CHD and heterotaxy based on investigation of the 990 individuals with CHD in these combined cohorts.
Table 1.
Rare heterozygous missense variants in FOXJ1 identified in individuals with congenital heart defect phenotypes
| Case ID | Phenotype | cDNA change | Protein change | gnomAD Genomes AF |
Individual prediction programsa | Ensemble prediction algorithmsa | ACMG interpretation | |
|---|---|---|---|---|---|---|---|---|
| REVEL | CADD | Varsome | ||||||
| 23519 | Dextrocardia | c.1085C>T | p.Ser362Leu | 0.000669294 | −/−/−/−/+/− | 0.30 | 16.9 | Benign |
| HTX-11-037 | Malrotation of the gut, biliary atresia, gall bladder abnormalities, polysplenia, SVC abnormality, AVC defect, ASD, aortic coarctation | c.782C>T | p.Ala261Val | 0.000138892 | −/−/−/−/+/− | 0.15 | 16.0 | Benign |
| HTX-11-052 | Abdominal situs inversus, dextrocardia, duodenal atresia, AVC defect, VSD, DORV, pulmonic atresia | c.322C>T | p.Pro108Ser | 2.92185E-05 | −/−/+/−/+/+ | 0.29 | 22.4 | Likely benign |
| HTX-11-131 | Abdominal situs inversus, dextrocardia, sacral agenesis, TAPVR, AVC defect | c.871A>T | p.Thr291Ser | 0.000503583 | −/−/+/−/+/+ | 0.16 | 16.7 | Benign |
| PT_X7GE7E9N | Myocardial dysfunction, coarctation of the aorta | c.1111G>A | p.Asp371Asn | 0.0000534 | −/−/+/+/+ | 0.46 | 23.2 | Benign |
| PT_N7MM7C1H | Mitral atresia, unobstructed totally anomalous pulmonary venous return, pulmonary stenosis (subvalvar), mitral atresia, HTX, pulmonary stenosis (valvar) | c.842G>A | p.Arg281Gln | 0.00000854 | +/−/+/+/+ | 0.29 | 21.3 | VUS |
| PT_EZ5RMAEC | D-loop TGA with intact ventricular septum, right aortic arch with mirror-image branching pattern, circumflex from right coronary in d-loop TGA | c.677G>A | p.Arg226His | 0.0000839 | −/−/+/−/+ | 0.28 | 22.3 | Benign |
| PT_2X1FM3E5 | Hypoplastic left heart syndrome, ASD (secundum) | c.611G>A | p.Arg204Gln | 0.000129 | −/+/+/−/+ | 0.52 | 24.5 | Benign |
| PT_M7E5VB09 | DORV, situs inversus totalis, D-loop TGA, mitral atresia, hypoplastic left ventricle (subnormal cavity volume), VSD, pulmonary atresia congenital, ASD, HTX | c.107T>C | p.Leu36Pro | NA | +/−/+/+/+ | 0.96 | 34.0 | Likely pathogenic |
| PT_6JZBA60G | Right aortic arch, anomalous origin of right coronary artery from pulmonary artery, tetralogy of Fallot | c.7G>C | p.Glu3Gln | NA | −/−/+/−/+ | 0.27 | 23.7 | VUS |
aSIFT/PolyPhen HVAR/MutationTaster/MutationAssesor/FATHMN MKL Coding.
AVC, atrioventricular canal; SVC, superior vena cava; TAPVR, total anomalous pulmonary venous return; HTX, heterotaxy syndrome; TGA, transposition of the great arteries; HLHS, hypoplastic left heart syndrome; ASD, atrial septal defect; DORV, double outlet right ventricle; VSD, ventricular septal defect; NA, not available.
Looping and structural heart defects, and abnormal position of the great arteries in Foxj1 null embryos
During development, the embryonic heart starts as a linear and symmetric tube localized alongside the ventral midline of the embryo. The tube bends rightwards forming an outer C-shape curvature known as C-loop or dextral loop, followed then by an additional bending adopting a complex helical shape, named the ‘S-shaped’ loop. These looping stages are crucial as they bring sections of the heart tube close together for the definitive positioning of the atrioventricular canal and the common atrium, as well as for the establishment of the normal left–right configuration of the heart ventricles (25). Heart looping is an important step during cardiac morphogenesis since many CHD cases are caused by abnormal looping (25,26). Defects in the left–right axis including sinistral or mirror-image hearts (dextrocardia) have been reported in Foxj1 null embryos (13,14,27). However, it is unknown whether Foxj1 null hearts display another type of looping defect as well as structural defects and abnormal positioning of the great arteries. Examination of Foxj1 null embryonic 14.5–17.5 hearts revealed more complex looping defects (Fig. 4 and Table 2). Out of 82 Foxj1 null embryos analyzed; 48 embryos exhibited normal or dextral looped hearts (levocardia; 58.5%) (Fig. 4B) and 34 embryos (41.5%) presented clear heart looping defects (Fig. 4C–F). Heart looping defects were classified according to Ware et al. (28). Phenotypic characterization of heart looping defects in Foxj1 null embryos included inverse or sinistral looping (dextrocardia; 24.4%) (Fig. 4C), ventral looping (heart tube loops forward in the frontal plane; 8.5%) (Fig. 4D), a combined ventral/sinistral looping (4.9%) and no looping or single ventricle heart (3.7%) (Fig. 4E and F, respectively). The position of the outflow tract (OFT) was also examined in Foxj1 null embryonic hearts. The OFT in wild-type as well as ~42% of Foxj1 null embryonic hearts is normally positioned on the right side (Fig. 4A and D, Table 3). In addition, the OFT in Foxj1 null hearts was also positioned on the left (Fig 4B, C and E), middle, right middle and left/middle (Fig. 4F and data not shown). Interestingly, abnormal remodeling of the OFT caused by embryonic laterality defects has been linked to unusual positions of the great arteries and some other cardiac malformations (29).
Figure 4.

Looping defects are diverse in Foxj1 null hearts. Representative gross images of (A) wild-type (WT) and (B–F) Foxj1 null embryonic 14.5–17.5 hearts displaying either normal (A, B) or abnormal heart looping (C–F). Hearts with normal or dextral looping (levocardia), in which the heart tube undergoes a rightward curve (A, B). Abnormal looping in Foxj1 null hearts included inverse or sinistral looping (dextrocardia), in which the heart undergoes a leftward curve (C), ventral looping, in which the heart tube loops forward in the frontal plane (D), a combined ventral and sinistral looping (E) and unlooped or single ventricle heart (F). Foxj1 null hearts also displayed abnormal positioning of the outflow tract—left-sided position (B, C and E) or middle position (F) among others. LV, left ventricle; OFT, outflow tract; RV, right ventricle.
Table 2.
Percent of embryonic 14.5–17.5 Foxj1 null hearts with looping defects; n = 34/82
| Heart looping defects in Foxj1 null hearts (%) | ||
|---|---|---|
| 1. Dextral or normal looping (levocardia) | 58.5% | |
| 2. Abnormal looping | 41.5% | |
| 2.1. Sinistral or inverse looping (dextrocardia) | 24.4% | |
| 2.2. Ventral looping | ||
| 2.2.1. Ventral only | 8.5% | |
| 2.2.2. Ventral and sinistral | 4.9% | |
| 2.3. Unlooped or single ventricle | 3.7% | |
Table 3.
Gross anatomic position of the outflow tract (OFT) of Foxj1 null hearts at embryonic days 14.5–17.5
| Position of the OFT | Foxj1 null hearts (n = 81) |
|---|---|
| Right | 41.9% (34) |
| Right/middle | 7.4% (6) |
| Middle | 17.3% (14) |
| Left/middle | 4.9% (4) |
| Left | 28.5% (23) |
Histological analysis of a subset (n = 20) of embryonic Foxj1 null hearts with (n = 17) or without (n = 3) looping defects demonstrated abnormal hearts in all embryos (Supplementary Material, Tables S1–S3). The analyses unveiled a wide range of complex cardiac defects including AVSD (Fig. 5B and Supplementary Material, Table S1), VSD (Fig. 5C and D), atrial septal defects (ASD; Fig. 5D), DORV (Fig. 5E), double inlet left ventricle (DILV; Fig. 5F) as well as atria isomerism (Fig. 5G and Supplementary Material, Table S2), inverted atria (Fig. 5H) or hearts with an abnormal, very enlarged atrium (Fig. 5I). It is important to note that the majority of Foxj1 null hearts examined histologically displayed combined defects. Also, different subtypes of VSDs were observed such as inlet, outlet, perimembranous and muscular.
Figure 5.

Foxj1 null hearts exhibit a broad range of cardiac defects. Transverse histological sections stained with hematoxylin and eosin of (A) wild-type (WT) and (B–I) Foxj1 null embryonic hearts with or without looping defects. A variety of cardiac defects were present in Foxj1 null hearts such as (B) atrioventricular septal defects (AVSD), (C, D) ventricular septal defects (VSD, shown by arrow), (D) atrial septal defects (ASD), (E) double outlet right ventricle (DORV, shown by arrows), (F) double inlet left ventricle (DILV), as well as hearts with (G) atrial isomerism, (H) inverted atria and (I) a very enlarged right atrium. Ao, Aorta; LA, left atrium; LV, left ventricle; PT, pulmonary trunk; RA, right atrium; RV, right ventricle.
Abnormal positions of the great arteries were also revealed by histological analysis in some embryonic Foxj1 null hearts (Fig. 6 and Supplementary Material, Table S3). The normal arrangement of the great arteries, in which the aorta is posterior and to the right of the pulmonary trunk, was observed in wild-type as well as in some Foxj1 null hearts (Fig. 6A and data not shown). Foxj1 null hearts also display aberrant position of the great arteries: the aorta is posterior and to the left of the pulmonary trunk (Fig. 6B), the aorta is anterior and to the right of the pulmonary trunk (Fig. 6C), the aorta is posterior (right behind) of the pulmonary trunk (Fig. 6D) and the aorta is to the right, side by side to the pulmonary trunk (Fig. 6E). Thus, Foxj1 is involved in cardiogenesis and Foxj1 loss-of-function in mice causes a wide range of looping and structural defects as well as mispositioning of the great arteries.
Figure 6.

Abnormal position of the great arteries in Foxj1 null hearts. Transverse histological sections stained with hematoxylin and eosin of (A) wild-type (WT) and Foxj1 null embryonic hearts (B–E). Normal arrangement of the great arteries was observed in WT hearts (A), in which the aorta is posterior and to the right of the pulmonary trunk. Foxj1 null hearts exhibited aberrant position of the great arteries in which the aorta is posterior and to the left of the pulmonary trunk (B), the aorta is anterior and to the right of the pulmonary trunk (C), the aorta is posterior to the pulmonary trunk (D) and the aorta is side by side to the pulmonary trunk (E). A, Anterior; Ao, aorta; L, left; LA, left atrium; P, posterior; PT, pulmonary trunk; R, right; RA, right atrium.
Discussion
The genetic causes of CHD can be diverse and complex with monogenic-CHD cases representing a small percentage of the overall genetic CHD causes (1,3). The genetic causes of isolated CHD are often not identified. However, pathogenic variants affecting single genes that are crucial for heart development and function are known causes of disease in a subset of CHD cases (1,3). In addition, isolated CHD cases are often characterized by incomplete penetrance, with variants in CHD-associated genes inherited from unaffected parents (30). On the other hand, pathogenic variants in motile cilia genes have been shown to be an important cause of isolated laterality defects, heterotaxy, primary cilia dyskinesia (PCD; a disorder characterized by mucociliary clearance defects accompanied in some cases by laterality defects, abnormal sperm motility and sporadic hydrocephalus), or the combination of heterotaxy and PCD (1,31,32).
In contrast to previously reported pathogenic variants in FOXJ1 detected in patients with either isolated hydrocephalus (c.287C>T; p.Thr96Arg, c.826C>T; p.Gln276*, c.967delG; p.Glu323Serfs*10) or with obstructive hydrocephalus along with PCD and situs inversus (c.868_871dup; p.Thr291Lysfs*12, c.826C>T; p.Gln276*, c.939delC; p.Ile314*Sers*19 and c.929_932delACTG; p.Asp310Glyfs*22) (19,33,34), the FOXJ1 c.784_799dup; p.Glu267Glyfs*12 variant described here resulted in an isolated case of complex CHD since no hydrocephalus, left–right asymmetry defect or heterotaxy was present in our affected proband. De novo pathogenic variants in FOXJ1 (c.826C>T; pGln276*, c.929_932delACTG; p.Asp310Glyfs*22) were detected in patients diagnosed with an isolated VSD or ASD but combined with obstructive hydrocephalus, mucociliary clearance disorder in addition to situs inversus for the individual with ASD (19,34). Isolated CHD, particularly AVSD and D-TGA, have been reported in patients with pathogenic variants in cilia genes (35). Interestingly, prediction of de novo variants and rare recessive forms of CHD by a gene-based burden analysis revealed that chromatin-modifying genes, but cilia-related genes, were not significantly enriched for de novo variants (36), suggesting that de novo variants in cilia genes are not a common cause of CHD.
The onset of obstructive hydrocephalus in most FOXJ1-related cases has been detected during the first weeks of life, although case reports have documented it occurring later such as in a 3-month-old infant as well as in a 54-year-old individual (19,34). It is interesting to speculate that some variants may substantially diminish but not abolish ciliary function, resulting in phenotypic variability. The sparse cilia found in the respiratory epithelium of our affected proband suggests the possibility of a later development of a chronic respiratory illness or mucociliary clearance disorder. No investigation of ciliary function was possible before the patient died. We note that a reduced number of motile cilia on respiratory epithelial cells and mislocalized basal bodies were detected in individuals carrying de novo mutations in FOXJ1 (c.901G>T; p.Glu301*, c.868_871dup; p.Thr291Lysfs*12, c.826C>T; p.Gln276*, c.967delG; p.Glu323Serfs*10). As a result, these individuals experienced frequent respiratory tract infections, persistent cough and bronchiectasis (19,34). It is important to point out that our proband was a heterozygous carrier for the FOXJ1 (c.784_799dup; p.Glu267Glyfs*12) variant and thus retained an intact FOXJ1 allele, which could address the formation of some cilia in the respiratory epithelium. In contrast, our experimental models more closely mimic a homozygous individual, where we either tested FOXJ1 or the FOXJ1 variant alone providing more drastic results.
The novel CHD-linked FOXJ1 damaging variant c.784_799dup; p.Glu267Glyfs*12 as well as the majority of previously reported de novo and novel FOXJ1 deleterious variants are located in exon 3 (19,33,34). FOXJ1 is a 421 amino acid transcription factor containing a proline-rich region in the N-terminal region, a forkhead/winged helix DNA-binding domain between residues 119 and 219, and four regions rich in acidic amino acid residues (A1–A4) localized upstream (A1) and downstream (A2–A4) of the DNA-binding domain (Fig. 7) (37–39). These rich acidic amino acid regions, of which the downstream A2 and A3 are the most potent regions for transcriptional activity, are important for transcriptional activation (38). Importantly, all the FOXJ1 damaging variants referred to above are localized within or between the A2 and A3 regions.
Figure 7.

FOXJ1 pathogenic variants are localized on critical transactivation domains. Schematic representation of the 421 amino acids long FOXJ1 protein, depicting a proline-rich region (orange striped box), the winged helix DNA-binding domain (blue box) and four regions rich in acidic amino acid residues (A1–A4, blue striped boxes) localized upstream (A1) and downstream (A2–A4) of the DNA-binding domain. The novel c.784_799dup; p.Glu267Glyfs*12 pathogenic variant is inside an empty red rectangle and previously described FOXJ1 pathogenic variants (19,33,34) are inside empty black rectangles. The likely pathogenic variant c.107T>C; p.Leu36Pro and the variants of unknown significance (c.842G>A; p.Arg281Gln and c.7G>C; p.Glu3Gln) reported in this study are inside empty green rectangles.
Individuals carrying pathogenic variants in motile cilia genes (i.e. DNHA5, DNAH11, HEATR2 and CCDC151 among several others) have been diagnosed with isolated laterality defects or heterotaxy (1,31,32). However, no individuals with pathogenic variants in FOXJ1 have been reported with heterotaxy. Interestingly, a novel likely pathogenic FOXJ1 variant (c.107T>C; p.Leu36Pro) was identified in one participant of the Kids First PCGC cohort diagnosed with heterotaxy syndrome (situs inversus with complex CHD). Unlike our case, the likely pathogenic variant (c.107T>C; p.Leu36Pro) is localized in the N terminus, close to the A1 region of FOXJ1 (Fig. 7) (37–39). Overall, in a cohort of nearly 1000 CHD participants which was at least partially enriched for left–right or laterality cases, one likely pathogenic variant and two VUSs were identified. This suggests that variants in FOXJ1 are not a common cause of heterotaxy syndrome or severe isolated CHD. Because the patient cohorts analyzed in this study did not include more mild isolated CHD defects such as atrial septal defects, future studies should evaluate a complete spectrum of CHD severity. Because larger clinical gene panels and exome or genome sequencing are increasingly used for diagnosis in CHD patients, it is important to consider variants in FOXJ1.
Foxj1 loss-of-function mouse models recapitulate the phenotypes previously described in individuals with FOXJ1 pathogenic variants as Foxj1 null animals display hydrocephalus, situs inversus and total absence of cilia on epithelial cells of the airways as well as in the oviduct and sperm flagella (13,14,18,27). The structural cardiac defects have not been well described in Foxj1 null mice perhaps in part because of late embryonic or early neonatal lethality in those with severe CHD (13,14). Here, we demonstrate that in addition to the inverse (sinistral) looping observed in isolated dextrocardia (13,14,27), Foxj1 null hearts exhibit other looping defects as well as complex structural defects and abnormal position of the great arteries.
In summary, the forkhead-domain transcription factor FOXJ1 is a key regulator of motile cilia biogenesis in vertebrates and alterations, or ablation of the gene disrupts ciliogenesis which can lead to laterality defects, mucociliary clearance defects, hydrocephalus as well as isolated CHD.
Materials and Methods
Clinical exome sequencing and analysis in an individual with FOXJ1 c.784_799dup
The initial clinical exome sequencing was completed using a clinical laboratory improvement amendments (CLIA) and International Standard Organization (ISO) accredited commercial genetic testing laboratory (PreventionGenetics, Marshfield, WI). Genomic DNA was extracted and coding regions of targeted genes plus ~10 bases of non-coding DNA flanking each exon were captured using Agilent Clinical Research exome hybridization probes. Sequencing of captured DNA was performed in a NovaSeq 6000 platform (Reversible Dye Terminator platform; Illumina, San Diego, CA, USA) using 150 by 150 bp paired ends reads. Quality control metrics were >98% of target bases were covered at >20× and mean coverage of target bases >120×. The in-house software Titanium-Exome was utilized for data analysis and interpretation. Briefly, the output data from the NovaSeq 6000 was converted to FASTQs by Illumina Bcl2Fastq v2.19.0.316 and aligned by BWA mem. Variant calls were made by the Genome Analysis Toolkit (GATK) Haplotype caller and annotated using an in-house software and SnpEff. Variants were filtered and annotated using VarSeq (www.goldenhelix.com) and common benign and low-quality variants were filtered from analysis. For Sanger sequencing, amplification of targeted regions was achieved with PCR. After purification of the PCR amplicons, cycle sequencing was carried out using the ABI Big Dye Terminator v.3.1 kit (Applied Biosystems). PCR products were resolved by electrophoresis on an ABI 3730xl capillary sequencer (Applied Biosystems), and cycle sequencing was performed separately in both directions (forward and reverse).
The data were obtained following family consent for independent review. We used the Exomiser tool for this independent analysis given its strengths for filtering candidate gene variants from next-generation sequencing data based on the patient’s phenotype using human phenotype ontology (HPO) codes (40). We used the HiPHIVE algorithm for Exomiser to leverage prioritizing variants based on known human and animal genotype–phenotype correlations, gene network analysis (interactome) and expected variant impact. For HPO codes, we used the following to describe the patient: third-degree atrioventricular block (HP:0001709), DORV (HP:0001719), D-TGA (HP:0031348), secundum ASD (HP:0001684) and VSD (HP:0001629). Our independent analysis confirmed the clinical laboratory findings.
CHD patient cohorts, exome sequencing and variant analysis
Two cohorts were examined to identify potentially damaging FOXJ1 variants in patients with CHD. The first cohort was composed of samples with heterotaxy, and related CHD (n = 279) collected at Cincinnati Children’s Hospital Medical Center (CCHMC) and Indiana University School of Medicine (IUSM) under protocols approved by the respective center’s Institutional Review Boards (CCHMC IRBs #2011-2751, #2011-2479 and IUSM IRB #1403871897). DNA from whole blood or saliva were extracted using standard procedures. Demographics, detailed phenotype including cardiac diagnosis, three-generation pedigrees and previous clinical genetic testing information were collected and curated in a REDCap database. Exome sequencing for this cohort was performed using the Nimblegen sequence capture kit (SeqCap EZ Human Exome 2.0) in an Illumina HiSeq2500 sequencer at CCHMC in a CLIA-approved laboratory. Data are deposited in dbGAP under the accession phs001814.v1.p1. Alignment, preprocessing and variant calling were performed using GATK best practices at the CCHMC Bioinformatics Core.
The second cohort that consisted of 711 trios (proband with CHD and both parents) was obtained from the Gabriella Miller Kids First study of the Pediatric Cardiac Genetics Consortium (Kids First PCGC, phs001138.v3.p2). Individual-level phenotype and genotype data for each proband with CHD were downloaded via an approved controlled-access request from dbGAP (project #23292) and the Kids First data portal. The Kids First PCGC cohort had whole-genome sequencing performed at the Broad Institute followed by GATK alignment and HaplotypeCaller processing as per the Kids First alignment workflow (https://github.com/kids-first/kf-alignment-workflow). Pre-processed gvcfs were downloaded from the Kids First data portal onto the Indiana University High Performance Slate-project space. GATK best practices were used for joint calling variants across participants.
We used Golden Helix SNP and Variation Suite v8.8 (Golden Helix, Bozeman, MT) for variant filtering and annotation in both CHD cohorts. Variants were annotated with RefSeq Genes and assessed for non-synonymous variants including missense, frameshift, splice site, stop loss, stop gain, small in-frame insertions and deletions, and damaging non-coding variants. Multiple bioinformatics functional prediction algorithms including SIFT, Polyphen2 HVAR, MutationTaster, MutationAssessor, FATHMM and FATHMM MKL Coding, CADD and REVEL were used to prioritize variants. Population allele frequencies were obtained from gnomAD and variants were restricted to an allele frequency <0.001. Finally, prioritized candidate variants were interpreted using the American College of Medical Genetics and Genomics (ACMG) guidelines.
FOXJ1 and FOXJ1 c.784_799dup plasmids
A pCMV3-untagged vector containing the cDNA ORF of FOXJ1 (HG20823-UT) was purchased from SinoBiological (Wayne, PA, USA). The pCMV3-FOXJ1 was transformed into One Shot TOP10 chemically competent Escherichia coli cells (Invitrogen; Waltham, MA) according to manufacturer’s instructions and plated onto LB-ampicillin (100 μg/ml) agar plates. Plates were incubated overnight at 37°C and individual bacterial clones selected and grown further in LB broth containing ampicillin (100 μg/ml). Plasmid DNA isolation was achieved with the QIAprep Spin Miniprep and QIAfilter Plasmid Maxi kits (Qiagen, Germantown, MD, USA) following the vendor’s recommendations. Sanger sequencing verified the FOXJ1 DNA sequence by using the following primers: T7: 5′ TAA TAC GAC TCA CTA TAG GG 3′; reverse BGH: 5′ TAG AAG GCA CAG TCG AGG 3′; FOXJ1 FW: 5′ ACT CGT ATG CCA CGC TCA TC 3′; FOXJ1 RV: 5′ CCT TTG AGG GGT TCC AGC TC 3′.
The pCMV3-FOXJ1 c.784_799dup plasmid was generated from the pCMV3-FOXJ1 plasmid using the Q5 Site Directed Mutagenesis kit (New England Biolabs, Ipswich, MA) with primers designed with the NEBaseChanger webtool: FW: 5′ tgc agg cgG GCT GGG GTG CAG GCG AG 3′ and RV: 5′ ccc cag ccC GCC TCC CCG GTG GCC TC 3′. PCR reactions were performed as follows: initial denaturation at 98°C for 30 s, 25 cycles of amplification (denaturation at 98°C for 10 s, annealing at 72°C for 30 s and extension at 72°C for 7 min 20 s) and a final extension at 72°C for 2 min. The pCMV3-FOXJ1 c.784_799dup was then transformed, plated and sequenced as described previously.
To generate FOXJ1 and FOXJ1 c.784_799dup polyA tail-mRNAs, the pCMV3-FOXJ1 and pCMV3-FOXJ1 c.784_799dup plasmids were linearized using the NotI-HF restriction enzyme (New England Biolabs), and mRNAs produced with the mMESSAGE mMACHINE T7 Ultra kit (Invitrogen) per manufacturer’s directions. mRNA concentration was assessed with the Qubit RNA BR fluorometric assay kit (Invitrogen) and the size for both FOXJ1 mRNA with and without a polyA tail was verified on a 1% (w/v) agarose gel alongside the Ambion RNA Millennium markers (ThermoFisher Scientific).
Xenopus laevis embryo production and injections
Xenopus laevis males (LM00715MX) and females (LM00535) were obtained from Nasco (Fort Atkinson, WI, USA). Females were injected with the chorionic gonadotropin Chorulon (Merck, NJ, USA) into their lymph sacks to stimulate oocyte release. Males were euthanized, and testes removed and minced for in vitro fertilization. Embryos were de-jellied with l-cysteine (Sigma-Aldrich, MO, USA) and then injected at the 2-cell stage with either the FOXJ1 mRNA (171 pg/cell; 342 pg/embryo) or the FOXJ1 c.784_799dup mRNA (178 pg/cell; 356 pg/embryo) diluted in Ficoll PM400 (GE Healthcare, IL, USA) using a micro-injector (Harvard Apparatus). Uninjected embryos served as experimental controls. Embryos developed at room temperature in MMR buffer (22 mm NaCl, 0.5 mm KCl, 0.5 mm CaCl2, 0.25 mm MgCl2 and 1.25 mm HEPES) and then were randomly selected at stage 26 (based on the Theiler staging (41)) to be either fixed in 4% (v/v) paraformaldehyde (PFA) or snapped frozen for qRT-PCR experiments. All buffers and protocols were adapted from Cold Spring Harbor Laboratory (NY, USA) and Xenbase (http://www.xenbase.org). Xenopus laevis were housed and cared for according to established protocols approved by the IUSM, Institutional Animal Care and Use Committee (IACUC).
Immunofluorescence
Lung paraffin sections
Lung sections from a 7-day-old unaffected control and from the proband with the FOXJ1 variant were obtained from the Department of Pathology and Laboratory Medicine, IUSM. Slides were deparaffinized and rehydrated through an ethanol gradient before undergoing heat-based antigen retrieval using Tris–EDTA buffer (10 mm Tris, 1 mm EDTA, pH 9.0). Sections were incubated in blocking solution [TBS (137 mm NaCl, 2.7 mm KCl, 25 mm Tris) containing 1% (v/v) goat serum, 3% (w/v) bovine serum albumin (BSA), 0.1% (v/v) Tween 20] for at least 2 h prior to incubation overnight at 4°C with anti-acetylated tubulin (T6793; Millipore Sigma, St. Louis, MO, USA; 1:500 dilution). Slides were then washed twice with blocking solution followed by an incubation with A488 goat anti-mouse antibody (A11001; ThermoFisher Scientific; 1:200 dilution). Sections were washed twice with blocking solution and then twice with phosphate-buffered saline (PBS) prior to a 5 min incubation with 300 nM DAPI (Vector Laboratories). After three washes with PBS, slides were mounted with Vectashield Vibrance mounting antifade media and imaged under a Leica DM4 B Fluorescence microscope with a K 5 camera (Leica, Wetzlar, Germany).
Xenopus
Fixed embryos were washed with PBS and permeabilized in PBS-T [PBS containing 0.1% (v/v) Triton X-100] on ice for 5 min. Embryos were then incubated with blocking buffer [PBS-T containing 3% (w/v) BSA and 1% (v/v) goat serum] for 3 h followed by an incubation with the primary antibody mouse anti-acetylated tubulin (T6793, Sigma-Aldrich; 1:250 dilution in blocking buffer) overnight at 4°C with gentle rocking. Embryos were washed three times with the blocking buffer for 1 h each at room temperature with gentle rocking followed by an overnight incubation at 4°C with the secondary antibodies Alexa Fluor 594 Phalloidin (A12381; ThermoFisher Scientific; 1:40 dilution) and the Alexa Fluor 488 goat anti-mouse (A-11001; ThermoFisher Scientific; 1:500 dilution) diluted in blocking buffer with gentle rocking. Embryos were washed as described previously, transferred to PBS and the skin carefully removed with forceps. Skin pieces were mounted on slides using 0.12 mm spacers (Sigma-Aldrich) with the Vectashield Vibrance antifade mounting medium containing DAPI (Vector Laboratories). Slides were imaged as described previously.
RNA extraction and real-time PCR
Total RNA was extracted from pools of Xenopus embryos (an average of 21 embryos/pool) at stage 26 using the Qiagen RNeasy Plus mini kit (Qiagen). RNA quality and quantity were assessed spectrophotometrically with the NanoDrop One (ThermoFisher Scientific). RNA was reverse transcribed to cDNA via the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). qPCR was performed on n = 8 non-injected controls, n = 4 FOXJ1 mRNA and n = 4 FOXJ1 c.784_799dup mRNA injected embryos using an Applied Biosystems 7500 system and the following TaqMan assay probes used: human FOXJ1 (Hs00230964_m1) and a custom design odc1 (APFVTXE) which served as endogenous control (Applied Biosystems). Each biological sample was run in triplicate and PCR reactions were carried out following protocols for the Duplex TaqMan Gene Expression assays. Statistical analysis was performed on average ΔCt values with the GraphPad Prism software (version 9.2.0). P-values were derived from a one-way analysis of variance (ANOVA) followed by the Tukey’s test for multiple comparisons and P-values < 0.05 were considered significant. The data were graphed as fold change values calculated with the 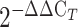 method (42).
method (42).
Cell culture, transfection, western blot and luciferase assay
Human Embryonic Kidney (HEK) 293 cells were cultured in high glucose Dulbecco’s modified Eagle medium containing l-glutamine (Gibco), supplemented with 10% (v/v) heat-inactivated fetal bovine serum (HyClone) and 10000 U/ml penicillin and 10 mg/ml streptomycin (Gibco) at 37°C in a humidified 5% (v/v) CO2 incubator. Cells were cultured without antibiotics prior to transfections.
Western blot
Cells were cultured in 60 mm plates until 70–80% confluency and transfections carried out using Lipofectamine 2000 (Invitrogen, ThermoFisher Scientific) according to vendor’s instructions. Cells were transfected with 8 μg of either the empty vector pHM6, or the expression constructs FOXJ1 or FOXJ1 c.784_799dup for 24 h. An untransfected plate was also included as an additional negative control. Cells were then collected and lysed in Pierce RIPA buffer (Thermo Scientific) containing a protease inhibitor cocktail (Cell Signaling Technologies, MA, USA). Protein concentration was determined from the supernatant using the Bradford assay (43) with BSA (Bio-Rad Laboratories, CA, USA) as standard. Total protein (20 μg) was loaded and separated under reducing conditions using one-dimensional Mini-Protean TGX 10% (w/v) precast gels and Tris–glycine–SDS buffer (Bio-Rad Laboratories). Proteins were transferred electrophoretically to PVDF 0.45 μm membranes (Thermo Scientific). Membranes were blocked for 1 h in 5% (w/v) BSA dissolved in TBS-T [10 mm Tris pH 7.6, 0.9% (w/v) NaCl and 0.1% (v/v) Tween-20] followed by an overnight incubation at 4°C with a rabbit polyclonal antibody recognizing FOXJ1 (PA5-85557; Invitrogen; 1:1000 dilution in blocking buffer). Membranes were washed and incubated for 1 h at room temperature with the corresponding horseradish peroxidase-conjugated antibody (1:10 000 dilution). After washing, blots were developed with the Clarity Western ECL substrate kit (Bio-Rad Laboratories). β-Actin (D6A8; Cell Signaling Technologies; 1:1000 dilution in blocking buffer) served as loading control.
Luciferase assay
Cells were cultured in 12-well plates at a concentration of 3 × 105 cells/ml 24 h prior to transfection. The pcDNA3.1-HA-RFX2, pGL3B-AP140-1 and pGL3B-AP-3′-AE1 plasmids (10) were a generous gift from Dr Hoogewijs (University of Fribourg, Switzerland). Plasmids were transformed and DNA isolated as described previously. Lipofectamine 2000 (ThermoFisher Scientific) served as transfection reagent following vendor’s instructions and transfections were set up as follows: the AP140-1 reporter plasmid combined with either the FOXJ1 or the FOXJ1 c.784_799dup expression construct (700 ng each), and the AP-3′-AE1 reporter plasmid along with either HA-RFX2 or the FOXJ1 or the FOXJ1 c.784_799dup expression constructs or their combination (500 ng each). Each transfection reaction received 50 ng of the pGL4.73 Renilla transfection control, and the pHM6-empty vector served to equalize the total amount of transfected DNA as well as a control. After 6 h, transfected cells were resuspended in fresh media and re-plated in solid white flat-bottomed plates in four technical replicates (Corning, NY, USA). Luciferase activity was assessed 24 h post-transfection with the Dual-Glo Luciferase assay system (Promega, Madison, WI, USA) and luminescence measured in a Synergy H4 Microplate reader (BioTek, Winooski, VT, USA). Firefly luminescence was normalized to Renilla luminescence. Results were derived from at least three independent experiments and the data analyzed using ANOVA followed by the Tukey’s test for multiple comparisons (GraphPad Prism software; version 9.2.0).
Foxj1 mouse line and embryo collection
The Foxj1CreERT2::GFP (Stock No: 027012; Foxj1tm1.1(cre/ERT2/GFP)Htg) knock-in/knock-out mice was purchased from The Jackson Laboratory repository (Bar Harbor, ME, USA). This mouse line expresses GFP and tamoxifen inducible Cre recombinase in epithelial cells with motile cilia (18). Genomic DNA was extracted from either ear clips (mice) or yolk sacs (embryos) and used for genotyping with the following primers: WT FW: 5′ GCA GAT GGA GAG AGG TGG AG 3′; common RV: 5′ CTT GGC GTT GAG AAT GGA GA 3′; mutant FW: 5′ ATT GCA TCG CAT TGT CTG AG 3′. Mice were housed in the IUSM animal facility accredited by the association for assessment and accreditation of laboratory animal care (AAALAC), and experiments were approved by IACUC.
For timed pregnancies, noon of the day of plug observation was designated as 0.5 day post-coitum (dpc). Pregnant females were euthanized by CO2 inhalation followed by cervical dislocation. Embryos were collected at 14.5–17.5 dpc, fixed in 4% (v/v) PFA diluted with PBS overnight at 4°C and then dissected under a Nikon SMZ18 zoom stereomicroscope (Nikon, NY, USA). On gross inspection, heart looping phenotypes were classified as dextral (normal), sinistral (inverse), ventral or single ventricle (unlooped) based on Ware et al. (28). Gross heart images were taken with a Nikon DS-Ri2 16MP digital camera (Nikon).
Histology
Heart phenotypes were confirmed by histological sections. Hearts were paraffin embedded as described elsewhere (44) and 8 μm transverse serial sections were obtained beginning at the OFT above the heart and ending below the ventricles and mounted. After deparaffination, sections were stained with hematoxylin and eosin and imaged on a Nikon Eclipse E400 microscope with a Nikon digital sight DS-Fi2 camera and NIS Elements imaging software (version 5.11; Nikon).
Supplementary Material
{kind=link}
Acknowledgements
We are very grateful to Gina Londre and Dr Brett Deml from PreventionGenetics in Marshfield, Wisconsin for Exome and Sanger sequencing analyses. We are also thankful to Dr Jaffar Khan from the Department of Pathology and Laboratory Medicine, IUSM, for the unaffected control and the lung sections from the patient with the FOXJ1 pathogenic variant. Finally, we thank Dr David Hoogewijs from University of Fribourg, Fribourg, Switzerland for the pcDNA3.1-HA-RFX2, pGL3B-AP140-1 and pGL3B-AP-3′-AE1 plasmids. The authors acknowledge the Indiana University Pervasive Technology Institute for providing supercomputing, visualization and data storage resources that have contributed to the research results reported within this paper.
Conflict of Interest statement: The authors declare no conflicts of interest.
Contributor Information
Maria B Padua, Department of Pediatrics, Herman B Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Benjamin M Helm, Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN 46202, USA; Department of Epidemiology, Indiana University Fairbanks School of Public Health, Indianapolis, IN 46202, USA.
John R Wells, Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Amanda M Smith, Department of Pediatrics, Herman B Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Helen M Bellchambers, Department of Pediatrics, Herman B Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Arthi Sridhar, Department of Pediatrics, Herman B Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Stephanie M Ware, Department of Pediatrics, Herman B Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202, USA; Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Funding
National Institute of Health (P01 HL 134599 to S.M.W.) and in part by the Lilly Endowment, Inc., through its support for the Indiana University Pervasive Technology Institute.
References
- 1. Pierpont, M.E., Brueckner, M., Chung, W.K., Garg, V., Lacro, R.V., McGuire, A.L., Mital, S., Priest, J.R., Pu, W.T., Roberts, A. et al. (2018) Genetic basis for congenital heart disease: revisited: a scientific statement from the American Heart Association. Circulation, 138(21), e653–e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Glessner, J.T., Bick, A.G., Ito, K., Homsy, J., Rodriguez-Murillo, L., Fromer, M., Mazaika, E., Vardarajan, B., Italia, M., Leipzig, J. et al. (2014) Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ. Res., 115(10), 884–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cowan, J.R. and Ware, S.M. (2015) Genetics and genetic testing in congenital heart disease. Clin. Perinatol., 42(2), 373–393 ix. [DOI] [PubMed] [Google Scholar]
- 4. Jin, S.C., Homsy, J., Zaidi, S., Lu, Q., Morton, S., DePalma, S.R., Zeng, X., Qi, H., Chang, W., Sierant, M.C. et al. (2017) Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet., 49(11), 1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tan, S.Y., Rosenthal, J., Zhao, X.Q., Francis, R.J., Chatterjee, B., Sabol, S.L., Linask, K.L., Bracero, L., Connelly, P.S., Daniels, M.P. et al. (2007) Heterotaxy and complex structural heart defects in a mutant mouse model of primary ciliary dyskinesia. J. Clin. Invest., 117(12), 3742–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li, Y., Klena, N.T., Gabriel, G.C., Liu, X., Kim, A.J., Lemke, K., Chen, Y., Chatterjee, B., Devine, W., Damerla, R.R. et al. (2015) Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature, 521(7553), 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fliegauf, M., Benzing, T. and Omran, H. (2007) When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol, 8(11), 880–893. [DOI] [PubMed] [Google Scholar]
- 8. Yoshiba, S. and Hamada, H. (2014) Roles of cilia, fluid flow, and Ca2+ signaling in breaking of left-right symmetry. Trends Genet., 30(1), 10–17. [DOI] [PubMed] [Google Scholar]
- 9. Haider, S., Gamperl, M., Burkard, T.R., Kunihs, V., Kaindl, U., Junttila, S., Fiala, C., Schmidt, K., Mendjan, S., Knofler, M. et al. (2019) Estrogen signaling drives ciliogenesis in human endometrial organoids. Endocrinology, 160(10), 2282–2297. [DOI] [PubMed] [Google Scholar]
- 10. Koay, T.W., Osterhof, C., Orlando, I.M.C., Keppner, A., Andre, D., Yousefian, S., Suarez Alonso, M., Correia, M., Markworth, R., Schodel, J. et al. (2021) Androglobin gene expression patterns and FOXJ1-dependent regulation indicate its functional association with ciliogenesis. J. Biol. Chem., 296, 100291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Patir, A., Fraser, A.M., Barnett, M.W., McTeir, L., Rainger, J., Davey, M.G. and Freeman, T.C. (2020) The transcriptional signature associated with human motile cilia. Sci. Rep., 10(1), 10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lohmussaar, K., Oka, R., Espejo Valle-Inclan, J., Smits, M.H.H., Wardak, H., Korving, J., Begthel, H., Proost, N., van de Ven, M., Kranenburg, O.W. et al. (2021) Patient-derived organoids model cervical tissue dynamics and viral oncogenesis in cervical cancer. Cell Stem Cell, 28(8), 1380–1396 e1386. [DOI] [PubMed] [Google Scholar]
- 13. Brody, S.L., Yan, X.H., Wuerffel, M.K., Song, S.K. and Shapiro, S.D. (2000) Ciliogenesis and left-right axis defects in forkhead factor HFH-4-null mice. Am. J. Respir. Cell Mol. Biol., 23(1), 45–51. [DOI] [PubMed] [Google Scholar]
- 14. Chen, J., Knowles, H.J., Hebert, J.L. and Hackett, B.P. (1998) Mutation of the mouse hepatocyte nuclear factor/forkhead homologue 4 gene results in an absence of cilia and random left-right asymmetry. J. Clin. Invest., 102(6), 1077–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stubbs, J.L., Oishi, I., Izpisua Belmonte, J.C. and Kintner, C. (2008) The forkhead protein Foxj1 specifies node-like cilia in Xenopus and zebrafish embryos. Nat. Genet., 40(12), 1454–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu, X., Ng, C.P., Habacher, H. and Roy, S. (2008) Foxj1 transcription factors are master regulators of the motile ciliogenic program. Nat. Genet., 40(12), 1445–1453. [DOI] [PubMed] [Google Scholar]
- 17. Alten, L., Schuster-Gossler, K., Beckers, A., Groos, S., Ulmer, B., Hegermann, J., Ochs, M. and Gossler, A. (2012) Differential regulation of node formation, nodal ciliogenesis and cilia positioning by Noto and Foxj1. Development, 139(7), 1276–1284. [DOI] [PubMed] [Google Scholar]
- 18. Muthusamy, N., Vijayakumar, A., Cheng, G., Jr. and Ghashghaei, H.T. (2014) A knock-in Foxj1(CreERT2::GFP) mouse for recombination in epithelial cells with motile cilia. Genesis, 52(4), 350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wallmeier, J., Frank, D., Shoemark, A., Nothe-Menchen, T., Cindric, S., Olbrich, H., Loges, N.T., Aprea, I., Dougherty, G.W., Pennekamp, P. et al. (2019) De novo mutations in FOXJ1 result in a motile ciliopathy with hydrocephalus and randomization of left/right body asymmetry. Am. J. Hum. Genet., 105(5), 1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Breeze, R. and Turk, M. (1984) Cellular structure, function and organization in the lower respiratory tract. Environ. Health Perspect., 55, 3–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stubbs, J.L., Davidson, L., Keller, R. and Kintner, C. (2006) Radial intercalation of ciliated cells during Xenopus skin development. Development, 133(13), 2507–2515. [DOI] [PubMed] [Google Scholar]
- 22. Hoogewijs, D., Ebner, B., Germani, F., Hoffmann, F.G., Fabrizius, A., Moens, L., Burmester, T., Dewilde, S., Storz, J.F., Vinogradov, S.N. et al. (2012) Androglobin: a chimeric globin in metazoans that is preferentially expressed in mammalian testes. Mol. Biol. Evol., 29(4), 1105–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Quigley, I.K. and Kintner, C. (2017) Rfx2 stabilizes Foxj1 binding at chromatin loops to enable multiciliated cell gene expression. PLoS Genet., 13(1), e1006538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu, Y., Hu, X., Li, Z., Wang, M., Li, S., Wang, X., Lin, X., Liao, S., Zhang, Z., Feng, X. et al. (2016) Transcription factor RFX2 is a key regulator of mouse spermiogenesis. Sci. Rep., 6, 20435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manner, J. (2009) The anatomy of cardiac looping: a step towards the understanding of the morphogenesis of several forms of congenital cardiac malformations. Clin. Anat., 22(1), 21–35. [DOI] [PubMed] [Google Scholar]
- 26. Kirby, M.L. and Waldo, K.L. (1995) Neural crest and cardiovascular patterning. Circ. Res., 77(2), 211–215. [DOI] [PubMed] [Google Scholar]
- 27. Zhang, M., Bolfing, M.F., Knowles, H.J., Karnes, H. and Hackett, B.P. (2004) Foxj1 regulates asymmetric gene expression during left-right axis patterning in mice. Biochem. Biophys. Res. Commun., 324(4), 1413–1420. [DOI] [PubMed] [Google Scholar]
- 28. Ware, S.M., Harutyunyan, K.G. and Belmont, J.W. (2006) Zic3 is critical for early embryonic patterning during gastrulation. Dev. Dyn., 235(3), 776–785. [DOI] [PubMed] [Google Scholar]
- 29. Bajolle, F., Zaffran, S., Kelly, R.G., Hadchouel, J., Bonnet, D., Brown, N.A. and Buckingham, M.E. (2006) Rotation of the myocardial wall of the outflow tract is implicated in the normal positioning of the great arteries. Circ. Res., 98(3), 421–428. [DOI] [PubMed] [Google Scholar]
- 30. Sifrim, A., Hitz, M.P., Wilsdon, A., Breckpot, J., Turki, S.H., Thienpont, B., McRae, J., Fitzgerald, T.W., Singh, T., Swaminathan, G.J. et al. (2016) Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet., 48(9), 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gabriel, G.C., Young, C.B. and Lo, C.W. (2021) Role of cilia in the pathogenesis of congenital heart disease. Semin. Cell Dev. Biol., 110, 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kennedy, M.P., Omran, H., Leigh, M.W., Dell, S., Morgan, L., Molina, P.L., Robinson, B.V., Minnix, S.L., Olbrich, H., Severin, T. et al. (2007) Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation, 115(22), 2814–2821. [DOI] [PubMed] [Google Scholar]
- 33. Jin, S.C., Dong, W., Kundishora, A.J., Panchagnula, S., Moreno-De-Luca, A., Furey, C.G., Allocco, A.A., Walker, R.L., Nelson-Williams, C., Smith, H. et al. (2020) Exome sequencing implicates genetic disruption of prenatal neuro-gliogenesis in sporadic congenital hydrocephalus. Nat. Med., 26(11), 1754–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shapiro, A.J., Kaspy, K., Daniels, M.L.A., Stonebraker, J.R., Nguyen, V.H., Joyal, L., Knowles, M.R. and Zariwala, M.A. (2021) Autosomal dominant variants in FOXJ1 causing primary ciliary dyskinesia in two patients with obstructive hydrocephalus. Mol. Genet. Genomic Med., 9(7), e1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Versacci, P., Pugnaloni, F., Digilio, M.C., Putotto, C., Unolt, M., Calcagni, G., Baban, A. and Marino, B. (2018) Some isolated cardiac malformations can be related to laterality defects. J Cardiovasc. Dev. Dis., 5(2), 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Watkins, W.S., Hernandez, E.J., Wesolowski, S., Bisgrove, B.W., Sunderland, R.T., Lin, E., Lemmon, G., Demarest, B.L., Miller, T.A., Bernstein, D. et al. (2019) De novo and recessive forms of congenital heart disease have distinct genetic and phenotypic landscapes. Nat. Commun., 10(1), 4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clevidence, D.E., Overdier, D.G., Peterson, R.S., Porcella, A., Ye, H., Paulson, K.E. and Costa, R.H. (1994) Members of the HNF-3/forkhead family of transcription factors exhibit distinct cellular expression patterns in lung and regulate the surfactant protein B promoter. Dev. Biol., 166(1), 195–209. [DOI] [PubMed] [Google Scholar]
- 38. Lim, L., Zhou, H. and Costa, R.H. (1997) The winged helix transcription factor HFH-4 is expressed during choroid plexus epithelial development in the mouse embryo. Proc. Natl. Acad. Sci. U. S. A., 94(7), 3094–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hackett, B.P., Brody, S.L., Liang, M., Zeitz, I.D., Bruns, L.A. and Gitlin, J.D. (1995) Primary structure of hepatocyte nuclear factor/forkhead homologue 4 and characterization of gene expression in the developing respiratory and reproductive epithelium. Proc. Natl. Acad. Sci. U. S. A., 92(10), 4249–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smedley, D., Jacobsen, J.O., Jager, M., Kohler, S., Holtgrewe, M., Schubach, M., Siragusa, E., Zemojtel, T., Buske, O.J., Washington, N.L. et al. (2015) Next-generation diagnostics and disease-gene discovery with the exomiser. Nat. Protoc., 10(12), 2004–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nieuwkoop, P.D. and Faber, J. (1994) Normal Table of Xenopus laevis (Daudin) : A Systematical and Chronological Survey of the Development from the Fertilized Egg till the End of Metamorphosis. Garland Pub, New York, Vol. 252, p. 210 leaves of plates. [Google Scholar]
- 42. Livak, K.J. and Schmittgen, T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods, 25(4), 402–408. [DOI] [PubMed] [Google Scholar]
- 43. Bradford, M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 44. Haaning, A.M., Quinn, M.E. and Ware, S.M. (2013) Heterotaxy-spectrum heart defects in Zic3 hypomorphic mice. Pediatr. Res., 74(5), 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.